

Abstract
High‐dimensional cytometry represents an exciting new era of immunology research, enabling the discovery of new cells and prediction of patient responses to therapy. A plethora of analysis and visualization tools and programs are now available for both new and experienced users; however, the transition from low‐ to high‐dimensional cytometry requires a change in the way users think about experimental design and data analysis. Data from high‐dimensional cytometry experiments are often underutilized, because of both the size of the data and the number of possible combinations of markers, as well as to a lack of understanding of the processes required to generate meaningful data. In this article, we explain the concepts behind designing high‐dimensional cytometry experiments and provide considerations for new and experienced users to design and carry out high‐dimensional experiments to maximize quality data collection.
Keywords: Analysis, experimental design, flow cytometry, high‐dimensional data, mass cytometry
High‐dimensional cytometry represents an exciting new era of immunology research; however, the transition from low‐ to high‐dimensional cytometry requires a change in the way users think about experimental design and data analysis. We explain the concepts behind designing high‐dimensional cytometry experiments and provide considerations for new and experienced users to design and carry out high‐dimensional experiments.

INTRODUCTION
Since 2010, the ability of immunologists to study high‐dimensional data has become increasingly possible. Both mass and spectral flow cytometry have led to an increase in the number of parameters that can be measured on, or in, a single cell. High‐dimensional cytometry experiments allow users to collect details of the expression of high numbers of proteins, thus providing enormous amounts of data. In contrast to conventional flow cytometry, mass cytometry couples specific antibodies to elemental isotopes, rather than fluorochromes, significantly reducing issues caused by spectral overlap and allowing for routine analysis of more than 40 parameters.1 Similarly, advances in spectral flow cytometry have enabled the measurement of up to 40 parameters per cell.2
Collectively, these new technologies have vastly increased the amount of data that can be collected in immunology experiments. While analysis software has also significantly progressed, for many immunologists, the adjustment to dealing with so much data has been difficult. The full potential of high‐dimensional data requires changes in experimental design and, in particular, analysis plans. Our conventional gating approaches are not always accurate or appropriate—for an example, an analysis of T‐cell populations in cancer 2013 (Figure 1a; low dimension) to an analysis of T‐cell populations in cancer in 2019 (Figure 1b; high dimension). There is a strong temptation to include as many parameters as possible, without consideration of how noisy or irrelevant the data could be or how significant differences between test groups will be determined.
Figure 1.
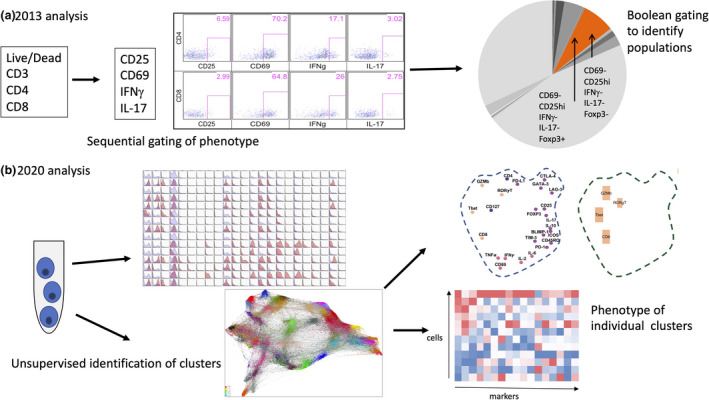
Comparison of analysis methods for low‐ and high‐dimensional cytometry data sets. (a) A typical analysis pathway with low‐dimensional cytometry performed in 2013. (b) A new analysis pathway with high‐dimensional cytometry, using the same type of samples as in a, performed in 2020. IFN, interferon; IL, interleukin; TNF, tumor necrosis factor.
This paper is designed for immunologists who are using, or plan to use, high‐dimensional cytometry data in research. The goal of this paper is to help users not only to design experiments but, more importantly, to design appropriate analysis strategies to ensure that their experiments provide high‐quality, relevant and meaningful data. These strategies include the use of clustering tools to group similar cells together for analysis, and tools that allow visualization of relationships between cells. High‐dimensional cytometry data are often underutilized, and this article provides guidelines for both new and existing users to plan experiments from design to output.
We will provide an overview of experimental considerations and then, in more detail, explain considerations for data analysis that have not been consistently required for analysis of cytometry data using small numbers of parameters. We will not provide a comprehensive overview of available software, but rather broadly explain the key concepts of high‐dimensional cytometry data analysis, illustrated with examples (Box 1).
BOX 1. I already know how to analyze cytometry data, why should I read this?
For a new user of high‐dimensional cytometry, the pathway for good analysis is shown in Figure 2. Each step is essential to maximize the collection and interpretation of high‐quality data. There are multiple traps users fall into:
(1) Poorly defined research questions
A specific research question guides the design of the experiment and also the analysis. It ensures that the appropriate number and type of parameters are chosen, including good controls. Without a specific question, users tend to incorporate as many markers as possible, overly excited by the opportunity for discovery. However, unless additional markers are well selected (e.g. known lineage markers), this approach makes it difficult to set boundaries for what is or is not a “real” population, leading to the creation of multiple potential subpopulations. If 2 of the 10 000 cells coexpress two markers, is it a new cell type?
(2) Planning experiments without incorporating biological knowledge
A preliminary gating and/or exclusion strategy is essential. To study B‐cell populations it is important to have a fool‐proof way to define both what a B cell is and what a B cell is not. Planning a serial gating or selection strategy directs the user toward the answer to the research question, rather than down a rabbit hole of irrelevant or biologically impossible results.
(3) Failing to adjust experimental design for multiple types of experiments
With high‐dimensional cytometry, there is scope to combine all previous experimental panels, for example, measurement of surface proteins, intracellular cytokines and phosphorylated proteins. However, it is important to remember that expression, stimulation, preservation and regulation of different proteins can all vary, and a one‐size‐fits‐all approach can be problematic.
Defining a research question
As for all research, defining a research question drives both experimental design and data analysis. This is particularly true for high‐dimensional experiments—the lure of multiple parameters can override experimental common sense. Extraneous markers can lead to nonsense data (have all markers been validated in the target population?) or noisy data (are all markers relevant?). An example may be the inclusion of a dendritic cell activation marker to a T‐cell‐based panel. The key to successful high‐dimensional research is the design of both an experimental and analysis plan to maximize the usefulness of data, and this plan must be driven by a research question (Figure 2). This is, of course, sensible advise for all experiments, but the potential downstream impact of poor design in high‐dimensional experiments is exponentially greater, especially at the analysis stage. As a first step, it is important to have a clear idea of the question or questions that are to be addressed, and to have a solid understanding of the biology of the experimental system, especially for exploratory research. There are many research questions that might motivate researchers to use high‐dimensional cytometry, which may be quite distinct from using traditional lower‐dimensional cytometry, and these feed into differences in experimental design and analysis.
Figure 2.
