


Abstract
Immunotherapy targeting the Programmed Death (PD‐1) receptor/ligand (L) “checkpoint” rapidly gains ground in the treatment of many cancer types. To increase treatment scope and efficacy, predictive biomarkers and rational selection of co‐treatments are required. To meet these demands, we must understand PD‐1 function in detail. We here outline recent insights into the regulation of the CD8+ T cell response by PD‐1. The prevailing view has been that blockade of PD‐1/ligand (L) interaction “reinvigorates” cytotoxic T lymphocytes (CTL) that were rendered dysfunctional in the tumor microenvironment (TME). However, this review stresses that tumors continuously communicate with adjacent draining lymph nodes (LNs) and that the PD‐1 checkpoint also operates during T cell priming. We clarify the role of the PD‐(L)1 system at the T cell/DC interface, where it regulates T cell receptor (TCR) signaling and CD28 costimulation and thus controls activation of tumor‐specific T cells. We also highlight the importance of CD4+ T cell help during priming, which allows DCs to provide other costimulatory and cytokine signals required for optimal CTL differentiation and likely avoidance of a dysfunctional state. Therefore, we pose that PD‐(L)1 blockade should exploit LN function and be combined with “help” signals to optimize CTL efficacy.
Keywords: immunotherapy, cytotoxic T cell, PD‐1, priming, exhaustion
Novel insights into exhaustion and PD‐1 signaling during T‐cell priming have challenged the classical view on the mechanisms underlying PD‐(L)1‐targeted therapy. This review integrates the latest findings on anti‐PD‐(L)1 responsive T cells, such as differentiation state, location, and molecular interactions, into a comprehensive model of PD‐(L)1 blockade effects in cancer.

Introduction
Immunotherapy is becoming the fourth pillar in cancer treatment next to surgery, chemotherapy and radiotherapy. Its aim is to activate a tumor‐specific cytotoxic T lymphocyte (CTL) response that eradicates all tumor mass, regardless of metastatic spread. CTL responses generally are polyclonal, harboring T cells with different antigenic specificities. Therefore, immunotherapy can be effective against genetically heterogeneous, disseminated tumors, where (targeted) chemotherapies fail. Nevertheless, tumors can display inherent or acquired resistance to immunotherapy [1].
This review focuses on the current mainstay of cancer immunotherapy in the clinic: monoclonal antibodies (mAbs) that block the co‐inhibitory receptor PD‐1 or its ligand PD‐L1 [1, 2]. This therapy proved to evoke durable responses in a proportion of patients with stage IV melanoma, non‐small cell lung cancer (NSCLC), and mismatch repair‐deficient colon cancer [2]. These cancer types all have a high tumor mutational burden (TMB) and consequently express neoantigens [3]. Such cancers potentially evoke a T cell response and then present as “T cell inflamed” or “hot” tumors with a T cell‐rich TME with an IFN‐driven signature [4]. The dramatic and long‐lasting responses observed in previously untreatable cancer patients have led to application of PD‐L1‐targeted therapy in earlier disease stages. In recent years, neo‐adjuvant immunotherapy, prior to surgical resection of the primary tumor has produced remarkable responses, also in cancers that were previously not recognized as antigenic [5]. In neo‐adjuvant treatment, the untouched tumor serves as a reservoir of tumor antigens, that can apparently be tapped into to generate systemic, anti‐tumor CTL responses by PD‐(L)1 blockade.
Since PD‐L1‐targeted immunotherapy is successful in only a fraction of patients, is costly and may be accompanied by immune‐related adverse events [6], predictive biomarkers are urgently needed. At present, key potential biomarkers are neoantigen load, T cell infiltrate, PD‐L1 expression, and/or an IFN‐γ signature in the TME [3, 7]. A recent meta‐analysis indicates that high TMB only predicts responsiveness to PD‐L1 blockade in about 25% of all cancers types where high TMB correlates with CD8+ T cell infiltration of the tumor. In a wide variety of other cancer types including triple negative breast cancer and prostate cancer, high TMB did not correlate with CD8+ T cell infiltration and overall response rates to PD‐L1 blockade [8]. These data clearly indicate that presence of recognizable antigens in the tumor does not equate immunogenicity and point to important additional requirements for evoking an effective anti‐tumor CD8+ T cell response, such as DC activation and CTL functionality in terms of tumor penetration, as well as undefined requirements for CD8+ T cells to respond to PD‐L1‐targeted therapy.
In addition, PD‐L1 targeting needs to be combined with other approaches to increase efficacy. There are hundreds of clinical trials ongoing that test combinations with radiotherapy, conventional or targeted anticancer drugs, or antibodies to other co‐inhibitory or costimulatory receptors. To select the most promising combination therapies, we need maximal information on the mechanism of action of PD‐L1 targeted therapy. Recently, several discoveries have been made regarding molecular and cellular mechanisms underlying the response to PD‐L1 checkpoint blockade, which are very important for research into biomarkers and candidate targets for combination therapy. We will present this new information and combine it with new data on the mechanism of CTL priming into a comprehensive model of anti‐PD‐L1 action that can help guide further translational research and clinical decision making.
The CD8+ T cell population that is targeted by PD‐L1 inhibition
In chronic infection and cancer, CD8+ T cell populations have been defined that express PD‐1 and other co‐inhibitory receptors and display impaired proliferative and cytotoxic capacities. Classically, cells with this phenotype have been termed “exhausted” and PD‐L1 targeted immunotherapy was thought to help “reinvigorate” these exhausted cells [9, 10]. However, the view on T cell exhaustion has dramatically changed in recent years, based on transcriptome data and lineage tracing that makes use of TCR sequences as clonal markers. It is now established that exhausted CD8+ T cells are not derived from previously active effector cells. Rather, CD8+ T cells can attain a reversible, “predysfunctional” state early after infection or tumorigenesis that may progress into an irreversible, terminally exhausted state [10, 11] (Fig. 1). There are different types of nomenclature for the predysfunctional state of CD8+ T cells and different markers, as outlined in, e.g., [12]. The common features of this state are deficient effector functionality, expression of PD‐1 and other co‐inhibitory receptors and, as recently recognized, expression of the transcription factor TCF‐1 [11, 12].
Figure 1.

Predysfunctional PD‐1+ CD8+ T cells in the TME. In the TME, a pool of PD‐1+ predysfunctional CD8+ T cells exists that undergoes self‐renewal. PD‐1 on the predysfunctional CD8+ T cells can interact with PD‐L1 on tumor cells or on cDCs. These interactions impair tumor cell killing, limiting tumor clearance and antigen availability. Furthermore, these interactions help drive the cells into an irreversibly exhausted state. Color codes: grey = tumor cell; blue = cDC; orange = predysfunctional CD8+ T cell; red = exhausted CD8+ T cell.
In chronic infection with lymphocytic choriomeningitis virus (LCMV), the predysfunctional pool of virus‐specific CD8+ T cells proved to rely on TCF‐1 and to co‐express PD‐1, CXCR5, and SLAMF6 [13, 14]. This population proliferates and constantly replenishes a short‐lived pool of CD8+ T cells that are truly exhausted, i.e., irreversibly, epigenetically fixed in a non‐functional state [15, 16, 17]. Also in mouse models of cancer, predysfunctional CD8+ T cells were found in the TME that could become irreversibly exhausted [16, 18, 19]. Likewise, in the TME of various human cancers, TCR tracing argues for progressive differentiation of predysfunctional into terminally exhausted CD8+ T cells [20, 21, 22, 23, 24, 25]. In human kidney, prostate and bladder tumors, TCF‐1+ predysfunctional CD8+ T cells were found in proximity of MHC class II+, CD11c+ cells, most likely DCs, denoting cellular niches that resembled T cell zones in secondary lymphoid organs. The presence of such niches in the TME positively correlated with long‐term patient survival after surgery [24].
