

Abstract
Hybridizing species provide a powerful system to identify the processes that shape genomic variation and maintain species boundaries. However, complex histories of isolation, gene flow and selection often generate heterogeneous genomic landscapes of divergence that complicate reconstruction of the speciation history. Here, we explore patterns of divergence to reconstruct recent speciation in the erato clade of Heliconius butterflies. We focus on the genomic landscape of divergence across three contact zones of the species H. erato and H. himera. We show that these hybridizing species have an intermediate level of divergence in the erato clade, which fits with their incomplete levels of reproductive isolation. Using demographic modeling and the relationship between admixture and divergence with recombination rate variation, we reconstruct histories of gene flow, selection and demographic change that explain the observed patterns of genomic divergence. We find that periods of isolation and selection within populations, followed by secondary contact with asymmetrical gene flow are key factors in shaping the heterogeneous genomic landscapes. Collectively, these results highlight the effectiveness of demographic modeling and recombination rate estimates to disentangling the distinct contributions of gene flow and selection to patterns of genomic divergence.
Keywords: Admixture, selection, species boundaries, reproductive isolation, heterogeneous divergence
Introduction
Disentangling the factors that drive genomic divergence is necessary for advancing our understanding of speciation. Targets of selection, for example, may be responsible for adaptive differences between species and their number, distribution, and effect on gene flow along the genome define the architecture of species boundaries. In population genomic studies, targets of selection are expected to show elevated divergence between species (Wolf and Ellegren 2017). These highly divergent loci often reflect local adaptation and/or incompatibilities between species, and can be considered the loci that define the species (Wu 2001; Ravinet et al. 2017). Natural selection thus acts to produce local genomic “barriers” to gene flow between hybridizing species (Arias et al. 2016; Tavares et al. 2018; Edelman et al. 2019; Martin et al. 2019). In contrast, the rest of the genome, which is not under such selective pressures, is often expected to be exchanged more freely between the species (i.e., gene flow). However, genetic variation at these latter genomic regions can be greatly influenced by neutral demographic processes (i.e., population size and migration) and the indirect effects of nearby targets of selection. Local recombination rates can further influence the extent to which selection impacts these nearby targets of selection or linked genetic variation, which collectively results in highly heterogeneous patterns of divergence (Charlesworth 2009; Aeschbacher et al. 2017; Burri 2017; Wolf and Ellegren 2017; Martin et al. 2019; Stankowski et al. 2019). Thus, to make correct inferences about the contribution of selection to the evolution of barrier loci, it is important to consider all processes that can shape the genomic landscape of divergence.
Heliconius butterflies have several hybridizing species that provide a powerful system to study the evolution of species barriers. Heliconius erato represents an adaptive radiation from Central and South America of about 25 geographic color pattern morphs. These color pattern morphs have evolved to signal their unpalatability to bird predators and mimic locally frequent phenotypes (Mallet and Joron 1999; Chouteau et al. 2016). From within the H. erato radiation, two populations are known to show reproductively isolating traits beyond the ecological post-mating isolation from selection on warning coloration: H. himera from Andean valleys in Ecuador is known to have pre-mating isolation based on mate choice but little or no post-mating reproductive barriers (McMillan et al. 1997; Merrill et al. 2014) and H. e. chestertonii occurring in Colombia is known to have both pre-mating isolation based on mate choice and post-mating isolation resulting from sex-chromosome linked incompatibilities (Muñoz et al. 2010; Van Belleghem et al. 2018). Despite mate preference between H. erato and H. himera, they are known to hybridize across at least three geographically distinct contact zones. Heliconius himera is found in dry forest areas of southern Ecuador and northern Peru (Jiggins et al. 1996). It comes into contact with H. e. cyrbia on the western slopes of the Ecuadorian Andes and with H. e. favorinus and H. e. emma on the eastern slopes of the Andes, both areas with wet forest (Figure 1). Hybridization is ongoing and hybrids are easily identifiable by their wing color patterns. In Ecuador, hybrids compose approximately 5% of the population in the contact zone (McMillan et al. 1997), and, although poorly characterized, hybridization is known to occur in the other contact zones.
Figure 1. Geographical distribution, population structure and phylogeny of the focal populations in relation to the Heliconius erato radiation.

(A) Maximum likelihood tree built using FastTree and using only autosomal sites from 121 whole genome resequenced individuals (see Figure S1 for the uncollapsed tree). Nodes in the tree that represent the major clades within H. erato (east and west of Andes) obtained high support (= 1) from the Shimodaira-Hasegawa test. (B) Principal Component Analysis (PCA) of the focal samples (colored points) among all the available whole genome data for the H. erato radiation (black points). (C) We sampled H. himera, H. e. cyrbia, H. e. emma and H. e. favorinus from two separate geographic areas each, indicated as North, South, East or West. The distribution of H. himera (red) covers dry valleys in the Andes of South Ecuador and North Peru. In the North, H. himera (N) comes into contact with a western H. e. cyrbia (S) population. In the South, H. himera comes into contact with an eastern H. e. emma (W) and H. e. favorinus (W) population. These contact zones are indicated with parenthesis.
Here, we focus on reconstructing the history of divergence between the two hybridizing Heliconius butterfly species Heliconius erato and Heliconius himera, and the extent to which isolation and selection have shaped their genomic divergence. First, we start by characterizing the genome-wide divergence patterns among several of the hybridizing H. erato populations that vary in their degree of reproductive isolation (RI), which we compare to patterns of divergence across three contact zones between H. erato and H. himera. We use these data to explore the relationship between RI and genomic divergence and test whether the H. erato × H. himera comparisons show the expected patterns of divergence based on the degree of RI between the species, when compared to other populations that show weaker or greater degrees of RI. Placing H. erato × H. himera in the spectrum of RI across the erato clade allows us to better understand the relationship between incomplete barriers and the realized intermediate patterns of genomic divergence. To understand this, we construct demographic models of their evolutionary history from genome-wide SNP data that we use to explore the extent that isolation and selection have shaped genomic divergence. For example, we use coalescent simulations informed by the demographic models to test whether selection can explain the observed heterogeneity of divergence and admixture across the genome. Specifically, the coalescent simulations explore the genome-wide impact of selection on linked sites. Selection on an allele within one population will increase divergence between populations and the amount of genomic sites linked to this pattern will be dependent on the local recombination rate (Kaplan et al. 1989). Hence, we expect a negative association between recombination rate and divergence across the genome (Keinan and Reich 2010; Brandvain et al. 2014; Aeschbacher et al. 2017; Stankowski et al. 2019). This pattern, however, is not indicative of a barrier locus, because such pattern could arise from selective sweeps within populations even between geographically isolated populations. Instead, if the species continue to hybridize and selection acts against introgression at many sites throughout the genome, we may expect a positive association between recombination rate and admixture. This is because neutral alleles in regions with a high recombination rate will be more likely to escape selection at nearby barrier loci and introgress (Barton and Bengtsson 1986; Schumer et al. 2018; Martin et al. 2019). This latter pattern would not be expected if they diverged in isolation (Nachman and Payseur 2012; Aeschbacher et al. 2017). Thus, we can use the relationships of recombination rate with divergence and admixture to infer the relative importance of selection at many loci during speciation.
In contrast to the relatively high rates of current hybridization between the H. erato and H. himera species pair, which fit well with intermediate levels of both RI and genomic divergence, our demographic models suggest that a period of reduced migration rates and subsequent admixture have contributed to the observed heterogeneous patterns of genomic divergence. Moreover, coalescent simulations that explore a scenario of recent and strong selective events on linked sites in the absence of gene flow best fit the observed relationship of genomic divergence, admixture, and recombination rate in H. erato and H. himera. Despite this history of isolation, we also show that the current species boundary between H. himera and H. erato is porous and that signals of recent admixture are asymmetrical, suggesting higher gene flow from H. himera into H. erato. We suggest that this asymmetrical signal of gene flow may be the result of either a polygenic architecture of the species boundaries that restrict introgression into H. himera or adaptive introgression of H. himera alleles into H. erato. Overall, our results highlight the contribution of both isolation and selection to the heterogeneous genomic divergence patterns between the species pair H. erato and H. himera and help us understand how demographic and selective processes contribute to speciation.
Methods
Genomic sampling
We obtained whole genome resequence data for a total of 122 Heliconius butterflies with a coverage between 15 to 30X (112 previously published and 10 H. e. cyrbia from North Ecuador newly sequenced; Table S1). These include H. e. cyrbia to the north (North Ecuador, n = 10) and south (South Ecuador H. himera contact zone, n = 4), H. e. emma to the west (Peru H. himera contact zone, n = 4) and east (Peru H. e. favorinus contact zone, n = 7) and H. e. favorinus to the west (Peru H. himera contact zone, n = 4) and east (Peru H. e. emma contact zone, n = 8). We used these populations to study admixture patterns with H. himera to the north (Ecuador H. e. emma/favorinus contact zone, n = 5) and south (Peru H. e. cyrbia contact zone, n = 4). Additionally, samples from H. e. petiverana (Mexico, n = 5), H. e. demophoon (Panama, n = 10), H. e. hydara (Panama, n = 6 and French Guiana, n = 5), H. e. erato (French Guiana, n = 6), H. e. amalfreda (Suriname, n = 5), H. e. notabilis (Ecuador Heliconius erato lativitta contact zone, n = 5 and Ecuador H. e. etylus contact zone, n = 5), H. e. etylus (Ecuador, n = 5), H. e. lativitta (Ecuador, n = 5), H. e. phyllis (Bolivia, n = 4), H. e. venus (Colombia, n = 5) and H. e. chestertonii (Colombia, n = 7) were used for contact zone divergence analysis and population structure visualization as well as samples of H. hermathena (Brazil, n = 3) as an outgroup to root the phylogenetic inference and polarize site frequency spectra. All data have been previously published (Van Belleghem et al. 2017, 2018), apart from the ten H. e. cyrbia from North Ecuador. Genotypes were obtained as in Van Belleghem et al. (2018), see Supplemental Methods for details.
