

Abstract
Background and Purpose
Activation of astrocytes contributes to synaptic remodelling, tissue repair and neuronal survival following traumatic brain injury (TBI). The mechanisms by which these cells interact to resident/infiltrated inflammatory cells to rewire neuronal networks and repair brain functions remain poorly understood. Here, we explored how TLR4‐induced astrocyte activation modified synapses and cerebrovascular integrity following TBI.
Experimental Approach
To determine how functional astrocyte alterations induced by activation of TLR4 pathway in inflammatory cells regulate synapses and neurovascular integrity after TBI, we used pharmacology, genetic approaches, live calcium imaging, immunofluorescence, flow cytometry, blood–brain barrier (BBB) integrity assessment and molecular and behavioural methods.
Key Results
Shortly after a TBI, there is a recruitment of excitable and reactive astrocytes mediated by TLR4 pathway activation with detrimental effects on post‐synaptic density‐95 (PSD‐95)/vesicular glutamate transporter 1 (VGLUT1) synaptic puncta, BBB integrity and neurological outcome. Pharmacological blockage of the TLR4 pathway with resatorvid (TAK‐242) partially reversed many of the observed effects. Synapses and BBB recovery after resatorvid administration were not observed in IP3R2−/− mice, indicating that effects of TLR4 inhibition depend on the subsequent astrocyte activation. In addition, TBI increased the astrocytic‐protein thrombospondin‐1 necessary to induce a synaptic recovery in a sub‐acute phase.
Conclusions and Implications
Our data demonstrate that TLR4‐mediated signalling, most probably through microglia and/or infiltrated monocyte–astrocyte communication, plays a crucial role in the TBI pathophysiology and that its inhibition prevents synaptic loss and BBB damage accelerating tissue recovery/repair, which might represent a therapeutic potential in CNS injuries and disorders.
Keywords: astrocytes, BBB breakdown, microglia/infiltrated monocytes, synaptic remodelling, TLR4 antagonism, traumatic brain injury
Abbreviations
- BBB
blood–brain barrier
- d.a.i
day(s) after injury
- GFAP
glial fibrillary acidic protein
- hi
high level of expression
- Iba‐1
ionized calcium‐binding adapter molecule‐1
- int
intermediate level of expression
- IP3R2
inositol 1,4,5‐trisphosphate receptor type 2
- neg
negative expression
- NSS
neurological severity score
- PSD‐95
post‐synaptic density‐95
- S100β
S100 calcium binding protein β
- SSC‐A
side scatter area
- TBI
traumatic brain injury
- TLR4
toll‐like receptor 4
- TLRs
toll‐like receptors
- TSP‐1
thrombospondin‐1
- VGLUT1
vesicular glutamate transporter‐1
- WT
wild‐type
What is already known
Astrocytes and microglia participate in the early cerebral and synaptic response after traumatic brain injury.
TLR4 antagonism exerts neuroprotection in acute brain injuries.
What does this study add
Astrocyte activation contributes to initial synaptic loss, BBB breakdown and later synaptic remodelling.
TLR4‐expressing microglia/infiltrating monocytes mediate astrocyte activation following traumatic brain injury (TBI).
What is the clinical significance
Inhibition of astrocyte activation through TLR4 antagonism could be used for acute treatment of TBI.
1. INTRODUCTION
Traumatic brain injury (TBI) leads to a loss of function due to cell death and neuronal dysfunction in the surrounding and remote brain areas (Carron et al., 2016; Villapol et al., 2014). Recent molecular to behaviour data indicate that functional recovery after TBI strongly depends on a complex network of distinct cell types including neurons, astrocytes and inflammatory cells in order to activate cell repair mechanisms, control acute inflammatory response and neurovascular integrity and initiate synaptic remodelling.
Astrocytes respond to a vast array of CNS insults and diseases (Burda et al., 2016; Clarke et al., 2018) and are a major determinant of injuries' outcome due to their role in synaptic transmission, plasticity and neurovascular functions (Béchade et al., 2013; Clark et al., 2015; Kim et al., 2016; Nikolakopoulou et al., 2016; Pascual et al., 2012; Perez et al., 2017; Villapol et al., 2014). Emerging evidence indicate that astrocytes exert either a neuroprotective or a neurotoxic role depending on the insult type or brain disease. In this way, astrocytic neuroprotective effects were observed in the control of the injury core following brain injury in vivo, whereas detrimental effects on synaptic number and debris phagocytosis were reported after ischaemia or infection in both in vivo and in vitro (Liddelow et al., 2017; Shinozaki et al., 2017). The transformation of quiescent astrocytes to a neuroprotective or neurotoxic phenotype depends on the microglia activation and release of a specific combination of cytokines and molecules of the complement component (Liddelow et al., 2017). Therefore, microglia is key to determine astrocyte fate by inducing reactive astrogliosis and by regulating astrocytic immune reactions (Donat et al., 2017). On the other hand, activated astrocytes‐derived factors also trigger microglial activation and control their cellular functions (Jha et al., 2018). In this way, microglia and astrocytes establish a bidirectional communication that is determinant for the fate of both cell types as well as for the function of neuronal networks.
Microglia and astrocytes express receptors capable of recognizing endogenous molecules released from damaged tissues and cells, which allow them to be rapidly activated upon injury. The best described of these receptors are the toll‐like family of receptors (TLRs) that serve to amplify pro‐inflammatory mediator release. To date, 10 and 12 functional TLRs have been identified in humans and mice, respectively. Among them, TLR4 subtype has an important role in the progression of brain damage as mediator of blood–brain barrier (BBB) integrity and production of pro‐inflammatory factors (Jiang et al., 2018; Yao et al., 2017). In the healthy rodent CNS, TLR4s are predominantly expressed in microglia (Bennett et al., 2016; Cahoy et al., 2008; Holm et al., 2012; Zhang et al., 2014, 2016). However, this selective expression is still debatable as reports also show TLR4 in cultured astrocytes and neurons after injury (Du et al., 2017; Fellner et al., 2013). Nonetheless, it is clear that TLR4 is expressed in endothelial (Nagyoszi et al., 2010) and peripheral immune cells that enter into the brain parenchyma after damage (Frik et al., 2018).
TLR4 inhibition has been shown beneficial on the TBI progression most probably through modulation of microglial pro‐inflammatory genes expression in vivo (Jiang et al., 2018; Yao et al., 2017). However, in vitro data show that astrocyte activation in response to TLR4 stimulation is only achievable in the presence of microglia and that absence of functional microglia completely abolished the expression of molecular markers for astrocyte activation (Holm et al., 2012). Therefore, it is plausible that inhibition of TLR4 pathway ameliorates TBI outcome due to an indirect effect of astrocyte and its interaction with inflammatory cell types.
Here, we investigated how functional alterations of astrocytes induced by TLR4 pathway regulate synapses and neurovascular unit after TBI. Our data show that, in response to the damage, there is a TLR4‐mediated recruitment of excitable and reactive astrocytes surrounding the injury, with detrimental effects on synapses, BBB and neurological outcome at 24 h post‐injury. We also found that TLR4 activation shortly after TBI increases the release of the astrocytic‐protein thrombospondin‐1 (TSP‐1) that serves to partially recover synapses few days after injury. Therefore, TLR4‐mediated astrocyte activation plays an important role in the remodelling of neuronal networks by modulating synaptic number and controlling BBB damage, which may have important pathophysiological relevance for long‐term plasticity.
2. METHODS
2.1. Animals
All animal experiments were carried out in accordance with the International Council for Laboratory Animal Science and the European Union 2010/63/EU Guidelines. Animal studies are reported in compliance with the ARRIVE guidelines (Percie du Sert et al., 2020) and with the recommendations made by the British Journal of Pharmacology (Lilley et al., 2020). The experimental protocol was approved by the Ethical Committee for Animal Research at the Hospital Nacional de Parapléjicos (Toledo, Spain, 25‐OH/2018) and by the Institutional Ethics Committee of Universidad Autónoma de Madrid, Spain (PROEX 109/18). All experiments were performed on male and female C57BL/6J wild‐type (WT) mice (RRID:IMSR_JAX:000664) and IP3R2 knockout adult mice (IP3R2−/− [RRID:MGI:3713675]) (generously donated by Dr. J. Chen, University of California, San Diego, CA, USA). All mice were aged between 8 and 16 weeks and weighing 25–35 g. IP3R2 knockout mice (Li et al., 2005) were backcrossed with the C57BL/6J substrain for at least six generations to achieve an IP3R2 knockout line with a C57BL/6J background. Heterozygous IP3R2 mice were mate to get IP3R2 knockout and littermate controls. Homozygous null IP3R2 mice were then mate to generate a homozygous colony. Mice were genotyped by PCR analysis as described by Li et al. (2005) from DNA extracted from tail clip showing to detect specific bands of 200 and 400 bp corresponding to IP3R2 WT gene and IP3R2 transgene, respectively. C57BL6/J mice were housed in the Universidad Autónoma de Madrid, Medicine School (Madrid, Spain) and IP3R2−/− mice were housed at the Hospital Nacional de Parapléjicos' Animal Facility (Toledo, Spain). All animals were group‐housed in a pathogen‐free and controlled temperature environment under a 12‐h light/dark cycle, with access to food and water ad libitum. All efforts were made to minimize the number of animals used and their suffering and applying the 3Rs regulation.
2.2. Traumatic brain injury (TBI) model
Closed head injury adapted from Flierl et al. (2009) was used as a model of TBI in mice. Briefly, 1 h before the procedure, animals were treated with buprenorphine of 0.05 mg/kg subcutaneously for analgesia. Animals were then anaesthetized with isoflurane 5% and placed on a hard platform to minimize dissipation of energy. Closed head injury was produced by a 50‐g weight dropped from a height of 34 cm along a metal rod connected to a tip of 5 mm of diameter that hits the mice on the right hemisphere at 1 to 1.5 ML, 0 to −1 AP. After trauma, mice were immediately subjected to post‐traumatic resuscitation (if necessary) and oxygen (1.5 L O2·min‐1) administration until regular breath was restored. Mice were subjected to closed head injury and, after post‐traumatic resuscitation, animals were randomly divided into the different experimental groups. At the end of the experiment, animals were killed by cervical dislocation. Animals were excluded from endpoint analyses if neurological severity score (NSS) was ≥9 one hour after brain trauma.
2.3. Histology, immunofluorescence and microscopy imaging
Mice were perfused with PBS; brains were excised and incubated in 4% paraformaldehyde for 24 h and then in sucrose 30% until sunk before freezing in an embedding Tissue‐Tek optimal cutting temperature compound (Cat# 4583, Sakura, FineTek) on dry ice. Then 18‐ to 20‐μm‐thick coronal slices of frozen brains were cut with a cryostat (CM 1100; Leica), mounted on Fisher brand Superfrost/Plus microscope slides and stored at −20°C. Nissl staining was performed with cresyl violet (Sigma‐Aldrich Cat# C5042) as described in Min et al. (2015). For immunofluorescence staining, slides were first incubated during 30 min at 36°C, then washed 3× in PBS and blocked in PBS buffer containing 10% goat serum (Jackson ImmunoResearch Cat# 005‐000‐121 [RRID:AB_2336990], West Grove, PA, USA) and 0.1% Triton X‐100 (Sigma Aldrich Cat# X100, St Louis, MO, USA) for 1 h. For glial fibrillary acidic protein (GFAP) and ionized calcium‐binding adapter molecule‐1 (Iba‐1) staining, sections were incubated overnight at 4°C with the primary antibodies (Abs): 1:400 rabbit anti‐GFAP (Millipore Cat# AB5804 [RRID:AB_2109645]) and 1:1000 rabbit anti‐Iba1 (Wako Cat# 019‐19741 [RRID:AB_839504]). For synaptic puncta staining (post‐synaptic density‐95 [PSD‐95] and vesicular glutamate transporter‐1 [VGLUT1]), sections were incubated for 2 days at 4°C with the primary antibodies: 1:500 rabbit anti‐PSD‐95 (Invitrogen Cat# 51‐6900 [RRID:AB_87705]) and 1:2500 guinea pig anti‐VGLUT1 (EMD Millipore Cat# AB5905 [RID:AB_2301751]). After incubation with primary Abs, sections were washed 3× in PBS and further incubated for 3 h at room temperature with the respective secondary antibodies goat anti‐guinea pig Alexa Fluor 488 (1:500, ThermoFisher Cat# A‐11073 [RRID:AB_2534117]), goat anti‐rabbit Alexa Fluor 546 (1:500, ThermoFisher Cat# A‐11035 [RRID:AB_2534093]) and goat anti‐rabbit Alexa Fluor 594 (1:500, ThermoFisher Cat# A‐11012 [RRID:AB_2534079]). After incubation in the secondary antibodies, sections were washed 4× in PBS for 15 min each and mounted using small drops of VECTASHIELD mounting medium with DAPI (Vector Laboratories Inc., Cat# H‐1000, [RRID:AB_2336789]) and then covering the slides with coverslips. All images of immunostained brains were collected on a Leica SP5 confocal microscope with a 63× oil immersion objective (NA = 1.35) either at the Microscopy Facility of the Hospital de La Princesa (Madrid, Spain) or at the Microscopy Facility of the Hospital Nacional de Parapléjicos (Toledo, Spain).
