

Abstract
Co-signaling receptors for the T cell receptor (TCR) are important therapeutic targets, with blockade of co-inhibitory receptors such as PD-1 now central in immuno-oncology. Advancing additional therapeutic immune modulation approaches requires understanding ligand regulation of other co-signaling receptors. One poorly understood potential therapeutic target is TIM-3 (T cell immunoglobulin and mucin domain containing-3). Which of TIM-3's several proposed regulatory ligands is/are relevant for signaling is unclear, and different studies have reported TIM-3 as a co-inhibitory or co-stimulatory receptor in T cells. Here, we show that TIM-3 promotes NF-κB signaling and IL-2 secretion following TCR stimulation in Jurkat cells, and that this activity is regulated by binding to phosphatidylserine (PS). TIM-3 signaling is stimulated by PS exposed constitutively in cultured Jurkat cells, and can be blocked by mutating the PS-binding site or by occluding this site with an antibody. We also find that TIM-3 signaling alters CD28 phosphorylation. Our findings clarify the importance of PS as a functional TIM-3 ligand, and may inform the future exploitation of TIM-3 as a therapeutic target.
Keywords: phosphatidylserine, phosphorylation/dephosphorylation, receptors, signalling, T-cells, TIM-3
Introduction
The functional outcome when an antigen engages the T cell receptor (TCR) depends on the activity of a wide range of ‘co-signaling receptors’ in T cells [1], which can be stimulatory (like CD28) or inhibitory (like CTLA-4 and PD-1). Co-stimulatory receptors promote T cell activity and play roles in priming naïve T cells or forming memory T cells. Conversely, co-inhibitory receptors restrain T cell activity and are important for immunological homeostasis — preventing autoimmunity under normal circumstances but also allowing tumors to evade immune responses in cancer. Both classes of co-receptor offer important opportunities in immunotherapy, including suppression of co-stimulatory receptor signaling in autoimmunity [2] and suppression of co-inhibitory receptors — or immune checkpoint blockade (ICB) — in cancer [3,4]. The key regulatory ligands are known for most co-signaling receptors currently targeted therapeutically, as is the designation of the receptor as co-inhibitory or co-stimulatory [5]. An important exception to this is TIM-3, or T cell immunoglobulin and mucin domain containing-3, for which multiple ligands have been proposed and both co-stimulatory and co-inhibitory activities have been described [6–8]. Nonetheless, pre-clinical studies have indicated that antibody blockade of TIM-3, in combination with PD-1 blockade, may be a promising therapeutic approach in cancer [9,10]. Several TIM-3 antibodies are now in clinical trials [11], underlining the need to understand this co-receptor mechanistically.
TIM-3 was first identified as a marker of CD4 T helper 1 (TH1) cells and CD8 cytotoxic T (TC1) cells [12], and was later identified on exhausted T cells in chronic viral infection and cancer [13–15]. Early in vivo studies led to the suggestion of a co-inhibitory signaling role for TIM-3 in T cells [16,17]. Indeed, blocking TIM-3 engagement in mice with antibodies or soluble TIM-3 extracellular domain was found to increase TH1 cell proliferation, and TIM-3 deficient mice showed defects in immune tolerance. Very recent studies, however, have revealed that the inhibitory effects of TIM-3 on anti-tumor immunity actually originate in dendritic cells, and not T cells [18]. In fact, most published in vitro studies in T cells indicate a co-stimulatory rather than inhibitory function for TIM-3 in TCR signaling [6,19–22] — although experimental support for co-inhibitory signaling has also been reported [23,24]. TIM-3 does not have a definable intracellular ITIM (immunoreceptor tyrosine-based inhibitory motif) or ITSM (immunoreceptor tyrosine-based switch motif), motifs that normally characterize co-inhibitory receptors and recruit SH2 domain-containing phosphatases to reduce T cell signaling [25].
Adding further to the complexity of understanding TIM-3, several different regulatory ligands have been reported. The first was the lectin family member galectin-9 [26], which has two β-galactoside-binding carbohydrate-recognition domains. Galectin-9 is thought to induce T cell death by binding to carbohydrates on TIM-3, although other work has refuted this [27,28]. The glycoprotein CEACAM1/CD66a and the alarmin HMGB1 have also been reported as TIM-3 ligands [8], but their mechanism and relevance are not yet clear. Another major TIM-3 ligand is the membrane phospholipid phosphatidylserine (PS), exposed on the surface of cells undergoing apoptosis and other processes [29,30], including T cell activation [31,32]. PS was initially suggested as a TIM-3 ligand based on homology between TIM-3 and the known PS receptor TIM-4 [33]. Crystallographic and binding studies have since confirmed that TIM-3 binds PS [34], and TIM-3 can also facilitate binding to and engulfment of apoptotic cells (efferocytosis) by macrophages like its relatives TIM-1 and TIM-4 [34–36]. Importantly, however, the role played by PS binding in modulating TIM-3 function in T cells has not been elucidated — although it was recently reported that the epitopes bound by immunomodulatory TIM-3 antibodies all overlap with the PS-binding site on TIM-3 [37].
Here, we explored the importance of PS in regulating the effects of TIM-3 on TCR signaling, using a Jurkat cell model. We asked whether PS is a key regulatory ligand for TIM-3's co-receptor function, beyond its role in promoting the engulfment of apoptotic cells when TIM-3 is expressed on macrophages. We found that the co-stimulatory effect of TIM-3 on TCR signaling in Jurkat cells requires the TIM-3 extracellular region, suggesting ligand-dependent regulation. Furthermore, we showed that TIM-3's co-stimulatory signaling is blocked by mutations that prevent PS binding or by an antibody that binds the PS-binding site. Thus, endogenous PS in this culture system appears to promote TIM-3 effects on TCR signaling. Our findings argue that TIM-3 function as a co-receptor specifically depends on PS binding, which may have important mechanistic implications for therapeutic targeting of this receptor.
Materials and methods
Cell culture
The NF-κB/Jurkat/GFPTM Transcriptional Reporter cell line was obtained from System Biosciences, and Raji B cells were obtained from the American Type Culture Collection. NF-κB reporter Jurkat cells and Raji cells were cultured in RPMI-1640 media supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. HEK293 LTV cells (Cell Biolabs Inc.), used to generate lentivirus, were cultured in DMEM supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. Human peripheral blood mononuclear cells (PBMCs) were a generous gift from the laboratory of Susan Kaech (Yale University). PBMCs were cultured in RPMI-1640 media supplemented with 10% FBS, 2 mM GlutaMAX (ThermoFisher Scientific), 50 μM beta-mercaptoethanol, 100 U/ml penicillin, and 100 μg/ml streptomycin. NF-κB reporter Jurkat, Raji, HEK293 LTV, and PBMCs were cultured at 37°C with 5% CO2 in a humidified environment. Expi293F cells used for protein expression (ThermoFisher Scientific) were cultured in Expi293 media (ThermoFisher Scientific) at 37°C in a humidified environment containing 8% CO2 while shaking.
Plasmids
pCDEF3-hTIM-3 (Addgene plasmid # 49212) contained the natural TIM-3 variant L119, which was corrected to R119 by site-directed mutagenesis. Full-length human TIM-3 (R119) was then sub-cloned into pcDNA3.1(+) using Gibson assembly [38]. Human PD-1 in pENTR223 [39] was provided by Aaron Ring at Yale University. The lentivirus transfer plasmid was provided by the laboratory of Mandar Muzumdar at Yale University and contained AmpR, PuroR under the SV40 promoter, and the gene of interest under the PGK promoter [40]. cDNA fragments encoding human TIM-3 and human PD-1 were sub-cloned into the lentivirus transfer plasmid. Lentivirus envelope and packaging plasmids pMD2.G and pCMV delta R8.2 were from Addgene (plasmids # 12259 and 12263). Plasmids containing human TIM-1 and human TIM-4 were obtained from Origene. For expression of extracellular regions, cDNA fragments encoding the ECRs of human TIM-3, TIM-1, and TIM-4 were sub-cloned into pcDNA3.1(+) using Gibson assembly, introducing a C-terminal hexa-histidine tag.
TIM-1–3, TIM-4-3, and PD-1/TIM-3 chimeras were constructed in the lentivirus transfer vector using Gibson assembly. The TIM-3F40A and TIM-3I96A/M97A variants were generated by QuikChange site-directed mutagenesis (Agilent) in pcDNA3.1 (+) for ECR protein expression and in the lentivirus transfer vector for cellular studies. Sequencing was performed to validate all plasmids before use.
Antibodies
Cells were stained for flow cytometry analysis with phycoerythrin-conjugated forms of anti-TIM-3 (R & D Systems, catalog #FAB2365P), anti-PD-1 (BioLegend, catalog #329905), anti-TIM-1 (BioLegend, catalog #353903), anti-TIM-4 (BioLegend, catalog #354003), anti-PD-L1 (also labeled with Dazzle 594: BioLegend, catalog #329731), or with an unlabeled TIM-3 primary antibody (R & D Systems, catalog #AF2365) using a phycoerythrin-conjugated anti-goat secondary antibody for detection (R & D Systems, catalog #F0107). PBMCs were stained with phycoerythrin-Dazzle 594-labeled anti-CD3 (BioLegend, catalog #317345), PerCP/Cy5.5TM-labeled anti-CD8 (BioLegend, catalog #301031), BB515-labeled anti-CD4 (BD Biosciences, catalog #564420), and phycoerythrin-labeled anti-TIM-3 (R & D Systems, catalog #FAB2365P). Western blot analysis was performed with anti-Zap70 pY319 (Cell Signaling Technology, catalog #2701), anti-LAT pY220 (Cell Signaling Technology, catalog #3584), anti-PLCγ pY783 (Cell Signaling Technology, catalog #14008), anti-ERK1/2 pT202/pY204 (Cell Signaling Technology, catalog #9106), anti-Grb2 (Cell Signaling Technology, catalog #3972), anti-CD28 pY218 (Sigma–Aldrich, catalog #SAB4504133), anti-CD28 pY191 (Cell Signaling Technology, catalog #16399), anti-AKT pT308 (Cell Signaling Technology, catalog #2965), anti-TIM-3 (R & D Systems, catalog #AF2365), anti-TIM-3 (Abcam, catalog #ab241332), and anti-PD-1 (Cell Signaling Technology, catalog #86163). For TCR stimulation, anti-CD3 clone OKT3 (BioLegend, catalog #317326) and anti-CD28 clone CD28.2 (BioLegend, catalog #302943) were used. For functional studies, anti-TIM-3 clone F38.2E2 (ThermoFisher Scientific, catalog #16-3109-85) was used.
Lentivirus transduction
Lentiviruses were generated by co-transfecting HEK293 LTV cells with lentivirus transfer, envelope, and packaging plasmids using the Mirus Bio TransIT-Lenti Transfection Reagent, according to the manufacturer's protocol. Briefly, HEK293 LTV cells were cultured to 80–95% confluency. Plasmids were mixed with Opti-MEM and TransIT-Lenti Transfection Reagent and were incubated at room temperature for 10 min before being added to HEK293 LTV cells and incubating at 37°C, 5% CO2. After 48 h, the virus-containing medium was collected and filtered through a 0.22 μm filter. Filtered virus suspension was then added to Jurkat cells (2 × 105 cells), and cells were transduced with lentivirus by spinfection (spinning at 800×g for 30 min at 32°C) [41] in sealed aerosol containment lids. Three days after transfection, the medium was replaced and cells were selected with puromycin (1 μg/ml) for 2 days. After selection, the medium was replaced again. Following selection and recovery, receptor expression was verified by flow cytometry. Cells were washed in FACS Buffer: 10% FBS plus 0.1% NaN3 in phosphate-buffered saline (PBS), 0.22 μm filtered. They were then stained with antibodies in the dark. Cells were rewashed and then analyzed on a FACSMelody (Becton Dickinson), with a four-color set up (488 nm and 561 nm lasers with 527/32, 700/54, 582/15, and 613/18 filter sets).