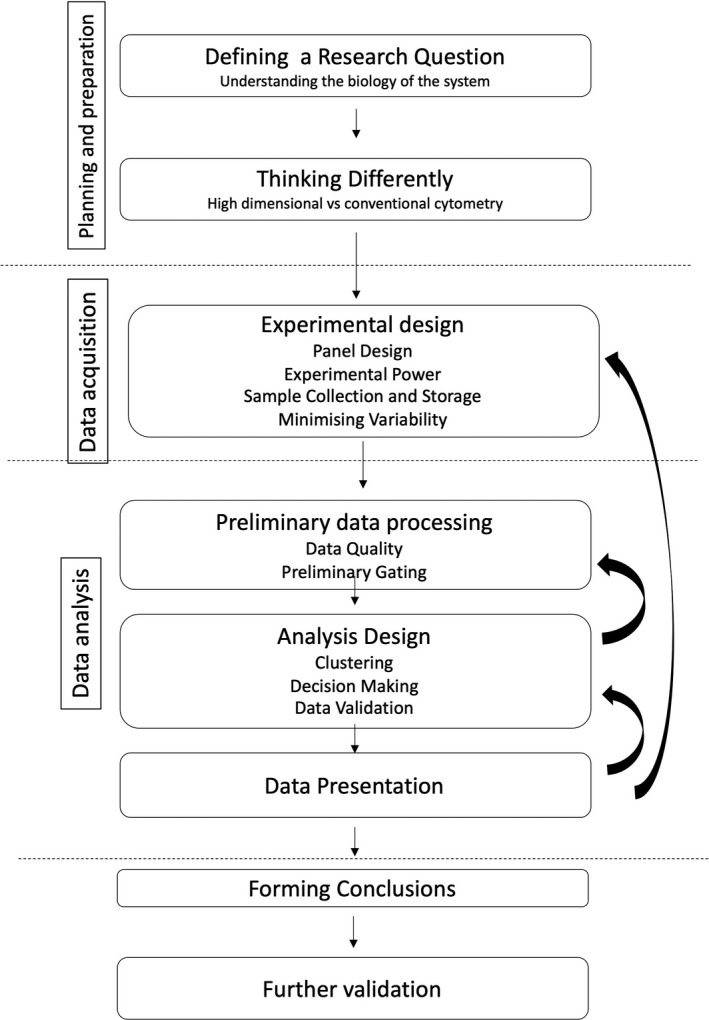
Planning a high‐dimensional cytometry experiment: steps involved in both experimental and analysis design.
Among others, some strengths of high‐dimensional cytometry include the ability to
examine the characteristics of a large number of cell subsets within a single sample
investigate heterogeneity within a known cell population
identify changes in cellular populations that correlate with clinical states
assess the similarity of target cell populations with known reference populations
determine changes in the physiological state of cell populations
identify intermediate states or branch points in developmental pathways
We have reviewed the now large number of research papers using high‐dimensional cytometry for immunology studies, and found that research questions can be broadly divided into four categories: (1) measurement of phenotype and proportion of cells of interest, for example, quantifying tumor infiltrating T cells in cancer3 (2) discovery of new, possibly rare, cells, for example, innate lymphoid cell subpopulations in infection4; (3) comparisons of data sets, for example, cancer patients treated with immune checkpoint inhibitors5 and (4) developmental trajectory analysis.6
While the details of individual high‐dimensional experiments are as diverse as the scientific questions underlying them, some general principles can help guide design. As with most experiments, a typical goal of high‐dimensional cytometry studies is to determine the effect of an intervention (e.g. drug treatment, gene deletion, infection) on measurable readouts such as the absolute number or proportion of a specific cell type, or levels of marker expression. With these in mind, we will discuss experimental design as well as analysis planning strategies applicable and/or appropriate for all these research questions.
STEP ONE: GETTING USED TO THINKING DIFFERENTLY
High‐dimensional cytometry is not conventional cytometry with extra spaces. It requires a change in thinking from the researcher to use it correctly and to its full potential—in a large parameter panel, serial gating of more than 40 markers is not practical or easily translatable into results. Our experience is that a change in thinking is essential to help researchers avoid the temptation of charging into a badly designed experiment that can waste time, money and, potentially, samples. Many projects still benefit from multiple conventional flow cytometry panels, or simply do not require multiple parameters, such as a clearly defined research question. Consider the following scenario: mucosal associated invariant T cells (MAIT cells) have been defined in the literature as CD8+CD161+Vα7.2+.7 A low‐dimensional study analyzing the frequency of MAIT cells in two groups could incorporate additional phenotypic or functional markers to compare the cells found in both groups. A high‐dimensional study analyzing the phenotype of MAIT cells could incorporate a large number of phenotypic and functional markers and use analysis tools to identify multiple MAIT cell populations, possibly with new combinations of phenotype or function, and to determine the relative frequency of these potentially rare populations between the two groups. Conversely, high‐dimensional experiments are often exploratory, an approach which is difficult to reconcile with the traditional training of formulating a specific hypothesis and locking in analyses and plans for statistical testing ahead of time. Instead, high‐dimensional experiments are often used as hypothesis‐generating data, and these newly developed hypotheses are then tested in independent experiments. This approach to science can be difficult for immunologists to accept, because traditional immunology has had a targeted approach, although in reality, high‐dimensional immunology is no different from genome‐wide association studies or studies sequencing large populations of microbes. Poor quality of low‐dimensional data can be worked around with manual gating on simple populations; however, achieving high‐quality data is essential to fully explore the potential results and hypotheses that arise from a high‐dimensional experiment.
STEP TWO: EXPERIMENTAL DESIGN
Defining a research question
Experimental design does not take place in isolation, but instead is part of an integrated approach, incorporating factors such as panel design and data analysis methods at a very early stage of planning, and must be driven by a research question or theme. Thinking ahead to data analysis is, of course, important during the planning of any experiment, but high‐dimensional approaches often have requirements that are distinct from typical low‐dimensional approaches with which most researchers may be familiar—for example, phenotyping cells can be done by looking at all markers simultaneously, using clusters of “like” cells, rather than studying the expression of each marker one at a time on a variety of cell types. A further level of complication is that practical considerations such as the number and types of samples available, or sample collection and storage methods, may constrain what can be achieved.
Changing the thinking about panel design
Time invested in the development of a high‐dimensional panel can ultimately determine the success or failure of a study, so it is important to allow sufficient resources to carefully and systematically develop one that meshes with downstream analysis approaches. In addition, the careful balancing of panel design considerations over a large number of channels means that in contrast to lower‐dimensional cytometry, high‐dimensional panels are typically more difficult to change in an ad hoc manner between experiments. The key factors that need to be considered in the panel design process will already be familiar to researchers who have experience with low‐dimensional cytometry. These include antigen density, interchannel signal spillover as well as differential sensitivity of individual channels. The principles of panel design are well described in the literature,8, 9, 10 so will not be discussed in detail here.
A first consideration is to include sufficient and appropriate markers to identify all major subsets in the sample, or at least enough markers to exclude them from analysis. While manual gating generally requires clear separation of populations on bivariate plots, high‐dimensional analysis techniques effectively integrate signals from many dimensions simultaneously. Markers may not discriminate populations by themselves but can still collectively contribute to separation of populations. Thus, the decision to add more markers must include a good justification for their inclusion and a strategy to remove them from analysis steps if necessary.
With a modular panel strategy, a restricted set of antibodies can be used for core functions, typically broad immunophenotyping and identification of major populations, and different sets of antibodies can be dropped into the remaining channels depending on specific requirements. Such requirements might include detailed phenotyping of individual lineages, such as regulatory T cells in colorectal cancer,3 or investigation of physiological changes within populations, such as measurement of phosphorylated signaling proteins in response to drug treatments.11
The degree of modularity that is chosen largely depends on long‐term practical plans as much as specific experimental questions. For example, if the aim is to develop a panel that comprehensively identifies all immune cell types in multiple tissues, then these may use most or all of the available channels with minimal thought to leaving free channels that can be used for other purposes. Alternatively, if the strategy is to tailor high‐dimensional cytometry to multiple models and research questions, then it may be more useful to use a smaller panel core that can be added to depending on the needs of individual experiments.