2.4. Sholl analysis of microglial morphology
Z‐stack confocal images from 20‐μm brains sections that were immunostained for Iba‐1 and DAPI were collected at 1‐μm intervals using a 63× Olympus objective as described before. Images obtained from TBI ipsilateral region were collected at ~200 μm from the damaged area to avoid necrotic damage in TBI mice. Images were obtained from three fields from each slice with two slices per mouse. Consecutive Z‐stack images of the Iba‐1 channel were converted to a maximum intensity projection using Fiji (RRID:SCR_002285). Images were then transformed to binary by thresholding to include microglial processes. Microglia were then isolated by manually removing surrounding processes with the eraser tool. Next, we used a line segment tool to draw a line from the centre of each soma to the tip of its longest process and used the Sholl analysis plugin (Ferreira et al., 2014) to define the first shell to be around 6 μm outside of the cell body and set each step to be 2‐μm size in order to determine intersections at each Sholl radius. Incomplete microglia or microglia with processes close to the margin of the field of view were avoided. From these data, we determined the number of primary branches (Np, the number of branches that originated from the microglia soma). Once Sholl analysis was finished, microglia cells were manually counted in the 63× image stack to determine the mean number of microglia per field and normalized by the total number of cells that were measured using the Particle Analysis plugin in ImageJ in the DAPI channel.
2.5. Synaptic puncta analysis
The number of colocalized synaptic puncta (VGLUT1/PSD‐95) was quantified as previously described (Ippolito & Eroglu, 2010). Briefly, 5‐μm‐thick confocal Z‐stacks (optical section depth of 0.33 μm; 15 sections/Z‐stack; imaged area measuring 11,272 μm2) of the synaptic zone in primary somatosensory cortex were imaged. Maximum projections of three consecutive optical sections were generated from the original Z‐stack. The number of pre‐, post‐ and colocalized synaptic puncta was quantified by using the Puncta Analyser plugin (developed by Barry Wark and provided by Cagla Eroglu) for ImageJ software (RRID:SCR_003070). Images were analysed by a researcher blinded to the groups. Three field of views from two sections per mouse were analysed, averaged and counted as n = 1. Synaptic puncta was defined as the normalized synaptic density as the ratio of synapses in the surrounding TBI area to that in the homotopic non‐injured contralateral cortex to correct for any endogenous differences in the case of transgenic mice. In the case of sham animals in which TBI was not performed, normalization was measured as the ratio of synapses in the right hemisphere to the left hemisphere.
2.6. qRT‐PCR
Total mRNA from mice cerebral cortex was obtained by TRIzol method (10296‐028, Invitrogen, CA, USA). cDNA was synthetized using the iScript cDNA synthesis kit (1708891, Bio‐Rad, CA, USA) and qRT‐PCR was performed with Power SYBR Green Master Mix (RR420L, Takara, Japan) in the QuantStudio 5 PCR system (Applied Biosystems, CA, USA). The target genes and the specific primers are the following: TNF‐α (forward, 5′‐GACGTGGAACTGGCAGAAGA‐3′; reverse, 5′‐ACTGATGAGAGGGAGGCCAT‐3′) and pro‐IL‐1β (forward, 5′‐GAAGAGCCCATCCTCTGTGA; reverse, 5′‐TTCATCTCGGAGCCTGTAG). Data were normalized to the expression of the housekeeping gene B2M and calculated by the Δ/Δ Ct method.
2.7. Calcium imaging acquisition and analysis
Animals were injected intraperitoneally (i.p.) with sulforhodamine 101 (SR101; 100 mg·kg‐1, Invitrogen Cat# S359) and left in the cage with food and water for 1–2 h (Perez‐Alvarez et al., 2014). After this period, animals were anaesthetized with pentobarbital (50 mg·kg‐1, i.p.) and decapitated. Brains were quickly removed and placed in ice‐cold artificial CSF (ACSF) containing 124 NaCl, 2.69 KCl, 1.25 KHP2O4, 2 MgSO4, 26 NaHCO3, 2 CaCl2 and 10 glucose, pH 7.3 and gassed with 95% O2/5% CO2. Slices (350 μm) were incubated during >1 h at room temperature (21–24°C) in ACSF. After that, the Ca+2 indicator Fluo‐4‐AM (Molecular Probes Cat# F14201, Eugene, OR; 2–5 μl of 2‐mM dye) was dropped over primary somatosensory cortex, attaining a final concentration of 2–10 μM and 0.01% of pluronic acid F‐127 (Sigma‐Aldrich Cat# P2443), for 15–20 min at room temperature (Nimmerjahn et al., 2004). In these conditions, most of the cells loaded were astrocytes, as confirmed by their electrophysiological properties in Perez‐Alvarez et al. (2014). Slices were then transferred to an immersion recording chamber and superfused with gassed ACSF in an epifluorescence Olympus BX50WI microscope. In some experiments, tetrodotoxin (1 μM, Tocris Cat# 1069) was included in the bath solution. Ca+2 levels in astrocytes specifically loaded with sulforhodamine 101 were monitored using a mono charge‐coupled device (CCD) monochrome camera attached to the microscope and controlled using SCIght software (Scientifica). Cells were illuminated with a CoolLED pE‐100 fluorescent excitation system and images were acquired every 1–1.5 s. Astrocytic Ca+2 signals were analysed offline using SARFIA, a custom‐written suite of macros running in Igor Pro (WaveMetrics [RRID:SCR_000325], Dorostkar et al., 2010). Astrocyte Ca+2 level was recorded from the astrocytes cell body and proximal ramifications, and Ca+2 variations were estimated as changes in the fluorescence signal over the baseline. Prior to analysis, images were registered to correct movements in the X and Y directions. Movies were rejected if the plane of focus altered significantly during imaging acquisition. Regions of interest containing both soma and proximal ramifications were chosen using a filtering algorithm based on a Laplace operator and segmented by applying a threshold, as described in detail in Dorostkar et al. (2010), Rosa et al. (2015) and Rosa et al. (2016). This algorithm defined most or all of the regions of interest that an experienced observer would recognize by eye. Individual region of interest responses were then normalized as the relative change in fluorescence (∆F/F), smoothed by binomial Gaussian filtering and analysed to detect activity using custom‐made scripts based on a first derivative detection algorithm. A threshold set at ~1.5–2 times the SD of the time derivative trace was used to detect changes in fluorescence within the regions of interest. The reliability of this algorithm to detect calcium activity was first tested by comparing the results with manual activity detection. In this paper, the fluorescent intensity of regions of interest is reported as the average intensity across all pixels within its area. Fluorescent responses are reported as normalized increases as follows:
where F is the instantaneous fluorescence induced by a spontaneous activity and F o is the baseline fluorescence. The Ca2+ oscillation frequency was obtained from the number of Ca2+ transients occurring in 6 to 15 astrocytes in the field of view during 1 min of recording.
2.8. Dissociation of cerebral tissue into single cell suspension
Mice were anaesthetized with pentobarbital (50 mg·kg‐1, i.p.) and perfused with PBS. The brain was quickly removed and placed into cold Hank balanced salt solution (+Ca/Mg) medium (Gibco, Cat# 14175‐053). The ipsilateral hemisphere was dissected out and mechanically dissected through a 100‐μm cell strainer. Tissue suspension was centrifuged at 286 g for 5 min at 4°C. Pellet was enzymatically digested in collagenase (0.16 μg/ml) (Sigma‐Aldrich, Cat# C9891) for 1 h at 37°C. The cell suspension was filtered through a 70‐μm filter with DNAse (666 U/ml) (Sigma‐Aldrich), resuspended cell pellet in 25% of Percoll density gradient and centrifuged at 521 g for 20 min at 18°C. The myelin coat was aspirated and cells were collected from the interface, washed once with Hank balanced salt solution and processed for flow cytometry.
2.9. Flow cytometry
After erythrocyte lysis in ACK lysis buffer (8.29 g/L NH4Cl, 1 g/L KHCO3 and 1‐mM EDTA in distilled H2O at pH 7.4: Panreac), isolated brain cells were resuspended in staining buffer (PBS supplemented with 10% FBS, 25‐mM HEPES buffer and 2% P/S: Gibco) and their Fc receptors were blocked for 10 min at 4°C with anti‐CD16/CD32 antibodies (clone2.4G; 10 μg/ml: BD Biosciences, Cat# 553142 [RRID:AB_394657]). After blocking, cells were labelled for 30 min at 4°C in the dark with the corresponding antibodies in staining buffer. The primary antibodies used were CD45 (Clone 30‐F11, FITC, 0.6 μg/ml, BioLegend Cat# 103108 [RRID:AB_312973]), CD11b (Clone M1/70, PercP‐Cy5.5, 4 μg/ml, BD Biosciences, Cat# #550993 [RRID:AB_394002]), Ly‐6C (clone AL‐21, APC‐Cy7, 4 μg/ml, BD Biosciences Cat# 560596 [RRID:AB_1727555]), Ly‐6G (clone 1A8, BV510, 4 μg/ml, BD Biosciences Cat# 740157 [RRID:AB_2739910]), F4/80 (clone BM‐8, eFluor450, 4 μg/ml, Invitrogen Cat# 48‐4801‐82 [RRID:AB_1548747]), CD11c (clone N418, APC, 4 μg/ml, Invitrogen Cat# 17‐0114‐82 [RRID:AB_469346]) and CD284 (TLR4, clone SA15‐21, PE, 2.5 μg/ml BioLegend Cat# 145404 [RRID:AB_2561871]). After staining, cells were washed in PBS and samples were acquired on a BD FACSCanto II cytometer (BD Biosciences). Data analysis was carried out with FlowJo 10.6.2 software (Tree Star Inc., Ashland, OR, USA [RRID:SCR_008520]). Cell‐type‐matched fluorescence minus one (FMO) controls were used to determine the positivity of each antibody. The gating strategy employed to quantify frequencies of infiltrating and resident immune cells is shown in Figure 7b. After cellular aggregates and debris exclusion, overall immune cells were identified based on expression of the leukocyte common antigen CD45. Different subpopulations were identified on CD45+ gated cells by differences in the expression levels of CD11b and CD45: microglia was defined as CD11bintCD45int; lymphocytes were defined as CD11bneg CD45hi; and infiltrating myeloid cells were defined as CD11bhiCD45hi. The myeloid subpopulation (CD11bhiCD45hi) was further gated to quantify neutrophils (Ly‐6Ghi) and Ly‐6G negative cells (Ly‐6Gneg). F4/80+ cells were gated on Ly‐6Gneg subpopulation in order to distinguish macrophages. Finally, early infiltrating monocytes were identified as Ly‐6C+ in F4/80+ cells. TLR4 expression was analysed on resident (microglia) and infiltrating immune cells (lymphocytes, neutrophils and infiltrating monocytes).
FIGURE 7.
