

Abstract
The aim of this study was to identify a high‐affinity BODIPY peptidomimetic that targets the prostate‐specific membrane antigen (PSMA) as a potential bimodal imaging probe for prostate cancer. For the structure‐activity study, several BODIPY (difluoroboron dipyrromethene) derivatives with varying spacers between the BODIPY dye and the PSMA Glu‐CO‐Lys binding motif were prepared. Corresponding affinities were determined by competitive binding assays in PSMA‐positive LNCaP cells. One compound was identified with comparable affinity (IC50=21.5±0.1 nM) to Glu‐CO‐Lys‐Ahx‐HBED‐CC (PSMA‐11) (IC50=18.4±0.2 nM). Radiolabeling was achieved by Lewis‐acid‐mediated 19F/18F exchange in moderate molar activities (∼0.7 MBq nmol−1) and high radiochemical purities (>99 %) with mean radiochemical yields of 20–30 %. Cell internalization of the 18F‐labeled high‐affinity conjugate was demonstrated in LNCaP cells showing gradual increasing PSMA‐mediated internalization over time. By fluorescence microscopy, localization of the high‐affinity BODIPY‐PSMA conjugate was found in the cell membrane at early time points and also in subcellular compartments at later time points. In summary, a high‐affinity BODIPY‐PSMA conjugate has been identified as a suitable candidate for the development of PSMA‐specific dual‐imaging agents.
Keywords: bimodal imaging, PET, BODIPY, PSMA, fluorescence imaging
Have it both ways: By structure–activity relationship studies, a bimodal imaging probe combining fluorescence with nuclear imaging based on BODIPY dyes with high affinity for the prostate‐specific membrane antigen (PSMA) has been identified. The bimodal conjugate demonstrated PSMA‐specific uptake in prostate cancer cells with accumulation in the cell membrane at early time points and additional accumulation in subcellular compartments at later time points.
Introduction
Optical fluorescence imaging represents an integral part of modern clinical oncology and provides numerous advantages for cancer detection, staging, characterization or therapy response assessment, including a high spatial resolution, a high portability as well as a detailed molecular profiling.[1] Because of its inherent properties, such as real‐time readout, the production of high‐resolution images, an easy complementation with other imaging modalities and the possibility to detect multiple signals concomitantly, fluorescence imaging is also suitable for tumor resection in targeted surgery.[2, 3] With the emergence of optical fluorescent imaging modalities as well as advanced in vivo or in vitro microscopy techniques in oncology and biomedical research, various fluorescent probes have been developed for targeting biological structures.[4, 5] Besides a large variety of sensors based on fluorescent proteins,[4, 6, 7, 8] a fluorescent probe typically consists of small organic fluorophores[9, 10] or lanthanide complexes,[11, 12] which allows in combination with a tumor‐specific vector the design of highly sensitive targeting probes for several cancer entities.[13] Among the large set of highly fluorescent probes in oncology and cancer research,[14, 15, 16] the class of sensors based on the 4,4‐difluoro‐4‐bora‐3a,4a‐diaza‐s‐indacene (difluoroboron dipyrromethene=BODIPY) scaffold given in Scheme 1A shows great potential for imaging applications.[4, 17, 18] Since the first report of BODIPY dyes by Treibs and Kreuzer in 1968,[19] these dyes have become more and more popular in imaging techniques due to their ease of synthesis and their beneficial spectroscopic properties. BODIPY dyes show resistance towards self‐aggregation and have high photostability, high molar absorption coefficients and fluorescence quantum yields as well as emission wavelengths in the visible spectral region with narrow absorption and emission bands and a small Stokes shift.[18, 20, 21, 22, 23] Moreover, synthetic BODIPY core modifications enable the facile variation of the photophysical, spectroscopic and chemical properties, improving the biological compatibility.[17, 18, 20, 21, 24] BODIPY‐based sensors for biomedical research were successfully used for specific imaging of subcellular compartments, as sensitive imaging probes for various tumor‐related receptors or overexpressed target proteins as well as for screening to identify key biomolecules for selective imaging and visualization of biological processes.[4, 25, 26, 27, 28] Furthermore, the BODIPY core can be modified to facilitate singlet oxygen generation for the potential use in photodynamic therapy.[29] However, fluorescence imaging suffers from limited tissue penetration depths, a fact that still prevents the use of this technology for whole‐body‐scanning in humans.[2, 30] One possible solution of this limitation is the combination of fluorescence imaging with nuclear imaging techniques, such as single photoemission computed tomography (SPECT) or positron emission tomography (PET), or anatomical imaging modalities such as magnetic resonance imaging (MRI).[31, 32, 33] PET is a non‐invasive whole‐body imaging modality, that detects pairs of gamma rays, a secondary product of the positron annihilation.[2, 34, 35] A combination of fluorescence with nuclear imaging would thus allow imaging from the whole‐body down to the subcellular level, making the development of dual‐modality probes an active field of research.[34] A major advantage of the BODIPY scaffold is the possibility to introduce the positron emitting radionuclide fluorine‐18 by Lewis‐acid assisted isotopic 19F/18F exchange combining a fluorescent and a nuclear probe in a single entity.[34, 36, 37, 38] In this respect, the potential of BODIPY dyes for the development of dual‐imaging probes has recently been reviewed.[37]
Scheme 1.

A) difluoroboron dipyrromethene (BODIPY) scaffold. B) BODIPY dyes 1 with a carboxylic acid functionality for bioconjugation and its corresponding NHS ester 2 used in this study.
Here, we report on the development of hybrid PET and optical probes based on the BODIPY scaffold targeting the prostate‐specific membrane antigen (PSMA) for potential use as dual imaging probes for prostate cancer (PCa). PSMA, also known as glutamate carboxypeptidase type II (GCP II), is a 750‐residue type II transmembrane glycoprotein that has become a valuable molecular target for PCa imaging and therapy due to its selective upregulation in PCa lesions, such as lymph node as well as bone metastases.3, 39 PSMA is also a prognostic factor for disease recurrence.[44] With the emergence of radiolabeled urea‐based probes in nuclear medicine, imaging of PCa by PET/CT (computed tomography) has become increasingly important for staging, restaging, and treatment selection and had a tremendous impact on the diagnosis, treatment planning, therapy monitoring as well as for endoradiotherapy of PCa patients.[45, 46, 47, 48, 49, 50, 51, 52] Additionally, radiolabeled bimodal PSMA inhibitors are currently the subject of intensive research with promising results in several preclinical studies towards hybrid imaging of PCa.[53, 54] More recently, the (sub)cellular fate of peptidomimetic PSMA inhibitors was studied using stimulated emission depletion (STED) nanoscopy.[55] PSMA‐specific internalization of bimodal PSMA inhibitors is mediated via clathrin‐dependent endocytosis and a homogenous distribution of the PSMA inhibitors in the cytoplasm was found over time.
To the best of our knowledge, a radiolabeled BODIPY‐PSMA conjugate for dual‐imaging of PCa has not been described to date. Alquicer et al. reported on the fluorescent probe TMR‐X‐Lys‐urea‐Glu comprising a BODIPY fluorophore for the identification of new GCP II targeting ligands by high‐throughput fluorescence polarization assays.[56] More recently, Son et al. have reported on a PSMA conjugate comprising the NIR dye BODIPY650/665 with about ten‐fold lower affinity than the potent PSMA inhibitor ZJ‐43. The BODIPY‐PSMA conjugate showed specific uptake in PSMA‐expressing cells and allowed visualization of PSMA‐positive tumors in a mouse model by optical imaging.[57] However, radiolabeling with fluorine‐18 and/or small‐animal PET imaging were not reported.
To identify a high‐affinity BODIPY‐PSMA conjugate, we synthesized a small series of peptidomimetic BODIPY‐PSMA conjugates with various spacer entities to establish a structure‐activity relationship. Simultaneously, we also optimized the chemical syntheses due to the sensitivity of the BODIPY scaffold to acidic reaction conditions. We furthermore report on the effect of the peptidomimetic PSMA‐binding motif on the spectroscopic properties of the BODIPY dye 1 (Scheme 1B), the radiofluorination of corresponding BODIPY‐PSMA conjugates 3–6, and their biological evaluation in the PSMA‐expressing LNCaP cell line with regards to binding affinity and cell internalization. This way, a BODIPY‐PSMA conjugate could be identified that might be a suitable candidate for the development of PSMA‐specific dual‐imaging agents.
Results and Discussion
Bioconjugate syntheses
For the preparation of the series of BODIPY‐PSMA conjugates, a mix of solid‐phase and solution‐based chemistry was performed to take advantage of the rapid and efficient coupling reactions on solid support while avoiding the acid‐catalyzed decomposition of the BODIPY dye (Scheme 2). First, the Glu‐CO‐Lys PSMA binding motif was synthesized on solid support according to literature methods with minor modifications.[45] Here, 1,1’‐carbonyldiimidazole (CDI) instead of triphosgene was used for the build‐up of the Glu‐CO‐Lys moiety.[58] Additional amino acid spacers were consequently introduced stepwise by standard Fmoc‐protected peptide chemistry using HATU (O‐(7‐Azabenzotriazol‐1‐yl)‐N,N,N’,N’‐tetramethyluronium‐hexafluorphosphate)/DIPEA (N,N‐diisopropylethylamine) to prepare intermediates for conjugation of the BODIPY dye 1 in solution.[50] For the structure activity relationships studies, four different spacer entities were prepared. To gain insight into the optimal distance between the Glu‐CO‐Lys moiety and the BODIPY dye 1, either no spacer, one or two 6‐aminohexanoic acid residues were introduced. For the fourth derivative, a 2‐naphthylalanin‐aminomethylcyclohexylic acid (2‐Nal‐Amc) spacer similar to the spacer of PSMA‐617 was used.[50] It is well‐known that BODIPY dyes are prone to acidic conditions,[59, 60, 61, 62] making them incompatible with conditions required during solid phase peptide synthesis, e. g. acid‐catalyzed deprotection and cleavage from the resin using trifluoroacetic acid (TFA). To circumvent the formation of side products, corresponding Glu‐CO‐Lys conjugates were thus deprotected and cleaved from the resin using a cocktail of TFA/H2O/triisopropylsilane (95/2.5/2.5, % v/v) prior to the conjugation of the BODIPY dye.
