

Summary
Unveiling the distance effect between different sites in multifunctional catalysts remains a major challenge. Herein, we investigate the distance effect by constructing a dual-site distance-controlled tandem catalyst with a five-layered TiO2/Pt/TiO2/Ni/TiO2 tubular nanostructure by template-assisted atomic layer deposition. In this catalyst, the Ni and Pt sites are separated by a porous TiO2 interlayer, and the distance between them can be precisely controlled on the subnanometer scale by altering the thickness of the interlayer, while the inner and outer porous TiO2 layers are designed for structural stability. The catalyst exhibits superior performance for the tandem hydrazine hydrate decomposition to hydrogen and subsequent nitrobenzene hydrogenation when the Ni and Pt site distance is on the subnanometer level. The performance increases with the decrease of the distance and is better than the catalyst without the TiO2 interlayer. Isotopic and kinetic experiments reveal that the distance effect controls the transfer of active hydrogen, which is the rate-determining step of the tandem reaction in a water solvent. Reduced Ti species with oxygen vacancies on the TiO2 interlayer provide the active sites for hydrogen transfer with -Ti-OH surface intermediates via the continuous chemisorption/desorption of water. A smaller distance induces the generation of more active sites for hydrogen transfer and thus higher efficiency in the synergy of Ni and Pt sites. Our work provides new insight for the distance effect of different active sites and the mechanism of intermediate transfer in tandem reactions.
Keywords: distance effect, dual sites, hydrogen transfer, interlayer, tandem catalyst
Graphical Abstract
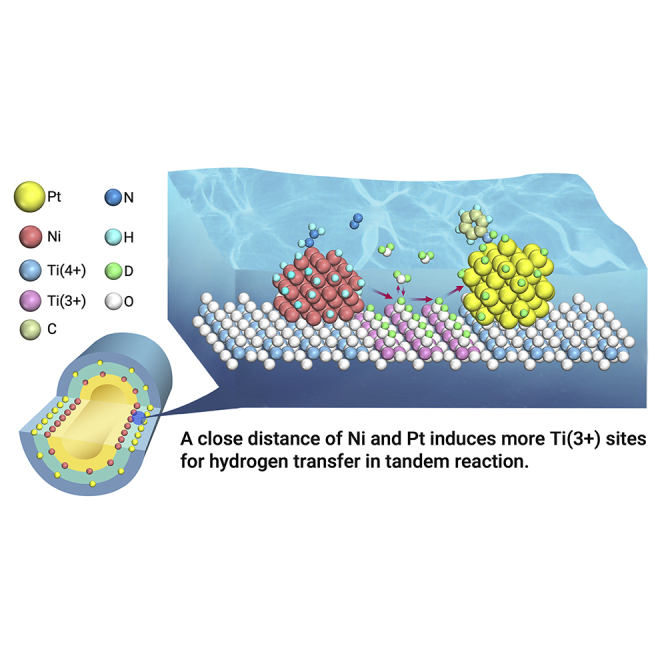
Public Summary
-
•
The distance effect is an interesting and important topic in catalysis
-
•
The distance of Ni-Pt dual sites is precisely controlled in subnanometer scale on a TiO2/Pt/TiO2/Ni/TiO2 five-layer catalyst by ALD
-
•
The distance controls the water-assisted hydrogen transfer, determining the overall efficiency of the tandem reaction
-
•
A close distance in subnanometer induces more active sites for hydrogen transfer and efficient synergy of Ni and Pt sites
Introduction
“One-pot” tandem catalysis, in which multiple catalysts and reagents are combined in a single reactor for precisely staged catalytic steps, can reduce energy loss, waste, and time, resulting in economic and environmental benefits in the synthesis of chemicals.1 It is highly desirable to design highly efficient multifunctional heterogeneous catalysts for tandem catalysis.2, 3, 4 In a tandem catalyst, multiple functional sites are combined to accommodate the same reaction conditions and realize the couple of individual reactions with high efficiency in the transfer of active intermediates between functional sites. Currently, it is still challenging to determine the distance effect on the transfer mechanism of intermediates due to the limitations in precisely controlling the distance between different active sites at the subnanoscale by traditional methods.