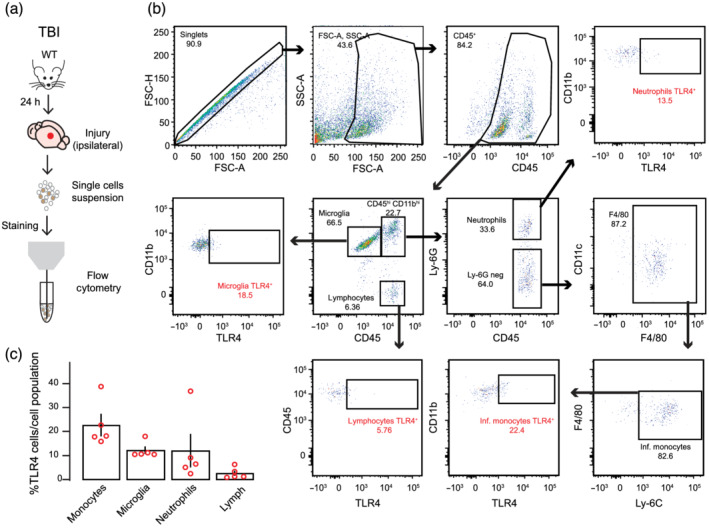
Infiltrating monocytes and resident microglia exhibits TLR4 receptors in injured cortex. (a) Schematic of the experimental protocol. (b) Flow cytometry analysis of immune cells gated on CD45+ from a representative TBI ipsilateral cortex. (c) Quantification of the percentage of cell types expressing TLR4 receptors of each cell population among CD45+ cells (microglia: CD11bintCD45int; lymphocytes: CD11bnegCD45hi; neutrophils: CD11bhiCD45hiLy‐6Ghi; and infiltrating monocytes: CD11bhiCD45hiLy‐6G‐F4/80 + Ly‐6Chi). Open dots on scatterplots are representative of each ipsilateral brain of n = 5 mice and depict the % of TLR4+ cells exhibiting extracellular receptor expression within each population
2.10. Neurological behavioural assessment (NSS)
The severity of the injury is based on the assessment of motor and neurobehavioural functions at 1 and 24 h after trauma using the NSS evaluation, modified from Flierl et al. (2009). The score consists of the evaluation of 10 parameters, described in Table 1. Animals with a score of 9–10 points at the 1‐h test were killed considering it an endpoint criterion. After the TBI, animals were kept in an isolated cage with access to food and water. One hour later, NSS was performed and points were counted. Animals returned to their cages until the next day when NSS was performed once more.
TABLE 1.
Parameters evaluated in neurological severity score evaluation
| Task | Description |
|---|---|
| Exit circle | Exit a circle of 30 cm of diameter in 2 min |
| Seeking behaviour | Interest in the environment and movement |
| Motor skills |
Straight walk Monoparesis or hemiparesis |
| Startle reflex | Response to a loud hand clap |
| Beam balancing | Ability to balance on a surface of 10 × 10 mm |
| Beam walk |
Cross a 30‐cm‐long beam of 3‐cm width Cross a 30‐cm‐long beam of 2‐cm width Cross a 30‐cm‐long beam of 1‐cm width |
| Round stick balancing | Ability to balance on a 5‐mm round stick |
Note: Success in each task is considered 0 points; failure is considered 1 point.
2.11. Pharmacological treatment
TAK242 (Resatorvid; Sigma‐Aldrich Cat# 614316) was used as an antagonist of TLR4 receptors. TAK242 was diluted in saline containing 3% DMSO (3 mg·kg‐1; Sigma‐Aldrich). Mice were randomly divided in three groups: (I) sham in which mice were subjected to anaesthesia but no injury was performed; (II) saline in which mice were subjected to TBI and treated with 0.9% NaCl saline solution; and (III) TAK242 in which mice were subjected to TBI and treated with the TLR4 inhibitor TAK242. TAK242 or saline were i.p. injected immediately following the 1‐h NSS. We were aware that TLR4 pathway is rapidly activated after a brain injury; however, in order to perform our behaviour neurological assessment without the influence of remaining levels of anaesthesia, TAK242 was only administered after NSS was performed (i.e., after 1 h).
2.12. BBB integrity
To evaluate the permeability of the blood–brain vessels and brain oedema, 2% Evans Blue tracer (Sigma‐Aldrich Cat# E2129) diluted in saline was i.p. injected following the 1‐h NSS evaluation. Animals returned to their cages and killed 24 h after trauma and brains were extracted and sectioned in 2‐mm slices using a mouse brain slicer. A total of four slices were used to analyse the affected volume in the entire cerebrum. Cerebellum was not included. The slices were scanned and the total area of Evans Blue extravasation was measured in each slice using ImageJ and cubic volume (mm3) was calculated.
2.13. ELISA
S100β levels were measured in plasma using a specific ELISA kit (MyBioSource, Cat# MBS2500369) following the manufacturer's instructions. Blood was collected in the sacrifice of mice at 24 h after trauma in EDTA‐treated tubes that were centrifuged at 5000 rpm during 10 min to obtain plasma.
2.14. Immunoblotting
Mice brain cortex was lysed in ice‐cold lysis buffer (1% Nonidet P‐40, 10% glycerol, 137‐mM NaCl, 20‐mM Tris–HCl, pH 7.5, 1 g/ml leupeptin, 1‐mM PMSF, 20‐mM NaF, 1‐mM sodium pyrophosphate and 1‐mM Na3VO4). Proteins (30 μg) from brain cell lysate were resolved by SDS–PAGE and transferred to Immobilon‐P membranes (Millipore Corp.). Membranes were incubated with anti‐TSP1 (1:500, NeoMarkers Cat# MS‐1066‐P1 [RRID:AB_64247]) or anti‐β‐actin (1:50,000, Sigma‐Aldrich Cat# A3854 [RRID:AB_262011]). Appropriate peroxidase‐conjugated secondary antibodies (1:10,000; Santa Cruz Biotechnology) were used to detect proteins by enhanced chemiluminescence. Different band intensities corresponding to immunoblot detection of protein samples were quantified using the Scion Image program [RRID:SCR_008673]. Immunoblots correspond to a representative experiment. Immunoblots correspond to a representative experiment that was repeated four or five times with similar results. All immuno‐related procedures involved comply with the editorial on immunoblotting and immunohistochemistry (Alexander et al., 2018).
2.15. Data and statistical analysis
All the data and statistical analyses comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). Mice were randomly assigned to each treatment group. Data analysis of GFAP quantification, Sholl analysis and synaptic puncta were performed by an experimenter blinded to the experimental group (i.e. data were collected and named as a sequence of random numbers by one researcher; thus, another researcher analysed the data completely blind to the group). All collected data were first tested with a Shapiro–Wilk test for normality and Levene's test for equality of variance. No exclusion criteria were used in the statistical analysis to remove possible outliers. Group measurements are expressed as mean ± SEM. To compare two groups, statistical analyses were performed using unpaired two‐tailed Student's t‐test. Differences between more than two groups were determined by one‐way ANOVA followed by Bonferroni's post hoc test when there was one independent factor, or two‐way ANOVA followed by Bonferroni's post hoc test when there were two independent factors. Post hoc tests were run only if F achieved P < .05 and there was no significant variance inhomogeneity. The threshold for statistical significance was P < .05 throughout. Sample sizes subjected to statistical analysis at least 5 animal per groups (n = 5), where n = number of independent values. Statistical differences were performed using Statistical and/or GraphPad Prism 7 (RRID:SCR_002798). Graphs and figures were made using Igor Pro (WaveMetrics) and Adobe Illustrator (RRID:SCR_010279). All measurements were undertaken in at least three technical replicates.
2.16. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in the IUPHAR/BPS Guide to PHARMACOLOGY http://www.guidetopharmacology.org (Harding et al., 2018) and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander et al., 2019).
3. RESULTS
3.1. TLR4 antagonism reduces astrogliosis and microgliosis after traumatic brain injury (TBI)
We first determined how TLR4 pathway regulated astrocyte reactivity and the induction of pro‐inflammatory microglia in the closed head injury mice model of TBI that simulates a biomechanics similar to human TBI (Flierl et al., 2009). For that, we blocked TLR4 receptors by treating TBI animals with the selective TLR4 antagonist TAK242 (3 mg·kg‐1; Figure 1a) or vehicle 1 h after injury. Cresyl violet staining showed substantial cortical damage at 1 day after injury (1 d.a.i.; Figure 1a) following a moderate unilateral injury over the primary somatosensory and motor cortical areas. The cortical damage was composed of an injury core (primary damage, absence of Nissl bodies; Figure 1a″ Region 1) surrounded by a peri‐impact area (secondary damage, loss of several Nissl bodies; Figure 1a″ Region 2) with no overt subcortical damage. TBI and sham brains were then immunostained against GFAP or Iba‐1 to mark reactive astrocyte or microglia, respectively (Figure 1b–d) and quantification was taken from 200 to 400 μm from the injury core (Figure 1a″ Region 3) to avoid the area with ongoing neuronal death. Increased number of GFAP+ astrocytes was observed in the ipsilateral cortex, whereas only few GFAP+ cells were found in sham or contralateral cortex (Figure 1c), corroborating previous reports showing absence of astrogliosis in cortical areas with no apparent damage (Shinozaki et al., 2017). TAK242 treatment significantly reduced GFAP+ cells. To examine by which degree microglia entered in a pro‐inflammatory state after closed head injury, we analysed their morphology in two ways. First, we counted the number of microglia displaying pro‐inflammatory phenotype, that is, large cell bodies and shorter and thicker projections (hypertrophic type) or enlarged cell bodies with multiple short processes forming thick bundles (bushy type), or non‐inflammatory phenotype, that is, ramified with small cell bodies and thin projections (Figure 1b). The decreased number of the ramified phenotype with consequent increase in the pro‐inflammatory one observed in the TBI ipsilateral cortex was significantly reversed by TAK242 treatment (Figure 1d). We then performed a Sholl analysis to provide a more accurate measurement of microglia activation. Figure 1e summarizing the Sholl analysis plots of distinct groups shows that the microglia branching profile in the region nearest the damaged tissue was shifted to the left, indicating a decrease in branching as a function of distance from the soma and therefore microglia activation. Figure 1f shows the effect of TAK treatment on the number of microglia primary branches calculated by the Sholl analysis. The results of two‐way ANOVA revealed a significant effect of hemisphere, treatment and interaction, indicating that TAK treatment reduces microglia activation after TBI.
FIGURE 1.