Scheme 2.

Synthetic procedure for the preparation of BODIPY‐PSMA bioconjugates 3–6 using a combination of solid‐phase and solution‐based chemistry.
For our studies, we selected the BODIPY derivative 1 as fluorophore, shown in Scheme 1B, as it is readily synthesized and offers a carboxylic acid group allowing covalent conjugation of a targeting vector via peptide bond formation. Compound 1 was prepared according to the literature.[63, 64] By minor modifications of the reaction conditions and the workup process, the yield could be increased from 21 % in the original synthesis to 55 % in this work.[63] Compound 1 was also transformed into the corresponding NHS ester 2 to obtain a BODIPY derivative that can readily be coupled to a suitable targeting vector without the addition of a coupling agent (Scheme 1B). By dilution of 2 in acetone and slow evaporation of the solvent at ambient temperature, orange crystals were obtained, which were suitable for X‐ray analysis (CCDC number: 2054523).
A graphical representation of the crystal structure of 2 is given in Figure 1 and corresponding crystallographic data is provided in the Supporting Information. Structural features are comparable to literature reports with BODIPY moiety formed by three conjugated heterocyclic rings (the central six‐membered ring with two adjacent five‐membered rings) being almost planar.[63] Conjugation of 1 to the Glu‐CO‐Lys binding motifs with different spacer entities was performed in solution using two different coupling methods. Either compound 1 together with HATU or the NHS derivative 2 were reacted with corresponding PSMA conjugates in the presence of DIPEA. This way, BODIPY‐PSMA conjugates 3–6 were obtained in overall yields of 37–44 % after purification by semi‐preparative HPLC, respectively. No significant differences in terms of coupling efficiency between compound 1 and HATU vs. NHS ester 2 were noted. Final bioconjugates 3–6 were fully characterized by one‐ and two‐dimensional 1H, 13C, 19F‐NMR spectroscopy, mass spectrometry, fluorescence spectroscopy, and analytical HPLC. Corresponding data are provided in the Supporting Information.
Figure 1.
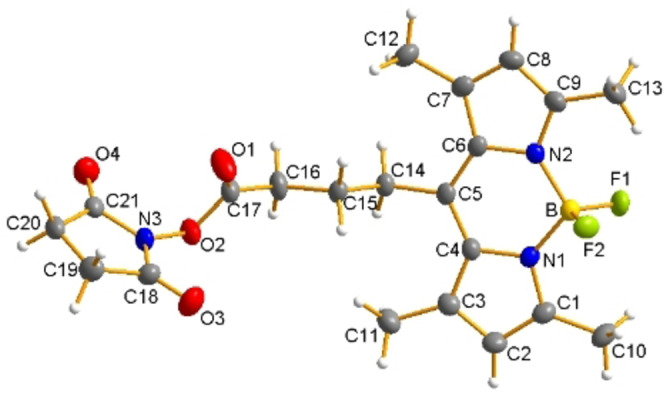
ORTEP representation of 2 with thermal ellipsoids drawn at 50 % probability level.
Fluorescence spectroscopy measurements
To investigate if conjugation of compound 1 to the targeting vectors impacted the fluorescence properties, additional fluorescence spectroscopy measurements of 3–6 in acetonitrile/water (1 : 1, v/v) were performed (Table 1). As can be seen from the results in Table 1 and Figure S27, the PSMA‐targeting peptidomimetics did not alter the absorption or the emission maxima of 3–6, with wavelengths in the range of λabs=494 nm to λabs=496 nm and λem=504 nm to λem=506 nm, respectively. We also examined the fluorescence quantum yield ϕFl and the fluorescence lifetime τFl. Conjugation of 1 to the PSMA ligands did not impact the fluorescence lifetime of 3–6 being in the same nanosecond range as for compound 1 (Table 1/Figure S27+S28). A faint increase of the fluorescence quantum yields of 3–6 compared to 1 could be the result of the transformation of the carboxylic acid group into an amide circumventing a potential negative charge in close proximity to the dipyrromethene core.
Table 1.
Spectroscopic data of 1 and BODIPY‐PSMA conjugates 3–6 in acetonitrile/water (1 : 1, v/v).
|
Compd |
λabs [nm] |
λexc [nm] |
λem [nm] |
ϕFl [%] |
τFl [ns] |
|---|---|---|---|---|---|
|
1 |
494 |
496 |
505 |
75±5 |
6.3±0.1 |
|
3 |
495 |
496 |
506 |
81±5 |
5.9±0.1 |
|
4 |
496 |
496 |
506 |
86±5 |
6.1±0.1 |
|
5 |
496 |
496 |
506 |
81±5 |
6.2±0.1 |
|
6 |
496 |
495 |
504 |
81±5 |
6.1±0.1 |
Studies on BODIPY stability
During the establishment of the synthetic routes of the BODIPY‐PSMA conjugates 3–6 and the radiolabeling experiments, we observed the decomposition of the BODIPY fluorophore under acidic conditions. For example, when performing the syntheses entirely on solid support and cleaving corresponding BODIPY‐PSMA conjugates from the resin using a TFA/H2O/triisopropylsilane (95/2.5/2.5, % v/v) cocktail, we isolated fractions by HPLC that eluted prior to the product peaks and that differed by a mass difference of m/z=‐48 in the mass spectrometry compared to the intact compounds confirming loss of the BF2‐core (Figure S25). To investigate the stability of the BODIPY dye under these conditions in more detail, compound 1 was incubated in 95 % TFA and samples were measured by fluorescence spectroscopy for up to 24 h (Figure 2A+B). While the absorption spectra of 1 in TFA only showed a small blue‐shift of maxima from λabs=496 nm to λabs=488 nm (Figure 2A), a significant change of corresponding emission spectra was noted. After a few seconds of the incubation process a slight red shift from λem=509 nm to λem=515 nm is observed (Figure 2B), probably due to the initial coordination of a proton to a nitrogen atom of the BODIPY core. Within 2 h a steady decrease of the emission intensity was noted, presumably as a result of the proton‐mediated loss of the BF2 moiety. At 24 h, the emission intensity was quenched completely, while the absorption intensity was still observable. The plot of the emission intensity vs. time gave the decay curve in Figure S34. From a tentative biexponential fit, two kinetic constants for decomposition/fading with k=0.168±0.011 min and k=0.018±0.006 min were obtained. Corresponding half‐lives of 1 in TFA were calculated to τ1/2=4.1±0.3 min and τ1/2=37.9±9.4 min. Our results are in agreement with results from Rumyantsev, who postulated a two‐step dissociation process of BODIPY complexes with a rapid protonation of the BODIPY core followed by a slow release of BF2 unit.[60, 61] We therefore confirm that proton coordination finally results in the release of the BF2 unit, while the chromophore remained intact. This finding is corroborated by the observation that fluorescence can widely be restored after 24 h upon addition of BF3‐diethyletherate (data not shown). The TFA‐mediated loss of the BF2 entity of compound 1 was also confirmed by analytical HPLC and MS (Figures S33B–S34). A similar finding was observed in the HPLC chromatograms of the 18F‐labeling reactions (vide infra).
Figure 2.
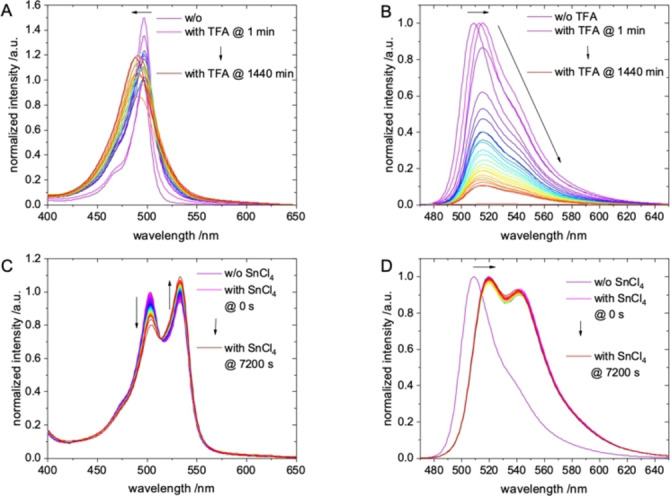
Normalized A) absorption and B) emission spectra (λexc=460 nm) of 1 in 95 % TFA at different time points. Normalized C) absorption and D) emission spectra (λexc=460 nm) of 1 in 1 M SnCl4 in acetonitrile at different time points.
