

SUMMARY
CD8 T cells play an essential role in defense against viral and bacterial infections and in tumor immunity. Deciphering T cell loss of functionality is complicated by the conspicuous heterogeneity of CD8 T cell states described across experimental and clinical settings. By carrying out a unified analysis of over 300 ATAC-seq and RNA-seq experiments from 12 studies of CD8 T cells in cancer and infection, we defined a shared differentiation trajectory towards dysfunction and its underlying transcriptional drivers and revealed a universal early bifurcation of functional and dysfunctional T cell states across models. Experimental dissection of acute and chronic viral infection using scATAC-seq and allele-specific scRNA-seq identified state-specific drivers and captured the emergence of similar TCF1+ progenitor-like populations at an early branch point, at which functional and dysfunctional T cells diverge. Our atlas of CD8 T cell states will facilitate mechanistic studies of T cell immunity and translational efforts.
Graphical Abstract
eTOC Blurb
Pritykin et al. reveal a shared path of T cells towards dysfunction and the early bifurcation of functional and dysfunctional T cell states from a TCF1+ progenitor by implementing a unified analysis of transcriptional and epigenetic states in T cells in cancer and infection.
INTRODUCTION
Upon completing their differentiation in the thymus, mature naïve T lymphocytes enter the periphery and recirculate through secondary lymphoid organs, where, upon an encounter with a cognate antigen in the presence of co-stimulatory molecules, they become activated, expand, and differentiate into effector or memory T cells. These cells then take up residence in lymphoid and non-lymphoid organs where they exert their immune functions. In contrast, chronic or suboptimal antigenic stimulation, e.g. in the absence of co-stimulation, can result in a state of hypo-responsiveness or anergy (Murphy and Weaver, 2016). Over the last decade, this simple textbook view has evolved into a markedly more nuanced and complex picture of T cell differentiation with a plethora of seemingly distinct states emerging from a large number of studies in mice and man (Fan and Rudensky, 2016, Jameson and Masopust, 2018, Kumar et al., 2018). CD8 T cells, whose function is essential for defense against viral and bacterial infections and for tumor immunity, serve as a case study in this regard. Phenotypic and functional analyses of CD8 T cells in acute and chronic viral infections, cancer, transplantation and “self” tolerance in both experimental animal models and in human patients have offered numerous descriptions of activated effector, long-lived central and short-lived effector memory cells and their precursors, as well as an array of CD8 T cell states with perturbed functionalities dubbed anergic, exhausted, and reversibly or irreversibly dysfunctional. Recent characterization of a small subset of exhausted/dysfunctional cells, named “stem cell-like” or progenitor cells, capable of self-renewal, adds further complexity to the topography of CD8 T cell activation and differentiation (McLane et al., 2019, Hashimoto et al., 2018, Blank et al., 2019).
Studies of CD8 T cell dysfunctional states, besides being highly significant for understanding of basic mechanisms of adaptive immunity, have attracted particular attention due to the realization that prevention or reversal of CD8 T cell dysfunction can serve as a potent strategy for the treatment of both solid organ and hematologic malignancies and chronic infections. Inefficient mobilization of endogenous CD8 T cell responses or a failure to engage them in cancer patients in response to checkpoint blockade inhibitors, as well as disease resistance or relapse following adoptive CD8 T cell therapies including CAR (chimeric antigen receptor)-T cells, have been attributed to a large degree to dysfunction or exhaustion of tumor- and virus-specific CD8 T cells (Ribas and Wolchok, 2018, Sharma et al., 2017, Kallies et al., 2019). Transcriptional and chromatin features associated with these states have been extensively explored through the analyses of epigenomes and transcriptomes of isolated subsets of functional and dysfunctional CD8 T cells using DHS-, ATAC-, and RNA-seq and through single cell transcriptomics and proteomics (Pauken et al., 2016, Sen et al., 2016, Scott-Browne et al., 2016, Philip et al., 2017, Mognol et al., 2017, Chen et al., 2019a, Scharer et al., 2017, Utzschneider et al., 2016, Man et al., 2017, Miller et al., 2019, Brummelman et al., 2018, Bengsch et al., 2018, Yao et al., 2019, Chen et al., 2019c, Beltra et al., 2020, Yu et al., 2017). These studies have significantly advanced the knowledge of CD8 T cell differentiation and highlighted pronounced changes in T cell chromatin states. However, the remarkable heterogeneity of CD8 T cell states revealed by these genome-wide analyses in diverse experimental and clinical settings poses a major problem of distinguishing between common vs. context-specific features of differentiation towards dysfunction and underlying regulatory mechanisms. For example, in chronic viral infection, T cells encounter antigen in an inflammatory and stimulatory context and have been described as progressing through an effector state prior to differentiating to a state often called exhaustion (Wherry and Kurachi, 2015). Meanwhile, a two-step process of differentiation from a reversible to an irreversible dysfunctional state was reported in studies including our own in the setting of early tumorigenesis, where naïve tumor-specific T cells encounter antigen in a non-inflammatory setting that may result in inadequate priming or activation (Philip et al., 2017, Philip and Schietinger, 2019). Therefore, in addition to inconsistent terminology (i.e. exhaustion vs. dysfunction), it is unclear how to reconcile different models of progression to dysfunction (the term we will use here) and how these differentiation programs give rise to two distinct states at late time points of chronic antigen exposure – self-renewing dysfunctional progenitors and terminally dysfunctional cells.
A vexing obstacle in addressing these issues has been the inability to directly compare genome-wide data from different studies due to technical sources of variation, including sample preparation, sequencing quality, batch effects, and cell numbers, making meaningful integration of massive amounts of data generated in mouse and human studies problematic. To address this challenge, we carried out a uniform reprocessing and a statistically principled batch effect correction approach to over 300 chromatin accessibility (ATAC-seq) and gene expression (RNA-seq) datasets generated in twelve independent studies of CD8 T cell states observed across experimental mouse models of acute and chronic infection and tumors. Our analysis revealed a universal signature of chromatin accessibility changes in the progression to terminal dysfunction in both tumors and chronic infection, implying early commitment to a dysfunctional fate in all settings of chronic antigen exposure. The chromatin state observed at early time points during the development of dysfunction was similar to that of dysfunctional progenitor cells found in late time points in infection and tumor models. Motif-based regression modeling of this unified chromatin accessibility compendium enabled inference of state-specific transcription factor activities and implicated new factors in the progression to terminal dysfunction. This bulk-level T cell state analysis suggested a universal early bifurcation of functional and dysfunctional T cell activation states across models of cancer and chronic infection.
We further characterized this early branch point by carrying out single-cell analysis of CD8 T cell populations in the context of acute and chronic lymphocytic choriomeningitis virus (LCMV) infection and observed a TCF1+ progenitor-like population resembling the memory precursor effector cell (MPEC) population in acute infection. Regression modeling of scATAC-seq clusters enabled refined association of T cell functional states with the activity of transcription factors, whose causal role was established through a comprehensive scRNA-seq atlas in T cell populations from F1 hybrid mice combining evolutionary distant genomes of laboratory and wild-derived mouse strains.
Together, these results provide new insights into the early emergence of a progenitor-like population in response to chronic antigen stimulation that appears to precede the establishment of dysfunction-committed progenitor cells, elucidating recent reports (Utzschneider et al., 2020, Chen et al., 2019c) with rich single cell characterizations. Our unified atlas of CD8 T cell chromatin and expression states across mouse models and single-cell analyses of the bifurcation between functional and dysfunctional T cell responses will provide a valuable resource to the community and facilitate further mechanistic and translational studies.
RESULTS
Dysfunctional T cells in tumors and chronic infection share a common epigenetic and transcriptional state space
We collected 166 chromatin accessibility (ATAC-seq) datasets (Figure 1A, Table S1) from 10 recent studies on CD8 T cell function in mouse models of infection and cancer. These encompassed T cells in settings of acute bacterial infection, acute and chronic viral infection, and tumor-infiltrating lymphocytes in hepatocarcinoma and melanoma models, including adoptively transferred endogenous and CAR-T cells, and with and without treatment with anti-PD1 immunotherapy; memory precursor cells, tissue-resident memory cells, and progenitor T cell populations isolated from various models of chronic antigen stimulation were included (Pauken et al., 2016, Sen et al., 2016, Scott-Browne et al., 2016, Scharer et al., 2017, Mognol et al., 2017, Philip et al., 2017, Chen et al., 2019a, Miller et al., 2019, Milner et al., 2017, Yu et al., 2017). We performed a uniform processing of these data to construct a high-resolution atlas of 129,799 reproducible chromatin accessibility peaks across CD8 T cell states; these peaks were further split into 221,054 subpeaks and associated to genes (Methods, Table S2).
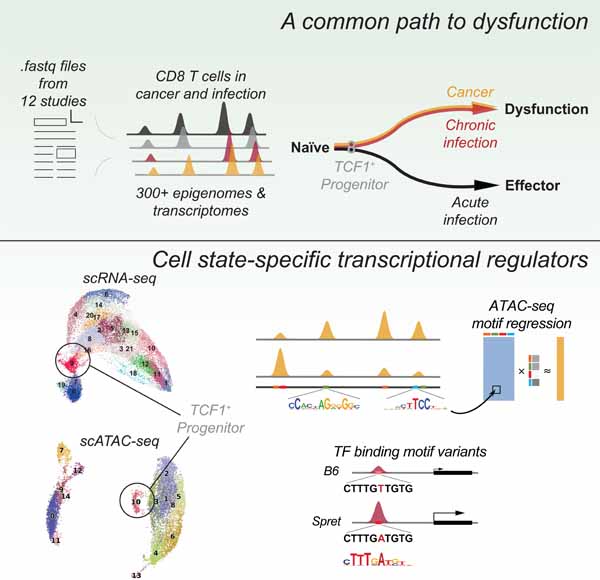
Figure 1. Dysfunctional CD8 T cells in tumors and chronic infection share a common chromatin state space.
A. Snapshot of the ATAC-seq compendium near the Pdcd1 locus (light grey bars, peaks, dark grey bars, peak summit regions). B. PCA of batch-effect corrected ATAC-seq signal in peak summit regions (functional cell state, color; data source, shape). C. First principal component (PC1) of PCA for batch-effect corrected ATAC-seq signal in dysfunctional T cells from different studies (shown separately below) for peaks differentially accessible between progenitor and terminally dysfunctional cells in chronic LCMV infection. D. Differential accessibility and differential expression between progenitor and terminally dysfunctional T cells in chronic LCMV infection and in melanoma, and between early and late states of dysfunction in hepatocarcinoma progression. Left: batch-effect corrected ATAC-seq signal log2 fold change for peaks of significantly differentially accessible genes; right: log2 fold change of RNA-seq gene expression (color for significantly decreased/increased individual peaks or genes, FDR < 0.05). See also Figures S1, S2 and Tables S1-S3.
Expectedly, principal component analysis (PCA) of peak read counts clustered samples by data source (Figure S1A). We therefore applied a generalized linear model (GLM) accounting for data source and functional state (naïve, functional, dysfunctional, see Methods). After this batch effect correction, distances between functionally related samples from different studies decreased (Figure S1B), and samples readily clustered into broad functional categories regardless of the data source (Figure 1B). A more conservative GLM correction that did not explicitly model differences between functional and dysfunctional cells produced similar results (Figure S1C). Furthermore, naïve cells across studies clustered together in the PCA plot, while effector and memory cells (as well as pre-activated cells injected in melanoma-bearing mice, but ignorant of the tumor antigen) formed a cluster of functional cells. Interestingly, cells profiled at day 4 in acute infection (Scharer et al., 2017) were positioned between clusters of naïve and functional cells, suggesting their intermediate state. Strikingly, the chromatin states of dysfunctional tumor-infiltrating T cells from different tumor models and T cells in chronic viral infection formed a distinct cluster, suggesting a universal program of T cell dysfunction across models and immune challenges. A similar analysis of 136 gene expression (RNA-seq) data sets from eight studies (Pauken et al., 2016, Scott-Browne et al., 2016, Man et al., 2017, Utzschneider et al., 2016, Miller et al., 2019, Mognol et al., 2017, Philip et al., 2017, Chen et al., 2019a) showed consistent results (Figure S1D, Tables S1, S2). Differential expression between functional and dysfunctional cells was significantly correlated with differential accessibility (Figure S1E). This confirmed that dysfunctional T cells are epigenetically and transcriptionally similar in the settings of chronic infection and across tumor models.
Genes with the strongest differential accessibility between functional and dysfunctional cells at their promoter, intronic, and nearby intergenic peaks (Methods) included well-known markers of T cell activation, cytotoxicity, adhesion, and apoptosis, as well as those encoding key transcription factors, cytokines and cytokine receptors, and other cell surface and intracellular proteins (Figure S1F, Table S2). Consistent with global differential accessibility and expression correlation (Figure S1E), differential accessibility of individual genes was often associated with significant differential expression (Figure S1F). Differential accessibility between naïve or memory and dysfunctional CD8 T cells in mouse models was significantly associated with orthologous changes in human donors and cancer patients, suggesting generalizability to the human context (Figure S1G, Table S3, Methods).
Thus our integrative accessibility and expression analyses across our compendium (Table S2) identified a high confidence universal epigenetic and transcriptional gene signature of T cell dysfunction.
T cell temporal progression in tumors and chronic infection mirrors the state change from progenitor to terminal dysfunction
Dysfunctional T cells profiled as early as day 5–8 after antigen encounter in tumor or chronic viral infection models clustered closely with terminally dysfunctional cells profiled at day 22–35 rather than with effector or memory cells (Figure 1B, Figure S1D). Cells characterized as progenitor dysfunctional cells in different studies also clustered with terminally dysfunctional cells. This suggested that T cells adopt a dysfunctional rather than an effector chromatin state as early as at day 5 after antigen encounter.
We next compared chromatin states of different dysfunctional T cell subsets across models and immune challenges. Differentially accessible peaks between progenitor and terminally dysfunctional cells, including endogenous or transferred CAR-T cells in melanoma or dysfunctional cells in chronic viral infection, were consistent between models and also displayed concordant changes between early (d5–8) and late (d22–35) states of dysfunction in chronic viral infection and hepatocarcinoma tumorigenesis (Figure 1C, Figure S2A-C, Table S2). Changes in accessibility between early and late dysfunctional states, and between progenitor and terminally dysfunctional states, correlated with changes in gene expression (Figure S2D). Importantly, T cells at early time points in all mouse models were similar in bulk chromatin state to dysfunctional progenitor cells, a subpopulation sorted from late time points in chronic viral infection or tumors. Differential accessibility between progenitor and terminally dysfunctional CD8 T cells in the melanoma mouse model was significantly associated with that in TILs from melanoma patients (Sade-Feldman et al., 2018), again supporting generalizability to human data (Figure S2E, Table S3, Methods). These analyses suggested a common axis of differentiation of T cell dysfunction across models and immune challenges.
Many individual genes displayed similar expression and chromatin accessibility changes across models along this common differentiation axis (Figure 1D, Table S2). For example, genes encoding markers of terminal dysfunction Entpd1 (Cd39), 2B4 (Cd244), Cd38 were significantly more accessible in terminal dysfunction and late tumor-specific dysfunction, while genes encoding progenitor cell markers Cxcr5, Slamf6, IL7Ra chain were significantly more accessible in progenitor and early dysfunctional cells. Additional genes encoding cell surface proteins, such as CD9, CD200, TNFRSF25 (DR3), CD83, CD69, were significantly more accessible and expressed in progenitor cells and could serve as candidate progenitor cell markers. The locus of the transcription factor (TF) Id2 was more accessible in terminal/late dysfunction, while Id3, Tcf7, Lef1, Nfkb1, Pou2f2, Pou6f1 were more accessible in progenitor or early dysfunctional cells, implicating multiple TFs in the establishment and maintenance of these states. In most cases, differential accessibility at individual genes in early/progenitor versus late/dysfunctional states was associated with differential expression, consistent with overall correlation of differential expression and accessibility (Figure S2D). However, our joint analysis did recover context-specific features, including accessibility and expression patterns associated with T cell activation in chronic viral infection compared to tumor contexts (Figures S2D).