Genomic divergence and phylogenetic relationships
We estimated levels of relative divergence (FST) (Hudson et al. 1992) between populations in nonoverlapping 50 kb windows using Python scripts and egglib (De Mita and Siol 2012). For this analysis, we only considered windows for which at least 10% of the positions were genotyped for at least 75% of the individuals within each population. On average 96% of windows met this criterion. To discern population structure among the H. erato and individuals, we performed principal component analysis (PCA) using EIGENSTRAT SmartPCA (Price et al. 2006). For this analysis, we only considered autosomal biallelic sites that had coverage in all individuals, were at least 1000 bp apart and excluding the Z chromosome (164,398 SNPs). Using autosomal biallelic sites that had coverage in all individuals and including the outgroup H. hermathena (4,927,152 SNPs), we used FastTree v2.1 (Price et al. 2010) to infer an approximate maximum likelihood phylogeny using the default parameters.
Measure of reproductive isolation
We correlated measures of Reproductive Isolation (RI) with patterns of genome-wide divergence between hybridizing pairs of H. erato color pattern morphs, H. himera and H. e. chestertonii. RI was estimated for 13 hybridizing pairs from the H. erato clade using information on isolating barriers from existing literature and the methods of Sobel and Chen (2014). For each pair, the strength of post-mating barriers due to selection on individual color pattern loci was determined using selection coefficients based on cline widths for the color pattern loci (Mallet 1986, 1993; Mallet et al. 1990; Jiggins et al. 1996; Shaak 2015), pre-mating isolation due to sexual selection was obtained from previous mate preference studies (Jiggins et al. 1996; Muñoz et al. 2010; Merrill et al. 2014), and post-mating isolation estimates due to hybrid inviability that has been reported for H. e. chestertonii and H. e. venus was obtained from Muñoz et al. (2010). For more details on estimates of each of the Reproductive Isolation (RI) values, see Supplemental Methods. To estimate Total Isolation (TI) for each hybridizing pairs, we used the RI values for each individual barrier and the TI calculator from Sobel and Chen (2014). We have included a version of the Sobel and Chen (2014) RI calculator in the supplementary information, to show the values of RI for each barrier used to estimate TI (Table S2).
Demographic modeling
To infer the demographic history of the H. erato and H. himera populations, we used the joint site-frequency spectra (JSFS) of five population comparisons and a modified version of ∂a∂i v1.7 (Gutenkunst et al. 2009). The five population comparisons included the three H. erato and H. himera hybrid zones plus a comparison of the H. erato populations east and west of the Andes and between H. e. emma and H. e. favorinus. Models tested include four basic scenarios in addition to 22 extensions of the basic models that allowed for independent assessment of additional selective and demographic parameters, including a growth rate parameters (g), heterogeneity in Ne across the genome (2N), heterogeneous migration rates (2m) (Rougeux et al. 2017) (Table S3).
To check for model convergence, a total of 20 independent optimizations were performed for each model on each population comparison. When running these models, consistency in the likelihood scores generally increased as the best optimized parameters from previous steps were incorporated into subsequent steps. To score the models and account for overparameterization, the Akaike Information Criterion (AIC: Burnham and Anderson 2004) was used and parameters from the top five scoring runs for each model were averaged. The models with the best average AIC score were retained for each comparison. The highest and lowest optimized parameter values in the top five replicate runs for each model were used to construct intervals to estimate uncertainty. For details on SNP filtering, demographic model extensions, and transformation of parameter estimates to absolute units, see Supplemental Methods.
Admixture statistics
We estimated admixture proportions for 50 kb non-overlapping windows using the fd statistic (Martin et al. 2015a). This statistic is based on the ABBA-BABA test or Patterson’s D statistic which measures an excess of derived allele sharing between sympatric non-sister taxa in a tree of three populations and an outgroup with the relationship (((P1, P2), P3), O) (Durand et al. 2011; for details see Supplemental Methods). The populations included as P1, P2, and P3 are indicated in the figures. Heliconius hermathena samples were consistently used as the outgroup taxa (O).
To quantify the distribution of tracks in the genome with high fd in the hybrid zones, we modeled a loess regression to the fd data in R (span = 0.005, degrees of freedom = 2). We obtained tracks of multiple adjacent 50 kb windows with high fd where a loess fit rose above an fd threshold of 0.062 for the H. himera – H. e. cyrbia and H. himera - H. e. emma comparisons and above an fd threshold of 0.071 for H. himera – H. e. favorinus comparisons. While these fd thresholds represent a low absolute proportion of admixture for that window, we note that a single H. erato sample carrying an allele with ancestry from H. himera would result in an admixture proportion of maximally 0.125 if 4 samples are used in each population for the admixture test. Further, as fd estimates generally give a substantial underestimate of expected admixture proportions (~0.5 lower, see Martin et al. 2015a), we used half of the expected admixture proportion for a single individual (0.063) as a threshold for admixture in our loess fit regression. While this is likely an approximation, visual inspection between of the extracted high fd tracks shows that these generally match well to sharp increases in fd at multiple adjacent 50 kb windows. To understand whether the fd tracks result from single introgressed haplotypes rather than from multiple smaller haplotypes introgressed in multiple individuals, we compared the fd signals to relative divergence patterns between H. himera and single individuals of H. erato. For this, we used the measure da, which subtracts the average nucleotide diversity of both populations from pairwise nucleotide differences (dXY) between populations (Nei and Li 1979). These analyses allowed us to address whether the length of fd tracks could be explained by introgression of multiple smaller haplotypes from H. himera into H. erato. If we found that track lengths of low divergence, when using single H. erato individuals, corresponded to the tracks of high fd, this would suggest that the high fd tracks can be explained by introgression of single long haplotypes from H. himera into H. erato, rather than resulting from the introgression of several tightly linked haplotypes in multiple individuals.
Admixture directionality
By expanding the four-taxon D statistic to a five-taxon scenario, it is possible to obtain information on the directionality of admixture (i.e., donor versus recipient population). A set of statistical measures that use a five-taxon symmetric phylogeny to infer both the taxa involved in and the direction of admixture are called the DFOIL statistics (Pease and Hahn 2015; for details see Supplemental Methods). We assessed admixture directionality in 50 kb non-overlapping windows in the three H. himera contact zones with H. erato populations using the available dfoil software (www.github.com/jbpease/dfoil). Samples from the H. himera North and South populations were specified as the P1 and P2 group, whereas samples from the considered H. erato populations were specified as P3 and P4. Heliconius hermathena was used as the outgroup taxon (ID hermathena_13 in Table S1). The DFOIL statistics were calculated between each possible combination of available ingroup taxa (i.e., one sample for each taxon group); 800 combinations for H. himera – H. e. cyrbia, 500 for H. himera – H. e. emma and 480 for H. himera – H. e. favorinus. Among these sample combinations, significant DFOIL signatures (χ2 goodness-of-fit test) were counted and used to obtain heterogeneous patterns of admixture directionality along the genome.
Recombination rate estimates
We estimated fine-scale variation in population recombination rate (ρ = 4Ner; r = probability of recombination per generation per bp) along the H. erato chromosomes from linkage-disequilibrium in population genetic data using LDhelmet v1.7 (Chan et al. 2012). Individual genotypes were phased from thirteen H. erato populations (i.e. H. e. cyrbia, H. e. venus, H. e. demophoon, H. e. hydara (Panama), H. e. emma, H. e. etylus, H. e. lativitta, H. e. notabilis, H. e. favorinus, H. e. phyllis, H. e. erato, H. e. hydara (French Guiana) and H. e. amalfreda) using Beagle v4.1 (Browning and Browning 2016) with default parameters. To reduce the potential effect of locus-specific changes in effective population size (Ne) on population recombination rate (ρ) estimates (e.g., due to population specific selective sweeps or background selection), we estimated ρ for each H. erato population separately and obtained averages across50 kb interval (see Supplemental Methods for details).
Divergence and admixture simulations
To compare patterns in our data to expectations, we simulated genealogies near a selected locus. Genealogies were simulated using the coalescent simulator msms (Ewing and Hermisson 2010) and from these genealogies 10 kb sequences were simulated (For details see Supplemental Methods). The genealogies were sampled at 100 kb increments from the selected locus. This was achieved by using an infinite recombination sites model and changing the position of the selected locus in increments of 10 neutral locus units (i.e., 10 × 10 kb) away from the sampled locus. Divergence (FST) was calculated as in Hudson et al. (1992) and admixture (fd) was calculated as in Martin et al. (2016) using Python scripts and egglib v3 (De Mita & Siol, 2012). We investigated the correlation between FST, fd and recombination rate at a distance of 500 kb from a selected locus but similar expectations are obtained from wide range of distances to the selected locus. Simulations were run with 100 replicates for each parameter combination. Pseudocode to run the msms command lines are provided in Table S4.
Results
Phylogenetic relations
As previously observed in Van Belleghem et al. (2017), phylogenetically, two distinct clades can be recognized within the H. erato radiation: a clade west of the Andes and a clade east of the Andes (Figure 1A). The clade west of the Andes also includes populations from Panama and Mexico. The newly obtained sequences of the H. e. cyrbia population from North Ecuador are phylogenetically indistinguishable from the South Ecuador populations. Heliconius himera and H. e. chestertonii form distinct genetic clusters, separated from geographically closely related H. erato populations (Figure 1B). We find increased divergence between H. himera and H. erato from the western Andes slopes compared to H. erato from eastern Andes slopes. As seen in the PCA and phylogenetic inference, this results from a deeper split between H. himera and H. e. cyrbia from the western Andes slope compared to H. himera and the H. erato that are found east of the Andes, including H. e. emma and H. e. favorinus (Figure 1A, B). This is consistent with previous phylogenetic studies that placed H. himera nested within the H. erato clade and not as a sister species to H. erato (Supple et al. 2015; Van Belleghem et al. 2017).
Reproductive isolation and genomic landscapes of divergence
Correlations of RI with genome-wide estimates of relative divergence (FST) show that divergently selected color patterns between hybridizing color pattern morphs of H. erato are likely not sufficient to drive genome-wide increases in divergence when pre- or post-mating barriers are absent (Figure 2A). In these hybridizing H. erato morphs, there are only narrow peaks of divergence largely centered over the loci known to be responsible for color pattern differences (Figure 2B). In contrast, genomic divergence was highest and elevated genome-wide between H. e. venus and H. e. chestertonii from Colombia, where both mate preference and hybrid inviability have been reported (Muñoz et al. 2010). As predicted, the H. erato and H. himera comparisons showed intermediate levels of divergence that corresponded to their relatively stronger, but incomplete levels of reproductive isolation. Across all three studied contact zones between H. erato and H. himera, divergence has increased to the extent that FST peaks near the known color pattern loci WntA (chr 10), cortex (chr 15), and optix (chr 18) are largely not detectable (Figure 2B). However, there are clear differences in the degree of divergence between the species among the three contact zones, with the H. himera and H. erato contact zones on the eastern Andes (H. e. emma and H. e. favorinus) showing lower overall genomic divergence than in the western Andes contact zone. Also as expected, we observe a notable increase in divergence on the Z chromosome relative to the autosomes (Figure 2) that appears to be accentuated with stronger reproductive isolation. This is in line with previous work on H. e. chestertonii, which suggested Z chromosome divergence was inflated by an important barrier to gene flow (Van Belleghem et al. 2018).