18F‐labeling, stability measurements and log Doct/PBS determination
Next, the bioconjugates 3–6 were labeled at room temperature with 18F via Lewis‐acid assisted 19F/18F isotopic exchange in dry acetonitrile using tetraethylammonium‐[18F]fluoride ([18F]F‐TEA) and SnCl4 within 15 min incubation.[34, 38] A representative scheme of the radiolabeling process is given in Scheme 3 for compound 4. Optimization of the labeling conditions included variation of the reaction temperature and the amount of SnCl4. The highest radiochemical yields (RCYs) were achieved when a 100‐fold molar excess of SnCl4 compared to the labeling precursor was used. An increase of the labeling temperature to 37 °C or 95 °C or higher amounts of SnCl4 (e. g. 500 eq.) did not result in an increase of RCYs. Under optimized labeling conditions, [18F]F‐3, [18F]F‐4, [18F]F‐5 and [18F]F‐6 were obtained in radiochemical yields of 20–30 % and radiochemical purities (RCP) of >99 %. The mean molar activities A m were ∼0.7 MBq ⋅ nmol−1. Following the labeling reactions, the radiolabeled products [18F]F‐3, [18F]F‐4, [18F]F‐5 and [18F]F‐6 were purified by C18 SepPak cartridges and reconstituted in saline (5 % EtOH) for further experiments. While the RCPs were >99 %, an additional peak in the UV/vis trace of the HPLC chromatograms of [18F]F‐3, [18F]F‐4, [18F]F‐5 and [18F]F‐6 was observed, which matched the HLPC trace of the BF2‐free conjugates. This finding is exemplarily shown for [18F]F‐4 in Figure 3.
Scheme 3.

Representative radiolabeling of BODIPY‐PSMA conjugate 4 by Lewis acid mediated isotopic 19F/18F exchange.
Figure 3.
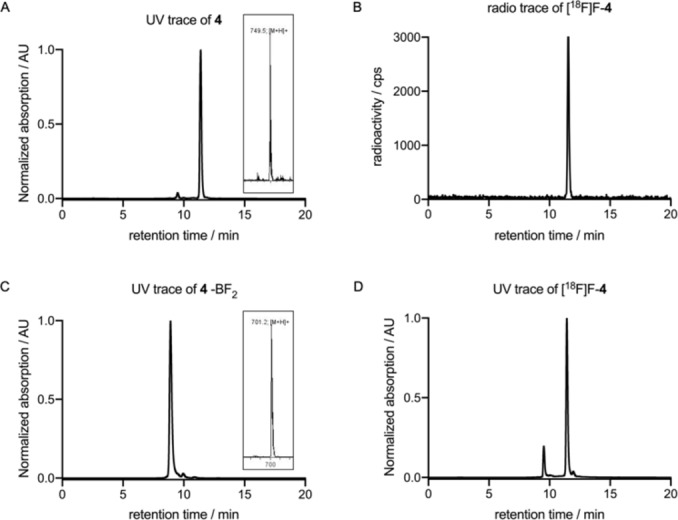
A) Representative UV/vis HPLC chromatogram of 4 (insert: MS spectrum of 4). B) Radio‐HPLC chromatogram of [18F]F‐4. C) Representative UV/vis HPLC chromatogram of 4 without BF2 (insert: MS spectrum of 4 ‐BF2). D) UV/vis HPLC chromatogram of [18F]F‐4 showing loss of the BF2 entity during radiolabeling by Lewis acid mediated isotopic 19F/18F isotopic exchange.
To gain more insight, the impact of the Lewis acid SnCl4, again on the decomposition of the precursor 1, was investigated in acetonitrile by UV/vis and fluorescence spectroscopy (Figure 2C+D). The corresponding emission spectra showed a bathochromic shift of the maximum from λem=509 nm to λem=520 nm compared to intact 1 immediately after dissolving in the SnCl4 solution (Figure 2D). Moreover, a second emission band with λem=545 nm was noted, which can unambiguously be traced back to an excitation maximum at λexc=530 nm (Figure S29). A similar observation was made for corresponding absorption spectra with an initial red shift and formation of a second absorption band varying slightly over the course of the measurements (Figure 2C). These changes might be attributed to the formation of an intermediate between the Lewis acid and the fluorine atoms of 1.[36] Another possible explanation could be the substitution of fluoride with chloride or even an insertion of SnClx. At least, both the pronounced spectral shift and the maintained fluorescence hint to distinctly disturbed, but still intact chromophore with a bridging heteroatom between the two pyrrole moieties. In summary, the acidic conditions for radiolabeling, mainly due to the Lewis acid SnCl4 and subsequent aqueous work‐up, also resulted in the partial loss of the BF2 core, which occurred to 5 % to 10 % depending on the reaction conditions. When higher amounts of SnCl4 were used, formation of by‐products increased. These impurities, however, could easily be separated from the 18F‐labeled bioconjugates by HPLC. And even more important, once formed, no degradation or formation of (radioactive) by‐products of HPLC‐purified [18F]F‐4 was observed in phosphate buffered saline (PBS) at pH 7.4 for 2 h, and the RCP was still >97 % after this incubation time, indicating high stability of the radiotracer.
An important indicator for the pharmacokinetics and target affinity is the lipophilicity of PSMA radiotracers. Thus, partition coefficients log D oct/PBS between 1‐octanol and PBS were determined for compounds [18F]F‐3‐6. Corresponding log D oct/PBS values of [18F]F‐3‐6 and [68Ga]Ga‐PSMA‐11 together with corresponding HPLC retention times are listed in Table 2. In the series of fluorine‐18 labeled compounds [18F]F‐3‐6, log D oct/PBS values increased with increasing spacer lengths. The results are furthermore corroborated by the analytical RP‐HPLC retention times. Compound [18F]F‐3 with no spacer was the most hydrophilic compound, while the introduction of additional Ahx spacers resulted in a gradual increase of the lipophilicity for [18F]F‐4 and [18F]F‐5, respectively. As expected, compound [18F]F‐6 exhibited the highest lipophilicity due to the lipophilic 2‐NaI‐Amc spacer. In comparison to the clinical gold standard [68Ga]Ga‐PSMA‐11, all BODIPY‐PSMA were more lipophilic.
Table 2.
Analytical HPLC retention times and log D oct/PBS values of bioconjugates [18F]F‐3‐6 together with the reference compound [68Ga]Ga‐PSMA‐11.
|
Compound |
tR [min] |
log D oct/PBS |
|---|---|---|
|
[18F]F‐3 |
11.26 |
−2.26±0.02 |
|
[18F]F‐4 |
11.39 |
−1.99±0.11 |
|
[18F]F‐5 |
11.50 |
−1.35±0.01 |
|
[18F]F‐6 |
12.96 |
−1.14±0.06 |
|
[68Ga]Ga‐PSMA‐11 |
n.d. |
−2.91[a] |
[a] From ref. [53]
In vitro characterization
In order to identify a BODIPY‐PSMA conjugate with high target affinity, a small series of compounds was prepared with differing spacer lengths and composition. To establish a structure‐activity relationship, the binding affinities of BODIPY‐PSMA conjugates 3–6 were thus determined in the PSMA‐positive human prostate cancer cell line LNCaP by a competitive binding assay as previously reported using [177Lu]Lu‐PSMA‐617 as radioligand.[65] Glu‐CO‐Lys‐Ahx‐HBED‐CC, the uncomplexed precursor of [68Ga]Ga‐PSMA‐11, was included as the control.[50] Corresponding IC50 values of bioconjugates 3–6 and Glu‐CO‐Lys‐Ahx‐HBED‐CC are summarized in Table 3.
Table 3.
PSMA binding affinities of the bioconjugates 3–6 and the reference Glu‐CO‐Lys‐Ahx‐HBED‐CC.
|
Compound |
IC50 [nM] |
|---|---|
|
3 |
187.6±0.5 |
|
4 |
21.5±0.1 |
|
4 ‐ BF2 |
>1000 |
|
5 |
96.4±0.2 |
|
6 |
124.0±0.2 |
|
Glu‐CO‐Lys‐Ahx‐HBED‐CC |
18.4±0.2 |
In the series of BODIPY‐PSMA conjugates, compound 4 with an Ahx spacer exhibited the highest affinity with an IC50=21.5±0.1 nM being comparable to the control PSMA‐11 with IC50=18.4±0.2 nM. Using the Cheng‐Prusoff equation, an inhibitory constant of K i=19.6 nM for 4 was calculated, which is comparable to the K i of natGa‐Glu‐CO‐Lys‐Ahx‐HBED‐CC (natGa‐PSMA‐11) with 12.0 nM.[45] This could be explained by the structural analogy between 4 and Glu‐CO‐Lys‐Ahx‐HBED‐CC. As discussed above, both ligands are using an Ahx linker between the PSMA binding motif and the radiolabel. Furthermore, the metal chelator HBED‐CC and the BODIPY dye do both possess lipophilic aromatic ring systems,[45] which are required for an optimal interaction with the S1 pocket and the S1 accessory pocket of the PSMA active site.[40, 66, 67] The BODIPY scaffold might additionally interact with the arene‐binding site close to the entrance funnel of the internal PSMA cavity.[66, 68] In contrast, the binding affinity significantly decreased for compound 3 (IC50=187.6±0.5 nM) with a shorter distance between the Glu‐CO‐Lys PSMA binding motif and the BODIPY dye, most likely due to steric hindrance preventing optimal accommodation of the conjugate into the PSMA binding pocket. Further elongation of the spacer by two Ahx moieties as for compound 5 also resulted in lower affinity (IC50=96.4±0.2 nM), indicating that this distance is most likely too long for optimal interaction of the lipophilic BODIPY dye with the lipophilic accessory pocket of PSMA. Surprisingly, compound 6 with a 2‐Nal‐Amc spacer similar to PSMA‐617 showed lower affinity with a IC50=124.0±0.2 nM compared to PSMA‐11. In case of 6, the low affinity might be a result of its high overall lipophilicity (Table 2). The 2‐Nal‐Amc‐spacer has been found to bind nicely into the hydrophobic arginine patch of the S1 accessory pocket and the distance between the PSMA binding motif and the radiolabel/fluorescent tag for the 2‐Nal‐Amc‐spacer appears to be optimal for PSMA‐617. However, in combination with the BODIPY scaffold the overall lipophilicity of compound 6 is substantially increased resulting in a significant reduction of binding affinity (Table 3).[50, 66, 69] Because of the partial release of BF2 during the isotopic 19F/18F‐exchange reaction, we additionally determined the binding affinity of the BF2‐free analog of 4 because this side product might compete with the bimodal probe in terms of PSMA binding. Interestingly, the binding affinity of BF2‐free 4 (4 ‐ BF2) diminished completely with an IC50 value of >1 μM indicating that BF2‐free side product of 4 will not interfere with binding of the radiotracer [18F]F‐4 to PSMA.
