

Summary
The controlled generation of reactive oxygen species (ROS) to selectively epoxidize styrene is a grand challenge. Herein, cyano-group-modified carbon nitrides (CNCYx and CN-Ty) are prepared, and the catalysts show better performance in regulating ROS and producing styrene oxide than the cyano-free sample. The in situ diffuse reflectance infrared and density functional theory calculation results reveal that the cyano group acts as the adsorption and activation site of oxygen. X-ray photoelectron spectroscopy and NMR spectrum results confirm that the cyano group bonds with the intact heptazine ring. This unique structure could inhibit H2O2 and ⋅OH formation, resulting in high selectivity of styrene oxide. Furthermore, high catalytic activity is still achieved when the system scales up to 2.7 L with 100 g styrene under solar light irradiation. The strategy of cyano group modification gives a new insight into regulating spatial configuration for tuning the utilization of oxygen-active species and shows potential applications in industry.
Keywords: epoxidation, cyano modification, carbon nitride, ROS, steric hindrance
Graphical Abstract
Public summary
-
•
Surface group modification is a significant strategy to improve catalyst activity
-
•
Cyano groups are incorporated on the surface of carbon nitride via copolymerization
-
•
Cyano group reactive sites allow high selectivity of styrene oxide for aerobic oxidation
-
•
The system can be scaled up under solar light irradiation
Introduction
Selective epoxidation of styrene by reactive oxygen species (ROS) originating from dioxygen is an economically important atomic reaction, and the styrene oxide products are versatile intermediates for the synthesis of perfumes, drugs, epoxy resins, and sweeteners and even for the chemical fixation of CO2.1, 2, 3 The ROS mainly include superoxide anion radical (O2⋅−, Scheme 1, Equation 3),4 hydrogen peroxide (H2O2, Equation 4, or the intermediate peroxyl species, -O-O-),5 hydroxyl radical (⋅OH, Equation 5),6 and singlet oxygen (1O2, Equations 8, 9, and 10, the energy transfer from the excited state of the sensitizer or O2⋅− oxidation product).7,8 Among all the ROS, -O-O- and 1O2 react with styrene via [2 + 2] photochemical cycloaddition and generate styrene oxide as the product.9,10 Yet, H2O2 and ⋅OH induce the olefin bond break and overoxidation reaction of styrene (Scheme 1, Equations 1 and 2),9,11 resulting in a low yield of styrene oxide (<50%) in most catalytic systems (16 catalytic systems are summarized in Table S1). Progress has been made on thermocatalysis and photocatalysis to achieve high epoxidation efficiency. For example, the transition-metal species, especially cobalt-based materials,12, 13, 14 WO3/TiO2 composite,15 CuNP/TiN2 photocatalysts,16 VLB120 enzyme,17 etc., could drive the selective epoxidation of various alkenes with O2 successfully. However, the toxicity of heavy metals and problem of enzyme recycling limit their application. Thus, it is desirable to design metal-free heterogeneous catalysts with high stability and recyclability. Moreover, a deep understanding of the ROS transformation mechanism is critical for catalyst engineering.
Scheme 1.

Proposed Reaction Pathways for the Oxidation of Styrene (Equations 1 and 2) and Oxygen Activation (Equations 3, 4, 5, 6, 7, 8, 9, and 10)
Polymeric carbon nitride has been widely investigated in the activation of molecular oxygen and in driving the corresponding aerobic oxidation reactions.4,5,7,18, 19, 20, 21, 22 It has been reported that the heptazine ring structure in carbon nitride can promote the two-electron reduction of oxygen and adjust H2O2 formation (Scheme 1, Equation 4).23,24 In addition, carbon nitride has exhibited high performance in ⋅OH generation (Equation 5) for photodegradation of organic contaminants.25 However, these ROS are not favorable for the photocatalytic epoxidation of styrene reactions. A large amount of low-value benzaldehyde and benzoic acid are still formed, although various transition-metal cations (Fe3+, Mn2+, Co2+, Ni2+, and Cu2+) are loaded.26 Surface modification of carbon nitride to manipulate the oxygen activation pathway seems to be a promising strategy for ROS regulation.27 For instance, the modification of carbon nitride with C-O-C, C-O, NO2, and C=O as surface groups can enhance 1O2 generation and suppress the production of H2O2 and ⋅OH.7 However, the role of functional groups and the harsh preparation conditions using reflux treatment with concentrated sulfuric/nitric acid limit its application. To date, there is no report on controlling ROS evolution with carbon nitride and then promoting the selectivity of styrene oxide. Thus, it is significantly important in seeking an eco-friendly approach to introduce suitable surface groups to regulate ROS evolution and uncover the reaction mechanism simultaneously.
