

Abstract
Background:
The presence of co-existent neuronal antibodies (neuronal-IgG) in patients with myelin oligodendrocyte glycoprotein immunoglobulin (MOG-IgG1) is not yet well understood.
Objectives:
To investigate the co-existence of a broad range of neuronal-IgG in MOG-IgG1+ patients.
Methods:
MOG-IgG1+ patients were tested for 17 neuronal-IgGs in CSF and serum in including; NMDA-R-IgG, AMPA-R-IgG, GABAB-R-IgG, LGI1-IgG, CASPR2-IgG, GABAA-R-IgG, GAD65-IgG, mGLUR1-IgG, DPPX-IgG, CRMP5-IgG, amphiphysin-IgG, PCA1,2,Tr -IgGs, ANNA1,2,3 -IgGs.
Clinical and radiological features of MOG-IgG1+ with neuronal antibodies were compared to a control cohort of MOG-IgG1+ patients without neuronal-IgGs.
Results:
A total of 376 MOG-IgG1+ patients underwent testing for neuronal-IgGs.
Serum testing for neuronal-IgGs (113 adults, 142 children) identified two children with NMDA-R-IgG (0.7%), CASPR2-IgG (0.7%), and two adults with LGI1-IgG (0.9%) and GABAA-R-IgG (0.9%).
CSF testing for neuronal-IgGs (97 adults, 169 children) identified seven children (4%) and seven adults (7%) with NMDA-R-IgG, and one adult with GABAA-R-IgG (1%).
The MOG-IgG1+/NMDA-R-IgG+ patients had a median age of 17 (range 2–39). Features associated with MOG-IgG1+/NMDA-R-IgG+ included; encephalopathy (p=0.001), seizures (p=0.045), leptomeningeal enhancement (p=0.045).
Conclusions:
NMDA-R-IgG was the most frequently detected neuronal-IgG to co-exist with MOG-IgG1. MOG-IgG1+/NMDA-R-IgG+ patients most often presented with encephalopathy and seizures. Testing for MOG-IgG1 and NMDA-R-IgG may be warranted in patients with encephalopathy and inflammatory demyelinating syndromes.
Keywords: MOG-IgG1, demyelination, autoimmune encephalitis, biomarkers, NMDA-R-IgG, neuronal antibodies
INTRODUCTION
Immunoglobulin G antibodies to myelin-oligodendrocyte-glycoprotein (MOG-IgG1) are found in adults and children with a spectrum of inflammatory central nervous system (CNS) disorders termed ‘MOG-IgG associated disorders’ (MOGAD) including; optic neuritis, myelitis, acute disseminated encephalomyelitis (encompassing autoimmune encephalopathy and encephalitis).1,2,3,4,5
As there is a spectrum of inflammatory CNS clinical phenotypes associated with MOG-IgG1, it plausible that MOG-IgG1 may co-exist with other neuronal antibodies (neuronal-IgGs) more often in certain clinical phenotypes, such as ADEM compared to optic neuritis. A study of anti-N-methyl-D-aspartate receptor (NMDA-R) encephalitis cohorts identified a few patients also seropositive for MOG-IgG1 with demyelination.6,7 It is unknown whether MOG-IgG1 co-exists with other neuronal-IgGs associated with autoimmune encephalitis such as α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPA-R-IgG), leucine-rich, glioma inactivated 1 (LGI1-IgG), contactin-associated protein-like 2 (CASPR2-IgG), gamma-aminobutyric acid-A and -B receptors (GABAA-R-IgG, GABAB-R-IgG), glutamic acid decarboxylase (GAD-65-IgG), dipeptidyl-peptidase–like protein 6 IgG (DPPX-IgG), metabotropic glutamate receptor 1 (mGLUR1-IgG), anti-neuronal nuclear antibody types 1,2,3 -IgGs (ANNA-1,2,3 -IgG), purkinje cell cytoplasmic antibody type 1,2, Tr -IgGs (PCA-1,2,Tr -IgG), amphiphysin IgG, collapsin response-mediator protein-5 IgG (CRMP5-IgG).8,9,10,11,12
Whether there are immunological differences between MOGAD clinical phenotypes or the presence of co-existent neuronal-IgGs in subgroups of MOGAD is not yet well elucidated. In this study we firstly sought to evaluate whether MOG-IgG1 co-existed with neuronal-IgGs and to evaluate the clinical phenotype of these patients compared to MOG-IgG1 patients without neuronal-IgGs.
MATERIALS and METHODS
Study design, participants and data collection
This is a retrospective observational study approved by the Institutional Review Board of Mayo Clinic. The Mayo Clinic Neuroimmunology Laboratory clinical and serological database was interrogated to identify MOG-IgG1 seropositive patients with neuronal-IgG testing. A waiver of consent for clinical data was obtained as part of serological test validation. Written consent was obtained for MRI imaging included for publication. Included MRIs were reviewed by the treating neurologist, neurologists (AK, SJP) and neuroradiologist (KNK).
Cohort selection
Patients with serum positive for MOG-IgG1 between 2008–2019 were selected with a minimum dataset including: type of samples tested, patient name or identifier, patient date of birth, collection date. The minimum dataset did not include MRI. From these, MOG-IgG1 positive patients with serum and/or CSF tested for neuronal-IgGs were selected (Supplemental figure e-1 and table e-1). The age of the first MOG-IgG1 positive test was used to stratify the cohort into children and adults (≥18 years).
A control cohort of Mayo Clinic MOGAD patients were identified by searching for patients with CSF testing that was negative for neuronal-IgGs, with available clinical and radiological data and consent for research participation (n=33).
MOG-IgG1 detection
All MOG-IgG testing was performed by the Mayo Clinic Neuroimmunology Laboratory using live flow cytometric assay described previously.13 A MOG-IgG1 binding index value ≥2.5 was considered positive. Titration was performed using serial dilution (1:20, 1:40, 1:100, 1:1000, 1:10000).