The difficulty with exploratory analyses
Given the exploratory nature of high‐dimensional cytometry experiments, researchers may uncover new or novel outcomes that were not part of the original study design, and there is often still a question as to whether the results are “real” or not, for example, the identification of CD25+ myeloid cell population in colorectal cancer.12 With many populations measured simultaneously, apparent changes in abundance of a numerically minor population may reflect an interesting biological phenomenon; however, the changes could also represent a statistical artifact. A difference in population frequency between two groups can be significantly influenced by changes in the number and selection of parameters included in the analysis. Alternatively, because of the increased numbers of markers included in high‐dimensional panels, antigens may be found on populations that, according to traditional thinking, should not express them. Ideally, these findings should then be verified using independent methodologies and/or data sets, particularly if they are puzzling or unexpected. One useful approach, if collection of samples is not a limiting factor, is to split total experimental subjects into two separate cohorts: one exploratory and the other for confirmation. Any changes found in the exploratory cohort can be confirmed in the (typically smaller) confirmation cohort, using a more focused panel. This obviously requires a decision at the experimental design stage, or the collection of a completely new set of samples. Unexpected antigen expression can be confirmed using a range of methods on reserved sample aliquots in focused experimental repeats. These could include incorporation of extra staining controls (e.g. fluorescence‐minus‐one or isoclonic controls), different antibody clones/fluorochromes or different technologies such as sorting for functional studies or microscopic approaches such as imaging flow cytometry to examine staining patterns (for example, to identify doublets or binding of membrane fragments). Finally, it is important to be able to use the same acquisition and analysis strategies in multiple experiments by the same user and for similar experiments by other users. Reporting of analysis parameters is not only good bookkeeping, but speaks to transparency and reproducibility which are essential for promoting good science.
The importance of determining experimental power
A critical step during the planning stage is to ensure that the experiment is likely to have adequate statistical power. In cytometry experiments, the process of estimating statistical power differs subtly but importantly from many other technology platforms as a result of errors arising during sampling because the number of events per population (or cluster node) of interest may only contain small numbers of cells. Under these conditions, Poisson statistics describe the relationship between the frequency of a population of interest and the number of cells required for a given level of precision of a measurement of that cell population. In turn, this dictates the number of total events from a sample that need to be acquired with larger cell numbers in a population leading to greater ability to determine differences between experimental groups.13, 14, 15 Key parameters influencing power that are under control of the investigator are group sample size and the number of events collected per sample (an important practical note here is to incorporate cell losses during processing, acquisition and preliminary gating). By contrast, parameters that are outside the control of the investigator, but which should be incorporated when estimating power, include the relative abundance of the rarest population of interest, between‐subject variability of populations (either abundance or staining intensity) and intrapopulation marker staining variability (if testing for effects on marker expression). In practice, these can often be difficult to determine but can be estimated either from prior experience with the biological system, in pilot experiments,16 or by examining published studies. An integrated model of the effect of group size, population variance and cell number on statistical power for single‐cell experiments has recently been described, and a web interface is available.17
The impact of sample collection and storage on experiment quality
The practicalities of sample collection and processing can have important effects on outcomes and need to be balanced carefully against experimental considerations. For example, cultured cells, peripheral blood or bone marrow samples are easily processed with minimal cell loss, whereas preparations from solid tissues risk loss of antigens during enzymatic digestion and loss of important cell populations.18 Solid tissue preparations may also be prone to clogging by instrument fluidics, and the extra processing time needed can make batching of large numbers of samples difficult. Rapid processing on collection, followed by immediate staining and analysis on the cytometer is generally ideal and may be possible for in vitro or animal‐based studies. Examples include in vitro studies of the effects of drugs on apoptotic pathways19 and animal studies investigating cellular heterogeneity within populations.20 In many cases, however, rapid processing is not possible, and some form of storage is needed so that sample processing and data collection can be both batched and matched to instrument availability. Similarly, freezing of samples after staining is possible if, for example, the instrument is at another site, or has a catastrophic issue and is unavailable (Box 2).21
BOX 2. Sample considerations: peripheral blood mononuclear cells as an example.
The balancing procedure between experimental and practical considerations is most clearly illustrated by the use of peripheral blood mononuclear cells (PBMCs) in human clinical studies, particularly for the study of lymphocyte populations.
Consideration 1: cryopreservation
For many clinical studies, the preparation and freezing of PBMCs are typically performed. When cryopreserving peripheral blood and cells such as tumor‐infiltrating lymphocytes, it is important to consider the viability of immune cells after defrosting. It is recommended to optimize the cryopreservation process and media to obtain maximum cell viability for all populations; this may include standardization of freezing and defrosting processes (including blood storage temperature) and timeframes, use of commercial freezing media and always counting live cells before and after preservation.21
Consideration 2: cell populations
Initial processing and storage of whole blood may be simpler than with PBMCs, but unless granulocytes are of specific interest, the benefits may be outweighed by the increased instrument acquisition time caused by granulocyte burden, given they are the major white blood cell component in the blood.
Consideration 3: practicality
Importantly, blood samples may often be collected in the field under less‐than‐ideal conditions, or by busy health‐care staff whose main priority is their patient. In these situations, ideal sample processing may need to be traded for speed and simplicity, and a number of cell preservation systems are commercially available to aid this. These systems may only be suitable for whole blood, and because they contain fixatives, they can affect the staining of certain antigens, particularly chemokine receptors.22 The increasing availability of new fixatives and reagents for preserving whole blood may resolve some of these issues, as well as the availability of freeze‐dried antibody cocktails for real‐time staining of blood in the field.
The use of batching and barcoding to minimize variability
An important factor leading to the success of high‐dimensional experiments is minimization of technical variability and artifact creation. Minimization of technical variability decreases the chance of false results arising from, for example, batch effects, and maximizes the likelihood of detecting real differences between experimental groups. There are many small adjustments to experimental procedures that can be performed which help minimize technical variability, from steps including sample collection and processing, staining to acquisition on the cytometer,23 and only those with particular relevance to high‐dimensional cytometry studies are presented here. Key among these is the concept of batching. For small‐scale experiments, all samples can often be processed, and data are acquired in a single run or batch. Running as a single batch minimizes the variability associated with sample preparation and staining, as well as with day‐to‐day variation in instrument responsiveness. The use of cell barcoding approaches further decreases intrabatch variability.24 With barcoding, the expansion of available channels with high‐dimensional instruments allows several (typically up to six) channels to be dedicated to combinations—or barcodes—that identify individual samples (e.g. anti‐CD45 antibodies labeled with In‐115 or Bi‐209 to barcode two individual donors). Once barcoded, samples are pooled together for staining and data acquisition on the cytometer, before the pooled data are deconvolved in silico into their original sample identities. Barcoding has been implemented in both flow cytometry25, 26 and mass cytometry.27, 28, 29
The difficulty of large studies and data sets
Many experiments—typically clinical studies—are simply too large for samples to be processed and run as a single batch, or even at a single site, and require additional measures to decrease the associated variability. The challenges associated with very large‐scale studies, such as the Human Immunology Project, have been described by multiple groups and a number of steps can be taken to improve data quality.30, 31 In particular, use of either lyophilized (flow cytometry)31, 32 or frozen (mass cytometry)33 aliquots of antibody cocktails has been shown to decrease batch‐related variability. A key approach that has been widely used in omics studies, which is applicable to cytometry, is to minimize the influence of confounding factors by using a balanced design, where experimental groups are distributed across batches.34, 35 In its most extreme form, it may be possible to use randomized complete block design approaches,36, 37 in which similar numbers of samples from experimental groups are randomly assigned to each run batch. Despite best efforts, some degree of batch‐associated variability inevitably occurs in multiday or multisite studies, and computational approaches have recently been described that can minimize the impact. These approaches typically use repeated measures of a control sample that can be stained and analyzed as part of each batch run and normalized (Box 3).38, 39
BOX 3. Statistical analyses.
A detailed discussion of how to plan appropriate statistical analyses is beyond the scope of this article, but readers are referred to a useful review by Skinner.40 The use of statistical tests is dependent on both the research question and the user’s data set. However, it is vital to consider statistical approaches that allow meaningful interpretation of the data and that enhance the validity and utility of the results. Furthermore, it is never too early to consider what statistical test(s) should be used for a given data set, even before the first sample data are acquired.
Preliminary data processing—assessing data quality and reliability
Perhaps one of the most important steps in analysis is the initial assessment of data quality. Three of the most common factors that influence data quality are (1) technical issues during acquisition, (2) debris in the sample and (3) staining quality.