Expression of TIM-3 on peripheral blood mononuclear cells
After overnight recovery, thawed PBMCs were transferred to a 24-well plate coated with 1 μg/ml αCD3, and soluble αCD28 (1 μg/ml) was added to the culture to promote outgrowth of T cells. Cells were maintained between 0.25 and 0.5 × 106 cells per cm2 with αCD3/αCD28 stimulation for 7 days to promote expression of TIM-3 [42]. After 7 days, PBMCs were washed in FACS Buffer (10% FBS plus 0.1% NaN3 in PBS, 0.22 μm filtered). PBMCs were then stained with labeled anti-CD3, anti-CD8, anti-CD4, and anti-TIM-3. Cells were washed again in FACS Buffer before analysis on a FACSMelody. PBMCs were first subjected to doublet discrimination before analyzing for expression of cell surface markers, as shown in Supplementary Figure S1A–E.
NF-κB reporter assay
NF-κB GFP reporter Jurkat cells were resuspended in RPMI-1640 media with 0% FBS and were serum starved for 4 h. After starvation, cells were counted and 5 × 105 cells were plated in a V-bottom 96-well plate and stimulated with αCD3/αCD28 (1 μg/ml each, or 0.5 μg/ml for αCD28), with additional treatments as stated in figure legends. In experiments investigating effects of anti-TIM-3 (F38.2E2), cells were pre-treated with TIM-3 antibody for 1 h before stimulation with αCD3/αCD28. Cells were returned to the incubator at 37°C, 5% CO2 for 16 h. Cells were then washed in FACS Buffer, stained with antibodies, and rewashed prior to analysis using the FACSMelody. Data were analyzed using FlowJo software. GFP signal was quantified for single cells, as determined through SSC-H vs. SSC-W and FSC-H vs. FSC-W gates.
Analysis of IL-2 secretion
NF-κB GFP reporter Jurkat cells were resuspended in RPMI-1640 media with 0% FBS and were serum starved for 4 h. After starvation, cells were counted and 5 × 105 cells were plated in a V-bottom 96-well plate. Cells were stimulated with 1 μg/ml αCD3 plus 0.5 or 1 μg/ml αCD28, and returned to the incubator at 37°C, 5% CO2, for 16 h. In experiments analyzing the effect of anti-TIM-3 (F38.2E2), cells were pre-treated with anti-TIM-3 for 1 h before stimulation with αCD3/αCD28. In experiments using Raji B cell-based stimulation, Raji B cells were loaded with 30 ng/ml Staphylococcal enterotoxin E (Toxin Technology Inc., catalog #ET404) for 30 min at 37°C and were washed in serum-free RPMI-1640 media to remove excess toxin. Jurkat NF-κB GFP reporter cells were serum starved in RPMI-1640 media with 0% FBS for 3 h. After starvation, 2 × 105 Jurkat cells were mixed with 105 SEE-loaded Raji B cells per well in a 96-well U-bottom plate, as described [43]. The plate was centrifuged for 1 min at 300×g to initiate cell contact, and the plate was returned to the incubator at 37°C, 5% CO2, for 6 h. Following antibody or cell-based stimulation, medium was collected for analysis of IL-2 secretion by ELISA (R & D Systems, cat #D2050), following the IL-2 Quantikine ELISA manual. Experiments were performed in technical duplicate and biological triplicate.
Western blot analysis
NF-κB GFP reporter Jurkat cells were serum starved and stimulated as above, with additional treatments as stated in figure legends. Where wortmannin was used (in Figure 2C), cells were pre-treated with the drug for 1 h before stimulation. Cells were pelleted by centrifugation (800×g for 1.5 min) in the last 1.5 min of stimulation. The supernatant was removed, and cells were lysed in ice-cold Lysis Buffer (Cell Signaling Technology) with freshly added cOmplete protease inhibitor (Roche) and PhosSTOP phosphatase inhibitor (Roche). Lysates were clarified by centrifugation (10 000×g for 10 min at 4°C), and clarified lysate was isolated. Lysates were mixed with NuPAGE LDS Sample Buffer (Invitrogen) plus dithiothreitol (DTT) and were boiled for 8 min. Samples were then analyzed by SDS–PAGE using NuPAGE 4–12% Bis-Tris gels. Gels were transferred to 0.22 μm nitrocellulose membranes using the Xcell Surelock Electrophoresis system (ThermoFisher Scientific). To probe multiple proteins simultaneously without requiring stripping and reprobing, the multistrip Western blotting procedure was used [44], in which horizontal strips are excised from the nitrocellulose membrane corresponding to molecular mass guides to allow probing of one blot with several antibodies. Membranes were blocked with 4% BSA in Tris-buffered saline plus Tween20 (TBS-T), probed with primary antibody for 1 h to overnight, and then probed with HRP-tagged secondary antibodies for 1 h. Detection was by enhanced chemiluminescence using SuperSignal Western Pico PLUS Chemiluminescent substrate (ThermoFisher Scientific), with visualization and quantitation using a Kodak Image Station (Kodak Scientific).
Figure 2. TIM-3 modulates CD28 phosphorylation after αCD3/αCD28 stimulation.
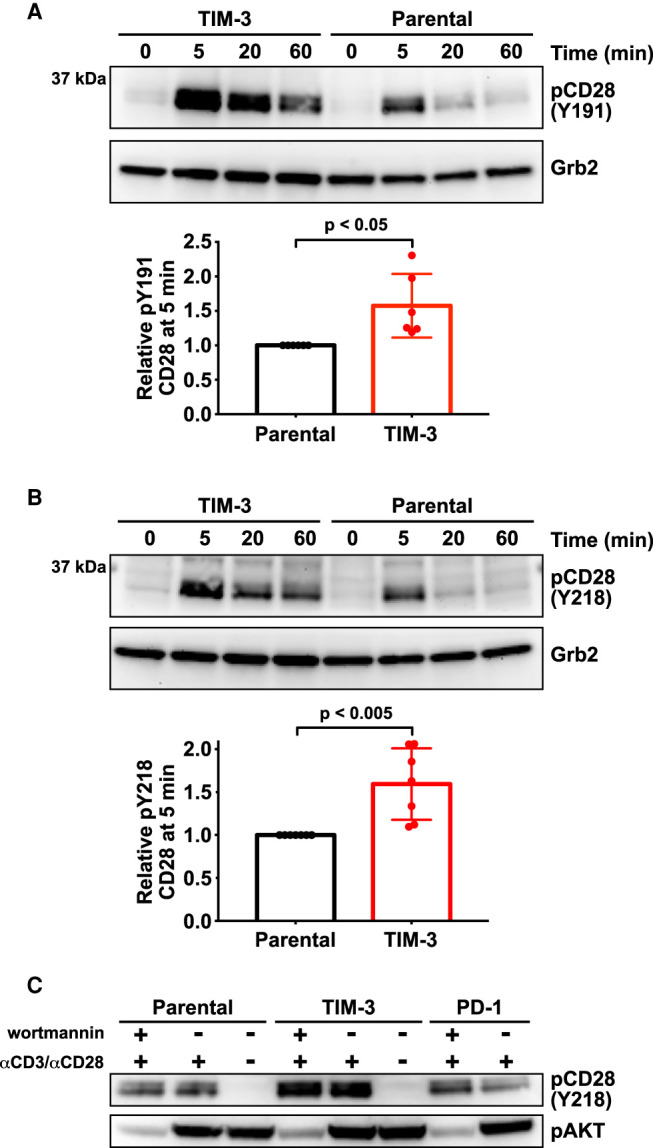
Changes in CD28 phosphorylation following TCR activation with αCD3/αCD28 as in Figure 1 were monitored by Western blotting using phospho-specific antibodies. (A) Representative Western blot of pY191 in CD28 for TIM-3+ and parental NF-κB reporter Jurkat cells. Grb2 was used as a loading control as described [44]. Quantification bar graphs indicate mean band intensity ± SD (n = 6 biological repeats). Two-tailed, unpaired Student's t-test was used to determine P-values. (B) Representative Western blot of pY218 in CD28 for TIM-3+ and parental NF-κB reporter cells, with quantitation below the blot. Grb2 was used as a loading control. Bars indicate mean band intensity ± SD for the 5 min time point (seven biological repeats) with P-values determined by two-tailed, unpaired Student's t-test. (C) Parental, TIM-3+, and PD-1-expressing NF-κB reporter Jurkat cells were stimulated for 5 min with αCD3/αCD28 (1 μg/ml each) with- or without the PI3K inhibitor, wortmannin (0.2 μM) to show that PTEN deficiency does not account for the effect of TIM-3 expression on CD28 phosphorylation.
Detection of annexin V staining by flow cytometry
PerCP-CyTM5.5 or FITC-labeled annexin V were obtained from BioLegend (catalog #640936 and 640905). As specified by the manufacturer, cells were washed twice with ice-cold PBS and then resuspended in 10 mM HEPES pH 7.5, 140 mM NaCl, 2.5 mM CaCl2 (binding buffer). Annexin V-PerCP-Cy5.5 or -FITC was added to each sample and incubated for 15 min in the dark. Additional binding buffer was added to each sample, followed by analysis using FACSMelody (Becton Dickinson) and FlowJo software.
Detection of annexin V staining by widefield fluorescence microscopy
As detailed above, Jurkat NF-κB GFP reporter cells were washed twice with ice-cold PBS and resuspended in annexin V binding buffer (10 mM HEPES pH 7.5, 140 mM NaCl, 2.5 mM CaCl2). Annexin V-AlexaFluor® 647 (BioLegend catalog #640911) was added to cells and incubated for 15 min in the dark. Cells were fixed in 4% paraformaldehyde in PBS with 2.5 mM CaCl2 for 15 min. Annexin V-stained cells were cytospun onto microscope slides at 800 RPM for 5 min. Cells were mounted using ProLongTM Diamond Antifade Mountant with DAPI (ThermoFisher Scientific, catalog #P36966) per the manufacturer's instructions. Images were collected with a Nikon Eclipse Ti2 widefield fluorescence microscope using a Nikon Plan Apo λ 40 × 0.95 N.A. objective. Excitation was provided by a Sola light engine with DAPI or Cy5 filter cube sets. Emission was collected by an sCMOS pco.edge camera. Images were analyzed with FIJI software.