In addition to the competitive binding studies, we also investigated the internalization of [18F]F‐4 into LNCaP cells for up to 2 h. To determine PSMA‐specific cell internalization, additional blocking experiments were performed using 2‐(phosphonomethyl)‐pentanedioic acid (2‐PMPA), a highly specific PSMA inhibitor.[53] A graphical representation of the PSMA‐mediated internalization and PSMA‐specific cell‐surface binding of [18F]F‐4 is given in Figure 4. Given P values apply to differences between blocked and unblocked experiments. As expected, the PSMA‐mediated internalized fraction of [18F]F‐4 increased gradually over time. At 15 min and 30 min, [18F]F‐4 was internalized to 2.24 % and 2.17 % (P<0.0057), respectively, which further increased to 3.3 % at 60 min (P<0.0015). The maximum was reached at 120 min with 3.83 % of [18F]F‐4 being internalized (P<0.0002). In contrast, the specific surface bound fraction of [18F]F‐4 remained essentially constant over the course of the experiment with ∼0.5 %.
Figure 4.
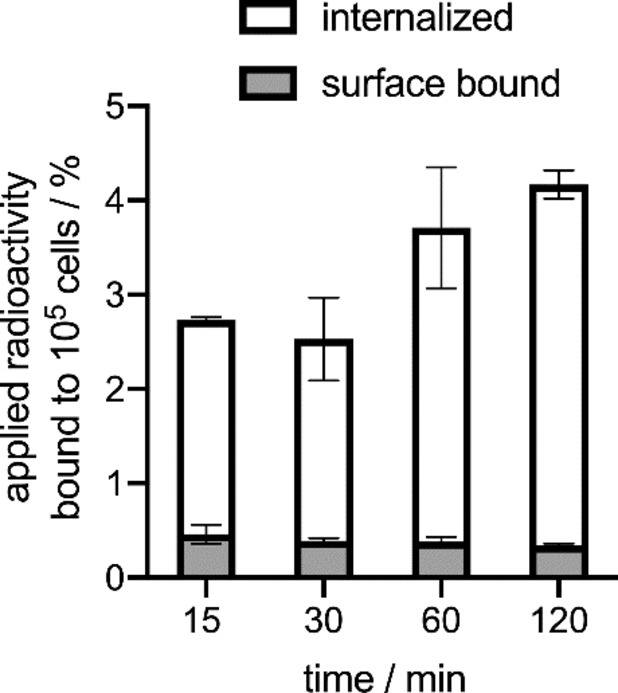
PSMA‐specific surface bound and internalized fractions of [18F]F‐4 in LNCaP cells. Values are given as mean±SD. Results are from two independent experiments in triplicate.
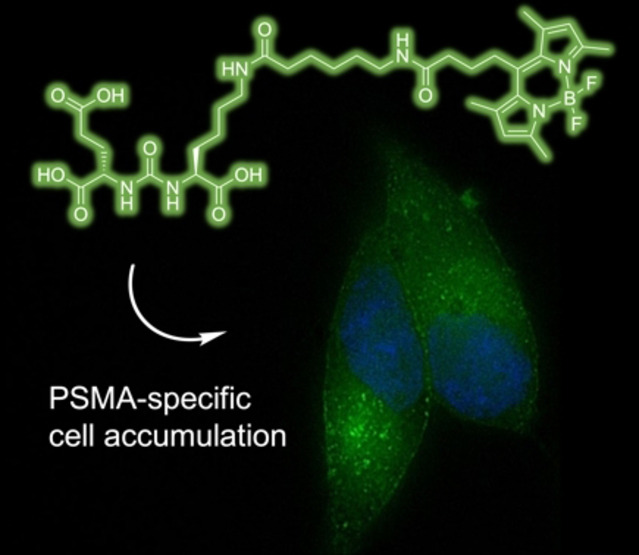
The specific cell surface binding and internalization of 4 in LNCaP cells at different time points was also visualized by fluorescence microscopy using a structured illumination microscope (SIM) (Figure 5). PSMA‐mediated uptake of compound 4 into LNCaP cells was confirmed by fluorescence microscopy demonstrating gradual internalization over the period of 1 h. At 15 min, a significant accumulation of 4 was found in the cell membrane of the LNCaP cells, in which the large extracellular domain of the expressed PSMA is located (Figure 5A). At later time points (30 min and 1 h), an increasing number of intracellular fractions of high intensity was noted (Figure 5B+C), being in concordance with the results of the cell internalization experiment using [18F]F‐4. Internalization of compound 4 occurs presumably via clathrin‐coated pits and subsequent endocytosis after interaction with PSMA.[70] Moreover, our results are in line with a recent study by Eder et al., which investigated the subcellular fate of bimodal PSMA inhibitors in more detail by STED nanoscopy.[55] At 4 °C, accumulation of 4 in the cell membrane was observed. To further demonstrate the specificity of the surface binding and internalization of 4, we also used 2‐PMPA for blocking studies (Figure 5E). After incubation with excess 2‐PMPA, neither specific accumulation in the membrane nor internalization were observed, indicating PSMA‐specific membrane accumulation and cellular internalization. The visible intracellular signals in the blocking study are caused by the autofluorescence of the cell compartments as can be seen from the negative control of fixed cells without addition of compound 4 (Figure 5F).
Figure 5.
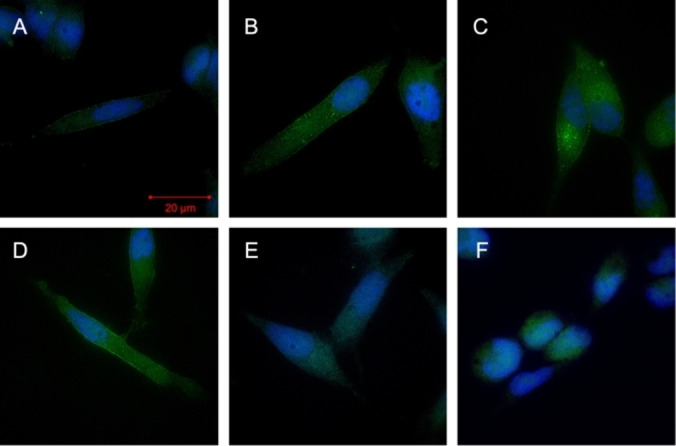
PSMA‐mediated uptake of 4 into LNCaP cells, confirmed by SIM‐fluorescence microscopy over time at 25 °C at A) 15 min, B) 30 min, C) 1 h, D) 1 h at 4 °C, E) 1 h+2‐PMPA, F) DAPI control.
Conclusion
In this work, we successfully introduced a synthetic strategy for the preparation of four BODIPY‐labeled PSMA‐targeting vectors using a combination of standard solid‐phase‐ and liquid synthesis techniques. The introduction of a peptidomimetic PSMA‐targeting moiety in this context did not alter fluorescent properties of the dye and afforded bioconjugates with high fluorescence quantum yields as well as fluorescence lifetimes equal to the BODIPY carboxylic acid 1. The ligands 3–6 were radiolabeled by Lewis acid‐assisted isotopic 19F/18F exchange in high radiochemical purities >99 % with moderate molar activities. In competitive cell binding and internalization experiments, a BODIPY‐PSMA conjugate was identified with comparable PSMA affinity to Glu‐CO‐Lys‐Ahx‐HBED‐CC, the precursor of clinical imaging standard [68Ga]Ga‐PSMA‐11. Cellular localization of 4 was also studied by fluorescence microscopy. Altogether, results make compound 4 a promising candidate for further development of PSMA‐specific dual‐imaging agents and support its future evaluation in vivo.