Technical issues during acquisition. Procedural or instrument technical issues during the data acquisition process can have the potential to introduce considerable variation in the data. For example, clogs and back‐pressure issues on a fluorescent flow cytometer affect the sheath flow rate, impacting the time to travel between lasers. As a result, the signals are no longer matched by the delay electronics and the resulting data, when viewed relative to time, are shown as an unstable signal. Similarly, across all platforms, clogs reduce detection of events, which again proves evident when viewing the time parameter as a histogram. Filtering samples, as close as possible to the time of acquisition, is a very sensible idea.
Debris in the sample. Does the data file look like it has a lot of debris (indicative in flow cytometry as forward and side scatter low, and in mass cytometry as DNA low/negative)? This may be a reflection on poor sample quality to begin with or mishandling during experimental procedures. This should flag that subsequent analysis may be subpar and potentially misleading.
Staining quality. Data must be assessed for quality: does the staining look like it worked (strong signal, consistent with previous experiments or published profiles)? Is there good separation between populations of cells positive for the marker of interest, with minimal background on populations which should be negative for the marker? While there are new computational approaches to help identify data quality, for example, FlowAI,41 they cannot completely replace a human eye and experience to manually check anything that appears unusual.
Preprocessing data—getting from the cytometer to the plots
Once initial data quality has been assessed, it is often necessary to export the events of interest for further analysis to a new .fcs file. This is an integral part of the process of adapting raw data to data ready for analysis and is referred to as “preprocessing.” Preprocessing may be as simple as excluding any internal control beads (such as EQ beads in the case of mass cytometry) or identifying live single events (achieved using a viability dye and singlet length/width relationships of scatter parameters for flow cytometry or DNA and event length parameters for mass cytometry). Further gating to specific populations of interest (such as T cells by gating on CD3+ events) may also be implemented if the downstream analysis aims only to focus on a defined population, and it is useful at this stage to debarcode events.
Most cytometry analyses include a scaling step to allow for comparison between multiple parameters that exist in different orders of magnitude. Scaling not only allows for direct comparison between markers, but normalizing data to a comparable dimension also ensures that all parameters are weighted equally. This may be a problem in immunology, as there is an argument that not all markers should be weighted equally.
The method of scaling often varies between analyses. Some analyses transform the raw values to bring them within a similar range—while inverse hyperbolic sine (Arcsinh) transformation is widely used as a viable “default,” it is best to keep both the underlying biology of the sample and the research question in mind and consider other options too. Other analyses use relative scaling that scale each parameter individually and give a range of low to high within each parameter (such as min–max normalization). Both of these approaches are suitable when there is a full range of expression for each parameter in the data set. However, “artificial” highs and lows can be created if a parameter is expressed by all cells or by no cells (https://github.com/ImmuneDynamics/Spectre42). Without additional validation (usually manual gating), false findings may not be recognized.
It is important to realize the importance of decisions made in these preprocessing steps, as implications are carried through all subsequent analyses. For example, exclusion of doublets might actually obscure meaningful biology to your individual research scenario, as is the case for platelets associated with monocytes.43
STEP THREE: ANALYSIS DESIGN
The analysis of high‐dimensional data must involve a strategy that incorporates the research question and the experimental design. Using one or more different tools, always in combination with the expert understanding of investigators, ultimately allows the findings to be presented in a clear and statistically valid way. Multiparameter data sets cannot be fully analyzed using a conventional approach focused on assessing one marker at a time (e.g. serial gating or Boolean gating). Applied to a high‐dimensional data set, and depending on the expert knowledge of the user, this approach can be cumbersome and insensitive as the user moves through gates using a binary inclusion–exclusion model to identify populations based only on one or two markers. Once the dimensionality of the data increases (in this case, each dimension is the expression of a selected surface marker in the panel) many more potential populations can be identified that need to be investigated and included in the analysis—our low‐dimensional‐capable brains need extra help in deciphering patterns. Doing this in any fully manual way becomes impossible at a certain point and algorithmic approaches become extremely useful (Box 4).
BOX 4. Frequently asked questions for new users.
Time. Do not underestimate time and effort of analysis (the general rule is 20% of time spent on experimental design and 80% on analysis design).
Expertise. Who will do the analysis? Are the skills available in‐house or by collaboration? Is it possible to have a dedicated student or postdoc who is motivated and has time to learn analysis approaches? Can analysis be outsourced to a bioinformatics group or an external provider? Is the available bioinformatics support specialized in cytometry data (which is very different from sequencing data)?
Tools. What tools and software are needed and are they available? Analysis packages are constantly updated, but there are many standalone programs, with in‐built and R plugins, web interfaces and R‐based pipelines and integrated packages.
Data management. For many users, high‐dimensional analysis can require multiple steps in multiple platforms, especially if they do not have substantial skills in R. It is vital to have an organized structure, including storage of data in a standardized format, and record keeping plan to keep track of all of the analysis steps.
It is necessary to cover some basic concepts in high‐dimensional data analysis and how it can differ from approaches to low‐dimensional data analysis. The first important concepts are dimensionality reduction and clustering, and these are inter‐related ideas. Dimensionality reduction algorithms reduce the appearance of complexity by creating an artificial space that achieves spread across the data attributable to parameters of greatest variance. Large amounts of numerical cytometric data can then be represented on a single two‐dimensional plot where individual cells can be seen. Because the cells are spread out on the greatest axes of diversity, similar cells tend to group together and their relationship to other cells in the experiment can then be easily visualized (although a limitation of dimensionality reduction is that distances are not always well‐preserved in low dimensions). Expression patterns of markers can be colored to identify populations manually, and multiple plots can be presented to illustrate population dynamics—for example, general changes in patterns of cell types, or marker expression, because of an experimental treatment. Thus, dimensionality reduction is a mechanism to improve visualization of data, allowing the user to see both common and rare cells at the single‐cell level. Summaries of the data (e.g. average expression of a cluster) can also be useful for overall visualization of the entire data set.
Clustering algorithms reduce complexity by combining cells into groups (i.e. clusters) in which the individual cells are similar in high‐dimensional space and can therefore be treated similarly for computational purposes. Cells can then be studied as groups with varying levels of relatedness to each other and relationships between clusters can be visualized by methods such as minimum‐spanning trees or force‐directed layouts (see the “Data analysis presentation—convincing others your data are real” section). Clustering and other similar categorization approaches also help with statistical analysis because the data from the clusters themselves, once validated by expert inspection, can be used as individual groups. This can take the form of such parameters as median marker level expression or population size, and can be fed into automated pipelines, algorithms or visualization plots that assist with identifying significant differences between treatments. One widely used presentation approach is to perform clustering and dimensionality reduction in parallel to map cluster identities, using color, onto a dimensionality reduction plot. This allows identification of populations through clustering, then presentation of the data at the single‐cell level using dimensionality reduction.
The second important concept is cyclical analysis. The nature of high‐dimensional data makes it impractical to assess every possible combination of markers. More importantly, it is impossible to incorporate and understand every potential outcome prior to the analysis—it cannot be determined whether a rare population justifies future investigation if the rare population has never been described. For many new users, this cyclical analysis approach may appear to be “milking” the data to get a desired result and could also introduce confirmation bias. In fact, an iterative approach actually improves the data quality. Traditionally, with low‐dimensional data, the use of sequential biaxial plots allows the user to obtain a quick overview of the basics of the data—event number, major populations or any distinct differences between data sets. However, with high‐dimensional data, many of the important findings are hidden, either by virtue of being a rare population or by having a complex phenotype. Validation by conventional manual gating is always recommended. Revisiting and replotting high‐dimensional data multiple times and in multiple ways refine the interpretation of the data, ultimately improving the quality. It is very easy to continue down this path indefinitely, so when interpreting the results, it is best to approach them with the research question in mind and focus on the best means to answer the desired question.
How to understand clusters
To attempt to answer the research question, there are many platforms available to researchers, covered elsewhere.44, 45, 46 However, the most common first decision is how to look at everything at once.