Importantly, PD‐L1 blockade was recently found to reinvigorate predysfunctional, but not terminally exhausted CD8+ T cells. Predysfunctional cells respond to PD‐L1 blockade by proliferation. This was demonstrated in mouse models of chronic LCMV infection [13, 26, 27] and cancer [16, 28, 29]. Moreover, in melanoma patients, an increased fraction of TCF‐1+ predysfunctional CD8+ tumor‐infiltrating lymphocytes is a positive predictor for a clinical response to PD‐1 targeted therapy [16, 30]. It is now clear that predysfunctional CD8+ T cells are in an early stage of CTL effector differentiation and still have the ability to proliferate and further differentiate. They do this to a certain extent upon PD‐1 blockade [11, 12].
PD‐L1 on DCs is important for the response to PD‐L1‐targeted therapy
Initially, a reductionist model was prevalent in the cancer immunotherapy field, in which PD‐1 on the CTL finds its ligand on tumor cells and thus imposes a brake on tumor cell killing in the TME (Fig. 1). This view was likely inspired by the landmark study of Iwai et al. who discovered that deliberate PD‐L1 expression in tumor cells inhibits anti‐tumor immunity [31]. PD‐1 has two ligands, PD‐L1 and PD‐L2, with distinct expression patterns. PD‐L1 is expressed on lymphocytes and myeloid cells and is upregulated under conditions of immune activation, e.g., by type I and type II IFN. In addition, PD‐L1 is expressed on non‐hematopoietic cells, such as vascular endothelium and pancreatic islands. PD‐L2 expression is more restricted and found on activated DCs, macrophages, mast cells, and peritoneal B1 cells [32]. Furthermore, both PD‐L1 and PD‐L2 are expressed on placental trophoblasts, indicating an important role in fetal tolerance [33]. PD‐1 sets signaling thresholds during T cell selection in the thymus and plays a role in peripheral tolerance, as judged by the auto‐immune phenotypes of genetically deficient mice [32].
Due to the reductionist model, clinical studies initially focused on PD‐L1 expression on tumor cells as biomarker for PD‐L1 targeted therapy. However, in early phase I studies with anti‐PD‐L1 mAb Atezolizumab (MPDL3280A), it was already noted that clinical responses in multiple cancer types correlated with expression of PD‐L1 on either tumor cells, or tumor‐infiltrating immune cells [34]. Immunohistochemistry identified three typical expression patterns of PD‐L1 in human cancer with expression on either tumor cells or immune cells, or both [35]. Mechanistic studies were performed in mouse models to assess the relative importance of PD‐L1 expression on either cell type. For this purpose, predominant tumor models are s.c. implanted MC38 or CT26 colon carcinoma cells that have a high mutational load, are “hot” and responsive to PD‐L1‐targeted therapy. Genetic deletion of PD‐L1 in tumor cells and/or in the background of the host proved that spontaneous tumor rejection was suppressed by PD‐L1 in both compartments [35]. Strikingly, the response to PD‐L1 targeted therapy was not affected by genetic deletion of PD‐L1 in MC38 tumor cells, while it was abolished by PD‐L1 deletion in the hematopoietic compartment of the host [36, 37]. In agreement, another group found that effective anti‐PD‐L1 therapy required PD‐L1 expression by the host in MC38, 1D8, and B16‐F10 tumor models [38]. Genetic deletion identified PD‐L1 on DCs (CD11c+ cells) as critical for the response of MC38 to anti‐PD‐L1 therapy [39]. These findings were refined by comparing the therapeutic response to PD‐L1‐deficient MC38 cells in mice that were PD‐L1 deficient in either conventional (c)DCs (Clec9a+ cells) or macrophages (LysM+ cells). PD‐L1 on cDCs proved most important for the response to anti‐PD‐L1 therapy, even though MC38 tumors harbor large amounts of PD‐L1+ macrophages [40]. In human melanoma, a correlation was suggested between PD‐L1 expression on DCs and the response to PD‐L1 targeted therapy [38].
The importance of cDCs, particularly cDC1s, in the response to PD‐L1 targeting therapy is supported by distinct lines of investigation. In the MC38 tumor model, response to anti‐PD‐1 treatment was lost upon genetic elimination of cDC1s, i.e., in a Batf3 −/− background [41]. In B16 mouse melanoma, the modest therapeutic effect of anti‐PD‐L1 mAb also depended on Batf3 proficiency. Promoting the generation of DCs from progenitors by Flt3 ligand together with DC‐activating compound poly‐IC, synergized with anti‐PD‐L1 therapy [42]. Moreover, anti‐PD‐1 treatment was found to induce IL‐12b production by tumor‐resident cDC1s, which proved important for tumor regression [43]. The cDC1s made IL‐12b in response to IFN‐γ that was produced by other cells in the TME upon anti‐PD‐1 therapy. Direct application of IL‐12 into the tumor increased IFN‐γ production, suggesting effector T cell and/or NK cell activation. The authors also present data from melanoma patients that were treated with ImmunoPulse tavokinogene telseplasmid, an IL‐12 encoding plasmid that was electroporated into melanoma lesions on the skin. IL‐12 delivery upregulated multiple genes encoding CTL effector molecules, supporting the idea that this important DC‐derived cytokine can promote CTL function in the TME [43]. CXCL9 production by intratumoral cDC1s has also been implicated in the response to anti‐PD‐1 therapy in the MC38 model, with the data supporting a model in which CXCL9 facilitates communication of these cDC1s with CD8+ T cells in the tumor. CXCL9 levels in blood positively correlated with the response to anti‐PD‐1 in a small cohort of melanoma patients [44]. In renal cell carcinoma and NSCLC patients treated with Atezolizumab, a cumulative expression score based on cDC1 signature genes XCR1, BATF3, IRF8, FLT3 in the tumor was associated with improved survival, also arguing in favor of a role for cDC1s in the response to PD‐L1‐targeted therapy [45]. The cumulative data change the reductionist model, highlighting that the response to PD‐L1‐targeted therapy primarily depends on altered T cell/cDC communication (Fig. 1). The special role of the cDC1 in invigorating the anti‐tumor CD8+ T cell response upon anti‐PD‐L1 treatment ties in with its well‐documented specialization in antigen cross‐presentation and cross‐priming of CD8+ T cells, which is key for anti‐tumor immunity [46].
PD‐L1‐targeted therapy impacts T cell priming
In the cancer immunotherapy field, the prevailing view has been that PD‐L1 blockade acts within the TME. However, it has long been known that PD‐1 regulates CD8+ T cell priming [47]. Earlier studies showed that PD‐1 is upregulated on T cells within 24 h after initial activation [48] and that PD‐1 ligands are expressed on activated DCs [32]. In fact, the original study that defined PD‐1 function already demonstrated that PD‐L1 is expressed on APCs and that PD‐1 inhibits both TCR‐ and CD28 signals [49]. PD‐1 signaling was shown to limit the TCR‐driven motility arrest of CD4+ T cells on cognate DC [50], which can be attributed to a later demonstrated negative impact on the formation of stable synaptic contacts needed for effector T cell differentiation [51]. In agreement, in a mouse model of acute virus infection PD‐1 constrained effector differentiation of CD8+ T cells during priming [52]. Clinical data pointed out that PD‐L1 targeted immunotherapy can also be effective in patients that do not express PD‐L1 in the TME [53]. Collectively, these data illuminate that PD‐L1 blockade may facilitate de novo priming of (tumor‐specific) CTLs in tumor‐draining lymph nodes (tdLNs).