Figure 2. Reproductive isolation and divergence among Heliconius erato populations.
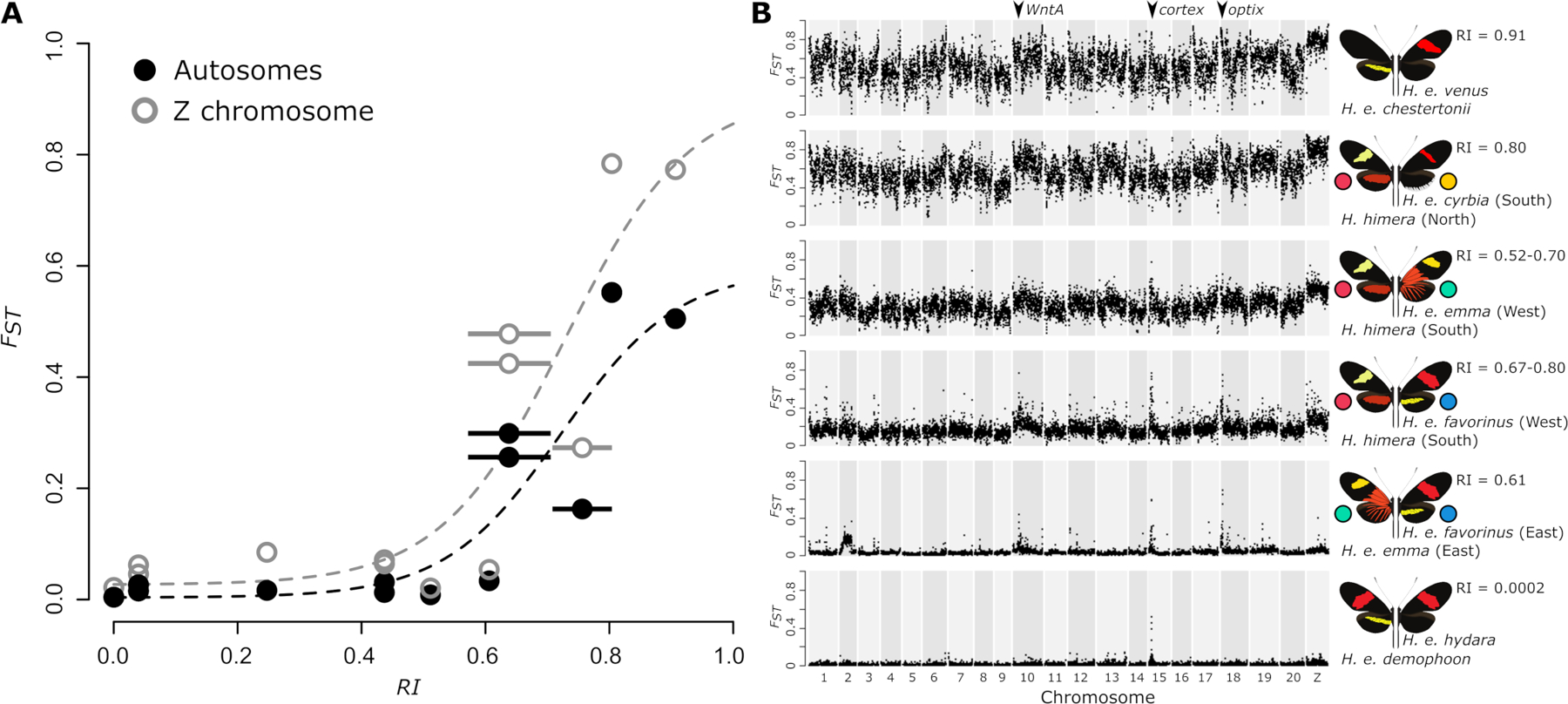
(A) Genome-wide averages of relative divergence (FST) show a sharp increase with increasing measures of reproductive isolation between parapatric H. erato populations. The measure of reproductive isolation (RI) was obtained by weighting ecological (RIec), pre-isolation (RIpre) and post-isolation (RIpost) components using the method of Sobel and Chen (2014) (Table S2). Horizontal bars indicate a range of RI estimates based on uncertainties in RI components. Dashed lines indicate a non-linear least-squares fit for autosome (black) and Z chromosome (gray) relation between FST and RI. Higher relative divergence on the Z chromosome can be observed for the more divergent parapatric comparisons, however, incompatibilities that are potentially Z-linked have only been suggested for H. e. chestertonii crosses (Muñoz et al. 2010; Van Belleghem et al. 2018). (B) Plots of relative divergence (average FST in 50 kb windows) between parapatric H. erato populations along the genome. Plots are ordered according to the measure of reproductive isolation (RI). Colored circles match color codes used for the focal populations in this study. Divergence peaks on chromosome 10, 15 and 18 correspond to the divergently selected color pattern genes WntA (affecting forewing band shape), cortex (affecting yellow hindwing bar), and optix (affecting red color pattern elements), respectively (Van Belleghem et al. 2017).
Demographic modeling
We compared the estimated joint site-frequency spectrum (JSFS) for the three geographically distinct contact zones between H. erato and H. himera to 26 alternative demographic scenarios that varied in split times, migration rates and population sizes. All three H. erato and H. himera contact zones best fit models that included secondary contact (SC) after a period of isolation without gene flow (Figure S2–3; Table S5). Figure 3 presents a composite of the best model for each pairwise H. erato and H. himera hybrid zone comparison plus a comparison of the H. erato populations east and west of the Andes and between H. e. emma and H. e. favorinus (parameter estimates of best pairwise ∂a∂i models is shown on Table S6). We also performed demographic modeling using geographic population subsets as a check to make sure that additional geographic population structure does not change our main results (Table S5). For all three zones we found support for asymmetrical migration. In each case, migration rates were predominantly in one direction, with on average 0.5 to 0.6 migrants per generation moving from H. erato into H. himera, compared to 0.07 to 0.13 moving from H. himera into H. erato. This result would be consistent with the effective migration rates being driven by the marked population size differences between the two species (Figure 3B).
Figure 3. Composite of best pairwise Secondary Contact (SC) demographic models of the H. himera and H. erato population history using ∂a∂i.

(A) Joint Site Frequency Spectra (JSFS) for data and best model (see Table S5 and Figure S2–3 for AIC values (Akaike Information Criterion)). (B) Reconstruction of historical demography of H. himera and H. erato populations using models with best AIC scores. All best models included a period of isolation and secondary contact. Arrows indicate effective migration rates (2Nam). Migration from H. himera into H. erato is indicated in orange, migration from H. erato into H. himera is indicated in blue. (C) Table with parameter ranges obtained from five best scoring models out of twenty runs. Na = ancestral population size, N1 = Size of population 1, N2 = size of population 2, g1 = growth coefficient of population 1, g2 = growth coefficient of population 2, ls = linked selection, Q = proportion of the genome with a reduced effective size due to linked selection (ls), m1<−2 = migration from population 2 into 1, m2<−1 = migration from populations 1 into 2, tsplit = split time, tSC = time of secondary contact, P = proportion of the genome evolving neutrally.
Nearly all the models that included exponential population growth (G) best fit the JSFS, with the exception of the H. himera and H. e. emma comparison. Estimates of ancestral and contemporary population sizes suggest strong expansions in H. himera and H. erato. These inferred changes in population sizes broadly fit previous results obtained from pairwise sequentially Markovian coalescent (PSMC) analysis (Van Belleghem et al. 2018), which suggested an overall population growth in H. erato east and west of the Andes in the past 1 Mya and size reduction for H. himera in the past 200 Kya (Figure S4). However, we found that estimates of contemporary population sizes varied greatly depending on the population comparison (Figure 3B, C), a result possibly explained by unaccounted population structure and difficulties in estimating growth (G). For H. e. favorinus, estimates of contemporary population size were generally much smaller than the ancestral population. This result fits the observation that H. e. favorinus is a smaller Andean population of the “postman” color pattern (i.e., red forewing band). Collectively, the models support a history that includes periods of allopatry, followed by lineage specific changes in population size that coincide with more recent gene flow.
To investigate if the JSFS contained evidence of selection driving patterns of divergence across the contact zones, we incorporated heterogeneity in population size (2N) and migration rate (2M) into the models, similar to Rougeux et al. (2017) and Tine et al. (2014), respectively. The 2N model allows heterogeneity in population size estimates across loci that result from the differences in allelic variation caused by linked selection (ls = effective population size of locus relative to neutral loci; Q = proportion of the genome affected by ls). We found that all contact zones between H. erato and H. himera well supported 2N models, suggesting the effect of linked selection in shaping patterns of genomic variation and divergence between the species. The strongest ls was observed for the population comparisons of H. erato East and West (ls = 0.10; Q = 0.35) and H. himera and H. erato West (ls = 0.15; Q = 0.90) and the lowest observed between H. e. emma and H. e. favorinus (ls = 0.07; Q = 0.04) (Figure 3C, Table S6).
Selection on locally adapted alleles will also result in regions containing these variants showing much lower rates of migration compared to the rest of the genome. The 2M models allow for this type of heterogeneity in migration rates across the genome. Both contact zones in the eastern Andes supported these models for H. himera and H. erato, suggesting that these populations have genomic regions with much lower rates of introgression than other parts of the genome.