Spatial isolation of complementary functional sites with an appropriate distance affords control over the reaction sequence and paths in tandem catalysis. For example, in a mixture of zeolite Y and alumina binder, the selectivity in cracking large hydrocarbons for high-quality diesel production is optimized when Pt nanoparticles are located on the binder with nanoscale separation rather than directly on zeolite Y with no separation.5 In some cases, intermediates without enough stability may be transformed into new useful products by quickly entering a subsequent catalytic cycle before decomposition.2 For example, the highly selective formation of olefins from syngas is enabled by a physical mixture of ZnCrOx and MSAPO zeolite through the generation of a C∗ (CH2CO) intermediate on ZnCrOx followed by the intermediate transfer and transformation in zeolite to olefins.6 Similar coupling approaches of oxides (or metal) and zeolites were utilized in CO2 hydrogenation to liquid fuels7 or olefins8 and syngas to isoparaffin,9 dimethyl ether,10 or aromatics.11,12 Although the isolation of different active sites and distance play a key role in a high-efficiency tandem catalyst, the transfer of intermediates and the distance effect are not clear due to the limited methodologies in distance control between functional sites in a tandem catalyst.
Atomic layer deposition (ALD) is a high-level film growth technology that relies on two sequential self-limiting surface reactions.13,14 The unique self-limiting character makes ALD capable of constructing nanomaterials with high conformity, good spatial uniformity, and atomic thickness control of films.15,16 In recent decades, advanced catalysts were synthesized by precisely controlling the size, interface, and pore structures via ALD at the atomic and molecular levels.17, 18, 19, 20, 21, 22, 23, 24 Moreover, ALD strategies have been developed to design and synthesize well-defined catalysts with complex functional structures, such as confined catalysts,25, 26, 27 sandwich catalysts,28 catalysts with spatially separated metal structures,29,30 tube-in-tube catalysts,31 and encapsulated catalysts.21 The confinement effect, the function of the metal-oxide interface, and the synergy effect have been well investigated and revealed in these catalytic systems.
Herein, the distance effect is investigated by developing a well-defined tubular five-layer tandem catalyst (TiO2/Pt/TiO2/Ni/TiO2) via template-assisted ALD to precisely tailor the distance between the metal sites on the subnanometer scale (Scheme 1). Pt and Ni nanoparticle layers were embedded in and separated by a porous titanium oxide layer. The distance between the Ni and Pt layers can be precisely controlled by altering the ALD cycle number of the TiO2 interlayer. The distance effect and function of the interlayer on hydrogen transfer were evaluated using hydrazine hydrate (N2H4·H2O) decomposition to hydrogen and subsequent nitrobenzene hydrogenation as the probe tandem reaction. In particular, the Ni and Pt layers catalyze only hydrazine hydrate decomposition and nitrobenzene hydrogenation, respectively. Compared with TiO2/Pt/Ni/TiO2 with no interlayer, tube-in-tube TiO2/Pt|Ni/TiO2, single-metal TiO2/Pt/TiO2 and TiO2/Ni/TiO2, and a mixture of TiO2/Pt/TiO2 and TiO2/Ni/TiO2, the TiO2/Pt/TiO2/Ni/TiO2 five-layer catalyst exhibits higher activity in the tandem reaction. Moreover, the catalytic activity decreases with increasing interlayer thickness and is optimized over TiO2/Pt/10TiO2/Ni/TiO2 (0.6 nm distance). The enhanced activity is ascribed to the enhancement of active hydrogen transfer between two metal layers. The water solvent participates in the key step of hydrogen transfer on the TiO2 interlayer.
Scheme 1.

