

Abstract
A 63-year-old diabetic woman presented to the outpatient clinic with a 1-week history of abdominal pain. On complete evaluation, she was diagnosed to have essential thrombocythemia. Abdominal imaging revealed portal vein thrombosis with a large splenic infarct. The patient was started on anticoagulant, antiplatelet and cytoreductive therapy. In view of persistent high platelet count, plasma apheresis was done, following which the patient’s platelet counts were reduced. Essential thrombocythemia has a high rate of complications, resulting in significant morbidity and mortality. Few cases of this disease and its treatment have been described in the literature, especially pertaining to the Indian scenario. Further studies are needed to establish a multidisciplinary algorithm for its diagnosis and to elucidate the guidelines for the successful treatment of the condition.
Keywords: haematology (drugs and medicines), haematology (incl blood transfusion), malignant and benign haematology, medical management, portal vein
Background
Myeloproliferative neoplasms (MPNs) are a group of disorders characterised by an increase in the number of red blood cells, white blood cells (WBCs) or platelets produced by the bone marrow as a result of a stem cell mutation. They include polycythemia vera, chronic idiopathic myelofibrosis, chronic myeloid leukaemia, chronic neutrophilic leukaemia, chronic eosinophilic leukaemia, hypereosinophilic syndrome, mast cell disease and essential thrombocythemia (ET).
ET is a rare MPN that manifests as overproduction of platelets (>450×109/L). There is abnormal platelet number and bone marrow megakaryocyte morphology. The incidence of ET is 1.0 to 2.5 individuals per 100 000 per year according to the western literature, and true prevalence in the Indian subcontinent is not known.1 Despite being a rare disease, it has life-threatening implications. Thus, early diagnosis and prompt treatment are of paramount importance in reducing the morbidity and mortality due to ET.
We aim to describe the presenting symptoms, diagnostic algorithm, imaging and management of this rare case in order to bring to light the multidisciplinary approach involved in the diagnosis as well as the importance of prompt recognition and treatment of this potentially life-threatening condition.
Case presentation
A 63-year-old woman presented to the outpatient clinic with reports of upper abdominal pain for 1 week. She also reported of low-grade, intermittent fever and chest pain for 1 day. The abdominal pain was mild, ill-defined and located in the epigastric and left hypochondriac region. There was no radiation of the pain. The chest pain is mild, retrosternal and non-radiating. The patient also reported of weakness and general fatiguability for the past month. She was on regular medication for Diabetes Mellitus and hypertension for the past 10 years.
General physical examination was within normal limits. Abdominal examination revealed mild tenderness in the epigastric and left hypochondriac region. The rest of the systemic examinations were normal.
Investigations
Laboratory investigations (table 1) revealed abnormally high platelet counts. Peripheral blood smear also showed markedly increased platelet counts with few giant platelets. With the suspicion of MPN, a bone marrow biopsy was done.
Table 1.
Laboratory investigations
| Lab investigations | Value | Normal range |
| Erythrocyte sedimentation rate | 14 mm/hour | 0–22 mm/hour |
| Haemoglobin | 121 g/L | 130–170 g/L |
| Platelet count | 1030 x 109/L | 150–400 x 109 /L |
| White cell count | 13.2 x 109/L | 4–11 x 109/L |
| Serum iron | 102 µg/dL | 33–193 µg/dL |
| Serum ferritin | 200.4 ng/mL | 30–400 ng/mL |
| Aspartate transaminase | 25 IU/L | 5–40 IU/L |
| Alanine transaminase | 29 IU/L | 5–40 IU/L |
| Alkaline phosphatase | 138 U/L | 40–130 U/L |
| Blood urea nitrogen | 30 mg/dL | 16–45 mg/dL |
| Serum creatinine | 0.9 mg/dL | 0.5–1.0 mg/dL |
| Uric acid | 4.9 mg/dL | 2.3–6.6 mg/dL |
| Glycated haemoglobin | 6.5 % | Less than 5.7 % |
| Troponin T | 0.004 ng/mL | Less than 0.020 ng/dL |
| C reactive protien | 2.3 mg/L | 0–5 mg/L |
Bone marrow biopsy revealed normal erythroid and myeloid elements with an increase in megakaryocytes. Megakaryocytes were characteristically large with hyperlobated nuclei favouring the diagnosis of ET (figure 1). Bone marrow iron stores were normal, eliminating iron deficiency as a cause for the condition.