Tox and Ikzf2, whose heightened expression was previously associated with terminal dysfunction, were indeed significantly more accessible and highly expressed in terminal vs. early T cell dysfunction during hepatocarcinoma tumorigenesis; however, both loci had many peaks significantly more accessible in progenitor cells, and Ikzf2 was significantly overexpressed in progenitor cells both in chronic LCMV infection and melanoma, suggesting the activity of these TFs in progenitor cells (Figure 1D).
Thus, our analysis identified universal markers and TFs associated with T cell differentiation towards dysfunction across models and organisms.
Coordinated activity of many TFs characterize functional and dysfunctional T cell states
We next sought to identify TFs associated with different T cell activation and differentiation states, particularly the progenitor cells. We analyzed TF targets, as predicted by TF binding motifs, and used negative binomial regression with ridge regularization to predict absolute levels of chromatin accessibility (ATAC-seq read coverage) in peaks from TF motif occurrences for each sample (Figure 2A, Figure S3A,B, Methods). For this analysis, we focused on 105 motifs for TFs expressed in CD8 T cells after grouping TFs with indistinguishable motifs. Using coefficients of the regression models, we estimated the effect on accessibility of each TF in each sample. This allowed us to map chromatin accessibility profiles into a lower dimensional inferred transcription factor activity space, largely preserving the relationships between samples (Figure S3C).
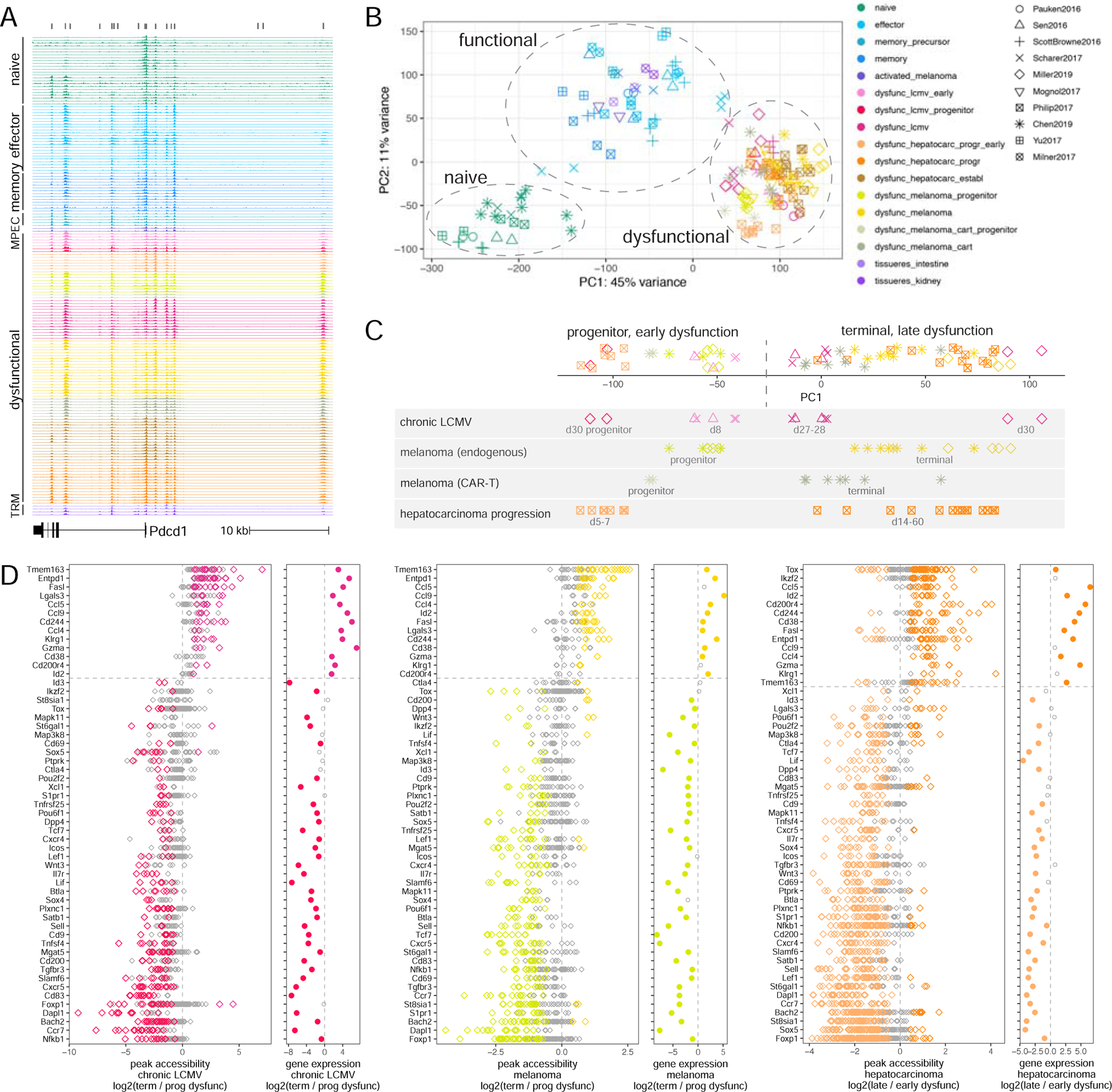
Figure 2. Predictive modeling identifies transcription factors associated with progenitor dysfunctional CD8 T cells.
A. Schematic of the negative binomial GLM to infer TF associations with chromatin accessibility. B. Inferred TF motif coefficients (with the highest variance across conditions, z-score row normalized) from each sample, consolidated between replicates across studies. C. Inferred TF motif coefficients with the highest variance across dysfunctional states (z-score row normalized). D. Network of predicted regulatory interactions between TFs in progenitor dysfunctional T cells (binding motifs not available for TOX and IKZF2, yellow). See also Figure S3.
In order to identify TFs most strongly associated with T cell functional states, we consolidated the most variable inferred TF activities across replicates and studies (Figure 2B). For example, as expected, the Eomes/Tbx21 motif was associated with low accessibility in naïve cells and high accessibility in all antigen-experienced cells, and the Lef1/Tcf7 motif with high accessibility in naïve cells and low accessibility in terminally dysfunctional cells (Figure 2B). Nr4a, Nfkb, Nfat, Pou, AP1 family motifs were strongly associated with high accessibility in early dysfunctional and progenitor cells as opposed to terminally dysfunctional, naïve or functional cells, while Zfp143 and Zfp523 motifs were associated with high accessibility in terminal dysfunction (Figure 2B,C); this included both previously (Chen et al., 2019a, Philip et al., 2017, Doering et al., 2012, Im et al., 2016, Beltra et al., 2020) and newly identified factors. Interestingly, Ctcf motifs were associated with differential accessibility between conditions (Figure 2B,C), suggesting changes in chromatin looping between T cell functional states (Johanson et al., 2019, Splinter et al., 2006); consistently, we observed a significant relationship between differential accessibility and differential expression when associating ATAC-seq peaks to genes using chromatin loops defined using published Hi-C data in naïve CD8 T cells (He et al., 2016) instead of our default approach (Figure S3D, Methods). Overall, motif-based regression modeling of ATAC-seq data was a powerful approach for associating candidate TFs with T cell functional states.
Inferred TF activities across all antigen-experienced cells were highly correlated, potentially suggesting coordinated regulatory programs in different functional states; importantly, correlated TFs generally had different motifs and were bound at different sites (Figure S3E). Many TFs expressed in progenitor dysfunctional cells had multiple predicted binding sites in loci encoding other TFs, including Tox and Ikzf2 for which we did not have a motif, and thus could potentially regulate their expression (Figure 2D). This suggested that the coordinated and hierarchically organized activity of a broad range of TFs may be required for establishing and maintaining T cell functional states, as opposed to a single “master regulator”.
Single-cell chromatin accessibility analysis reveals the early emergence of a progenitor-like T cell population
Our integrative analysis of bulk ATAC-seq data found an early (d5–8) divergence of CD8 T cells between responses to acute and chronic immune challenges (Figure 1B) and showed that plastic and reprogrammable cells early (d7–8) in the development of T cell dysfunction were more similar to immunotherapy-responsive progenitor dysfunctional cells identified at later (d20–35) time points than to terminally dysfunctional cells (Figure 1C). To better characterize chromatin states of the early divergence between functional and dysfunctional fates, we performed single-cell chromatin accessibility (scATAC-seq) analysis of the total splenic CD8 T cell compartment in mice at day 7 (d7) upon infection with LCMV Armstrong (Arm), resulting in acute infection, and LCMV clone 13 (Cl13), resulting in chronic infection (Figure 3A).
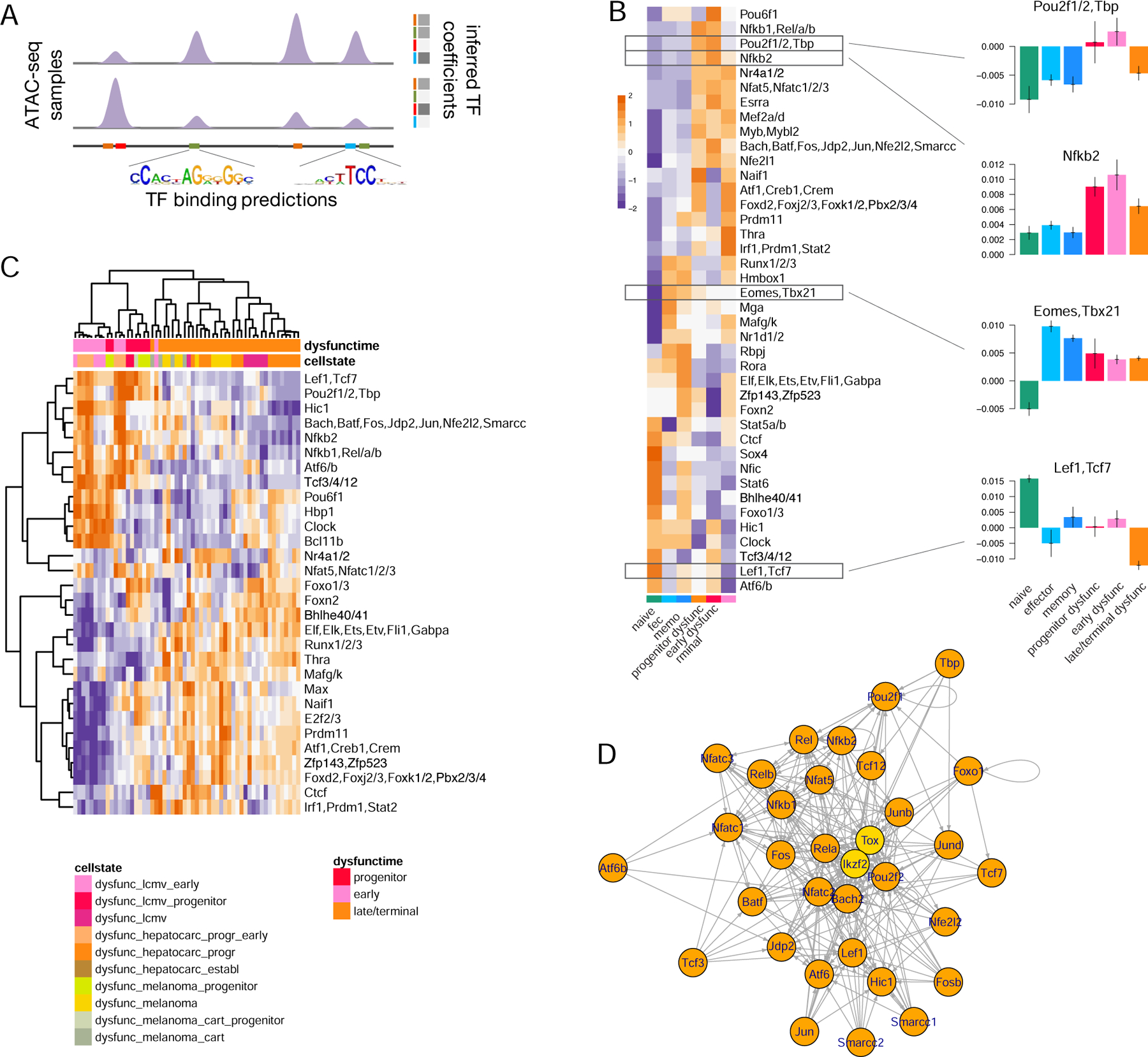
Figure 3. scATAC-seq analysis reveals heterogeneity and overlapping but divergent CD8 T cell responses to acute and chronic immune challenges.
A. Experimental setup. B. UMAP of TF-IDF-transformed scATAC-seq data. C. Louvain clustering of scATAC-seq data. D. Single-cell heatmap showing the naïve cell signature from bulk ATAC-seq data. E. Association by ssGSEA of batch-effect corrected bulk ATAC-seq data with normalized cluster-aggregated scATAC-seq signal in each of the two samples (z-score row normalized). F. Fraction of cells in each sample that belong to each cluster (restricted to clusters 1–8, 10, 13). G. Inferred TF motif coefficients (with the highest variance, z-score row normalized) for cluster-aggregated scATAC-seq signal (for clusters 1–8, 10, 13) in each of the two samples. H. Progenitor dysfunctional signature derived from bulk ATAC-seq data scored in scATAC-seq data for cells in clusters 1–8, 10, 13 (single-cell heatmap, top, violin plot for cluster-aggregated signal, bottom). I. Genome browser tracks for selected peaks. Bulk ATAC-seq for progenitor and terminally dysfunctional cells, and for terminal effector and memory precursor cells. Cluster-aggregated single-cell ATAC-seq for clusters 1–8, 10. J. Progenitor dysfunctional and memory precursor cell signatures derived from bulk ATAC-seq data scored in cells from cluster 10 in acute and chronic infection (Mann-Whitney U test). See also Figures S4, S5 and Table S4.
We first constructed a combined atlas of 189,281 chromatin accessibility peaks including both the bulk ATAC-seq data peak atlas and 59,482 newly identified scATAC-seq peaks (Table S4). Quantification and filtering yielded 4,767 and 5,865 cells, with 12,598 and 13,195 median reads per cell, initially isolated from Arm and Cl13 infected mice, respectively. Normalization using term frequency-inverse document frequency (TF-IDF), dimensionality reduction using PCA followed by uniform manifold approximation and projection (UMAP), and Louvain clustering (Methods) suggested that d7 responses to infection with the two LCMV clones were heterogeneous and overlapping (Figure 3B,C, Figure S4A,B).
To characterize functional features of d7 cell populations, we scored scATAC-seq data with epigenetic signatures derived from bulk ATAC-seq data (Figure 3D, Figure S4C,D, Methods) and used ssGSEA to associate scATAC-seq cell clusters with the bulk ATAC-seq data compendium (Figure 3E, Methods). We found that cells in clusters 0, 7, 9, 11, 12, 14 were likely a mixture of naïve and background-activated memory cells not specific to the LCMV antigen, while clusters 1–6, 8, 10, 13 likely consisted of LCMV responding cells. Some clusters showed no strong bias between LCMV clones, but we also found differences e.g. in clusters 1 and 6 (Figure 3E,F, Figure S4E), suggesting a clear divergence between responses to Arm and Cl13 as early as d7 post infection.
To validate and refine our bulk TF analyses (Figure 2, Figure 1D), we applied the same motif-based predictive modeling to the pseudo-bulk signal aggregated from each scATAC-seq cluster, separately for acute and chronic infection. This identified TFs most strongly associated with functional states in early response to acute or chronic infection (Figure 3G, Figure S4F).
Cells in cluster 10, when compared with bulk ATAC-seq data, were most similar to progenitor dysfunctional cells profiled at late chronic infection (Figure 3E,H, Methods), suggesting the presence of a progenitor-like state early in infection by Cl13 (Utzschneider et al., 2020). Surprisingly, cluster 10 also contained a small number of cells from acute infection that were similar to memory precursor cells (Joshi et al., 2007, Vodnala et al., 2019) based on comparison with bulk ATAC-seq data (Figure 3F,H, Figures S4E, S5A,B). Cluster 10 cells from acute and chronic infection displayed similar genome-wide chromatin accessibility profiles (and thus clustered together), TF activities via enrichment of Lef1/Tcf7, Nr4a, Nfkb, Nfat, Pou, and AP1 family motifs (Figure 3G), and accessibility at genes important for T cell activation and function, including progenitor marker Cxcr5 and dysfunction marker Tox (Figure 3I, Figure S5A-C), consistent with bulk ATAC-seq analysis (Figure 1D, Figure 2B,C). This shared cluster suggested that progenitor-like cells, by analogy to memory precursor cells, are established as early as at d7, as recently reported (Miller et al., 2019, Beltra et al., 2020, Utzschneider et al., 2020). Furthermore, employing epigenetic signatures from mice enabled a characterization of progenitor dysfunctional T cells in human tumors using scATAC-seq data (Satpathy et al., 2019) (Figure S5D-G, Methods). However, we also observed differential accessibility between the progenitor-like cells in chronic and acute infection (Figure 3J, Figure S5C) and potentially different TF activity (Figure 3G), suggesting an even earlier divergence between functional and dysfunctional fates.