Schematic Illustration of the Synthesis Process of the TiO2/Pt/TiO2/Ni/TiO2 Distance-Controlled Dual-Site Tandem Catalyst by ALD.
For control experiments, TiO2/Ni/TiO2 without a Pt layer and TiO2/Pt/TiO2 without a Ni layer are prepared by omitting Pt ALD and Ni ALD, respectively. In addition, a TiO2/Pt|Ni/TiO2 catalyst with a tube-in-tube structure is synthesized using polyimide as an interlayer followed by calcination in air and reduction in hydrogen.
Results
Synthesis and Characterization of the Distance-Controlled Dual-Site Tandem Catalyst
To investigate the distance effect of dual sites in the tandem reaction, we designed a five-layer catalyst with a TiO2/Pt/yTiO2/xNi/TiO2 structure as an example. It was prepared by sequentially depositing an inner TiO2 layer (300 cycles), NiO (x cycles, x = 300), a TiO2 interlayer (y cycles, y = 0, 10, 20, 30, 50, and 100), Pt (20 cycles), and an outer TiO2 layer (300 cycles) by the template-assisted ALD method using carbon nanofibers (CNFs) as templates, followed by calcination in air and reduction in H2/Ar. The inner and outer TiO2 layers (with a fixed thickness) are used to maintain the structural stability of the catalyst, since the thickness of the TiO2 interlayer is limited, particularly for distance control at the subnanometer level. The TiO2 interlayer is used to separate the Ni and Pt sites. The distance between the Ni and Pt sites can be precisely controlled by changing the ALD cycle number (y) of the TiO2 interlayer with an average growth rate of 0.06 nm/cycle. In addition, other catalysts were also synthesized by the same method for comparison. For instance, TiO2/Ni/TiO2 without a Pt layer, TiO2/Pt/TiO2 without a Ni layer, and TiO2/Pt/Ni/TiO2 without a TiO2 interlayer were prepared by omitting the ALD process for the Pt, Ni, and TiO2 interlayers, respectively. In addition, a TiO2/Pt|xNi/TiO2 catalyst with a tube-in-tube structure was prepared with polyimide as the interlayer followed by calcination in air and reduction in H2/Ar, and the movable inner tube results in an undefined distance and possible instant contact between two metal sites.
As revealed by transmission electron microscopy (TEM) (Figures 1 and S1), all the catalysts show a similar sandwiched structure. A 3.0 nm TiO2 interlayer is observed for TiO2/Pt/50TiO2/Ni/TiO2 (Figures 1A and 1B), which is in accordance with the deposition rate of TiO2 (0.06 nm/cycle). However, the thickness of the TiO2 interlayer between Pt and Ni is too thin to be observed for TiO2/Pt/10TiO2/Ni/TiO2 and TiO2/Pt/30TiO2/Ni/TiO2. The Pt particles (0.4 wt%, ICP-OES) and Ni particles (1.4 wt%, ICP-OES) are not clearly visible due to their low content, small particle size, and reduced contrast due to these TiO2 layers. Furthermore, a cross-sectional slice of TiO2/Pt/10TiO2/Ni/TiO2 along the direction perpendicular to the tube was prepared by focused ion beam milling. The cross-section of the five layers of TiO2/Pt/10TiO2/Ni/TiO2 is clearly observed in Figures 1C and 1D. The electronic structure of the TiO2 interlayer and the inner and outer layers on this slice was also characterized by electron energy loss spectroscopy (EELS) measurements (Figure 1E). Typically, the inner and outer TiO2 layers exhibit two similar L2 and L3 peaks of Ti (4+) species at 464.9 and 459.6 eV, while an obvious redshift (0.5 eV) of Ti-L2 and Ti-L3 peak of reduced Ti(3+) species are detected for the region at the TiO2 interlayer.32,33 In addition, the high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and energy dispersive X-ray (EDX) spectrometry images of TiO2/Pt/50TiO2/Ni/TiO2 further indicate the distinguished five-layer structure and the homogeneous distribution of Ti, Ni, and Pt compositions (Figure 1F). These results confirm that the distance between the Ni and Pt dual sites can be precisely controlled depending on the designed five-layer catalyst structure.