Figure 1.
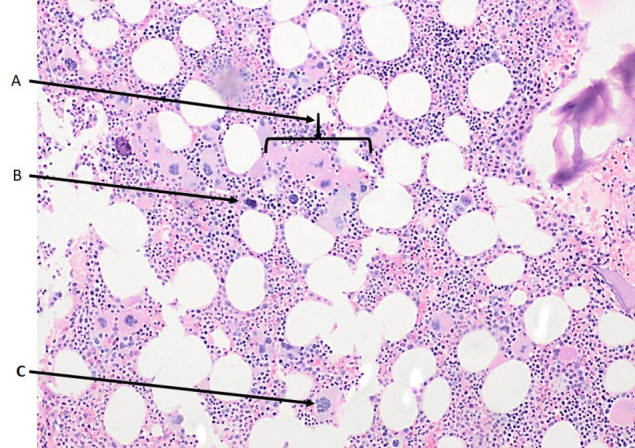
Bone marrow biopsy photomicrograph (20×) showing an increased number of megakaryocytes (A), dyspoietic (B) and hyperlobated (C) megakaryocytes favouring the diagnosis of essential thrombocythemia.
Molecular studies (reverse transcription PCR) for JAK2/MPL/CAL-R were conducted and JAK-2 (V617F) mutation was positive, further strengthening the evidence for the above diagnosis.
Ultrasound abdomen showed splenomegaly with an ill-defined anechoic area in the splenic parenchyma. Contrast-enhanced CT of the abdomen revealed portal vein thrombosis with a large well-defined hypodense lesion in the mid and lower poles of the spleen suggestive of a splenic infarct (figure 2).
Figure 2.
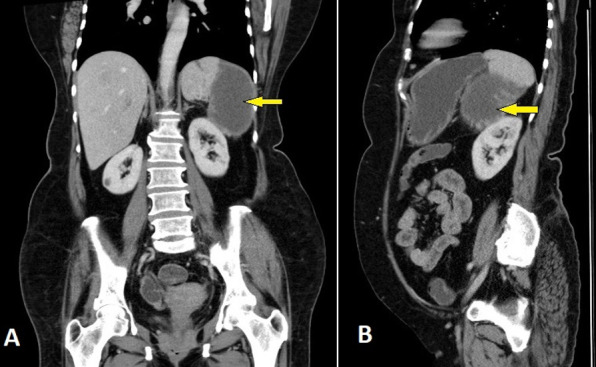
Contrast-enhanced computed tomography of the abdomen (A: coronal, B: sagittal views) showing a large well-defined hypodense lesion in the mid and lower poles of the spleen suggestive of splenic infarct (arrows).
International Prognostic Score for Thrombosis in ET (IPSET) score, calculated based on the above findings, indicated high-risk disease (presence of thrombosis, age >60 years, JAK2 mutation positive).
The ECG, echocardiography and cardiac biomarker (troponin T) were within normal limits ruling out a cardiac cause for her chest pain.