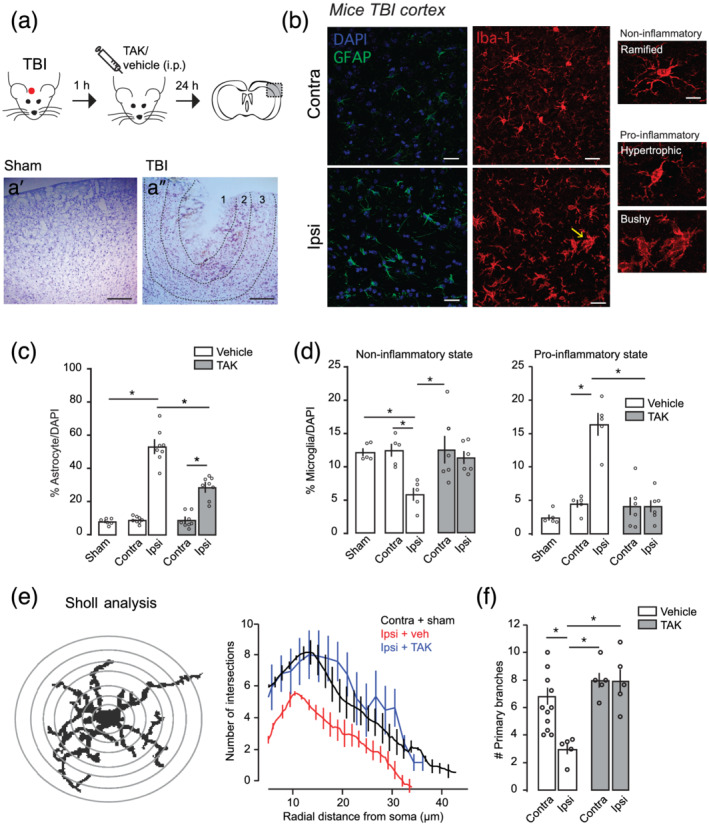
TLR4 antagonism reduces astrogliosis and microgliosis caused by traumatic brain injury. (a) Top: Schematic diagram showing the experimental strategy for the injury, pharmacological treatment and brain slice (adapted from Paxinos & Franklin, 2019). TAK242 (TAK‐242; TAK) or vehicle were i.p. injected 1 h after injury. Dotted black box represents the area in the somatosensory (S1) cortex where functional and anatomical experiments are performed. Images in a′ and a″ are from the area delimited in the dotted black rectangle. Nissl staining tissues showing gross morphological damage 24 h after sham (a′) or traumatic brain injury (TBI) (a″) injury. Scale bar: 150 μm. Dotted lines in a″ delimit Region 1 (~200 μm), which represents the injury core, Region 2 (~150–250 μm), which represents the peri‐injured area and Region 3 representing the ‘area of interest’ (~200–400 μm) in which all the experiments were performed. (b) High magnification confocal images of somatosensory cortex immunostained with a marker for astrocytes (GFAP, green) or microglia (Iba‐1, red) in contralateral and ipsilateral S1 cortex. On the right, insets from individual microglia cells displaying distinct phenotypes. Scale bar: 5 μm. Yellow arrow represents the ‘bushy’ microglia phenotype represented on the right. Sections were counterstained with DAPI (blue) to illustrate nuclei. (c) GFAP+ cells quantification in the primary somatosensory cortex from sham (n = 6) and TBI mice treated with vehicle (n = 8) or TAK242 (n = 8; 3 mg·kg‐1). (d) Iba1+ cells quantification in the primary somatosensory cortex from sham (n = 5) and TBI (n = 5 veh, n = 6 TAK) mice according to the microglial inflammatory phenotype. (e) Left: Schematic illustration of Sholl analysis performed to determine microglial phenotype. Right: Number of intersections of the microglia branches with the designed circles and distance from soma in each group (sham + contralateral vehicle = 27 cells per 11 mice; TBI veh, n = 12 cells per five mice; TBI TAK242, n = 21 cells per five mice). (f) Quantification of primary branches of microglial cells in the S1 cortex after TBI treatment with vehicle or TAK242 (3 mg·kg‐1) from the same sample in (e). Values are mean ± SEM. Two‐way ANOVA, Bonferroni's post hoc test, *P < .05
By quantifying microglia‐released cytokines in TBI brains (Figure 2), we also found that increased levels of TNF‐α and IL‐1β after TBI (Figure 2b,c) were reversed by TAK242 administration, corroborating previous findings showing that TLR4 regulates the inflammatory response of microglia. Together, these data indicate that the inhibition of TLR4 pathway after our TBI model modulates both microglia activation and astrocyte reactivity.
FIGURE 2.
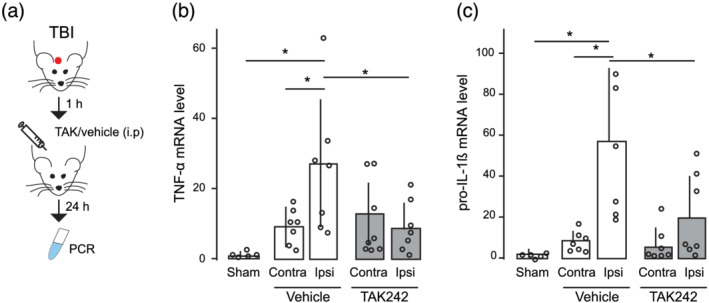
Traumatic brain injury (TBI)‐induced pro‐inflammatory cytokines are reversed by TAK242 (TAK) treatment. (a) The schematic diagram shows the experimental strategy and the timing of the pharmacological treatment. TAK242 or vehicle were i.p. injected 1 h after the injury. mRNA levels of (b) TNF‐α (sham = 5, vehicle‐treated TBI = 7, TAK‐treated TBI = 8) and (c) pro‐IL‐1β genes (sham = 5, vehicle‐treated TBI = 7, TAK‐treated TBI = 7). Values are mean ± SEM. One‐way ANOVA, Bonferroni's post hoc test, *P < .05
3.2. Acute TBI recruits cortical astrocytes with somatic Ca2+ transients via TLR4 activation
Reactive astrocytes are known to result in a spectrum of morphological and molecular changes that influence injury progression and recovery (Nikolakopoulou et al., 2016); however, little is known about the changes in functional activity of reactive astrocytes that could affect further synaptic stability and neuronal networks. Using epifluorescence microscope imaging, we examined spontaneous somatic Ca2+ transients from astrocytes loaded with the astrocytic marker sulforhodamine 101 and the intracellular calcium dye Fluo4‐AM localized at 200–400 μm of the damaged area of TBI and sham mice (Figure 3a,b). Spontaneous Ca2+ transients were defined as a change in fluorescence intensity relative to baseline. The proportion of ‘active astrocytes’ showing one or more Ca2+ transients during the 1‐min recording period were markedly increased in the ipsilateral TBI cortex at 1 d.a.i. (Figure 3d). These effects were independent of neuronal activity, as tetrodotoxin bath application did not prevent the increase in the active astrocytic population. In addition, no changes in the frequency of astrocytic spontaneous Ca2+ transients were observed (Figure 3d; one‐way ANOVA, F 4,25 = 1.81), suggesting that acute TBI leads to functional changes in cortical astrocytes observed as an increase in the number of responsive cells nearby the injury rather than an intrinsic increase in the astrocytic Ca2+ transients or extension of intracellular Ca2+ network. We next tested to which degree the recruitment of active astrocytes in the peri‐injured area was dependent on TLR4 pathway (Figure 3c). TAK242 treatment drastically decreased the number of active astrocytes in the ipsilateral cortex with no changes in the Ca2+ oscillation frequency (Figure 3d). These results indicate that TLR4 activation plays a crucial role in triggering anatomical and functional changes in cortical astrocytes following TBI.
FIGURE 3.
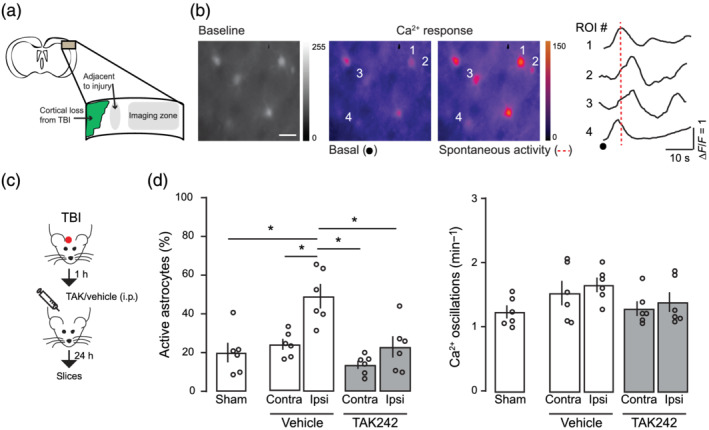
Traumatic brain injury recruits active astrocytes through TLR4 activation. (a) Schematic view of the Ca2+ imaging experiments showing the cortical area where astrocyte activity is registered. (b) Left: Fluorescent intensities of a traumatic brain injury (TBI) primary somatosensory cortex loaded with Fluo4‐AM (1 μg·μl‐1) and heat maps of maximal ∆F/F before and during a spontaneous activity indicated in the rightmost figure. Colour scale indicates normalized changes in fluorescence during spontaneous activity. Scale bar: 10 μm. Right: Time course of spontaneous Ca2+ oscillations from astrocytes indicated on the left. (c) Schematic diagram showing the experimental strategy for the injury and the pharmacological treatment. (d) Left: Proportion of active astrocytes that showed at least one Ca2+ event during a 1‐min recording period in sham (n = 6 animals per 432 regions of interest (ROI)), vehicle (n = 6 animals, contra: 432 regions of interest and ipsilateral (ipsi): 480 regions of interest) or TAK242 (n = 6, contra: 336 regions of interest and ipsi: 720 regions of intererst) group. Right: Corresponding frequency of Ca2+ transients. Values are mean ± SEM. One‐way ANOVA, Bonferroni's post hoc test, *P < .05
3.3. TLR4‐induced astrocyte reactivity affects BBB damage after TBI
Next, we investigated the consequences of the TLR4‐dependent recruitment of astrocytes in the TBI pathophysiology. We first examined the BBB integrity, which is highly regulated by astrocytes and disrupted following TBI (Jha et al., 2018; Liauw et al., 2008; Min et al., 2015; Villapol et al., 2014). S100β is an astrocytic marker released into the plasma following BBB disruption (i.e., the higher the severity of BBB disruption, more S100β in serum is found). One‐way ANOVA followed by bonferroni post hoc test showed increased amount of S100β in the serum collected 1 d.a.i, indicating elevated astrocyte activity and/or damage (Figure 4a). TAK242 treatment decreased ~0.4‐fold S100β levels, corroborating a role for TLR4 activation in the astrocyte modulation. We then injected Evans Blue dye intraperitoneally in TBI mice and assessed BBB integrity by measuring the amount of dye leakage into the brain parenchyma (Figure 4b). Dye leakage was mostly detected around the injury in the ipsilateral cortex, with very low leakage levels in the contralateral cortex (Figure 4b,c). Blockage of TLR4 pathway decreased dye staining as showed by the two‐way ANOVA showing significant effects for hemisphere, treatment and interaction. To test whether the effect of TLR4 on BBB integrity was directly related to astrocytes, we used the inositol 1,4,5‐trisphosphate receptor type 2 knockout mice (Itpr2 −/−, herein referred to as IP3R2−/−). IP3R2 is selectively expressed in astrocytes and its genetic deletion abolishes somatic Ca2+ transients in these mice (Li et al., 2005). Two‐way ANOVA showed that ipsilateral dye leakage was also found to significantly increased after TBI in IP3R2−/− animals (Figure 4c; hemisphere) and it was even significantly higher when compared with WT extravasation, indicating that decreased astrocyte activity enhances BBB rupture following TBI. TAK242 treatment in IP3R2−/− mice did not lessen Evans Blue extravasation (Figure 4c) and it was significantly different from values obtained in TAK‐treated WT mice (avg ± SEM: WT, 23.7 ± 4.55; IP3R2−/−, 53.67 ± 10.05; unpaired Student's t‐test), indicating that TLR4 pathway partially regulates BBB rupture through modulation of astrocyte activity.
FIGURE 4.