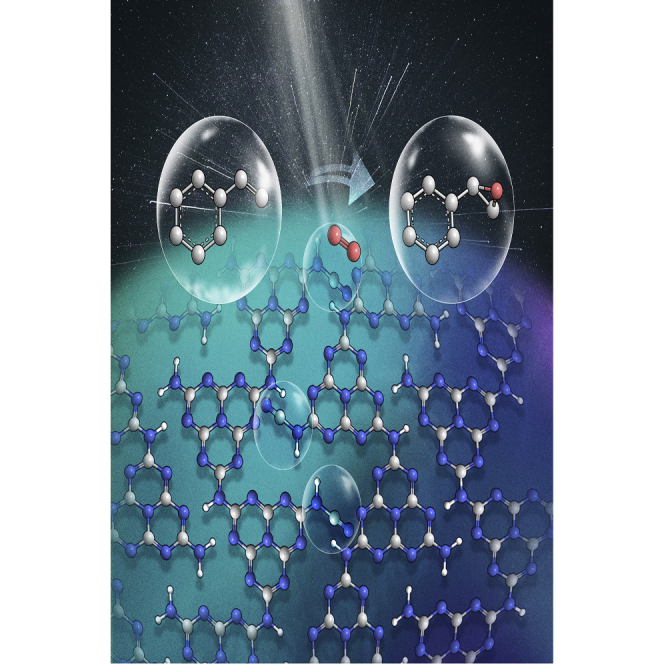
Herein, we report a simple and environmentally friendly method to introduce a surface cyano group onto carbon nitride by co-pyrolyzing trithiocyanuric acid with thiourea (Figure 1A). The cyano-group-modified samples CNCY and CN-T have special spatial structures that can control the transfer of oxygen and inhibit the generation of H2O2 or ⋅OH, achieving a higher styrene oxide yield than the cyano-free sample. The 13C NMR spectrum reveals that the cyano groups bond with the intact heptazine ring. And this unique structure reduces the steric resistance of styrene as attacking the adsorbed 1O2 or -O-O-, which could inhibit the side reaction pathway and prompt the formation of epoxy products. Various characterization techniques and density functional theory (DFT) calculations were used to investigate the reaction mechanism. During the pyrolysis of thiourea, the generated SCN− species28,29 react with the terminal -NH2 forming -C≡N groups, which is the same as the thiocyanuric acid pyrolysis process reported before.4,30 Compared with the traditional method whereby excessive thiocyanate (KSCN, NaSCN, etc.) (Scheme S1) is added, this novel strategy with cyano group introduction avoids using alkali-metal ions.31 This is the first report on the use of thiourea as the cyano group precursor, aimed at preparing a unique space configuration and in turn controlling the ROS evolution. Other precursors, such as dicyandiamide or melamine, are also applicable, which opens broader application of our strategy for cyano group introduction.
Figure 1.

Structure Characterization of Cyano-Group-Modified Carbon Nitride
(A) Proposed cyano introduction mechanism via trithiocyanuric acid and thiourea co-pyrolysis.
(B) XRD patterns of CN, CNCY, and CN-T.
(C) FTIR spectra of CN, CNCY, CN-T, and CN-T0.7.
(D and E) (D) N 1s and (E) C 1s XPS spectra of CN, CNCY, and CN-T.
Results and Discussion
Crystal Structure Changes after Cyano Group Modification and Correlation with the Selectivity Control
The X-ray diffraction (XRD) patterns of cyano-group-modified samples CNCY (prepared using trithiocyanuric as precursor) and CN-T (prepared using trithiocyanuric and thiourea as precursors) show a weaker in-plane peak (100) at 12.8° and interlayer stacking (002) peak at 27.5° compared with those of the cyano-free sample CN (prepared using melamine as precursor) (Figure 1B),32,33 indicating decreased crystallinity after cyano group introduction. The morphology of CN, CNCY, and CN-T was analyzed by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). As shown in Figures S3 and S4, a porous and sheet-like morphology is observed after cyano group introduction. The specific surface area and pore volume of CNCY and CN-T are similar (Figure S5 and Table S2), and are larger than those of the CN sample, in turn providing more active sites and promoting the catalytic reaction.33
The molecular structure change of the samples was studied by Fourier transform infrared (FTIR) spectroscopy. Compared with the CN sample, a new peak around 2,100–2,250 cm−1, which can be identified as the cyano group,31 was observed for CNCY and CN-T. The peak area of the cyano group increases with thiourea addition; however, excessive thiourea will decrease the peak area of the cyano group (Figure 1C). The controlled sample CNth with thiourea as precursor was then prepared, yet exhibits low cyano peak intensity in the FTIR spectroscopy (Figure S6). We speculate that superfluous SCN− fragments from pyrolysis with thiourea will polymerize with each other. Thus, the cyano group can be introduced only by maintaining the appropriate concentration of SCN− fragments in the polymerization process.
The chemical composition and structure of the obtained cyano-group-modified samples were studied by X-ray photoelectron spectroscopy (XPS). The presence of N 1s and C 1s signals of CNCY and CN-T samples indicates the successful introduction of a cyano group in the heptazine. The three peaks centered at 398.6, 400.1, and 401.3 eV (Figure 1D) are attributed to the C-N=C, N-(C)3, and C-N-Hx groups in the heptazine framework, respectively.34 The relative peak area ratios of C-N-H with the total area decrease from 3.8% to 3.4% and 3.0% (calculation method is shown in Table S3) for CN, CNCY, and CN-T samples, respectively, indicating that some C-N-H groups were replaced. It has been reported that the C 1s binding energy of -C≡N at 286.4 eV is similar to that of C-NHx, and the introduction of cyano groups led to the generation of amino (-NH2) groups and an enhanced peak intensity at 286.4 eV.35 However, there is no obvious enhancement in the peak area (Figure 1E) and the peak area ratios between C-NHx and -C≡N and the N-C=N of the three samples were all 0.03 (calculation method is shown in Table S4). These results demonstrate that the cyano group is introduced by replacing the C-N-H groups, while the structure of heptazine remains unchanged. In addition, the absence of sulfur signal (Figure S7) in CNCY and CN-T indicates that all the sulfurs are removed from the samples.
Figure S8 gives the solid-state 13C NMR spectra of CN, CNCY, and CN-T. It can be seen that all samples present two strong signal groups at 156.2 and 164.3 ppm, corresponding to the C(1) atoms of N=C-N(NHx) and the C(3 or 5) atoms of N=C-N2 in the heptazine units, respectively.36 For CN-T, two new peaks at 123.5 and 162.6 ppm can also be clearly observed, which can be assigned to the C(17) atoms of -C≡N groups and the neighbor C(1′) atoms, respectively. In heptazine, the C(1′) atoms at 162.6 ppm are linked to the terminal -NH2 groups.37 It has been reported that a large number of hydrogen bonds exist between the strands of polymeric carbon nitride units,34 and these hydrogen bonds will passivate the NMR peak type. The -NH-C≡N could break the hydrogen bond structure, leading to the emergence of a sharp peak at 162.6 ppm. Therefore, we can safely conclude that the -C≡N group was bonded with the intact heptazine. As we reported previously, the cyano group content can be obtained by titration (Figure S9 and Table S5; the detailed methods are shown in the Supplemental information).30 A good linear relationship between the cyano group content and the yield of styrene oxide was found (Figure 2A), indicating that cyano groups probably act as the active sites in this reaction. The content of cyano carbon in CN-T is only 0.83 wt% based on the cyano group content (1.8 wt%), which might explain its low signal intensity in 13C NMR spectra.