Indeed, in the MC38 tumor model, newly primed CD8+ T cells were found to contribute to the treatment response. Tang et al. established that s.c. implantation of MC38 cells raises an IFN‐γ‐producing T cell response in the tdLNs. Entrapment of newly primed T cells in the tdLN with the drug FTY720 prohibited the response to anti‐PD‐L1 therapy [36]. Extending these findings, Fransen et al. showed that s.c. implantation of MC38 cells leads to increased PD‐L1 expression on myeloid cells in tdLNs, but not in non‐tdLNs. It also leads to CD8+ T cell priming in the tdLNs, which is increased by PD‐1 blockade. Either FTY720 treatment, or surgical resection of the tdLNs abrogated tumor infiltration by newly primed CD8+ T cells and anti‐PD‐1 mediated tumor control [54]. These studies clarify that T‐cell inflamed tumors communicate with their tdLNs to invite new T cell priming, which is constrained by the PD‐L1 checkpoint. The link between these compartments is formed by migratory cDCs that deliver tumor antigen and signals that dictate the T cell response. Participation of the tdLNs in the response to PD‐L1 blockade was also demonstrated in a mouse model with mesothelioma tumor cell lines that were implanted intrapleurally. This tumor type was shown to raise predysfunctional (“progenitor exhausted”) PD‐1+ CD4+ and CD8+ T cells in mediastinal tdLNs. Selective inhibition of PD‐L1 in tdLNs by low dose, intrapleural injection of the mAb increased the frequency and proliferation of intratumoral CD8+ T cells and mediated tumor control, which relied on PD‐L1 expression in the tdLN by cDC1s and cDC2s, but not macrophages. Thereby, this study supports the concept that PD‐L1 blockade promotes priming of tumor‐specific T cells (Fig. 2). The authors furthermore suggest that in human stage II melanoma, PD‐1/PD‐L1 proximity in tdLNs, rather than in the tumor, is a prognosticator for early distant recurrence following surgery [55].
Figure 2.

The PD‐1 checkpoint in T cell priming and effector stages. Upon killing of tumor cells, antigen can be taken up by cDCs and subsequently be transported via the lymph to tdLNs. Here, the cDCs prime naive T cells which leave the tdLN via the blood and enter the tumor. If these cells are properly differentiated into effector CTLs, they can kill more tumor cells, increasing antigen availability. However, interactions between PD‐1 and its ligands may limit efficacy of CTLs by limiting their priming and activity in the TME. Color codes: beige = naive CD8+ T cell; blue = cDC; orange = predysfunctional CD8+ T cell; grey = tumor cell.
These findings imply that the clinical response to PD‐L1 targeted therapy may be visualized in the peripheral blood, where after therapy certain newly primed T cell clones should be expanded. Indeed, in a cohort of 29 advanced stage NSCLC patients, 70% responded by an increase in dividing (Ki‐67+) PD‐1+ CD8+ T cells in the blood upon PD‐1‐targeted therapy [56]. More detailed information has recently come from TCR‐based tracing of T cell clones. In a neo‐adjuvant trial with anti‐PD‐1 treatment of stage II NSCLC patients, TCR sequencing was performed on serial samples of peripheral blood, as well on tumor and normal lung samples at the endpoint. Interestingly, high intratumoral TCR clonality was associated with reduced residual tumor mass at time of surgery and tumor responsiveness was correlated to high TCR clonality, with clonotypes shared between tumor and peripheral blood [57]. In basal cell carcinoma, single cell transcriptomics coupled to TCR sequencing revealed that particularly in the intratumoral CD8+ T cell population, changes in TCR repertoire took place upon PD‐1 targeted therapy. T cell clones that were present in the TME pre‐treatment and displayed a terminally exhausted phenotype did not contribute to the response. Also, the main responders were not TCF‐1+ predysfunctional CD8+ T cells that were present pretreatment, but new clonotypes with an exhausted phenotype that were found in the tumor posttreatment. These data suggest that upon PD‐1 targeted therapy, newly primed CD8+ T cells entered into the tumor and turned into exhausted cells in the TME [58]. An independent analysis of the same data lent support to this conclusion and emphasized the sharing of T cell clones between tumor and blood [59]. Together, these data change the view on the mode of action of PD‐(L)1 targeted therapy: Rather than acting exclusively in the TME by “reinvigorating” tumor‐resident predysfunctional CD8+ T cells, it can instead or in addition, promote CD8+ T cell priming in tdLNs [60].
PD‐1 checkpoint controls TCR and CD28 signaling during T cell‐DC communication
CTLA‐4 and PD‐1 are both termed “checkpoints” for T cell activation and considered co‐inhibitory receptors, but their mechanisms of action are distinct. CTLA‐4's function is to inhibit CD28 co‐stimulation of conventional T cells (Tconvs). CTLA‐4 is constitutively expressed on regulatory CD4+ T cells (Tregs) and inducibly expressed on activated CD4+ and CD8+ Tconvs. CTLA‐4 inhibits CD28 co‐stimulation of Tconvs by binding and downregulating the CD28 ligands CD80 and/or CD86 from the plasma membrane of DCs by transendocytosis [61]. Tregs attenuate in this way the costimulatory state of migratory DCs that present autoantigens. CTLA‐4‐deficient mice develop dramatic auto‐immune symptoms, even under germ‐free conditions, supporting that CTLA‐4 is essential to maintain peripheral tolerance to self‐antigen. Deletion of CTLA‐4 from Tregs likewise precipitates autoimmunity [62]. Autoimmune symptoms resulting from PD‐1 deficiency are much milder, alluding to the distinct functions of CTLA‐4 and PD‐1 [32].
In terms of signal transduction, CTLA‐4 and PD‐1 are very different. CTLA‐4 has tyrosines in its cytoplasmic tail that were long thought to represent immunoreceptor tyrosine‐based inhibitory motifs (ITIMs). However, CTLA‐4 does not have bona fide ITIMs, but internalization motifs that allow rapid endocytosis from the plasma membrane. CTLA‐4 is constitutively recycled from the plasma membrane, befitting its role in transendocytosis of CD80 and CD86 [61]. The cytoplasmic tail of PD‐1 on the other hand, contains two well‐defined ITIMs that upon tyrosine phosphorylation by Src family kinases recruit the tyrosine phosphatase SHP‐2 [51, 63, 64, 65, 66]. Both the TCR/CD3 complex and CD28 activate the T cell by virtue of tyrosine kinases that phosphorylate many signal transduction molecules. By bringing SHP‐2 in proximity, PD‐1 negates these signals [51]. In the cancer immunotherapy field, PD‐1 was largely viewed as an inhibitor of the TCR/CD3 signal, but reinvigoration of dysfunctional CD8+ T cells by PD‐1 blockade proved to critically depend on CD28 [67]. In the same year, a biochemical study using reconstituted lipid vesicles revealed that the PD‐1/SHP‐2 complex dephosphorylates CD28 with preference over CD3 components [63]. Subsequently, knowledge was advanced by mass spectrometry studies showing that in the cellular context, PD‐1 prefers SHP‐2 over SHP‐1 but can function with either and dephosphorylates CD28, and the LAT and SLP72 signaling adaptors [64]. In conclusion, CTLA‐4 exclusively inhibits CD28 co‐stimulation, while PD‐1 inhibits both TCR/CD3 signaling and CD28 co‐stimulation (Fig. 3A). It follows from these findings that both “checkpoints” can act in priming and effector phases of the T cell response, particularly at the T cell/DC interface. It still needs to be resolved how combined inhibition of both “checkpoints” can synergize in stimulating the T cell response [68].