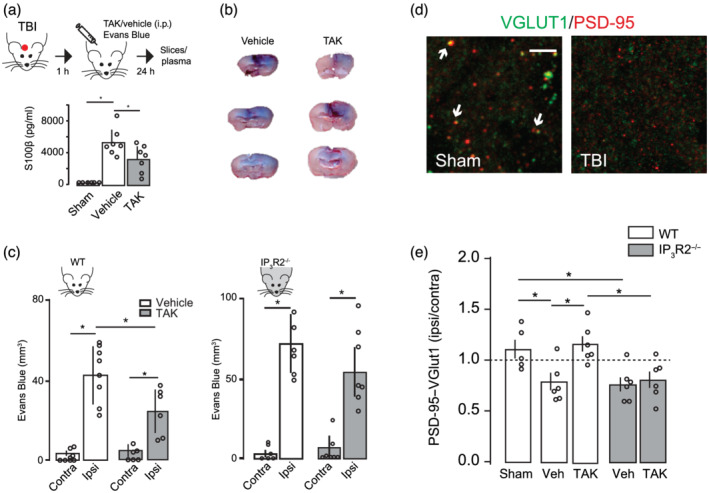
TLR4 antagonism improves synaptic recovery and prevents blood–brain barrier damage after traumatic brain injury (TBI). (a) Top: Schematic diagram showing the experimental strategy. Bottom: Bar graph showing the effect of vehicle or TAK242 (TAK) administration on the levels of S100β in plasma (sham = 5; TBI veh = 7; TBI TAK = 7). (b) Illustration of three distinct coronal sections through the TBI centre showing the Evans Blue distribution throughout a vehicle or TAK242‐treated mouse brain. (c) Blood–brain barrier integrity assessed by Evans Blue extravasations (mm3) 24 h after TBI in the contralateral and ipsilateral brain of WT mice (vehicle, n = 8; TAK242 , n = 6) or IP3R2−/− mice (vehicle, n = 6; TAK242, n = 7). (d) Representative confocal images of synapses (white arrowheads) indicated by colocalization of pre‐synaptic (VGLUT1, green) and post‐synaptic (PSD‐95, red) markers in sham and TBI ipsilateral S1 cortex. Scale bar: 5 μm. (e) Quantitative analysis showing the number of synaptic puncta 24 h after TBI in WT (sham, n = 5; TBI veh, n = 6; TBI TAK242, n = 6) and IP3R2−/− (grey bars) mice (TBI veh, n = 6; TBI TAK242, n = 6). Data represented as the ratio of synapses (ipsi/contra) in which values < 1 mean synaptic loss in the ipsilateral (ipsi) cortex. Values are mean ± SEM. One‐way ANOVA, Bonferroni's post hoc test, *P < .05 for (a) and (e). Two‐way ANOVA, Bonferroni's post hoc test, *P < .05 for (c)
3.4. Changes in the number of synaptic puncta density in the primary somatosensory cortex after TBI
Microglia and astrocytes influence neuronal transmission through the regulation of synapses and the modulation of neuronal network (Araque et al., 2014; Béchade et al., 2013; Perea et al., 2009; Wake et al., 2009). To further investigate the pathophysiological consequences of TLR4 pathway activation following TBI, we quantified the synaptic density surrounding the injured cortex by double immunostaining for pre‐ and post‐synaptic proteins, that is, the number of colocalized pre‐synaptic VGLUT1 and post‐synaptic PSD‐95 puncta, at 1 d.a.i. We defined the normalized synaptic density as the synaptic ratio in the surrounding TBI area to that in the homotopic non‐injured contralateral cortex to correct for any endogenous differences in the case of transgenic mice. Our results showed that ipsilateral cortex had significantly less PSD‐95/ VGLUT1 punctas compared with sham (Figure 4d,e). TAK242 treatment partially reduced the synaptic loss, indicating that blockage of TLR4 pathway shortly after TBI either restores synaptic level or inhibits synaptic loss. We next quantified the number of synaptic puncta in TBI brain from IP3R2−/− mice to test whether the neuroprotective effect on synapses observed following TAK242 treatment was exerted by TLR4‐induced astrocyte activation. IP3R2−/− mice exhibited decreased number of synaptic punctas following TBI, indicating that decreased astrocyte activity did not prevent synaptic loss after TBI. In addition, a partial synaptic recovery was not observed after pharmacological treatment of IP3R2−/− mice with TAK242 (Figure 4e), suggesting that modulation of TLR4 pathway after TBI only affects synapses in the presence of somatic active astrocytes.
3.5. TSP‐1 astrocytic release regulates synaptic number in the sub‐acute phase of TBI
Because the mechanisms controlling microglia and astrocyte activation are known to have distinct phases from hours to days after injury (Villapol et al., 2014), we determined the anatomical and functional changes of TLR4‐induced astrocyte activation 7 d.a.i (Figure 5a). Calcium imaging experiments from TBI cortex revealed a similar number of active astrocytes and Ca2+ oscillation frequency as compared with those sham or contralateral cortex (Figure 5b). Increased number of GFAP+ astrocytes was still observed 7 d.a.i. (Figure 5c), but such increase was significantly smaller than those found 1 d.a.i (unpaired t‐test). Second, we determined the changes in microglia morphology by using both phenotype counting and Sholl analysis (Figure 5d). No differences were observed neither in the number of non‐inflammatory or pro‐inflammatory microglia (one‐way ANOVA, F 2,12 = 1.21) as well as Sholl analysis compared with sham 7 d.a.i. (unpaired t‐test primary branches). Finally, we determined the levels of synaptic colocalization and found similar synaptic number in TBI mice to those sham (Figure 5e), indicating a synaptic recovery in a sub‐acute phase. The later corroborates previous anatomical and electrophysiological findings showing that after ischaemic stroke or TBI in mice, there is an initial synaptic loss and decreased neuronal excitability followed by its gradual homeostatic recovery (Carron et al., 2016; Ding et al., 2011; Johnstone et al., 2013; Johnstone et al., 2015; Liauw et al., 2008). Altogether, our data indicate that the recovery of microglial non‐inflammatory phenotype and astrocyte activity 7 d.a.i. is accompanied by a recovery in the number of synapses.
FIGURE 5.
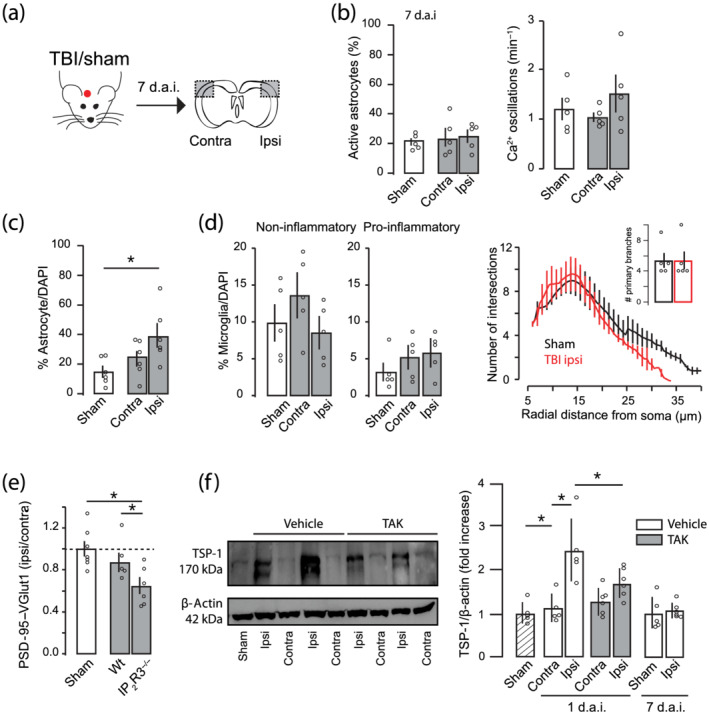
Sub‐acute recovery of astrocyte activity and synaptic density 7 days after traumatic brain injury (TBI). (a) Schematic diagram showing the experimental strategy. (b) Percentage of active astrocytes (left) and number of Ca2+ oscillations (right) in contralateral and ipsilateral cortex of sham (n = 5) or TBI mice (n = 5) 7 days after the injury. (c) Percentage of GFAP+ astrocytes in contralateral and ipsilateral cortex of TBI mice 7 days after injury (n = 6). (d) Quantitative assessment of microglial phenotypes (Iba‐1 staining) in the S1 cortex from sham and TBI mice. Sholl analysis of microglia morphology and primary branches (inset; sham = 5, TBI = 5). (e) Quantitative analysis of synaptic puncta density 7 days after TBI in WT and IP3R2−/− (sham, n = 7; WT, n = 5; IP3R2−/−, n = 6). (f) Left, original (unedited) western blot of TSP‐1 and actin from sham, two vehicles (Veh) and two TAK242 treated mice (TAK). Right, semi‐quantitative analysis of TSP‐1 immunoreactivity of TSP‐1 after TBI (for 24 h mice: sham, n = 5; vehicle, n = 5; TAK242, n = 6; for 7 days after injury (d.a.i.): sham = 5; TBI ipsilateral (ipsi) = 6). Values are mean ± SEM. One‐way ANOVA, Bonferroni's post hoc test, *P < .05
We next sought to identify the potential molecular mechanism that causally links astrocyte activation to synaptic regulation at 7 d.a.i. We focused on TSP‐1, which is an astrocyte‐secreted soluble protein that is necessary and sufficient to promote synaptic formation during neuronal development or following brain damage (Christopherson et al., 2005; Liauw et al., 2008). Increased TSP‐1 expression was found in the ipsilateral cortex at 1 d.a.i., which returned to basal levels at 7 d.a.i. (Figure 5f). Blockage of TLR4 signalling significantly reduced TSP‐1 expression to similar levels to sham or contralateral tissues, indicating that TLR4 activation controls TSP‐1 release from astrocytes following TBI. To investigate whether the transient up‐regulation of TSP‐1 protein within the first few hours was linked to the synaptic remodelling at 7 d.a.i, we used the IP3R2−/− mice known to have reduced release of TSP‐1 after injuries (Kim et al., 2016). Synaptic puncta number in IP3R2−/− TBI brain remained significantly decreased at 7 d.a.i. (Figure 5e), indicating the important role of astrocyte activation in the homeostatic regulation of synapses in a sub‐acute phase through the release of TSP‐1.
3.6. Blockage of TLR pathway improves functional outcome after TBI
Synaptic damage is thought to be a major contributor to neurological symptoms and behavioural deficits following brain injuries. Given the significant effect on synapses and BBB integrity, we explored the possibility that TLR4‐induced astrocyte activation might also affect neurological behaviour associated with TBI. We therefore investigated whether TAK242 treatment improved functional outcome by subjecting animals to a battery of behavioural tests specifically developed to assess motor and neurobehavioural outcome in TBI mice: NSS test (Figure 6; Flierl et al., 2009). One‐hour following TBI, mice had an NSS score around 7 points, indicating high functional deficits. Twenty‐four hours later, the performance of TBI mice gradually recovered reaching values close to 5 (Figure 6b vehicle). TAK242‐treated mice at 1 h after injury exhibited a significantly better NSS score 24 h later when compared with those animals treated only with the vehicle and interaction. To determine whether the improvement of neurological deficits after TAK was dependent on astrocyte activation, we used IP3R2−/− mice. Two‐way ANOVA showed no effect for treatment, time and interaction, indicating that IP3R2−/− mice did not display neurological improvement after TAK242 treatment. These data demonstrate that blockage of the TLR4‐induced astrocyte activation has beneficial effects on early neurological motor tests following TBI.
FIGURE 6.
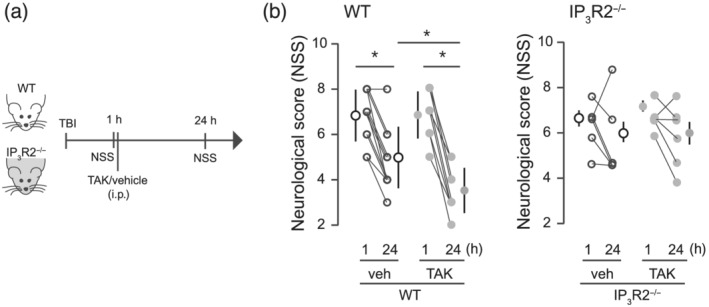
Neurological and behavioural outcome is improved by TAK242 treatment in WT but not in IP3R2 mice. (a) Schematic diagram showing the experimental strategy. (b) Effects of TLR4 pharmacological blockade on neurological severity score (NSS) in WT (vehicle, n = 12; TAK, n = 9) and IP3R2−/− mice (vehicle, n = 6; TAK, n = 6). NSS was evaluated at 1 and 24 h after the injury. Values are mean ± SEM. Two‐way ANOVA, Bonferroni's post hoc test, *P < .05
In order to determine the cell target of the TAK242 pharmacological inhibition, we used immune cell characterization by flow cytometry to analyse the presence of peripheral/resident immune cells exhibiting TLR4 receptors in the ipsilateral cortex of TBI mice (Figure 7). At 1 d.a.i., TLR4 expression was mainly observed in myeloid cells, being infiltrating monocytes and microglia the two main subpopulations showing the highest percentage of TLR4+ signal. These data pointed to a crucial role of resident/infiltrated myeloid cells in the observed pharmacological effect of TAK242 on BBB, synapse remodelling and behavioural outcome 24 h after TBI.