Figure 2.

Catalytic Performance Test of Cyano-Group-Modified Carbon Nitride
(A) Styrene oxide yield as a function of cyano content.
(B) Time profile of the conversion and the selectivity of styrene oxide over CN-T and CN. Reaction conditions: 0.04 mmol of styrene, 2 mL of acetonitrile, 10 mg of catalyst, 1 atm air, 400 mW/cm2 white LED light irradiation, 60 °C, 4 h (for A) and12 h (for B). Conv., conversion; Sel., selectivity for styrene oxide.
Structure-Activity Relationship after Cyano Group Introduction
The results from various temperatures, light intensities, and wavelengths indicate that the epoxidation of styrene is primarily driven by photoinduced electrons (Figures S10 and S11). For semiconductor photocatalysis, the generation and separation efficiency of photogenerated carriers can directly affect the catalytic performance.38 The transient photocurrent, electrochemical impedance spectroscopy, steady-state photoluminescence (PL), and time-resolved PL spectra results all demonstrate that the recombination efficiency of charge carriers for the cyano-modified sample is lower than for the bulk carbon nitride (Figure S12). Yet, in our system, the improvement of charge separation efficiency shows no influence on the selectivity of the products.
In the dynamic experiments, there is only a slight change in the styrene oxide selectivity over CN and CN-T as the reaction is prolonged. Furthermore, at the same conversion rate, CN-T shows higher styrene oxide selectivity than the cyano-free sample (Figure 2B). These results indicate that the selectivity is mainly dependent on the structure of active sites (major factor) rather than the enhancement of photoelectric performance (minor factor). The photoelectric performance of carbon nitride can be significantly improved by heteroatom doping, such as with phosphorus, without changing the terminal group of the sample.39 In the controlled experiment, the phosphorus-doped sample CN-P (prepared using melamine as precursor and 0.25 g ammonium phosphate as doping source), which shows better photoelectric performance than CN, was prepared and investigated (Figure S13A and Table S6). The conversion of styrene was increased, whereas the selectivity of styrene oxide was not improved. Furthermore, CNMT (prepared using 10 g melamine mixed with 0.3 g thiourea as precursor) and CNDT (prepared using 10 g dicyandiamide mixed with 0.3 g thiourea as precursor) were prepared. These samples show higher cyano peak area in the FTIR spectra and better catalytic performance and selectivity for styrene oxide than bulk carbon nitride (CN and CND) (Figure S13). These results further confirm that the cyano group could improve the selectivity of the styrene oxide product.
The band structures of the catalysts were studied and determined by combining the results of UV-Vis spectra, Kubelka-Munk formula, and Mott-Schottky electrochemical test (Figure S14). The CB and VB positions of all samples make them able to activate O2 forming O2⋅− species (Scheme 1, Equation 3), whereas they are unable to directly oxidize water to ⋅OH (Equation 7) (Figure 3A).40 Electron paramagnetic resonance (EPR) is a powerful technique to monitor catalytically relevant species that contain unpaired electrons.41 The in situ EPR spectra in Figure 3B show that the ⋅OH signal intensity27 is the highest for the CN sample and reduces with the increase in cyano group content in CNCYx and CN-T. It is probable that the ⋅OH is formed through the decomposition of H2O2 as follows from Equation 5. The content of H2O2 decreases as the cyano groups increase and a trace of H2O2 was detected for CNCY or CN-T (Figure S15; the H2O2 content was determined by iodometry as shown in the Supplemental information). These results indicate that the introduced cyano groups successfully inhibit the generation of H2O2 and ⋅OH species, while O2⋅−, 1O2, and -O-O- species are probably the main ROS that participate in the epoxidation reaction.
Figure 3.

Characterization of Reactive Oxygen Species
(A) Band structure of CN, CNCY, and CN-T.
(B) In situ EPR spectra. Room-temperature EPR spectra were measured after in situ illumination for 5 min by using DMPO as trap agent.
(C–E) Insitu FTIR spectra of CN-T with the addition of O2 or dual addition of O2 and styrene to observe the formation of ROS (peroxy species) and the reaction pathway on the catalyst surface. The FTIR spectra were obtained in a sealed in situ reaction cell and the dosages of the styrene and oxygen were 20 μL and 5 mL (1 atm), respectively.
(F) Dioxygen adsorption mode on cyano-modified sample, located above (i) N7-C17 or (ii) N7-N18.
To identify the individual role of ROS involved in this study, O2⋅− and 1O2 scavengers were adopted in the reaction system. The reaction was completely terminated after adding 0.05 eq of tetrazolium blue (NBT, O2⋅− scavenger, the dosage is comparable to styrene)4 (Figure S16A). Meanwhile, the conversion rate (Figure S16B) dropped by 45% after adding 9,10-diphenylanthracene42 as a 1O2 trapping agent. This indicates that O2⋅− is the initial active species that can convert to other ROS. In general, 1O2 is generated through an energy transfer process from the exited carbon nitride via dipole-dipole (Förster processes) or direct electron-exchange interactions (Dexter processes).43 Adding NBT can terminate the whole reaction, which indicates that 1O2 is formed via the O2⋅− oxidation process (Equation 10) rather than a conventional energy transfer process.8 We speculate that this transformation path improves the utilization of photogenerated charge carriers, because it consumes electrons and holes subsequently. Only 27% of 1O2 is observed with the cyano-free sample, which illustrates the low utilization efficiency of photogenerated charge carriers. As previously reported, O2⋅− could be adsorbed on the surface of carbon nitride and reduced to -O-O- through a fast two-electron reduction reaction.23,24 Thus the remaining 55% of O2⋅− in the cyano-group-modified sample probably evolves into -O-O- species.