Figure 3.

Impact of PD‐L1 on TCR/CD28 signaling at the T cell/DC interface. (A) PD‐L1 expressed on activated DCs binds to PD‐1 on CD8+ T cells, thereby inhibiting the TCR/CD28 signal during priming. Co‐stimulation via CD28 is also impaired by the CTLA‐4 mediated downregulation of its ligand CD80. (B) When PD‐L1 forms a heterodimer with CD80 on the cell surface of the DC, signaling through PD‐1 is abrogated, but signaling through CD28 can still take place. Moreover, the CD80;PD‐L1 dimer can no longer be downregulated by CTLA‐4, resulting in an increase of CD28 co‐stimulation.
Recent data add another layer of complexity on the mode of action of PD‐L1 inhibition. It turns out that PD‐L1 can bind CD80 on the membrane of the same cell (in cis) [69]. This ability to heterodimerize is unique to the CD80;PD‐L1 pair and not shared with CD86 and/or PD‐L2 [70, 71]. The CD80;PD‐L1 dimer cannot bind to PD‐1, nor can it be downregulated by CTLA‐4 [70, 71]. Strikingly, the CD80;PD‐L1 heterodimer can bind to CD28 and as a consequence, it induces T cell co‐stimulation while negating co‐inhibition (Fig. 3B). Primary CD4+ and CD8+ T cells were shown to escape from PD‐1‐mediated inhibition by virtue of CD80;PD‐L1 interactions in a study comparing wild‐type DCs and those with mutations disrupting the CD80;PD‐L1 interaction. Accordingly, mice with the same mutations had an attenuated anti‐tumor response upon therapeutic vaccination [71]. Reportedly, activated cDC1s and cDC2s express PD‐L1 [39, 45, 71]. At first glance, this contradicts the role of activated cDCs in T cell priming, but co‐expression of PD‐L1 with CD80 will allow their heterodimerization, which favors CD28 co‐stimulation. These new insights emphasize that we need to pay more attention to the expression patterns of CD80, CD86, PD‐L1, and PD‐L2 on cDC1s and cDC2s, since these dictate the ability of cDCs to co‐stimulate Tconvs via CD28 and to relieve CTLA‐4‐based suppression by Tregs. We also need to reconsider the potential mechanisms underlying synergistic effects of combined CTLA‐4 and PD‐L1 blockade in the clinic [68] and be aware that clinical targeting of PD‐1 or PD‐L1 may not have the same effects on the T cell response.
It is also very important to consider the effects of checkpoint inhibition on Treg responses. It is well established that PD‐1 engagement on Tconvs can drive their conversion into peripherally induced (p)Tregs [72] and in this context, PD‐1 blockade is expected to promote Tconv responses. However, it has recently been established in mice genetically deficient for PD‐1 in Foxp3‐expressing cells, that PD‐1 constrains Treg activation, proliferation and suppressive activity, at least in part via the PKB/AKT pathway [73]. This finding indicates that clinical PD‐1 blockade may lead to increased Treg‐mediated suppression of Tconv responses. Accordingly, a pioneering study in human NSCLC and gastric cancer combined with work in the MC‐38 mouse model confirms these findings and provides evidence that it is the ratio of PD‐1+ Tconvs versus PD‐1+ Tregs in the TME that predicts responsiveness to PD‐(L)1 blockade rather than the frequency of PD‐1+ Tconvs as such [74].
The PD‐1 checkpoint in two‐step priming
In this section, we would like to present a model that clarifies how reinvigoration of predysfunctional tumor‐specific CD8+ T cells by PD‐L1 blockade links to PD‐L1 checkpoint function in T cell priming. We have recently provided evidence that predysfunctional CD8+ T cells likely have been primed in absence of CD4+ T cell help [12]. CD4+ T cell help is essential for full differentiation of CD8+ T cells into short‐lived effector and effector/memory cells [75, 76, 77]. We discovered that predysfunctional CD8+ T cells in mouse models of chronic LCMV infection and cancer have a “helpless” gene expression signature [12], as we defined in CD8+ T cells that were primed by vaccination in absence of CD4+ T cell help [75]. An independent study supports this notion [78]. In our vaccination model, helpless CD8+ T cells displayed the typical predysfunctional phenotype and had limited efficacy against a s.c. implanted tumor, due to reduced cytotoxic and migratory potential and expression of PD‐1, BTLA and other co‐inhibitory receptors. Our hypothesis based on these data is that CD8+ T cells primed in absence of CD4+ T cell help have not yet completed their effector differentiation pathway and therefore present as “predysfunctional.” However, they are not exhausted and can still complete their effector differentiation pathway, provided that they receive the stimuli that equate “help” [12] (Fig. 4).
Figure 4.
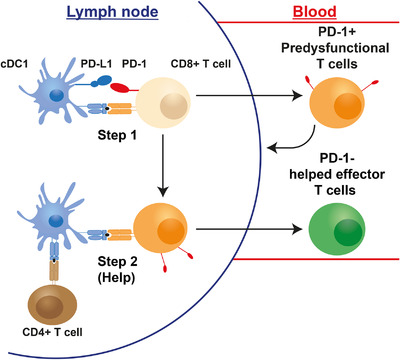
Proposed connection between help, optimal CTL priming and the PD‐1 checkpoint. Naive CD8+ T cells are activated by (migratory) cDC1s during the first step of priming. The PD‐1 axis inhibits TCR/CD28 signaling and thereby prevents clonal expansion. CD8+ T cells may leave the LN at this point, but are in an early stage of effector differentiation with limited cytotoxic and migratory abilities and expression of PD‐1 and other co‐inhibitory receptors. We propose that this “helpless” state equals the “predysfunctional” state identified in many studies. When appropriate activating signals are available, such as type I IFN, the CD8+ T cells undergo a second step of priming wherein help is delivered by activated CD4+ T cells via LN‐resident cDC1s. This leads to optimal effector differentiation of CD8+ T cells into potent CTLs that lack PD‐1 expression. Color codes: beige = naive CD8+ T cell; blue = cDC; orange = unhelped/predysfunctional CD8+ T cell; brown = CD4+ T helper cell; green = optimally primed/helped CTL.
To explain how helpless/predysfunctional CD8+ T cells may emerge from the priming process in LNs, we refer to recent insights from intravital imaging. In secondary lymphoid organs, individual T cells move from one DC to the other, until they are arrested by a TCR‐mediated “stop” signal on the DC that presents their cognate antigen. Among DC subsets, migratory cDCs deliver self‐antigens at steady state and may deliver foreign antigen from infected or transformed tissue, while LN‐resident cDCs pick up antigen locally. Data argue that CD4+ and CD8+ T cells are initially activated independent from each other by migratory cDC1 and cDC2 subsets. After this first step of priming, a second step of priming takes place on LN‐resident cDC1s [79, 80, 81]. In this interaction, CD4+ T cell help for CD8+ T cells is delivered [77]. Upon cognate contact with the CD4+ T cell, the LN‐resident cDC1 gains the ability to optimize the CD8+ T cell response, in terms of clonal expansion, effector and effector/memory differentiation. Key signals include interaction between CD40 ligand on the CD4+ T cell and CD40 on the cDC1, which promotes its expression of IL‐12, CD80/CD86 and CD70. CD27 co‐stimulation of CD8+ T cells via CD70 is a very important effector pathway of CD4+ T cell help [75, 77]. Whether CD4+ T cell help is delivered depends on chemokines such as XCL‐1 that bring the cells together and on type I IFN that is likely delivered by plasmacytoid DCs [82]. Type I IFN has long been known to promote CTL responses and optimizes the cross‐presentation and costimulatory ability of cDC1s [83].