Admixture directionality
To investigate the impact of hybridization on genomic divergence, we explored patterns of admixture along chromosomes for the three contact zones between H. erato and H. himera. We were interested in both the patterns of admixture across our three replicate hybrid zones and inferring the direction of admixture. To determine if patterns of admixture were similar across the three contact zones, we used the fd statistic which indicates admixture proportions (proportion of sites that are shared between taxa) within genomic windows (Martin et al. 2015a). Across several chromosomes we observed long tracks of increased fd, particularly in the hybrid zone between H. himera and H. e. cyrbia west of the Andes (Figure 4A). These high fd tracks included a total of 37.97 Mb and had an average length of 2.53 Mb (sd = 3.31 Mb) (Figure S5). High fd tracks included 35.67 Mb between H. himera and H. e. emma (average length: 1.00 Mb ± 1.34 Mb) and 29.49 Mb between H. himera and H. e. favorinus (average length: 1.23 Mb ± 1.017 Mb) and were distributed more evenly along the chromosomes (Figure 4A). In general, there was a lack of overlap between genomic regions with high fd in the different hybrid zone comparisons. We found 4.48 Mb (6.5 %) of high fd tracks were shared between the H. himera/H. e. cyrbia and H. himera/H. e. emma hybrid zone, 3.83 Mb (6.0 %) were shared between the H. himera/H. e. cyrbia and H. himera/H. e. favorinus hybrid zone and 8.04 Mb (14.1 %) were shared between the H. himera/H. e. emma and H. himera/H. e. favorinus hybrid zone.
Figure 4. Admixture (fd) and admixture directionality (DFOIL) between H. himera and H. erato in three contact zones.
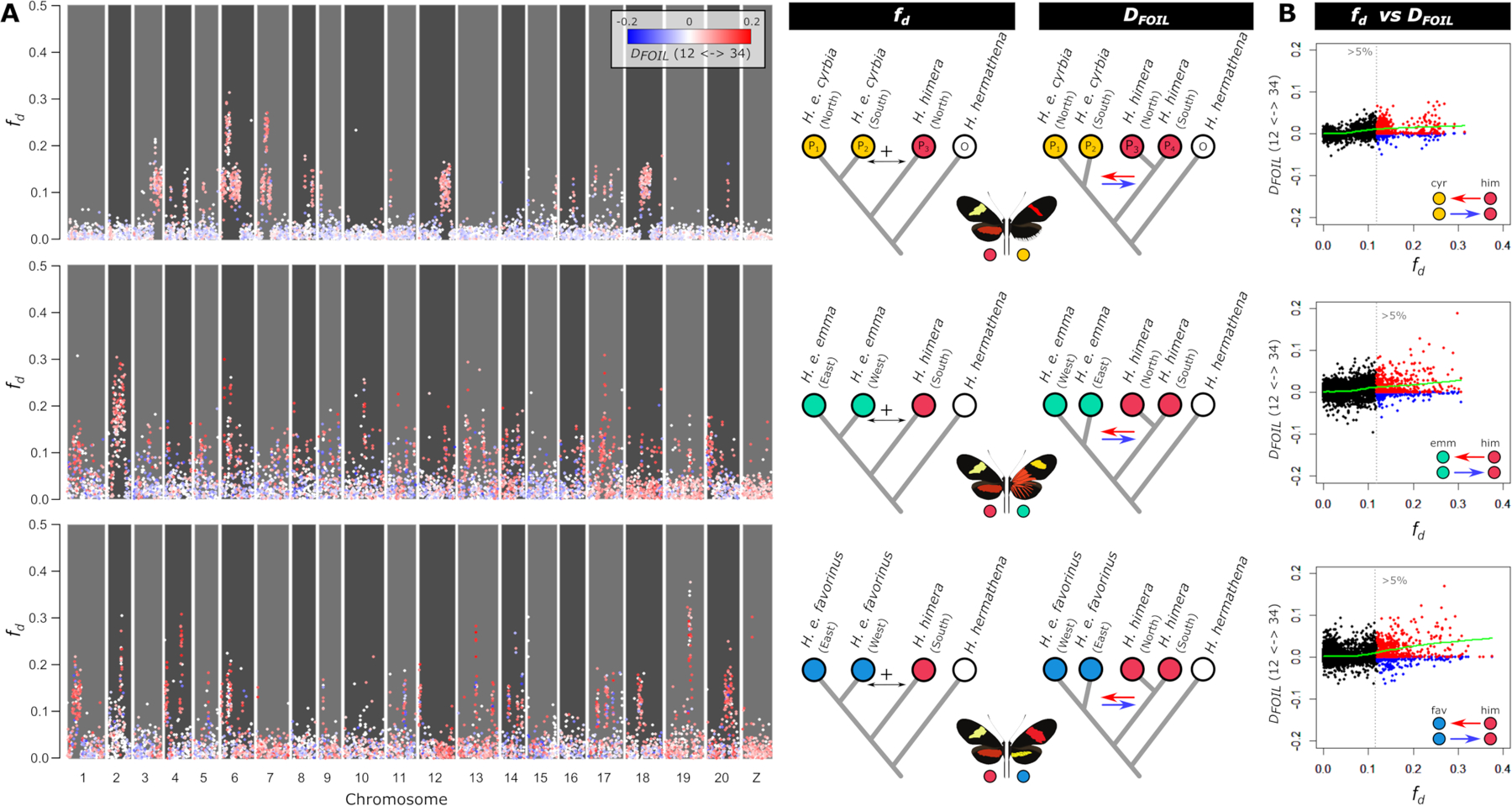
(A)Points show admixture (fd) values, whereas coloring shows directionality (DFOIL) in 50 kb non-overlapping windows for the contact zones H. himera – H. e. cyrbia (top), H. himera – H. e. emma (middle) and H. himera – H. e. favorinus (bottom). Blue indicates predominant admixture from H. erato into H. himera (12 -> 34), whereas red indicates predominant admixture from H. himera into H. erato (12 <- 34) based on the DFOIL tests. (B) Summary of admixture versus directionality with points above the 95% quantile indicated in blue (12 -> 34) and red (12 <- 34) demonstrates that the majority of windows with high fd indicate admixture from H. himera into H. erato. The green line indicates a loess fit of the data. Colored circles match color codes in Figure 1.
To determine the directionality of introgression across the genome we used a five-taxon DFOIL-statistic (Pease and Hahn 2015). This test considers all available taxa (i.e., two populations of H. himera and two populations of H. erato) and calculates all possible four-taxon D-statistic comparisons to infer admixture as well as directionality of admixture. While the DFOIL-statistic is calculated using a single genome for each taxon, we performed this test on all possible combinations of available samples and represented the DFOIL signal as the proportion of significant comparisons (Figure 4A). Comparing fd and DFOIL results revealed that among loci that show strong evidence of admixture (high fd), there is a relative paucity of loci in H. himera individuals carrying H. erato alleles (Figure 4B). This result contrasts with the demographic modeling which suggested higher rates of gene flow from H. erato into H. himera (Figure 3) and likely results from differences in temporal dynamics measured by each method, with ∂a∂i models fitting older signals of admixture that are potentially driven by a higher effective population size of H. erato compared to H. himera. This asymmetry suggests H. erato is more porous to introgressed alleles from H. himera than vice-versa. Consistent with a well characterized “large X(Z) effect” in speciation, on the sex (Z) chromosome there are only a few loci that show signals of admixture (high fd) and again the DFOIL tests only show signals of introgression of H. himera alleles into H. erato on the Z chromosome (Figure 4A).
Because long tracks of increased fd may result from either introgression of long haplotypes or introgression at multiple tightly linked loci, we next investigated divergence signals between single H. erato individuals from hybrid zones with the H. himera populations (Figure S6). In all three contact zones, we find that many of the high fd tracks correspond to tracks of reduced divergence between a single H. erato individual and H. himera. This is in line with the high fd tracks being largely driven by H. erato individuals carrying a haplotype with H. himera ancestry.
Association of divergence and admixture with recombination rate
Because the effect of selection on linked sites will depend on recombination rate, differences in the relationships between recombination rate and relative divergence (FST) and between recombination rate and admixture (fd) can be used to determine how selection may have shaped genomic divergence between H. erato and H. himera. We first explored this relationship of recombination rate, relative divergence, and admixture across the optix locus. The optix locus controls red wing pattern differences among H. erato color pattern morphs, as well as between H. erato and H. himera and is a target of strong selection (Figure 5A) (Counterman et al. 2010; Supple et al. 2015; Van Belleghem et al. 2017). We found recombination rate (ρ) estimates were markedly lower upstream, compared to downstream of the optix gene (Figure 5C). Such sharp decreases in recombination rate near chromosome ends have been observed in other Heliconius species (Martin et al. 2019) and likely explain the decrease in recombination rate upstream of the optix gene. To test if selection at the optix locus has produced the expected negative relationship of recombination rate with divergence and positive relationship of recombination rate and admixture, we visualized these relationships 500 kb from the center of the peak of the divergence at optix (Figure 5D) so they could be directly compared to the simulation results, which were also sampled 500 kb from the target of selection (see ‘Divergence and admixture simulations’). As expected, we observe a negative relationship between recombination rate and divergence (FST) but no obvious correlation between recombination rate and admixture (fd).
Figure 5. Divergence (FST), admixture (fd) and recombination rate (ρ) near the red color pattern gene optix.

(A) The optix gene is located near the start of chromosome 18 and has been demonstrated to control the expression of red color pattern elements in Heliconius wings (Reed et al. 2011). (B) Relative divergence (FST) and admixture (fd) comparisons performed between H. himera, H. e. cyrbia, H. e. emma and H. e. favorinus. Colored circles match color codes in Figure 2. (C) Lines show FST, fd and recombination rate (ρ) calculated in 50 kb non-overlapping windows. The green line in the bottom plot shows a loess fit of the recombination rate. Gene models including the location of the optix gene are presented at the top. (D) Relationship between FST and recombination rate (ρ) 500 kb left and right from the center of the optix regulatory sequence divergence peak. The relation between fd and recombination rate (ρ) is near zero and not shown.
To understand how recombination rate variation, selection and admixture have contributed to patterns of genome-wide divergence, we next compared recombination rates with divergence and admixture across the whole genome. In line with expected patterns of linked selection, we found a significant negative association between recombination rate (ρ) and relative divergence (FST) between H. himera and H. erato populations (Figure 5A) but no association with admixture (fd) (Figure 6B).We also observed a positive association between the proportion of coding sequence and relative divergence, further suggesting the importance of linked selection for genome divergence (Figure S7). Additionally, we found a significant negative relationship between FST and fd in the H. himera – H. e. cyrbia and H. e. emma hybrid zones, which indicates that admixture can partly explain reduced FST (Figure 6C).
Figure 6. Correlations of divergence and admixture proportions with recombination rate in the three H. himera – H. erato contact zones.