The formation of -O-O- species and the function of cyano groups in this process were confirmed by in situ FTIR spectra measurements. Under illumination, the cyano-group-modified sample exhibits a clear vibration peak around 892 cm−1 (-O-O-)23,44,45 under an oxygen atmosphere (Figure 3C). Meanwhile, the intensity of the cyano group increases gradually with the above peak (Figure 3D, bottom), which can be attributed to the electron transfer between the O2 and the cyano group and the change in dipole moment of the cyano group.25,46 The role of the cyano group as an oxygen transformation site is confirmed by adding 20 μL of styrene into the reaction cell. In the first 20 min, the intensity of the cyano group remains almost unchanged, yet the twisted vibration (990 cm−1) and out-of-plane oscillation (910 cm−1) of =CH2 is decreasing (Figure 3E).47 As time goes on, the intensity of the cyano groups increases (Figure 3D, top) with the increase in the -O-O- (890 cm−1) vibration peak (Figure 3E). These results indicate that the cyano group is the active site for ROS evolution, which could anchor the -O-O- and then facilitate the attack of styrene. The interaction between 1O2 and the cyano groups was studied by DFT calculation. In the cyano-free sample, according to the work reported and our calculation results, 1O2 also prefers to be adsorbed at the N4-C1 site (Figure S17 and Table S7).24 However, the optimized configuration was changed after cyano modification. As shown in Figure 3F, (i) and (ii) are more stable than the other five configurations (Figure S18 and Table S8). This indicates that the interaction of 1O2 with a cyano-group-modified sample is the strongest when the 1O2 is located at the N7-C17 (Figure 3F, i) or N7-N18 (Figure 3F, ii) site with a relative energy of −39.3 and −39.1 kJ/mol, respectively. It is only −28.6 kJ/mol when the 1O2 is located at N4-C1.
According to the above analysis, we can safely conclude that the cyano groups bond with the heptazine ring and act as the optimal adsorption site for -O-O- and 1O2 species. This unique configuration will change the reaction pathway. Traditionally, the -O-O- and 1O2 species are easily adsorbed at C1-N4 (inset of Figure 3F) in the cyano-free sample CN.24 However, owing to the steric hindrance caused by the rigid plane heptazine ring units, it is difficult for the activated styrene to attack the -O-O- or 1O2 species at C1-N4. Furthermore, the 1,4-adsorption mode of -O-O- (i–iii in Scheme 2) in cyano-free CN is favorable for the attack of protons (at positions 2 and 6) forming H2O2.5 Once the -O-O- and 1O2 are adsorbed at the cyano group site, the steric hindrance will be drastically reduced, which will be beneficial for generating olefin oxidation intermediates. Meanwhile, after cyano group introduction, the terminal adsorption mode formed will increase the offensive distance of protons to form H2O2, and also provide a retrospective interpretation of why a trace of H2O2 was detected with cyano-group-modified carbon nitride. Thus, the cyano groups bonded with the heptazine ring (structure B, Table 1) is favorable for enhancing the influence of spatial configuration on product selectivity. For comparison, cyano-group-modified carbon nitrides with different spatial configurations were prepared (structures C and D, post-heating treatment,48 copolymerization with NaCl,49 KOH etching35 and post-treatment using NaBH4;50 structure B, KSCN coheating31). As shown in Table 1, only the samples with B-type structure can increase the yield of styrene oxide. This demonstrates that the spatial configuration determines the reaction selectivity.
Scheme 2.

Proposed Reaction Mechanism for Epoxidation of Styrene over Cyano-Group-Free and Cyano-Group-Modified Carbon Nitride
Table 1.
Photocatalytic Activity over Various Carbon Nitride Catalysts under Visible Lighta
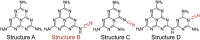 | ||||
|---|---|---|---|---|
| Entry | Sample | Yield (%) | Sample structure | Reference |
| 1 | CN | 19.2 | A | this paper |
| 2 | CN-T | 65.7↑ | B | this paper |
| 3 | CN(KSCN) | 61.3↑ | B | Lau et al.31 |
| 4 | CN | 26.4 | A | Niu et al.48 |
| 5 | CNQ680 | 23.7 | C | Niu et al.48 |
| 6 | Bulk g-C3N4 | 32.3 | A | Yuan et al.49 |
| 7 | Ribbon-like g-C3N4 | 15.9 | C | Yuan et al.49 |
| 8 | g-C3N4 | 36.5 | A | Yu et al.35 |
| 9 | g-C3N4-0.01 | 35.1 | C | Yu et al.35 |
| 10 | CN | 20.3 | A | Liu et al.50 |
| 11 | DCN-200 | 20.1 | D | Liu et al.50 |
The reaction conditions were as follows: 0.04 mmol of styrene, 2 mL of acetonitrile, 10 mg of catalyst, 1 atm air, 400 mW/cm2 white LED light irradiation, 60°C, 24 h. Samples in entries 3–11 were synthesized referring to the literature methods (details shown in the Supplemental information).
Catalyst Stability and Reaction Scale-up under Solar Irradiation
These cyano-group-modified samples exhibit high stability. There is no obvious change in XRD, UV, or FTIR spectra after recycling five times (Figures S19A–S19C). The yield of styrene oxide remains unchanged above 65% (Figure S19D, diamond), which is superior to the catalytic performance of iron, cobalt-based, and zeolites (summarized in Table S1). With isobutyraldehyde as reductant the yield of styrene oxide further increases to 85% (Figure S19D, star), since the isobutyraldehyde can promote the conversion of the olefin oxidation intermediate to the styrene oxide products.10 As shown in Table S9, the cyano-group-modified photocatalyst also exhibits good performance on electron-rich group-containing, aromatic, conjugated, and linear alkenes.