Neuronal antibody detection
Neuronal-IgGs were tested on clinically validated assays including; rodent tissue indirect immunofluorescence (IFA), commercial cell based assay (CBA) employing human embryonic kidney 293 cells transfected with plasmids expressing the respective antigens (NMDA-R, AMPA-R, GABAB-R, CASPR2, LGI1, mGluR1, DPPX [Euroimmun, Lubeck, Germany], immunoprecipitation assay (IPA) (GAD65), tissue immunofluorescence or Western blot (CRMP5, amphiphysin, PCA1,2,Tr, ANNA1,2,3). Positivity was determined by repeat testing (≥2 times per sample). Serum GAD65-IgG positivity was restricted to ≥20 nmol/L in serum, associated with higher specificity for neuronal autoimmunity.11
Clinical-anatomical syndrome criteria
The patients were evaluated as to whether they met the following clinical criteria: ADEM14,15, NMDA-R-IgG encephalitis14 and the 2015 seronegative neuromyelitis optica spectrum disorder diagnostic criteria.16
Statistical analyses
Two tailed Fisher exact tests were used to compare the frequency of detected co-existent neuronal-IgGs. Two tailed Fisher exact tests and Wilcoxun rank sum test were used to compare the clinical and radiological factors of MOG-IgG1 cohorts with and without NMDA-R-IgG in CSF. P values less than 0.05 were deemed statistically significant.
RESULTS
Study Cohort
There were 376 MOG-IgG1 positive patients who were included who underwent testing for neuronal-IgGs in serum and/or CSF. This included 157 adults (90 females [57%]) with median age of 40 years (interquartile range [IQR] 30–53) and 219 children (113 females [52%]) with a median age of 8 years (IQR 5–11). The median MOG-IgG1 titer was 1:100 (IQR 1:40–1:1000) in both children and adults.
NMDA-R-IgG was the most frequently detected co-existing neuronal-IgG in adults and children
Among 169 MOG-IgG1 positive children evaluated in CSF, seven (4%) were positive for NMDA-R-IgG. Among 97 MOG-IgG1 positive adults evaluated in CSF, seven (7%) were positive for NMDA-R-IgG and one was positive for GABAA-R-IgG (1%) (Supplementary eTable 1).
Among 142 MOG-IgG1 positive children evaluated in serum, two (1.4%) were positive for NMDA-R-IgG (1, [0.7%]) and CASPR2-IgG (1, [0.7%]). Among 113 MOG-IgG1 positive adults evaluated in serum, two had neuronal-IgGs including; LGI1-IgG (1, [0.9%]), and GABAA-R-IgG (1, [0.9%]) (Supplementary eTable 1).
NMDA-R-IgG was more likely to co-exist with MOG-IgG1 than either of CASPR2-IgG, LGI1-IgG or GABAA-R-IgG (p<0.001).
Autoimmune encephalopathy was the predominant clinical phenotype for dual-positive MOG-IgG1 and NMDA-R-IgG
The median age of the 14 patients (six females, eight males) dual-positive for MOG-IgG1 and NMDA-R-IgG was 17 (range 2–39 years). Clinical, serological and radiological data were available for 11/14 patients (Table 1).
Table 1:
Clinical, serological and radiological features of dual-positive MOG-IgG1+/NMDA-R-IgG+ patients
| No/Age^/Sex | MOG-IgG1 titer | Coexistent antibodies# | Timing of antibody detection | Clinical-anatomical phenotype of first attack | Clinical-anatomical phenotype of subsequent attacks | Attac ks (n) | Time to second attack | Enceph alo-pathy | Seizure s | NMDA-R-IgG encephalitis clinical features | ADEM criteria | NMDA-R encephali tis criteria | NMO SD criteri a | CSF OCB (≥4 CSF restrict ed) | MRI |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1/2/M | 1:20a | NMDA-R-IgG IFA+ (no titer) & CBA+ | MOG-IgG1 (1st attack, age 2) NMDA-R-IgG+/MOG-IgG1- (2nd attack age 7) | Multifocal meningoencephalitis (meningeal, cortical, diencephalon involvement) | Multifocal meningoencephalitis (meningeal, cortical, diencephalon involvement), ON, cervical TM | 2 | 5 years | Y | Y | Cognitive and behavioral abnormalities | Y (1st attack) | Y (2nd attack) | N | N | Age 2: T2 hyperintensity in left caudate, left thalamus, left basal ganglia. Diffuse leptomeningeal enhancement and patchy cortical T2/FLAIR hyperintensity. Age 7: Bilateral optic neuritis. Multiple enhancing lesions in supratentorial white matter. Periventricular enhancement right occipital horn. Enhancing lesions-central/dorsal cord C2–3, right hemicord C4, central cord C6–7. |
| 2/6/F | 1:20b | NMDA-R-IgG IFA+(1:64) & CBA+ | NMDA-R-IgG+/MOG-IgG1+ (3rd & 4th attacks) | ON | ON, Multifocal meningoencephalitis (meningeal, cortical, diencephalon involvement), cervical TM | 5 | 4 years | Y | N | Aphasia, agitation, perseveration, mutism, choreoathetoid movements, visual and auditory hallucinations, psychosis (attacks 3–5) | Y (3rd, 4th, 5th attacks) | Y (3rd, 4th, 5th attacks) | N | ND | Age 13: T2 hyperintensity in insular cortex and left frontal operculum. Left frontoparietal unilateral leptomeningeal enhancement. Age 18: Leptomeningeal enhancement of right hemisphere, T2/FLAIR hyperintensity right temporal lobe. Patchy T2 signal in the ventral cord C4-C6 Age 23: T2 hyperintensity in inferior left temporal lobe |
| 3/12/F | 1:1000 | NMDA-R-IgG CBA+ | Simultaneous MOG-IgG1+/NMDA-R-IgG+ (onset) | Encephalitis (diencephalon) | NA | 1 | NA | Y | Y | Choreiform movements, oral dyskinesias and tongue biting | Y (1st attack) | Y (1st attack) | N | NA | Age 12: T2 hyperintensity in the left anterior basal ganglia. |
| 4/14/F | 1:100 | NMDA-R-IgG IFA+ & CBA+ | Simultaneous MOG-IgG1+/NMDA-R-IgG+ (4 weeks from onset) | Multifocal meningoencephalitis | NA | 1 | NA | Y | Y | Aphasia, visual hallucinations, insomnia | Y (1st attack) | Y (1st attack) | N | Y | Age 14: Leptomeningeal and gyral enhancement around splenium corpus and callosum |