We have found that CD8+ T cells primed in absence of CD4+ T cell help can go from the LN into the circulation, but then display the helpless/predysfunctional phenotype that includes PD‐1 cell surface expression. In contrast, cells that have received help have downregulated PD‐1 [75]. Thus, we propose a model in which the PD‐1 axis operates transiently during T cell priming to inhibit CD28 co‐stimulation and resulting T cell clonal expansion, likely at the first step of priming. It is intuitive that clonal expansion should only take place optimally after the second step of priming, since in this way, only a limited number of LN‐resident cDC1s are needed to relay CD4+ T cell help. In tumor settings, PD‐1 expression on CD8+ T cells in the blood enriches for tumor‐specificity [84], in line with the idea that tumors likely prime helpless CTLs, because essential signals such as activated CD4+ T cells and/or type I IFN may be lacking [83].
Several preclinical studies in chronic LCMV infection and tumor models have shown that helpless/predysfunctional PD1+ CD8+ T cells are the population that is reinvigorated by PD‐(L)1 blockade [13, 16, 26]. In line with the idea that they receive CD28 co‐stimulation, reinvigorated cells undergo proliferation and transiently gain effector functions that can contribute to tumor control, but they do not undergo full CTL effector differentiation. Instead, they retain expression of inhibitory receptors and eventually turn into terminally exhausted cells [16, 85]. At present, there are no data that argue in favor of a transition of predysfunctional CD8+ T cells into terminally differentiated, short‐lived effector CTL upon PD‐L1‐targeting therapy [11]. Reinvigoration likely takes place on migratory cDC1s that present antigen from the tumor. The interaction can take place within the TME, and/or in the tdLN (Fig. 5).
Figure 5.

Proposed effects of PD‐L1 targeted therapy in tdLNs and tumor. The effects of PD‐L1 blockade are threefold: (1) TCR/CD28 signaling is enabled during priming of naive (tumor‐specific) T cells, inducing proliferation of these cells. However, PD‐L1 blockade alone is not sufficient to allow the second step of priming in which help signals are delivered (dotted lines). Therefore, (2) PD‐L1 blockade in the tdLN increases the pool of (tumor‐specific) predysfunctional T cells. These cells will exit the tdLN and potentially reach the tumor, even though they have limited migratory ability. (3) In the TME, PD‐L1 blockade will also allow predysfunctional T cells to proliferate and transiently improve their cytotoxic capacities. However, ultimately these “reinvigorated” T cells will become exhausted. When “help mimicking” signals such as CD40 or CD27 agonism are added to PD‐L1 blockade, the second step of priming will be recapitulated and helped effector CTLs will be generated that lack PD‐1 expression and can more effectively kill the tumor cells. CD4+ T cell help (or its replacing signals) also allows for generation of CTL memory. Color codes: beige = naive CD8+ T cell; blue = cDC; orange = unhelped/predysfunctional CD8+ T cell; brown = CD4+ T helper cell; green = optimally primed/helped CTL; red = exhausted CD8+ T cell; grey = tumor cell.
Conclusions and future perspectives
The remarkable effects of PD‐L1 checkpoint blockade have put immunotherapy firmly on the map as a treatment modality for cancers that cannot be controlled by surgery or radiotherapy alone. As we reviewed here, PD‐L1 blockade transiently activates predysfunctional CD8+ T cells by enabling TCR signaling and CD28 co‐stimulation at the interface with cDCs, either in the tumor or in the tdLNs. These new insights argue that sparing tdLNs during surgery is important in adjuvant immunotherapy. The recent finding that PD‐L1 blockade can likewise promote Treg responses, brings Tregs into the equation when considering effects of PD‐1 blockade on T cell priming and effector stages. Furthermore, we propose that the restoration of CTL function by PD‐L1 blockade is temporary and incomplete, because other essential costimulatory and cytokine signals are still lacking. For adequate display of CTL effector function and generation of memory, the full spectrum of CD4+ T cell help signals should be provided to predysfunctional CD8+ T cells (Fig. 5). CD4+ T cell help signals can be replaced by combining PD‐L1 blockade with CD27 agonism [77, 86, 87, 88]. Alternatively, CD40 agonism will instruct cDCs to provide all required costimulatory signals and cytokines [77]. In addition, enlarging the pool of cDC1s by the differentiation factor Flt3L, in combination with DC activation signals can be a powerful approach to break anti‐PD‐1 resistance [89]. A constraint in all of these approaches may be that tumors prime strong Treg responses, which reduce the ability of tdLN to prime tumor‐specific conventional T cells [90, 91]. Detailed knowledge on the division of labor between CD80, CD86, PD‐L1, and PD‐L2 and their impact on Treg versus Tconv priming should help to allow for new interventions that do not break tolerance to self‐antigen but do overrule Treg activity and promote responses to tumor antigens.
Author contributions
Jannie Borst wrote the paper, Julia Busselaar provided insights for text and figures and edited the paper, D.M.T.B. made the figures and edited the paper, F.O. provided insights and edited the paper.
Conflict of interest
The authors declare no commercial or financial conflict of interest.
Abbreviations
- c
conventional
- CTL
cytotoxic T lymphocytes
- LCMV
lymphocytic choriomeningitis virus
- mAb
monoclonal antibody
- NSCLC
non‐small cell lung cancer
- PD
programmed Death
- Tconv
conventional T cells
- TCR
T cell receptor
- td
tumor‐draining
- TME
tumor microenvironment
Acknowledgments
This work was financially supported by grant 11079 from the Dutch Cancer Society (KWF) to Jannie Borst. We thank Elselien Frijlink for helpful discussions.