The scale-up reaction driven by solar light was conducted to investigate the performance of the catalyst for industrial application. Figure 4 depicts the scaled-up reaction (styrene was up to 100 g, which was 25,000-fold quality increase compared with the lab-scale reactions) under solar light irradiation. The reaction temperature and solar light intensity ranged from 25 °C to 35 °C and 13 to 85 mW/cm2, respectively. The conversion rate could reach above 99% with a selectivity of >82% in the scaled-up reaction under identical irradiation time (Figure 4B). It should be noted that the conversion rate will decrease on rainy or cloudy days; this result implies that it will be easier to realize the practical application of photocatalysis in drought areas or those with long sunshine time.
Figure 4.

Characterization of Catalytic Performance under Solar Light Irradiation
(A) Photograph of up-scaled reaction with volume of 2.7 L.
(B) Amplification reaction with 100 g styrene and the temperature and light intensity change curve. Catalytic setup was placed on a rooftop (latitude 37°51′ north, longitude 112°32′ east) so that the reactor would be directly irradiated by the sun for the duration of the experiment. Reaction conditions: 100 g of styrene, 2,500 mL of acetonitrile, 200 mL of isobutyraldehyde, 10 g of catalyst, bubble with 1.5 L of oxygen every day, May 28 to June 11, 2020, 25 °C–35 °C, light intensity 13–85 mW/cm2. Conv., conversion; Sel., selectivity.
Based on the above discussion, a possible mechanism is proposed (Scheme 2). The catalyst has an appropriate CB position to activate oxygen forming O2⋅−, of which 45% could convert to 1O2. The 1O2 is preferentially adsorbed on the cyano group, facilitating the attack of styrene. The remaining 55% of the O2⋅− can convert to -O-O- via a fast two-electron reduction process, and the -O-O- could also be adsorbed on the cyano group. The styrene is more easily absorbed in the hole-enriched region, such as 2 and 6 in Scheme 2A,24 and transfer an electron to the VB of the catalyst. The activated styrene will react with the -O-O- or 1O2 to generate a four-membered ring intermediate. In the action of another styrene molecule, two molecules of styrene oxide are finally formed. This process is similar to the transition-metal activation process discussed in the introduction, which can avoid the bond-breaking reaction and then increase the selectivity of styrene oxide.
Conclusions
In conclusion, we successfully designed and synthesized cyano-group-modified carbon nitrides CNCYx and CN-Ty for selective epoxidation of styrene. The cyano-group-modified sample achieved higher styrene yield than the bulk carbon nitride using O2 as oxidant. Solid-state 13C NMR and XPS spectra proved that the cyano group bonded with the heptazine ring. The in situ FTIR spectra and DFT calculations demonstrated that cyano groups could selectively adsorb peroxyl and 1O2 species. This structure could also inhibit the formation of H2O2 and ⋅OH species as well as reducing the steric hindrance beneficial to the interaction between the active site and styrene. Thus, the special spatial structure, which could adjust the transformation of ROS, led to the enhanced photocatalytic epoxidation of styrene for cyano-group-modified carbon nitride. The cyano-group-modified sample can be applied to a variety of substrates and the catalytic system can be scaled up to 2.7 L under solar irradiation. This surface-group-modification strategy highlights that a surface-functionalized carbon nitride photocatalyst could be prepared via a simple and green route, which opens up a new window to design and fabrication of highly active metal-free photocatalysts for solar ray-driven selective controlled organic synthesis.
Materials and Methods
Materials
All chemicals were purchased from Aladdin and used as received without further purification.
Catalyst Preparation
Cyano-Modified Samples (CN-Ty)
Typically, y g (y = 0.1, 0.3, 0.5, and 0.7) of thiourea and 10 g of trithiocyanuric acid were dissolved in 100 mL of deionized water under vigorous stirring. The deionized water was evaporated at 95°C to afford a yellow block mixture powder. The mixture powder was collected and heated in a crucible loaded in a muffle furnace at 550°C for 4 h with a ramp rate of 3°C/min. The samples were named as CN-Ty. The CN-T0.3 sample showed highest catalytic performance (Figure S1) and was also named as CN-T.
Cyano-Modified Samples (CNCYx)
The samples were prepared by the thermal polymerization method as we reported previously.4 Typically, m g of melamine and n g of trithiocyanuric acid (m + n = 10, x = n/(m + n)) were dissolved in 50 mL of deionized water under vigorous stirring. The deionized water was evaporated at 95°C to afford a yellow block mixture powder. After that the mixture powder was treated following the above calcination procedure. Among the samples, CNCY1.00 exhibited the highest catalytic performance in the epoxidation reaction and was abbreviated as CNCY (Figure S1).
Bulk Carbon Nitride (CN)
The bulk carbon nitride was prepared by using melamine as precursor, following the above calcination procedure. The sample was named as CN. Other samples mentioned in this study were prepared by the reported methods (details shown in the Supplemental information).
The above light-yellow product was ground thoroughly into a fine powder using a pestle and mortar prior to various characterizations.