| 5/15/F | 1:100 | NMDA-R-IgG CBA+ | NMDA-R-IgG+ at onset, MOG-IgG1+ (8 weeks from onset) | Multifocal meningoencephalitis (meningeal, cortical, diencephalon involvement), ON | Multifocal encephalitis (cortical, diencephalon, brainstem involvement) | 2 | 4 weeks | Y | Y | Combative, agitation | Y (1st attack) | Y (1st attack) | N | N | Age 15: T2 hyperintensity in right hypothalamus, chiasm and right optic tract. Right optic disc enhancement. 2nd attack: T2 hyperintense lesions in the left thalamus, right frontal subcortical and left pons. |
| 6/18/F | 1:1000 | NMDA-R-IgG CBA+ | Simultaneous MOG-IgG1+/NMDA-R-IgG+ (6 weeks from onset) | Multifocal encephalitis with cortical, diencephalic involvement | Multifocal encephalitis with cortical, diencephalic and brainstem involvement | 2 | 6 weeks | Y | N | Gait and truncal ataxia | Y (1st attack) | Y (1st attack) | N | N | Age 18: Gyral swelling in the left parietal region and sulcal FLAIR hyperintensity. 2nd attack: Bilateral T2/FLAIR hyperintensity in deep nuclei and corticospinal tracts, internal capsule and brainstem |
| 7/19/M | 1:40 | NMDA-R-IgG IFA+ (1:8) & CBA+ | Simultaneous MOG-IgG1+/NMDA-R-IgG+ (2nd attack, 13 years from onset) | Multifocal encephalitis with diencephalic involvement | Multifocal encephalitis with diencephalic, brainstem involvement | 2 | 12 years | Y | N | Deja vu, auditory and visual hallucinations | Y (1st and 2nd attack) | Y (2nd attack) | N | Y | Age 19: T2/FLAIR hyperintensities in right periventricular and subcortical white matter Age 31: Punctate and curvilinear enhancement right ventral pons. Punctate lesions in pons and midbrain. T2 hyperintensity and enhancement of posterior limb of internal capsule. |
| 8/27/F | 1:40 | NMDA-R-IgG CBA+ AQP4-IgG CBA+ 1:100 | Simultaneous MOG-IgG1+/NMDA-R-IgG+/AQP4-IgG+ (onset) | Cervical LETM, right optic neuritis | NA | 1 | NA | N | N | NA | N | N | Y | Age 27: T2/FLAIR changes in right temporal lobe, right thalamus, optic tract and chiasm without enhancement. C2-C6 lesion with enhancement. | |
| 9/33/M* | 1:100 | NMDA-R-IgG IFA+ (no titer) & CBA+ | NMDA-R-IgG+ (onset) MOG-IgG1+ (4 weeks from onset) | Multifocal meningoencephalitis (meningeal, cortical, diencephalon involvement) | Multifocal meningoencephalitis, ON, TM | 2 | 3 weeks | Y | Y | Visual and auditory hallucinations, paranoia | Y (1st attack) | Y (1st attack) | N | NA | Age 33: Right temporal lobe enhancement, leptomeningeal enhancement and cortical edema, upper brainstem enhancement. 2nd attack: Enhancing lesions in mesial temporal lobes, subcortical cerebral hemispheres, bilateral middle cerebellar peduncles left cerebellum. Enhancement of the optic nerves and nerve sheaths bilaterally. Small enhancing lesions in the cervical and thoracic spine. |
| 10/33/M | 1:100 | NMDA-R-IgG IFA+ (1:64) & CBA+ GFAP-IgG IFA+ (1:64) & CBA+c | Simultaneous MOG-IgG1+/NMDA-R-IgG+/GFAP-IgG+ (4 months from onset) | Multifocal meningoencephalitis (meningeal, cortical involvement) | Cervical LETM | 2 | 8 weeks | Y | Y | Aphasia | Y (1st attack) | Y (1st attack) | N | N | Age 33: T2 hyperintensity and leptomeningeal enhancement along the left temporal, frontal, parietal and suprasylvian sulci. 2nd attack: T2/FLAIR hyperintensity in the left basal ganglia and the left inferior cerebellar peduncle. Cervical spine T2 hyperintensity and expansion C1-C3 and C6-C7 level with enhancement |
| 11/39/M | 1:100 | NMDA-R-IgG CBA+ (CSF) | Simultaneous MOG-IgG1+/NMDA-R-IgG+ (3 months from onset) | Multifocal meningoencephalitis with leptomeningeal, cortical, diencephalic, brainstem involvement. Left optic neuritis. | NA | 1 | NA | Y | N | Agitation, disinhibition | Y (1st attack) | Y (1st attack) | N | Y | Age 39: Patchy leptomeningeal and cortical enhancement of the brainstem, midbrain, right cerebellar hemisphere, vermis, bilateral temporal and frontal lobes. T2 hyperintensity in cerebellum, brainstem, and supratentorial brain. Multiple enhancing nodules along the anterior and posterior surfaces of the spinal cord. Enhancement of the distal left optic nerve. |
Abbreviations: CBA= cell-based assay, IFA = immunofluorescence assay, ON = optic neuritis, TM = transverse myelitis, LETM = longitudinally extensive transverse myelitis. ND= not done, NA= not applicable or available
Age at onset of disease
All patients with NMDA-R-IgG positivity were tested in CSF
Patient tested positive for MOG-IgG1 twice, 4 years apart. Positive at 1:20, titration not completed.
Patient tested positive for MOG-IgG1 at disease onset 1:20, titration not completed. Subsequent serum testing in 5 years from onset was negative for MOG-IgG1.
GFAP-IgG positive in CSF
Case described by Carroll et al in Practical Neurology: case reports. November/December 2019.
Clinical-anatomical phenotypes at first attack included; multifocal meningoencephalitis/encephalitis (9), optic neuritis (3), transverse myelitis (1). Clinical-anatomical phenotypes at subsequent attacks included; multifocal meningoencephalitis/encephalitis (6), optic neuritis (3), transverse myelitis (4) (Table 1).
MOGAD clinical phenotypes throughout the disease course of included; ADEM (10), myelitis (5), optic neuritis (5 [4 monophasic, 1 recurrent]). NMDA-R-IgG encephalitis clinical features throughout the diseases course included; encephalopathy (10), seizures (6), auditory and visual hallucinations (4), speech dysfunction (3), movement disorder (3) Deja-vu (1), psychosis (1).