References
- 1.Sharma, P., Hu‐Lieskovan, S., Wargo, J. A. and Ribas, A., Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017. 168: 707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ribas, A. and Wolchok, J. D., Cancer immunotherapy using checkpoint blockade. Science. 2018. 359: 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schumacher, T. N. and Schreiber, R. D., Neoantigens in cancer immunotherapy. Science. 2015. 348: 69–74. [DOI] [PubMed] [Google Scholar]
- 4.Palucka, A. K. and Coussens, L. M., The basis of oncoimmunology. Cell. 2016. 164: 1233–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Topalian, S. L., Taube, J. M. and Pardoll, D. M., Neoadjuvant checkpoint blockade for cancer immunotherapy. Science. 2020. 367: eaax0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang, Y., Kong, D., Wang, C., Chen, J., Li, J., Liu, Z., Li, X., et al. A systematic review and meta‐analysis of immune‐related adverse events of anti‐PD‐1 drugs in randomized controlled trials. Technol. Cancer Res. Treat. 2020. 19: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gibney, G. T., Weiner, L. M. and Atkins, M. B., Predictive biomarkers for checkpoint inhibitor‐based immunotherapy. Lancet Oncol. 2016. 17: e542–e551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McGrail, D. J., Pilié, P. G., Rashid, N. U., Voorwerk, L., Slagter, M., Kok, M., Jonasch, E., et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann. Oncol. 2021: Advance online publication. DOI: 10.1016/j.annonc.2021.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wherry, E. J. and Kurachi, M., Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015. 15: 486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blank, C. U., Haining, W. N., Held, W., Hogan, P. G., Kallies, A., Lugli, E., Lynn, R. C., et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019. 19: 665–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van der Leun, A. M., Thommen, D. S. and Schumacher, T. N., CD8+ T cell states in human cancer: insights from single‐cell analysis. Nat. Rev. Cancer. 2020. 20: 218–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Busselaar, J., Tian, S., van Eenennaam, H. and Borst, J., Helpless priming sends CD8+ T cells on the road to exhaustion. Front. Immunol. 2020. 11: 592569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Utzschneider, D. T., Charmoy, M., Chennupati, V., Pousse, L., Ferreira, D. P., Calderon‐Copete, S., Danilo, M., et al. T Cell Factor 1‐expressing memory‐like CD8+T cells sustain the immune response to chronic viral infections. Immunity. 2016. 45: 415–427. [DOI] [PubMed] [Google Scholar]
- 14.Wu, T., Ji, Y., Moseman, E. A., Xu, H. C., Manglani, M., Kirby, M., Anderson, S. M., et al. The TCF1‐Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci. Immunol. 2016. 1: eaai8593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen, Z., Ji Z., Ngiow, S. F., Manne, S., Cai, Z., Huang, A. C., Johnson, J., et al. TCF‐1‐centered transcriptional network drives an effector versus exhausted CD8 T cell‐fate decision. Immunity. 2019. 51: 840–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller, B. C., Sen, D. R., Al Abosy, R., Bi, K., Virkud, Y. V., LaFleur, M. W., Yates, K. B., et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019. 20: 326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beltra, J. ‐. C., Manne, S., Abdel‐Hakeem, M. S., Kurachi, M., Giles, J. R., Chen, Z., Casella, V., et al. Developmental relationships of four exhausted CD8+ T cell subsets reveals underlying transcriptional and epigenetic landscape control mechanisms. Immunity. 2020. 52: 825–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schietinger, A., Philip, M., Krisnawan, V. E., Chiu, E. Y., Delrow, J. J., Basom, R. S., Lauer, P., et al. Tumor‐specific T cell dysfunction is a dynamic antigen‐driven differentiation program initiated early during tumorigenesis. Immunity. 2016. 45: 389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Philip, M., Fairchild, L., Sun, L., Horste, E. L., Camara, S., Shakiba, M., Scott, A. C., et al. Chromatin states define tumour‐specific T cell dysfunction and reprogramming. Nature. 2017. 545: 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng, C., Zheng, L., Yoo, J. K., Guo, H., Zhang, Y., Guo, X., Kang, B., et al. Landscape of infiltrating T cells in liver cancer revealed by single‐cell sequencing. Cell. 2017. 169: 1342–1356. [DOI] [PubMed] [Google Scholar]
- 21.Brummelman, J., Mazza, E. M. C., Alvisi, G., Colombo, F. S., Grilli, A., Mikulak, J., Mavilio, D., et al. High‐dimensional single cell analysis identifies stem‐like cytotoxic CD8+ T cells infiltrating human tumors. J. Exp. Med. 2018. 215: 2520–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo, X., Zhang, Y., Zheng, L., Zheng, C., Song, J., Zhang, Q., Kang, B., et al. Global characterization of T cells in non‐small‐cell lung cancer by single‐cell sequencing. Nat. Med. 2018. 24: 978–985. [DOI] [PubMed] [Google Scholar]
- 23.Zhang, L., Yu, X., Zheng, L., Zhang, Y., Li, Y., Fang, Q., Gao, R., et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature. 2018. 564: 268–272. [DOI] [PubMed] [Google Scholar]
- 24.Jansen, C. S., Prokhnevska, N., Master, V. A., Sanda, M. G., Carlisle, J. W., Bilen, M. A., Cardenas, M., et al. An intra‐tumoral niche maintains and differentiates stem‐like CD8 T cells. Nature. 2019. 576: 465–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li, H., van der Leun, A. M., Yofe, I., Lubling, Y., Gelbard‐Solodkin, D., van Akkooi, A. C. J., van den Braber, M., et al. Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell. 2019. 176: 775–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Im, S. J., Hashimoto, M., Gerner, M. Y., Lee, J., Kissick, H. T., Burger, M. C., Shan, Q., et al. Defining CD8+ T cells that provide the proliferative burst after PD‐1 therapy. Nature. 2016. 537: 417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He, R., Hou, S., Liu, C., Zhang, A., Bai, Q., Han, M., Yang, Y., et al. Follicular CXCR5‐expressing CD8+ T cells curtail chronic viral infection. Nature. 2016. 537: 412–416. [DOI] [PubMed] [Google Scholar]
- 28.Siddiqui, I., Schaeuble, K., Chennupati, V., Fuertes Marraco, S. A., Calderon‐Copete, S., Pais Ferreira, D., Carmona, S. J., et al. Intratumoral Tcf1+PD‐1+CD8+ T cells with stem‐like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity. 2019. 50: 195–211. [DOI] [PubMed] [Google Scholar]
- 29.Kurtulus, S., Madi, A., Escobar, G., Klapholz, M., Nyman, J., Christian, E., Pawlak, M., et al. Checkpoint blockade immunotherapy induces dynamic changes in PD‐1 − CD8 + tumor‐infiltrating T cells. Immunity. 2019. 50: 181–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sade‐Feldman, M., Yizhak, K., Bjorgaard, S. L., Ray, J. P., de Boer, C. G., Jenkins, R. W., Lieb, D. J., et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell. 2018. 175: 998–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iwai, Y., Ishida, M., Tanaka, Y., Okazaki, T., Honjo, T. and Minato, N., Involvement of PD‐L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD‐L1 blockade. Proc. Natl. Acad. Sci. U. S. A. 2002. 99: 12293–12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keir, M. E., Butte, M. J., Freeman, G. J. and Sharpe, A. H., PD‐1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008. 26: 677–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Philips, E. A., Garcia‐España, A., Tocheva, A. S., Ahearn, I. M., Adam, K. R., Pan, R., Mor, A., et al. The structural features that distinguish PD‐L2 from PD‐L1 emerged in placental mammals. J. Biol. Chem. 2020. 