Experimental Section
General
Chemicals and solvents were purchased from Sigma‐Aldrich, Carbolution, Iris Biotech, Carl Roth, ABX advanced biochemical compounds GmbH and TCI Europe, and used as received. Fluorine‐18 was obtained from Life Radiopharma Fcon GmbH (Holzhausen a. d. Heide, Germany). Solvents used were dried using common laboratory methods. All air‐sensitive reactions were carried out under a nitrogen atmosphere. NMR spectra were recorded in CD3OD or CDCl3 at room temperature using an Avance II spectrometer from Bruker BioSpin (400, 376 or 100 MHz). Chemical shifts are given in parts per million (ppm) and are reported relative to trimethylsilane (TMS). Coupling constants are reported in Hertz (Hz). The multiplicity of the NMR signals is described as follows: s=singlet, d=doublet, t=triplet, q=quartet, m=multiplet. 1H and 13C spectra were referenced to residual protonated solvents, 19F spectra were measured with external standard TFA. High performance liquid chromatography (HPLC) was performed on a Shimadzu LC‐20AT system equipped with a Shimadzu SPD‐20 A UV detector (UV detection at 490 nm) and a Raytest Ramona radiation detector (Raytest GmbH, Straubenhardt, Germany) in series. A Prontosil column (250×4 mm) was used for analytical HPLC. The solvent system was A=H2O (0.1 % TFA) and B=acetonitrile (0.1 % TFA). The gradient was 0–1 min 5 % B, 1–15 min 5–95 % B, 15 −17 min 95 % B at a flow rate of 1 mL ⋅ min−1. Semi‐preparative HPLC was performed on a Techlab HPLC system in combination with a VDSpher 100 C18‐E column (250×20 mm), with A=H2O (0.1 % TFA) and B=acetonitrile (0.1 % TFA) as the solvent system. Gradients used for semi‐preparative HPLC, the corresponding retention times, synthesis yields, and characterization data for each compound are given below. Samples were lyophilized using a Christ Alpha 1–2 LD plus lyophilizer. Analytical thin‐layer chromatography (TLC) was performed on 0.2 mm silica gel 60 with fluorescence indicator pre‐coated aluminium sheets (40×80 mm, Macherey Nagel). Column chromatography was carried out on silica gel 60 (230‐400 mesh particle size, technical grade, Sigma Aldrich). High resolution mass spectrometry ((+)‐HR‐ESI‐MS) was performed on an AB Sciex API 5500 QTRAP spectrometer. For low resolution mass spectrometry ((+)‐LR‐ESI‐MS) an Advion expression CMS system was used. UV/Vis spectroscopy was performed using a JASCO V‐650 absorption spectrometer. Fluorescence‐ and excitation spectra were recorded with a JASCO fluorescence spectrophotometer (DP‐6500). Unless otherwise stated, all measurements were carried out using polystyrene disposable cuvettes (1 cm path length, Sarstedt) with an 1 : 1 (v/v) acetonitrile/water mixture as the solvent system and for each measurement micromolar concentrations were used. For the determination of fluorescence quantum yields, a Hamamatsu Photonics C11347 – Absolute PL quantum yield spectrometer was used with 0.8 mL clear flat base vials (8 mm crimp, Thermo Scientific). The lifetime measurements were carried out applying a tunable fiber laser (FemtoFiber pro TVIS, TOPTICA Photonics, λ exc=490 nm) with a self‐made TCSCP electronic. A bandpass filter (HQ 525/50, AHF analysis technique) was used for the filtration of the laser light. The detection occurred with a combination of a single photon‐avalanche detector (PDM100ct SPAD, Micro Photon Device) and a photon counting device (PicoHarp300, Pico Quant). The IRF (instrumental response function) was measured with a diluted colloidal LUDOX solution (LUDOX TM 50, Sigma Aldrich, λ em=470 nm, IRF (FWHM) ∼100 ps) with an optical density filter of 1.0. Fluorescence lifetimes were analyzed with the Sympho Time 64 software (Pico Quant).[71] All instruments measuring radioactivity were calibrated and maintained in accordance with previously reported routine quality‐control procedures.[72] Radioactivity was measured using an Activimeter ISOMED 2010 (Nuklear‐Medizintechnik, Dresden, Germany). For accurate quantification of radioactivity, experimental samples were counted for 1 min on a calibrated Perkin Elmer (Waltham, MA, USA) 2480 Automatic Wizard Gamma Counter by using a dynamic energy window of 400–600 keV for fluorine‐18 (511 keV emission). Statistical analyses (Student's t‐test, confidence interval 95 %) were performed using GraphPad Prism Version 7.0.
Syntheses
Compound 1
4‐(4,4‐Difluoro‐1,3,5,7‐tetramethyl‐4‐bora‐3a,4a‐diaza‐s‐inda‐cene‐8‐yl)‐butyric acid was synthesized according to literature procedure with minimal modifications during the workup.[63, 64] Yield: 55 %. 1H‐NMR (400 MHz, CDCl3): δ=6.07 (s, 2H, 2xCH), 3.01–3.05 (m, 2H, CH2), 2.56 (t, 3 J=7.28 Hz, 2H, CH2), 2.52 (s, 6H, 2xCH3), 2.43 (s, 6H, 2xCH3), 1.94‐2.02 (m, 2H, CH2) ppm. 13C‐NMR (100 MHz, CDCl3): δ=177.8, 154.3, 144.6, 140.3, 131.4, 121.8, 33.9, 27.3, 26.5, 16.3, 14.4 ppm. 19F‐NMR (377 MHz, CDCl3): δ=‐146.55 (m, 2F, BF2) ppm. ESI‐MS calcd m/z for C17H20N2O2BF2 −: 333.16, found: 333.16.
Compound 2
4‐(4,4‐Difluoro‐1,3,5,7‐tetramethyl‐4‐bora‐3a,4a‐diaza‐s‐inda‐cene‐8‐yl)‐butyric‐N‐hydroxy‐succinimide‐ester was synthesized according to the literature.[63, 73] Yield: quant. 1H‐NMR (400 MHz, CDCl3): δ=6.06 (s, 2H, 2xCH), 3.05–3.09 (m, 2H, CH2), 2.83 (bs, 4H, 2xCH2), 2.79 (t, 3 J=7.28 Hz, 2H, CH2), 2.50 (s, 6H, 2xCH3), 2.42 (s, 6H, 2xCH3), 2.04–2.06 (m, 2H, CH2) ppm. 13C‐NMR (100 MHz, CDCl3): δ=171.1, 168.9, 167.7, 154.3, 144.0, 140.4, 131.3, 121.8, 60.3, 30.8, 26.9, 26.1, 25.5, 16.3, 14.4, 14.1 ppm. 19F‐NMR (377 MHz, CDCl3): δ=‐146.46 (m, 2F, BF2) ppm. ESI‐MS calcd m/z for C21H24N3O4BF2 +: 431.25, found: 431.26. Crystallographic data are given in the Supporting Information.
Preparation of resin immobilized PSMA‐binding motif Linker conjugation and resin cleavage
The preparation of the 2‐chlorotritylchlorid resin‐immobilized PSMA‐binding motif Glu‐CO‐Lys as well as the conjugation of the respective spacers were performed according to the literature with minor modifications using standard Fmoc‐chemistry starting with Fmoc‐L–Lys(Alloc)‐OH.[45, 50] The activation of L‐glutamic acid‐di‐tert‐butyl ester for urea‐coupling was carried out with 1,1’‐carbonyldiimidazole.[58]
General procedures for the synthesis of PSMA‐BODIPY bioconjugates 3–6
The final bioconjugates were prepared in solution.
Route A
In a typical reaction, a solution of 1 (1 eq.) and HATU (0.9 eq.) containing N,N'‐diisopropylethylamine (2 eq.) in dry DMF (1 mL) was added to a solution of the crude precursor (1 eq.) in dry DMF (1 mL). The dark orange solution was stirred for 2 h at 25 °C quenched with water (0.1 % TFA) and purified by semi‐preparative HPLC.
Route B
For the NHS‐Ester coupling, a solution of 2 (1 eq.) and N,N'‐diisopropylethylamine (2 eq.) in dry DMF (1 mL) was added to a solution of the amino acid precursors (1 eq.) at room temperature. The dark orange solution was stirred for 2 h at 25 °C quenched with water (0.1 % TFA) and purified by semi‐preparative HPLC.
Compound 3
According to general procedure starting with resin immobilized Glu‐CO‐Lys (100 mg, 0.082 mmol). Gradient: 50 % to 60 % B in 15 min at a flow rate of 7 mL ⋅ min−1. Retention times: tR (semi‐preparative HPLC): 11.75 min, tR (analytical HPLC): 11.26 min, Yield: 20 mg (0.031 mmol, 38 %) of an orange solid. 1H‐NMR (400 MHz, CD3OD): δ=6.14 (s, 2H, 2xCH), 4.29 (2xdd, 3 J cis=5.02 Hz, 3 J trans=8.53 Hz, 3 J cis=4.77 Hz, 3 J trans=8.28 Hz, 2H, 2xCCH) 3.19 (td, 2 J=1.25 Hz, 3 J=7.03 Hz, 2H, CH2), 3.02–3.07 (m, 2H, CH2), 2.46 (s, 6H, 2xCH3), 2.44 (s, 6H, 2xCH3), 2.38–2.43 (m, 4H, 2xCH2), 2.11–2.17 (m, 1H, CH), 1.83–1.95 (m, 4H, 2xCH, CH2), 1.62‐1.69 (m, 1H, CH), 1.51–1.57 (m, 2H, CH2), 1.41–1.48 (m, 2H, CH2) ppm. 13C‐NMR (100 MHz, CD3OD): δ=176.6, 175.0, 155.1, 154.9, 154.1, 147.2, 142.6, 132.6, 122.9, 54.1, 53.7, 40.2, 37.2, 31.2, 30.0, 29.4, 29.0, 28.7, 23.9, 16.7, 14.6 ppm. 19F‐NMR (377 MHz, CD3OD) δ=‐148.29 (m, 2F, BF2) ppm. LR‐ESI‐MS calcd m/z for C29H41N5O8BF2 +: 636.3, found: 636.5.
Compound 4
According to general procedure starting with resin immobilized Glu‐CO‐Lys‐Ahx (100 mg, 0.068 mmol). Gradient: 50 % to 60 % B in 15 min at a flow rate of 7 mL ⋅ min−1. Retention times: tR (semi‐preparative HPLC): 11.80 min, tR (analytical HPLC): 11.39 min, Yield: 20 mg (0.027 mmol, 39 %) of an orange solid. 1H‐NMR (400 MHz, CD3OD): δ=6.14 (s, 2H, 2xCH), 4.24–4.33 (2xdd, 2x 3 J cis=5.02 Hz, 2x 3 J trans=8.53 Hz, 2H, 2xCH), 3.18 (t, 3 J=6.78 Hz, 2H, CH2), 3.13 (t, 3 J=6.78 Hz, 2H, CH2), 3.03–3.08 (m, 2H, CH2), 2.45 (s, 12H, 4xCH3), 2.36–2.41 (m, 4H, 2xCH2), 2.15–2.21 (m, 3H, CH, CH2), 1.84–1.95 (m, 4H, 2xCH, CH2), 1.58–1.66 (m, 3H, CH, CH2), 1.48–1.55 (m, 4H, 2xCH2), 1.33–1.42 (m, 4H, 2xCH2) ppm. 13C‐NMR (100 MHz, CD3OD): δ=176.6, 176.3, 175.0, 160.3, 155.4, 147.4, 147.1, 142.6, 132.8, 122.9, 54.1, 53.3, 40.5, 40.2, 37.2, 37.1, 33.3, 31.2, 30.2, 30.0, 29.4, 29.0, 28.7, 27.7, 26.8, 24.1, 16.7, 14.6 ppm. 19F‐NMR (377 MHz, CD3OD) δ=−148.01 (m, 2F, BF2) ppm. LR‐ESI‐MS calcd m/z for C35H52N6O9BF2 +: 749.4, found: 749.7.