By complementing our bulk ATAC-seq analysis across models, scATAC-seq analysis characterized the early divergence between CD8 T cell chromatin states in response to acute and chronic immune challenges, identified progenitor-like subpopulations in acute and chronic infection, and implicated cell-state specific TFs.
Single-cell transcriptional analysis confirms a progenitor/precursor T cell population in early response to both acute and chronic infection
To further explore the heterogeneity and temporal progression of CD8 T cell responses at single-cell resolution and to validate our state-specific TF predictions through allele-specific analyses, we performed single-cell RNA-seq analysis of CD8 T cells from hybrid mice generated upon breeding C57Bl/6J and SPRET/EiJ mice [F1(B6xSpret)] at three time points during acute and chronic LCMV infection (Figure 4A, Figure S6). This approach takes advantage of widespread natural genetic variation (~40 million SNPs) between the evolutionary distant parental genomes and assessing its effect on gene expression and chromatin accessibility. Virus-specific T cells were profiled before infection (d0) and upon activation at day 7 and day 40 (d40) during the acute LCMV infection and activated CD62L-negative cells were profiled at day 7 and day 35 (d35) during the chronic LCMV infection. Cells isolated from chronically infected B6 mice at d35 were used as a control. Bulk ATAC-seq profiling of CD8 T cells at d0, d7, and d60 upon acute LCMV infection in F1(B6xSpret) mice (van der Veeken et al., 2019) confirmed their consistency with the counterparts from B6 mice (Figure S7A).
Figure 4. scRNA-seq analysis uncovers phenotypic heterogeneity of CD8 T cell response and cell-state specific transcription factor expression.
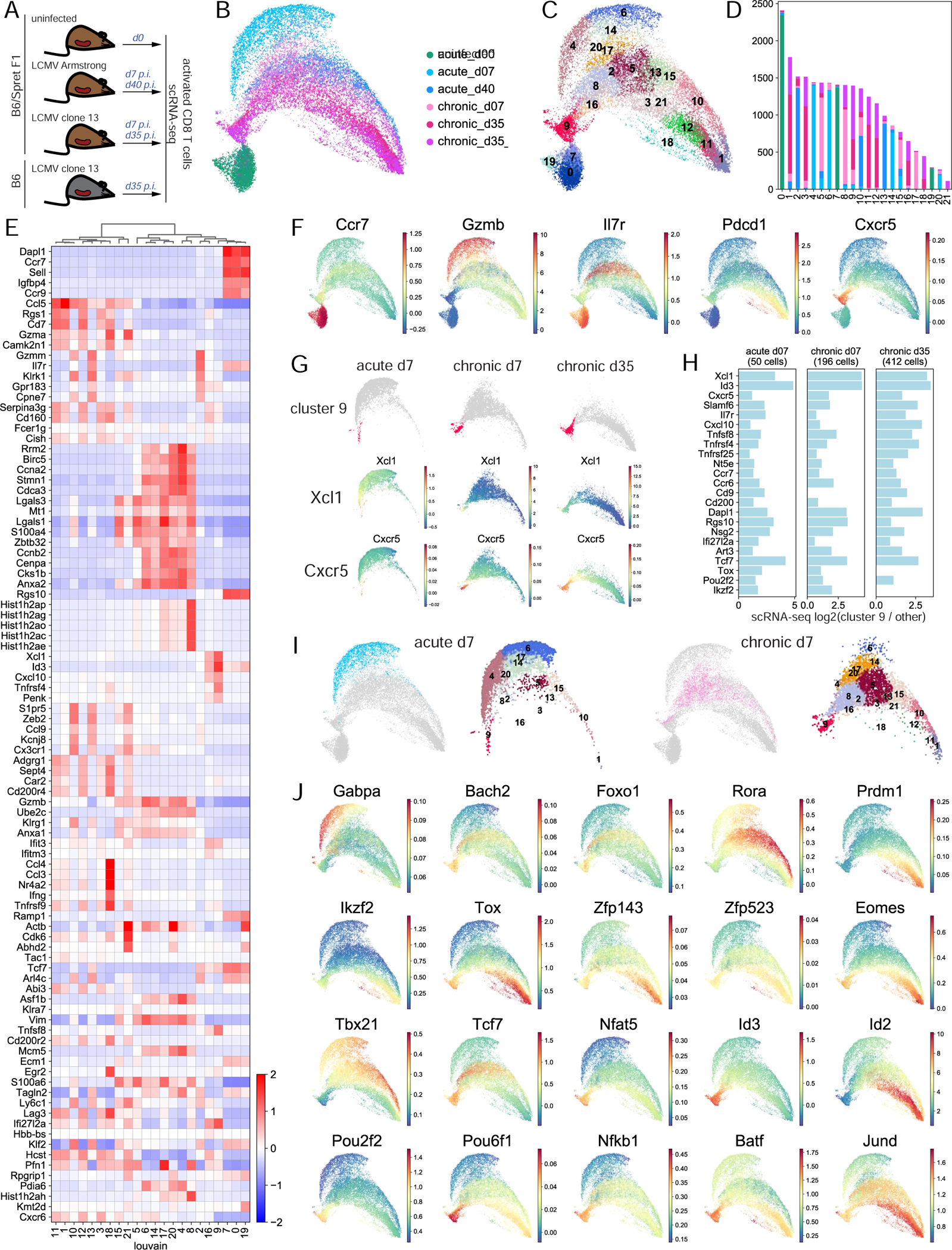
A. Experimental setup. B. UMAP of normalized scRNA-seq data. C. Louvain clustering of scRNA-seq data. D. Barplot of the number of cells in each cluster from each sample. E. Cluster-aggregated scRNA-seq gene expression in each cluster for differentially expressed genes between clusters (z-score row normalized). F. MAGIC-imputed gene expression for selected genes. G. MAGIC-imputed gene expression in individual samples. H. Log2 fold change of expression in individual samples of selected genes in cluster 9 cells vs. all other cells. I. Cells at d7 in acute and chronic infection separately and their cluster composition, within overall UMAP. J. MAGIC-imputed expression of selected genes encoding TFs. See also Figures S6-S11 and Table S5.
After scRNA-seq filtering steps (Methods), our data set consisted of 9,822 genes profiled in 24,400 cells, ranging from 3,419 to 4,824 cells per sample, with median 1,147 expressed genes per cell. UMAP embedding and Louvain clustering revealed a well-separated naïve state and diverging phenotypic arms in acute and chronic responses with heterogeneity within each sample and gradients of expression of well-known T cell function markers potentially reflecting differentiation trajectories (Figure 4B-D, Figure S7B,C). Cells at d35 in chronic infection from B6 and F1(B6xSpret) mice were highly similar (Figure 4B-D, Figure S7D,E). Dimension reduction using other well-known methods produced similar results (Figure S7F-H).
We used a combination of complementary approaches to functionalize the scRNA-seq data (Figure 4E,F, Figures S8, S9A, Table S5, Methods). Our analysis identified cell clusters encompassing archetypal CD8 T cell subsets including naïve T cells, memory precursor cells, short-lived effector cells, central memory, effector memory, terminally dysfunctional cells and their precursors. This cellular spectrum is similar to but more detailed than previously published scRNA-seq datasets of CD8 T cell responses in acute or chronic infection (Chen et al., 2019c, Miller et al., 2019, Yao et al., 2019, Kurd et al., 2020, Milner et al., 2020), showing that these cell states are consistently generated across independent studies and in mice of distinct genetic backgrounds.
Consistent with our scATAC-seq analysis (Figure 3), our scRNA-seq analysis identified a subpopulation of cells (cluster 9) (Figure 4G) resembling the previously characterized progenitor dysfunctional cells based on the enrichment of signature genes such as Id3, Tcf7, Slamf6, and Cxcr5 (Figure 4E-H, Figure S9B) and comparison with bulk RNA-seq from sorted subpopulations (Figure S9A). Many genes significantly more accessible and expressed in progenitor dysfunctional cells in bulk RNA-seq analysis and bulk and single-cell ATAC-seq analysis (Figure 1D, Figure 3I, Figure S5) were enriched in scRNA-seq cluster 9. Thus, cluster 9 consisted of progenitor and progenitor-like cells with a mixed gene expression profile resembling naïve, recently activated, dysfunctional, and memory cells.
Finally, we wanted to validate the similarity of progenitor-like cells in early chronic infection and memory precursor cells, their counterpart cells in acute infection, as observed in scATAC-seq analysis (Figure 3E-I, Figures S4E, S5A,B). Indeed, scRNA-seq cluster 9 contained cells from all non-naïve samples (Figure 4D, Figure S9C). Despite the limited resolution of differential expression analysis for small subsets of cells, many critical marker genes were consistently overexpressed in this cluster when considered independently in d7 acute, d7 chronic, or d35 chronic infection, including TFs and putative drivers of chromatin accessibility changes such as Tox, Pou2f2, and Ikzf2; the most overexpressed gene in this cluster, Xcl1; previously described progenitor markers Cxcr5 and Slamf6; and memory precursor marker Il7r (Figure 4G,H, Figure S9D). This again confirmed the emergence of progenitor-like cells early in chronic infection, together with a small number of similar cells in acute infection that clustered with progenitor dysfunctional cells, consistent with recent reports (Chen et al., 2019b, Utzschneider et al., 2020). We independently confirmed the presence of this CD8 T cell subpopulation at d7 in acute infection by reanalyzing three recently published scRNA-seq data sets (Yao et al., 2019, Kurd et al., 2020, Chen et al., 2019c) and verified enrichment of genes Tox, Ikzf2, Cxcr5 in this subpopulation (Figure S10). Trajectory analysis using RNA velocity, although noisy, suggested that progenitor cells arise from less differentiated cells that are only sparsely captured in our data set (Figure S11).
Thus, our scRNA-seq analysis provided a comprehensive atlas of CD8 T cell functional and dysfunctional states and confirmed the existence of progenitor/progenitor-like cell populations with similar transcriptional profiles in acute and chronic infection.
scRNA-seq characterizes overlapping but divergent CD8 T cell responses to acute and chronic viral infection and cell-state specific TF expression
We next sought to characterize the divergence of TF drivers of acute and chronic immune responses using our scRNA-seq data set. Consistent with our scATAC-seq analysis (Figure 3), cells were already highly heterogeneous and markedly different transcriptionally at d7 in acute and chronic infection (Figure 4I, Table S5). Thus, although acute infection predominantly drives CD8 T cell response along the effector cell trajectory, a small fraction of responding cells differentiate towards memory or even potentially along the dysfunctional trajectory. Conversely, chronic infection infrequently drives cells into an effector cell trajectory, consistent with previously reports (Chen et al., 2019c). Together, our observations suggest that both chronic and acute infection give rise to cells at similar functional, activation, and differentiation states, but these cells accumulate at different proportions depending on the challenge.
Cell-state specific enrichment of TF expression (Figure 4J, Figure S7C) was suggestive of cell state-specific function, consistent with observations from bulk and single-cell ATAC-seq data (Figure 1D, Figure 2B,C, Figure 3G). For example, we identified Gabpa as effector-specific, Rora as memory-specific, factors Tcf1, Nr4a1, Nfat5, Id3, Pou2f2, Pou6f1, Nfkb1, Batf as specific for progenitor dysfunctional state, and Zfp143, Zfp523 as specific for terminal dysfunction. Thus our scRNA-seq data supports the cell-state specific expression of TF drivers described in our bulk and single cell ATAC analyses.
Allele-specific analysis validates cell state-specific transcription factor activity
To complement our observations of cell-state specific TF activities from bulk and single-cell ATAC-seq and RNA-seq data analyses, we exploited our scRNA-seq profiling in F1 hybrid mice via allele-specific analysis of gene expression and TF binding, following and extending our previous studies (van der Veeken et al., 2019, van der Veeken et al., 2020). Indeed, we identified thousands of genes with significant allele-specific expression in many scRNA-seq clusters (Figure S12A), including many genes involved in T cell activation and function (Figure 5A). Most genes were consistently imbalanced in their expression towards B6 or Spret allele across clusters, while a few genes, including Gzmk or Fas, were significantly imbalanced in a cluster-dependent manner. Allelic imbalance of gene expression between B6 and Spret was correlated with differential expression between B6 and F1(B6xSpret) mice, supporting the accuracy of our allelic imbalance estimates (Figure S12B).
Figure 5. Allele-specific scRNA-seq analysis reveals cis-regulation of gene expression by transcription factors.
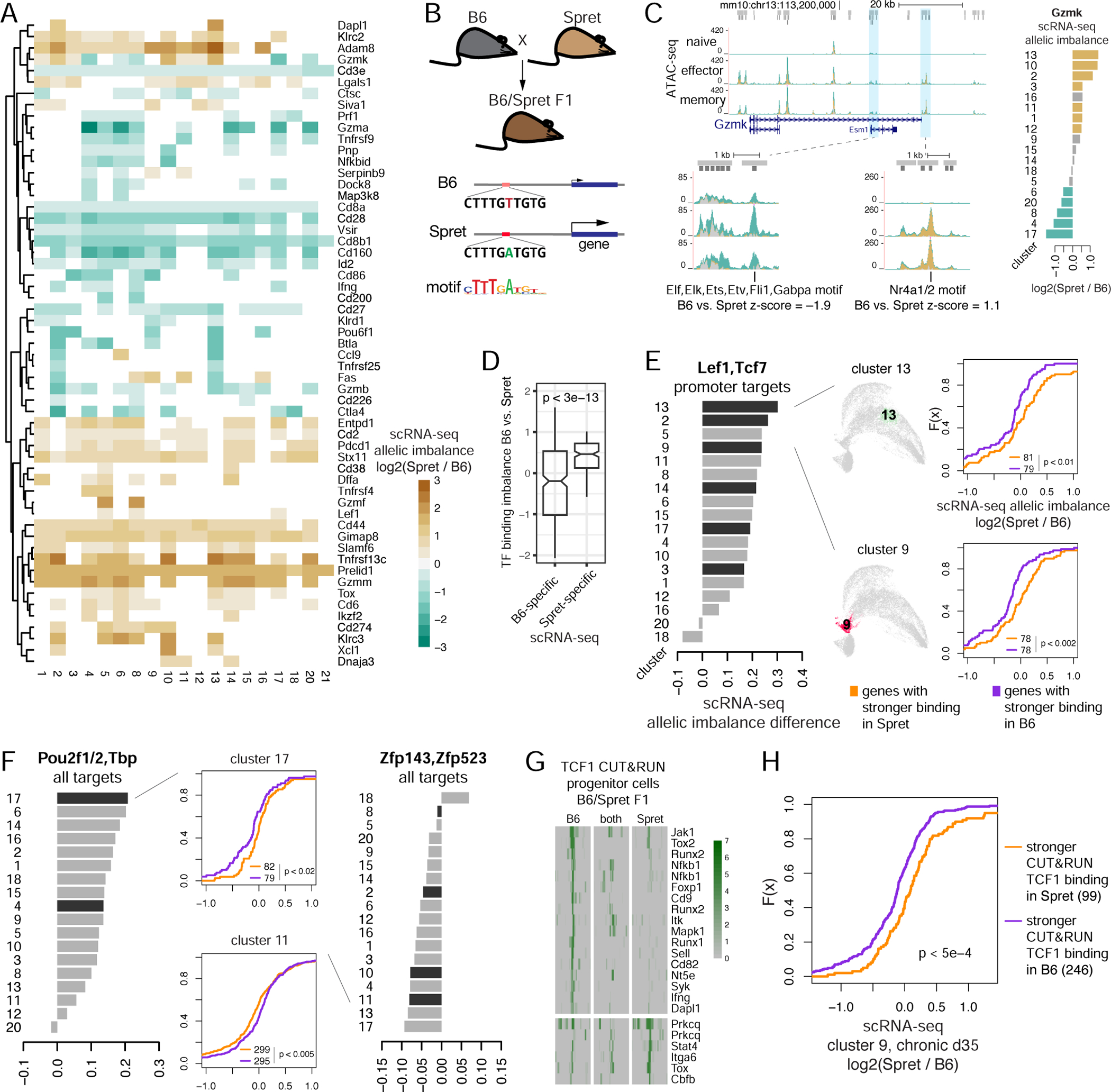
A. Allelic imbalance of gene expression in scRNA-seq data clusters (color, p < 0.05, Mann-Whitney U). B. Schematic for analysis of association between allelic imbalance of motif-based TF binding and gene expression. C. Left: ATAC-seq from B6/Spret F1 mice at Gzmk locus and examples of peaks with B6-specific (green), Spret-specific (brown) and ambiguous (gray) ATAC-seq signal and allele-specific predicted TF binding. Right: allelic specificity of Gzmk expression with significant B6-specific (green) and Spret-specific (brown) expression highlighted. D. Predicted allelic imbalance of TF binding (difference between TF motif log odds scores, z-score normalized) for genes with significant allele-specific expression in any of the scRNA-seq clusters (for genes with a single imbalanced motif). E-F. Allele-specific expression analysis of scRNA-seq data. CDF plots: allelic imbalance between B6 and Spret of normalized cluster-aggregated scRNA-seq signal, for genes predicted to be bound more strongly in B6 or Spret using sequence motif analysis in promoter peak summit regions. Barplots: summary of the above analysis for each TF motif over clusters (black bars, Kolmogorov-Smirnov p < 0.05). G. Examples of allele-specific TCF1 CUT&RUN binding sites in progenitor dysfunctional cells in the established chronic infection. H. Allelic imbalance between B6 and Spret of normalized scRNA-seq signal in cluster 9 at d35 in chronic infection, for genes bound by TCF1 more strongly in B6 or Spret as measured by CUT&RUN in progenitor dysfunctional cells. See also Figures S12, S13 and Table S6.