Clustering involves grouping cells together based on their phenotype and can be useful for identifying major cell subsets. Phenotypic markers (such as lineage markers) that identify cell types of interest are included in the staining panel, and only these are typically selected as clustering parameters. However, the use of lineage‐specific markers is very experiment dependent; some decisions may result in loss of novel or rare populations. A number of clustering tools are available, and each has their own advantages and disadvantages depending on the context of the data (for examples of appropriate clustering tools for different types of experiments, see Weber and Robinson47 and Liu et al.48). The result of the clustering process is that each cell is assigned to a group. These groups of cells usually represent biologically meaningful populations, for example, T cells (CD3+CD19–) or B cells (CD19+CD3–). Clustering can be unsupervised, whereby the computer algorithm determines similarity between cell types based on coexpression of molecules; supervised, where the user can guide the algorithm by creating subgroups first (e.g. all CD3+ events are T cells), or somewhere in between. Clustering can be useful for quick identification of subsets, which can then be easily quantified and compared between groups. Clustering is, therefore, more prone to batch and experimental variability, and therefore, the resulting clusters need to be validated (most commonly achieved with manual gating). A limitation of most analysis pipelines is that the default readout is the median expression of a marker within clusters, when in reality, there is a spread of expression, for example, a mixture of cells both high and low/negative would be averaged to report an average intensity—this would only be realized with validation, a process that always depends on the level of expert biological knowledge.
Interpreting cluster visualizations
The two most common readouts of clusters are abundance of individual clusters between samples and the cluster phenotype. There are several visualization tools that can achieve this; for a summary, see Saeys et al.49 This approach provides information on variability as well as the heterogeneity in cells between samples. Force‐directed visualizations, for example, present each cell as an individual dot that is then colored by the cluster to which that cell belongs. It also provides information on the heterogeneity of each cluster—tight clusters represent more homogenous populations than loose clusters. The nature of these visualizations means that the user can also observe how the clusters group into meta‐clusters. For example, three populations of activated CD4+ T cells may all group together distantly from regulatory T cell populations, but all lie within the larger group of CD4+ T‐cell clusters.
Overclustering and underclustering
Overclustering occurs when true single populations are coerced into multiple clusters by the cluster analysis algorithm. The result of overclustering is the possible appearance of more populations than are predicted to actually exist within the data set. Figure 3 shows an example of the same data set arranged into different numbers of clusters—low to high. Determining which is the correct cluster number to use ultimately resides with the researchers and their knowledge of both the biology and the data. Underclustering occurs when single populations are merged that are clearly mixed populations, for example, if CD4+ and CD8+ T cells were contained within a single cluster. These problems reinforce the need for the user to interrogate the data, rather than assuming an algorithm knows what immune cells are and how to meaningfully differentiate cells based on marker expression values. This interrogation includes cyclical analysis, exploration of variance and validation of findings with manual gating.
Figure 3.
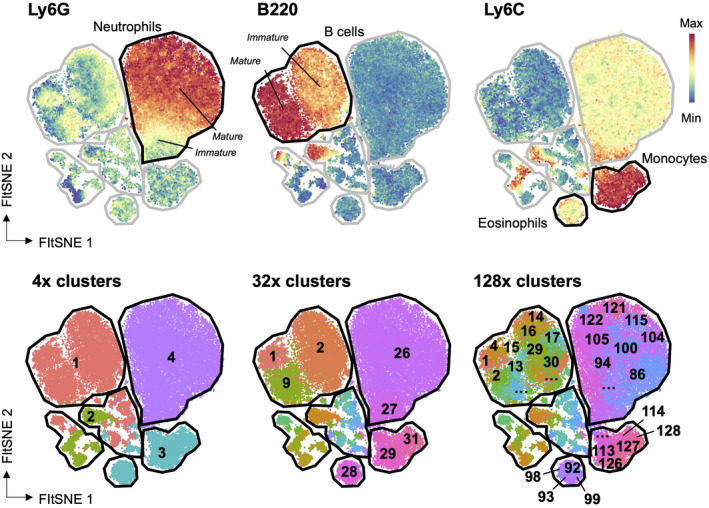
Overclustering and underclustering. Mouse hematopoietic cells were analyzed by dimensionality reduction and clustering. Expression of individual markers identifying populations is shown in the top row on a dimensionality reduction plot, and in the bottom row clusters are projected onto the same plot. Use of too few clusters (the left panel) fails to identify real populations, whereas too many clusters (the right panel) leads to identification of heterogeneity that is not biologically relevant.
Overclustering and underclustering can falsely demonstrate heterogeneity in the data and can lead to problems with downstream analyses. Direct comparisons between individual clusters may not be representative of actual changes in the population, if each cluster only represents a fraction of a true population. Analyses typically include cluster number validation steps, such as elbow point validation or cluster silhouette index (see Glossary) that attempt to predict the actual cluster number and avoid overclustering. Other approaches allow the user to define the number of clusters, and this often leads to overclustering. For example, the clustering algorithm FlowSOM, by default, intentionally overclusters by using a 10 × 10 self‐organizing map, thereby creating 100 clusters.50
Downsampling
Downsampling is commonly used for high‐dimensional data analysis to reduce the necessary computing power required for these complex approaches. It can also counter the problem of “saturation” in dimensionality reduction plots, when the visualization of data is impaired because there are too many cells. Downsampling usually involves random sampling from the initial data set, for example, using 10% of the original sample. However, it is possible that the downsampling includes an unknown bias, for example, overselection of cells from a diseased cohort, rather than an equal selection of cells from the diseased and healthy cohorts. One approach is to deliberately overcluster and then downsample so that cells are sampled from each cluster. Consider the research question and biology (How many subsets are expected to be identified? Does each sample contain the same number of cells?) when deciding the clustering approach.
What to look for and why?
Just as the analysis of high‐dimensional data is cyclical, so too is the experimental design. To determine what to look for, the original panel needs to include all the parameters that will be needed to make the analysis decisions. Starting out with an important or large experiment is not advised—practicing the entire process from experimental design to analysis and data readouts will reveal the cyclical process of developing a meaningful experiment.
A large number of parameters are needed to inform the analysis process: Which markers determine clusters (CD3, CD19)? Which markers are coexpressed and validate phenotypes (FOXP3, CD25)? If detecting new biomarkers is the primary goal, using markers that best discriminate between samples can be more valuable than visualizing differences in all possible populations. As always, the research question informs which markers are the most valuable, taking into account variance between samples.
Validating the data—how to take back control from the computer overlords
It is essential to validate findings generated using analysis approaches that involve data manipulation. The best approach is to return to the raw data in cytometry platforms, such as FlowJo, and verify that expression values are above background and that the predicted populations can be identified using conventional manual gating.
The most informative visualization is to simply show traditional gating strategies of specific marker expression on a population of interest. This is appropriate to answer very basic questions such as “how does expression of cytokine x change on population y between treatment groups?”. These analyses can usually be done using conventional cytometry and substantial amounts of information from the high‐dimensional data may be missed. These simple analyses can still be incorporated into high‐dimensional data, for example, showing expression on a newly discovered cluster of cells, rather than a predetermined population. Conversely, high‐dimensional analyses can also be performed on previously acquired data (e.g. on 7–14‐parameter panels), and many software programs have incorporated commonly used algorithms to allow this, without requiring the user to have extensive coding skills (Box 5).
BOX 5. Classifiers.
One downside to algorithms is their reliance on a complete data set: that is, if more data are generated later, the algorithms must be rerun to include everything. This in turn will provide slightly different results, such as a change in cluster number or different dimensionality reduction plots. However, with classifiers, it is possible to assign new data sets to previously acquired results without having to rerun all algorithms.