At the onset of their disease, 9/11 fulfilled the criteria for ADEM14,15 and 7/11 fulfilled the diagnostic criteria for NMDA-R-IgG encephalitis14 (2 patients with meningoencephalitis did not have testing for NMDA-R-IgG at onset (patients 1 and 7). The majority of the patients (10/11) had at least one attack of multifocal meningoencephalitis/encephalitis (Table 1). One patient met the NMOSD criteria with longitudinally extensive myelitis and optic neuritis (patient 8) (Table 1).
During their disease course, the MRI brain scans were abnormal in all patients. MRI radiological features are described in detail in table 1 and included predominantly T2/FLAIR hyperintense signal with mild patchy contrast enhancement involving the anatomical areas; diencephalon (10), cortices (8), brainstem or cerebellum (6), cervical or thoracic cord (5), optic nerve or chiasm (4). Leptomeningeal and/or cortical gadolinium contrast enhancement was observed in 6 patients (Table 1) (Figure 1).
Figure 1: MRI features of dual-positive MOG-IgG1/NMDA-R-IgG patients.
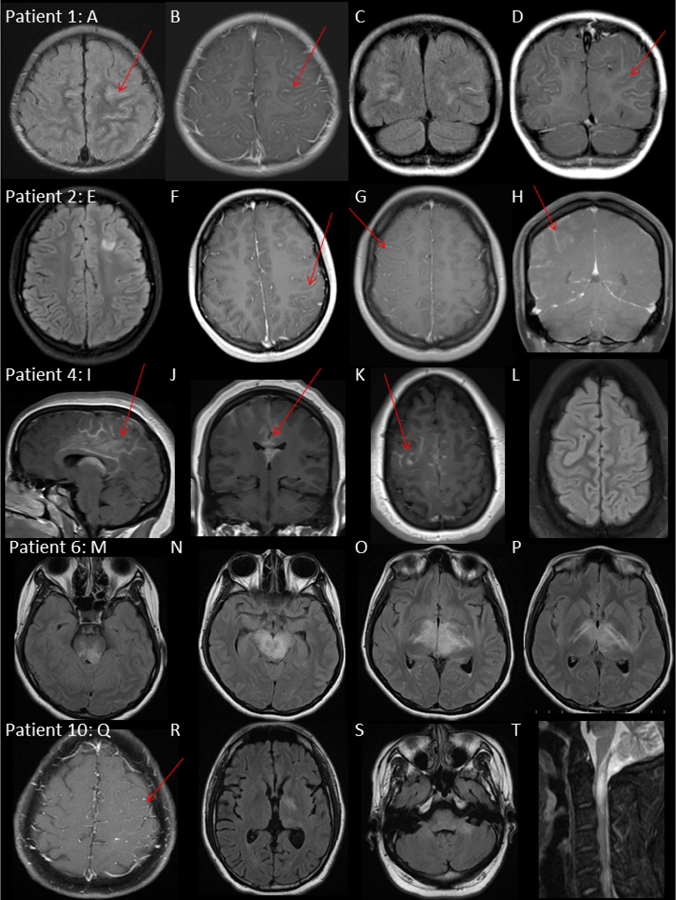
Patient 1, male 2 years.
MRI brain: Axial T2/FLAIR (A) T1 Gd+ (B), coronal T2/FLAIR (C) T1 Gd+ (D) at first attack demonstrating bilateral frontoparietal sulcal T2/FLAIR hyperintensity, diffuse leptomeningeal enhancement and patchy cortical T2/FLAIR hyperintensity.
Patient 2, female 13 years.
MRI brain: Axial T2/FLAIR (E) and T1 Gd+ (F) at third attack age 13, demonstrating focal T2/FLAIR hyperintensity deep to left superior frontal sulcus and in left frontal opercular cortex with left unilateral leptomeningeal enhancement and focus of parenchymal enhancement. Axial T1 Gd+ (G) coronal T1 Gd+(H) at fourth attack age 18, demonstrating right unilateral hemisphere leptomeningeal enhancement and resolution of left frontoparietal leptomeningeal and parenchymal signal abnormalities.
Patient 4, female 14 years
MRI brain: Sagittal T1 Gd+ (I), Coronal T1 Gd+ (J) and Axial T1 Gd+ (K) and Axial T2/FLAIR at first attack demonstrating bilateral (right>left) leptomeningeal enhancement around the splenium corpus and callosum.
Patient 6, female 18 years, Axial T2/FLAIR (M), (N), (O), (P) at second attack 4 weeks from onset. T2/FLAIR hyperintensity of internal capsule, bilateral thalami, midbrain and pons.
Patient 10, male 33 years.
MRI brain: Axial T1 Gd+ (Q) at first attack demonstrating left unilateral leptomeningeal enhancement. Axial T2/FLAIR (R), (S) at second attack 8 weeks later demonstrating T2/FLAIR hyperintensity of the left basal ganglia and bilateral cerebellar peduncles
MRI cervical spine: Sagittal T2 STIR (T), demonstrating cervical myelitis with T2 hyperintensity and cord enlargement from medulla to C4 and associated patchy enhancement (not shown).
Preceding history of infection was identified in two patients (upper respiratory tract infection, mycoplasma pneumonia). No patients had malignancy identified.
Serological features of dual-positive MOG-IgG1 and NMDA-R-IgG patients
The median MOG-IgG1 serum titer was 1:100 (range 1:20–1:1000). NMDA-R-IgG was positive in CSF on CBA in 14 patients and tissue IFA in seven (titer range 1:4–1:64) (Table 1). Serology at first attack was positive for MOG-IgG1 in nine (Patients 2 and 7 not tested at onset) and NMDA-R-IgG in eight (Patients 1, 2, 7 not tested at onset). In the eight patients tested at onset, seven were tested within six weeks of disease onset.
Seroconversion to MOG-IgG1 negative was seen in one patient at second attack, five years from onset (patient 1). All patients had CSF pleocytosis (median cell count 68 [range 9–474 cells/hpf], oligoclonal bands were present in 3/7 patients tested. A third glial antibody was detected in two patients; GFAP-IgGα was detected in CSF (patient 10), AQP4-IgG was detected in serum (patient 8).
Disease course and outcomes of the MOG-IgG1 and NMDA-R-IgG dual-positive patients
The median follow up from disease onset was 15 months (range 2 months-19 years). Seven patients had recurrent attacks (64%). Four patients had their second attack within 3 months of their first attack. Three patients had neurological attacks several years apart (range 4–13 years).