To gain evidence for the causal role of TFs in regulating gene expression, we looked for an association between allele-specific TF binding, as predicted by motif analysis, and allele-specific expression (Figure 5B,C). For each TF motif match in the B6 or Spret sequence around an ATAC-seq peak summit, we calculated the TF motif binding imbalance as the difference in motif log-odds scores between B6 and Spret, z-score-normalized. Many genes, e.g. Gzmk (Figure 5C), had many TFs with predicted binding imbalance at their promoter and enhancer ATAC-seq peaks. We expected that genes with only a few potential TF regulators would yield stronger association between allele-specific TF binding and allele-specific expression. Accordingly, when we restricted to genes with a single TF with predicted allele-specific binding, this allele-specific binding was concordant with allele-specific expression (Figure 5D). Considering genes with at most 20 ATAC-seq peaks, we found that imbalanced binding of certain TF motifs was significantly associated with gene expression imbalance. For example, genes with stronger TCF1/LEF1 promoter binding to the Spret allele were significantly more imbalanced in their expression towards Spret than genes with stronger binding of the same motif in B6 (Figure 5E). This suggests that TCF1/LEF1 binding at promoters is associated with activation of expression. Overall, this approach associated TF binding with activation or repression of gene expression for multiple factors (Figure 5F, Figure S12C-E).
TCF1 was reported as a critical marker of progenitor dysfunctional cells and is required for their generation during chronic infection (Im et al., 2016, Utzschneider et al., 2016). To experimentally validate that TCF1 binding is associated with activation of gene expression, we mapped 3,325 TCF1 bound sites by CUT&RUN analysis in progenitor dysfunctional cells from F1(B6xSpret) hybrid mice at d35 upon Cl13 infection (Figure S13A, Table S6). Direct TCF1 binding sites were strongly enriched for the TCF1 motif (Figure S13B) and included many T cell activation genes and progenitor markers (Figure S13C,D), including some targets with allele-specific TCF1 binding such as Tox, Tox2, Ifng, Nfkb1 (Figure 5G, Figure S13E). Consistent with previous observations and our predictions (Figure 2), accessibility levels at nearly all TCF1 targets were much higher in progenitor cells than in terminally dysfunctional cells (Figure S13F). Surprisingly, these accessibility changes were not associated with changes in expression of nearby genes (Figure S13F). However, allele-specific TCF1 binding was significantly associated with allele-specific expression of nearby genes in progenitor dysfunctional cells in established chronic infection (Figure 5H), consistent with a causal role for TCF1 in activation of gene expression. Furthermore, allele-specific motif enrichment analysis in CUT&RUN-defined TCF1 binding sites revealed TCF1 binding co-factors, such as Runx and Nfkb family factors (Figure S13G), consistent with allele-agnostic co-factor analysis (Figure S13B).
Thus our allele-specific analysis in the hybrid genome revealed cis-regulatory effects of TF binding on gene expression in CD8 T cells that are missed with standard approaches.
Progenitor cells differentiate to terminal dysfunction under acute viral challenge
Adoptive transfer experiments using sorted progenitor dysfunctional cells have demonstrated their potential to self-renew and give rise to terminally dysfunctional cells in models of melanoma and chronic infection (Miller et al., 2019, Im et al., 2016, Beltra et al., 2020). Since single cell chromatin and expression analyses showed that dysfunctional progenitors resemble progenitor-like cells at the plastic stage of T cell dysfunction development as well as the MPEC population in acute infection, we asked whether progenitor cells retain the potential to give rise to effector cells. We therefore transferred FACS sorted progenitor dysfunctional cells isolated from chronically infected mice on d35 post infection into congenically marked recipients infected with LCMV Arm (Figure 6A,B, Methods). The transferred cells were profiled at d7 post infection using flow cytometry and scRNA-seq. Flow cytometry revealed that the transferred CD8 T cells proliferated and persisted (Figure 6C,D). Furthermore, scRNA-seq analysis showed that these cells exhibited heterogeneous gene expression states (Figure 6E-I). Despite the acute infection setting, the phenotypic space defined by transferred cells after expansion most strongly overlapped with that of cells in chronic infection settings. Clustering analysis of the combined scRNA-seq data (Figure 6F,G) confirmed that the expanded transferred cells were most similar in cluster composition and cluster-wise differential expression to cells at d7 in chronic infection (Figure 6I, Table S5). The expanded population lacked effector cells (clusters t3, t10, t11, Figure 6F,I) suggesting the transferred population did not contain naïve or memory cells. Importantly, progenitor cells persisted upon transfer, confirming their self-renewal capacity (cluster t4, Figure 6H). Consistent with a recent report (Utzschneider et al., 2020), our single cell analyses suggest that the progenitor cells observed in chronic infection (d35) were already committed to a dysfunctional fate.
Figure 6. Progenitor CD8 T cells from chronic infection transferred to acute infection are committed to dysfunction.
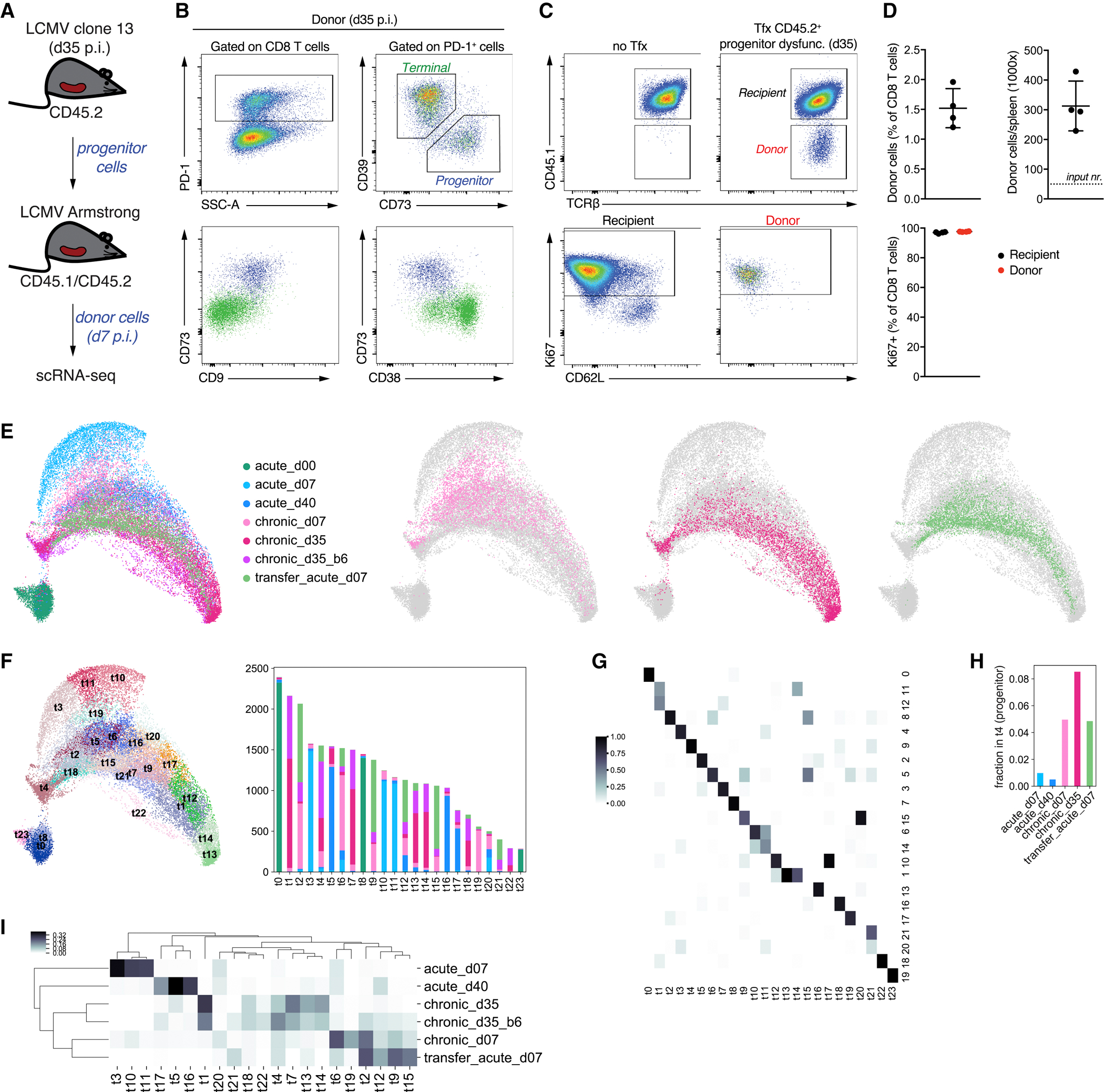
A. Setup for the adoptive cell transfer experiment. B-C. Flow cytometry of selected genes for progenitor cells before transfer (B) and after transfer and expansion (C) in acute infection. D. Quantification of flow cytometry results for donor cells after transfer and expansion. E. Left: UMAP of all scRNA-seq data including the expanded donor cells; right: UMAP showing individual samples within the overall map. F. New Louvain clustering of all scRNA-seq data, with the barplot showing the number of cells in each cluster from each sample. G. Comparison of new clustering with the previous one (Figure 4C). Shown is a fraction of cells in clusters t0–t23 that belong to previously obtained clusters 0–21. H. Fraction of cells in each sample that belong to the progenitor cluster t4. I. Cluster composition of samples. Heatmap showing for each cluster what fraction of cells (excluding naïve) that cluster occupies in each sample. See also Table S5.
DISCUSSION
Our unified analysis of bulk ATAC-seq and RNA-seq data across diverse studies of T cell response to chronic antigen exposure defined a universal program of progression to terminal dysfunction. By joint analysis of data from numerous resources, we gained statistical power to robustly identify individual chromatin accessible sites, patterns of accessibility and expression changes common across experimental models, and TFs whose binding motifs explain global accessibility patterns in distinct chromatin states in T cell differentiation.
Given the universality of T cell dysfunction program across mouse models, we then focused on the LCMV infection as a model to elucidate the early divergence in chromatin and transcriptional states between dysfunctional and functional T cell responses at single cell resolution using scATAC-seq and scRNA-seq. We found that a subset of antigen-specific T cells in chronic LCMV infection are appropriately activated and become effectors at day 7, while most progress along a dysfunctional trajectory. Conversely, a small number of cells in acute LCMV infection had progressed to a differentiated effector memory state, and potentially also to a dysfunctional state by day 7. These findings, consistent with previous smaller-scale scRNA-seq studies of functional or dysfunctional T cells, enable a more nuanced comparison of acute vs. chronic immune responses, where T cells can commit to multiple fates at differing frequencies.
In our single-cell analyses we found a TCF1+ progenitor-like cell population emerging as early as day 7 post infection in both chronic and acute infection. This population formed the shared cluster across the two infection settings in both scATAC-seq and scRNA-seq data and therefore displayed the strongest transcriptional and epigenomic similarity among all the cells profiled, in contrast with recent analysis of sorted populations (Utzschneider et al., 2020). Accordingly, in both settings we found the same genes enriched in expression and chromatin accessibility at multiple promoter and enhancer peaks in this population, including Tcf7, Xcl1, Cxcl10, Ccr7, Slamf6, Id3, and Cxcr5, but also Tox and Ikzf2, which had previously been associated with terminally dysfunctional T cells. Notably, multiple lines of evidence implicated several TFs as strongly associated with the progenitor and progenitor-like population (Figures 1D, 2B,C, 3G, 4J, 5E,F, Figures S5B, S9B), including the previously described TCF1 and the newly identified OCT2 (encoded by Pou2f2). However, differential accessibility analysis and motif regression modeling, despite being supported by analysis of relatively small cell numbers, identified subtle differences between cells from chronic and acute samples in this progenitor-like subpopulation, with cells from acute infection potentially representing an IL7Rhi MPEC subset and those from chronic infection forming a nascent progenitor reservoir (Figure 3G,J, Figure S5C). These findings may suggest an earlier divergence between progenitor and MPEC populations at the level of chromatin state and gene expression.
The presence of cells from both chronic and acute viral infection in the scRNA-seq cluster containing TCF1+ PD1+ progenitor cells and in the scATAC-seq progenitor-like cluster led us to test the plasticity of the progenitor cell population. Using an adoptive cell transfer experiment together with scRNA-seq profiling and mapping these data to the comprehensive scRNA-seq atlas of CD8 T cell responses that we constructed, we confirmed that progenitor cells isolated from an established chronic viral infection were committed to a dysfunctional fate. While transferred progenitor cells could proliferate in a new host under acute immune challenge, they displayed the single cell phenotypic profile of a chronic immune response. These results are consistent with earlier adoptive T cell transfer studies showing that by day 15 post antigen encounter, T cells that had responded to chronic LCMV infection lost their ability to form functional memory under acute LCMV challenge (Angelosanto et al., 2012) and with similar experiments documenting loss of plasticity for tumor-specific T cells in an autochthonous liver cancer model (Schietinger et al., 2016). However, these earlier studies also confirmed that at early time points in settings of chronic antigen stimulation, antigen-specific T cells – or at least a subpopulation of these cells – retained the potential to mount a functional response and form memory cells. Our single cell analyses of T cells under chronic immune challenge suggest this transient plasticity resides within the early progenitor-like population, consistent with recent phenotypic studies (Utzschneider et al., 2020).
Trajectory analysis using RNA velocity remains noisy. Further studies of the origins and regulatory mechanisms of maintenance and differentiation potential of the progenitor and progenitor-like cell populations are warranted, for example using heritable bar codes for tracking cell fates within a TCR clonal population over time (Wagner and Klein, 2020) and at time points earlier than d7 upon T cell activation. Performing these studies in F1 hybrid mice can further help interrogate regulatory mechanisms of gene expression by linking allele-specific analysis of TF motif occurrences (or allele-specific TF occupancy) to allelic imbalance of target gene expression.
In conclusion, our data sets and analyses lay the groundwork for resolving the fundamental questions of T cell differentiation programs. We expect that our unified T cell atlas will provide a valuable resource both for basic T cell biologists and for cancer immunologists seeking to therapeutically target the transcriptional programs underlying progression to fixed dysfunction.