Differential diagnosis
Clonal MPNs such as polycythemia vera and primary myelofibrosis are important differential diagnoses that can be ruled out by bone marrow biopsy and genetic testing. In ET, bone marrow biopsy shows megakaryocyte proliferation, atypical nuclei and rarely an increase in reticulin fibres (myelofibrosis), while polycythemia vera is characterised by trilineage hypercellularity. Reactive thrombocytosis may be a result of infection, inflammation, haemolysis or malignancies, each of which can be ruled out by specific diagnostic tests. Spurious thrombocytosis is due to erroneous counting, resulting from faulty laboratory techniques or investigator error. Cryoglobulin crystals, cytoplasmic fragments of circulating leukemic cells and bacteria can be mistaken for platelets, leading to thrombocytosis. Furthermore, portal vein thrombosis is often seen in prothrombotic states, including antiphospholipid antibody syndrome and coagulation pathway abnormalities.1
Treatment and outcome
The patient was admitted to the medical ward. Anticoagulants (enoxaparin 60 mg, subcutaneous, two times per day) antiplatelets (aspirin 75 mg/day) and cytoreductive therapy (hydroxyurea 500 mg three times per day) were initiated soon after the diagnosis of ET. Despite cytoreductive therapy, there was a serial rise in platelet count. On day 5 of admission, her platelet count was 16 lakhs cells/µL and hence platelet apheresis was performed. Repeat platelet count post first cycle of apheresis was 13 lakhs cells/µL and hence one more cycle of apheresis was done. Following the second cycle, the platelet count decreased to 6 lakhs cells/µL. She had no fever after admission and her other symptoms were reduced gradually. Injection enoxaparin was changed over to an oral anticoagulant (dabigatran). She was discharged on day 14 of admission with the advice to continue hydroxyurea (500 mg two times per day), anticoagulant, antiplatelet, antihypertensive and antidiabetic medications.
Follow-up
At follow-up in the outpatient clinic 4 weeks after discharge, she reported no symptoms and her platelet count was within normal limits (2.56 lakhs cells/µL). Her repeated haemoglobin was 12 g/dL and WBC was 5900 cells/µL. Repeated ultrasound abdomen showed a significant reduction in the splenic infarct.
Discussion
ET is a rare MPN that manifests as overproduction of platelets (>450×109/L). There is an abnormal platelet number and bone marrow megakaryocyte morphology. The disorder is assumed to be caused by JAK-STAT pathway activation, leading to the unregulated proliferation of haematopoietic stem cells.2
Although ET is an uncommon disease, its clinical implications warrant immediate treatment, particularly in high-risk cases. The clinical features of ET range from being asymptomatic to life-threatening thrombosis and haemorrhage.
Our patient presented with abdominal pain, chest pain and fever, which are non-specific symptoms of a myriad of underlying diseases. The increase in platelets on the differential count and the negative cardiac biomarker panel pointed towards infectious, neoplastic or reactive causes of thrombocytosis. Imaging revealed splenic infarcts, which could account for the fever, abdominal pain and thrombocytosis. In all cases of thrombocytosis with unclear aetiology, JAK2 mutation testing should be done to identify myeloproliferative disorders such as ET. If positive, a bone marrow biopsy is also warranted. Our case satisfies the WHO 2016 diagnostic criteria for ET by fulfilling four major criteria.3
When the platelet counts rise above 1 million/μL, as in our case, acquired Von Willebrand disease should be considered. It is postulated to be due to increased in vivo adsorption onto malignant cells.3 Thus, our patient should further undergo tests to detect Von Willebrand factor levels and factor VIII levels despite the normal activated partial thromboplastin time (aPTT) value. This is because if aspirin is given in such patients, it can lead to significant bleeding. Additionally, in patients with ET, diagnostic studies for congenital or acquired prothrombotic states should also be done as these states increase the risk of thrombosis and can markedly increase the mortality in ET. Furthermore, it should be noted that high platelet counts may be associated with both bleeding and thrombosis.4 5