Structural Characterization
Powder XRD patterns were collected using a Bruker D8 Advanced diffractometer operating with a Cu Kα X-ray source (λ = 1.5405 Å). The scan range was from 5° to 60° with the scan rate of 5°/min. FTIR spectra were obtained by a Bruker Tensor II spectrometer in a KBr pellet and the frequency range was 4,000–600 cm−1. SEM was performed using a Hitachi S-4800 microscope with an acceleration voltage of 5 kV. Morphology was assayed using TEM (Tecnai G2 F20 S-Twin) with an accelerating voltage of 200 kV. The specific surface areas were analyzed by a nitrogen adsorption-desorption instrument (TriStar II 3020). Prior to measurement, all the photocatalysts were degassed under evacuation at 120°C for 10 h. The pore size distribution was obtained by desorption isotherms using the Barret-Joyner-Halenda method. UV-Vis diffuse reflectance spectra were obtained on a Shimadzu UV 3600 spectrophotometer using BaSO4 as the reference. XPS was conducted on a USA Thermo ESCALAB 250 spectrometer using a monochromate Al Kα X-ray radiation source (200 W). The binding energy correction was referenced to C 1s peak (284.6 eV) arising from surface hydrocarbons. The spectra deconvolution was carried out using the XPS PEAK41 software package. The solid NMR spectra were performed using a Bruker AV-III 600 MHz wide-bore spectrometer equipped with a 4 mm double-resonance probe. Steady-state PL spectra were performed on a Hitachi F-7000 FL spectrophotometer at room temperature with an excitation wavelength of 365 nm. Time-resolved PL decay curves were recorded on a FLS920 fluorescence lifetime spectrophotometer (Edinburgh Instruments, UK) under excitation of 365 nm and probed at 460 nm. EPR measurements were obtained using a Bruker model EMXPLUS 10/12 spectrometer. The samples were prepared as follows: 1 mg of the catalyst was dispersed in 1 mL of acetonitrile by sonication; 10 μL of the above mixture was mixed with 10 μL of DMPO acetonitrile solution (1 mg/μL). The EPR spectra were measured after in situ illumination for 5 min. The in situ diffuse reflectance infrared experiments were performed on Bruker Tensor II FTIR spectrometer (Figure S2).
Acknowledgments
This work was supported by the National Natural Science Foundation of China (21773284), the Key Laboratory of Interface Science and Engineering in Advanced Materials, the Ministry of Education (KLISEAM201903), and the Hundred Talents Program of the Chinese Academy of Sciences and Shanxi Province.
Declaration of Interests
The authors declare that they have no conflicts of interest to this work.
Published Online: February 5, 2021
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.xinn.2021.100089.
Contributor Information
Xianmo Gu, Email: guxm@sxicc.ac.cn.
Zhanfeng Zheng, Email: zfzheng@sxicc.ac.cn.
Supplemental Information
References
- 1.Lin Y., Pan X., Qi W., et al. Nitrogen-doped onion-like carbon: a novel and efficient metal-free catalyst for epoxidation reaction. J. Mater. Chem. A. 2014;2:12475–12483. doi: 10.1039/C4TA01611D. [DOI] [Google Scholar]
- 2.Alassmy Y.A., Pescarmona P.P. The role of water revisited and enhanced: a sustainable catalytic system for the conversion of CO2 into cyclic carbonates under mild conditions. ChemSusChem. 2019;12:3856–3863. doi: 10.1002/cssc.201901124. [DOI] [PubMed] [Google Scholar]
- 3.Hu T.D., Jiang Y., Ding Y.H. Computational screening of metal-substituted HKUST-1 catalysts for chemical fixation of carbon dioxide into epoxides. J. Mater. Chem. A. 2019;7:14825–14834. doi: 10.1039/c9ta02455g. [DOI] [Google Scholar]
- 4.Tan H., Gu X., Kong P., et al. Cyano group modified carbon nitride with enhanced photoactivity for selective oxidation of benzylamine. Appl. Catal. B. Environ. 2019;242:67–75. doi: 10.1016/j.apcatb.2018.09.084. [DOI] [Google Scholar]
- 5.Kofuji Y., Isobe Y., Shiraishi Y., et al. Carbon nitride–aromatic diimide–graphene nanohybrids: metal-free photocatalysts for solar-to-hydrogen peroxide energy conversion with 0.2% efficiency. J. Am. Chem. Soc. 2016;138:10019–10025. doi: 10.1021/jacs.6b05806. [DOI] [PubMed] [Google Scholar]
- 6.Cui Y., Ding Z., Liu P., et al. Metal-free activation of H2O2 by g-C3N4 under visible light irradiation for the degradation of organic pollutants. Phys. Chem. Chem. Phys. 2012;14:1455–1462. doi: 10.1039/C1CP22820J. [DOI] [PubMed] [Google Scholar]
- 7.Wang H., Jiang S., Chen S., et al. Enhanced singlet oxygen generation in oxidized graphitic carbon nitride for organic synthesis. Adv. Mater. 2016;28:6940–6945. doi: 10.1002/adma.201601413. [DOI] [PubMed] [Google Scholar]
- 8.Nosaka Y., Nosaka A.Y. Generation and detection of reactive oxygen species in photocatalysis. Chem. Rev. 2017;117:11302–11336. doi: 10.1021/acs.chemrev.7b00161. [DOI] [PubMed] [Google Scholar]
- 9.Al-Ajlouni A.M., Espenson J.H. Epoxidation of styrenes by hydrogen peroxide as catalyzed by methylrhenium trioxide. J. Am. Chem. Soc. 1995;117:9243–9250. doi: 10.1021/ja00141a016. [DOI] [Google Scholar]
- 10.Frimer A.A. The reaction of singlet oxygen with olefins: the question of mechanism. Chem. Rev. 1979;79:359–387. doi: 10.1021/cr60321a001. [DOI] [Google Scholar]