Limitations of the Study
Sorted populations subjected to bulk profiling are still a mixture of the purer subpopulations we characterized with single-cell analysis, complicating interpretation of the bulk analysis. While our batch effect correction succeeded in finding strong similarities and some differences between models, generating all data in a controlled manner within the same lab remains the gold standard for differential analyses. We limited our single-cell analyses to the LCVM model to enable comparison of acute and chronic responses; however, similar single-cell analyses in a tumor model would directly address whether the underlying distribution over T cell states differs in cancer vs. chronic infection. T cell differentiation is a particularly difficult setting for computational trajectory analysis, with subpopulations that rapidly expand or collapse and where overall differences between functional states can be subtle. While we provide results of RNA velocity analysis, we believe that direct experimental determination with cell tagging approaches will be needed to resolve differentiation trajectories.
STAR Methods
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Christina Leslie (cleslie@cbio.mskcc.org).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The code for computational analysis and additional data visualizations are available at https://bitbucket.org/leslielab/cd8t_atlas. New scATAC-seq, scRNA-seq and CUT&RUN data are available at NCBI GEO under the accession number GSE164978. Previously published datasets reanalyzed in this study are listed in Table S1.
Experimental model and subject details
Mice
Animals were housed at the Memorial Sloan Kettering Cancer Center (MSKCC) animal facility under specific pathogen free (SPF) conditions on a 12-hour light/dark cycle under ambient conditions with free access to food and water. All studies were performed under protocol 08–10-023 and approved by the MSKCC Institutional Animal Care and Use Committee. Mice used in this study had no previous history of experimentation or exposure to drugs. Male Spret/EiJ mice were purchased from Jackson laboratory and bred to female CD45.1 mice on a C57BL/6 genetic background. Adult (at least 8 weeks old) male and female F1 offspring were used for experiments as indicated.
Method details
Construction of ATAC-seq peak atlas
ATAC-seq data from multiple previous studies were downloaded from GEO using fastq-dump from NCBI SRA Toolkit (https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=toolkit_doc). Functional annotation of samples (naïve, effector, memory, dysfunctional, etc.) was obtained from each study (Table S1). Reads were aligned to the mouse genome mm10.GRCm38 using bowtie2 v2.3.4.3 (Langmead and Salzberg, 2012). Uniquely aligned reads were extracted using SAMtools v1.9 (Li et al., 2009). Peaks were called using MACS2 v2.1.1.20160309 (Zhang et al., 2008). For each data source, peaks were called using all samples from all replicates combined. Then IDR (Li et al., 2011) was used to identify reproducible peaks between at least some pair of replicates of the same condition. The union of all such peaks formed peak library for each study. Then peaks identified in each study were all combined into a common list, by taking the union of all genomic positions covered by peaks from different studies using function reduce(c()) in R (R v3.4.0 was used in all our analysis except when otherwise noted). Peaks were further split into subpeaks around summits of signal, using a custom script based on the signal processing package https://bitbucket.org/leslielab/biosignals. Peak summit regions were then filtered based on ATAC-seq read coverage of 150bp regions centered at the summits, by including only the peak summit regions with at least 100 reads in at least one sample and at least 5 reads on average across all samples within any of the three categories (naïve, functional, and dysfunctional). Peaks that included at least one of such summit regions were included in the final atlas. This resulted in an atlas of 129799 chromatin accessibility peaks for CD8 T cells. Peaks were annotated using GENCODE vM14 (Frankish et al., 2019). Each peak was associated with the closest gene, if this gene was within 50Kb in genomic coordinates. Peaks were annotated by applying the following sequence of rules: peaks were classified as promoter peaks if within 2Kb from a transcription start site of any annotated transcript; otherwise as exonic if overlapping with any exon of any annotated transcript; otherwise as intronic if within a gene body of any annotated gene; or finally as intergenic if within 50Kb of a gene. All the remaining peaks were left unclassified. Reads from each sample were counted in peaks and 150bp genomic regions centered around peak summits using Rsubread v1.32.4 (Liao et al., 2019). For visualization, bigWig files and count matrices in bins around peak summits were produced using deepTools (Ramirez et al., 2016). Snapshots of ATAC-seq signal at selected genomic regions were obtained from UCSC Genome Browser (Kent et al., 2002).
Batch effect correction for ATAC-seq
DESeq2 v1.22.2 (Love et al., 2014) was used to fit multi-factorial models to ATAC-seq read counts in peaks or peak summits. The main model included two factors where one factor represented the source of the data, and the other factor represented the functional annotation of the sample, broadly defined as naïve, functional, and dysfunctional. For PCA analysis and visualization, batch effect-corrected values were used. To do the batch effect correction, log2FC values associated with the data source factor were extracted from the model for all peaks and used to correct the original counts, by dividing them by the exponentiated log2FC values.
The alternative variant of the model included a two-valued functional annotation factor, where one value was for naïve samples, and the other for all antigen-experienced cells, including all functional and dysfunctional cells. This alternative variant of the model produced results similar to the main model (Figure S1C).
In order to determine whether memory precursor cell samples and tissue-resident memory cell samples were similar to functional or dysfunctional cells, a variant of this alternative model with a two-valued functional annotation factor was used. For this, differentially accessible peaks were detected between functional and dysfunctional cells but excluding memory precursor and tissue-resident memory cell samples. Then PCA analysis for all samples was run on ATAC-seq read counts restricted to these differentially accessible peaks, revealing that memory precursor and tissue-resident memory cells are more similar to other functional effector and memory cells.
Differential accessibility analysis
DESeq2 v1.22.2 was used to perform differential accessibility analysis between selected pairs of conditions. For this, original counts were used, and the model included factors for functional annotation, cell state, data source, and/or type of immune challenge, where appropriate. For each gene, overall differential accessibility of its peaks between a certain pair of cell states was assessed as follows. A Mann-Whitney U test was used to compare the vector of all peak accessibility log2 fold changes between these two cell states with the vector of log2 fold changes only for the peaks associated with the gene. Then the resulting p-values (one for each gene) were adjusted for multiple hypothesis testing (multiple tested genes) using q-value. This analysis was visualized with a scatter plot where the x-axis shows the log2 fold change of gene expression, the y-axis shows mean accessibility log2 fold change of the peaks associated with a gene, black indicates significant differential expression (FDR < 0.05), and red and blue indicate significant overall differential accessibility of peaks associated with a gene (q < 0.01), only for significantly differentially expressed genes. For functional analysis, most significantly differentially accessible genes (q < 0.01) were selected, split into those with positive and negative average differential accessibility across gene’s peaks, and functional enrichment assessed for these genes using gprofiler2 with the background of all expressed genes (Raudvere et al., 2019). Genes significantly more accessible in terminally dysfunctional cells in chronic infection as compared with terminally dysfunctional cells in the tumors (Figures S2D) were most significantly enriched for GO terms such as “immune system process” (rank 1, p < 3e-19) and “T cell activation” (rank 4, p < 6e-14). Genes significantly more accessible in endogenous than in CAR-T dysfunctional cells (Figures S2D) were often associated with more differentiated dysfunction as evidenced by higher accessibility at loci of important dysfunction marker genes (Tox, Havcr2, Entpd1, Tigit).
Transcription factor motif analysis
Transcription factor (TF) binding motifs for Mus musculus were downloaded from CIS-BP version 1.02 (Weirauch et al., 2014) via the web interface (compressed archive Mus_musculus_2016_06_01_2–46_pm.zip). For the DNA sequences in 150bp-wide regions around peak summits, script findMotifsGenome.pl from HOMER suite (Heinz et al., 2010) was run with parameters ‘‘mm10 -len 8,10,12 -size given -S 100 -N 1000000 -bits -p 10 -cache 1000’’ in order to identify the significance of presence of each motif in the sequences of the peaks as compared with the background sequences. We limited the analysis to motifs corresponding to expressed TFs, defined as those with at least 200 library-size normalized RNA-seq reads in at least one condition in at least one study. We focused only on the motifs present in at most 50% of the peak summit region sequences. Furthermore, we detected de novo motifs using HOMER in the same sequences and associated them with TFs by similarity with the CIS-BP motifs using script compareMotifs.pl with parameters “-reduceThresh 0.7 -matchThresh 0.9”. The most significant motif per TF, either from the database or identified de novo, was selected for further analysis (with potentially multiple TFs associated with the same motif), if it had HOMER p-value < 0.001. This resulted in 113 motifs. Furthermore, we merged motifs that had correlation of motif occurrences in the peak summits above 0.75. This resulted in the list of 105 motifs corresponding to 204 TFs for further analysis. FIMO version 4.11.2 (Grant et al., 2011) was used to search for motif matches in 150bp regions around the peak summits. Matches with p-value < 1e–3 were chosen as significant.
Predictive modeling of transcription factor activity
To infer cell-state specific TF activities, we performed a supervised modeling of chromatin accessibility data based on TF motif occurrences. We formed a feature matrix that consisted of TF motif match predictions in regions around peak summits. Each value in the matrix was a sum of FIMO log-odds scores of a TF motif occurrence in a peak summit region. For each ATAC-seq sample, we performed negative binomial generalized linear regression modeling of the batch-effect corrected chromatin accessibility values y in peak summit regions using this feature matrix, with ridge regularization, using function cv.glmreg() from R package mpath (Wang et al., 2014) with parameters family = “negbin”, alpha = 0, theta = 1, nfolds = 5, maxit = 20000, thresh = 1e-5. We limited the regression analysis to peak summit regions with at least 10 batch-effect corrected reads on average across all samples. We identified a hyperparameter multiplier a of the ridge regularization penalty term using 5-fold cross-validation; for this we ran the regression with 30 values of a formed by multiplying the mean of y by a vector of values from 10−8 to 103 (with equidistant log10 values) and choosing the value of a that maximizes the Spearman correlation between the observed and predicted values of y (Figure S3B). Then we used the coefficients of this regression as a proxy to TF activity scores and used them for downstream analysis; for this, we limited our downstream analysis to results of regression only for those ATAC-seq samples where the fit converged for at least 4 values of the hyperparameter a.
RNA-seq data analysis
RNA-seq data from multiple previous studies were downloaded from GEO using fastq-dump. Functional annotation of samples (naïve, effector, memory, dysfunctional, etc.) was obtained from each study. Reads were aligned to the mouse genome mm10.GRCm38 using hisat2 v2.1.0 (Kim et al., 2019). Reads from each sample were counted in genes annotated by GENCODE vM14 using Rsubread v1.32.4. Batch effect correction and differential expression analysis were performed the same as for ATAC-seq data.
Comparison of ATAC-seq and RNA-seq using Hi-C contacts
Published Hi-C data from naïve CD8 T cells (He et al., 2016) was used to define associations of ATAC-seq peaks to genes and compare it with our default peak-to-gene association (see Construction of ATAC-seq peak atlas), using a computational experiment comparing ATAC-seq and RNA-seq data. HiC read pairs were processed using HiC-Pro pipeline (version 2.11.1) (Servant et al., 2015). Reads were trimmed to 30nt and mapped to the mm10 reference genome. We used valid pairs output in cis from HiC-Pro that were mapped uniquely to the genome. Valid pairs from replicates were merged and binned into 10kb bins. HiC-DC (Carty et al., 2017) was used to assign statistical significance to each interaction bin. Significant chromatin loops between 10Kb bins were defined at q-value < 0.05 and with support by more than 20 Hi-C valid pairs, resulting in 22,337 significant loops. Median distance between loop ends was 180Kb, and only 5% of loops had distance between ends shorter than 50Kb. For control, non-significant loops were defined as those with q-value between 0.1 and 0.2 and supported by not more than 10 Hi-C valid pairs, resulting in 118,497 loops. Peaks were associated to genes using these loops as follows. For a loop between 10Kb-long genomic regions A and B, a peak was associated to a gene if the peak was within 5Kb window from A and another peak at a promoter of the gene was within 5Kb from B. Then differential gene expression (RNA-seq log2FC) between naïve and effector cells was compared between genes associated with at least one significantly more accessible peak in naïve than in effector cells and in effector than in naïve cells (FDR < 0.05), for different methods of association of peaks to genes: default associations, associations based on significant loops, and associations based on non-significant loops (Figure S3D). The same analysis was performed for comparison between naïve and dysfunctional cells (Figure S3D).
Single-cell ATAC-seq experiments
Four male C57BL/6J mice per group were infected with either LCMV Armstrong (2×105 p.f.u. via intraperitoneal injection) or LCMV Clone 13 (2×106 p.f.u. via retroorbital injection). On day 7 post-infection, total CD8 T cells from pooled spleens were double sorted by flow cytometry and prepared for single cell ATAC-seq analysis.
scATAC-seq was performed using Chromium instrument (10x Genomics) and Single Cell ATAC Reagent Kits (Chemistry v1). The suspension of cells was processed following the User Guide (CG000168 Rev A). Briefly, cells were lysed in bulk, washed and resulting nuclei suspension treated with ATAC reagents provided in the kit. Approximately, 8000 nuclei per sample were loaded onto microfluidics Chip E and encapsulated with DNA barcodes and reaction mix. Following emulsion-PCR the resulting material was purified and subjected to 12-cycles of indexing PCR. The indexed scATAC-Seq libraries were double-size selected using SPRI beads and sequenced on an Illumina NovaSeq 6000 instrument (Read 1 – 50 cycles, i7 index – 8 cycles, i5 index – 16 cycles, and Read 2 – 50 cycles) at 180M reads per sample.
Single-cell ATAC-seq data preprocessing, dimensionality reduction, clustering
The data was preprocessed with cellranger-atac from 10X. Then using the BAM files with read alignments for the two samples produced by cellranger-atac, peaks were detected using MACS2. An extended list of peaks was formed by combining the previously constructed bulk ATAC-seq peaks with the non-overlapping newly identified peaks. The cellranger-atac tool was then rerun with this extended peak list as input to obtain a count matrix for each of the two samples. Cells with a log library size less than 3.5 and peaks that were accessible in less than 4 cells were excluded from the analysis. The count matrix was then binarized and transformed using term frequency-inverse document frequency (TF-IDF, scikit-learn package v0.20.3, https://scikit-learn.org/) for normalization, followed by PCA and UMAP for dimensionality reduction and visualization. Cells were clustered by the Louvain clustering method.
Comparison of scATAC-seq with bulk ATAC-seq data compendium
Signature peak sets for progenitor, naïve, effector, MPEC, and terminal dysfunction were derived from bulk ATAC-seq analysis (Table S2) and then used to score cells by taking the average normalized counts of the peak set and subtracting the average normalized counts of a reference peak set. The naïve cell signature was defined as peaks significantly more accessible in naïve cells in naïve vs. functional and naïve vs. dysfunctional cell comparisons (using thresholds baseMean > 50, FDR < 0.001, log2 fold change < −2 for both comparisons). The effector cell signature was defined as peaks significantly more accessible in effector cells in naïve vs. effector (baseMean > 50, FDR < 0.01, log2 fold change > 1), effector vs. memory (baseMean > 10, FDR < 0.05, log2 fold change < 0), and effector vs. early dysfunctional (baseMean > 10, FDR < 0.05, log2 fold change < 0) cell comparisons. The memory cell signature was defined as peaks significantly more accessible in memory cells in effector vs. memory (baseMean > 50, FDR < 0.001, log2 fold change > 0.5) and memory vs. terminally dysfunctional (baseMean > 50, FDR < 0.001, log2 fold change < −2) cell comparisons. The MPEC signature was defined as peaks significantly more accessible in MPEC vs. effector cells (baseMean > 50, FDR < 0.001, log2 fold change > 1). The progenitor dysfunctional cell signature was defined as peaks significantly more accessible in progenitor vs. terminally dysfunctional cells (baseMean > 100, FDR < 0.001, log2 fold change < −2). The terminally dysfunctional cell signature was defined as peaks significantly more accessible in dysfunctional cells in naïve vs. dysfunctional (baseMean > 10, FDR < 0.05, log2 fold change > 0), memory vs. terminally dysfunctional (baseMean > 10, FDR < 0.05, log2 fold change > 0), progenitor vs. terminally dysfunctional (baseMean > 50, FDR < 0.01, log2 fold change > 1), effector vs. early dysfunctional (log2 fold change > 0) cell comparisons. Analysis and visualizations were performed using python package scanpy v1.4.4 (Wolf et al., 2018).