All patients were treated with acute immunotherapy (intravenous (IV)/oral corticosteroids (11), IV immunoglobulin (7), plasma exchange (3). Long term immunotherapy was commenced in 10 patients and included; rituximab (9 [treatment duration range 6 months – 2 years]), Oral prednisone (4 [treatment duration range 6 weeks – 3 months]), IVIG (3 [treatment duration range 3–9 months]), mycophenolate mofetil (1, [treatment duration 5 months]). No patients on these long term immunotherapies had an attack. One patient treated with azathioprine for only one month (due to non-compliance) had a relapse (patient 2) (Table 2).
Table 2:
Immunotherapy and clinical outcomes of dual-positive MOG-IgG1+/NMDA-R-IgG+ patients
| No/Age^/Sex | Acute IT | Long term IT, durationa | Attacks on long term IT | Follow up | Neurocognitive outcomes | Motor outcomes | Visual outcomes |
|---|---|---|---|---|---|---|---|
| 1/2/M | IVMP & oral steroids | RTX (1 course, 6 months)b | No | 8.7 years | Forgetfulness, attention deficit disorder | Normal | Normal |
| 2/6/F | IVMP, IVIG, PLEX | Oral prednisone (3 months)c IVIG (3 months)c RTX (1 course, 6 months)c AZA (1 month, non-compliant)d RTX (2 years)e |
Attack on Azathioprine (inadequately treated). No attacks on rituximab. | 19.3 years | Emotional lability, low mood | Normal | Normal |
| 3/12/F | IVMP, IVIG | RTX (1 course, 6 months) | No | 1.3 years | Poor performance at school | Normal | Normal |
| 4/14/F | IVMP, oral steroids, IVIG | IVIG (4 months) RTX (1 course, 6 months) | No | 8 months | Pre-existing learning and behavioral problems. Status has returned to pre-morbid state. | Normal | Normal |
| 5/15/F | IVMP, IVIG | Oral prednisone (2 months)b RTX (1 course, 6 months)b |
No | 1.6 years | Mild attention, executive function, and anxiety | Normal | Left APD |
| 6/18/F | IVIG, IVMP, oral prednisone (3 weeks) | Oral prednisone (6 weeks)b MMF (5 months)b IVIG (9 months)b |
No | 11 months | Pre-existing autism and non-verbal communication. Status has returned to pre-morbid state. | Mild pyramidal signs with minimal disability, no gait aid | Normal |
| 7/19/M | IVMP, PLEX | RTX (18 months)b | No | 12.5 years | Mood swings, compulsive behaviors | Normal | Normal |
| 8/27/F | IVMP, IVIG, PLEX | RTX (1 course, 6 months) | No | 2 months | No deficits | Mild pyramidal weakness with minimal disability, no gait aid | OD 20/24, OS 20/50 |
| 9/33/M* | IVMPb, IVIGb | Oral prednsione (2 months)b RTX (1 course, 6 months)b |
No | 1.1 years | Mild deficits in attention, aspects of executive functioning, and nonverbal memory | Normal | Normal |
| 10/33/M | IVMP | RTX (10 months)b | No | 1.1 years | Difficulty with repetition of complex sentences and word finding pauses | Normal | Normal |
| 11/29/M | IVMP | Nil | NA | 1.3 years | Mild cognitive deficits (critical thinking, memory and attention) | Normal | Normal |
Abbreviations: IVMP= intravenous methylprednisolone. IVIV = intravenous immunoglobulin, RTX = rituximab, PLEX = plasma exchange, MMF = mycophenolate mofetil, AZA = azathioprine.
Age at onset of disease
Case described by Carroll et al in Practical Neurology: case reports. November/December 2019.
IT commenced at first attack unless otherwise marked.
IT commenced at 2nd attack
IT commenced at 4th attack
IT switch to azathioprine during period of disease stability
IT commenced at 5th attack
Neurocognitive symptoms at last follow up included; poor attention, depression, labile mood, poor attention, compulsive behaviors and poor school performance and were seen in 72% (8) (Table 2). Of the patients with myelitis (5), two had residual pyramidal signs and none had a gait aid at last follow up. Of the patients with optic neuritis (5), one had residual relative afferent pupillary defect and one had visual acuity deficits at last follow up.
Comparison of dual-positive MOG-IgG1+/NMDA-R-IgG+ patients to MOG-IgG1+ patients without neuronal-IgGs
The demographics, clinical and radiological features of dual-positive patients were compared to a control group of MOGAD patients with CSF testing that was negative for NMDA-R-IgG in table 3. NMDA-R-IgG co-existence was associated with encephalopathy (p=0.001), seizures (p=0.045), leptomeningeal and/or cortical enhancement (p=0.045).
Table 3:
Comparison of clinical and radiological factors of MOG-IgG1+ with and without NMDA-R-IgG
| Variable | MOG-IgG1+, NMDA-R-IgG- (N=33) | MOG-IgG1+, NMDA-R-IgG+ (N=11) | Total (N=44) | p value |
|---|---|---|---|---|
| Sex | 1.0001 | |||
| Male | 14 (42.4%) | 5 (45.5%) | 19 (43.2%) | |
| Female | 19 (57.6%) | 6 (54.5%) | 25 (56.8%) | |
| Age | 0.2842 | |||
| Median | 27.4 | 18.0 | 22.2 | |
| IQR | 10.6, 50.7 | 13.0, 30.0 | 11.1, 40.6 | |
| MRI brain leptomeningeal enhancement | 0.0451 | |||
| Absent | 27 (81.8%) | 5 (45.5%) | 32 (72.7%) | |
| Present | 6 (18.2%) | 6 (54.5%) | 12 (27.3%) | |
| Transverse myelitis | 0.4891 | |||
| Absent | 13 (39.4%) | 6 (54.5%) | 19 (43.2%) | |
| Present | 20 (60.6%) | 5 (45.5%) | 25 (56.8%) | |
| Optic neuritis | 0.1691 | |||
| Absent | 10 (30.3%) | 6 (54.5%) | 16 (36.4%) | |
| Present | 23 (69.7%) | 5 (45.5%) | 28 (63.6%) | |
| Encephalopathy | 0.0011 | |||
| Absent | 22 (66.7%) | 1 (9.1%) | 23 (52.3%) | |
| Present | 11 (33.3%) | 10 (90.9%) | 21 (47.7%) | |
| Seizures | 0.0451 | |||
| Absent | 27 (81.8%) | 5 (45.5%) | 32 (72.7%) | |
| Present | 6 (18.2%) | 6 (54.5%) | 12 (27.3%) |
Fisher’s Exact Test for Count Data
Wilcoxun Rank rank sum test
Clinical phenotype of MOG-IgG1+ patients in whom other neuronal-IgGs were detected to co-exist
Three patients were identified with other neuronal-IgGs co-existing with MOG-IgG1 including; GABAA-R-IgG, CASPR2-IgG and LGI1-IgG. MOG-IgG1 (titer 1:1000) and CASPR2-IgG were identified in a 10 year old child presenting with ascending paralysis and intractable seizures. MRI demonstrated multifocal hemispheric and brainstem lesions and the patient improved with IV methylprednisolone (IVMP) (table 4).