On genetic testing, our patient demonstrated a positive JAK2 mutation. This mutation is not specific for ET and a bone marrow biopsy should be done to exclude other disorders associated with this mutation. Furthermore, according to previously published studies, the risk of thrombosis is increased in patients with JAK2 mutation, increasing the evidence for prompt rapid treatment.6 Studies also showed that in patients presenting with idiopathic Budd-Chiari syndrome (BCS) and portal venous thrombosis (PVT), V617F JAK2 was found in 20% of the cases, which strengthens the association between ET and PVT/BCS. Thus, JAK2 mutations should be tested in all patients presenting with idiopathic BCS or PVT, for the early detection of ET and the prevention of its life-threatening complications.7 8
Treatment for ET is based on whether the patient is very low, low, intermediate or high-risk for thrombosis. This can be calculated using the IPSET-thrombosis model, based on patient age, history of thrombosis, presence of JAK2 V617F mutation and cardiovascular risk factors.9 Very low and low-risk patients can be treated with conservative treatment or low-dose aspirin. However, intermediate and high-risk categories mandate cytoreduction therapy, which could be pharmacological or non-pharmacological.10
Our patient was prescribed hydroxyurea but continued to demonstrate increasing platelet counts, warranting a more rapidly acting therapy, platelet apheresis. The indications for platelet apheresis have not yet been fully described. Current recommendations suggest the initiation of cytoreduction in the setting of ET with further cardiovascular risk factors (hypertension and T2DM in our patient) or symptomatic complications. Furthermore, age >60 years, prior thrombotic event and platelet counts >1500×109 /L are absolute indications for cytoreduction. Although cytoreduction can be achieved pharmacologically (hydroxyurea/anagrelide), rapidly evolving complications warrant prompt treatment with plasma apheresis.11 Our patient showed rapidly rising platelet counts of over 16 lakhs, which is a definite indication for platelet apheresis. Following two rounds of therapy, the platelet count was reduced significantly, which is strong evidence of the efficacy and utility of platelet apheresis in the acute setting.
Recent studies have shown that platelet apheresis may decrease prothrombotic Factor VIII, fibrinogen, antithrombin, protein C, protein S levels as well as Von Willebrand factor levels. It has also been demonstrated that this procedure reduces the number of morphologically abnormal platelets.11 Studies have also shown that non-splenectomised patients demonstrate a more rapid decrease in platelet numbers following plateletpheresis than postsplenectomy patients, possibly due to rapid platelet mobilisation from the splenic pool.12 13 In our patient, although splenectomy may be indicated due to the presence of splenic infarcts, prior plateletpheresis should be done to avoid thrombocytosis following the procedure.
One major drawback of plasma apheresis is that it causes a transient rise in platelet-derived growth factors as a result of a rapid reduction in platelet counts. This could initiate a prothrombotic state further aggravating the condition.11 However, in our patient, the benefits outweigh the risks.
Based on the above evidence, the American Society for Apheresis recommends plateletpheresis as a category II (ie, second-line therapy) in symptomatic primary thrombocytosis.14
Our case not only strengthens the established guidelines for management but also sheds light on the potential inclusion of platelet apheresis as first-line treatment in the acute setting. However, the high cost and need for special equipment limit its widespread use, especially in developing countries.
Patient’s perspective.
I was actively doing all the household activities before I got sick. I was very scared when I saw my blood report (high platelet count). I was also very anxious about the procedure (platelet apheresis). But now I am very happy to see my platelet count back to normal and I can do my daily activities like before without any symptoms.
Learning points.
Patients with essential thrombocythemia (ET) can present with a wide range of non-specific symptoms.
Bone marrow biopsy and testing for JAK2 mutation are paramount in identifying ET. Thus, it should be done routinely in all patients presenting with thrombocytosis.
Hydroxyurea proved to be ineffective in the acute setting. This strengthens existing evidence stating that platelet apheresis is the procedure of choice in order to rapidly reduce platelet counts.
Thrombocytosis can be an independent risk factor for thrombosis.
Acknowledgments
We acknowledge the Department of Radiology for providing CECT abdomen images and the Department of Immunohematology & Blood Transfusion (IHBT) for performing platelet apheresis.