BAM files were generated with aligned scATAC-seq reads corresponding to cells from each cluster, further split between the two samples. Pseudobulk counts were obtained from these BAM files for the extended peak list using Rsubread v1.32.4. Limited only to clusters 1–6, 8, 10, 13 that contained activated virus-specific cells, differential accessibility analysis was then run using these pseudobulk counts for each of the clusters, within each of the two samples, vs. all the remaining clusters split between the two samples. The most significantly differentially open peaks formed a signature set of peaks for each cluster split between the two samples. Then ssGSEA (Barbie et al., 2009) was run for these signatures against batch-effect corrected library-size normalized bulk ATAC-seq counts using the package GSVA v1.30.0 (Hanzelmann et al., 2013).
Isolation of cells for scRNA-seq
For isolation of CD8 T cells responding to acute infection, B6/Spret F1 mice, where B6 stands for C57BL/6J and Spret stands for SPRET/EiJ, were infected with 2×105 p.f.u. of LCMV Armstrong via intraperitoneal injection or left uninfected. Splenocytes were stained with a cocktail of fluorescent antibodies, NP396 tetramer, and viability dye to mark dead cells, and sorted using flow cytometry. Naïve CD8 T cells were isolated from pooled spleens of 2 uninfected mice as CD44-CD62L+ CD8 T cells. Activated and memory effector cells were isolated as NP396+ CD8 T cells from pooled splenocytes of 2 or 3 mice on day 7 or day 40 post-infection, respectively and used as input for scRNA-seq analysis.
For isolation of cells responding to chronic infection, B6/Spret F1 or B6 mice were infected with LCMV clone 13 by retroorbital injection of 4×106 p.f.u. of virus. Mice were depleted of CD4 T cells by injection of 200ug aCD4 antibody (BioXCell cat: BE0003–1) one day prior to and one day post-infection. Cells were isolated as live CD62L- CD8 T cells from spleens of 3 pooled mice per group and used as input for scRNA-seq analysis.
Adoptive transfer of progenitor dysfunctional cells
Four C57BL/6J mice were infected with LCMV Clone 13 (2×106 p.f.u. via retroorbital injection). On day 35 post-infection, splenocytes were pooled and stained using a cocktail of fluorescently labeled antibodies and Ghostdye Violet 510 (Tonbo cat: 13–0870-T100) to distinguish dead cells. Dysfunctional progenitor cells were isolated by flow cytometry-based sorting as PD1+, CD73+, CD39- CD8 T cells. This population was also CD9+ and CD38 low. 50,000 dysfunctional precursor cells/recipient were transferred into 4 CD45.1/CD45.2 mice via retroorbital injection. Recipients were infected with LCMV Armstrong (2×105 p.f.u. via intraperitoneal injection). On day 7 post-infection donor cells were re-isolated based on expression of congenic markers and prepared for scRNA-seq.
Single-cell barcoding, library preparation and sequencing
Single-cell suspensions were loaded on 10x Genomics Chromium instrument and encapsulated in microfluidic droplets with barcoded DNA hydrogel beads and RT reagents from Chromium Single Cell 3’ Reagent kit (v3). The cDNA synthesis/barcoding was performed following manufacturer’s instructions: 53°C for 45 min followed by heat inactivation at 85°C for 5 min. The barcoded-cDNA was purified and PCR-amplified and prepared for sequencing according to the Single Cell 3′Reagent kit 3 User Guide (CG000183; Rev B). The DNA sequencing was performed on the Illumina NovaSeq 6000 instrument (R1 read – 26 cycles, R2 read – 70 cycles or higher, and index read – 8 cycles), aiming for ~200 million reads per ~5,000 single cells.
Single-cell RNA-seq data preprocessing, dimensionality reduction, clustering
We analyzed scRNA-seq data from the six samples labeled “acute_d00”, “acute_d07”, “acute_d60”, “chronic_d07”, “chronic_d35”, “chronic_d35_b6”. Reads were aligned to the combined B6 and Spret genome using hisat2 v2.1.0. For this, files Mus_musculus.GRCm38.dna.toplevel.fa.gz and Mus_spretus_spreteij.SPRET_EiJ_v1.dna.toplevel.fa.gz with genomic sequences of B6 and Spret mice, respectively, were obtained from NCBI FTP server, and hisat2 index for the chromosomes from both files was constructed. Using the BAM files of scRNA-seq read alignment, UMI counts for each gene in B6 and Spret genomes were obtained using a custom script, by overlapping read alignments with exonic annotations of genes in either B6 or Spret and counting UMI corrected using the method from seqc (Azizi et al., 2018). For this, files Mus_musculus.GRCm38.91.gtf.gz and Mus_spretus_spreteij.SPRET_EiJ_v1.86.gtf.gz with gene annotations for B6 and Spret genomes, respectively, were obtained from NCBI FTP server. The resulting read count tables were used for downstream analysis in scanpy v1.3.7. Cells with less than 100 genes with positive counts or cells with more than 40000 total positive UMI counts were filtered out, and genes with less than 500 positive counts across cells from all samples were filtered out. Cells with high expression of B cell genes (likely contamination) were filtered out (15 cells). Only protein-coding genes were included in the analysis, and furthermore ribosomal genes were excluded from the analysis. Based on inspection of the distribution of the number of genes detected per cell in each sample, cells from sample “acute_d00” with log10 gene count less than 2.85, from sample “acute_d07” with log10 gene count less than 2.9, from sample “acute_d60” with log10 gene count less than 2.9, from sample “chronic_d07” with log10 gene count less than 2.9, from sample “chronic_d35” with log10 gene count less than 2.8, from sample “chronic_d35_b6” with log10 gene count less than 2.8 were excluded from the analysis. Then the count matrix was filtered again to include only cells with at least 500 genes and genes with at least 100 counts. This resulted in a read count table of 9822 genes in 24400 cells across the six samples.
Counts for 9124 genes present in both B6 and Spret annotations were used for normalization, dimensionality reduction, and clustering. For normalization, the total count in each cell was calculated, excluding the top 50 genes with the highest total count across all cells, and then each count divided by the total count per cell and multiplied by the median of total counts per cell. For dimensionality reduction and visualization, principal component analysis (PCA) was run for the normalized counts, the first 50 principal components (PCs) were selected, and the kNN (nearest neighbor) graph was built for k = 50 nearest neighbors per cell using Euclidian metric. The kNN graph was then used to construct three-dimensional uniform manifold approximation and projection (UMAP) with default parameters. The data was also visualized with t-distributed stochastic neighbor embedding (tSNE) using function “TSNE” from package sklearn.manifold with perplexity = 150 and otherwise default parameters applied to the first 50 PCs of normalized count matrix; with diffusion maps and force-directed atlas applied to the kNN graph. Louvain clustering was then applied to the kNN graph with resolution = 1.9 and otherwise default parameters. For visualization of gene expression, imputation algorithm MAGIC (Van Dijk et al., 2018) was applied to the normalized count matrix using package magic with parameters a=15, k=30, knn_dist=‘euclidean’, n_pca=50, random_state=0, t=3.
To assess the similarity of samples “chronic_d35” and “chronic_d35_b6”, kNN graph analysis was performed. For each cell c and a sample S, the distance between c and S was calculated as the average distance in the kNN graph between c and cells from S, measured as the number of edges in the shortest path. This value was calculated for each cell c from samples “chronic_d35” and “chronic_d35_b6” against each of the samples, and shown as a boxplot (Figure S7E).
Overview of functional annotation of scRNA-seq data
We used a combination of complementary approaches in order to functionalize the scRNA-seq data. First, unbiased differential expression analysis identified genes enriched in each cluster (Figure 4E, Figure S8A,B, Methods). Second, visualizing the expression of the T cell signature genes (Figure 4F, Figure S7C, S8C) further helped to interpret the clusters and characterize subsets of cells. Clusters 0, 7, 19, dominated by cells profiled before infection (Figure S8D), were enriched for naïve T cell marker genes Ccr7, Ccr9, Lef1, Tcf7. Gzmb was highly expressed in clusters 4, 6, 14, all dominated by cells at d7 upon acute infection, consistent with effector function. Cell proliferation and survival related genes Birc5, Stmn1, Cdca3 were enriched in clusters 4, 6, 8, 14, 17, 20, which were dominated by cells from early response (d7) to acute or chronic infection. Clusters 2, 10, 13 displayed high Il7r expression and were dominated by cells from acute infection at d40, which were mostly memory cells. Clusters 1, 3, 11, 12, 13, 21 were dominated by cells at d35 in chronic infection and enriched for markers of dysfunction Pdcd1, Lag3, Entpd1, and Cd38. Clusters 5, 15 were dominated by cells from chronic infection at d7 and enriched for Lgals1, Lgals3, Mt1, Mt2. Cluster 18 consisted of dysfunctional cells enriched for Pdcd1, Lag3, and Entpd1 that also overexpressed Ifng, Ccl3, Ccl4, and Nr4a2 and thus were also highly activated. Clusters 9 and 16 overexpressed markers of progenitor cells Cxcr5, Slamf6, Tcf7, and Id3. Finally, association of our bulk RNA-seq compendium (Figure S1D) with scRNA-seq cluster-specific expression profiles using ssGSEA largely confirmed our cluster characterizations (Figure S9A).
Differential expression analysis of scRNA-seq data
Differential expression analysis between cells from groups A and B was performed as follows. Let xG and yG be vectors of normalized scRNA-seq expression values of the gene G in individual cells from groups A and B, respectively. Then log2 fold change (log2FC) of expression for G is estimated as log2((YG + c) / (XG + c)) where XG = mean(xG), YG = mean(yG) are means of normalized expression of G in cells from groups A and B, respectively, and c is a corrective value defined as median over all values Mg where Mg is the mean expression of a gene g across all cells from all samples. Only genes with the absolute log2FC above a certain threshold were then tested for significance. For each such a gene G, the significance of the log2FC for G was estimated with the Mann-Whitney U test applied to normalized counts xG and yG for G in individual cells from groups A and B; the Bonferroni correction for multiple hypothesis testing was then applied to these p-values.
Using this approach, differential expression analysis was performed for each cluster vs. cells from all other clusters, testing for significance of all absolute log2FC values above 0.5. The same analysis was also repeated when excluding all naïve cells defined as cells from sample “acute_d00” or clusters 0, 7, 19. Differential expression analysis was also performed for each pairwise comparison between clusters. Furthermore, for each sample, differential expression analysis was performed for each cluster vs. cells from all other clusters when restricting the analysis only to cells from that sample.
Comparison of scRNA-seq with bulk RNA-seq data compendium
The most significantly differentially expressed genes in each scRNA-seq cluster (adjusted p < 0.001, log2FC > 0.8), defined as the union of at most 100 genes from the comparison of the cluster with all other clusters and at most 100 genes from the same comparison restricted to non-naïve cells, formed a signature set of genes for this cluster. ssGSEA was then run for these signatures against batch-effect corrected library-size normalized bulk RNA-seq read counts using package GSVA v1.30.0.
Analysis of scRNA-seq data from adoptive cell transfer experiment
The six main scRNA-seq samples were combined with the sample “transfer_acute_d07” from the progenitor dysfunctional cells extracted from the established chronic infection and adoptively transferred and expanded under acute infection. The extended data set was preprocessed in the same manner as the main data set, resulting in a count matrix for 9274 genes in 28447 cells, including 4047 cells from the transfer sample. The analysis of this extended count matrix was performed in the same manner as with the main six samples, resulting in clusters t0–t23. This cell clustering, restricted to the cells from the main six samples, was compared with the previously obtained clusters, suggesting cluster t4 consisted of the progenitor and progenitor-like cells. The cluster composition of each sample, calculated as fraction of cells in each sample that belongs to each cluster, was also compared between samples, suggesting the sample “transfer_acute_d07” was most similar to the sample “chronic_d07”.
Allele-specific scRNA-seq expression
For allele-specific analysis, scRNA-seq sequencing reads were re-aligned in allele-specific manner as previously described (van der Veeken et al., 2019, Crowley et al., 2015). Briefly, the genetic variants of Spret mice were obtained from the mouse genome project (Keane et al., 2011). A pseudo-Spret genome was built by modifying the reference genome with SNPs, insertions and deletions found in the wild-derived inbred strain. The RNA-seq reads were aligned to the reference and pseudo Spret genomes in parallel using STAR (Dobin et al., 2013) with the following parameter setting: with the following parameter settings: “STAR --runMode alignReads --readFilesCommand zcat --outSAMtype BAM --outBAMcompression 6 --outFilterMultimapNmax 1 --outFilterMatchNmin 30 --alignIntronMin 20 --alignIntronMax 20000 --alignEndsType Local”. Next, the genomic coordinates of pseudo-genome aligned reads were converted back to the corresponding B6 coordinates. To determine the allelic origins of the reads, the mapping scores of the two alignments of each read were compared. We retained the alignment with the highest score and generated the final BAM files. In cases where the diploid genome alignment produced identical scores for both genomes, one of the alignments was selected randomly.
Spret- and B6-specific and ambiguous UMI counts of gene expression for each cell were obtained with custom scripts, in the same manner as in the main analysis (see Single-cell RNA-seq data analysis). Further analysis was restricted only to cells and genes selected for the main scRNA-seq analysis. Library-size normalization for Spret- and B6-specific and ambiguous read counts was obtained applying the same scaling for each cell as in the main analysis. The allelic imbalance of expression of each gene in each scRNA-seq cluster was defined as log2((w1 + 0.5 * w2) / (w0 + 0.5 * w2)), where w0 and w1 are B6- and Spret-specific total library-size normalized counts for this gene in this cluster, respectively, and w2 is the ambiguous count for this gene in this cluster. Significance of the allelic imbalance was assessed by a Mann-Whitney U test applied to all B6-specific vs. all Spret-specific library-size normalized gene expression estimates for this gene over cells in this cluster.
Allele-specific predicted TF binding
Spret sequences of bulk ATAC-seq peak summit regions, as defined above (see Construction of ATAC-seq peak atlas), were obtained by introducing genetic variants between B6 and Spret into B6 sequences of these regions. To estimate allelic specificity of predicted TF binding, FIMO was run for Spret sequences and rerun for B6 sequences of the peak summit regions with the same motif collection as described previously (see Transcription factor motif analysis) with a relaxed p-value threshold of 5e-3. All matches at p < 1e-4 in either B6 or Spret sequence were selected, and allelic imbalance of predicted TF binding for each motif in each peak summit region was estimated as the difference between the total FIMO log odds score for all matches of this motif in Spret and in B6. Then the distribution of these values for this motif across all peaks was z-score-normalized.
Analysis of the association of allelic imbalance of predicted TF binding and of scRNA-seq gene expression was performed for each TF motif and each scRNA-seq cluster as follows. All predicted TF motif binding sites with substantially stronger predicted match at Spret or B6 sequence were selected as those with z-score above 0.2 or below −0.2, respectively. Then the scRNA-seq allelic imbalance in this cluster was compared between genes nearby B6-specific and Spret-specific predicted binding sites, with significance assessed by Kolmogorov-Smirnov test.
CUT&RUN experiments
Terminal and progenitor dysfunctional cells were isolated from the spleens of 4 male B6/Spret F1 mice or B6 mice on day 35 post-infection, based on expression of cell surface markers (see Adoptive transfer of progenitor dysfunctional cells). Cells from 2 mice were pooled to generate biological duplicates with approximately 70,000 cells per replicate. CUT&RUN libraries were prepared as described (Skene and Henikoff, 2017) with the modifications described below. Because Concanavalin-A (ConA) is a well-known T cell mitogen, we avoided the use of ConA-coated beads for cell isolation and handling. 70k cells per replicate were collected in a V-bottom 96 well plate by centrifugation and washed in antibody buffer (buffer 1 (1x permeabilization buffer from eBioscience Foxp3/Transcription Factor Staining Buffer Set diluted in nuclease free water, 1X EDTA-free protease inhibitors, 0.5mM spermidine) containing 2mM EDTA). Cells were incubated with TCF1 antibody for 1h on ice. After 2 washes in buffer 1, cells were incubated with pA/G-MNase at 1:4000 dilution in buffer 1 for 1h at 4 degrees. Cells were washed twice in buffer 2 (0.05% (w/v) saponin, 1X EDTA-free protease inhibitors, 0.5mM spermidine in PBS) and resuspended in calcium buffer (buffer 2 containing 2mM CaCl2) to activate MNase. Following a 30 minute incubation on ice, 2x stop solution (20mM EDTA, 4mM EGTA in buffer 2) was added and cells were incubated for 10 minutes in a 37 degree incubator to release cleaved chromatin fragments. Supernatants were collected by centrifugation and DNA was extracted using a Qiagen MinElute kit.