Protein expression and purification
As specified in the Expi293 Expression System manual, Expi293F cells were cultured to reach a density of 4.5–5.5 × 106 cells/ml on the day of transfection, and seeded at 3 × 106 cells/ml. A transfection mixture containing DNA vector (1 μg per ml of culture to be infected), Expifectamine reagent (ThermoFisher Scientific), and Opti-MEM was then incubated at room temperature for 10 min and subsequently added to the culture. Cells were returned to the humidified incubator at 37°C, 8% CO2. Expi293 enhancers (ThermoFisher Scientific) were added 18–22 h after transfection, and the culture was harvested 4–6 days later by centrifugation (1000 RPM). Culture supernatant was collected and diafiltered against 4 times the culture volume with 10 mM HEPES pH 8, 150 mM NaCl (TIM Buffer 1). Nickel affinity chromatography was performed on the diafiltered supernatant with Ni-NTA agarose (Qiagen), eluting fractions with increasing concentrations of imidazole in TIM Buffer 1. Protein-containing fractions were pooled, diluted to 50 mM NaCl, filtered (0.22 μm), and injected onto a Fractogel TMAE column (EMD Millipore) equilibrated in 25 mM HEPES pH 8, 50 mM NaCl. Protein was eluted using a gradient from 50 to 700 mM NaCl in 25 mM HEPES pH 8. Peak fractions (∼150 mM NaCl) were collected, concentrated using a 10 kDa MWCO centrifugal filter (EMD Millipore), filtered (0.22 μm), and applied to a Superose 12 10/300 column (Cytiva Life Sciences) for size exclusion chromatography (SEC) in TIM Buffer 2 (10 mM HEPES pH 7.6, 150 mM NaCl). Peak fractions were collected, concentrated using a 10 kDa MWCO centrifugal filter (EMD Millipore) and filtered (0.22 μm). SDS–PAGE was performed to confirm protein size and purity as shown in Supplementary Figure S6. Protein concentration was determined by 280 nm absorbance using a NanoDrop spectrophotometer (ThermoFisher Scientific) with calculated extinction coefficients.
Vesicle preparation
Lipids were purchased from Avanti Polar Lipids in chloroform solution, including dioleoylphosphatidylcholine (DOPC), dioleoylphosphatidylserine (DOPS), dioleoylphosphatidic acid (DOPA), and dioleoylphosphatidylethanolamine (DOPE). Lipid solutions were combined at the appropriate molar ratios in a glass vial. Chloroform was blown off under a stream of nitrogen gas, fully drying the lipid mixture under vacuum. Lipid mixtures were rehydrated with 10 mM HEPES pH 7.6, 150 mM NaCl, vortexed to mix, and subjected to at least 10 freeze–thaw cycles, where the suspension was frozen in liquid nitrogen and thawed in a warm sonicating water bath, to generate unilamellar vesicles. Vesicle suspensions were stored at −20°C. Before use, thawed sonicated vesicles were extruded using a 100 nm filter membrane in an Avanti Mini Extruder.
Surface plasmon resonance studies
Surface plasmon resonance (SPR) analyses of protein–lipid interactions were performed using a Biacore 3000 instrument as described [45,46]. Lipid vesicles containing DOPC or the specified percentage (mole/mole) of lipid in a DOPC background were immobilized by flowing lipid vesicles across an L1 chip (Cytiva) in TIM Buffer 2 (10 mM HEPES pH 7.6, 150 mM NaCl). After immobilization, purified proteins were flowed across the chip at varying concentrations in the presence or absence of 1 mM CaCl2. Resonance units detected by the Biacore 3000 were corrected for background (DOPC) binding on a separate sensorchip surface, and were plotted against protein concentration. Curves were fit using the equation: RUmax = Bmax × [TIM]/(Kd + [TIM]), and apparent Kd was estimated from these curves.
Luminex analysis of TNFα secretion
For analysis of TNFα secretion, cells were starved for 4 h in serum-free RPMI-1640 media. 5 × 105 cells were then resuspended in fresh serum-free medium and plated in a V-bottom 96-well plate. Cells were stimulated with αCD3/αCD28 (1 μg/ml each) overnight at 37°C. The next day, the 96-well plate was centrifuged (300×g, 5 min), and supernatants from each sample were isolated for analysis. Components of the HCYTOMAG-60K Human Cytokine kit, including beads for detecting TNFα (Millipore Sigma), were warmed to room temperature and prepared according to the manufacturer's instructions, including standards with known TNFα concentrations. After adding assay buffer and standards or cell medium, plates were incubated for 2 h at room temperature with shaking and then washed, using a magnetic plate to recover the beads. After incubation with detection antibodies and streptavidin–phycoerythrin, beads were washed and the plate was analyzed using a Luminex MAGPIX® System. Standard curves were created and fit with a log5P curve using the Luminex analysis software, and this, in turn, was used to determine the TNFα concentration in each sample. Every sample contained three biological repeats with two technical repeats of each.
Quantification and statistical analysis
Experiments were completed with at least three biological repeats unless noted otherwise. For transcriptional reporter experiments, each independent experiment included three technical repeats. In addition to mean fluorescence intensity, the percent of GFP-positive cells and the median fluorescence intensity of GFP-positive cells were quantitated for transcriptional reporter experiments, to ascertain whether the analysis method influenced interpretation. In all cases, the result was the same as seen when assessing mean fluorescence intensity as done in the figures — lending confidence to our analysis and its qualitative conclusions. All quantitated values are represented as mean values ± standard deviation (SD). Statistical analysis was performed with two-tailed, unpaired Student's t-tests.
Results
TIM-3 promotes TCR signaling when overexpressed in Jurkat cells
In considering possible mechanisms of TIM-3 signaling [8] one incompletely answered question is whether TIM-3 functions in T cells as a co-inhibitory or co-stimulatory receptor. To establish a system for studying its regulation, we asked how TIM-3 overexpression affects TCR activation in Jurkat cells — a valuable cellular model for studying TCR signaling [47]. We first established that TIM-3 was undetectable by flow cytometry or Western blotting in parental Jurkat cells (Figure 1A) — in agreement with others [22] — and that it was not induced upon TCR stimulation. We then used lentiviral delivery of TIM-3 to generate stably expressing (TIM-3+) derivatives of an NF-κB/Jurkat/GFPTM transcriptional reporter cell line in which NF-κB activation downstream of TCR stimulation promotes GFP expression. Importantly, we confirmed that levels of TIM-3 seen following viral transduction of Jurkat NF-κB GFP Reporter cells were in the same range as those observed in human primary T cells (Supplementary Figure S1E).
Figure 1. Ectopic expression of TIM-3 enhances activation of Jurkat cells.

(A) Flow cytometry analysis of human TIM-3 expression in parental (black) and TIM-3 lentivirus-transduced NF-κB GFP reporter Jurkat cells (red), detected with phycoerythrin-conjugated TIM-3 antibody. The inset shows Western blot analysis of TIM-3 expression in the same cells as marked — with three biological repeats of each. (B) Representative histogram of NF-κB-driven GFP expression in stimulated NF-κB reporter cells (black), and those expressing TIM-3 (red) — compared with unstimulated parental (gray) and TIM-3-expressing (pink) cells. Stimulation employed 1 μg/ml αCD3 plus 1 μg/ml αCD28 for 16 h to mimic antigen binding. T cell activation was quantified by flow cytometry analysis of the GFP reporter of NFκB transcriptional activity. Enhanced T cell activation in TIM-3-expressing cells requires cell stimulation, with no change being observed in unstimulated cells. (C) Mean GFP fluorescence intensity (MFI) values for NF-κB GFP reporter assays in cells stimulated as in (B) relative to parental NF-κB reporter cells (black). Data represent the mean ± standard deviation (SD) for four biological repeats for parental and TIM-3-expressing cells (red) and two repeats for PD-1-expressing cells (blue). P-value for the TIM-3/parental comparison was determined with a two-tailed, unpaired Student's t-test. (D) ELISA analysis of IL-2 secretion into the medium of parental and TIM-3+ NF-κB reporter cells following stimulation as in (B). Data represent mean ± SD across three biological repeats, with P-value determined using a two-tailed, unpaired Student's t-test. (E) IL-2 production by parental and TIM-3-expressing Jurkat NF-κB/GFP reporter cells stimulated with SEE-loaded Raji B cells to mimic the formation of the immunological synapse during T cell activation. IL-2 levels were quantified by ELISA. Data represent mean ± SD for three biological repeats, with P-value determined using a two-tailed, unpaired Student's t-test.
Stimulating NF-κB reporter Jurkat cells with antibodies against CD3 and CD28 (αCD3/αCD28) to mimic antigen binding and TCR activation promoted NF-κB-dependent GFP expression as expected [48] (Figure 1B). The GFP expression signal was significantly increased by TIM-3 expression only after T cell stimulation (Figure 1B,C), suggesting a co-stimulatory role for TIM-3 in Jurkat cell activation consistent with the majority of previous studies [6,19–22] — but differing from two other reports [23,24]. In contrast, PD-1 overexpression in the same NF-κB/Jurkat/GFPTM cells had no significant effect on TCR-activated NF-κB signaling (Figure 1C), as expected in the absence of PD-L1 to engage PD-1 in these experiments (Supplementary Figure S2A). In parallel, ELISA assays showed that TIM-3 expression substantially increased IL-2 production induced by TCR activation with αCD3/αCD28 (Figure 1D), consistent with previous studies [22] and again supporting a co-stimulatory role for TIM-3. Importantly, the co-stimulatory role of TIM-3 was evident whether cells were stimulated with both αCD3 and αCD28 together or with 1 μg/ml αCD3 alone (Supplementary Figure S1F,G).
We also stimulated Jurkat cells using Staphylococcal enterotoxin E (SEE)-loaded Raji B cells as antigen-presenting cells to engage the TCR and other key signaling molecules through the formation of an immunological synapse (IS). TIM-3 appears to be recruited to the IS [49], and was recently reported to be recruited to the IS upon Raji B cell activation of Jurkat T cells in this way [21]. In agreement with our observations using antibody stimulation alone, TIM-3 expression also significantly enhanced IL-2 secretion induced by Raji cell activation of Jurkat cells (Figure 1E), showing that the ability of TIM-3 to promote T cell activation is preserved with IS formation. Together, these data support a robust co-stimulatory role of TIM-3 in Jurkat T cells when activated by antibodies or by cell-based methods — in common with the majority of previous studies [6,19–22].
Effects of TIM-3 overexpression on signaling responses to αCD3/αCD28
To investigate how TIM-3 expression might promote T cell signaling, we next used Western blotting to monitor phosphorylation of components associated with TCR activation after stimulation with αCD3/αCD28. Few differences could be detected when assessing phosphorylation of Zap70, PLCγ1, ERK1/2, or LAT in parental and TIM-3+ cells (Supplementary Figure S3). Some experiments did suggest that phosphorylation of Zap70 and LAT is slightly greater and more sustained in TIM-3 overexpressing cells (Supplementary Figure S3B,E), but this did not reach statistical significance. Levels and dynamics of phosphorylation of these proteins also appeared essentially unchanged by PD-1 overexpression (Supplementary Figure S3B–E).
One difference that did reach significance was the level of CD28 phosphorylation, which was modestly increased in TIM-3 overexpressing cells (Figure 2). This elevation was seen for phosphorylation of both Y191 (Figure 2A) and Y218 (Figure 2B). As expected [50], elevated CD28 phosphorylation was seen at both sites following αCD3/αCD28 stimulation of parental cells, but TIM-3 expression further increased the magnitude of this response by ∼50% — with no apparent elevation of baseline CD28 phosphorylation. Interestingly, Y191 phosphorylation also seemed to be significantly longer-lived in TIM-3+ cells than in parental cells (Figure 2A, Supplementary Figure S3F). Phosphorylation of Y218 in CD28 has been reported to be important for NF-κB activation by CD3/CD28 ligation in Jurkat and primary CD4 T cells [51], consistent with our results for NF-κB activation. Moreover, phosphorylation of all three distal tyrosines in the CD28 tail (including Y218) appears to be required for stimulation of IL-2 secretion by T cells [52]. Thus, alterations in the extent of CD28 phosphorylation and/or its dynamics may be important for the increased NF-κB signaling and IL-2 secretion seen when TIM-3 is overexpressed in Jurkat cells. Phosphorylation of Y191 in CD28 promotes recruitment of phosphoinositide 3-kinase (PI3K) to CD28 [53,54], and the consequences of altering its phosphorylation are likely blunted in Jurkat cells, which lack PTEN [55]. We did consider that the constitutively high levels of PI3K products seen in Jurkat cells might be responsible for the change in CD28 phosphorylation. However, as shown in Figure 2C, the effect of TIM-3 expression on levels of CD28 phosphorylation was maintained in the presence of the PI3K inhibitor wortmannin — indicating no dependence on PI3K activity.