- 11.Barak G., Sasson Y. Dual-function phase-transfer catalysis in the metal-assisted oxidation by hydrogen peroxide of styrene to benzaldehyde or acetophenone. J. Chem. Soc. Chem. Commun. 1987:1266–1267. doi: 10.1039/C39870001266. [DOI] [Google Scholar]
- 12.Sebastian J., Jinka K.M., Jasra R.V. Effect of alkali and alkaline earth metal ions on the catalytic epoxidation of styrene with molecular oxygen using cobalt(II)-Exchanged zeolite X. J. Catal. 2006;244:208–218. doi: 10.1016/j.jcat.2006.09.005. [DOI] [Google Scholar]
- 13.Rahman S., Santra C., Kumar R., et al. Highly active Ga promoted Co-HMS-X catalyst towards styrene epoxidation reaction using molecular O2. Appl. Catal. A Gen. 2014;482:61–68. doi: 10.1016/j.apcata.2014.05.024. [DOI] [Google Scholar]
- 14.Liu J., Meng R., Li J., et al. Achieving high-performance for catalytic epoxidation of styrene with uniform magnetically separable CoFe2O4 nanoparticles. Appl. Catal. B. Environ. 2019;254:214–222. doi: 10.1016/j.apcatb.2019.04.083. [DOI] [Google Scholar]
- 15.Caudillo-Flores U., Munoz-Batista M.J., Hungria A.B., et al. Toluene and styrene photo-oxidation quantum efficiency: comparison between doped and composite tungsten-containing anatase-based catalysts. Appl. Catal. B. Environ. 2019;245:49–61. doi: 10.1016/j.apcatb.2018.12.032. [DOI] [Google Scholar]
- 16.Huang Y., Liu Z., Gao G., et al. Stable copper nanoparticle photocatalysts for selective epoxidation of alkenes with visible light. ACS Catal. 2017;7:4975–4985. doi: 10.1021/acscatal.7b01180. [DOI] [Google Scholar]
- 17.van Schie M.M.C.H., Paul C.E., Arends I.W.C.E., Hollmann F. Photoenzymatic epoxidation of styrenes. Chem. Commun. 2019;55:1790–1792. doi: 10.1039/C8CC08149B. [DOI] [PubMed] [Google Scholar]
- 18.Zheng Y., Jiao Y., Chen J., et al. Nanoporous graphitic-C3N4@Carbon metal-free electrocatalysts for highly efficient oxygen reduction. J. Am. Chem. Soc. 2011;133:20116–20119. doi: 10.1021/ja209206c. [DOI] [PubMed] [Google Scholar]
- 19.Deng X., Cao H., Chen C., et al. Organotellurium catalysis-enabled utilization of molecular oxygen as oxidant for oxidative deoximation reactions under solvent-free conditions. Sci. Bull. 2019;64:1280–1284. doi: 10.1016/j.scib.2019.07.007. [DOI] [PubMed] [Google Scholar]
- 20.Xiong X., Wang Z., Zhang Y., et al. Wettability controlled photocatalytic reactive oxygen generation and Klebsiella pneumoniae inactivation over triphase systems. Appl. Catal. B. Environ. 2020;264:118518. doi: 10.1016/j.apcatb.2019.118518. [DOI] [Google Scholar]
- 21.Wang X., Zhou C., Shi R., et al. Supramolecular precursor strategy for the synthesis of holey graphitic carbon nitride nanotubes with enhanced photocatalytic hydrogen evolution performance. Nano Res. 2019;12:2385–2389. doi: 10.1007/s12274-019-2357-0. [DOI] [Google Scholar]
- 22.Wang Z.-T., Xu J.-L., Zhou H., Zhang X. Facile synthesis of Zn(II)-doped g-C3N4 and their enhanced photocatalytic activity under visible light irradiation. Rare Met. 2019;38:459–467. doi: 10.1007/s12598-019-01222-5. [DOI] [Google Scholar]
- 23.Shiraishi Y., Kanazawa S., Kofuji Y., et al. Sunlight-driven hydrogen peroxide production from water and molecular oxygen by metal-free photocatalysts. Angew. Chem. Int. Ed. 2014;53:13454–13459. doi: 10.1002/anie.201407938. [DOI] [PubMed] [Google Scholar]
- 24.Shiraishi Y., Kofuji Y., Sakamoto H., et al. Effects of surface defects on photocatalytic H2O2 production by mesoporous graphitic carbon nitride under visible light irradiation. ACS Catal. 2015;5:3058–3066. doi: 10.1021/acscatal.5b00408. [DOI] [Google Scholar]
- 25.Liu M., Gao M., Pei L., et al. Tailoring phenol photomineralization pathway over polymeric carbon nitride with cyano group multifunctional active sites. Appl. Catal. B. Environ. 2021;284:119710–119717. doi: 10.1016/j.apcatb.2020.119710. [DOI] [Google Scholar]
- 26.Ding Z., Chen X., Antonietti M., Wang X. Synthesis of transition metal-modified carbon nitride polymers for selective hydrocarbon oxidation. ChemSusChem. 2011;4:274–281. doi: 10.1002/cssc.201000149. [DOI] [PubMed] [Google Scholar]
- 27.Xu S., Zhou P., Zhang Z., et al. Selective oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid using O2 and a photocatalyst of Co-thioporphyrazine bonded to g-C3N4. J. Am. Chem. Soc. 2017;139:14775–14782. doi: 10.1021/jacs.7b08861. [DOI] [PubMed] [Google Scholar]
- 28.Wang S., Gao Q.Y., Wang J.C. Thermodynamic analysis of decomposition of thiourea and thiourea oxides. J. Phys. Chem. B. 2005;109:17281–17289. doi: 10.1021/jp051620v. [DOI] [PubMed] [Google Scholar]
- 29.Zhang G., Zhang J., Zhang M., Wang X. Polycondensation of thiourea into carbon nitride semiconductors as visible light photocatalysts. J. Mater. Chem. 2012;22:8083–8091. doi: 10.1039/c2jm00097k. [DOI] [Google Scholar]
- 30.Tan H., Kong P., Liu M., et al. Enhanced photocatalytic hydrogen production from aqueous-phase methanol reforming over cyano-carboxylic bifunctionally-modified carbon nitride. Chem. Commun. 2019;55:12503–12506. doi: 10.1039/C9CC06600D. [DOI] [PubMed] [Google Scholar]