CUT&RUN libraries were prepared using the Kapa Hyper Prep Kit (Kapa Biosystems KK8504) and Kapa UDI Adapter Kit (Kapa Biosystems KK8727) according to manufacturers protocol with the modifications described below. A-tailing temperature was reduced to 50 degrees to avoid melting of short DNA fragments and reaction time was increased to 1h to compensate for reduced enzyme activity as described by Liu et al. 2018. Following the adapter ligation step, 3 consecutive rounds of Ampure purification were performed using a 1.4x bead to sample ratio to remove excess unligated adapters while retaining short adapter-ligated fragments. Libraries were amplified for an average of 15 cycles using a 10 second 60 °C annealing/extension step to enrich for shorter library fragments. Following amplification, libraries were purified using 3 consecutive rounds of Ampure purification with a 1.2x bead to sample ratio to remove amplified primer dimers while retaining short library fragments. A 0.5x Ampure purification step was included to remove large fragments prior to sequencing.
Identification of transcription factor binding sites in CUT&RUN data
CUT&RUN reads from B6 mouse samples were aligned to the genome using bowtie2 v2.3.4.3. Similar to RNA-seq, paired-end CUT&RUN reads from F1 B6/Spret mouse samples were mapped to the diploid genome using STAR with the splicing alignment feature switched off. The command line was as follow: “STAR --runMode alignReads --genomeLoad NoSharedMemory --readFilesCommand zcat --outSAMtype BAM SortedByCoordinate --outFilterMultimapNmax 1 --outFilterMatchNmin 40 --outBAMcompression 6 --outFilterMatchNminOverLread 0.4 --seedSearchStartLmax 20 --alignIntronMax 1 --alignEndsType Local”. We only used the read pairs if their fragment length was between 50 to 500 bp.
For analysis, we used two control IgG samples and biological replicates for TCF1 CUT&RUN, including two samples from B6 mice and two samples from F1 B6/Spret mice. CUT&RUN and control reads were counted in 150bp windows around ATAC-seq peak summits. To estimate library sizes, control regions were obtained by shifting ATAC-seq peak summit regions by 2Kb in either direction, extending the shifted segments by 500bp preserving their center, and excluding those that overlapped with ATAC-seq peak summit regions, and then calculating control and CUT&RUN read counts in these control regions and using DESeq2 v1.22.2, as described previously (Konopacki et al., 2019). Differential CUT&RUN count analysis was then run using DESeq2, identifying 3325 TCF1 binding sites in the progenitor dysfunctional cells defined as ATAC-seq peak summit regions with significantly higher TCF1 CUT&RUN read counts than IgG control (adjusted p-value < 0.1). To produce heatmaps of TCF1 CUT&RUN signal around binding sites, bigWig files and count matrices in bins around binding sites were produced using deepTools (Ramirez et al., 2016).
Motif enrichment in TCF1 binding sites was estimated as log2FC of frequency of TCF1 binding sites with significant TCF1 motif match (FIMO p < 1e-4) over such frequency for all ATAC-seq peak summit regions, with significance of enrichment estimated using hypergeometic test followed by Bonferroni correction for multiple hypothesis testing.
Allele-specific analysis of CUT&RUN data
For the TCF1 binding sites, allelic imbalance of TCF1 binding was defined as log2((w1 + 0.5 * w2) / (w0 + 0.5 * w2)), where w0 and w1 are B6- and Spret-specific CUT&RUN counts, respectively, and w2 is the count of ambiguously aligned reads. For the CDF plots of allelic imbalance of TCF1 binding (Figure S13E), TCF1 binding sites with predicted stronger TCF1 motif batch in Spret or B6 were selected as those with TCF1 motif match imbalance z-score above 0.1 or below −0.1, respectively. For the allele-specific TCF1 co-factor analysis, Spret- or B6-specific TCF1 binding sites were detected as those with log2FC of CUT&RUN read count in Spret over B6 above 0.5 or below −0.5, respectively. Then for these sets of allele-specific TCF1 binding sites, for each motif, co-factor motif enrichment was calculated by comparing motif match imbalance z-scores against those in all ATAC-seq peak summit regions, with significance estimated using hypergeometric test. Motif match imbalance z-scores were then plotted as a boxplot (Figure S13G) for all the motifs that were detected as significant both for Spret- and B6-specific TCF1 CUT&RUN binding sites.
Construction of human ATAC-seq peak atlas and scATAC-seq count matrix
Bulk and single-cell ATAC-seq data for CD8 T cells in human donors and patients was obtained from three recent publications (Sade-Feldman et al., 2018, Philip et al., 2017, Satpathy et al., 2019). Bulk ATAC-seq data (Sade-Feldman et al., 2018, Philip et al., 2017) was processed in the same way as described above for mouse data. For scATAC-seq data (Satpathy et al., 2019), samples SU001_Tcell_Post2, SU001_Tcell_Post, SU006_Tcell_Pre, SU008_Tcell_Post, SU008_Tcell_Pre, SU009_Tcell_Post, SU009_Tcell_Pre were identified as those with large number of T cells profiled, according to metadata available at https://github.com/GreenleafLab/10x-scATAC-2019. Fragment data for these samples was obtained from GEO and mapped from hg19 to hg38 coordinates using liftOver tool from UCSC Genome Browser (Kent et al., 2002). Fragments associated with cells from T cell clusters 12–19 (Satpathy et al., 2019) were likely corresponding to CD8 T cells and therefore were selected for further analysis. Pseudo-reads of length 30nt were created for each start and end of the selected fragments, and MACS2 was run on this collection of pseudo-reads to identify accessibility peaks. These peaks were merged with those from bulk ATAC-seq data analysis using function reduce(c()) in R, resulting in a list of putative peaks. Overlaps of bulk ATAC-seq reads and scATAC-seq fragment ends (corresponding to Tn5 cut sites) in these peaks were counted using Rsubread and findOverlaps(), respectively. Then peaks with at least total count of 50 in any of the three datasets (two bulk ATAC-seq datasets and one scATAC-seq dataset) were selected, forming an atlas of 161,140 peaks. Of them, 40,449 or 36,241 peaks overlapped at least one mouse atlas peak or peak summit, respectively, when mapped to human genome using liftOver().
Analysis of human scATAC-seq data
Count matrices for each of the seven human scATAC-seq samples obtained as described above were merged together, binarized, and transformed using the TF-IDF transform, as done with the mouse scATAC-seq data. The nearest neighbor graph was computed using the top 50 PCs and batch corrected by patient (SU001, SU006, SU008, SU009) using the batch balanced k-nearest neighbor procedure (Polanski et al., 2020). Subsequent UMAP dimensionality reduction and Leiden clustering with a resolution of 0.8 were performed on the corrected kNN graph.
Comparison of differential accessibility between mouse and human
Differential accessibility in human bulk ATAC-seq data between naïve and dysfunctional cells, central memory and dysfunctional cells, and progenitor and terminally dysfunctional cells was performed in the same manner as for mouse data. For comparison, differentially accessible peak summit regions in mice between naïve and dysfunctional cells and between central memory and dysfunctional cells were selected as those with log2FoldChange > 1 and adjusted p-value < 0.001, between progenitor and terminally dysfunctional cells in melanoma as those with log2FoldChange > 0.5 and adjusted p-value < 0.001. The selected differentially accessible peaks were then mapped to human genome using liftOver() and overlapped with human peaks. For human scATAC-seq analysis, peak signatures obtained from mouse bulk ATAC-seq data were mapped to human genome using liftOver() and then scored in human scATAC-seq clusters, as described above for mouse scATAC-seq data analysis. For differential accessibility analysis between Leiden clusters 0 and 8, scATAC-seq counts were aggregated over all cells in these clusters in each sample separately, and then these pseudobulk counts compared between the two clusters treating each sample as a biological replicate (7 samples in each category) using DESeq2.
RNA velocity analysis
Spliced and unspliced read counts for each sample were obtained by running cellranger followed by velocyto (La Manno et al., 2018), using default mm10 genome. Then RNA velocity analysis for each sample was run using scvelo (Bergen et al., 2020). Only the cells that were used in the main scRNA-seq analysis after filtering were used for the velocity analysis. Naïve cells (from clusters 0, 7, 19) were excluded from analysis of each sample. Genes for the analysis of each sample were selected using scvelo.pp.filter_genes() with min_shared_counts=30. Furthermore, for each sample except “chronic_d35_b6”, only those genes were used in subsequent analysis that had Spearman correlation at least 0.8 between spliced counts and previously obtained F1 genome counts used in the main scRNA-seq analysis. RNA velocity analysis was run using function scvelo.tl.velocity() with mode=‘deterministic’, implementing the approach from the original single-cell RNA velocity publication (La Manno et al., 2018). Genes labeled as velocity genes by scvelo.tl.velocity() were further filtered to include only those with R2 of the fit (velocity_r2) above 0.3. This resulted in 36 genes for sample “acute_d07”, 27 genes for “acute_d40”, 72 genes for “chronic_d07”, 22 genes for “chronic_d35” and 141 genes for “chronic_d35_b6” that were used in constructing velocity vectors for velocity graph construction and projection to 2-dimensional UMAP. Vector map visualizations were obtained using scvelo.pl.velocity_embedding_grid() with parameters arrow_size=1, arrow_length=3, density=1, smooth=0.7, min_mass=3.
We suspect that the RNA velocity analysis in our data has a number of caveats. Insufficient sequencing coverage resulting in low read counts, particularly for unspliced reads, may have caused only a small number of genes to be selected for the velocity analysis. Furthermore, transcriptional differences across CD8 T cell functional and differentiation states captured in our scRNA-seq data may be smaller than those between different cell types previously analyzed with RNA velocity analysis (La Manno et al., 2018, Bergen et al., 2020). Finally, interpretation of the velocity vectors projected to 2-dimensional UMAP may be complicated, because of the oversimplification from reducing the data to only two dimensions and thus overlaying potential differentiation trajectories. Altogether, this may have contributed to noisier results of the analysis that are harder to interpret than those in previous publications (La Manno et al., 2018, Bergen et al., 2020). Nevertheless, we observe consistently across samples that velocity is directed towards cells on the right side of the UMAP, e.g. in cluster 1, which suggests these cells are more differentiated, consistent with their characterization as terminally dysfunctional.
Quantification and statistical analysis
All the details of quantifications and statistical analyses are fully described in the main text, figure legends and Method Details section.
Supplementary Material
Table S1. Published data used in the analysis (related to Figure 1). Details of published bulk ATAC-seq (A) and RNA-seq (B) datasets used in the analysis.
Table S2. Results of bulk ATAC-seq and RNA-seq analysis (related to Figure 1). A. ATAC-seq peaks. B. ATAC-seq peak summits. C-E. Differential accessibility for individual peak summits (C) and for peak summits consolidated over genes (D) and differential expression (E) between functional and dysfunctional cells. F-H. Differential accessibility for individual peak summits (F) and for peak summits consolidated over genes (G) and differential expression (H) between early and late dysfunctional cells. I-K. Differential accessibility for individual peak summits (I) and for peak summits consolidated over genes (J) and differential expression (K) between progenitor and terminally dysfunctional cells. L-Q. ATAC-seq signature peaks for naïve (L), effector (M), memory (N), MPEC (O), progenitor dysfunctional (P) and terminally dysfunctional (Q) cells derived from bulk ATAC-seq data analysis.
Table S3. Results of human ATAC-seq analysis (related to Figure 1). A. ATAC-seq peaks. B-D. Differential accessibility between naïve and dysfunctional cells (B), between memory and dysfunctional cells (C), and between progenitor and terminally dysfunctional cells (D).
Table S4. Results of scATAC-seq analysis (related to Figure 3). A. scATAC-seq peaks. B. scATAC-seq peak summits. CA-CO. Differential accessibility for cells in each cluster from LCMV Armstrong infection. DA-DO. Differential accessibility for cells in each cluster from LCMV clone 13 infection.
Table S5. Results of scRNA-seq analysis (related to Figures 4 and 6). AA-AV. Differential expression for each cluster as compared with cells from all other clusters (for clusters in Figure 4C). BA-BS. Differential expression for each non-naïve cluster as compared with all other non-naïve clusters (for clusters in Figure 4C). CA-CU. Within the sample “transfer_acute_d07” (adoptively transferred progenitor dysfunctional cells expanding in response to LCMV Armstrong infection), differential expression for each non-naïve cluster (for clusters in Figure 6F) as compared with the rest of the cells in the sample.
Table S6. Results of TCF1 CUT&RUN analysis (related to Figure 5).
KEY RESOURCES TABLE
Highlights.
Meta-analysis of chromatin states in T cells responding to cancer and infection
Chronically activated T cells follow a common path towards dysfunction
Cell-state specific transcription factors define T cell differentiation trajectories
Functional and dysfunctional T cells emerge from similar TCF1+ progenitor states
Acknowledgments
This study was supported by the NCI CSBC Research Center for Cancer Systems Immunology (U54 CA209975), the Alan and Sandra Gerry Metastasis and Tumor Ecosystems Center, NCI Cancer Center Support Grant (P30 CA008748), AACR-Bristol-Myers Squibb Immuno-oncology Research Fellowship (19–40-15-PRIT) and MSK-PICI-Cycle for Survival Fund (to Y.P.). We thank Lauren Fairchild for useful discussions and preliminary data analysis, Roshan Sharma and Manu Setty for advice on scRNA-seq analysis, and members of the Leslie, Rudensky, and Pe’er labs for discussion and suggestions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
A.Y.R. is an SAB member, has equity in Sonoma Biotherapeutics, RAPT Therapeutics, Surface Oncology, and Vedanta Biosciences, and is a co-inventor or has IP licensed to Takeda that is unrelated to the content of this study.