Table 4:
Clinical and paraclinical data for other detected neuronal antibodies with MOG-IgG1
| No | Age | Sex | MOG-IgG titer | Co-existent antibody | Timing | Clinical history |
|---|---|---|---|---|---|---|
| 1 | 10 | M | 1:1000 | CASPR2-IgG | CASPR2-IgG positive twice, 6 months apart. MOG-IgG1 positive at 7 months. | Presented with ascending paralysis, intractable seizures. MRI brain demonstrated bilateral hemispheric and brainstem T2 hyperintense lesions without enhancement with appearances consistent with ADEM. Follow up MRI brain demonstrated involvement of internal capsule, cerebral peduncles, midbrain. Treatment included IV steroids with clinical improvement. |
| 2 | 55 | F | 1:100 | LGI1-IgG | 0 days | NA |
| 3 | 59 | M | 1:20 | GABAA-R-IgG | 0 days | Presented with focal seizures, encephalopathy. MRI brain demonstrated T2 hyperintensity in bilateral temporal lobes. A thymoma was detected and removed. Treatment included IVMP and IVIG with initial improvement but persistent cognitive symptoms. At three months follow up he developed confusion, seizures and paranoia. Treatment included IVIG, IVMP, rituximab. Neurocognitive outcomes included impairment in immediate verbal recall, mild impairment in confrontation naming, verbal agility, and semantic fluency.* |
O’Connor et al GABAA receptor autoimmunity. Neurol Neuroimmunol Neuroinflamm May 2019.
MOG-IgG1 (titer 1:20) and GABAA-R-IgG were identified in a 59 year old who presented with seizures and encephalopathy. A thymoma was detected and surgically removed. Treatment included IVIG, IVMP with initial improvement but persistent cognitive symptoms. Three months later the patient developed confusion, seizures and paranoia and was treated with further IVIG, IVMP, plasma exchange and rituximab (Table 4).12
DISCUSSION
In this large cohort of MOG-IgG1 patients evaluated for neuronal-IgGs we have identified that NMDA-R-IgG was the most frequently detected co-existing neuronal-IgG. In addition to confirming previous reports of MOG-IgG1+ co-existing with NMDA-R-IgG+,6,7,17,18 this study has identified common clinical and radiological features of encephalopathy, seizures and leptomeningeal enhancement in these dual-positive patients. This study has also evaluated 17 other neuronal antibodies and found that MOG-IgG1 rarely co-existed with LGI1-IgG, CASPR2-IgG and GABAA-R-IgG and no other neuronal-IgGs commonly associated with paraneoplastic autoimmunity were identified to co-exist, suggesting a unique association with NMDA-R-IgG.
These dual-positive MOG-IgG1+/NMDA-R-IgG+ patients were a young cohort with a median age of 17. Unlike anti-NMDA-R encephalitis cohorts, these dual-positive MOG-IgG1+/NMDA-R-IgG+ patients did not have a female sex predominance.19 Clinical features described in both NMDA-R-IgG-encephalitis and MOGAD were seen in these patients including; encephalopathy, seizures, myelitis, optic neuritis. Ten of the dual-positive MOG-IgG1+/NMDA-R-IgG+ patients met both criteria for ADEM and anti-NMDA-R-encephalitis and the predominant clinical feature of these patients was encephalopathy.14
Dual-positivity with MOG-IgG1+/NMDA-R-IgG+ was more often associated with encephalopathy, seizures and leptomeningeal enhancement (+/− cortical enhancement) (clinical and radiological findings that often cluster together) when compared to MOGAD patients without NMDA-R-IgG. Encephalopathy in the setting of ADEM can be seen in approximately 20–30% of MOGAD3,20 (greater proportion in pediatric patients). Seizures and cortical/leptomeningeal encephalitis has been described in; MOGAD patients who are NMDA-R-IgG negative1,21, anti-NMDA-R-IgG encephalitis22 and GFAP-IgG meningoencephalitis23, thus these clinical and radiological findings are not specific for dual-positivity. Nonetheless, the frequency of these features in our dual-positive MOG-IgG1+/NMDA-R-IgG+ cohort, similar to other reports18,24 suggests that clinicians should consider serological screening for MOG-IgG1 and NMDA-R-IgG in patients presenting with encephalopathy, seizures and/or cortical/leptomeningeal encephalitis.
Similar to studies of MOGAD, our dual-positive MOG-IgG1+/NMDA-R-IgG+ patients had good visual and motor outcomes, with minimal disability in those with myelitis and optic neuritis.3,25 Neurocognitive symptoms were observed in 72% of the dual-positive MOG-IgG1+/NMDA-R-IgG+ patients, similar to reports of persistent neurocognitive symptoms in anti-NMDA-R encephalitis and other infectious and autoimmune encephalidities.26,27
The inciting events leading to dual-antibody positivity is unknown. NMDA-R-IgG autoimmunity is often associated with malignancy (most often teratoma). However, post-herpes simplex virus infection can occur, more often in children, with anti-NMDA-R encephalitis.28 Infection may also be a trigger for MOG-IgG1 associated disorders and two potential mechanisms have been proposed; CNS infection could lead to antigen leakage into the peripheral circulation or an immune response against MOG or molecular mimicry could activate T cells in the periphery.29 Similar to prior studies, dual-positive MOG-IgG1+/NMDA-R-IgG+ patients did not have teratoma, further suggesting a possible non-paraneoplastic cause for dual-autoimmunity in some cases.6
Additional glial antibodies were present in two patients with MOG-IgG1+/NMDA-R-IgG+. GFAP-IgGα was detected in CSF in one patient and has been previously demonstrated to co-exist with variable frequency in patients with NMDA-R-IgG encephalitis and can also occur in the para- and post-infectious setting.23,7 One patient had co-existent AQP4-IgG (1:100) and low-medium titer MOG-IgG (1:40), which we have previously described rarely occurs (most often with high titer AQP4-IgG and low titer MOG-IgG1).30 This patient had clinical features that could be seen in either AQP4-IgG NMOSD and MOGAD including cervical myelitis and optic chiasm involvement. MRI brain demonstrated involvement of the thalamus and temporal lobe (commonly seen in ADEM and encephalitis) and repeat CSF testing was positive for NMDA-R-IgG, however, there were no clinical features of encephalitis.