The TIM-3 extracellular region is required for its activity in Jurkat cells
We next chose to exploit the ability of TIM-3 to enhance NF-κB signaling in the GFP-reporter Jurkat cell line to investigate whether (and how) the extracellular region of TIM-3 regulates its co-stimulatory function, and to understand which of TIM-3's ligand(s) are important. Lee et al. [22] previously used a similar strategy to investigate signaling requirements of the TIM-3 cytoplasmic tail. They reported the surprising finding that deletion of the TIM-3 extracellular region did not abolish its ability to enhance αCD3/αCD28-induced NFAT activation [22]. As summarized below, our results contrast with this finding and suggest an important regulatory function for the extracellular region.
Since NF-κB signaling in the Jurkat reporter cell line was increased by expression of TIM-3, but not by expression of PD-1, we first asked whether PD-1/TIM-3 chimeras would preserve this effect — which they should if the TIM-3 intracellular region is sufficient for signaling. We replaced the TIM-3 extracellular region with that from PD-1, in ‘PTT’ or ‘PPT’ chimeras (Figure 3A) that retain either the TIM-3 (PTT) or PD-1 (PPT) transmembrane (TM) domain. After generating Jurkat reporter cells stably expressing these chimeras at the cell surface as assessed by flow cytometry (Supplementary Figure S2B) and at levels similar to full-length TIM-3 (Supplementary Figure S2E), we assessed their ability to enhance NF-κB activation by αCD3/αCD28. As shown in Figures 3B,C, PTT and PPT cells displayed only a very a slight increase in NF-κB activation compared with parental and PD-1 reporter cells, significantly less than seen in TIM-3+ cells. This result argues that the TIM-3 extracellular region does play an important role in its signaling effects in Jurkat cells. One possibility is that TIM-3 interacts constitutively with the TCR or other cellular components to influence the effects of TCR stimulation. Alternatively, it is possible — and perhaps more likely — that TIM-3 is engaged by one of its extracellular ligands in Jurkat cell cultures. Indeed, it was previously shown that a biotinylated form of the soluble TIM-3 extracellular region associates with the surface of CD4 T cells in flow cytometry studies [16,17], and we know that the TIM-3 ligand PS is exposed on the surface of activated T cells following antigen recognition [31] — consistent with this possibility. Interestingly, expression of PTT promoted NF-κB signaling to a slightly greater extent than PPT, possibly implicating the TIM-3 TM domain in specific interactions. Indeed, Kataoka et al. [21] recently reported that the TM domain plays a key role in recruiting TIM-3 to the IS and promoting co-stimulatory activity.
Figure 3. The TIM-3 extracellular region is required for co-stimulatory effects on T cell signaling.

(A) Schematic representation of PD-1/TIM-3 chimeric constructs. PTT contains the PD-1 extracellular region (ECR) plus transmembrane (TM) and intracellular regions of TIM-3. In PPT, only the intracellular region is replaced with that from TIM-3. (B) A GFP reporter was used to measure NF-κB transcriptional activity downstream of TCR activation in parental NF-κB reporter Jurkat cells (black) and the same cells engineered to stably express TIM-3 (red curve), PTT (purple solid curve), PPT (purple dotted curve), or PD-1 (blue curve), stimulated with 1 μg/ml αCD3 plus 1 μg/ml αCD28. Representative histograms of NF-κB-driven GFP expression are shown. (C) Relative mean GFP fluorescence intensity (MFI) from three biological repeats of the experiment shown in (B) is plotted, expressed as fold change above that measured for parental NF-κB GFP reporter cells. Bars are color coded as in (B) and represent mean MFI value (± SD) — with P-values determined by two-tailed, unpaired Student's t-test.
Phosphatidylserine exposed in Jurkat cell culture may engage TIM-3
Although the TIM-3 extracellular region could not be replaced with that from PD-1, chimeras using the TIM-1 or TIM-4 extracellular regions (which both bind PS) appeared to retain function (Supplementary Figure S4). We therefore next asked if TIM-3 co-stimulatory signaling could be modulated by adding its putative ligand, PS. We began by quantifying αCD3/αCD28-stimulated NF-κB activity in TIM-3+ cells after adding unilamellar vesicles containing high levels of PS. Vesicles containing 20% (mole/mole) DOPS in a background of DOPC had no effect on NF-κB activity in parental Jurkat cells, but also failed to modulate NF-κB activity in cells expressing TIM-3 to a statistically significant degree when compared with pure PC vesicles (Supplementary Figure S5A,B). We, therefore, hypothesized that Jurkat cells in culture might expose PS on their surface, which could explain the previously observed TIM-3 ligand expressed on CD4 T cells [16,17]. E. coli-derived recombinant murine TIM-3 IgV domain was previously shown to bind T cells, fibroblasts and macrophages from several species [56] — consistent with the recognition of a conserved component of these cells, such as PS. To test the hypothesis that PS on the surface of Jurkat cells promotes TIM-3 effects in our studies, we first asked whether adding excess annexin V — a known PS-binding protein — might block or sequester surface-exposed PS and suppress TIM-3-dependent elevation of NF-κB activity. Unfortunately, adding annexin V elevated TCR-driven NF-κB signaling independently of TIM-3 expression (Supplementary Figure S5C,D) — likely because of its reported effects on membrane structure [57], which are likely to alter TCR clustering and activity as seen when cholesterol levels are changed [58]. As a result, this experiment was uninterpretable.
Nonetheless, the classical use of annexin V as a PS probe did prove useful for assessing the exposure of this phospholipid on our Jurkat cells, revealing that PS is indeed exposed on their surface in our cultures — presumably as cells undergo apoptosis and/or activation, two processes known to lead to PS exposure on the cell surface [29,31,59]. We stained cells with fluorescently labeled (PerCP-CyTM5.5) annexin V, and flow cytometry analysis showed that both parental and virally-transduced TIM-3+ NF-κB reporter Jurkat cells expressed PS on their surface at readily detectable levels (Figure 4A). Interestingly, most cells showed only low levels of PS exposure (peak signal ∼400), but a small population had high levels of PS exposure (∼105) as described in previous studies of T cells [32]. As a secondary approach, we also imaged cells stained with fluorescently labeled (AlexaFluor® 647) annexin V prior to fixation, and demonstrated annexin V staining of both parental and TIM-3+ NF-κB reporter Jurkat cells (Figure 4B). These data suggest that sufficient PS may be exposed on Jurkat cells in our experiments to fully engage TIM-3 and promote its co-stimulatory signaling. Indeed, the fact that adding PS-containing vesicles had no further influence on NF-κB activity suggests that the PS levels in culture may be near saturation and/or may represent an optimal lipid composition for TIM-3 that is not recapitulated by our synthetic vesicles. These results, therefore, prompted us to ask whether (and how) PS functions as a TIM-3 ligand to modulate its co-stimulatory signaling in our experiments.
Figure 4. The TIM family ligand PS is present on Jurkat cells and binds TIM-3 with a ∼10 μM dissociation constant.
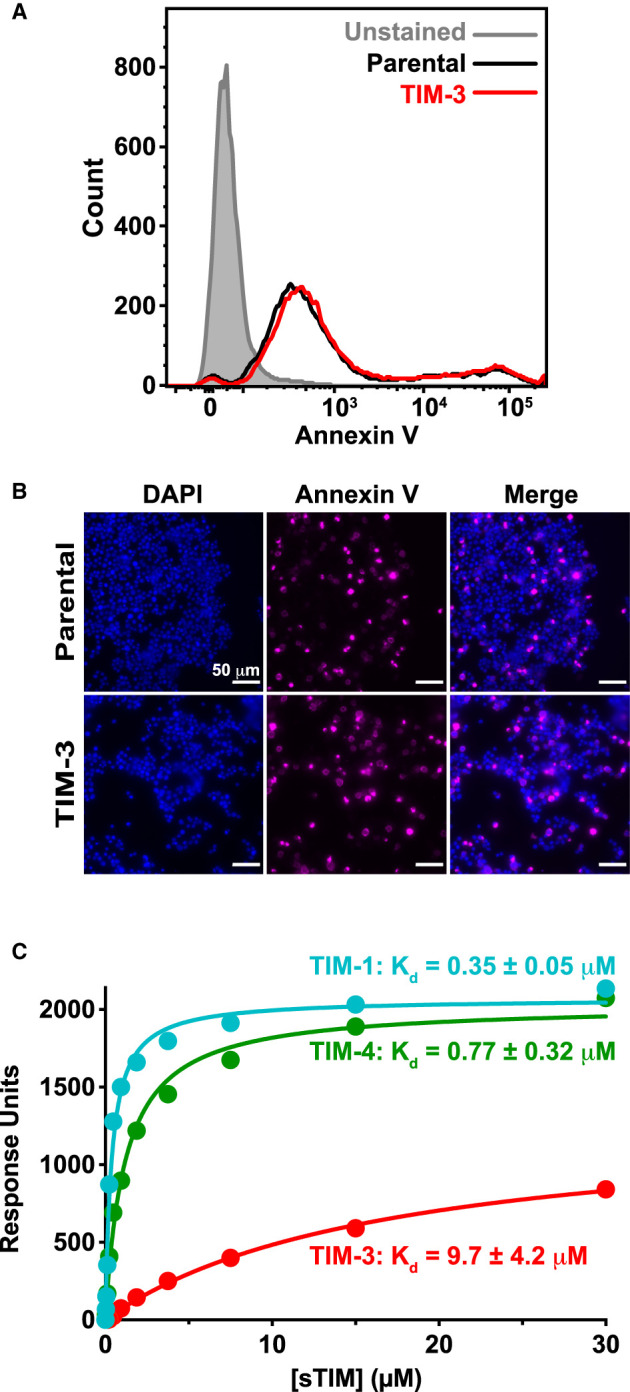
(A) PS exposure on Jurkat cells in culture, detected by staining with fluorescently-tagged (PerCP-CyTM5.5) annexin V, and quantified by flow cytometry analysis of parental NF-κB GFP reporter cells (black) and those expressing TIM-3 (red) — compared with unstained cells (gray shaded). A representative histogram from five biological repeats is shown. (B) Fluorescence imaging of parental and TIM-3+ NF-κB GFP reporter cells stained with DAPI (left panels) and fluorescently-tagged (AlexaFluor 647) annexin V (middle panels). Cells were stained with annexin V prior to fixation and staining with DAPI. (C) PS binding of sTIM-1 (teal), sTIM-3 (red), and sTIM-4 (green) was analyzed using SPR by flowing purified TIM ECRs over 20% DOPS/80% DOPC lipid vesicles immobilized on an L1 sensorchip. Binding was measured in the presence of 1 mM CaCl2. The curves indicate the fit of the data in Prism 9 to a one-site specific binding equation. Binding curves are representative of at least five independent experiments. Mean Kd and SD values are listed in the figure (n = 6 for sTIM-1, n = 11 for sTIM-3, and n = 5 for sTIM-4). Binding to additional lipids and effects of calcium are shown in Supplementary Figure S7.