(A) Relative divergence (FST) versus population recombination rate (ρ). (B) Admixture proportion (fd) versus population recombination rate (ρ). (C) Relative divergence (FST) versus admixture proportion (fd). Statistics were calculated in 50 kb non-overlapping windows. Recombination rates were calculated from each H. erato population separately and averaged over populations (see methods) and showed a genome-wide average of ρ equal to 0.071 (SD = 0.026; ρ = 4Ner). Colored circles match geographic distributions and contact zones in Figure 2.
Divergence and admixture simulations
We used a range of estimates of divergence times, population size changes, and migration from the demographic models to seed coalescent simulations and investigate expected patterns of recombination rate on divergence and admixture rates. Specifically, the simulations test the prediction whether selection (i.e., recent selective sweeps) and isolation (i.e., secondary contact) in the past million year of divergence between H. himera and H. erato can explain the observed relationships of recombination rate with relative divergence (FST) and admixture (fd).
From our demographic models, the split between H. himera and H. erato (East) was estimated between 215 and 527 thousand years ago. In our simulations this time point fits the split between (P1, P2) and P3, which we modeled at 2,000,000 generations (t2; Figure 7A). Assuming a generation time of 0.25 years, 2,000,000 generations match a split of 500 thousand years ago. After these populations split, migration (m) was restricted between P2 and P3 only, with symmetrical migration rates starting at time tm. In accordance with our demographic modeling results, we ran simulations by also varying the parameters ts (selection start time), m (migration rate) and ρ (population recombination). In Figure 7, we highlight two extreme scenarios of migration and selection start time which we denoted as “Isolation with Migration” (IM) and “Secondary Contact” (IM). As demonstrated by other studies, in a scenario of IM with long periods of migration and divergent selection, our simulations predicted a strong negative correlation between recombination rate and FST (Keinan and Reich 2010; Brandvain et al. 2014; Crawford et al. 2015) and a strong positive correlation between recombination rate and fd (Aeschbacher et al. 2017; Martin et al. 2019) (Figure 7C, left). The correlation pattern between recombination rate and FST reflects the degree of linkage disequilibrium between neutral sites and loci under divergent selection (Charlesworth 1998; Nachman and Payseur 2012). In contrast, our simulations show that the relationship between recombination rate and fd is reduced when migration is more recent or low (Figure 7C, left). For example, in a SC scenario that is characterized by a more recent onset of migration, the simulations show that FST values are generally high and fd values close to zero (Figure 7C, right). This results in the absence of a relationship between recombination rate and FST or fd in most SC scenarios when the onset of migration started less than 10,000 generations ago and in IM models with migration rates (2Nem) lower than 2. However, when selection in the genome is recent (~selective sweeps), a negative relationship between recombination rate and divergence, but not admixture, emerges in the SC scenario (Figure 7C). This relationship arises due to linked selection that reduces diversity within populations and increases the relative divergence, FST, between populations over larger genomic regions with lower recombination rate (Cruickshank and Hahn 2014). The relationship holds for lower selection strengths at relatively short distances from the selected site (s = 0.02; Figure S8).
Figure 7. Expected relationship of recombination rate with divergence (FST) and admixture (fd) near a divergently selected locus.

(A) The population tree shows the simulated scenario with the onset of divergent selection on a derived allele indicated in red and in which migration rate (m) and migration time (tm) between P2 and P3 and selection start time (ts) are varied. Left and right of the simulated scenario are parameter combinations for two extreme scenarios that both include linked selection; on the left a scenario with Isolation with Migration (IM) and on the right a scenario reflecting Secondary Contact (SC). (B) Effect of population recombination rate (ρ) on relative divergence (FST) and admixture (fd) near a divergently selected locus for the two simulated scenarios with parameter combinations as in panel A. The selected allele occurs at position 0. The dashed red line indicates a locus at 500 kb from the selected locus at which the relationship between ρ, divergence and admixture is assessed in panel C. (C) The effect of migration start time (tm) and selection start time (ts) on the relation between ρ, divergence and admixture. Apart from the respective parameters being evaluated, other parameters were fixed as in panel A, with the red lines indicating the exact parameter combinations as in panel A and B. For simulations with a lower selection coefficient (s = 0.02), see Figure S8.
The simulated secondary contact model is an extreme scenario with recent onset of strong migration (start migration << 100,000 generations). However, similar relationships between recombination rate, relative divergence and admixture arise in a scenario where secondary contact is older (>> 100,000 generations) but migration rates are low (Figure 7C). This latter scenario fits the observed demographic estimates for H. erato and H. himera, with low migration rates (2Nem < 0.2) estimated to start at 100,000 – 750,000 generations ago.
Finally, we note that the observed relationship between recombination rate and divergence may include the effects of genetic hitchhiking, selection against gene flow as well as background selection. While our genomic dataset does not allow us to differentiate background selection from genetic hitchhiking and selection against gene flow, our simulations suggest that the observed patterns can be explained by these latter two processes, and other studies suggest background selection may be too subtle to cause these patterns (Stankowski et al. 2019).
Discussion
By relating patterns of genomic divergence to ecological and other barriers to gene flow, our results provide a view into the genomic landscape among lineages with increasing degrees of reproductive isolation (Kronforst et al. 2013; Roux et al. 2016). In line with theoretical expectations of the speciation process (Barton and De Cara 2009; Nosil et al. 2017), the increase in divergence does not seem to be a linear process. Rather, there is a marked increase in divergence associated with assortative mating which has evolved between H. himera and H. erato, highlighting the global impacts that incomplete pre-mating barriers could have in the genome. We also observed a notable increase in divergence on the Z chromosome relative to the autosomes which supports a large effect of the Z chromosome in the genomic divergence during the early stages of speciation (Sperling 1994; Prowell 1998). Despite being able to reconstruct this progression of genomic divergence from increased reproductive isolation, it remains difficult to determine if genomic peaks of divergence result from barrier loci that are resistant to ongoing gene flow (heterogeneous gene flow), from recent selective sweeps in isolated populations (heterogeneous selection), or both. Nevertheless, from comparing the observed patterns of genomic divergence with demographic models and coalescent simulations, we can test various hypotheses of the architecture of the species boundaries between H. erato and H. himera that allow us to determine the extent isolation and selection contribute to the heterogeneous landscapes of divergence.
Architecture of the species boundaries
Our data favors a history of isolation accompanied with selection. From our coalescent simulations, we found that heterogeneity in levels of divergence (FST) may largely be explained by selection acting on alleles within isolated populations and is not representative of selection against gene flow. In contrast, observed signals of admixture (fd) and the length of high admixture tracks may reflect a snapshot from recent hybridization events. Although we note that the expectations of admixture track lengths may differ when there is selection against recombinants, we argue that the long length of high fd tracks compared to the linkage disequilibrium breakdown within 1–2 kb estimated in H. melpomene (Martin et al. 2016) together with single H. erato individuals harboring long haplotypes of H. himera ancestry (Figure S6) suggests these patterns may represent recent and independent admixture events at the three contact zones. The heterogeneous signal of admixture may thus be the result of a few generations of backcrossing of hybrid individuals even in the absence of selection. Nevertheless, whether introgressed haplotypes may become purged by selection or result from adaptive introgression is hard to predict with the current data. One possibility is that the multitude of loci with asymmetrical gene flow from H. himera into H. erato represents the genetic signal of a polygenic species barrier. These barriers could result from co-adapted loci in H. himera that cannot be replaced by H. erato alleles without fitness consequences. The inference that this pattern of asymmetrical gene flow results from a polygenic species barrier and not from mate choice directionality or demographic factors (e.g., larger effective population size of H. erato) is further strengthened by two observations. First, H. himera males have been shown to mate more frequently with F1 hybrids compared to H. erato males (McMillan et al. 1997), which should result in an opposite asymmetric admixture pattern than what we found (Latour et al. 2013), with more alleles introgressing from H. erato into H. himera. Second, the smaller effective population size estimates of H. himera should also result in the same opposite pattern of asymmetric admixture (Frantz et al. 2014). This latter expectation is observed in the demographic modeling results, which show greater migration of alleles from H. erato into H. himera and corresponds to their estimated differences in population size between the species (Figure 3B) but is not seen in the more recent signals of admixture as measured by fd. This discrepancy between the migration rates estimated from the demographic modeling and the fd statistic potentially results from differences in the temporal dynamics measured by each method, with ∂a∂i models fitting older signals of admixture.
The finding that selection at multiple loci may have resulted in a polygenic species boundary would add to a number of studies that illustrate how pervasive selection along the genome can lead to the evolution of polygenic architectures of species boundaries (Brandvain et al. 2014; Janousek et al. 2015; Aeschbacher et al. 2017; Berner and Roesti 2017; Martin et al. 2019). This is also consistent with evidence that many loci contribute to population divergence among hybridizing population in the H. erato clade (Lewis et al. 2020). However, it also possible that asymmetrical adaptive introgression could contribute to the observed patterns of divergence and admixture between H. erato and H. himera. Adaptive introgression has been well documented at color pattern loci across Heliconius (Dasmahapatra et al. 2012; Wallbank et al. 2016; Van Belleghem et al. 2017; Edelman et al. 2019) but larger genomic datasets are often needed to perform these more rigorous tests for signatures of selection and introgression (Edelman et al. 2019; Meier et al. 2020; Moest et al. 2020). We find very few genomic regions that show admixture across all three H. himera and H. erato contact zones, which would be expected if these regions represent random tracts of admixture from recent gene flow, but not if adaptive introgression were driving admixture of specific alleles with selective advantage. Therefore, it seems possible that the patterns reflect a polygenic species boundary driven by selection against gene flow. However, further data and analyses are needed to assess the possible contribution of adaptive introgression.