Classifiers “classify” cells and add them to preassigned groups (such as subsets or clusters). Commonly used classifiers include k‐nearest neighbor and decision trees/random forests.51 A set of user‐selected training data (such as data that have cluster numbers assigned) is provided to generate “rules” that the classifier can use to differentiate between these cells. For example, a decision tree defines a set of rules that the user can follow based on a previous decision. Instead of simultaneously assessing all available parameters in high dimensions, this method works by examining binary positive and negative staining patterns in a cascading way, allowing the placement of individual cells into groups. Random forests are a consensus made up of multiple decision trees. In contrast to decision trees, the most relevant output from a random forest is a plot that scores the value of each marker individually on its ability to discriminate the data set.
Once training is complete, the test data (such as newly acquired data) are assigned to each group based on these rules. In cytometry data, classifiers assign cells to groups that share a similar phenotype.52 This grouping can be greatly affected by batch/experimental variability, so being mindful of this, by checking that data sets match up before undertaking, would be advised. However, it is still a faster approach because the trained classifier is run on only the newly acquired data, rather than running an algorithm across both old and new data.
Data analysis presentation—convincing others your data are real
The last step in the analysis pathway is planning data presentation. Considering the data in terms of the broad research categories from the Introduction: (1) measurement of proportion and phenotype of cells of interest; (2) discovery of new, possibly rare, cells; (3) comparisons of data sets and (4) developmental trajectories; these categories can inform decisions on how to share the information. Returning to the individual research question is essential. Showing multiple plots of everything that has been collected obfuscates the findings, confuses readers and may also imply incorrect interpretation of the results.
There are several options available to communicate the results of the scientific study, including conventional heat maps, or Brick plots,12 and these have been reviewed elsewhere.45 Instead, this section focuses on the tools used to analyze the data and present this analysis to others.
Principal component analysis
Principal component analysis (PCA) is a linear dimensionality reduction algorithm. A PCA is capable of identifying parameters that contribute a large amount of variance across a given data set. Because a PCA is extremely fast, it can be useful to determine marker selection for further analyses, which assists in removing markers that do not provide large variation to the data. For cytometry, this can include the identification of markers that may differentiate between a large number of subsets. Downstream calculations, therefore, do not require as much computing power.
Once a PCA has reduced the number of dimensions based on their variance, it is possible to test whether the variance between samples (such as individual patients) can explain the difference between groups (such as healthy versus disease). This makes it possible to identify the parameters (such as cluster or marker expression) that contribute to the variance and in turn provide differentiation between groups. For example, Lugli et al.53 used clustering and PCA to assist in the differentiation of age groups based on an individual’s T‐cell compartment.
tSNE/viSNE and UMAP
T‐distributed stochastic neighbor embedding (tSNE)/viSNE and uniform manifold approximation and projection (UMAP) are nonlinear dimensionality reduction algorithms that are commonly used to visualize the heterogeneity of cell subsets across a data set.54, 55 Dimensionality reduction algorithms maintain cells independently of each other (rather than forming clusters or groups). They distinguish between subsets and highlight changes that may occur across groups, but their appearance can be misleading: a subset of highly homogenous cells group very tightly together on a plot, while a more heterogenous subset of cells may appear to be a larger population but does not necessarily make up the majority of cells on the plot. Using density plots can clarify these differences. As a reader, it is important to know which markers were used to generate the plots, as well as the overall distribution of cells. Coloring the plots by cluster can help verify a clustering algorithm. These types of plots should only be used for data visualization.
Minimum spanning tree
Minimum spanning trees (MSTs) or variants of this such as spanning‐tree progression analysis for density‐normalized events (SPADE)56 provide a simple overview of cluster relationships, that is, how related each cluster is to another. This approach can be useful for an indication of the number of populations within a lineage of cells, for example. A limitation of MSTs is that the cluster relationships may not be meaningful without some biological context. This issue highlights the need for validation and cyclical analysis. Different MST tools have different limitations; for example, in SPADE, nodes that are close to each other are related but nodes that are distant from each other are not necessarily unrelated. Similar to UMAP/tSNE, MST plots are most useful for visualizing the data set.
Force‐directed plots
Force‐directed plots, such as single‐cell analysis by fixed force‐ and landmark‐directed (SCAFFoLD),57 arrange the data using “forces” that are created by the user based on existing biological data. Force‐directed plots are, therefore, more useful for analyzing single cells than clusters. Cytometry data are represented in two dimensions, using nodes, representing markers (e.g. cell type) and edges (lines), representing “relatedness” of nodes. Nodes repel each other and edges attract each other; therefore, the plot accurately reflects relatedness of populations. The result is that similar data points eventually reside in close proximity, whereas dissimilar data points are distant.
A useful analogy to integrate all of these approaches can be as simple as planning a meal. Opening the fridge allows you to see everything that you have (UMAP/tSNE). Organizing the fridge allows you to group similar items together, for example, meat, vegetables, dairy (clustering and MST/force directed plots). Quantifying how much of each food type you have and whether you have enough variety to make a meal requires a detailed analysis of each cluster (Box 6).
BOX 6. Potential pitfalls for new (and experienced) users.
Inadequate controls
A serious problem that comes with revisiting existing data is the lack of relevant controls for the current analyses. The importance of proper experimental design (including the use of appropriate controls) has been discussed in the literature,23 but how can this be achieved for analytical techniques that were not known during the preparation stage? It is even more important in these situations to make sure the data are critically assessed for usability, particularly if they are lacking adequate controls. Data can still be run, but it is important to understand the limitations of the existing data set when interpreting the results, such as a lack of batch controls affecting the resulting computations.
Data quality
The reason humans are better at pattern recognition than computers is because computers are much more sensitive and precise when considering data. To humans, cells that are similar (but not identical) are considered the same. Batch variability (from human, experimental or instrumental error) therefore becomes a serious issue when using automated analyses. Humans can control for these errors through checks at each point of analysis (e.g. using manual gating to shift a gate if a signal dropped between experimental runs). There are algorithms available that are capable of normalizing data (either between samples or experimental batches), but the resulting data or conclusion must be confirmed in a suitable manner (such as inspecting with manual gating).
Inconsistency in human versus computer analyses
Humans are at best capable of thinking in three dimensions, yet we commonly settle for two‐dimensional plots to represent data. This greatly limits the comprehension of data in more than two dimensions (all flow cytometry data will include forward‐ and side‐scatter parameters). Gating through 10 two‐dimensional plots is not equivalent to computing in 20 dimensions: it is still only two dimensions. When using algorithms that compute in high‐dimensional space, it is easier to find differences between populations of cells, for the same reasons two‐dimensional plots can provide clearer separation of subsets than one‐dimensional histograms, although this does not guarantee statistically significant differences. The most common approach to validate data is manual gating, as humans can naturally control for many issues that will negatively influence algorithms. However, it is not always possible to reproduce the findings of an algorithm by manual gating (even if the data are of high quality). Although algorithms have been developed to generate minimum gating strategies, including Hypergate58 and GateFinder,59 in these situations, it is important to understand the context of the work, particularly the markers that are being expressed on the cells. Does the finding make sense biologically? Are there further experiments that could be done to support this finding? Are the same results reproduced when a different algorithm is used? It is important to question the output of algorithms, but it is just as important to be critical of the validation.
Concluding thoughts
High‐dimensional cytometry continues to evolve, but the steps involved in making the most of the technology remain the same. Fundamentals, such as framing a clear research question, logical panel construction and careful experimental design, allow high‐quality high‐dimensional data to be generated. The means by which data are analyzed are also changing. These include increased understanding of molecular interactions and expression of molecules in cell subsets, as well as standardized gating strategies. Large changes between groups are likely to be noticed by humans, as well as by computers, but it is subtle changes that algorithms are able to identify that make their use worthwhile. Any set of data can be analyzed an almost infinite number of ways: gates can be readjusted countless times to result in different outcomes. For this reason, it is not possible to find all changes by using a single method of analysis. Running previous data through a clustering algorithm will undoubtedly reveal new differences, because of its ability to process in a higher number of dimensions. The question is whether the resulting changes are real and/or relevant. Validation of results is essential but can reveal new messages that were previously missed. This is most efficient when answering a specific research question (either the same or new), to provide a more focused approach (Box 7).