Phospholipid binding by human TIM extracellular regions
Before attempting to modulate PS binding by TIM-3 in our cellular experiments, we wanted to understand its PS-binding properties and compare them with those of TIM-1 and of TIM-4, the original PS receptor [33]. We expressed the complete soluble TIM protein extracellular regions (sTIM proteins), containing the IgV and mucin domains, in Expi293 cells and used SPR to measure their binding to membranes containing 20% (mole/mole) DOPS in a background of DOPC as described [45]. Importantly, all sTIM proteins expressed at high levels, showed good solubility and monodispersity in SEC, and could be purified readily to >90% as assessed using Coomassie stained gels (Supplementary Figure S6A–D). All sTIM proteins bound to 20% PS membranes in SPR studies, and displayed rapid dissociation following the end of the injection, indicating an absence of aggregation on the membrane surface (Supplementary Figure S6E–G). sTIM-3 bound to membranes containing 20% PS with an apparent dissociation constant (Kd, app) of 9.7 ± 4.2 μM in the presence of 1 mM CaCl2 (Figure 4C). The human sTIM-1 and sTIM-4 proteins bound significantly more strongly to the same membranes under identical conditions, with Kd, app values of 0.35 ± 0.05 μM and 0.77 ± 0.32 μM, respectively (Figure 4C). The relative affinities that we observed here reflect what has been reported for murine TIM proteins using ELISA-type assays [34,35] and other approaches [36], with sTIM-3 binding PS membranes 12–27-fold more weakly than sTIM-1 or sTIM-4. The actual Kd, app values that we report are substantially weaker than those suggested using ELISA methods (which were in the nM range), as is typical for multivalent systems when comparing results from direct binding studies such as SPR with those from ELISA assays [46,60]. Our Kd, app values are in the same range as those measured for other PS-binding proteins, pleckstrin homology domains and other peripheral membrane proteins [45,61,62], and indeed, for recognition of the prototypical T cell co-receptors CD28 and PD-1 for their respective ligands, CD80/86 [63] and PD-L1 [64].
In additional, more comprehensive, binding studies (Supplementary Figure S7) we found that the presence of Ca2+ ions increases the PS-binding affinities of sTIM-1 and sTIM-4 by ∼10-fold, but increases the affinity of sTIM-3 for PS by just ∼2-fold (Supplementary Figure S7A, Supplementary Table S1) — again consistent with previous work [36]. We also asked whether the sTIM proteins can bind other phospholipids known to become exposed on the cell surface during cell death, namely phosphatidic acid (PA) and phosphatidylethanolamine (PE) [65,66]. sTIM-1, sTIM-3, and sTIM-4 all bound to 20% PA-containing membranes (Supplementary Figure S7C–E), but saturated at a lower level than on membranes containing the same level of PS. Interestingly, the inclusion of 5% PA also enhanced binding of all sTIM proteins to membranes containing 20% PS (Supplementary Figure S7F–H), such that sTIM-3 bound more strongly to membranes containing 20% PS plus 5% PA than to membranes containing 20% or 25% PS (Supplementary Figure S7I). Only sTIM-1 showed significant binding to membranes containing the zwitterionic phospholipid PE (Supplementary Figure S7C). This is consistent with a previous report [67] that also indicated that PE on the surface of apoptotic cells promotes their TIM-1-mediated phagocytosis.
Mutations that impair TIM-3 binding to PS reduce its impact on T cell signaling
Reasoning that co-stimulatory signaling by TIM-3 upon TCR activation in Jurkat cells is promoted when TIM-3 binds cell surface PS in our experiments, we next mutated TIM-3 to reduce its PS-binding affinity. Our goal was to ask whether such mutations diminish the co-stimulatory effect of TIM-3 on TCR signaling. Guided by the crystal structure of the IgV-like domain in the murine TIM-3 extracellular region complexed with a short acyl-chain PS [34], we substituted F40 of TIM-3 (mature human protein numbering) with alanine in one variant (F40A), and replaced both I96 and M97 with alanine in another (I96A/M97A). The corresponding residues in murine TIM-3 (W41/S42, and L99/M100 in PDB entry 3KAA) ‘sandwich’ the bound PS (Figure 5A). DeKruyff et al. [34] showed previously that mutating L99 and M100 of murine TIM-3 greatly impaired PS binding. They also showed that mutating W41 in mTIM-3 abrogated PS binding, and that the W41 side-chain occupies a very similar location to that of the F40 side-chain in hTIM-3 (Figure 5A).
Figure 5. Mutated TIM-3 variants with impaired PS-binding lose co-stimulatory signaling.

(A) Details of the PS-binding site in murine TIM-3 (PDB: 3KAA — blue) overlaid on human TIM-3 (PDB: 6TXZ — red), revealing key interacting residues [34,82]. Inset at the right shows the PS-binding pocket, where short-chain PS is sandwiched by L99/M100 and W41 in mTIM-3 (I96/M97 and F40 in human TIM-3). A calcium ion (gray sphere) also sits in the binding pocket and interacts with the negatively charged PS headgroup. (B) PS binding of wild-type sTIM-3 (solid red curve), sTIM-3F40A (solid magenta curve), and sTIM-3I96A/M97A (dotted magenta curve) were analyzed using SPR by flowing the purified sTIM-3 variants over 20% DOPS/80% DOPC lipid vesicles immobilized on an L1 chip (in the presence of 1 mM CaCl2). Binding curves are representative of at least three independent experiments. Kd values for sTIM-3F40A and sTIM-3I96A/M97A were too high to measure, both appearing to exceed ∼200 μM (although PS binding was detectable). (C) A GFP reporter was used to measure NF-κB transcriptional activity downstream of TCR activation with 1 μg/ml αCD3 plus 0.5 μg/ml αCD28 for 16 h. Representative histogram of NF-κB-driven GFP expression is shown for parental NF-κB GFP reporter cells (black), and those expressing TIM-3WT (solid red curve), TIM-3F40A (solid magenta curve), and TIM-3I96A/M97A (dotted magenta curve) variants. (D) Relative mean GFP fluorescence intensity (MFI), expressed as fold change over parental cells within each experiment, was determined across eight biological replicates for parental NF-κB GFP reporter cells (black) or those expressing TIM-3WT (solid red line, open bar), TIM-3F40A (solid magenta line, open bar and data points), or TIM-3I96A/M97A (dotted magenta line, gray filled bar and data points). Means ± SD are plotted, with P-values determined by two-tailed, unpaired Student's t-tests. (E) ELISA analysis of IL-2 secreted into the cell culture medium by parental NF-κB GFP reporter cells, and those expressing, TIM-3WT (solid red line, open bar), TIM-3F40A (solid magenta line, open bar and data points), or TIM-3I96A/M97A (dotted magenta line, gray filled bar and data points) following stimulation by αCD3/αCD28, as described in (C). Means ± SD are plotted for three biological repeats. P-values comparing TIM-3WT with the mutated TIM-3 variants were determined using two-tailed, unpaired Student's t-tests. (F) Analysis of TNFα secretion into the cell culture medium was performed using a Luminex assay. Cells were stimulated with αCD3/αCD28 (1 μg/ml each) for 16 h. Mean values ± SD are plotted for parental (black line, open bar), TIM-3WT (solid red line, open bar), TIM-3F40A (solid magenta line, open bar and points), and TIM-3I96A/M97A (dotted magenta line, gray filled bar and data points). Two-tailed, unpaired Student's t-tests were used to compare TIM-3WT with mutated TIM-3 variants for three biological repeats. (G) SEE-loaded Raji B cells were used to stimulate NF-κB GFP reporter Jurkat cells to recapitulate the formation of an immunological synapse. IL-2 production following Raji B cell stimulation was determined by ELISA analysis for parental NF-κB GFP reporter cells (solid black line, open bar) and cells expressing TIM-3WT (solid red line, open bar), TIM-3F40A (solid magenta line, open bar and data points), or TIM-3I96A/M97A (dotted magenta line, gray filled bar and data points). Mean ± SD are plotted for three biological repeats, with P-values comparing TIM-3WT with the mutated TIM-3 variants determined using two-tailed, unpaired Student's t-tests.
We first used SPR to show that these mutations reduce (but do not abolish) PS binding. Both sTIM-3F40A and sTIM-3I96A/M97A displayed substantially reduced PS binding (Figure 5B), reaching only 8% and ∼30% of saturation respectively at the highest protein concentrations tested. We then generated NF-κB reporter Jurkat cells stably expressing full-length TIM-3 variants with these mutations. Having used Western blotting and flow cytometry to confirm that the mutated variants are robustly expressed at the cell surface (Supplementary Figure S2E,F), we assessed NF-κB signaling and IL-2 secretion after αCD3/αCD28 stimulation of the TCR. Cells expressing the mutated TIM-3 variants showed significantly reduced TCR-induced NF-κB activation compared with that seen for cells expressing wild-type TIM-3 (Figure 5C,D), although the effect of TIM-3 was not completely abolished. Moreover, cells expressing the mutated variants secreted significantly less IL-2 into their medium compared with cells expressing wild-type TIM-3 after stimulation with αCD3/αCD28 (Figure 5E). TNFα secretion was also significantly reduced (Figure 5F). Importantly, similar results were observed when SEE-loaded Raji B cells were used to activate the parental or TIM-3 variant-expressing Jurkat cells. Cells expressing the mutated TIM-3 variants produced significantly less IL-2 than wild-type TIM-3-expressing cells (Figure 5G), although the diminution was less than seen with αCD3/αCD28 stimulation — possibly reflecting some restoration of (residual) PS binding avidity by the mutated variants when TIM-3 is clustered in ISs [21,49]. Together, these results demonstrate that reducing the ability of TIM-3 to bind PS diminishes its ability to promote TCR signaling, supporting the hypothesis that TIM-3 is engaged with (or saturated by) PS in our Jurkat cell experiments to promote co-stimulatory TIM-3 signaling.
A TIM-3 antibody that blocks PS binding also diminishes T cell signaling
One concern with such mutational studies is that the mutations might impair TIM-3 function indirectly — through misfolding, for example. We, therefore, also investigated the consequences of blocking PS binding with a TIM-3 antibody. Several TIM-3 antibodies are now in clinical trials in lung cancer and other malignancies [11]. TIM-3 antibodies that have shown efficacy in preclinical studies in mice have all been shown to block PS and CEACAM1 (but not galectin-9) binding [37]. The same study also showed that the human TIM-3 monoclonal antibody F38.2E2 blocks PS binding, by interacting with the loops that contain F40 and I96/M97 shown in Figure 5A, and which we mutated for the studies described above. As an orthogonal approach to test the importance of PS binding for TIM-3 signaling, we explored the effect of F38.2E2 on co-stimulatory TIM-3 signaling, assessing both NF-κB activation and IL-2 secretion following TCR stimulation in our Jurkat reporter cell assay. As CEACAM1 is not endogenously expressed in Jurkat cells [68,69], any observed effects from F38.2E2 treatment should be specific to blocking the PS-binding ability of TIM-3.