Different histories can generate similar heterogeneous divergence patterns
Although the genomic landscape of divergence between hybridizing taxa reflects the history of selection and demographic changes they have experienced, different histories can generate strikingly similar heterogeneous patterns. This can greatly limit our ability to make inferences about the evolutionary processes driving genomic change (Ravinet et al. 2017). For example, in Heliconius melpomene there is, at first sight, a similar heterogeneous landscape of divergence to those we report here for H. erato and H. himera, despite known differences in their evolutionary histories (Quek et al. 2010; Martin et al. 2013). Heliconius melpomene and H. erato are co-mimics and experience similar strong selective pressures on wing color patterns throughout their distributions. In the H. melpomene clade, H. melpomene comes into contact and hybridizes with H. cydno in Panama and occasionally with H. timareta in Ecuador and northern Peru. Although the genomic landscapes in the erato and melpomene clades appear similar at first sight, correlation analyses reveal potential differences in the relative importance of admixture. The H. melpomene comparisons showed strong correlations of recombination rate with divergence as well as admixture, which supports a long history of divergent selection with gene flow (Martin et al. 2019) and agrees with our coalescent simulations. In contrast, H. himera and H. erato comparisons showed recombination rate correlated with divergence, but not admixture. We propose two possible explanations for this difference between the Heliconius clades. First, there may be differences in the genetic architecture of the species barriers, such that there are fewer or more tightly clustered barrier loci in the H. erato and H. himera comparisons, thereby limiting the ability for a correlation between recombination rate and admixture to arise. However, the genetic architectures of adaptive divergence (e.g., color pattern) are strikingly similar in the erato and melpomene clades, and in melpomene, loci responsible for mate preference and adaptive traits (e.g., color pattern) have been demonstrated to be tightly linked (Jiggins et al. 2001; Kronforst et al. 2006; Merrill et al. 2019). Therefore, differences in the number or having too few barrier loci is likely insufficient to explain this difference in admixture patterns between the H. erato - H. himera and H. melpomene – H. cydno/timareta populations. Alternatively, there may not have been sufficient migration events and time for selection to remove foreign alleles after introgression and thus for a correlation between recombination rate and admixture to arise between H. erato and H. himera. In this latter case, genetic differences that accumulated in periods of isolation would have had more profound effects on the divergence landscape of H. erato and H. himera whereas gene flow has likely been more continuous throughout the divergence history of the species pair in the H. melpomene clade (Martin et al. 2015b, 2019). These differences in the history of divergence among the co-mimetic species highlights the power of approaches like those applied here to resolve the roles of different evolutionary processes in generating seemingly similar heterogeneous patterns of genomic divergence.
Supplementary Material
Table S 1. Samples used in this study. Samples from H. himera populations and H. erato populations that come into contact with H. himera are indicated in color. H. himera North, light red; H. himera South, dark red; H. e. cyrbia North, light yellow, H. e. cyrbia South, dark yellow; H. e. emma East, light green; H. e. emma West dark green; H. e. favorinus East, light blue; H. e. favorinus West, dark blue. Earthcape IDs are available through https://heliconius.ecdb.io.
Table S 2. Hybrid zone characteristics between H. erato and H. himera populations. Major color pattern (CP) loci include the genes optix (~red), WntA (~forewing band shape) and cortex (~yellow hindwing bar and white fringes) Reproductive isolation (RI) was estimated as a combination of ecological isolation (RIec) due to divergent selection on color patterns, pre-mating isolation (RIpre) due to mate preference, and post-mating isolation (RIpost) due to hybrid sterility.
Table S 3. 2D δaδi model descriptions.
Table S 4. Bash pseudocode to run msms (Ewing and Hermisson 2010) simulations.Note that mutation rate is only used by seq-gen when simulating the sequences, population size is used to scale times, selection and migration proportions.
Table S 5. AIC weights dadi models. AIC scores for each model are averages of the top 5 runs. Shaded cells indicate lowest AIC values.
Table S 6. Best model parameters for best model class as well as alternative model classes. Split times are given as Ttotal+TAM in AM, and Ttotal+TSC in SC. Although all best fitting demographic models include secondary contact (SC), several alternative models including AM (Ancient Migration) and IM (Isolation and Migration) fit relatively well to the observed JSFS (Figure 3A; Figure S4–5). However, in these models, migration rates generally scale proportionately with migration duration compared to the SC models, indicating that our interpretations on migration rates should not be affected by the uncertainty of finding a best demographic model.
Figure S 1. FastTree of samples used in this study. Node support values are based on the Shimodaira–Hasegawa test.
Figure S 2. Average AIC (Akaike Information Criterion) of all tested δaδi models for twenty runs. SI = Strict Isolation, IM = Isolation and Migration, AM = Ancestral Migration, SC = Secondary Contact, G = exponential growth, 2N = heterogeneous effective population size (with 2 classes of loci shared by the two populations = Hill-Robertson effects), and 2m = 2 migration rates.
Figure S 3. Average AIC (Akaike Information Criterion) of all tested δaδi models for twenty runs using subpopulations. SI = Strict Isolation, IM = Isolation and Migration, AM = Ancestral Migration, SC = Secondary Contact, G = exponential growth, 2N = heterogeneous effective population size (with 2 classes of loci shared by the two populations = Hill-Robertson effects), and 2m = 2 migration rates.
Figure S 4. Inference of historical effective population size changes using pairwise sequentially Markovian coalescent (PSMC) analysis. Lines and shading in the left panel represent the average effective population size and 95% CI, respectively, estimated from PSMC analyses on individual genome samples as shown in the panels on the right. The PSMC estimates are scaled using a generation time of 0.25 years and a mutation rate of 2e-9. Data adopted from Van Belleghem et al. (2018).
Figure S 5. Loess fit to admixture (fd) between H. himera and H. erato in three contact zones. Points show admixture (fd) values, whereas coloring shows directionality (DFOIL) in 50 kb non-overlapping windows for the contact zones H. himera ¬– H. e. cyrbia (top), H. himera – H. e. emma (middle) and H. himera – H. e. favorinus (bottom) as in Figure 3. Blue indicates predominant admixture from H. erato into H. himera (12 <- 34), whereas red indicates predominant admixture from H. himera into H. erato (12 -> 34) based on the DFOIL tests. Green lines indicate the Loess fit.
Figure S 6. Comparison of admixture (fd) signals with relative divergence (da) of single H. erato individuals to H. himera. Panels with black points show the fd signals between contact zones of H. himera with H. e. cyrbia (top), H. e. emma (middle) and H. e. favorinus (bottom). Panels with colored points show relative divergence da, calculated from pairwise sequence differences minus the average nucleotide diversity of both populations, between H. himera and a single individual of a hybridizing H. erato population. Green rectangles are high fd tracks as identified from a Loess fit to continues windows of fd larger than 0.06 for the H. e. cyrbia and H. e. emma contact zones and 0.07 for the H. e. favorinus contact zone.
Figure S 7. Correlations of divergence and admixture proportions with recombination rate and gene density in H. himera – H. erato contact zones. (A) Relative divergence (FST) versus gene density and recombination rate (ρ). (B) Admixture proportion (fd) versus gene density and recombination rate (cM/Mb). (C) Relative divergence (FST) versus admixture proportion (fd). Statistics were calculated in 50 kb non-overlapping windows. Colored circles match color codes in Figure 2.
Figure S 8. Expected relationship of recombination rate with divergence (FST) and admixture (fd) near a divergently selected locus (s = 0.02). (A) The population tree shows the simulated scenario with the onset of divergent selection on a derived allele indicated in red and in which migration rate (m) and migration time (tm) between P2 and P3 and selection start time (ts) are varied. Left and right of the simulated scenario are parameter combinations for two extreme scenarios that both include linked selection; on the left a scenario with Isolation with Migration (IM) and on the right a scenario reflecting Secondary Contact (SC). (B) Effect of population recombination rate (ρ) on relative divergence (FST) and admixture (fd) near a divergently selected locus for the two simulated scenarios with parameter combinations as in panel A. The selected allele occurs at position 0. The dashed red line indicates a locus at 500 kb from the selected locus at which the relationship between ρ, divergence and admixture is assessed in panel C. (C) The effect of migration start time (tm) and selection start time (ts) on the relation between ρ, divergence and admixture. Apart from the respective parameters being evaluated, other parameters were fixed as in panel A, with the red lines indicating the exact parameter combinations as in panel A and B.
Acknowledgments
This work was funded by NSF grant (DEB 1257689) to BAC and RP and NSF EPSCoR RII Track-2 FEC (OIA 1736026) to RP and BAC. For sequencing and computational resources, we thank the University of Puerto Rico, the Puerto Rico INBRE Grant P20 GM103475 from the National Institute for General Medical Sciences (NIGMS), a component of the National Institutes of Health (NIH); and awards 1010094 and 1002410 from the EPSCoR program of the NSF. We thank the Ecuadorian Ministerio del Ambiente (No. 005-13 ICFAU- DNB/MA), Peruvian Ministerio de Agricultura and Instituto Nacional de Recursos Naturales (201-2013-MINAGRI-DGFFS/DGEFFSS) and Autoridad Nacional De Licencias Ambientales-ANLA in Colombia (Permiso Marco 0530) for permission to collect butterflies. We thank Simon Martin for sharing his code and useful discussions that shaped this paper. We thank Nicola Nadeau for access to sequence data for northern H. erato cyrbia samples, which was funded by her UK Natural Environment Research Council (NERC) fellowship (NE/K008498/1). We thank Matthew Streisfeld and the reviewers for their exceptional help in improving this manuscript.
Footnotes
Conflict of Interest: The authors have no conflict of interest to declare.
Data accessibility
For GenBank accession numbers of whole genome resequence data see Table S1. Scripts and data for RI calculations and genomic analysis are available at https://github.com/StevenVB12/Genomics. Scripts for ∂a∂I inference are available at https://github.com/jmcole003/2018_herato_demography.