BOX 7. Glossary.
Abundance : relative or absolute size of a population.
Arcsinh transformation: a method to transform data to reduce unfair weighting when comparing datapoints with high versus low values.
Barcoding: labeling of individual samples with a combination of parameters (barcode) that allow subsequently mixed samples to be stained and acquired on a cytometer as a single mixed sample. Debarcoding of data allows analysis of the original individual samples.
Batching: coordinated sample processing and acquisition of groups of samples or runs to minimize between‐sample variability.
Bivariate plots: graphs showing expression of two parameters.
Classifier(s): algorithms that assign data points (such as individual cells) to predetermined groups (such as cell subsets or clusters). Classifiers can be established on a training data set and validated on a testing data set.
Cluster: a group of individual cells put together based on similarity of parameters.
Cluster silhouette index: a method used to determine consistency within clusters.
Dimensionality reduction: a calculation that summarizes the data in a smaller number of dimensions than the original data; useful for visualization.
Elbow point validation: a method of determining the variance in multiple clusters to determine the number of clusters required to interpret the data set. As the number of clusters increases, the summed variance decreases; the elbow point is where the summed variance plateaus—at this point, a higher number of clusters would provide no additional benefit.
Scaling: the process of transforming values (e.g. expression values) to create a standardized range across multiple datapoints, while still retaining the variance.
Supervision: the guidance provided to a clustering algorithm to generate useful clustering data. Unsupervised analyses weigh all parameters equally; supervised analyses make use of rules, usually based on previously established data, to create clusters.
Variance: the spread of values for a given parameter; for example, the range of the expression within a marker.
Weight: the impact of a value on the result of the clustering—high‐weighted parameters have more impact on the number and composition of clusters than low‐weighted parameters.
CONFLICT OF INTEREST
The authors have no conflicts of interest to disclose.
AUTHOR CONTRIBUTION
Felix MD Marsh‐Wakefield: Conceptualization; Formal analysis; Investigation; Methodology; Resources; Validation; Visualization; Writing‐original draft; Writing‐review & editing. Andrew J Mitchell: Conceptualization; Formal analysis; Methodology; Visualization; Writing‐original draft; Writing‐review & editing. Samuel E Norton: Formal analysis; Investigation; Methodology; Validation; Writing‐review & editing. Thomas Myles Ashhurst: Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Writing‐original draft; Writing‐review & editing. Julia KH Leman: Formal analysis; Methodology. Joanna M Roberts: Conceptualization; Formal analysis; Funding acquisition; Writing‐review & editing. Jessica E Harte: Methodology. Helen M McGuire: Conceptualization; Data curation; Funding acquisition; Investigation; Methodology; Supervision; Validation; Visualization; Writing‐original draft; Writing‐review & editing. Roslyn A Kemp: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Project administration; Resources; Writing‐original draft; Writing‐review & editing.
ACKNOWLEDGMENTS
We thank the Australia and New Zealand Society for Immunology and the Australasian Cytometry Society for providing funding to host a workshop to create this work. FM‐W, TMA and HMM are supported by the International Society for the Advancement of Cytometry (ISAC) Marylou Ingram Scholars program. We thank Adam Girardin and the Dunedin Colorectal Cohort for the data shown in Figure 1. We thank Nic West, Justin Tirados, Rachel Hannaway and Jayden O’Brien for review of the manuscript.
REFERENCES
- 1.Spitzer MH, Nolan GP. Mass cytometry: Single cells, many features. Cell 2016; 165: 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Park LM, Lannigan J, Jaimes MC. OMIP‐069: Forty‐color full spectrum flow cytometry panel for deep immunophenotyping of major cell subsets in human peripheral blood. Cytometry A 2020; 97: 1044–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Norton SE, Ward‐Hartstonge KA, McCall JL, et al. High‐dimensional mass cytometric analysis reveals an increase in effector regulatory T cells as a distinguishing feature of colorectal tumors. J Immunol 2019; 202: 1871–1884. [DOI] [PubMed] [Google Scholar]
- 4.Huhn O, Ivarsson MA, Gardner L, et al. Distinctive phenotypes and functions of innate lymphoid cells in human decidua during early pregnancy. Nat Commun 2020; 11: 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Subrahmanyam PB, Dong Z, Gusenleitner D, et al. Distinct predictive biomarker candidates for response to anti‐CTLA‐4 and anti‐PD‐1 immunotherapy in melanoma patients. J Immunother Cancer 2018; 6: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bendall SC, Davis KL, el Amir AD, et al. Single‐cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell 2014; 157: 714–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Godfrey DI, Koay HF, McCluskey J, Gherardin NA. The biology and functional importance of MAIT cells. Nat Immunol 2019; 20: 1110–1128. [DOI] [PubMed] [Google Scholar]
- 8.Ferrer‐Font L, Pellefigues C, Mayer JU, Small SJ, Jaimes MC, Price KM. Panel design and optimization for high‐dimensional immunophenotyping assays using spectral flow cytometry. Curr Protoc Cytom 2020; 92: e70. [DOI] [PubMed] [Google Scholar]
- 9.Mahnke YD, Roederer M. Optimizing a multicolor immunophenotyping assay. Clin Lab Med 2007; 27: 469–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mair F, Tyznik AJ. High‐dimensional immuno‐phenotyping with fluorescence‐based cytometry: A practical guidebook. Methods Mol Biol 2019; 2032: 1–29. [DOI] [PubMed] [Google Scholar]
- 11.Bendall SC, Simonds EF, Qiu P, et al. Single‐cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 2011; 332: 687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Norton SE, Leman JKH, Khong T, et al. Brick plots: an intuitive platform for visualizing multiparametric immunophenotyped cell clusters. BMC Bioinformatics 2020; 21: 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hedley BD, Keeney M. Technical issues: Flow cytometry and rare event analysis. Int J Lab Hematol 2013; 35: 344–350. [DOI] [PubMed] [Google Scholar]
- 14.Lambert C, Yanikkaya Demirel G, Keller T, et al. Flow cytometric analyses of lymphocyte markers in immune oncology: A comprehensive guidance for validation practice according to laws and standards. Front Immunol 2020; 11: 2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rosenblatt JI, Hokanson JA, McLaughlin SR, Leary JF. Theoretical basis for sampling statistics useful for detecting and isolating rare cells using flow cytometry and cell sorting. Cytometry 1997; 27: 233–238. [DOI] [PubMed] [Google Scholar]
- 16.Gaudilliere B, Fragiadakis GK, Bruggner RV, et al. Clinical recovery from surgery correlates with single‐cell immune signatures. Sci Transl Med 2014; 6: 255ra131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liang S, Willis J, Dou J, et al. Sensei: how many samples to tell evolution in single‐cell studies? bioRxiv (pre‐print). 2020. 10.1101/2020.05.31.126565 [DOI] [Google Scholar]
- 18.Botting RA, Bertram KM, Baharlou H, et al. Phenotypic and functional consequences of different isolation protocols on skin mononuclear phagocytes. J Leukoc Biol 2017; 101: 1393–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teh CE, Gong JN, Segal D, et al. Deep profiling of apoptotic pathways with mass cytometry identifies a synergistic drug combination for killing myeloma cells. Cell Death Differ 2020; 27: 2217–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sierro F, Evrard M, Rizzetto S, et al. A Liver Capsular Network of Monocyte‐Derived Macrophages Restricts Hepatic Dissemination of Intraperitoneal Bacteria by Neutrophil Recruitment. Immunity 2017; 47: 374–388.e6. [DOI] [PubMed] [Google Scholar]