F38.2E2 addition caused a modest, but significant, dose-dependent decrease in αCD3/αCD28-stimulated NF-κB signaling in cells expressing TIM-3, but not in parental cells (Figure 6A–E). The inhibitory effect of F38.2E2 treatment became saturated at ∼1 μg/ml antibody (Figure 6B,E). Importantly, 10 μg/ml F38.2E2 also reduced (by ∼50%) the amount of IL-2 secreted by TIM-3 expressing cells upon TCR activation (Figure 6F). Since F38.2E2 is known to block PS binding, these data provide additional support for the argument that TIM-3 engagement by PS is important for its co-stimulatory signaling activity in this system. Inhibition is only partial, but F38.2E2 blocks ∼50% of the TIM-3-dependent increase in NF-κB signaling or IL-2 secretion, bringing the relative MFI from ∼1.0 to <0.7 for TIM-3+ cells compared with a value of ∼0.35 for parental cells (compare Figure 6D,E), and reducing IL-2 levels from ∼500 to ∼300 pg/ml (compared with ∼40 pg/ml for parental cells). One possible explanation for the fact that the observed inhibition is partial is that ligands other than PS may also be important — such as galectin-9, binding of which to TIM-3 is not blocked by F38.2E2. Alternatively, the partial nature of the response could reflect the need for F38.2E2 to compete with a high level of PS on cells in culture, which might be clustered as suggested on activated T cells [31] in a way that enhances its avidity for TIM-3 binding. As another alternative, F38.2E2 antibody treatment itself could cross-link or cluster TIM-3 in a way that promotes its co-stimulatory function. In either case, taken together with the mutational and chimeric receptor studies, these data strongly support a role for extracellular PS binding in regulating TIM-3 co-stimulatory signaling when overexpressed in Jurkat cells, and suggest that inhibition of PS binding may be an important component of the function of TIM-3 antibodies currently being studied in the clinic — if they achieve their effects by binding T cells.
Figure 6. TIM-3 antibody treatment selectively reduces co-stimulatory signaling.

(A) Representative histograms of NF-κB-driven GFP expression in parental NF-κB GFP reporter Jurkat cells. Cells were starved for 4 h — including a 1 h pre-treatment with anti TIM-3 (F38.2E2) at the concentrations marked — and were then stimulated with 1 μg/ml αCD3 plus 0.5 μg/ml αCD28 for 16 h or left unstimulated (gray). GFP expression was analyzed by flow cytometry, and quantitation is shown in (D). (B) Representative histograms for TIM-3-expressing NF-κB GFP reporter Jurkat cells, stimulated as in (A), with from 0 μg/ml to 10 μg/ml F38.2E2. Unstimulated TIM-3+ cells are shown in pink. Quantitation is shown in (E). (C) Representative histograms from TIM-3-expressing NF-κB GFP reporter Jurkat cells stimulated with 0 μg/ml (red) or 10 μg/ml F38.2E2 (blue), showing that 10 μg/ml F38.2E2 does reduce the level of NF-κB-driven GFP expression seen in stimulated TIM-3 cells. (D,E) Quantitation of data in (A) and (C) over three independent biological repeats, plotting mean values for relative mean GFP fluorescence intensity (MFI) and SD. P-values were determined with two-tailed, unpaired Student's t-tests. (F) IL-2 secreted into cell medium was measured by ELISA for parental NF-κB GFP reporter Jurkat cells (black bars) and TIM-3-expressing cells (red bars). Adding 10 μg/ml F38.2E2 reduces enhanced IL-2 production in TIM-3 expressing cells. Bars represent the mean concentration of IL-2 (± SD) for three biological replicates. P-values were determined with two-tailed, unpaired Student's t-tests.
Discussion
The co-stimulatory effect of TIM-3 on TCR signaling in Jurkat cells that we describe here is consistent with that seen in numerous previous studies [6,19–22], and we find that it requires the ability of the TIM-3 extracellular region to bind PS. These results argue that PS exposed on the surface of Jurkat cells in culture — which we confirm with annexin V staining — is sufficient to engage TIM-3 and promote its co-stimulatory signaling in this cell culture setting. The role of PS binding in regulating signaling effects of TIM-3 has remained largely unexplored [8,70,71]. PS binding to TIM-3 on macrophages, dendritic cells and fibroblasts has been shown to mediate their phagocytosis of apoptotic cells [34,35,72], whereas the same interaction only allows T cells to form conjugates with apoptotic cells — with no engulfment [34]. Our studies argue that, beyond this coupling function, PS binding to TIM-3 can regulate its activity as a TCR co-stimulatory receptor.
Several previous studies have reported the presence of a TIM-3 ligand on the surface of various cultured cells, from CD4 T cells [16,17] to fibroblasts to macrophages — and from several different species [56]. The wide cross-species reactivity observed supports the suggestion that this ligand may be PS rather than a protein, and Cao et al. [56] demonstrated that it is not galectin-9, the most well-studied potential TIM-3 ligand. Together with our results and existing evidence for PS binding by TIM-3 [34–36], it seems likely that most published studies of TIM-3 signaling in culture may have been performed with TIM-3 that was already engaged by a ligand (PS). Nonetheless, the prevailing hypothesis in the field is that galectin-9 is the key TIM-3 ligand and binds TIM-3 to trigger co-inhibitory signaling and thus T cell death [8,26,73]. However, as a carbohydrate-binding protein, galectin-9 is known to bind numerous other targets [74–77], and several studies have argued that key effects of galectin-9 on T cells are TIM-3-independent [28,77,78] — or alternatively that galectin-9 does not bind TIM-3 at all [27]. One interesting possibility is that PS is in fact the primary ligand for TIM-3, and that galectin-9 modulates PS binding and/or its consequences.
Our findings may alternatively suggest that the inhibitory signaling previously reported for TIM-3 [23,24] reflects displacement of an activating ligand — namely PS. Galectin-9 could exert its TIM-3-dependent effects by pulling TIM-3 into clusters that no longer bind PS, or bind it in a different way. Lee et al. [22], commented in studies of TIM-3 co-stimulatory signaling that the receptor initially augments T cell activation, but that rapid inhibition of T cell activation is seen when TIM-3 is cross-linked with an antibody. Interestingly, that antibody was 5D12, which has subsequently been shown to block PS binding to TIM-3 [37] — so this phenomenon could have arisen through reversal of PS stimulation. TIM-3 has also been reported to enhance DC and natural killer (NK) cell activation, as well as FcεR1 signaling in mast cells [19,79,80]. Moreover, stimulation of TIM-3 with PS was recently reported to promote TIM-3 phosphorylation in NK cells, leading to inhibition of cytokine secretion [81]. It, therefore, seems clear that TIM-3 can signal in several different contexts, possibly exerting distinct effects (and effects in different directions) through differential ligand engagement and/or phosphorylation of the tyrosines in its cytoplasmic tail.
Several groups are currently pursuing the development of therapeutic TIM-3 antibodies to promote anti-tumor immune responses, mostly in combination with PD-1 or PD-L1 antibodies — with the assumption that they will reverse a co-inhibitory signaling effect of TIM-3 [3,8,11]. Most TIM-3 antibodies under investigation have been shown to recognize the PS-binding site of TIM-3 [37,82], including Sym023 [83], BGB-A425 [84], ICAGN02390 [85], IBI104 [86], and LY3321367 [87]. Counterintuitively, our results suggest that these antibodies might actually impair T cell responses by blocking PS-induced co-stimulatory TIM-3 signaling — as indeed we observed for F38.2E2 in Figure 6. However, as mentioned in the Introduction, very recent studies have suggested that the inhibitory effect of TIM-3 on anti-tumor immunity actually originates from its effects on NLRP3 inflammasome activation in dendritic cells [18], and not its effects in T cells. In light of the diversity of its effects, it will clearly be important to understand how TIM-3 can signal distinctly in different contexts in order to apply existing and new TIM-3 antibodies effectively in the clinic.
To fully appreciate the functions and mechanisms of these complex immunomodulatory receptors, it will be important to identify both their intrinsic individual properties in model systems and the more complex functions that emerge in different cellular and physiological settings. Here, we have shown that PS-binding defective TIM-3 variants fail to provide co-stimulatory signals in a model T cell system, providing a useful tool for investigating precisely how PS-binding influences the more complex and varied functions of TIM-3 in T cells, NK cells, DCs, and other cells in different immune system settings. In the case of T cells, our data suggest that PS exposure in the environment of the T cell promotes function, which might — for example — serve as a mode of positive feedback regulation to maximize local specific responses. It would be interesting to know whether there is an optimal PS exposure level, beyond which one might speculate that TIM-3 signaling could begin to inhibit the TCR — perhaps once a certain level of cell death has been reached.
Acknowledgements
We thank members of the Lemmon and Ferguson laboratories, as well as Aaron Ring and Carla Rothlin for valuable discussions and comments.
Abbreviations
- DOPS
dioleoylphosphatidylserine
- DOPC
dioleoylphosphatidylcholine
- IS
immunological synapse
- NK
natural killer
- PA
phosphatidic acid
- PBMCs
peripheral blood mononuclear cells
- PBS
phosphate-buffered saline
- PE
phosphatidylethanolamine
- PS
phosphatidylserine
- SEC
size exclusion chromatography
- SEE
Staphylococcal enterotoxin E
- SPR
surface plasmon resonance
Data Availability
The data needed to evaluate this work are all included in the manuscript, and are available upon request.
Competing Interests
The authors declare that there are no competing interests associated with the manuscript.
Funding
This work was supported in part by NIH grant R35-GM122485 (to M.A.L.) and a PhRMA Foundation Predoctoral Fellowship to C.M.S.
CRediT Author Contribution
Mark A. Lemmon: Conceptualization, supervision, funding acquisition, investigation, methodology, writing — original draft, project administration, writing — review and editing. Courtney M. Smith: Conceptualization, data curation, supervision, funding acquisition, investigation, writing — original draft, writing — review and editing. Alice Li: Investigation. Nithya Krishnamurthy: Investigation.