References
- Aeschbacher S, Selby JP, Willis JH, and Coop G. 2017. Population-genomic inference of the strength and timing of selection against gene flow. Proc. Natl. Acad. Sci 114:7061–7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias CF, Van Belleghem S, and McMillan WO. 2016. Genomics at the evolving species boundary. Curr. Opin. Insect Sci 13:7–15. Elsevier Inc. [DOI] [PubMed] [Google Scholar]
- Barton N, and Bengtsson BO. 1986. The barrier to genetic exchange between hybridising populations. Heredity (Edinb). 57:357–376. [DOI] [PubMed] [Google Scholar]
- Barton NH, and De Cara MAR. 2009. The evolution of strong reproductive isolation. Evolution. 63:1171–1190. [DOI] [PubMed] [Google Scholar]
- Berner D, and Roesti M. 2017. Genomics of adaptive divergence with chromosome-scale heterogeneity in crossover rate. Mol. Ecol 26:6351–6369. [DOI] [PubMed] [Google Scholar]
- Brandvain Y, Kenney AM, Flagel L, Coop G, and Sweigart AL. 2014. Speciation and introgression between Mimulus nasutus and Mimulus guttatus. PLoS Genet. 10:e1004410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnham KP, and Anderson DR. 2004. Multimodel inference: Understanding AIC and BIC in model selection. Sociol. Methods Res 33:261–304. [Google Scholar]
- Burri R 2017. Interpreting differentiation landscapes in the light of long-term linked selection. Evol. Lett 1:118–131. [Google Scholar]
- Chan AH, Jenkins PA, and Song YS. 2012. Genome-wide fine-scale recombination rate variation in Drosophila melanogaster. PLoS Genet. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B 2009. Effective population size and patterns of molecular evolution and variation. Nat. Rev. Genet 10:195–205. [DOI] [PubMed] [Google Scholar]
- Charlesworth B 1998. Measures of divergence between populations and the effect of forces that reduce variability. Mol. Biol. Evol 15:538–543. [DOI] [PubMed] [Google Scholar]
- Chouteau M, Arias M, and Joron M. 2016. Warning signals are under positive frequency-dependent selection in nature. Proc. Natl. Acad. Sci 113:2164–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counterman BA, Araujo-Perez F, Hines HM, Baxter SW, Morrison CM, Lindstrom DP, Papa R, Ferguson L, Joron M, Ffrench-Constant RH, Smith CP, Nielsen DM, Chen R, Jiggins CD, Reed RD, Halder G, Mallet J, and McMillan WO. 2010. Genomic hotspots for adaptation: the population genetics of Müllerian mimicry in Heliconius erato. PLoS Genet. 6:e1000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford JE, Riehle MM, Guelbeogo WM, Gneme A, Sagnon N, Vernick KD, Nielsen R, and Lazzaro BP. 2015. Reticulate speciation and barriers to introgression in the Anopheles gambiae species complex. Genome Biol. Evol 7:3116–3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruickshank TE, and Hahn MW. 2014. Reanalysis suggests that genomic islands of speciation are due to reduced diversity, not reduced gene flow. Mol. Ecol 23:3133–3157. [DOI] [PubMed] [Google Scholar]
- Dasmahapatra KK, Walters JR, Briscoe AD, Davey JW, Whibley A, Nadeau NJ, Zimin AV, Adler S, Ahn S-J, Baker DA, Dalmay T, Gilbert LE, Gordon K, Heckel DG, and Hines HM. 2012. Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature 487:94–98. Nature Publishing Group. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Mita S, and Siol M. 2012. EggLib: Processing, analysis and simulation tools for population genetics and genomics. BMC Genet. 13:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand EY, Patterson N, Reich D, and Slatkin M. 2011. Testing for ancient admixture between closely related populations. Mol. Biol. Evol 28:2239–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelman NB, Frandsen PB, Miyagi M, Clavijo B, Davey J, Dikow R, García-accinelli G, Van Belleghem SM, Patterson N, Daniel E, Challis R, Kumar S, Moreira G, Salazar C, Counterman B, Papa R, Blaxter M, Reed RD, Kronforst M, Joron M, Jiggins CD, Mcmillan WO, Di Palma F, Blumberg AJ, Wakeley J, Jaffe D, and Mallet J. 2019. Genomic architecture and introgression shape a butterfly radiation. Science. 366:24174–24183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing G, and Hermisson J. 2010. MSMS: a coalescent simulation program including recombination, demographic structure and selection at a single locus. Bioinformatics 26:2064–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantz LAF, Madsen O, Megens HJ, Groenen MAM, and Lohse K. 2014. Testing models of speciation from genome sequences: Divergence and asymmetric admixture in Island South-East Asian Sus species during the Plio-Pleistocene climatic fluctuations. Mol. Ecol 23:5566–5574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutenkunst RN, Hernandez RD, Williamson SH, and Bustamante CD. 2009. Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet. 5:e1000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR, Slatkin M, and Maddison WP. 1992. Estimation of levels of gene flow from DNA sequence data. Genetics 132:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janousek V, Munclinger P, Wang L, Teeter KC, and Tucker PK. 2015. Functional Organization of the Genome May Shape the Species Boundary in the House Mouse. Mol. Biol. Evol 2:1208–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiggins CD, Mcmillan O, Neukirchen W, Mallet J, and Nw L. 1996. What can hybrid zones tell us about speciation? The case of Heliconius erato and H. himera (Lepidoptera: Nymphalidae). Biol. J. Linn. Soc 59:221–242. [Google Scholar]
- Jiggins CD, Naisbit RE, Coe RL, and Mallet J. 2001. Reproductive isolation caused by colour pattern mimicry. Nature 411:302–305. [DOI] [PubMed] [Google Scholar]
- Kaplan NL, Hudson RR, and Langley CH. 1989. The “hitchhiking effect” revisited. Genetics 123:887–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keinan A, and Reich D. 2010. Human population differentiation is strongly correlated with local recombination rate. PLoS Genet. 6:e1000886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronforst MR, Hansen MEB, Crawford NG, Gallant JR, Zhang W, Kulathinal RJ, Kapan DD, and Mullen SP. 2013. Hybridization reveals the evolving genomic architecture of speciation. Cell Rep. 5:666–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronforst MR, Young LG, Kapan DD, McNeely C, O’Neill RJ, and Gilbert LE. 2006. Linkage of butterfly mate preference and wing color preference cue at the genomic location of wingless. Proc. Natl. Acad. Sci. U. S. A 103:6575–6580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latour Y, Perriat-Sanguinet M, Caminade P, Boursot P, Smadja CM, and Ganem G. 2013. Sexual selection against natural hybrids may contribute to reinforcement in a house mouse hybrid zone. Proc. R. Soc. B Biol. Sci 281:20132733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JJ, Van Belleghem SM, Papa R, Danko CG, and Reed RD. 2020. Many functionally connected loci foster adaptive diversification along a neotropical hybrid zone. Sci. Adv 6:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet J 1986. Hybrid zones of Heliconius butterflies in Panama and the stability and movement of warning colour dines. Heredity. 56:191–202. [Google Scholar]
- Mallet J 1993. Speciation, raciation, and color pattern evolution in Heliconius butterflies: evidence from hybrid zones. Pp. 226–260 in Harrison RG, ed. Hybrid zone and the Evolutionary process. Oxford University Press, New York. [Google Scholar]
- Mallet J, Barton N, Lamas G, C JS, M MM, and Eeley H. 1990. Estimates of selection and gene flow from measures of cline width and linkage disequilibrium in Heliconius hybrid zones. Genetics 124:921–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet J, and Joron M. 1999. Evolution of diversity in warning color and mimicry: Polymorphisms, shifting balance and speciation. Annu. Rev. Ecol. Syst 30:201–233. [Google Scholar]
- Martin SH, Dasmahapatra KK, Nadeau NJ, Salazar C, Walters JR, Simpson F, Blaxter M, Manica A, Mallet J, and Jiggins CD. 2013. Genome-wide evidence for speciation with gene flow in Heliconius butterflies. Genome Res. 23:1817–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SH, Davey JW, and Jiggins CD. 2015a. Evaluating the use of ABBA-BABA statistics to locate introgressed loci. Mol. Biol. Evol 32:244–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SH, Davey JW, Salazar C, and Jiggins CD. 2019. Recombination rate variation shapes barriers to introgression across butterfly genomes. PLOS Biol. 17:e2006288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SH, Eriksson A, Kozak KM, Manica A, and Jiggins CD. 2015b. Speciation in Heliconius Butterflies : Minimal Contact Followed by Millions of Generations of Hybridisation. BioRxiv 1–24. [Google Scholar]
- Martin SH, Möst M, Palmer WJ, Salazar C, McMillan WO, Jiggins FM, and Jiggins CD. 2016. Natural selection and genetic diversity in the butterfly Heliconius melpomene. Genetics 203:525–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan WO, Jiggins CD, and Mallet J. 1997. What initiates speciation in passion-vine butterflies? Proc. Natl. Acad. Sci. U. S. A 94:8628–8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier JI, Salazar PA, Ku M, Davies RW, Dréau A, Aldás I, Power OB, Nadeau NJ, Bridle JR, Rolian C, Barton NH, Mcmillan WO, and Jiggins CD. 2020. Haplotype tagging reveals parallel formation of hybrid races in two butterfly species. bioRxiv 1–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill RM, Chia A, and Nadeau NJ. 2014. Divergent warning patterns contribute to assortative mating between incipient Heliconius species. Ecol. Evol 4:911–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill RM, Rastas P, Martin SH, Melo MC, Barker S, Davey J, McMillan WO, and Jiggins CD. 2019. Genetic dissection of assortative mating behavior. PLoS Biol. 17:e2005902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moest M, Van Belleghem SM, James J, Salazar C, Martin S, Barker S, Moreira G, Mérot C, Joron M, Nadeau N, Steiner F, and Jiggins C. 2020. Selective sweeps on novel and introgressed variation shape mimicry loci in a butterfly adaptive radiation. PLoS Biol. 18:e3000597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz AG, Salazar C, Castaño J, Jiggins CD, and Linares M. 2010. Multiple sources of reproductive isolation in a bimodal butterfly hybrid zone. J. Evol. Biol 23:1312–1320. [DOI] [PubMed] [Google Scholar]
- Nachman MW, and Payseur BA. 2012. Recombination rate variation and speciation: Theoretical predictions and empirical results from rabbits and mice. Philos. Trans. R. Soc. B Biol. Sci 367:409–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, and Li WH. 1979. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. U. S. A 76:5269–5273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosil P, Feder JL, Flaxman SM, and Gompert Z. 2017. Tipping points in the dynamics of speciation. Nat. Ecol. Evol 1:1–8. [DOI] [PubMed] [Google Scholar]
- Pease JB, and Hahn MW. 2015. Detection and polarization of introgression in a five-taxon phylogeny. Syst. Biol 64:651–662. [DOI] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, a Shadick N, and Reich D. 2006. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet 38:904–909. [DOI] [PubMed] [Google Scholar]