295: 4372–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herbst, R. S., Soria, J., Kowanetz, M., Fine, G. D., Hamid, O., Gordon, M. S., Sosman, J. A., et al. Predictive correlates of response to the anti‐PD‐L1 antibody MPDL3280A in cancer patients. Nature. 2014. 515: 563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lau, J., Cheung, J., Navarro, A., Lianoglou, S., Haley, B., Totpal, K., Sanders, L., et al. Tumour and host cell PD‐L1 is required to mediate suppression of anti‐tumour immunity in mice. Nat. Commun. 2017. 8: 14572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang, H., Liang, Y., Anders, R. A., Taube, J. M., Qiu, X., Mulgaonkar, A., Liu, X., et al. PD‐L1 on host cells is essential for PD‐L1 blockade–mediated tumor regression. J. Clin. Invest. 2018. 128: 580–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kleinovink, J. W., Marijt, K. A., Schoonderwoerd, M. J. A., van Hall, T., Ossendorp, F. and Fransen, M. F., PD‐L1 expression on malignant cells is no prerequisite for checkpoint therapy. Oncoimmunology. 2017. 6: e1294299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin, H., Wei, S., Hurt, E. M., Green, M. D., Zhao, L., Vatan, L., Szeliga, W., et al. Host expression of PD‐L1 determines efficacy of PD‐L1 pathway blockade–mediated tumor regression. J. Clin. Invest. 2018. 128: 805–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peng, Q., Qiu, X., Zhang, Z., Zhang, S., Zhang, Y., Liang, Y., Guo, J., et al. PD‐L1 on dendritic cells attenuates T cell activation and regulates response to immune checkpoint blockade. Nat. Commun. 2020. 11: 4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oh, S. A., Wu, D. ‐. C., Cheung, J., Navarro, A., Xiong, H., Cubas, R., Totpal, K., et al. PD‐L1 expression by dendritic cells is a key regulator of T‐cell immunity in cancer. Nat. Cancer. 2020. 1: 681–691. [DOI] [PubMed] [Google Scholar]
- 41.Sánchez‐Paulete, A. R., Cueto, F. J., Martínez‐López, M., Labiano, S., Morales‐Kastresana, A., Rodríguez‐Ruiz, M. E., Jure‐Kunkel, M., et al. Cancer immunotherapy with immunomodulatory anti‐CD137 and anti–PD‐1 monoclonal antibodies requires BATF3‐dependent dendritic cells. Cancer Discov. 2016. 6: 71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salmon, H., Idoyaga, J., Rahman, A., Leboeuf, M., Remark, R., Jordan, S., Casanova‐Acebes, M., et al. Expansion and activation of CD103+ dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD‐L1 and BRAF inhibition. Immunity. 2016. 44: 924–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garris, C. S., Arlauckas, S. P., Kohler, R. H., Zippelius, A., Weissleder, R. and Correspondence, M. J. P., Successful anti‐PD‐1 cancer immunotherapy requires T cell‐dendritic cell crosstalk involving the cytokines IFN‐g and IL‐12. Immunity. 2018. 49: 1148–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chow, M. T., Ozga, A. J., Servis, R. L., Frederick, D. T., Lo, J. A., Fisher, D. E., Freeman, G. J., et al. Intratumoral activity of the CXCR3 chemokine system is required for the efficacy of anti‐PD‐1 therapy. Immunity. 2019. 50: 1498–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mayoux, M., Roller, A., Pulko, V., Sammicheli, S., Chen, S., Sum, E., Jost, C., et al. Dendritic cells dictate responses to PD‐L1 blockade cancer immunotherapy. Sci. Transl. Med. 2020. 12: eaav7431. [DOI] [PubMed] [Google Scholar]
- 46.Gajewski, T. F. and Cron, K. R., CDC1 dysregulation in cancer: An opportunity for intervention. J. Exp. Med. 2020. 217: e20200816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Probst, H. C., McCoy, K., Okazaki, T., Honjo, T. and Van Den Broek, M., Resting dendritic cells induce peripheral CD8+ T cell tolerance through PD‐1 and CTLA‐4. Nat. Immunol. 2005. 6: 280–286. [DOI] [PubMed] [Google Scholar]
- 48.Baumeister, S. H., Freeman, G. J., Dranoff, G. and Sharpe, A. H., Coinhibitory pathways in immunotherapy for cancer. Annu. Rev. Immunol. 2016. 34: 539–573. [DOI] [PubMed] [Google Scholar]
- 49.Freeman, G. J., Long, A. J., Iwai, Y., Bourque, K., Chernova, T., Nishimura, H., Fitz, L. J., et al. Engagement of the PD‐1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000. 192: 1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fife, B. T., Pauken, K. E., Eagar, T. N., Obu, T., Wu, J., Tang, Q., Azuma, M., et al. Interactions between PD‐1 and PD‐L1 promote tolerance by blocking the TCR‐induced stop signal. Nat. Immunol. 2009. 10: 1185–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yokosuka, T., Takamatsu, M., Kobayashi‐Imanishi, W., Hashimoto‐Tane, A., Azuma, M. and Saito, T., Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 2012. 209: 1201–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ahn, E., Araki, K., Hashimoto, M., Li, W., Riley, J. L., Cheung, J., Sharpe, A. H., et al. Role of PD‐1 during effector CD8 T cell differentiation. Proc. Natl. Acad. Sci. 2018. 115: 4749–4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen, D. S. and Mellman, I., Elements of cancer immunity and the cancer‐immune set point. Nature. 2017. 541: 321–330. [DOI] [PubMed] [Google Scholar]
- 54.Fransen, M. F., Schoonderwoerd, M., Knopf, P., Camps, M. G., Hawinkels, L. J., Kneilling, M., van Hall, T., et al. Tumor‐draining lymph nodes are pivotal in PD‐1/PD‐L1 checkpoint therapy. JCI Insight. 2018. 3: e124507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dammeijer, F., van Gulijk, M., Mulder, E. E., Lukkes, M., Klaase, L., van den Bosch, T., van Nimwegen, M., et al. The PD‐1/PD‐L1‐checkpoint restrains T cell immunity in tumor‐draining lymph nodes. Cancer Cell. 2020. 38: 1–16. [DOI] [PubMed] [Google Scholar]
- 56.Kamphorst, A. O., Pillai, R. N., Yang, S., Nasti, T. H., Akondy, R. S., Wieland, A., Sica, G. L., et al. Proliferation of PD‐1+ CD8 T cells in peripheral blood after PD‐1‐targeted therapy in lung cancer patients. Proc. Natl. Acad. Sci. U. S. A. 2017. 114: 4993–4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang, J., Ji, Z., Caushi, J. X., El Asmar, M., Anagnostou, V., Cottrell, T. R., Chan, H. Y., et al. Compartmental analysis of T‐cell clonal dynamics as a function of pathologic response to neoadjuvant PD‐1 blockade in resectable non‐small cell lung cancer. Clin. Cancer Res. 2020. 26: 1327–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yost, K. E., Satpathy, A. T., Wells, D. K., Qi, Y., Wang, C., Kageyama, R., McNamara, K. L., et al. Clonal replacement of tumor‐specific T cells following PD‐1 blockade. Nat. Med. 2019. 25: 1251–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu, T. D., Madireddi, S., de Almeida, P. E., Banchereau, R., Chen, Y. ‐. J. J., Chitre, A. S., Chiang, E. Y., et al. Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature. 2020. 579: 274–278. [DOI] [PubMed] [Google Scholar]
- 60.Callahan, M. K. and Wolchok, J. D., Recruit or reboot? How does anti‐PD‐1 therapy change tumor‐infiltrating lymphocytes? Cancer Cell. 2019. 36: 215–217. [DOI] [PubMed] [Google Scholar]
- 61.Walker, L. S. K. and Sansom, D. M., Confusing signals: Recent progress in CTLA‐4 biology. Trends Immunol. 2015. 36: 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wing, K., Yamaguchi, T. and Sakaguchi, S., Cell‐autonomous and ‐non‐autonomous roles of CTLA‐4 in immune regulation. Trends Immunol. 2011. 32: 428–433. [DOI] [PubMed] [Google Scholar]