Supplementary Material
References
- 1.Chen, L. and Flies, D.B. (2013) Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 13, 227–242 10.1038/nri3405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Edner, N.M., Carlesso, G., Rush, J.S. and Walker, L.S.K. (2020) Targeting co-stimulatory molecules in autoimmune disease. Nat. Rev. Drug Discov. 19, 860–883 10.1038/s41573-020-0081-9 [DOI] [PubMed] [Google Scholar]
- 3.Andrews, L.P., Yano, H. and Vignali, D.A.A. (2019) Inhibitory receptors and ligands beyond PD-1, PD-L1 and CTLA-4: breakthroughs or backups. Nat. Immunol. 20, 1425–1434 10.1038/s41590-019-0512-0 [DOI] [PubMed] [Google Scholar]
- 4.Topalian, S.L., Taube, J.M. and Pardoll, D.M. (2020) Neoadjuvant checkpoint blockade for cancer immunotherapy. Science 367, eaax0182 10.1126/science.aax0182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ogawa, S. and Abe, R. (2019) Signal transduction via co-stimulatory and co-inhibitory receptors. Adv. Exp. Med. Biol. 1189, 85–133 10.1007/978-981-32-9717-3_4 [DOI] [PubMed] [Google Scholar]
- 6.Avery, L., Filderman, J., Szymczak-Workman, A.L. and Kane, L.P. (2018) Tim-3 co-stimulation promotes short-lived effector T cells, restricts memory precursors, and is dispensable for T cell exhaustion. Proc. Natl Acad. Sci. U.S.A. 115, 2455–2460 10.1073/pnas.1712107115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferris, R.L., Lu, B. and Kane, L.P. (2014) Too much of a good thing? Tim-3 and TCR signaling in T cell exhaustion. J. Immunol. 193, 1525–1530 10.4049/jimmunol.1400557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolf, Y., Anderson, A.C. and Kuchroo, V.K. (2020) TIM3 comes of age as an inhibitory receptor. Nat. Rev. Immunol. 20, 173–185 10.1038/s41577-019-0224-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fourcade, J., Sun, Z., Benallaoua, M., Guillaume, P., Luescher, I.F., Sander, C.et al. (2010) Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 207, 2175–2186 10.1084/jem.20100637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sakuishi, K., Apetoh, L., Sullivan, J.M., Blazar, B.R., Kuchroo, V.K. and Anderson, A.C. (2010) Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 207, 2187–2194 10.1084/jem.20100643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Acharya, N., Sabatos-Peyton, C. and Anderson, A.C. (2020) Tim-3 finds its place in the cancer immunotherapy landscape. J. Immunother. Cancer 8, e000911 10.1136/jitc-2020-000911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Monney, L., Sabatos, C.A., Gaglia, J.L., Ryu, A., Waldner, H., Chernova, T.et al. (2002) Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 415, 536–541 10.1038/415536a [DOI] [PubMed] [Google Scholar]
- 13.Jin, H.T., Anderson, A.C., Tan, W.G., West, E.E., Ha, S.J., Araki, K.et al. (2010) Cooperation of Tim-3 and PD-1 in CD8T-cell exhaustion during chronic viral infection. Proc. Natl Acad. Sci. U.S.A. 107, 14733–14738 10.1073/pnas.1009731107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones, R.B., Ndhlovu, L.C., Barbour, J.D., Sheth, P.M., Jha, A.R., Long, B.R.et al. (2008) Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J. Exp. Med. 205, 2763–2779 10.1084/jem.20081398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou, Q., Munger, M.E., Veenstra, R.G., Weigel, B.J., Hirashima, M., Munn, D.H.et al. (2011) Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood 117, 4501–4510 10.1182/blood-2010-10-310425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sabatos, C.A., Chakravarti, S., Cha, E., Schubart, A., Sanchez-Fueyo, A., Zheng, X.X.et al. (2003) Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat. Immunol. 4, 1102–1110 10.1038/ni988 [DOI] [PubMed] [Google Scholar]
- 17.Sanchez-Fueyo, A., Tian, J., Picarella, D., Domenig, C., Zheng, X.X., Sabatos, C.A.et al. (2003) Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat. Immunol. 4, 1093–1101 10.1038/ni987 [DOI] [PubMed] [Google Scholar]
- 18.Dixon, K.O., Tabaka, M., Schramm, M.A., Xiao, S., Tang, R., Dionne, D.et al. (2021) TIM-3 restrains anti-tumour immunity by regulating inflammasome activation. Nature 595, 101–106 10.1038/s41586-021-03626-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anderson, A.C., Anderson, D.E., Bregoli, L., Hastings, W.D., Kassam, N., Lei, C.et al. (2007) Promotion of tissue inflammation by the immune receptor Tim-3 expressed on innate immune cells. Science 318, 1141–1143 10.1126/science.1148536 [DOI] [PubMed] [Google Scholar]
- 20.Gorman, J.V., Starbeck-Miller, G., Pham, N.L., Traver, G.L., Rothman, P.B., Harty, J.T.et al. (2014) Tim-3 directly enhances CD8T cell responses to acute Listeria monocytogenes infection. J. Immunol. 192, 3133–3142 10.4049/jimmunol.1302290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kataoka, S., Manandhar, P., Lee, J., Workman, C.J., Banerjee, H., Szymczak-Workman, A.L.et al. (2021) The co-stimulatory activity of Tim-3 requires Akt and MAPK signaling and its recruitment to the immune synapse. Sci. Signal. 14, eaba0717 10.1126/scisignal.aba0717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee, J., Su, E.W., Zhu, C., Hainline, S., Phuah, J., Moroco, J.A.et al. (2011) Phosphotyrosine-dependent coupling of Tim-3 to T-cell receptor signaling pathways. Mol. Cell. Biol. 31, 3963–3974 10.1128/MCB.05297-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee, M.J., Woo, M.Y., Chwae, Y.J., Kwon, M.H., Kim, K. and Park, S. (2012) Down-regulation of interleukin-2 production by CD4(+) T cells expressing TIM-3 through suppression of NFAT dephosphorylation and AP-1 transcription. Immunobiology 217, 986–995 10.1016/j.imbio.2012.01.012 [DOI] [PubMed] [Google Scholar]
- 24.Tomkowicz, B., Walsh, E., Cotty, A., Verona, R., Sabins, N., Kaplan, F.et al. (2015) TIM-3 suppresses anti-CD3/CD28-induced TCR activation and IL-2 expression through the NFAT signaling pathway. PLoS One 10, e0140694 10.1371/journal.pone.0140694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daëron, M., Jaeger, S., Du Pasquier, L. and Vivier, E. (2008) Immunoreceptor tyrosine-based inhibition motifs: a quest in the past and future. Immunol. Rev. 224, 11–43 10.1111/j.1600-065X.2008.00666.x [DOI] [PubMed] [Google Scholar]
- 26.Zhu, C., Anderson, A.C., Schubart, A., Xiong, H., Imitola, J., Khoury, S.J.et al. (2005) The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 6, 1245–1252 10.1038/ni1271 [DOI] [PubMed] [Google Scholar]
- 27.Leitner, J., Rieger, A., Pickl, W.F., Zlabinger, G., Grabmeier-Pfistershammer, K. and Steinberger, P. (2013) TIM-3 does not act as a receptor for galectin-9. PLoS Pathog. 9, e1003253 10.1371/journal.ppat.1003253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Su, E.W., Bi, S. and Kane, L.P. (2011) Galectin-9 regulates T helper cell function independently of Tim-3. Glycobiology 21, 1258–1265 10.1093/glycob/cwq214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leventis, P.A. and Grinstein, S. (2010) The distribution and function of phosphatidylserine in cellular membranes. Annu. Rev. Biophys. 39, 407–427 10.1146/annurev.biophys.093008.131234 [DOI] [PubMed] [Google Scholar]
- 30.Shlomovitz, I., Speir, M. and Gerlic, M. (2019) Flipping the dogma - phosphatidylserine in non-apoptotic cell death. Cell Commun. Signal. 17, 139 10.1186/s12964-019-0437-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fischer, K., Voelkl, S., Berger, J., Andreesen, R., Pomorski, T. and Mackensen, A. (2006) Antigen recognition induces phosphatidylserine exposure on the cell surface of human CD8+ T cells. Blood 108, 4094–4101 10.1182/blood-2006-03-011742 [DOI] [PubMed] [Google Scholar]
- 32.Elliott, J.I., Surprenant, A., Marelli-Berg, F.M., Cooper, J.C., Cassady-Cain, R.L., Wooding, C.et al. (2005) Membrane phosphatidylserine distribution as a non-apoptotic signalling mechanism in lymphocytes. Nat. Cell Biol. 7, 808–816 10.1038/ncb1279 [DOI] [PubMed] [Google Scholar]
- 33.Miyanishi, M., Tada, K., Koike, M., Uchiyama, Y., Kitamura, T. and Nagata, S. (2007) Identification of Tim4 as a phosphatidylserine receptor. Nature 450, 435–439 10.1038/nature06307 [DOI] [PubMed] [Google Scholar]
- 34.DeKruyff, R.H., Bu, X., Ballesteros, A., Santiago, C., Chim, Y.L., Lee, H.H.et al. (2010) T cell/transmembrane, Ig, and mucin-3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J. Immunol. 184, 1918–1930 10.4049/jimmunol.0903059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakayama, M., Akiba, H., Takeda, K., Kojima, Y., Hashiguchi, M., Azuma, M.et al. (2009) Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood 113, 3821–3830 10.1182/blood-2008-10-185884 [DOI] [PubMed] [Google Scholar]
- 36.Tietjen, G.T., Gong, Z., Chen, C.H., Vargas, E., Crooks, J.E., Cao, K.D.et al. (2014) Molecular mechanism for differential recognition of membrane phosphatidylserine by the immune regulatory receptor Tim4. Proc. Natl Acad. Sci. U.S.A. 111, E1463–E1472 10.1073/pnas.1320174111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sabatos-Peyton, C.A., Nevin, J., Brock, A., Venable, J.D., Tan, D.J., Kassam, N.et al. (2018) Blockade of Tim-3 binding to phosphatidylserine and CEACAM1 is a shared feature of anti-Tim-3 antibodies that have functional efficacy. Oncoimmunology 7, e1385690 10.1080/2162402X.2017.1385690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gibson, D.G. (2011) Enzymatic assembly of overlapping DNA fragments. Methods Enzymol. 498, 349–361 10.1016/B978-0-12-385120-8.00015-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seiler, C.Y., Park, J.G., Sharma, A., Hunter, P., Surapaneni, P., Sedillo, C.et al. (2014) DNASU plasmid and PSI:Biology-Materials repositories: resources to accelerate biological research. Nucl. Acids Res. 42, D1253–D1260 10.1093/nar/gkt1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Muzumdar, M.D., Chen, P.Y., Dorans, K.J., Chung, K.M., Bhutkar, A., Hong, E.et al. (2017) Survival of pancreatic cancer cells lacking KRAS function. Nat. Commun. 8, 1090 10.1038/s41467-017-00942-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O'Doherty, U., Swiggard, W.J. and Malim, M.H. (2000) Human immunodeficiency virus type 1 spinoculation enhances infection through virus binding. J. Virol. 74, 10074–10080 10.1128/jvi.74.21.10074-10080.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mujib, S., Jones, R.B., Lo, C., Aidarus, N., Clayton, K., Sakhdari, A.et al. (2012) Antigen-independent induction of Tim-3 expression on human T cells by the common γ-chain cytokines IL-2, IL-7, IL-15, and IL-21 is associated with proliferation and is dependent on the phosphoinositide 3-kinase pathway. J. Immunol. 188, 3745–3756 10.4049/jimmunol.1102609 [DOI] [PubMed] [Google Scholar]
- 43.Xu, X., Hou, B., Fulzele, A., Masubuchi, T., Zhao, Y., Wu, Z.et al. (2020) PD-1 and BTLA regulate T cell signaling differentially and only partially through SHP1 and SHP2. J. Cell Biol. 219, e201905085 10.1083/jcb.201905085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aksamitiene, E., Hoek, J.B. and Kiyatkin, A. (2015) Multistrip Western blotting: a tool for comparative quantitative analysis of multiple proteins. Methods Mol. Biol. 1312, 197–226 10.1007/978-1-4939-2694-7_23 [DOI] [PubMed] [Google Scholar]