Boron difluoride‐free compound 4 ‐BF2
Compound 4 (10 mg, 0,013 mmol) was dissolved in 95 % TFA (2 mL) and stirred for 2 h at r.t. After incubation, the mixture was purified using a SepPak C18 cartridges (Waters) and eluted using 50 % aqueous ethanol. Retention time: tR (analytical HPLC): 8.91 min, Yield: quantitative. LR‐ESI‐MS calcd m/z for C35H53N6O9 +: 701.3, found: 701.2.
Compound 5
According to general procedure starting with resin immobilized Glu‐CO‐Lys‐Ahx‐Ahx (100 mg, 0.062 mmol). Gradient: 75 % to 95 % B in 15 min at a flow rate of 6 mL ⋅ min−1. Retention times: tR (semi‐preparative HPLC): 9.43 min, tR (analytical HPLC): 11.50 min. Yield: 20 mg (0.023 mmol, 37 %) of an orange solid. 1H‐NMR (400 MHz, CD3OD): δ=6.14 (s, 2H, 2xCH), 4.24–4.26 (m, 1H, CH), 4.31–4.32 (m, 1H, CH), 3.11–3.20 (m, 6H, 3xCH2), 3.02–3.06 (m, 2H, CH2), 2.46 (s, 6H, 2xCH3), 2.44 (s, 6H, 2xCH3), 2.36–2.40 (m, 4H, 2xCH2), 2.16–2.19 (m, 5H, CH, 2xCH2), 1.89–1.96 (m, 4H, 2xCH, CH2), 1.60–1.62 (m, 3H, CH, CH2), 1.48–1.53 (m, 8H, 4xCH2), 1.32–1.36 (m, 6H, 3xCH2) ppm. 13C‐NMR (100 MHz, CD3OD): δ=174.6, 174.5, 173.5, 173.4, 153.7, 140.8, 131.2, 131.1, 121.2, 120.1, 51.4, 38.0, 35.5, 35.4, 29.4, 28.0, 27.6, 27.1, 26.9, 26.7, 26.0, 25.6, 25.1, 24.8, 21.8, 14.6, 12.4 ppm. 19F‐NMR (377 MHz, CD3OD) δ=−148.72 (m, 2F, BF2) ppm. LR‐ESI‐MS calcd m/z for C41H63N7O10BF2 +: 862.5, found: 862.8.
Compound 6
According to general procedure starting with resin immobilized Glu‐CO‐Lys‐2‐Nal‐Amc (100 mg, 0.058 mmol). Gradient: 50 % to 95 % B in 15 min at a flow rate of 7 mL ⋅ min−1. Retention times: tR (semi‐preparative HPLC): 9.40 min, tR (analytical HPLC): 12.96 min. Yield: 25 mg (0.026 mmol, 44 %) of an orange solid. 1H‐NMR (400 MHz, CD3OD): δ=7.74–7.78 (m, 3H, 3xAr‐CH), 7.66 (s, 1H, Ar‐CH), 7.37–7.41 (m, 3H, 3xAr‐CH), 6.14 (s, 2H, 2xCH), 4.64–4.69 (dd, 3 J=6.72 Hz, 3 J=8.80 Hz, 1H, CH) 4.29–4.33 (dd, 3 J=4.89 Hz, 3 J=8.44 Hz, 1H, CH), 4.17–4.20 (dd, 3 J=4.52 Hz, 3 J=8.31 Hz, 1H, CH), 3.24 (2xd, 3 J=6.85 Hz, 3 J=6.48 Hz, 2H, CH2), 3.12 (t, 3 J=6.36 Hz, 2H, CH2), 2.99–3.04 (m, 4H, 2xCH2), 2.45 (2xs, 12H, 4xCH3), 2.36–2.41 (m, 5H, CH, 2xCH2), 2.09–2.18 (m, 2H, 2xCH), 1.86–1.94 (m, 3H, CH, CH2), 1.77–1.80 (m, 2H, 2xCH), 1.66–1.72 (m, 2H, 2xCH), 1.52–1.58 (m, 2H, 2xCH), 1.33–1.39 (m, 6H, 3xCH2), 0.89–0.98 (m, 2H, CH2) ppm. 13C‐NMR (100 MHz, CD3OD): δ=175.8, 173.2, 154.1, 153.6, 145.5, 140.8, 134.4, 133.3, 132.4, 130.8, 129.2, 128.6, 127.4, 126.7, 122.9, 121.1, 56.0, 54.0, 53.2, 46.3, 44.1, 39.7, 39.1, 38.8, 37.6, 36.7, 31.6, 29.4, 28.7, 28.4, 24.4, 16.7, 14.6 ppm. 19F‐NMR (377 MHz, CD3OD) δ=−148.01 (m, 2F, BF2) ppm. LR‐ESI‐MS calcd m/z for C50H65N7O10BF2 +: 972.48, found: 972.6.
Stability measurements
UV/vis and fluorescence measurements
A solution of 1 (73.4 mg, 0.22 mmol) in TFA (1 mL) was stirred for 2 h. Every minute, an aliquot of this solution (1 μL) was diluted with acetonitrile (3 mL) and analyzed. After 1 h, the measurement‐interval was expanded to 5 min. One additional measurement was performed after 24 h. To determine the effect of SnCl4, 1.22 mg (3.65 μmol) of 1 were dissolved in acetonitrile (200 μL) and 20 μL of this stock‐solution were then mixed with 1 mL of a 1 M SnCl4 solution in acetonitrile. The absorption and fluorescence spectra of the reaction mixture were recorded every 32 s (absorption scan or fluori scan program) in a Hellma optical glass cuvettes (OS®) over a period of 40 min.
HPLC studies
For analytical HPLC measurements of the dye stability, compound 1 (1 mg) was mixed with 95 % aqueous TFA (1 mL) and incubated at r.t. for 2 h. Fractions were collected manually and analyzed using mass spectroscopy.
Radiolabeling of BODIPY‐PSMA bioconjugates
Radiolabeling of the bioconjugates with [18F]F− was performed manually. [18F]fluoride for radiolabeling was separated from the irradiated [18O]H2O‐containing solution by passage through an anion‐exchange cartridge (Waters Accel Plus QMA Cartridge Light). The [18F]fluoride loaded anion exchange‐cartridge was transferred to a Wheaton‐vial, eluted with tetraethylammonium bicarbonate (TEAB) (25 mg) in acetonitrile/water (1 mL, 95 : 5), washed with acetonitrile (1 mL), and placed inside the heating block. [18F]F‐TEA was dried by azeotropic distillation with dry acetonitrile (3×2 mL) under a gentle stream of argon at 95 °C and stored under argon in dry acetonitrile. For a typical Lewis‐acid assisted isotopic 19F/18F exchange, 37 μL of a SnCl4 stock solution (1 M, in dry acetonitrile, 100 eq.) were added to 100 μL of the corresponding conjugate stock solution in dry acetonitrile, followed by the addition of 300 MBq [18F]F‐TEA in 100 μL dry acetonitrile. The mixture was incubated for 15 min at r.t. and quenched by addition of a sodium hydroxide solution (0.1 M, 4 mL) and purified by passage through a SepPak C18 cartridge (Waters). After rinsing with a sodium hydroxide solution (2 mL) and water (2 mL), the final product was eluted with 2 mL 50 % aqueous ethanol. Radiochemical purities were >99 % and the mean radiochemical yields (RCY) were 20–30 %. The mean molar activities for all compounds were A m ∼0,7 MBq ⋅ nmol−1.
In vitro characterization
Lipophilicity (log Doct/PBS) measurements
For log D oct/PBS measurements, 1–2 MBq of [18F]F‐3, [18F]F‐4, [18F]F‐5 and [18F]F‐6 in 5 μL 50 % aqueous ethanol were added to a mixture of phosphate buffered saline pH 7.4 (PBS) (495 μL) and octanol (500 μL). Samples were shaken for 30 min at r.t., centrifuged at 13,200 rpm for 5 min and 100 μL of each phase were counted using a gamma counter. Experiments were performed in triplicate.
Stability measurements
For each experiment, ∼5 MBq (10 μL) of [18F]F‐4 were added to phosphate buffered saline pH 7.4 (90 μL). At selected time points, 20 μL aliquots were taken from the solution and analyzed by HPLC. The percentage of intact [18F]F‐4 was calculated from the HPLC chromatograms. All experiments were performed in triplicate.
Cell culture
LNCaP cells (DSMZ, Braunschweig, Germany) were cultured at 37 °C in a 5 % CO2 atmosphere (RPMI medium containing 10 % fetal bovine serum, 1 % 10,000 U ⋅ mL−1 penicillin and 10,000 U ⋅ mL−1 streptomycin, 1 % sodium‐pyruvate 100 mM).