References
- Angelosanto JM, Blackburn SD, Crawford A & Wherry EJ (2012) Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection. Journal of virology, 86, 8161–8170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, Nainys J, Wu K, Kiseliovas V, Setty M, et al. (2018) Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell, 174, 1293–1308 e36. 10.1016/j.cell.2018.05.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E & Scholl C (2009) Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature, 462, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltra JC, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, Chen Z, Casella V, Ngiow SF, Khan O, Huang YJ, et al. (2020) Developmental Relationships of Four Exhausted CD8(+) T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity, 52, 825–841 e8. 10.1016/j.immuni.2020.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengsch B, Ohtani T, Khan O, Setty M, Manne S, O’brien S, Gherardini PF, Herati RS, Huang AC & Chang K-M (2018) Epigenomic-guided mass cytometry profiling reveals disease-specific features of exhausted CD8 T cells. Immunity, 48, 1029–1045. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergen V, Lange M, Peidli S, Wolf FA & Theis FJ (2020) Generalizing RNA velocity to transient cell states through dynamical modeling. Nat Biotechnol, 38, 1408–1414. 10.1038/s41587-020-0591-3 [DOI] [PubMed] [Google Scholar]
- Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, Lynn RC, Philip M, Rao A, Restifo NP, et al. (2019) Defining ‘T cell exhaustion’. Nat Rev Immunol 10.1038/s41577-019-0221-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummelman J, Mazza EMC, Alvisi G, Colombo FS, Grilli A, Mikulak J, Mavilio D, Alloisio M, Ferrari F, Lopci E, et al. (2018) High-dimensional single cell analysis identifies stem-like cytotoxic CD8(+) T cells infiltrating human tumors. J Exp Med, 215, 2520–2535. 10.1084/jem.20180684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carty M, Zamparo L, Sahin M, Gonzalez A, Pelossof R, Elemento O & Leslie CS (2017) An integrated model for detecting significant chromatin interactions from high-resolution Hi-C data. Nat Commun, 8, 15454. 10.1038/ncomms15454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Lopez-Moyado IF, Seo H, Lio CJ, Hempleman LJ, Sekiya T, Yoshimura A, Scott-Browne JP & Rao A (2019a) NR4A transcription factors limit CAR T cell function in solid tumours. Nature, 567, 530–534. 10.1038/s41586-019-0985-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Yu M, Zheng Y, Fu G, Xin G, Zhu W, Luo L, Burns R, Li QZ, Dent AL, et al. (2019b) CXCR5(+)PD-1(+) follicular helper CD8 T cells control B cell tolerance. Nat Commun, 10, 4415. 10.1038/s41467-019-12446-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Ji Z, Ngiow SF, Manne S, Cai Z, Huang AC, Johnson J, Staupe RP, Bengsch B, Xu C, et al. (2019c) TCF-1-Centered Transcriptional Network Drives an Effector versus Exhausted CD8 T Cell-Fate Decision. Immunity, 51, 840–855 e5. 10.1016/j.immuni.2019.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley JJ, Zhabotynsky V, Sun W, Huang S, Pakatci IK, Kim Y, Wang JR, Morgan AP, Calaway JD & Aylor DL (2015) Analyses of allele-specific gene expression in highly divergent mouse crosses identifies pervasive allelic imbalance. Nature genetics, 47, 353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M & Gingeras TR (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doering TA, Crawford A, Angelosanto JM, Paley MA, Ziegler CG & Wherry EJ (2012) Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity, 37, 1130–44. 10.1016/j.immuni.2012.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X & Rudensky AY (2016) Hallmarks of Tissue-Resident Lymphocytes. Cell, 164, 1198–1211. 10.1016/j.cell.2016.02.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, et al. (2019) GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res, 47, D766–D773. 10.1093/nar/gky955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant CE, Bailey TL & Noble WS (2011) FIMO: scanning for occurrences of a given motif. Bioinformatics, 27, 1017–8. 10.1093/bioinformatics/btr064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanzelmann S, Castelo R & Guinney J (2013) GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics, 14, 7. 10.1186/1471-2105-14-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto M, Kamphorst AO, Im SJ, Kissick HT, Pillai RN, Ramalingam SS, Araki K & Ahmed R (2018) CD8 T Cell Exhaustion in Chronic Infection and Cancer: Opportunities for Interventions. Annu Rev Med, 69, 301–318. 10.1146/annurev-med-012017-043208 [DOI] [PubMed] [Google Scholar]
- He B, Xing S, Chen C, Gao P, Teng L, Shan Q, Gullicksrud JA, Martin MD, Yu S, Harty JT, et al. (2016) CD8(+) T Cells Utilize Highly Dynamic Enhancer Repertoires and Regulatory Circuitry in Response to Infections. Immunity, 45, 1341–1354. 10.1016/j.immuni.2016.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H & Glass CK (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell, 38, 576–89. 10.1016/j.molcel.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, et al. (2016) Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature, 537, 417–421. 10.1038/nature19330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jameson SC & Masopust D (2018) Understanding Subset Diversity in T Cell Memory. Immunity, 48, 214–226. 10.1016/j.immuni.2018.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johanson TM, Chan WF, Keenan CR & Allan RS (2019) Genome organization in immune cells: unique challenges. Nat Rev Immunol, 19, 448–456. 10.1038/s41577-019-0155-2 [DOI] [PubMed] [Google Scholar]
- Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L & Kaech SM (2007) Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity, 27, 281–95. 10.1016/j.immuni.2007.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallies A, Zehn D & Utzschneider DT (2019) Precursor exhausted T cells: key to successful immunotherapy? Nature Reviews Immunology, 1–9. [DOI] [PubMed] [Google Scholar]
- Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, Heger A, Agam A, Slater G, Goodson M, et al. (2011) Mouse genomic variation and its effect on phenotypes and gene regulation. Nature, 477, 289–94. 10.1038/nature10413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM & Haussler D (2002) The human genome browser at UCSC. Genome Res, 12, 996–1006. 10.1101/gr.229102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C & Salzberg SL (2019) Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol, 37, 907–915. 10.1038/s41587-019-0201-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopacki C, Pritykin Y, Rubtsov Y, Leslie CS & Rudensky AY (2019) Transcription factor Foxp1 regulates Foxp3 chromatin binding and coordinates regulatory T cell function. Nat Immunol, 20, 232–242. 10.1038/s41590-018-0291-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar BV, Connors TJ & Farber DL (2018) Human T Cell Development, Localization, and Function throughout Life. Immunity, 48, 202–213. 10.1016/j.immuni.2018.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurd NS, He Z, Louis TL, Milner JJ, Omilusik KD, Jin W, Tsai MS, Widjaja CE, Kanbar JN, Olvera JG, et al. (2020) Early precursors and molecular determinants of tissue-resident memory CD8(+) T lymphocytes revealed by single-cell RNA sequencing. Sci Immunol, 5. 10.1126/sciimmunol.aaz6894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lonnerberg P, Furlan A, et al. (2018) RNA velocity of single cells. Nature, 560, 494–498. 10.1038/s41586-018-0414-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B & Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods, 9, 357–9. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R & Genome Project Data Processing, S. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25, 2078–9. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Brown JB, Huang H & Bickel PJ (2011) Measuring reproducibility of high-throughput experiments. The annals of applied statistics, 5, 1752–1779. [Google Scholar]
- Liao Y, Smyth GK & Shi W (2019) The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic acids research, 47, e47–e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W & Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol, 15, 550. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man K, Gabriel SS, Liao Y, Gloury R, Preston S, Henstridge DC, Pellegrini M, Zehn D, Berberich-Siebelt F, Febbraio MA, et al. (2017) Transcription Factor IRF4 Promotes CD8(+) T Cell Exhaustion and Limits the Development of Memory-like T Cells during Chronic Infection. Immunity, 47, 1129–1141 e5. 10.1016/j.immuni.2017.11.021 [DOI] [PubMed] [Google Scholar]
- Mclane LM, Abdel-Hakeem MS & Wherry EJ (2019) CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu Rev Immunol, 37, 457–495. 10.1146/annurev-immunol-041015-055318 [DOI] [PubMed] [Google Scholar]
- Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, Lafleur MW, Yates KB, Lako A, Felt K, Naik GS, et al. (2019) Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol, 20, 326–336. 10.1038/s41590-019-0312-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner JJ, Toma C, He Z, Kurd NS, Nguyen QP, Mcdonald B, Quezada L, Widjaja CE, Witherden DA, Crowl JT, et al. (2020) Heterogenous Populations of Tissue-Resident CD8(+) T Cells Are Generated in Response to Infection and Malignancy. Immunity, 52, 808–824 e7. 10.1016/j.immuni.2020.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner JJ, Toma C, Yu B, Zhang K, Omilusik K, Phan AT, Wang D, Getzler AJ, Nguyen T, Crotty S, et al. (2017) Runx3 programs CD8(+) T cell residency in non-lymphoid tissues and tumours. Nature, 552, 253–257. 10.1038/nature24993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mognol GP, Spreafico R, Wong V, Scott-Browne JP, Togher S, Hoffmann A, Hogan PG, Rao A & Trifari S (2017) Exhaustion-associated regulatory regions in CD8(+) tumor-infiltrating T cells. Proc Natl Acad Sci U S A, 114, E2776–E2785. 10.1073/pnas.1620498114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy K & Weaver C (2016) Janeway’s immunobiology, Garland Science. [Google Scholar]
- Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, Drake AM, Chen Z, Sen DR, Kurachi M, et al. (2016) Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science, 354, 1160–1165. 10.1126/science.aaf2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, Scott AC, Viale A, Lauer P, Merghoub T, et al. (2017) Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature, 545, 452–456. 10.1038/nature22367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip M & Schietinger A (2019) Heterogeneity and fate choice: T cell exhaustion in cancer and chronic infections. Curr Opin Immunol, 58, 98–103. 10.1016/j.coi.2019.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polanski K, Young MD, Miao Z, Meyer KB, Teichmann SA & Park JE (2020) BBKNN: fast batch alignment of single cell transcriptomes. Bioinformatics, 36, 964–965. 10.1093/bioinformatics/btz625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F & Manke T (2016) deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res, 44, W160–5. 10.1093/nar/gkw257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H & Vilo J (2019) g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res, 47, W191–W198. 10.1093/nar/gkz369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A & Wolchok JD (2018) Cancer immunotherapy using checkpoint blockade. Science, 359, 1350–1355. 10.1126/science.aar4060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, De Boer CG, Jenkins RW, Lieb DJ, Chen JH, Frederick DT, Barzily-Rokni M, et al. (2018) Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell, 175, 998–1013 e20. 10.1016/j.cell.2018.10.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satpathy AT, Granja JM, Yost KE, Qi Y, Meschi F, Mcdermott GP, Olsen BN, Mumbach MR, Pierce SE, Corces MR, et al. (2019) Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat Biotechnol, 37, 925–936. 10.1038/s41587-019-0206-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharer CD, Bally AP, Gandham B & Boss JM (2017) Cutting Edge: Chromatin Accessibility Programs CD8 T Cell Memory. J Immunol, 198, 2238–2243. 10.4049/jimmunol.1602086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schietinger A, Philip M, Krisnawan VE, Chiu EY, Delrow JJ, Basom RS, Lauer P, Brockstedt DG, Knoblaugh SE, Hammerling GJ, et al. (2016) Tumor-Specific T Cell Dysfunction Is a Dynamic Antigen-Driven Differentiation Program Initiated Early during Tumorigenesis. Immunity, 45, 389–401. 10.1016/j.immuni.2016.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott-Browne JP, Lopez-Moyado IF, Trifari S, Wong V, Chavez L, Rao A & Pereira RM (2016) Dynamic Changes in Chromatin Accessibility Occur in CD8(+) T Cells Responding to Viral Infection. Immunity, 45, 1327–1340. 10.1016/j.immuni.2016.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, Tsao HW, Godec J, Lafleur MW, Brown FD, et al. (2016) The epigenetic landscape of T cell exhaustion. Science, 354, 1165–1169. 10.1126/science.aae0491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servant N, Varoquaux N, Lajoie BR, Viara E, Chen C-J, Vert J-P, Heard E, Dekker J & Barillot E (2015) HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome biology, 16, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Hu-Lieskovan S, Wargo JA & Ribas A (2017) Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell, 168, 707–723. 10.1016/j.cell.2017.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ & Henikoff S (2017) An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife, 6, e21856. 10.7554/eLife.21856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splinter E, Heath H, Kooren J, Palstra RJ, Klous P, Grosveld F, Galjart N & De Laat W (2006) CTCF mediates long-range chromatin looping and local histone modification in the beta-globin locus. Genes Dev, 20, 2349–54. 10.1101/gad.399506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, Danilo M, Alfei F, Hofmann M, Wieland D, et al. (2016) T Cell Factor 1-Expressing Memory-like CD8(+) T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity, 45, 415–27. 10.1016/j.immuni.2016.07.021 [DOI] [PubMed] [Google Scholar]
- Utzschneider DT, Gabriel SS, Chisanga D, Gloury R, Gubser PM, Vasanthakumar A, Shi W & Kallies A (2020) Early precursor T cells establish and propagate T cell exhaustion in chronic infection. Nat Immunol, 21, 1256–1266. 10.1038/s41590-020-0760-z [DOI] [PubMed] [Google Scholar]
- Van Der Veeken J, Glasner A, Zhong Y, Hu W, Wang ZM, Bou-Puerto R, Charbonnier LM, Chatila TA, Leslie CS & Rudensky AY (2020) The Transcription Factor Foxp3 Shapes Regulatory T Cell Identity by Tuning the Activity of trans-Acting Intermediaries. Immunity, 53, 971–984 e5. 10.1016/j.immuni.2020.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Veeken J, Zhong Y, Sharma R, Mazutis L, Dao P, Pe’er D, Leslie CS & Rudensky AY (2019) Natural Genetic Variation Reveals Key Features of Epigenetic and Transcriptional Memory in Virus-Specific CD8 T Cells. Immunity, 50, 1202–1217 e7. 10.1016/j.immuni.2019.03.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dijk D, Sharma R, Nainys J, Yim K, Kathail P, Carr AJ, Burdziak C, Moon KR, Chaffer CL & Pattabiraman D (2018) Recovering gene interactions from single-cell data using data diffusion. Cell, 174, 716–729. e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vodnala SK, Eil R, Kishton RJ, Sukumar M, Yamamoto TN, Ha N-H, Lee P-H, Shin M, Patel SJ & Yu Z (2019) T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science, 363, eaau0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner DE & Klein AM (2020) Lineage tracing meets single-cell omics: opportunities and challenges. Nat Rev Genet, 21, 410–427. 10.1038/s41576-020-0223-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Ma S, Wang CY, Zappitelli M, Devarajan P & Parikh C (2014) EM for regularized zero-inflated regression models with applications to postoperative morbidity after cardiac surgery in children. Stat Med, 33, 5192–208. 10.1002/sim.6314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, Najafabadi HS, Lambert SA, Mann I, Cook K, et al. (2014) Determination and inference of eukaryotic transcription factor sequence specificity. Cell, 158, 1431–1443. 10.1016/j.cell.2014.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ & Kurachi M (2015) Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol, 15, 486–99. 10.1038/nri3862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf FA, Angerer P & Theis FJ (2018) SCANPY: large-scale single-cell gene expression data analysis. Genome Biol, 19, 15. 10.1186/s13059-017-1382-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao C, Sun HW, Lacey NE, Ji Y, Moseman EA, Shih HY, Heuston EF, Kirby M, Anderson S, Cheng J, et al. (2019) Single-cell RNA-seq reveals TOX as a key regulator of CD8(+) T cell persistence in chronic infection. Nat Immunol, 20, 890–901. 10.1038/s41590-019-0403-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B, Zhang K, Milner JJ, Toma C, Chen R, Scott-Browne JP, Pereira RM, Crotty S, Chang JT & Pipkin ME (2017) Epigenetic landscapes reveal transcription factors that regulate CD8+ T cell differentiation. Nature Immunology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol, 9, R137. 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Published data used in the analysis (related to Figure 1). Details of published bulk ATAC-seq (A) and RNA-seq (B) datasets used in the analysis.
Table S2. Results of bulk ATAC-seq and RNA-seq analysis (related to Figure 1). A. ATAC-seq peaks. B. ATAC-seq peak summits. C-E. Differential accessibility for individual peak summits (C) and for peak summits consolidated over genes (D) and differential expression (E) between functional and dysfunctional cells. F-H. Differential accessibility for individual peak summits (F) and for peak summits consolidated over genes (G) and differential expression (H) between early and late dysfunctional cells. I-K. Differential accessibility for individual peak summits (I) and for peak summits consolidated over genes (J) and differential expression (K) between progenitor and terminally dysfunctional cells. L-Q. ATAC-seq signature peaks for naïve (L), effector (M), memory (N), MPEC (O), progenitor dysfunctional (P) and terminally dysfunctional (Q) cells derived from bulk ATAC-seq data analysis.
Table S3. Results of human ATAC-seq analysis (related to Figure 1). A. ATAC-seq peaks. B-D. Differential accessibility between naïve and dysfunctional cells (B), between memory and dysfunctional cells (C), and between progenitor and terminally dysfunctional cells (D).
Table S4. Results of scATAC-seq analysis (related to Figure 3). A. scATAC-seq peaks. B. scATAC-seq peak summits. CA-CO. Differential accessibility for cells in each cluster from LCMV Armstrong infection. DA-DO. Differential accessibility for cells in each cluster from LCMV clone 13 infection.
Table S5. Results of scRNA-seq analysis (related to Figures 4 and 6). AA-AV. Differential expression for each cluster as compared with cells from all other clusters (for clusters in Figure 4C). BA-BS. Differential expression for each non-naïve cluster as compared with all other non-naïve clusters (for clusters in Figure 4C). CA-CU. Within the sample “transfer_acute_d07” (adoptively transferred progenitor dysfunctional cells expanding in response to LCMV Armstrong infection), differential expression for each non-naïve cluster (for clusters in Figure 6F) as compared with the rest of the cells in the sample.
Table S6. Results of TCF1 CUT&RUN analysis (related to Figure 5).
Data Availability Statement
The code for computational analysis and additional data visualizations are available at https://bitbucket.org/leslielab/cd8t_atlas. New scATAC-seq, scRNA-seq and CUT&RUN data are available at NCBI GEO under the accession number GSE164978. Previously published datasets reanalyzed in this study are listed in Table S1.