- 45.Moravcevic, K., Mendrola, J.M., Schmitz, K.R., Wang, Y.H., Slochower, D., Janmey, P.A.et al. (2010) Kinase associated-1 domains drive MARK/PAR1 kinases to membrane targets by binding acidic phospholipids. Cell 143, 966–977 10.1016/j.cell.2010.11.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Narayan, K. and Lemmon, M.A. (2006) Determining selectivity of phosphoinositide-binding domains. Methods 39, 122–133 10.1016/j.ymeth.2006.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abraham, R.T. and Weiss, A. (2004) Jurkat T cells and development of the T-cell receptor signalling paradigm. Nat. Rev. Immunol. 4, 301–308 10.1038/nri1330 [DOI] [PubMed] [Google Scholar]
- 48.Hayden, M.S., West, A.P. and Ghosh, S. (2006) NF-kappaB and the immune response. Oncogene 25, 6758–6780 10.1038/sj.onc.1209943 [DOI] [PubMed] [Google Scholar]
- 49.Clayton, K.L., Haaland, M.S., Douglas-Vail, M.B., Mujib, S., Chew, G.M., Ndhlovu, L.C.et al. (2014) T cell Ig and mucin domain-containing protein 3 is recruited to the immune synapse, disrupts stable synapse formation, and associates with receptor phosphatases. J. Immunol. 192, 782–791 10.4049/jimmunol.1302663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boomer, J.S. and Green, J.M. (2010) An enigmatic tail of CD28 signaling. Cold Spring Harb. Perspect. Biol. 2, a002436 10.1101/cshperspect.a002436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cook, J.A., Albacker, L., August, A. and Henderson, A.J. (2003) CD28-dependent HIV-1 transcription is associated with Vav, Rac, and NF-kappa B activation. J. Biol. Chem. 278, 35812–35818 10.1074/jbc.M302878200 [DOI] [PubMed] [Google Scholar]
- 52.Teng, J.M., King, P.D., Sadra, A., Liu, X., Han, A., Selvakumar, A.et al. (1996) Phosphorylation of each of the distal three tyrosines of the CD28 cytoplasmic tail is required for CD28-induced T cell IL-2 secretion. Tissue Antigens 48, 255–264 10.1111/j.1399-0039.1996.tb02643.x [DOI] [PubMed] [Google Scholar]
- 53.Prasad, K.V., Cai, Y.C., Raab, M., Duckworth, B., Cantley, L., Shoelson, S.E.et al. (1994) T-cell antigen CD28 interacts with the lipid kinase phosphatidylinositol 3-kinase by a cytoplasmic Tyr(P)-Met-Xaa-Met motif. Proc. Natl Acad. Sci. U.S.A. 91, 2834–2838 10.1073/pnas.91.7.2834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pagès, F., Ragueneau, M., Rottapel, R., Truneh, A., Nunes, J., Imbert, J.et al. (1994) Binding of phosphatidylinositol-3-OH kinase to CD28 is required for T-cell signalling. Nature 369, 327–329 10.1038/369327a0 [DOI] [PubMed] [Google Scholar]
- 55.Shan, X., Czar, M.J., Bunnell, S.C., Liu, P., Liu, Y., Schwartzberg, P.L.et al. (2000) Deficiency of PTEN in Jurkat T cells causes constitutive localization of Itk to the plasma membrane and hyperresponsiveness to CD3 stimulation. Mol. Cell. Biol. 20, 6945–6957 10.1128/mcb.20.18.6945-6957.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cao, E., Zang, X., Ramagopal, U.A., Mukhopadhaya, A., Fedorov, A., Fedorov, E.et al. (2007) T cell immunoglobulin mucin-3 crystal structure reveals a galectin-9-independent ligand-binding surface. Immunity 26, 311–321 10.1016/j.immuni.2007.01.016 [DOI] [PubMed] [Google Scholar]
- 57.Lin, Y.C., Chipot, C. and Scheuring, S. (2020) Annexin-V stabilizes membrane defects by inducing lipid phase transition. Nat. Commun. 11, 230 10.1038/s41467-019-14045-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang, W., Bai, Y., Xiong, Y., Zhang, J., Chen, S., Zheng, X.et al. (2016) Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature 531, 651–655 10.1038/nature17412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lemke, G. (2017) Phosphatidylserine is the signal for TAM receptors and their ligands. Trends Biochem. Sci. 42, 738–748 10.1016/j.tibs.2017.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heinrich, L., Tissot, N., Hartmann, D.J. and Cohen, R. (2010) Comparison of the results obtained by ELISA and surface plasmon resonance for the determination of antibody affinity. J. Immunol. Methods 352, 13–22 10.1016/j.jim.2009.10.002 [DOI] [PubMed] [Google Scholar]
- 61.Lemmon, M.A. (2008) Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell Biol. 9, 99–111 10.1038/nrm2328 [DOI] [PubMed] [Google Scholar]
- 62.Yeung, T., Gilbert, G.E., Shi, J., Silvius, J., Kapus, A. and Grinstein, S. (2008) Membrane phosphatidylserine regulates surface charge and protein localization. Science 319, 210–213 10.1126/science.1152066 [DOI] [PubMed] [Google Scholar]
- 63.Sansom, D.M. (2000) CD28, CTLA-4 and their ligands: who does what and to whom? Immunology 101, 169–177 10.1046/j.1365-2567.2000.00121.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zak, K.M., Grudnik, P., Magiera, K., Dömling, A., Dubin, G. and Holak, T.A. (2017) Structural biology of the immune checkpoint receptor PD-1 and its ligands PD-L1/PD-L2. Structure 25, 1163–1174 10.1016/j.str.2017.06.011 [DOI] [PubMed] [Google Scholar]
- 65.Baxter, A.A., Hulett, M.D. and Poon, I.K. (2015) The phospholipid code: a key component of dying cell recognition, tumor progression and host-microbe interactions. Cell Death Differ. 22, 1893–1905 10.1038/cdd.2015.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Emoto, K., Toyama-Sorimachi, N., Karasuyama, H., Inoue, K. and Umeda, M. (1997) Exposure of phosphatidylethanolamine on the surface of apoptotic cells. Exp. Cell Res. 232, 430–434 10.1006/excr.1997.3521 [DOI] [PubMed] [Google Scholar]
- 67.Richard, A.S., Zhang, A., Park, S.J., Farzan, M., Zong, M. and Choe, H. (2015) Virion-associated phosphatidylethanolamine promotes TIM1-mediated infection by Ebola, dengue, and West Nile viruses. Proc. Natl Acad. Sci. U.S.A. 112, 14682–14687 10.1073/pnas.1508095112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen, D., Iijima, H., Nagaishi, T., Nakajima, A., Russell, S., Raychowdhury, R.et al. (2004) Carcinoembryonic antigen-related cellular adhesion molecule 1 isoforms alternatively inhibit and costimulate human T cell function. J. Immunol. 172, 3535–3543 10.4049/jimmunol.172.6.3535 [DOI] [PubMed] [Google Scholar]
- 69.Donda, A., Mori, L., Shamshiev, A., Carena, I., Mottet, C., Heim, M.H.et al. (2000) Locally inducible CD66a (CEACAM1) as an amplifier of the human intestinal T cell response. Eur. J. Immunol. 30, 2593–2603 [DOI] [PubMed] [Google Scholar]
- 70.Dayoub, A.S. and Brekken, R.A. (2020) TIMs, TAMs, and PS- antibody targeting: implications for cancer immunotherapy. Cell Commun. Signal. 18, 29 10.1186/s12964-020-0521-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Freeman, G.J., Casasnovas, J.M., Umetsu, D.T. and DeKruyff, R.H. (2010) TIM genes: a family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol. Rev. 235, 172–189 10.1111/j.0105-2896.2010.00903.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhao, D., Guo, M., Liu, B., Lin, Q., Xie, T., Zhang, Q.et al. (2017) Frontline science: Tim-3-mediated dysfunctional engulfment of apoptotic cells in SLE. J. Leukoc. Biol. 102, 1313–1322 10.1189/jlb.3HI0117-005RR [DOI] [PubMed] [Google Scholar]
- 73.Zhu, C., Anderson, A.C. and Kuchroo, V.K. (2011) TIM-3 and its regulatory role in immune responses. Curr. Top. Microbiol. Immunol. 350, 1–15 10.1007/82_2010_84 [DOI] [PubMed] [Google Scholar]
- 74.Niki, T., Tsutsui, S., Hirose, S., Aradono, S., Sugimoto, Y., Takeshita, K.et al. (2009) Galectin-9 is a high affinity IgE-binding lectin with anti-allergic effect by blocking IgE-antigen complex formation. J. Biol. Chem. 284, 32344–32352 10.1074/jbc.M109.035196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bi, S., Hong, P.W., Lee, B. and Baum, L.G. (2011) Galectin-9 binding to cell surface protein disulfide isomerase regulates the redox environment to enhance T-cell migration and HIV entry. Proc. Natl Acad. Sci. U.S.A. 108, 10650–10655 10.1073/pnas.1017954108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cao, A., Alluqmani, N., Buhari, F.H.M., Wasim, L., Smith, L.K., Quaile, A.T.et al. (2018) Galectin-9 binds IgM-BCR to regulate B cell signaling. Nat. Commun. 9, 3288 10.1038/s41467-018-05771-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wu, C., Thalhamer, T., Franca, R.F., Xiao, S., Wang, C., Hotta, C.et al. (2014) Galectin-9-CD44 interaction enhances stability and function of adaptive regulatory T cells. Immunity 41, 270–282 10.1016/j.immuni.2014.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Oomizu, S., Arikawa, T., Niki, T., Kadowaki, T., Ueno, M., Nishi, N.et al. (2012) Galectin-9 suppresses Th17 cell development in an IL-2-dependent but Tim-3-independent manner. Clin. Immunol. 143, 51–58 10.1016/j.clim.2012.01.004 [DOI] [PubMed] [Google Scholar]
- 79.Gleason, M.K., Lenvik, T.R., McCullar, V., Felices, M., O'Brien, M.S., Cooley, S.A.et al. (2012) Tim-3 is an inducible human natural killer cell receptor that enhances interferon gamma production in response to galectin-9. Blood 119, 3064–3072 10.1182/blood-2011-06-360321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Phong, B.L., Avery, L., Sumpter, T.L., Gorman, J.V., Watkins, S.C., Colgan, J.D.et al. (2015) Tim-3 enhances FcεRI-proximal signaling to modulate mast cell activation. J. Exp. Med. 212, 2289–2304 10.1084/jem.20150388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tan, S., Xu, Y., Wang, Z., Wang, T., Du, X., Song, X.et al. (2020) Tim-3 hampers tumor surveillance of liver-resident and conventional NK cells by disrupting PI3K signaling. Cancer Res. 80, 1130–1142 10.1158/0008-5472.CAN-19-2332 [DOI] [PubMed] [Google Scholar]
- 82.Zhang, D., Jiang, F., Zaynagetdinov, R., Huang, H., Sood, V.D., Wang, H.et al. (2020) Identification and characterization of M6903, an antagonistic anti-TIM-3 monoclonal antibody. Oncoimmunology 9, 1744921 10.1080/2162402X.2020.1744921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lindsted, T., Gad, M., Grandal, M.V., Frölich, C., Bhatia, V.K., Gjetting, T.et al. (2018) Abstract 5629: Preclinical characterization of Sym023 a human anti-TIM3 antibody with a novel mechanism of action. Cancer Res. 78, 5629–5629 10.1158/1538-7445.Am2018-5629 [DOI] [Google Scholar]
- 84.Zhang, T., Xue, L., Zhang, J., Liu, Q., Ma, J., Zhang, Y.et al. (2017) Abstract 2628: BGB-A425: a humanized anti-human Tim-3 antibody that exhibits strong immune cell activation. Cancer Res. 77, 2628–2628 10.1158/1538-7445.Am2017-2628 [DOI] [Google Scholar]
- 85.Waight, J., Iyer, P., Breous-Nystrom, E., Riordan, C., Findeis, M., Underwood, D.et al. (2018) Abstract 3825: INCAGN02390, a novel antagonist antibody that targets the co-inhibitory receptor TIM-3. Cancer Res. 78, 3825–3825 10.1158/1538-7445.Am2018-3825 [DOI] [Google Scholar]
- 86.Kuang, Z., Li, L., Zhang, P., Chen, B., Wu, M., Ni, H.et al. (2020) A novel antibody targeting TIM-3 resulting in receptor internalization for cancer immunotherapy. Antib. Ther. 3, 227–236 10.1093/abt/tbaa022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Harding, J.J., Moreno, V., Bang, Y.J., Hong, M.H., Patnaik, A., Trigo, J.et al. (2021) Blocking TIM-3 in treatment-refractory advanced solid tumors: a phase Ia/b study of LY3321367 with or without an anti-PD-L1 antibody. Clin. Cancer Res. 27, 2168–2178 10.1158/1078-0432.CCR-20-4405 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data needed to evaluate this work are all included in the manuscript, and are available upon request.