- 21.Sumatoh HR, Teng KW, Cheng Y, Newell EW. Optimization of mass cytometry sample cryopreservation after staining. Cytometry A 2017; 91: 48–61. [DOI] [PubMed] [Google Scholar]
- 22.Sakkestad ST, Skavland J, Hanevik K. Whole blood preservation methods alter chemokine receptor detection in mass cytometry experiments. J Immunol Methods 2020; 476: 112673. [DOI] [PubMed] [Google Scholar]
- 23.Rybakowska P, Alarcon‐Riquelme ME, Maranon C. Key steps and methods in the experimental design and data analysis of highly multi‐parametric flow and mass cytometry. Comput Struct Biotechnol J 2020; 18: 874–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wagar LE. Live cell barcoding for efficient analysis of small samples by mass cytometry. Methods Mol Biol 2019; 1989: 125–135. [DOI] [PubMed] [Google Scholar]
- 25.Giudice V, Feng X, Kajigaya S, Young NS, Biancotto A. Optimization and standardization of fluorescent cell barcoding for multiplexed flow cytometric phenotyping. Cytometry A 2017; 91: 694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krutzik PO, Nolan GP. Fluorescent cell barcoding in flow cytometry allows high‐throughput drug screening and signaling profiling. Nat Methods 2006; 3: 361–8. [DOI] [PubMed] [Google Scholar]
- 27.Hartmann FJ, Simonds EF, Bendall SC. A universal live cell barcoding‐platform for multiplexed human single cell analysis. Sci Rep 2018; 8: 10770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mei HE, Leipold MD, Schulz AR, Chester C, Maecker HT. Barcoding of live human peripheral blood mononuclear cells for multiplexed mass cytometry. J Immunol 2015; 194: 2022–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zunder ER, Finck R, Behbehani GK, et al. Palladium‐based mass tag cell barcoding with a doublet‐filtering scheme and single‐cell deconvolution algorithm. Nat Protoc 2015; 10: 316–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leipold MD, Obermoser G, Fenwick C, et al. Comparison of CyTOF assays across sites: Results of a six‐center pilot study. J Immunol Methods 2018; 453: 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maecker HT, McCoy JP, Nussenblatt R. Standardizing immunophenotyping for the human immunology project. Nat Rev Immunol 2012; 12: 191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Finak G, Langweiler M, Jaimes M, et al. Standardizing flow cytometry immunophenotyping analysis from the human immunophenotyping consortium. Sci Rep 2016; 6: 20686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schulz AR, Baumgart S, Schulze J, Urbicht M, Grutzkau A, Mei HE. Stabilizing antibody cocktails for mass cytometry. Cytometry A 2019; 95: 910–916. [DOI] [PubMed] [Google Scholar]
- 34.Leek JT, Scharpf RB, Bravo HC, et al. Tackling the widespread and critical impact of batch effects in high‐throughput data. Nat Rev Genet 2010; 11: 733–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goh WWB, Wang W, Wong L. Why batch effects matter in omics data, and how to avoid them. Trends Biotechnol 2017; 35: 498–507. [DOI] [PubMed] [Google Scholar]
- 36.Casler MD. Blocking principles for biological experiments. In: Glaz B, Yeater K (eds). Applied statistics in agricultural, biological, and environmental sciences. American Society of Agronomy, Inc; Soil Society of America, Inc; Crop Science Society of America, Inc; 2018. [Google Scholar]
- 37.Yan L, Ma C, Wang D, et al. OSAT: a tool for sample‐to‐batch allocations in genomics experiments. BMC Genom 2012; 13: 689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schuyler RP, Jackson C, Garcia‐Perez JE, et al. Minimizing batch effects in mass cytometry data. Front Immunol 2019; 10: 2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van Gassen S, Gaudilliere B, Angst MS, Saeys Y, Aghaeepour N. CytoNorm: A normalization algorithm for cytometry data. Cytometry A 2020; 97: 268–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Skinner J. Statistics for Immunologists. Curr Protoc Immunol 2018; 122: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Monaco G, Chen H, Poidinger M, Chen J, de Magalhaes JP, Larbi A. flowAI: automatic and interactive anomaly discerning tools for flow cytometry data. Bioinformatics 2016; 32: 2473–2480. [DOI] [PubMed] [Google Scholar]
- 42.Ashhurst TM, Marsh‐Wakefield F, Putri GH, et al. Integration, exploration, and analysis of high‐dimensional single‐cell cytometry data using Spectre. bioRxiv (pre‐print). 2020. 10.1101/2020.10.22.349563 [DOI] [PubMed] [Google Scholar]
- 43.Majumder B, North J, Mavroudis C, Rakhit R, Lowdell MW. Improved accuracy and reproducibility of enumeration of platelet‐monocyte complexes through use of doublet‐discriminator strategy. Cytometry B Clin Cytom 2012; 82: 353–359. [DOI] [PubMed] [Google Scholar]
- 44.Keyes TJ, Domizi P, Lo YC, Nolan GP, Davis KL. A Cancer biologist's primer on machine learning applications in high‐dimensional cytometry. Cytometry A 2020; 97: 782–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kimball AK, Oko LM, Bullock BL, Nemenoff RA, van Dyk LF, Clambey ET. A beginner's guide to analyzing and visualizing mass cytometry data. J Immunol 2018; 200: 3–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Olsen LR, Leipold MD, Pedersen CB, Maecker HT. The anatomy of single cell mass cytometry data. Cytometry A 2019; 95: 156–172. [DOI] [PubMed] [Google Scholar]
- 47.Weber LM, Robinson MD. Comparison of clustering methods for high‐dimensional single‐cell flow and mass cytometry data. Cytometry A 2016; 89: 1084–1096. [DOI] [PubMed] [Google Scholar]
- 48.Liu X, Song W, Wong BY, et al. A comparison framework and guideline of clustering methods for mass cytometry data. Genome Biol 2019; 20: 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saeys Y, Van Gassen S, Lambrecht BN. Computational flow cytometry: helping to make sense of high‐dimensional immunology data. Nat Rev Immunol 2016; 16: 449–462. [DOI] [PubMed] [Google Scholar]
- 50.Van Gassen S, Callebaut B, Van Helden MJ, et al. FlowSOM: Using self‐organizing maps for visualization and interpretation of cytometry data. Cytometry A 2015; 87: 636–645. [DOI] [PubMed] [Google Scholar]
- 51.Eliot M, Azzoni L, Firnhaber C, et al. Tree‐based methods for discovery of association between flow cytometry data and clinical endpoints. Adv Bioinformatics 2009: 235320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abdelaal T, van Unen V, Hollt T, Koning F, Reinders MJT, Mahfouz A. Predicting cell populations in single cell mass cytometry data. Cytometry A 2019; 95: 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lugli E, Pinti M, Nasi M, et al. Subject classification obtained by cluster analysis and principal component analysis applied to flow cytometric data. Cytometry A 2007; 71: 334–344. [DOI] [PubMed] [Google Scholar]
- 54.el Amir AD, Davis KL, Tadmor MD, et al. viSNE enables visualization of high dimensional single‐cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol 2013; 31: 545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Becht E, McInnes L, Healy J, et al. Dimensionality reduction for visualizing single‐cell data using UMAP. Nat Biotechnol 2019; 37: 38–44. [DOI] [PubMed] [Google Scholar]
- 56.Qiu P, Simonds EF, Bendall SC, et al. Extracting a cellular hierarchy from high‐dimensional cytometry data with SPADE. Nat Biotechnol 2011; 29: 886–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Spitzer MH, Gherardini PF, Fragiadakis GK, et al. IMMUNOLOGY. An interactive reference framework for modeling a dynamic immune system. Science 2015; 349: 1259425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Becht E, Simoni Y, Coustan‐Smith E, et al. Reverse‐engineering flow‐cytometry gating strategies for phenotypic labelling and high‐performance cell sorting. Bioinformatics 2019; 35: 301–308. [DOI] [PubMed] [Google Scholar]
- 59.Aghaeepour N, Simonds EF, Knapp D, et al. GateFinder: projection‐based gating strategy optimization for flow and mass cytometry. Bioinformatics 2018; 34: 4131–4133. [DOI] [PMC free article] [PubMed] [Google Scholar]