Co-existence of other neuronal-IgGs with MOG-Ig1 were detected in three patients; CASPR2-IgG (serum)/high titer MOG-IgG1 (1:1000), LGI1-IgG (serum)/medium titer MOG-IgG1 (1:100) and GABA-A-IgG (CSF and serum)/low titer MOG-IgG1 (1:20) patients. Each of these neuronal-IgGs are known to be biomarkers of autoimmune encephalitis and two of these patients had a confirmed clinical phenotype of autoimmune encephalitis.10,12 These neuronal-IgGs have not previously been described to occur with MOG-IgG1. However, unlike NMDA-R-IgG (identified in 4% of our MOG-IgG1 cohort) as only one of each of these co-existent neuronal-IgGs occurred, a strong association between these (CASPR2-IgG, LGI1-IgG and GABAA-R-IgG) and MOG-IgG1 cannot be asserted from this study and false positivity cannot be excluded. In this study, no neuronal-IgGs typically seen in paraneoplastic autoimmunity (CRMP5-IgG, amphiphysin-IgG, PCA1,2,Tr -IgGs, ANNA1,2,3 -IgGs) were identified to co-exist with MOG-IgG1.
The strength of this study is in the large size of this MOG-IgG1 cohort comprehensively tested for a 17 neuronal-IgGs. This study is limited in the ability to evaluate treatments and outcomes in this small cohort of dual-positive MOG-IgG1+/NMDA-R-IgG+ patients. Clinical data was missing for three dual-positive MOG-IgG1+/NMDA-R-IgG+ patients, potentially contributing to selection bias of this cohort. Our comparison cohort was restricted to MOGAD patients who had completed negative neuronal IgG testing in CSF and therefore was limited in size. As our study cohort was selected for patients with both MOG-IgG1 and neuronal-IgG testing, there is likely selection bias impacting the proportion of neuronal-IgGs detected, therefore the frequency of detection should not be interpreted as seroprevalence of neuronal-IgGs within MOG-IgG1 positive patients.
We have demonstrated the most commonly detected neuronal-IgG to co-exist with MOG-IgG1 is NMDA-R-IgG. Testing for MOG-IgG1 and NMDA-R-IgG may be warranted in patients with encephalopathy and inflammatory demyelinating syndromes.
Supplementary Material
Acknowledgements:
We would like to thank Dr. Ilya Kister from New York University Lagone, who provided clinical data for a patient.
We would like to thank and acknowledge our administrative and staff; Ms. Mary Curtis, Ms. Sara Vinje, Ms. Jessica Sagen, Ms. Katie Doane, Ms. Cara Thomas. We would like to thank our neuroimmunology laboratory technical staff; Mr. John Schmeling, Ms. Nancy Peters, Ms. Vickie Mewhorter.
Funding:
We would like to acknowledge funding support from the NIH National Institute of Neurological Disorders and Stroke (R01NS113828).
Footnotes
Declaration of Conflicting Interests:
Amy Kunchok has received research support from Biogen in previous employment.
Eoin P. Flanagan is a site principal investigator in a randomized placebo-controlled clinical trial of Inebilizumab (A CD19 inhibitor) in neuromyelitis optica spectrum disorders funded by MedImmune/Viela Bio. He receives no personal compensation and just receives reimbursement for the research activities related to the trial.
Karl. N. Krecke, John J. Chen, J. Alfredo Caceres, Justin Dominick, Ian Ferguson, Revere Kinkel, John C. Probasco, Miguel Ruvalcaba, Kurt Sieloff, Jeremy Timothy report no disclosures.
Jonathan D. Santoro receives honoraria through GLG consulting and has received grant funding through the National Multiple Sclerosis Society.
Brian G. Weinshenker receives royalties from RSR Ltd, Oxford University, Hospices Civil de Lyon, and MVZ Labor PD Dr. Volkmann und Kollegen GbR for a patent of NMO-IgG as a diagnostic test for NMO and related disorders, served on adjudication committee for clinical trials in NMO conducted by MedImmune and Alexion, and consulted for Chugai and Mitsubishi Tanabe regarding clinical trials for NMO.
Andrew McKeon has patent applications pending for the following IgGs as biomarkers of autoimmune neurological disease: Septin-5 and MAP1B. He has consulted for Grifols, Medimmune, and Euroimmun; and received research support from Grifols, Medimmune, Alexion and Euroimmun but has not received personal compensation.
Sean J. Pittock reports grants, personal fees and non-financial support from Alexion Pharmaceuticals, Inc.; grants from Grifols, Autoimmune Encephalitis Alliance; grants, personal fees, non-financial support and other from MedImmune, Inc.; Dr. Pittock has a patent # 9,891,219 (Application#12–573942) “Methods for Treating Neuromyelitis Optica (NMO) by Administration of Eculizumab to an individual that is Aquaporin-4 (AQP4)-IgG Autoantibody positive”.