- 63.Hui, E., Cheung, J., Zhu, J., Su, X., Taylor, M. J., Wallweber, H. A., Sasmal, D. K., et al. T cell costimulatory receptor CD28 is a primary target for PD‐1‐mediated inhibition. Science. 2017. 355: 1428–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Celis‐Gutierrez, J., Blattmann, P., Zhai, Y., Jarmuzynski, N., Ruminski, K., Grégoire, C., Ounoughene, Y., et al. Quantitative interactomics in primary T cells provides a rationale for concomitant PD‐1 and BTLA coinhibitor blockade in cancer immunotherapy. Cell Rep. 2019. 27: 3315–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Patsoukis, N., Duke‐Cohan, J. S., Chaudhri, A., Aksoylar, H. ‐. I., Wang, Q., Council, A., Berg, A., et al. Interaction of SHP‐2 SH2 domains with PD‐1 ITSM induces PD‐1 dimerization and SHP‐2 activation. Commun. Biol. 2020. 3: 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Okazaki, T., Maeda, A., Nishimura, H., Kurosaki, T. and Honjo, T., PD‐1 immunoreceptor inhibits B cell receptor‐mediated signaling by recruiting src homology 2‐domain‐containing tyrosine phosphatase 2 to phosphotyrosine. Proc. Natl. Acad. Sci. U. S. A. 2001. 98: 13866–13871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kamphorst, A. O., Wieland, A., Nasti, T., Yang, S., Zhang, R., Barber, D. L., Konieczny, B. T., et al. Rescue of exhausted CD8 T cells by PD‐1–targeted therapies is CD28‐dependent. Science. 2017. 355: 1423–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wei, S. C., Levine, J. H., Cogdill, A. P., Zhao, Y., Anang, N., Andrews, M. C., Sharma, P., et al. Distinct cellular mechanisms underlie anti‐CTLA‐4 and anti‐PD‐1 checkpoint blockade. Cell. 2017. 170: 1120–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chaudhri, A., Xiao, Y., Klee, A. N., Wang, X., Zhu, B. and Freeman, G. J., PD‐L1 binds to B7‐1 only in cis on the same cell surface. Cancer Immunol. Res. 2018. 6: 921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhao, Y., Lee, C. K., Lin, C. H., Gassen, R. B., Xu, X., Huang, Z., Xiao, C., et al. PD‐L1:CD80 cis‐heterodimer triggers the co‐stimulatory receptor CD28 while repressing the inhibitory PD‐1 and CTLA‐4 pathways. Immunity. 2019. 51: 1059–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sugiura, D., Maruhashi, T., Okazaki, I. M., Shimizu, K., Maeda, T. K., Takemoto, T. and Okazaki, T., Restriction of PD‐1 function by cis‐PD‐L1/CD80 interactions is required for optimal T cell responses. Science. 2019. 364: 558–566. [DOI] [PubMed] [Google Scholar]
- 72.Wang, L., Pino‐Lagos, K., De Vries, V. C., Guleria, I., Sayegh, M. H. and Noelle, R. J., Programmed death 1 ligand signaling regulates the generation of adaptive Foxp3+CD4+ regulatory T cells. Proc. Natl. Acad. Sci. U. S. A. 2008. 105: 9331–9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tan, C. L., Kuchroo, J. R., Sage, P. T., Liang, D., Francisco, L. M., Buck, J., Thaker, Y. R., et al. PD‐1 restraint of regulatory T cell suppressive activity is critical for immune tolerance. J. Exp. Med. 2021. 218: e20182232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kumagai, S., Togashi, Y., Kamada, T., Sugiyama, E., Nishinakamura, H., Takeuchi, Y., Vitaly, K., et al. The PD‐1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD‐1 blockade therapies. Nat. Immunol. 2020. 21: 1346–1358. [DOI] [PubMed] [Google Scholar]
- 75.Ahrends, T., Spanjaard, A., Pilzecker, B., Bąbała, N., Bovens, A., Xiao, Y., Jacobs, H., et al. CD4+T cell help confers a cytotoxic T cell effector program including coinhibitory receptor downregulation and increased tissue invasiveness. Immunity. 2017. 47: 848–861. [DOI] [PubMed] [Google Scholar]
- 76.Ahrends, T., Busselaar, J., Severson, T. M., Bąbała, N., de Vries, E., Bovens, A., Wessels, L., et al. CD4+ T cell help creates memory CD8+ T cells with innate and help‐independent recall capacities. Nat. Commun. 2019. 10: 5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Borst, J., Ahrends, T., Bąbała, N., Melief, C. J. M. and Kastenmüller, W., CD4+ T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2018. 18: 635–647. [DOI] [PubMed] [Google Scholar]
- 78.Provine, N. M., Larocca, R. A., Aid, M., Penaloza‐MacMaster, P., Badamchi‐Zadeh, A., Borducchi, E. N., Yates, K. B., et al. Immediate dysfunction of vaccine‐elicited CD8 + T cells primed in the absence of CD4 + T cells. J. Immunol. 2016. 197: 1809–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Eickhoff, S., Gobel, A., Gerner, M. Y., Klauschen, F., Komander, K., Hemmi, H., Garbi, N., et al. Robust anti‐viral immunity requires multiple distinct T cell‐dendritic cell interactions. Cell. 2015. 162: 1322–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hor, J. L., Whitney, P. G., Zaid, A., Brooks, A. G., Heath, W. R. and Mueller, S. N., Spatiotemporally distinct interactions with dendritic cell subsets facilitates CD4+and CD8+T cell activation to localized viral infection. Immunity. 2015. 43: 554–565. [DOI] [PubMed] [Google Scholar]
- 81.Kitano, M., Yamazaki, C., Takumi, A., Ikeno, T., Hemmi, H., Takahashi, N., Shimizu, K., et al. Imaging of the cross‐presenting dendritic cell subsets in the skin‐draining lymph node. Proc. Natl. Acad. Sci. U. S. A. 2016. 113: 1044–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Brewitz, A., Eickhoff, S., Dähling, S., Quast, T., Bedoui, S., Kroczek, R. A., Kurts, C., et al. CD8+T cells orchestrate pDC‐XCR1+dendritic cell spatial and functional cooperativity to optimize priming. Immunity. 2017. 46: 205–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Flood, B. A., Higgs, E. F., Li, S., Luke, J. J. and Gajewski, T. F., STING pathway agonism as a cancer therapeutic. Immunol. Rev. 2019. 290: 24–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gros, A., Parkhurst, M. R., Tran, E., Pasetto, A., Robbins, P. F., Ilyas, S., Prickett, T. D., et al. Prospective identification of neoantigen‐specific lymphocytes in the peripheral blood of melanoma patients. Nat. Med. 2016. 22: 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hudson, W. H., Gensheimer, J., Hashimoto, M., Wieland, A., Valanparambil, R. M., Li, P., Lin, J. ‐. X., et al. Proliferating transitory T cells with an effector‐like transcriptional signature emerge from PD‐1+ stem‐like CD8+ T cells during chronic infection. Immunity. 2019. 51: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ahrends, T., Babala, N., Xiao, Y., Yagita, H., van Eenennaam, H. and Borst, J., CD27 agonism plus PD‐1 blockade recapitulates CD4+ T‐cell help in therapeutic anticancer vaccination. Cancer Res. 2016. 76: 2921–2931. [DOI] [PubMed] [Google Scholar]
- 87.Buchan, S. L., Fallatah, M., Thirdborough, S. M., Taraban, V. Y., Rogel, A., Thomas, L. J., Penfold, C. A., et al. PD‐1 blockade and CD27 stimulation activate distinct transcriptional programs that synergize for CD8+ T‐cell–driven antitumor immunity. Clin. Cancer Res. 2018. 24: 2383–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vitale, L. A., He, L. Z., Thomas, L. J., Wasiuk, A., O'Neill, T., Widger, J., Crocker, A., et al. Development of CDX‐527: a bispecific antibody combining PD‐1 blockade and CD27 costimulation for cancer immunotherapy. Cancer Immunol. Immunother. 2020. 69: 2125–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Oba, T., Long, M. D., Keler, T., Marsh, H. C., Minderman, H., Abrams, S. I., Liu, S., et al. Overcoming primary and acquired resistance to anti‐PD‐L1 therapy by induction and activation of tumor‐residing cDC1s. Nat. Commun. 2020. 11: 5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bauer, C. A., Kim, E. Y., Marangoni, F., Carrizosa, E., Claudio, N. M. and Mempel, T. R., Dynamic Treg interactions with intratumoral APCs promote local CTL dysfunction. J. Clin. Invest. 2014. 124: 2425–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Heeren, A. M., Koster, B. D., Samuels, S., Ferns, D. M., Chondronasiou, D., Kenter, G. G., Jordanova, E. S., et al. High and interrelated rates of PD‐L1+ CD14+ antigen‐presenting cells and regulatory T cells mark the microenvironment of metastatic lymph nodes from patients with cervical cancer. Cancer Immunol. Res. 2015. 3: 48–58. [DOI] [PubMed] [Google Scholar]