Competitive binding assay
The binding affinities of 3, 4, 5, 6 and boron difluoride‐free 4 were determined by a cell‐based competitive binding assay in the human prostate cancer cell line LNCaP with [177Lu]Lu‐PSMA‐617 as the radioligand as previously described.[65] Binding assays were performed in 24‐well plates precoated with poly‐L‐lysine. Briefly, each compound at different concentrations (0–10,000 nM) was incubated for 2 h at r.t. with [177Lu]Lu‐PSMA‐617 (30,000 cpm ⋅ well−1) and 2×105 LNCaP cells per well. After incubation, cells were washed three times with ice cold binding buffer and cell‐associated activity recovered by addition of 1 M NaOH. Radioactivity was measured by a gamma counter and data fitted using non‐linear regression (GraphPad Prism). Experiments were performed two times in triplicate.
Cell internalization assay
For cell internalization, LNCaP cells were seeded at a density of 106 cells per well in 6‐well plates and incubated overnight with RPMI‐medium with GlutaMAX containing 10 % fetal bovine serum, 1 % 10,000 U ⋅ mL−1 penicillin and 10,000 U ⋅ mL−1 streptomycin, and 1 % sodium‐pyruvate 100 mM. After 24 h, the medium was removed and the cells were washed with PBS. Approximately 5 pmol of [18F]F‐4 were added to the cells (in triplicates) to give a final volume of 1.5 mL PBS in each well followed by incubation for 1 h at 37 °C, 5 % CO2. To determine nonspecific membrane binding and internalization, excess of 2‐(phoshonomethyl)‐pentanedioic acid (2‐PMPA) (final concentration 1 μM) was added to the selected wells. At each time point, the internalization was stopped by removing the medium and washing the cells twice with ice‐cold PBS. To remove the receptor‐bound radioligand, an acid wash was carried out twice with a 0.1 M glycine buffer (pH 2.8) for 5 min on ice. Finally, cells were solubilized with 1 M NaOH. Radioactivity of cell fractions (supernatant, receptor‐bound and internalized) was measured by a Hidex gamma counter. Experiments were performed twice in triplicate.
Microscopy
For structured illumination microscopy (SIM), 105 cells were seeded on a poly‐L‐lysine coated coverslip (25 mm) in a 6‐well‐plate 24 h before incubation at 37 °C, 5 % CO2. After washing with PBS, the cells were incubated with 4 in RPMI‐medium (20 nM final concentration for each well) for 15 min, 30 min as well as 60 min at different temperatures. In a second series, incubation was repeated with a final concentration of 100 nM. Blocking studies were performed by incubation with 2‐PMPA (20 mM final concentration) for 30 min. After incubation, cells were washed with PBS and fixed with a paraformaldehyde‐solution (4 %, in PBS, containing 1.8 mM CaCl2 and 1.0 mM MgCl2) for 15 min at r.t. After fixation, two washing steps with PBS and water followed. Cell staining with 4‘,6‐diamidino‐2‐diphenylindole dihydrochloride (DAPI) was performed using 2 drops NucBlueTM Fixed Cell Stain ReadyProbesTM in a total volume of 2 mL water for 10 min. For mounting, 1 drop of Pro‐Long Diamond Antifade Mountant per coverslip was used and incubated for 5 min. The coverslips were stored at 4 °C for microscopy. The membrane uptake and internalization of 4 was measured using a SIM microscope (Carl Zeiss Elyra PS.1 microscope) using a 63×objective (nA 1.43). DAPI was excited by a 405 nm laser and 4 with a 488 nm laser. Raw data were processed for super resolution with Zen2012 software (Carl Zeiss) and postprocessed and analyzed with ImageJ v.1.53 for presentation.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
M.D.B. acknowledges financial support from the HOMFOR program of Saarland University. C.H. and G.J. thank the German Science Foundation for financial support (DFG; JU650/10‐1). Instrumentation and technical assistance for this work were provided by the Service Center X‐ray Diffraction, with financial support from Saarland University and German Science Foundation (project number INST 256/506‐1). The authors also thank Dr. Volker Huch and Dr. Bernd Morgenstern for their support in collection and analysis of the X‐ray diffraction data of the BODIPY‐NHS‐ester 2. Open access funding enabled and organized by Projekt DEAL.
T. Stemler, C. Hoffmann, I. M. Hierlmeier, S. Maus, E. Krause, S. Ezziddin, G. Jung, M. D. Bartholomä, ChemMedChem 2021, 16, 2535.
Dedicated to the memory of our colleague Dr. Harald Natter
References
- 1.Kosaka N., Ogawa M., Choyke P. L., Kobayashi H., Future Oncol. 2009, 5, 1501–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bu L., Shen B., Cheng Z., Adv. Drug Delivery Rev. 2014, 76, 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tipirneni K. E., Rosenthal E. L., Moore L. S., Haskins A. D., Udayakumar N., Jani A. H., Carroll W. R., Morlandt A. B., Bogyo M., Rao J., Warram J. M., Mol. Imaging Biol. 2017, 19, 645–655. [DOI] [PubMed] [Google Scholar]
- 4.Kowada T., Maeda H., Kikuchi K., Chem. Soc. Rev. 2015, 44, 4953–4972. [DOI] [PubMed] [Google Scholar]
- 5.Ntziachristos V., Bremer C., Weissleder R., Eur. Radiol. 2003, 13, 195–208. [DOI] [PubMed] [Google Scholar]
- 6.Zhang J., Campbell R. E., Ting A. Y., Tsien R. Y., Nat. Rev. Mol. Cell Biol. 2002, 3, 906–918. [DOI] [PubMed] [Google Scholar]
- 7.Wu B., Piatkevich K. D., Lionnet T., Singer R. H., Verkhusha V. V., Curr. Opin. Cell Biol. 2011, 23, 310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stiles B. M., Bhargava A., Adusumilli P. S., Stanziale S. F., Kim T. H., Rusch V. W., Fong Y., Surgery 2003, 134, 357–364. [DOI] [PubMed] [Google Scholar]
- 9.Luo S., Zhang E., Su Y., Cheng T., Shi C., Biomaterials 2011, 32, 7127–7138. [DOI] [PubMed] [Google Scholar]
- 10.Lavis L. D., Raines R. T., ACS Chem. Biol. 2008, 3, 142–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Teo R. D., Termini J., Gray H. B., J. Med. Chem. 2016, 59, 6012–6024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spangler C. M., Spangler C., Schäerling M., Ann. N. Y. Acad. Sci. 2008, 1130, 138–148. [DOI] [PubMed] [Google Scholar]
- 13.Paganin-Gioanni A., Bellard E., Paquereau L., Ecochard V., Golzio M., Teissié J., Radiol. Oncol. 2010, 44, 142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shea C. R., Chen N., Wimberly J., Hasan T., Cancer Res. 1989, 49, 3961–3965. [PubMed] [Google Scholar]
- 15.Vicente M. G., Curr. Med. Chem. Anti-Cancer Agents 2001, 1, 175–194. [DOI] [PubMed] [Google Scholar]
- 16.Falco J., Cavallo C., Vetrano I. G., de Laurentis C., Siozos L., Schiariti M., Broggi M., Ferroli P., Acerbi F., Front. Surg. 2019, 6, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Franke J. M., Raliski B. K., Boggess S. C., Natesan D. V., Koretsky E. T., Zhang P., Kulkarni R. U., Deal P. E., Miller E. W., J. Am. Chem. Soc. 2019, 141, 12824–12831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boens N., Leen V., Dehaen W., Chem. Soc. Rev. 2012, 41, 1130–1172. [DOI] [PubMed] [Google Scholar]
- 19.Treibs A., Kreuzer F.-H., Liebigs Ann. 1968, 718, 208–223. [Google Scholar]
- 20.Loudet A., Burgess K., Chem. Rev. 2007, 107, 4891–4932. [DOI] [PubMed] [Google Scholar]
- 21.Bittel A. M., Davis A. M., Wang L., Nederlof M. A., Escobedo J. O., Strongin R. M., Gibbs S. L., Sci. Rep. 2018, 8, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jeon S., Kim T.-I., Jin H., Lee U., Bae J., Bouffard J., Kim Y., J. Am. Chem. Soc. 2020, 142, 9231–9239. [DOI] [PubMed] [Google Scholar]
- 23.Schüller A., Goh G. B., Kim H., Lee J.-S., Chang Y.-T., Mol. Inf. 2010, 29, 717–729. [DOI] [PubMed] [Google Scholar]
- 24.Ni Y., Wu J., Org. Biomol. Chem. 2014, 12, 3774–3791. [DOI] [PubMed] [Google Scholar]
- 25.Nørager N. G., Jensen C. B., Rathje M., Andersen J., Madsen K. L., Kristensen A. S., Strømgaard K., ACS Chem. Biol. 2013, 8, 2033–2041. [DOI] [PubMed] [Google Scholar]
- 26.Vernekar S. K. V., Hallaq H. Y., Clarkson G., Thompson A. J., Silvestri L., Lummis S. C. R., Lochner M., J. Med. Chem. 2010, 53, 2324–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Waghorn P. A., Jones M. W., McIntyre A., Innocenti A., Vullo D., Harris A. L., Supuran C. T., Dilworth J. R., Eur. J. Inorg. Chem. 2012, 2012, 2898–2907. [Google Scholar]