4. DISCUSSION AND CONCLUSIONS
Here, we identified a fine‐tune interaction between astrocytes and microglia/infiltrated monocytes that control synaptic number and BBB integrity with pathophysiological relevance for long‐term plasticity in TBI. We show that acute TBI recruits active astrocytes surrounding the injury in a TLR4‐dependent manner (Figures 1, 2, 3) with detrimental effects on synapses, BBB and neurological outcome (Figures 4, 5, 6). On the other hand, TBI‐induced astrocyte activation mediates the release of the astrocytic protein TSP‐1 with beneficial effects on synaptic recovery in the subacute phase of the injury (Figures 5 and 6). Hence, pharmacological modulation of TLR4‐induced astrocyte activation slow down the damage progression and, in turn, may accelerate long‐term plasticity and rebuilding of neuronal networks in the damaged area following TBI.
Microglia immediately respond to tissue damage by polarizing into a pro‐inflammatory or into an alternative/anti‐inflammatory phenotype (Davis et al., 1994). Similarly, transcriptome analyses have identified two types of reactive astrocytes: A1 that up‐regulates genes described to be destructive to synapses, suggesting a detrimental role and A2 that increases neurotrophic factors and thrombospondins that promote synapse repair, suggesting a neuroprotective role (Liddelow et al., 2017; Shinozaki et al., 2017; Zamanian et al., 2012). Hence, the inflammatory response is not an isolated response of a single cell type but is composed of a set of different resident/infiltrated cells that, as a whole, offer an orchestrated and fine‐tuned response after damage. Our data evidenced that the closed head injury model of TBI induces a subtype of microglia and astrocyte with ‘harmful’ effects that might correspond to the pro‐inflammatory microglia and A1 astrocytes. The control of microglial inflammation exerts neuroprotective effects in several injury/disease models (Donat et al., 2017; Parada et al., 2019) and our data indicate that some of the acute detrimental effects of microglia after TBI, most probably through TLR4 pathway activation, is only achieved by a direct interaction with astrocytes. However, we cannot discard that some of the anti‐inflammatory effects of TLR4 blocker administration might be also mediated by modulation of the inflammatory responses of infiltrated monocytes. On the other hand, the acute activation of astrocytes seems to be needed to restore synaptic levels in a sub‐acute phase. Therefore, astrocyte activation has a dual role depending on the time point after injury: in an acute phase, its role is detrimental for synapses and BBB and, in the sub‐acute phase, is important for synaptic remodelling after TBI.
4.1. Acute TBI recruits active astrocytes
The induction of reactive astrocytes during the pathogenesis of brain injuries has been extensively reported by anatomical and molecular techniques (Kim et al., 2016). In contrast, only few studies have explored the functional changes in astrocytes following CNS injuries (Kim et al., 2016; Takatsuru et al., 2013). By using Ca2+ imaging from TBI brains, we demonstrate that active cortical astrocytes are recruited in the injured area within the first few hours following TBI. No changes in the frequency of the spontaneous responses were observed, suggesting that the astrocytic network is unaffected despite the increased extracellular glutamate observed at the synaptic cleft after injuries (Cantu et al., 2015). The recruitment of active astrocytes was abolished by blocking TLR4 pathway activation, with improvement in the synaptic number as well as BBB integrity. These data support the idea that TLR4‐induced astrocyte activation plays a detrimental role in the neuronal and vascular functions, and therefore, blockage of TLR4 pathway may be a potential therapeutic target for TBI.
4.2. Activation of TLR4 pathway regulates synaptic puncta following TBI
Microglia and astrocytes are known to control synapses and to modulate neuronal networks after brain lesions or diseases. For example, in in vivo models microglia activation of the complement cascade controls neuronal synaptic pruning (Tremblay et al., 2010; Wake et al., 2009), whereas microglia‐derived molecules regulate synaptic strength and neuronal activity (Béchade et al., 2013) by inducing synaptic facilitation (Clark et al., 2015) and increasing the frequency of spontaneous excitatory synaptic currents (Pascual et al., 2012). Independently of the mechanism used by microglia to control synaptic transmission, it is clear that microglia plays a crucial role in the control of neuronal networks. On the other hand, astrocytes also regulate synaptic transmission and neuronal homeostasis through several mechanisms (Araque et al., 2014; Navarrete et al., 2019; Rosa et al., 2015). In vivo models show that astrocytic release of TSP‐1/2 is associated with synapse formation and axonal outgrowth (Kim et al., 2016; Liauw et al., 2008), whereas enhanced astrocytic production and release of d‐serine or ephrins are detrimental to synapses (Nikolakopoulou et al., 2016; Perez et al., 2017). Here, we give new evidence that astrocyte regulates neuronal synapses through the activation of TLR4 pathway in inflammatory cells following TBI. TLR4 receptors are mainly implicated in the recognition of infectious pathogens or endogenous ligands released upon injury and are the main pathway to activate microglia after TBI. However, TLR4 is also expressed in other cell types such as endothelial and peripheral immune cells (monocytes, neutrophils, granulocytes and lymphocytes) (for more, see https://www.brainrnaseq.org/) that access the brain parenchyma following a robust BBB rupture (Nagyoszi et al., 2010). Given that infiltrated monocytes and microglia, but not neutrophils and lymphocytes, were the most prominent myeloid cell types expressing TLR4 under our experimental model, we cannot discard that some of the neuroprotective effects observed after systemic administration of TLR4 blocker might be mediated by modulation of the inflammatory responses via monocytes. On the other hand, infiltrating monocytes influence the proliferation of juxtavascular astrocytes located in the proximity of damaged area following traumatic injury (Bardehle et al., 2013), which could also affect synaptic remodelling through the modulation of astrocyte and microglia activation. Supporting our hypothesis, we found that synaptic loss is partially reversed by TLR4 pathway modulation in an astrocyte‐dependent manner, because IP3R2−/− mice fail to recover synapses after TLR4 receptor blockage. In addition, astrocytic‐protein TSP‐1 released following TBI seems to regulate the synaptic recovery in the sub‐acute phase of the injury. Given all these evidence, our results support that control of astrocyte activation through inhibition of TLR4 pathway in inflammatory cells plays an important role in the regulation of synapses following TBI.
The effect of pharmacological blockage of TLR4 observed in this study could also be attributed to a direct effect on astrocytes as several studies have shown TLR4 expression in cultured astrocytes. However, these data are currently contradictory with some reports showing that TLR4 is expressed in both microglia and astrocytes (Du et al., 2017; Fellner et al., 2013), whereas others showing that TLR4 response in astrocytes is only mediated by previous microglia activation (Holm et al., 2012). Nonetheless, several recent reports using cutting‐edge genetic techniques have shown a predominant expression of TLR4 in microglia, but not in astrocytes (Cahoy et al., 2008; Zhang et al., 2014, 2016). Our data using IP3R2−/− mice support the idea that the effect observed in this study is mediated by TLR4 pathway activation either in microglia or infiltrating peripheral cells.
4.3. Sub‐acute effects of TLR4‐induced astrocyte activation on synapses
Reactive astrocytes exert neuroprotective effects after TBI by limiting the extension of the injured core and by forming a glial scar (Shinozaki et al., 2017). Our data extend the beneficial effects of astrocytes to the synaptic remodelling in the TBI sub‐acute phase. By using anatomical quantification of PSD‐95/VGLUT1 puncta, we demonstrate a significant recovery of the initial synaptic loss a week following injury. This physiological mechanism seems to depend on the astrocyte activation as IP3R2−/− mice continue to exhibit reduced synaptic number 7 d.a.i. Our data also indicate that such synaptic regulation depends on TSP‐1, an astrocyte‐released protein that serves to control neuronal synapses after brain damage (Christopherson et al., 2005). In our experiments, TSP‐1 is transiently up‐regulated 1 d.a.i, but its effect on synapse is observed only a few days after injury. These data corroborate other studies suggesting that TSP‐1 effects may take a several day period to be observed (Christopherson et al., 2005), as it acts on the expression of proteins that participate in the formation of new synaptic proteins rather than regulating cell adhesion proteins to lock synapses into place (i.e., stabilizing role) (Liauw et al., 2008). So, while neuronal cortical synapses are rapidly lost (probably due to direct neuronal death), TLR4‐induced astrocyte activation increases TSP‐1 release in the first few hours to trigger the expression of neuronal synaptic proteins later on, leading to a partial synaptic recovery 7 d.a.i. Interestingly, the synaptic recovery can be speed‐up by an order of days after pharmacological TLR4 blockage. Hence, TLR4 inhibition of astrocyte activation works to slow down the progression of synaptic damage, thereby accelerating the recovery/repair of the surrounded damaged area.
4.4. Inhibition of astrocyte activation through TLR4 blockage improves BBB after TBI
Both microglia and astrocytes physically interact with brain endothelial blood vessels influencing BBB integrity (Burda et al., 2016). Here, we found that although TLR4 blockage attenuates BBB rupture after TBI, this effect is not observed in IP3R2−/− mice, indicating a direct effect of astrocyte and its interaction with inflammatory cells (Donat et al., 2017; Jha et al., 2018). The astrocytic detrimental effects on BBB after TBI could be explained by the release of vasoactive endothelial growth factor (Argaw et al., 2009) or apolipoprotein E (Bell et al., 2012) shown to exacerbate BBB damage. However, astrocytes are also known to release molecules that act beneficially on endothelia to reduce BBB permeability and to promote its repair including sonic hedgehog (Alvarez et al., 2011) and retinoic acid (Mizee et al., 2014). Such results divergence demonstrates that astrocytes are pivotal regulators of endothelial BBB properties that act to open, maintain or restore barrier functions via specific molecular mechanisms that remain to be investigated.
4.5. Conclusion
Our data provide new evidence showing a role of TLR4‐mediated astrocyte activation in the pathophysiology of brain injuries (Figure 8). We postulate that after a TBI, extracellular molecules that are rapidly released upon tissue damage activate TLR4 signalling to recruit active astrocytes in the peri‐injured area that in turn will affect neuronal excitability by modulating synaptic number in a time‐dependent manner. Shortly after the injury, astrocyte decreases synapses probably to slow down the neuronal network and to avoid further neurotoxic damage. At the same time, active astrocytes release TSP‐1 to promote synaptic recovery later on. After that, a homeostatic control of both microglia and infiltrated monocyte activity may take place followed by a decrease in reactive/active astrocytes. Moreover, this cell‐to‐cell interaction also exacerbates immediate BBB breakdown probably due to the release of astrocytic proteins and pro‐inflammatory cytokines known to have detrimental effects on BBB. Overall, our data show that decreasing TLR4 activation and the pro‐inflammatory microglia phenotype and infiltrated monocytes during the first few hours following a TBI could be used as a target for therapeutic treatments that may promote synaptic plasticity and improve cognitive function via astrocytic modulation.
FIGURE 8.
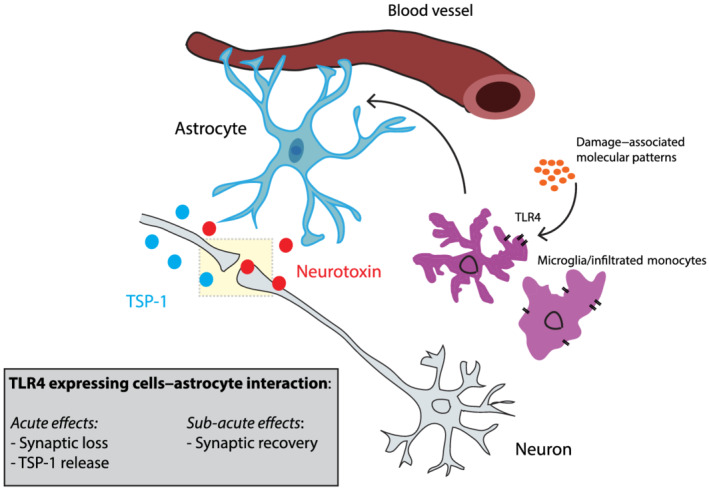
Schematic diagram of the microglia/infiltrated monocyte–astrocyte interaction following TBI. Control of astrocyte activation represents a key physiological mechanism to regulate both neuronal vasculature and synaptic number following TBI in a time‐dependent manner. Shortly after the injury, astrocyte activity decreases the number of synapses probably to slow down the neuronal network and to avoid further neurotoxic damage. At the same time, actived astrocytes release TSP‐1 to promote synaptic recovery later on. After this, a homeostatic control of microglia and infiltrated monocytes activity may take place followed by a decrease in actived astrocytes and neuronal excitability. Moreover, inflammatory cell types–astrocyte interaction also exacerbates immediate BBB breakdown probably due to the release of astrocytic proteins and pro‐inflammatory cytokines known to have detrimental effects on BBB
AUTHOR CONTRIBUTIONS
JMR and JE conceived and designed the study. JMR and VFA performed and analysed immunofluorescence experiments. VF performed and analysed Evans Blue extravasation experiments. JMR performed, analysed and wrote the scripts for Ca2+ imaging experiments. MCO, DC and VVS designed, performed, analysed and wrote the text about flow cytometry experiments. MN assisted with astrocyte imaging experiments and advised on calcium imaging analysis. APA, EFL and PNF assisted with IP3R2−/− experiments. VF and CD performed and analysed PCR experiments. All authors revised the manuscript. JMR and JE wrote the manuscript.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry and Animal Experimentation, and as recommended by funding agencies, publishers and other organizations engaged with supporting research.