Footnotes
Contributors: AMM wrote the draft of the manuscript. SPM revised the manuscript critically for important intellectual content. SS involved in patient management. SV involved in the histopathological diagnosis and provided the bone marrow biopsy photomicrograph. All the authors contributed to the literature review and approved the final manuscript for submission.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Ethics statements
Patient consent for publication
Consent obtained directly from patient(s)
References
- 1.Ashorobi D, Gohari P. Essential Thrombocytosis. 2021 May 1. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing, 2021. [PubMed] [Google Scholar]
- 2.Shallis RM, Podoltsev NA. Emerging agents and regimens for polycythemia vera and essential thrombocythemia. Biomark Res 2021;9:40. 10.1186/s40364-021-00298-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Awada H, Voso MT, Guglielmelli P, et al. Essential thrombocythemia and acquired von Willebrand syndrome: the Shadowlands between thrombosis and bleeding. Cancers 2020;12:1746. 10.3390/cancers12071746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pavithran K, Thomas M. Dural sinus thrombosis in essential thrombocythemia. Case Rep Clin Pract Rev 2003;4:212–3. [Google Scholar]
- 5.Cai X-Y, Zhou W, Hong D-F, et al. A latent form of essential thrombocythemia presenting as portal cavernoma. World J Gastroenterol 2009;15:5368–70. 10.3748/wjg.15.5368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tefferi A, Barbui T, vera P. Polycythemia vera and essential thrombocythemia: 2019 update on diagnosis, risk-stratification and management. Am J Hematol 2019;94:133–43. 10.1002/ajh.25303 [DOI] [PubMed] [Google Scholar]
- 7.de Suray N, Pranger D, Brenard R. Portal vein thrombosis as the first sign of a primary myeloproliferative disorder: diagnostic interest of the V617F Jak-2 mutation. A report of 2 cases. Acta Gastroenterol Belg 2008;71:39–41. [PubMed] [Google Scholar]
- 8.Karaköse S, Oruç N, Zengin M, et al. Diagnostic value of the JAK2 V617F mutation for latent chronic myeloproliferative disorders in patients with Budd-Chiari syndrome and/or portal vein thrombosis. Turk J Gastroenterol 2015;26:42–8. 10.5152/tjg.2015.5738 [DOI] [PubMed] [Google Scholar]
- 9.Haider M, Gangat N, Lasho T, et al. Validation of the revised International prognostic score of thrombosis for essential thrombocythemia (IPSET-thrombosis) in 585 Mayo clinic patients. Am J Hematol 2016;91:390–4. 10.1002/ajh.24293 [DOI] [PubMed] [Google Scholar]
- 10.Elhassan AM, Alsaud A, Yassin MA, et al. Thrombocytapheresis in patient with essential thrombocythemia: a case report. Case Rep Oncol 2020;13:675–9. 10.1159/000507651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boddu P, Falchi L, Hosing C, et al. The role of thrombocytapheresis in the contemporary management of hyperthrombocytosis in myeloproliferative neoplasms: a case-based review. Leuk Res 2017;58:14–22. 10.1016/j.leukres.2017.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Panlilio AL, Reiss RF. Therapeutic plateletpheresis in thombocythemia. Transfusion 1979;19:147–52. 10.1046/j.1537-2995.1979.19279160283.x [DOI] [PubMed] [Google Scholar]
- 13.Lee EJ, Schiffer CA. Evidence for rapid mobilization of platelets from the spleen during intensive plateletpheresis. Am J Hematol 1985;19:161–5. 10.1002/ajh.2830190208 [DOI] [PubMed] [Google Scholar]
- 14.Schwartz J, Padmanabhan A, Aqui N, et al. Guidelines on the use of therapeutic apheresis in clinical Practice-Evidence-Based approach from the writing Committee of the American Society for apheresis: the seventh special issue. J Clin Apher 2016;31:149–338. 10.1002/jca.21470 [DOI] [PubMed] [Google Scholar]