- 31.Lau V.W.H., Moudrakovski I., Botari T., et al. Rational design of carbon nitride photocatalysts by identification of cyanamide defects as catalytically relevant sites. Nat. Commun. 2016;7:12165–12174. doi: 10.1038/ncomms12165. https://www.nature.com/articles/ncomms12165#supplementary-information [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ruan L.-W., Zhu Y.-J., Qiu L.-G., et al. First principles calculations of the pressure affection to g-C3N4. Comp. Mater. Sci. 2014;91:258–265. [Google Scholar]
- 33.Cheng J., Hu Z., Lv K., et al. Drastic promoting the visible photoreactivity of layered carbon nitride by polymerization of dicyandiamide at high pressure. Appl. Catal. B. Environ. 2018;232:330–339. doi: 10.1016/j.apcatb.2018.03.066. [DOI] [Google Scholar]
- 34.Kang Y., Yang Y., Yin L.-C., et al. An amorphous carbon nitride photocatalyst with greatly extended visible-light-responsive range for photocatalytic hydrogen generation. Adv. Mater. 2015;27:4572–4577. doi: 10.1002/adma.201501939. [DOI] [PubMed] [Google Scholar]
- 35.Yu H., Shi R., Zhao Y., et al. Alkali-assisted synthesis of nitrogen deficient graphitic carbon nitride with tunable band structures for efficient visible-light-driven hydrogen evolution. Adv. Mater. 2017;29:1605148–1605156. doi: 10.1002/adma.201605148. [DOI] [PubMed] [Google Scholar]
- 36.Zhao D., Dong C.-L., Wang B., et al. Synergy of dopants and defects in graphitic carbon nitride with exceptionally modulated band structures for efficient photocatalytic oxygen evolution. Adv. Mater. 2019;31:1903545–1903554. doi: 10.1002/adma.201903545. [DOI] [PubMed] [Google Scholar]
- 37.Lotsch B.V., Döblinger M., Sehnert J., et al. Unmasking melon by a complementary approach employing electron diffraction, solid-state NMR spectroscopy, and theoretical calculations—structural characterization of a carbon nitride polymer. Chem. Eur. J. 2007;13:4969–4980. doi: 10.1002/chem.200601759. [DOI] [PubMed] [Google Scholar]
- 38.Savateev A., Ghosh I., König B., Antonietti M. Photoredox catalytic organic transformations using heterogeneous carbon nitrides. Angew. Chem. Int. Ed. 2018;57:15936–15947. doi: 10.1002/anie.201802472. [DOI] [PubMed] [Google Scholar]
- 39.Guo S., Deng Z., Li M., et al. Phosphorus-doped carbon nitride tubes with a layered micro-nanostructure for enhanced visible-light photocatalytic hydrogen evolution. Angew. Chem. Int. Ed. 2016;55:1830–1834. doi: 10.1002/anie.201508505. [DOI] [PubMed] [Google Scholar]
- 40.Huang S., Xu Y., Zhou T., et al. Constructing magnetic catalysts with in-situ solid-liquid interfacial photo-fenton-like reaction over Ag3PO4@NiFe2O4 composites. Appl. Catal. B. Environ. 2018;225:40–50. doi: 10.1016/j.apcatb.2017.11.045. [DOI] [Google Scholar]
- 41.Brückner A. In situ electron paramagnetic resonance: a unique tool for analyzing structure–reactivity relationships in heterogeneous catalysis. Chem. Soc. Rev. 2010;39:4673–4684. doi: 10.1039/B919541F. [DOI] [PubMed] [Google Scholar]
- 42.Corey E.J., Taylor W.C. A study of the peroxidation of organic compounds by externally generated singlet oxygen molecules. J. Am. Chem. Soc. 1964;86:3881–3882. doi: 10.1021/ja01072a062. [DOI] [Google Scholar]
- 43.Kovalev D., Fujii M. Silicon nanocrystals: photosensitizers for oxygen molecules. Adv. Mater. 2005;17:2531–2544. doi: 10.1002/adma.200500328. [DOI] [Google Scholar]
- 44.Zhang M., de Respinis M., Frei H. Time-resolved observations of water oxidation intermediates on a cobalt oxide nanoparticle catalyst. Nat. Chem. 2014;6:362–367. doi: 10.1038/nchem.1874. https://www.nature.com/articles/nchem.1874#supplementary-information [DOI] [PubMed] [Google Scholar]
- 45.Shiraishi Y., Kanazawa S., Sugano Y., et al. Highly selective production of hydrogen peroxide on graphitic carbon nitride (g-C3N4) photocatalyst activated by visible light. ACS Catal. 2014;4:774–780. doi: 10.1021/cs401208c. [DOI] [Google Scholar]
- 46.Nicu V.P., Autschbach J., Baerends E.J. Enhancement of IR and VCD intensities due to charge transfer. Phys. Chem. Chem. Phys. 2009;11:1526–1538. doi: 10.1039/B816151H. [DOI] [PubMed] [Google Scholar]
- 47.Masson J.F., Pelletier L., Collins P. Rapid FTIR method for quantification of styrene-butadiene type copolymers in bitumen. J. Appl. Polym. Sci. 2001;79:1034–1041. doi: 10.1002/1097-4628(20010207)79:63.3. [DOI] [Google Scholar]
- 48.Niu P., Qiao M., Li Y., et al. Distinctive defects engineering in graphitic carbon nitride for greatly extended visible light photocatalytic hydrogen evolution. Nano Energy. 2018;44:73–81. doi: 10.1016/j.nanoen.2017.11.059. [DOI] [Google Scholar]
- 49.Yuan B., Chu Z., Li G., et al. Water-soluble ribbon-like graphitic carbon nitride (g-C3N4): green synthesis, self-assembly and unique optical properties. J. Mater. Chem. C. 2014;2:8212–8215. doi: 10.1039/C4TC01421A. [DOI] [Google Scholar]
- 50.Liu G., Zhao G., Zhou W., et al. In situ bond modulation of graphitic carbon nitride to Construct p–n homojunctions for enhanced photocatalytic hydrogen production. Adv. Funct. Mater. 2016;26:6822–6829. doi: 10.1002/adfm.201602779. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.