ACKNOWLEDGEMENTS
This work was supported by grants from the Instituto de Salud Carlos III (ISCIII) (Programa Miguel Servet II Grants CPII19/00005; PI16/00735; PI19/00082 to JE; and PI18/00357 to DC, partially funded by FEDER ‐ European Union ‘Una manera de hacer Europa’) and Fundación Mutua Madrileña to JE; European Union's Horizon 2020 research and innovation programme under the H2020 Marie Skłodowska‐Curie Actions grant agreement no. 794926 and Stop Fuga de Cerebros Roche Pharma to JMR; and Ministerio de Ciencia e Innovación RTI2018‐094887‐B‐I00 and RYC‐2016‐20414 to MN and RYC2019‐026870‐I to JMR. DC, MCO, VVS and EFL are hired by SESCAM. We also thank Dr. Ju Chen (University of California, San Diego, CA, USA) and Dr. Gertrudis Perea (Cajal Institute, Madrid, Spain) for kindly providing IP3R2−/− animals and Dr. Juan Aguilar (HNP, Toledo, Spain) for technical and scientific support.
Rosa JM, Farré‐Alins V, Ortega MC, et al. TLR4 pathway impairs synaptic number and cerebrovascular functions through astrocyte activation following traumatic brain injury. Br J Pharmacol. 2021;178:3395–3413. 10.1111/bph.15488
Juliana M. Rosa and Víctor Farre‐Alins equally contributed to this study.
Funding information Ministerio de Ciencia e Innovación, Grant/Award Numbers: RTI2018‐094887‐B‐I00, RYC‐2016‐20414, RYC2019‐026870‐I; H2020 Marie Skłodowska‐Curie Actions, Grant/Award Number: 794926; Instituto de Salud Carlos III, Grant/Award Numbers: CPII19/00005, PI16/00735, PI18/00357, PI19/00082; Stop Fuga de Cerebros Roche Pharma, Grant/Award Number: SFC2017
Contributor Information
Juliana M. Rosa, Email: jmartinsd@sescam.jccm.es.
Javier Egea, Email: javier.egea@inv.uam.es.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Alexander, S. P. H., Kelly, E., Mathie, A., Peters, J. A., Veale, E. L., Armstrong, J. F., et al. (2019). The Concise Guide to PHARMACOLOGY 2019/20: Introduction and other protein targets. British Journal of Pharmacology, 176(Suppl 1), S1–S20. 10.1111/bph.14747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H., Roberts, R. E., Broughton, B. R. S., Sobey, C. G., George, C. H., Stanford, S. C., Cirino, G., Docherty, J. R., Giembycz, M. A., Hoyer, D., Insel, P. A., Izzo, A. A., Ji, Y., MacEwan, D. J., Mangum, J., Wonnacott, S., & Ahluwalia, A. (2018). Goals and practicalities of immunoblotting and immunohistochemistry: A guide for submission to the British Journal of Pharmacology . British Journal of Pharmacology, 175, 407–411. 10.1111/bph.14112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez, J. I., Dodelet‐Devillers, A., Kebir, H., Ifergan, I., Fabre, P. J., Terouz, S., et al. (2011). The hedgehog pathway promotes blood‐brain barrier integrity and CNS immune quiescence. Science (New York, N.Y.), 334(6063), 1727–1731. [DOI] [PubMed] [Google Scholar]
- Araque, A., Carmignoto, G., Haydon, P. G., Oliet, S. H. R., Robitaille, R., & Volterra, A. (2014). Gliotransmitters travel in time and space. Neuron, 81(4), 728–739. 10.1016/j.neuron.2014.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argaw, A. T., Gurfein, B. T., Zhang, Y., Zameer, A., & John, G. R. (2009). VEGF‐mediated disruption of endothelial CLN‐5 promotes blood‐brain barrier breakdown. Proceedings of the National Academy of Sciences of the United States of America, 106(6), 1977–1982. 10.1073/pnas.0808698106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardehle, S., Krüger, M., Buggenthin, F., Schwausch, J., Ninkovic, J., Clevers, H., Snippert, H. J., Theis, F. J., Meyer‐Luehmann, M., Bechmann, I., & Dimou, L. (2013). Live imaging of astrocyte responses to acute injury reveals selective juxtavascular proliferation. Nature Neuroscience, 16, 508–586. [DOI] [PubMed] [Google Scholar]
- Béchade, C., Cantaut‐Belarif, Y., & Bessis, A. (2013). Microglial control of neuronal activity. Frontiers in Cellular Neuroscience, 7(32), 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell, R. D., Winkler, E. A., Singh, I., Sagare, A. P., Deane, R., Wu, Z., Holtzman, D. M., Betsholtz, C., Armulik, A., Sallstrom, J., Berk, B. C., & Zlokovic, B. V. (2012). Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature, 485(7399), 512–516. 10.1038/nature11087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, M. L., Bennett, F. C., Liddelow, S. A., Ajami, B., Zamanian, J. L., Fernhoff, N. B., Mulinyawe, S. B., Bohlen, C. J., Adil, A., Tucker, A., Weissman, I. L., Chang, E. F., Li, G., Grant, G. A., Hayden Gephart, M. G., & Barres, B. A. (2016). New tools for studying microglia in the mouse and human CNS. Proceedings of the National Academy of Sciences of the United States of America, 113(12), E1738–E1746. 10.1073/pnas.1525528113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burda, J. E., Bernstein, A. M., & Sofroniew, M. V. (2016). Astrocyte roles in traumatic brain injury. Experimental Neurology, 275(Pt 3), 305–315. 10.1016/j.expneurol.2015.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy, J. D., Emery, B., Kaushal, A., Foo, L. C., Zamanian, J. L., Christopherson, K. S., Xing, Y., Lubischer, J. L., Krieg, P. A., Krupenko, S. A., Thompson, W. J., & Barres, B. A. (2008). A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 28, 264–278. 10.1523/JNEUROSCI.4178-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantu, D., Walker, K., Andresen, L., Taylor‐Weiner, A., Hampton, D., Tesco, G., & Dulla, C. G. (2015). Traumatic brain injury increases cortical glutamate network activity by compromising GABAergic control. Cerebral Cortex (New York, N.Y.:1991), 25(8), 2306–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carron, S. F., Alwis, D. S., & Rajan, R. (2016). Traumatic brain injury and neuronal functionality changes in sensory cortex. Frontiers in Systems Neuroscience, 10(47), 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopherson, K. S., Ullian, E. M., Stokes, C. C. A., Mullowney, C. E., Hell, J. W., Agah, A., Lawler, J., Mosher, D. F., Bornstein, P., & Barres, B. A. (2005). Thrombospondins are astrocyte‐secreted proteins that promote CNS synaptogenesis. Cell, 120(3), 421–433. 10.1016/j.cell.2004.12.020 [DOI] [PubMed] [Google Scholar]
- Clark, A. K., Gruber‐Schoffnegger, D., Drdla‐Schutting, R., Gerhold, K. J., Malcangio, M., & Sandkühler, J. (2015). Selective activation of microglia facilitates synaptic strength. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 35(11), 4552–4570. 10.1523/JNEUROSCI.2061-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, L. E., Liddelow, S. A., Chakraborty, C., Münch, A. E., Heiman, M., & Barres, B. A. (2018). Normal aging induces A1‐like astrocyte reactivity. Proceedings of the National Academy of Sciences of the United States of America, 115(8), E1896–E1905. 10.1073/pnas.1800165115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J., Alexander, S., Cirino, G., Docherty, J. R., George, C. H., Giembycz, M. A., Hoyer, D., Insel, P. A., Izzo, A. A., Ji, Y., & MacEwan, D. J. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology. England, 175(7), 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, E. J., Foster, T. D., & Thomas, W. E. (1994). Cellular forms and functions of brain microglia. Brain Research Bulletin, 34(1), 73–78. 10.1016/0361-9230(94)90189-9 [DOI] [PubMed] [Google Scholar]
- Ding, M.‐C., Wang, Q., Lo, E. H., & Stanley, G. B. (2011). Cortical excitation and inhibition following focal traumatic brain injury. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 31(40), 14085–14094. 10.1523/JNEUROSCI.3572-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donat, C. K., Scott, G., Gentleman, S. M., & Sastre, M. (2017). Microglial activation in traumatic brain injury. Frontiers in Aging Neuroscience, 9(208), 1–20. 10.3389/fnagi.2017.00208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorostkar, M. M., Dreosti, E., Odermatt, B., & Lagnado, L. (2010). Computational processing of optical measurements of neuronal and synaptic activity in networks. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 188, 141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, S., Dong‐Fang, Q., Chuan‐Xiang, C., Si, C., Chao, L., Zhoumeng, L., Wang, H., & Xie, W. B. (2017). Toll‐like receptor 4 mediates methamphetamine‐induced neuroinflammation through caspase‐11 signaling pathway in astrocytes. Frontiers in Molecular Neuroscience, 10(409), 1–17. 10.3389/fnmol.2017.00409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellner, L., Regina, I., Kathrin, S., Markus, R., Lars, K., Werner, P., Wenning, G. K., & Stefanova, N. (2013). Toll‐like receptor 4 is required for α‐synuclein dependent activation of microglia and astroglia. Glia, 61, 349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira, T. A., Blackman, A. V., Oyer, J., Jayabal, S., Chung, A. J., Watt, A. J., Sjöström, P. J., & Van Meyel, D. J. (2014). Neuronal morphometry directly from bitmap images. Nature Methods, 11(10), 982–984. 10.1038/nmeth.3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flierl, M. A., Stahel, P. F., Beauchamp, K. M., Morgan, S. J., Smith, W. R., & Shohami, E. (2009). Mouse closed head injury model induced by a weight‐drop device. Nature Protocols, 4(9), 1328–1337. 10.1038/nprot.2009.148 [DOI] [PubMed] [Google Scholar]
- Frik, J., Merl‐Pham, J., Plesnila, N., Mattugini, N., Kjell, J., Kraska, J., Gómez, R. M., Hauck, S. M., Sirko, S., & Götz, M. (2018). Cross‐talk between monocyte invasion and astrocyte proliferation regulates scarring in brain injury. EMBO Reports, 19, e45294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D., Sharman, J. L., Faccenda, E., Southan, C., Pawson, A. J., Ireland, S., Gray, A. J. G., Bruce, L., Alexander, S. P. H., Anderton, S., Bryant, C., Davenport, A. P., Doerig, C., Fabbro, D., Levi‐Schaffer, F., Spedding, M., Davies, J. A., & NC‐IUPHAR . (2018). The IUPHAR/BPS guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46(D1), D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm, T. H., Draeby, D., & Owens, T. (2012). Microglia are required for astroglial toll‐like receptor 4 response and for optimal TLR2 and TLR3 response. Glia, 60(4), 630–638. 10.1002/glia.22296 [DOI] [PubMed] [Google Scholar]
- Ippolito, D. M., & Eroglu, C. (2010). Quantifying synapses: An immunocytochemistry‐based assay to quantify synapse number. Journal of Visualized Experiments, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha, M. K., Jo, M., Kim, J. ‐H., & Suk, K. (2018). Microglia‐astrocyte crosstalk: An intimate molecular conversation. The Neuroscientist: A Review Journal Bringing Neurobiology, Neurology and Psychiatry, 1073858418783959. [DOI] [PubMed] [Google Scholar]