- 28.Lee J.-S., Kang N., Kim Y. K., Samanta A., Feng S., Kim H. K., Vendrell M., Park J. H., Chang Y.-T., J. Am. Chem. Soc. 2009, 131, 10077–10082. [DOI] [PubMed] [Google Scholar]
- 29.Kamkaew A., Lim S. H., Lee H. B., Kiew L. V., Chung L. Y., Burgess K., Chem. Soc. Rev. 2012, 42, 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hussain T., Nguyen Q. T., Adv. Drug Delivery Rev. 2014, 66, 90–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beltrán Hernández I., Rompen R., Rossin R., Xenaki K. T., Katrukha E. A., Nicolay K., van Bergen En Henegouwen P., Grüll H., Oliveira S., Mol. Imaging Biol. 2019, 21, 1079–1088. [DOI] [PubMed] [Google Scholar]
- 32.Hekman M. C. H., Rijpkema M., Bos D. L., Oosterwijk E., Goldenberg D. M., Mulders P. F. A., Boerman O. C., J. Nucl. Med. 2017, 58, 706–710. [DOI] [PubMed] [Google Scholar]
- 33.Huynh A. M., Müller A., Kessler S. M., Henrikus S., Hoffmann C., Kiemer A. K., Bücker A., Jung G., ChemMedChem 2016, 11, 1568–1575. [DOI] [PubMed] [Google Scholar]
- 34.Paulus A., Desai P., Carney B., Carlucci G., Reiner T., Brand C., Weber W. A., Eur. J. Nucl. Med. Mol. Imaging Res. 2015, 5, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paulus A., Maenen M., Drude N., Nascimento E. B. M., van Marken Lichtenbelt W. D., Mottaghy F. M., Bauwens M., PLoS One 2017, 12, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu S., Li D., Zhang Z., Surya Prakash G. K., Conti P. S., Li Z., Chem. Commun. 2014, 50, 7371–7373. [DOI] [PubMed] [Google Scholar]
- 37.Kwon Y.-D., Byun Y., Kim H.-K., Nucl. Med. Biol. 2021, 93, 22–36. [DOI] [PubMed] [Google Scholar]
- 38.Liu S., Lin T.-P., Li D., Leamer L., Shan H., Li Z., Gabbaï F. P., Conti P. S., Theranostics 2013, 3, 181–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davis M. I., Bennett M. J., Thomas L. M., Bjorkman P. J., Proc. Natl. Acad. Sci. USA 2005, 102, 5981–5986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barinka C., Rojas C., Slusher B., Pomper M., CMC 2012, 19, 856–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Silver D. A., Pellicer I., Fair W. R., Heston W. D., Cordon-Cardo C., Clin. Cancer Res. 1997, 3, 81–85. [PubMed] [Google Scholar]
- 42.Israeli R. S., Powell C. T., Corr J. G., Fair W. R., Heston W. D. W., Cancer Res. 1994, 54, 1807–1811. [PubMed] [Google Scholar]
- 43.Chang A. J., Autio K. A., Roach M., Scher H. I., Nat. Rev. Clin. Oncol. 2014, 11, 308–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ross J. S., Sheehan C. E., Fisher H. A. G., Kaufman R. P., Kaur P., Gray K., Webb I., Gray G. S., Mosher R., Kallakury B. V. S., Clin. Cancer Res. 2003, 9, 6357–6362. [PubMed] [Google Scholar]
- 45.Eder M., Schäfer M., Bauder-Wüst U., Hull W.-E., Wängler C., Mier W., Haberkorn U., Eisenhut M., Bioconjugate Chem. 2012, 23, 688–697. [DOI] [PubMed] [Google Scholar]
- 46.Afshar-Oromieh A., Avtzi E., Giesel F. L., Holland-Letz T., Linhart H. G., Eder M., Eisenhut M., Boxler S., Hadaschik B. A., Kratochwil C., Weichert W., Kopka K., Debus J., Haberkorn U., Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eder M., Neels O., Müller M., Bauder-Wüst U., Remde Y., Schäfer M., Hennrich U., Eisenhut M., Afshar-Oromieh A., Haberkorn U., Kopka K., Pharmaceuticals 2014, 7, 779–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Afshar-Oromieh A., Malcher A., Eder M., Eisenhut M., Linhart H. G., Hadaschik B. A., Holland-Letz T., Giesel F. L., Kratochwil C., Haufe S., Haberkorn U., Zechmann C. M., Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 486–495. [DOI] [PubMed] [Google Scholar]
- 49.Afshar-Oromieh A., Zechmann C. M., Malcher A., Eder M., Eisenhut M., Linhart H. G., Holland-Letz T., Hadaschik B. A., Giesel F. L., Debus J., Haberkorn U., Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Benešova M., Schäfer M., Bauder-Wüst U., Afshar-Oromieh A., Kratochwil C., Mier W., Haberkorn U., Kopka K., Eder M., J. Nucl. Med. 2015, 56, 914–920. [DOI] [PubMed] [Google Scholar]
- 51.Kratochwil C., Giesel F. L., Stefanova M., Benešova M., Bronzel M., Afshar-Oromieh A., Mier W., Eder M., Kopka K., Haberkorn U., J. Nucl. Med. 2016, 57, 1170–1176. [DOI] [PubMed] [Google Scholar]
- 52.Kratochwil C., Bruchertseifer F., Giesel F. L., Weis M., Verburg F. A., Mottaghy F., Kopka K., Apostolidis C., Haberkorn U., Morgenstern A., J. Nucl. Med. 2016, 57, 1941–1944. [DOI] [PubMed] [Google Scholar]
- 53.Baranski A.-C., Schäfer M., Bauder-Wüst U., Roscher M., Schmidt J., Stenau E., Simpfendörfer T., Teber D., Maier-Hein L., Hadaschik B., Haberkorn U., Eder M., Kopka K., J. Nucl. Med. 2018, 59, 639–645. [DOI] [PubMed] [Google Scholar]
- 54.Hensbergen A. W., Buckle T., van Willigen D. M., Schottelius M., Welling M. M., van der Wijk F. A., Maurer T., van der Poel H. G., van der Pluijm G., van Weerden W. M., Wester H.-J., van Leeuwen F. W. B., J. Nucl. Med. 2020, 61, 234–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Matthias J., Engelhardt J., Schäfer M., Bauder-Wüst U., Meyer P. T., Haberkorn U., Eder M., Kopka K., Hell S. W., Eder A.-C., Cancer Res. 2021, canres.1624.2020. [DOI] [PubMed] [Google Scholar]
- 56.Alquicer G., Sedlák D., Byun Y., Pavlíček J., Stathis M., Rojas C., Slusher B., Pomper M. G., Bartůněk P., Bařinka C., J. Biomol. Screening 2012, 17, 1030–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Son S.-H., Kwon H., Ahn H.-H., Nam H., Kim K., Nam S., Choi D., Ha H., Minn I., Byun Y., Bioorg. Med. Chem. Lett. 2020, 30, 126894. [DOI] [PubMed] [Google Scholar]
- 58.Hartwig S., Schwarz J., Hecht S., J. Org. Chem. 2010, 75, 772–782. [DOI] [PubMed] [Google Scholar]
- 59.Wang M., Graça M., Vicente H., Mason D., Bobadova-Parvanova P., ACS Omega 2018, 3, 5502–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Y. Marfin, S. Usoltsev, E. Rumyantsev, in BODIPY Dyes - A Privilege Molecular Scaffold with Tunable Properties, 2019, 66–90.
- 61.Rumyantsev E., Aleshin S., Desoki A., Marfin Y., Antina E., Russ. J. Inorg. 2013, 58, 596–601. [Google Scholar]
- 62.Yu M., Wong J. K.-H., Tang C., Turner P., Todd M. H., Rutledge P. J., Beilstein J. Org. Chem. 2015, 11, 37–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang D., Fan J., Gao X., Wang B., Sun S., Peng X., J. Org. Chem. 2009, 74, 7675–7683. [DOI] [PubMed] [Google Scholar]
- 64.Li Z., Mintzer E., Bittman R., J. Org. Chem. 2006, 71, 1718–1721. [DOI] [PubMed] [Google Scholar]
- 65.Läppchen T., Holland J. P., Kiefer Y., Bartholomä M. D., Eur. J. Nucl. Med. Mol. Imaging Radiopharm. Chem. 2018, 3, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Barinka C., Byun Y., Dusich C. L., Banerjee S. R., Chen Y., Castanares M., Kozikowski A. P., Mease R. C., Pomper M. G., Lubkowski J., J. Med. Chem. 2008, 51, 7737–7743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mesters J. R., Barinka C., Li W., Tsukamoto T., Majer P., Slusher B. S., Konvalinka J., Hilgenfeld R., EMBO J. 2006, 25, 1375–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kopka K., Benešová M., Bařinka C., Haberkorn U., Babich J., J. Nucl. Med. 2017, 58, 17–26. [DOI] [PubMed] [Google Scholar]
- 69.Benešová M., Bauder-Wüst U., Schäfer M., Klika K. D., Mier W., Haberkorn U., Kopka K., Eder M., J. Med. Chem. 2016, 59, 1761–1775. [DOI] [PubMed] [Google Scholar]
- 70.Fendler W. P., Ferdinandus J., Czernin J., Eiber M., Flavell R. R., Behr S. C., Wu I.-W. K., Lawhn-Heath C., Pampaloni M. H., Reiter R. E., Rettig M. B., Gartmann J., Murthy V., Slavik R., Carroll P. R., Herrmann K., Calais J., Hope T. A., J. Nucl. Med. 2020, 61, 1793–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Grüter A., Hoffmann M., Müller R., Wohland T., Jung G., Anal. Bioanal. Chem. 2019, 411, 3229–3240. [DOI] [PubMed] [Google Scholar]
- 72.Zanzonico P., J. Nucl. Med. 2008, 49, 1114–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hsu Y., Nandakumar M., Lai H., Chou T., Chu C., Lin C., Lo L., J. Org. Chem. 2015, 80, 8458–8463. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information