- Price MN, Dehal PS, and Arkin AP. 2010. FastTree 2 – Approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prowell D 1998. Sex linkage and speciation in Lepidoptera. Pp. 309–319 in Howard D and Berlocher S, eds. Endless forms. Species and speciation Oxford University Press, New York. [Google Scholar]
- Quek SP, Counterman BA, De Moura PA, Cardoso MZ, Marshall CR, McMillan WO, and Kronforst MR. 2010. Dissecting comimetic radiations in Heliconius reveals divergent histories of convergent butterflies. Proc. Natl. Acad. Sci. U. S. A 107:7365–7370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravinet M, Faria R, Butlin RK, Galindo J, Bierne N, Rafajlovic M, Noor MAF, Mehlig B, and Westram AM. 2017. Interpreting the genomic landscape of speciation: a road map for finding barriers to gene flow. J. Evol. Biol 30:1450–1477. [DOI] [PubMed] [Google Scholar]
- Reed RD, Papa R, Martin A, Hinas HM, Counterman BA, Pard-Diaz C, Jiggins CD, Chamberlain NL, Kronforst MR, Chen R, Nijhout HF, and McMillan WO. 2011. optix drives the repeated convergent evolution of butterfly wing pattern mimicry. Science. 333:1137–1141. [DOI] [PubMed] [Google Scholar]
- Rougeux C, Bernatchez L, and Gagnaire PA. 2017. Modeling the multiple facets of speciation-with-gene-flow toward inferring the divergence history of lake whitefish species pairs (Coregonus clupeaformis). Genome Biol. Evol 9:2057–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux C, Fraisse C, Romiguier J, Anciaux Y, Galtier N, and Bierne N. 2016. Shedding light on the grey zone of speciation along a continuum of genomic divergence. PLoS Biol. 14:e2000234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumer M, Xu C, Powell DL, Durvasula A, Skov L, Holland C, Blazier JC, and Sankararaman S. 2018. Natural selection interacts with recombination to shape the evolution of hybrid genomes. Science (80-. ). 660:656–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaak SG 2015. Selection dynamics in Heliconius hybrid zones and the origin of adaptive variation. Mississippi State University. [Google Scholar]
- Sobel JM, and Chen GF. 2014. Unification of methods for estimating the strength of reproductive isolation. Evolution (N. Y). 68:1511–1522. [DOI] [PubMed] [Google Scholar]
- Sperling FAH 1994. Sex-linked genes and species differences in Lepidoptera. Can. Entomol 126:807–818. [Google Scholar]
- Stankowski S, Chase MA, Fuiten AM, Rodrigues MF, Ralph PL, and Streisfeld MA. 2019. Widespread selection and gene flow shape the genomic landscape during a radiation of monkeyflowers. PLOS Biol. 17:e3000391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supple M, Papa R, Hines HM, McMillan WO, and Counterman BA. 2015. Divergence with gene flow across a speciation continuum of Heliconius butterflies. BMC Evol. Biol 15:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavares H, Whibley A, Field DL, Bradley D, Couchman M, Copsey L, Elleouet J, Burrus M, Andalo C, Li M, Li Q, Xue Y, Rebocho AB, Barton NH, and Coen E. 2018. Selection and gene flow shape genomic islands that control floral guides. Proc. Natl. Acad. Sci. U. S. A 115:11006–11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tine M, Kuhl H, Gagnaire PA, Louro B, Desmarais E, Martins RST, Hecht J, Knaust F, Belkhir K, Klages S, Dieterich R, Stueber K, Piferrer F, Guinand B, Bierne N, Volckaert FAM, Bargelloni L, Power DM, Bonhomme F, Canario AVM, and Reinhardt R. 2014. European sea bass genome and its variation provide insights into adaptation to euryhalinity and speciation. Nat. Commun 5:5770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Belleghem SM, Rastas P, Papanicolaou A, Martin SH, Arias CF, Supple MA, Hanly JJ, Mallet J, Lewis JJ, Hines HM, Ruiz M, Salazar C, Linares M, Moreira GR, Jiggins CD, Counterman BA, McMillan WO, and Papa R. 2017. Complex modular architecture around a simple toolkit of wing pattern genes. Nat. Ecol. Evol 1:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Belleghem SM, Salazar C, Jiggins CD, Baquero M, Papa R, Counterman BA, Mcmillan WO, and Martin SH. 2018. Patterns of Z chromosome divergence among Heliconius species highlight the importance of historical demography. Mol. Ecol 27:3852–3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallbank RWR, Baxter SW, Pardo-diaz C, Hanly J, Martin SH, Mallet J, and Dasmahapatra KK. 2016. Evolutionary novelty in a butterfly wing pattern through enhancer shuffling. PLoS Biol. 14:e1002353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf JBW, and Ellegren H. 2017. Making sense of genomic islands of differentiation in light of speciation. Nat. Rev. Genet 18:87–100. [DOI] [PubMed] [Google Scholar]
- Wu C 2001. The genic view of the process of speciation. J. Evol. Biol 14:851–865. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S 1. Samples used in this study. Samples from H. himera populations and H. erato populations that come into contact with H. himera are indicated in color. H. himera North, light red; H. himera South, dark red; H. e. cyrbia North, light yellow, H. e. cyrbia South, dark yellow; H. e. emma East, light green; H. e. emma West dark green; H. e. favorinus East, light blue; H. e. favorinus West, dark blue. Earthcape IDs are available through https://heliconius.ecdb.io.
Table S 2. Hybrid zone characteristics between H. erato and H. himera populations. Major color pattern (CP) loci include the genes optix (~red), WntA (~forewing band shape) and cortex (~yellow hindwing bar and white fringes) Reproductive isolation (RI) was estimated as a combination of ecological isolation (RIec) due to divergent selection on color patterns, pre-mating isolation (RIpre) due to mate preference, and post-mating isolation (RIpost) due to hybrid sterility.
Table S 3. 2D δaδi model descriptions.
Table S 4. Bash pseudocode to run msms (Ewing and Hermisson 2010) simulations.Note that mutation rate is only used by seq-gen when simulating the sequences, population size is used to scale times, selection and migration proportions.
Table S 5. AIC weights dadi models. AIC scores for each model are averages of the top 5 runs. Shaded cells indicate lowest AIC values.
Table S 6. Best model parameters for best model class as well as alternative model classes. Split times are given as Ttotal+TAM in AM, and Ttotal+TSC in SC. Although all best fitting demographic models include secondary contact (SC), several alternative models including AM (Ancient Migration) and IM (Isolation and Migration) fit relatively well to the observed JSFS (Figure 3A; Figure S4–5). However, in these models, migration rates generally scale proportionately with migration duration compared to the SC models, indicating that our interpretations on migration rates should not be affected by the uncertainty of finding a best demographic model.
Figure S 1. FastTree of samples used in this study. Node support values are based on the Shimodaira–Hasegawa test.
Figure S 2. Average AIC (Akaike Information Criterion) of all tested δaδi models for twenty runs. SI = Strict Isolation, IM = Isolation and Migration, AM = Ancestral Migration, SC = Secondary Contact, G = exponential growth, 2N = heterogeneous effective population size (with 2 classes of loci shared by the two populations = Hill-Robertson effects), and 2m = 2 migration rates.
Figure S 3. Average AIC (Akaike Information Criterion) of all tested δaδi models for twenty runs using subpopulations. SI = Strict Isolation, IM = Isolation and Migration, AM = Ancestral Migration, SC = Secondary Contact, G = exponential growth, 2N = heterogeneous effective population size (with 2 classes of loci shared by the two populations = Hill-Robertson effects), and 2m = 2 migration rates.
Figure S 4. Inference of historical effective population size changes using pairwise sequentially Markovian coalescent (PSMC) analysis. Lines and shading in the left panel represent the average effective population size and 95% CI, respectively, estimated from PSMC analyses on individual genome samples as shown in the panels on the right. The PSMC estimates are scaled using a generation time of 0.25 years and a mutation rate of 2e-9. Data adopted from Van Belleghem et al. (2018).
Figure S 5. Loess fit to admixture (fd) between H. himera and H. erato in three contact zones. Points show admixture (fd) values, whereas coloring shows directionality (DFOIL) in 50 kb non-overlapping windows for the contact zones H. himera ¬– H. e. cyrbia (top), H. himera – H. e. emma (middle) and H. himera – H. e. favorinus (bottom) as in Figure 3. Blue indicates predominant admixture from H. erato into H. himera (12 <- 34), whereas red indicates predominant admixture from H. himera into H. erato (12 -> 34) based on the DFOIL tests. Green lines indicate the Loess fit.
Figure S 6. Comparison of admixture (fd) signals with relative divergence (da) of single H. erato individuals to H. himera. Panels with black points show the fd signals between contact zones of H. himera with H. e. cyrbia (top), H. e. emma (middle) and H. e. favorinus (bottom). Panels with colored points show relative divergence da, calculated from pairwise sequence differences minus the average nucleotide diversity of both populations, between H. himera and a single individual of a hybridizing H. erato population. Green rectangles are high fd tracks as identified from a Loess fit to continues windows of fd larger than 0.06 for the H. e. cyrbia and H. e. emma contact zones and 0.07 for the H. e. favorinus contact zone.
Figure S 7. Correlations of divergence and admixture proportions with recombination rate and gene density in H. himera – H. erato contact zones. (A) Relative divergence (FST) versus gene density and recombination rate (ρ). (B) Admixture proportion (fd) versus gene density and recombination rate (cM/Mb). (C) Relative divergence (FST) versus admixture proportion (fd). Statistics were calculated in 50 kb non-overlapping windows. Colored circles match color codes in Figure 2.
Figure S 8. Expected relationship of recombination rate with divergence (FST) and admixture (fd) near a divergently selected locus (s = 0.02). (A) The population tree shows the simulated scenario with the onset of divergent selection on a derived allele indicated in red and in which migration rate (m) and migration time (tm) between P2 and P3 and selection start time (ts) are varied. Left and right of the simulated scenario are parameter combinations for two extreme scenarios that both include linked selection; on the left a scenario with Isolation with Migration (IM) and on the right a scenario reflecting Secondary Contact (SC). (B) Effect of population recombination rate (ρ) on relative divergence (FST) and admixture (fd) near a divergently selected locus for the two simulated scenarios with parameter combinations as in panel A. The selected allele occurs at position 0. The dashed red line indicates a locus at 500 kb from the selected locus at which the relationship between ρ, divergence and admixture is assessed in panel C. (C) The effect of migration start time (tm) and selection start time (ts) on the relation between ρ, divergence and admixture. Apart from the respective parameters being evaluated, other parameters were fixed as in panel A, with the red lines indicating the exact parameter combinations as in panel A and B.