- Jiang, H., Wang, Y., Liang, X., Xing, X., Xu, X., & Zhou, C. (2018). Toll‐like receptor 4 knockdown attenuates brain damage and neuroinflammation after traumatic brain injury via inhibiting neuronal autophagy and astrocyte activation. Cellular and Molecular Neurobiology, 38(5), 1009–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone, V. P. A., Wright, D. K., Wong, K., O'Brien, T. J., Rajan, R., & Shultz, S. R. (2015). Experimental traumatic brain injury results in long‐term recovery of functional responsiveness in sensory cortex but persisting structural changes and sensorimotor, cognitive, and emotional deficits. Journal of Neurotrauma, 32(17), 1333–1346. 10.1089/neu.2014.3785 [DOI] [PubMed] [Google Scholar]
- Johnstone, V. P. A., Yan, E. B., Alwis, D. S., & Rajan, R. (2013). Cortical hypoexcitation defines neuronal responses in the immediate aftermath of traumatic brain injury. PLoS ONE, 8(5), e63454. 10.1371/journal.pone.0063454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S. K., Hayashi, H., Ishikawa, T., Shibata, K., Shigetomi, E., Shinozaki, Y., Inada, H., Roh, S. E., Kim, S. J., Lee, G., Bae, H., Moorhouse, A. J., Mikoshiba, K., Fukazawa, Y., Koizumi, S., & Nabekura, J. (2016). Cortical astrocytes rewire somatosensory cortical circuits for peripheral neuropathic pain. The Journal of Clinical Investigation, 126(5), 1983–1997. 10.1172/JCI82859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X., Zima, A. V., Sheikh, F., Blatter, L. A., & Chen, J. (2005). Endothelin‐1‐induced arrhythmogenic Ca2+ signaling is abolished in atrial myocytes of inositol‐1,4,5‐trisphosphate (IP3)‐receptor type 2‐deficient mice. Circulation Research, 96(12), 1274–1281. 10.1161/01.RES.0000172556.05576.4c [DOI] [PubMed] [Google Scholar]
- Liauw, J., Hoang, S., Choi, M., Eroglu, C., Choi, M., Sun, G. ‐H., Percy, M., Wildman‐Tobriner, B., Bliss, T., Guzman, R. G., & Barres, B. A. (2008). Thrombospondins 1 and 2 are necessary for synaptic plasticity and functional recovery after stroke. Journal of Cerebral Blood Flow and Metabolism: Official Journal of the International Society of Cerebral Blood Flow and Metabolism, 28(10), 1722–1732. 10.1038/jcbfm.2008.65 [DOI] [PubMed] [Google Scholar]
- Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., Bennett, M. L., Münch, A. E., Chung, W. S., Peterson, T. C., Wilton, D. K., Frouin, A., Napier, B. A., Panicker, N., Kumar, M., Buckwalter, M. S., Rowitch, D. H., Dawson, V. L., Dawson, T. M., … Barres, B. A. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature, 541(7638), 481–487. 10.1038/nature21029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley, E., Stanford, S. C., Kendall, D. E., Alexander, S. P., Cirino, G., Docherty, J. R., George, C. H., Insel, P. A., Izzo, A. A., Ji, Y., Panettieri, R. A., Sobey, C. G., Stefanska, B., Stephens, G., Teixeira, M., & Ahluwalia, A. (2020). ARRIVE 2.0 and the British Journal of Pharmacology: Updated guidance for 2020. British Journal of Pharmacology, 177(16), 3611–3616. 10.1111/bph.15178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min, H., Hong, J., Cho, I. ‐H., Jang, Y. H., Lee, H., Kim, D., Yu, S. W., Lee, S., & Lee, S. J. (2015). TLR2‐induced astrocyte MMP9 activation compromises the blood brain barrier and exacerbates intracerebral hemorrhage in animal models. Molecular Brain, 10, 8–23. 10.1186/s13041-015-0116-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizee, M., Nijland, P., van der Pol, S. M., van Het Hof, B., Mebius, R., van Horseen, J., Reijerkerk, A., & de Vries, H. E. (2014). Astrocyte‐derived retinoic acid: A novel regulator of blood‐brain barrier function in multiple sclerosis. ActaNeuropathol, 128(5), 691–703. [DOI] [PubMed] [Google Scholar]
- Nagyoszi, P., Wilhelm, I., Farkas, A. E., Fazakas, C., Dung, N. T. K., Haskó, J., & Krizbai, I. A. (2010). Expression and regulation of toll‐like receptors in cerebral endothelial cells. Neurochemistry International, 57, 556–564. [DOI] [PubMed] [Google Scholar]
- Navarrete, M., Cuartero, M. I., Palenzuela, R., Draffin, J. E., Konomi, A., Serra, I., Colié, S., Castaño‐Castaño, S., Hasan, M. T., Nebreda, Á. R., & Esteban, J. A. (2019). Astrocytic p38α MAPK drives NMDA receptor‐dependent long‐term depression and modulates long‐term memory. Nature Communications, 10(2968). 10.1038/s41467-019-10830-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolakopoulou, A. M., Koeppen, J., Garcia, M., Leish, J., Obenaus, A., & Ethell, I. M. (2016). Astrocytic ephrin‐B1 regulates synapse remodeling following traumatic brain injury. ASN Neuro, 8(1), 1–18. 10.1177/1759091416630220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn, A., Kirchhoff, F., Kerr, J. N. D., & Helmchen, F. (2004). Sulforhodamine 101 as a specific marker of astroglia in the neocortex in vivo. Nature Methods, 1, 31–37. 10.1038/nmeth706 [DOI] [PubMed] [Google Scholar]
- Parada, E., Casas, A. I., Palomino‐Antolin, A., Gómez‐Rangel, V., Rubio‐Navarro, A., Farré‐Alins, V., Narros‐Fernandez, P., Guerrero‐Hue, M., Moreno, J. A., Rosa, J. M., Roda, J. M., Hernández‐García, B. J., & Egea, J. (2019). Early toll‐like receptor 4 blockade reduces ROS and inflammation triggered by microglial pro‐inflammatory phenotype in rodent and human brain ischaemia models. British Journal of Pharmacology, 176(15), 2764–2779. 10.1111/bph.14703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual, O., Ben Achour, S., Rostaing, P., Triller, A., & Bessis, A. (2012). Microglia activation triggers astrocyte‐mediated modulation of excitatory neurotransmission. Proceedings of the National Academy of Sciences of the United States of America, 109(4), E197–E205. 10.1073/pnas.1111098109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos, G., & Franklin, K. (2019). Paxinos and Franklin's the mouse brain in stereotaxic coordinates (5th ed.). San Diego: Elsevier Academic Press. [Google Scholar]
- Percie du Sert, N., Hurst, V., Ahluwalia, A., Alam, S., Avey, M. T., Baker, M., Browne, W. J., Clark, A., Cuthill, I. C., Dirnagl, U., Emerson, M., Garner, P., Holgate, S. T., Howells, D. W., Karp, N. A., Lazic, S. E., Lidster, K., MacCallum, C. J., Macleod, M., … Würbel, H. (2020). The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biology, 18(7), e3000410. 10.1371/journal.pbio.3000410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea, G., Navarrete, M., & Araque, A. (2009). Tripartite synapses: Astrocytes process and control synaptic information. Trends in Neurosciences, 32(8), 421–431. 10.1016/j.tins.2009.05.001 [DOI] [PubMed] [Google Scholar]
- Perez, E. J., Tapanes, S. A., Loris, Z. B., Balu, D. T., Sick, T. J., Coyle, J. T., & Liebl, D. J. (2017). Enhanced astrocytic d‐serine underlies synaptic damage after traumatic brain injury. The Journal of Clinical Investigation, 127(8), 3114–3125. 10.1172/JCI92300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Alvarez, A., Navarrete, M., Covelo, A., Martin, E. D., & Araque, A. (2014). Structural and functional plasticity of astrocyte processes and dendritic spine interactions. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 34, 12738–12744. 10.1523/JNEUROSCI.2401-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa, J. M., Bos, R., Sack, G. S., Fortuny, C., Agarwal, A., Bergles, D. E., Flannery, J. G., & Feller, M. B. (2015). Neuron‐glia signaling in developing retina mediated by neurotransmitter spillover. eLife, 4(eLife.09590). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa, J. M., Ruehle, S., Ding, H., & Lagnado, L. (2016). Crossover inhibition generates sustained visual responses in the inner retina. Neuron, 90, 308–319. 10.1016/j.neuron.2016.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinozaki, Y., Shibata, K., Yoshida, K., Shigetomi, E., Gachet, C., Ikenaka, K., Tanaka, K. F., & Koizumi, S. (2017). Transformation of astrocytes to a neuroprotective phenotype by microglia via P2Y1 receptor downregulation. Cell Reports, 19(6), 1151–1164. 10.1016/j.celrep.2017.04.047 [DOI] [PubMed] [Google Scholar]
- Takatsuru, Y., Eto, K., Kaneko, R., Masuda, H., Shimokawa, N., Koibuchi, N., & Nabekura, J. (2013). Critical role of the astrocyte for functional remodeling in contralateral hemisphere of somatosensory cortex after stroke. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 33(11), 4683–4692. 10.1523/JNEUROSCI.2657-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay, M.‐È., Lowery, R. L., & Majewska, A. K. (2010). Microglial interactions with synapses are modulated by visual experience. PLoS Biology, 8(11), e1000527. 10.1371/journal.pbio.1000527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villapol, S., Byrnes, K. R., & Symes, A. J. (2014). Temporal dynamics of cerebral blood flow, cortical damage, apoptosis, astrocyte‐vasculature interaction and astrogliosis in the pericontusional region after traumatic brain injury. Frontiers in Neurology, 5(82). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake, H., Moorhouse, A. J., Jinno, S., Kohsaka, S., & Nabekura, J. (2009). Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 29(13), 3974–3980. 10.1523/JNEUROSCI.4363-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, X., Shengwen, L., Wei, D., Pengjie, Y., Qian, J., Min, Z., Hu, F., & Zhang, H. (2017). TLR4 signal ablation attenuated neurological deficits by regulating microglial M1/M2 phenotype after traumatic brain injury in mice. Journal of Neuroimmunology, 310, 38–45. 10.1016/j.jneuroim.2017.06.006 [DOI] [PubMed] [Google Scholar]
- Zamanian, J. L., Xu, L., Foo, L. C., Nouri, N., Zhou, L., Giffard, R. G., & Barres, B. A. (2012). Genomic analysis of reactive astrogliosis. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 32(18), 6391–6410. 10.1523/JNEUROSCI.6221-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y., Chen, K., Sloan, S. A., Bennett, M. L., Scholze, A. R., O'Keeffe, S., Phatnani, H. P., Guarnieri, P., Caneda, C., Ruderisch, N., Deng, S., Liddelow, S. A., Zhang, C., Daneman, R., Maniatis, T., Barres, B. A., & Wu, J. Q. (2014). An RNA‐sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 34(36), 11929–11947. 10.1523/JNEUROSCI.1860-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y., Sloan, S. A., Clarke, L. E., Caneda, C., Plaza, C. A., Blumenthal, P. D., Vogel, H., Steinberg, G. K., Edwards, M. S. B., Li, G., Duncan, J. A.III, Cheshier, S. H., Shuer, L. M., Chang, E. F., Grant, G. A., Gephart, M. G. H., & Barres, B. A. (2016). Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron, 89(1), 37–53. 10.1016/j.neuron.2015.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.