References:
- 1.Ogawa R, Nakashima I, Takahashi T, et al. MOG antibody-positive, benign, unilateral, cerebral cortical encephalitis with epilepsy. Neurol Neuroimmunol neuroinflammation 2017; 4: e322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen JJ, Flanagan EP, Jitprapaikulsan J, et al. Myelin Oligodendrocyte Glycoprotein Antibody-Positive Optic Neuritis: Clinical Characteristics, Radiologic Clues, and Outcome. Am J Ophthalmol 2018; 195: 8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jurynczyk M, Messina S, Woodhall MR, et al. Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain 2017; 140: 3128–3138. [DOI] [PubMed] [Google Scholar]
- 4.Armangue T, Olivé-Cirera G, Martínez-Hernandez E, et al. Associations of paediatric demyelinating and encephalitic syndromes with myelin oligodendrocyte glycoprotein antibodies: a multicentre observational study. Lancet Neurol 2020; 19: 234–246. [DOI] [PubMed] [Google Scholar]
- 5.Brilot F, Dale RC, Selter RC, et al. Antibodies to native myelin oligodendrocyte glycoprotein in children with inflammatory demyelinating central nervous system disease. Ann Neurol 2009; 66: 833–842. [DOI] [PubMed] [Google Scholar]
- 6.Titulaer MJ, Höftberger R, Iizuka T, et al. Overlapping demyelinating syndromes and anti-N-methyl-D-aspartate receptor encephalitis. Ann Neurol 2014; 75: 411–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martinez-Hernandez E, Guasp M, García-Serra A, et al. Clinical significance of anti-NMDAR concurrent with glial or neuronal surface antibodies. Neurology. Epub ahead of print11March2020. DOI: 10.1212/WNL.0000000000009239. [DOI] [PubMed] [Google Scholar]
- 8.Hoftberger R, van Sonderen A, Leypoldt F, et al. Encephalitis and AMPA receptor antibodies: Novel findings in a case series of 22 patients. Neurology 2015; 84: 2403–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jeffery OJ, Lennon VA, Pittock SJ, et al. GABAB receptor autoantibody frequency in service serologic evaluation. Neurology 2013; 81: 882–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Irani SR, Pettingill P, Kleopa KA, et al. Morvan syndrome: Clinical and serological observations in 29 cases. Ann Neurol 2012; 72: 241–255. [DOI] [PubMed] [Google Scholar]
- 11.Pittock SJ, Yoshikawa H, Ahlskog JE, et al. Glutamic acid decarboxylase autoimmunity with brainstem, extrapyramidal, and spinal cord dysfunction. Mayo Clin Proc 2006; 81: 1207–1214. [DOI] [PubMed] [Google Scholar]
- 12.O’Connor K, Waters P, Komorowski L, et al. GABA A receptor autoimmunity. Neurol - Neuroimmunol Neuroinflammation 2019; 6: e552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waters PJ, Komorowski L, Woodhall M, et al. A multicenter comparison of MOG-IgG cell-based assays. Neurology 2019; 10.1212/WNL.0000000000007096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Graus F, Titulaer MJ, Balu R, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol 2016; 15: 391–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krupp LB, Tardieu M, Amato MP, et al. International Pediatric Multiple Sclerosis Study Group criteria for pediatric multiple sclerosis and immune-mediated central nervous system demyelinating disorders: revisions to the 2007 definitions. Mult Scler 2013; 19: 1261–7. [DOI] [PubMed] [Google Scholar]
- 16.Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015; 85: 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rojc B, Podnar B, Graus F. A case of recurrent MOG antibody positive bilateral optic neuritis and anti-NMDAR encephalitis: Different biological evolution of the two associated antibodies. J Neuroimmunol 2019; 328: 86–88. [DOI] [PubMed] [Google Scholar]
- 18.Zhou L, ZhangBao J, Li H, et al. Cerebral cortical encephalitis followed by recurrent CNS demyelination in a patient with concomitant anti-MOG and anti-NMDA receptor antibodies. Mult Scler Relat Disord 2017; 18: 90–92. [DOI] [PubMed] [Google Scholar]
- 19.Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: An observational cohort study. Lancet Neurol 2013; 12: 157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Waters P, Fadda G, Woodhall M, et al. Serial Anti-Myelin Oligodendrocyte Glycoprotein Antibody Analyses and Outcomes in Children with Demyelinating Syndromes. JAMA Neurol 2020; 77: 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Budhram A, Kunchok AC, Flanagan EP. Unilateral Leptomeningeal Enhancement in Myelin Oligodendrocyte Glycoprotein Immunoglobulin G-Associated Disease. JAMA Neurology; 77. Epub ahead of print2020. DOI: 10.1001/jamaneurol.2020.0001. [DOI] [PubMed] [Google Scholar]
- 22.Kim H, Ryu H, Kang JK. Anti-NMDA Receptor Antibody Encephalitis Presenting with Unilateral Non-convulsive Status Epilepticus in a Male Patient. J epilepsy Res 2015; 5: 17–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flanagan EP, Hinson SR, Lennon VA, et al. Glial fibrillary acidic protein immunoglobulin G as biomarker of autoimmune astrocytopathy: Analysis of 102 patients. Ann Neurol 2017; 81: 298–309. [DOI] [PubMed] [Google Scholar]
- 24.Taraschenko O, Zabad R. Overlapping demyelinating syndrome and anti-N-methyl-D-aspartate receptor encephalitis with seizures. Epilepsy Behav Reports 2019; 12: 100338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen JJ, Flanagan EP, Jitprapaikulsan J, et al. Myelin Oligodendrocyte Glycoprotein Antibody–Positive Optic Neuritis: Clinical Characteristics, Radiologic Clues, and Outcome. Am J Ophthalmol 2018; 195: 8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Finke C, Kopp UA, Prüss H, et al. Cognitive deficits following anti-NMDA receptor encephalitis. J Neurol Neurosurg Psychiatry 2012; 83: 195–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harris L, Griem J, Gummery A, et al. Neuropsychological and psychiatric outcomes in encephalitis: A multi-centre case-control study. PLoS One; 15. Epub ahead of print2020. DOI: 10.1371/journal.pone.0230436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Armangue T, Moris G, Cantarín-Extremera V, et al. Autoimmune post–herpes simplex encephalitis of adults and teenagers. Neurology 2015; 85: 1736–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reindl M, Di Pauli F, Rostásy K, et al. The spectrum of MOG autoantibody-associated demyelinating diseases. Nature Reviews Neurology 2013; 9: 455–461. [DOI] [PubMed] [Google Scholar]
- 30.Kunchok A, Chen JJ, McKeon A, et al. Coexistence of Myelin Oligodendrocyte Glycoprotein and Aquaporin-4 Antibodies in Adult and Pediatric Patients. JAMA Neurology. Epub ahead of print2019. DOI: 10.1001/jamaneurol.2019.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.