Figure 1.

Structural Characterization of the Catalyst.
(A and B) TEM images of TiO2/Pt/50TiO2/Ni/TiO2.
(C and D) TEM image of the cross-sectional slice of TiO2/Pt/10TiO2/Ni/TiO2 prepared by focused ion beam milling and the corresponding area for the EELS line scan.
(E) Selected Ti-L-edge EELS spectra recorded at positions 1–7 in (D).
(F) STEM image and STEM-EDX element mapping of the Ni-L-, Ti-L-, and Pt-L-edges of TiO2/Pt/50TiO2/Ni/TiO2.
The physicochemical properties of the catalysts were characterized by various technologies. First, N2 physical absorption revealed that the pore structure is similar for TiO2/Pt/yTiO2/Ni/TiO2 (y = 10–100) and TiO2/Pt/Ni/TiO2 (Figure S2; Table S1). The surface area and average Barrett-Joyner-Halenda pore diameter are approximately 35–36 m2/g and 3.6–3.7 nm, respectively, for different samples (Table S1). Nanopores are formed during calcination due to the crystallization of TiO2.28 Second, the loading of Ni (1.39–1.41 wt%) and Pt (0.39–0.41 wt%) is also similar for TiO2/Pt/yTiO2/Ni/TiO2, TiO2/Pt/TiO2, and TiO2/Ni/TiO2. Third, only diffraction peaks of the anatase phase and no diffraction peaks of Pt and Ni are observed in the X-ray diffraction (XRD) patterns of different catalysts due to the low content and high dispersion of these two metals (Figure S3). Fourth, the number of surface-active sites for hydrogen adsorption measured by hydrogen pulse adsorption at 30°C are also similar over different catalysts (Table S1). The similar pore structure, metal loadings, crystal structure, and the number of the active site for hydrogen adsorption are critically important for reliably revealing the distance effect and the functions of the interlayer.
X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectroscopy were conducted to illustrate the electronic structure of Pt and Ni species. In the Ni K-edge XANES spectra, the white lines for TiO2/Pt/yTiO2/Ni/TiO2 and TiO2/Ni/TiO2 are higher but rather similar to that of the Ni foil (Figure 2A), indicating the presence of both metallic Ni and Ni oxide species.34 The edge features of the Pt L3-edge for TiO2/Pt/yTiO2/Ni/TiO2, TiO2/Pt/Ni/TiO2, and TiO2/Pt/TiO2 are similar to those of the Pt foil, indicating the dominant metallic Pt (Figure 2B). Fourier transform of EXAFS spectra in r space and corresponding fitting results are shown in Figures 2C and 2D and Tables S2 and S3. The Pt-Ni bond from the alloy is observed only for TiO2/Pt/Ni/TiO2 without the TiO2 interlayer. Thus, reliable spatial isolation of the Ni and Pt layers is realized even by a subnanometer-thick TiO2 interlayer for TiO2/Pt/10TiO2/Ni/TiO2. Furthermore, the low coordination number of Ni and Pt species in the fitting results indicates the formation of small metallic Ni and Pt subnanoclusters,35 and the coordination structure of the two metals is nearly unchanged with the thickness of the interlayer.
Figure 2.

Electronic Structure of the Catalysts.
The normalized intensity of XANES spectra for (A) Ni K and (B) Pt L3. The normalized intensity of Fourier transform weighted EXAFS spectra for (C) Ni K and (D) Pt L3.
Catalytic Performance and Distance Effect of the Dual-Site Tandem Catalyst
On the basis of a precisely designed catalyst structure, the distance effect between Pt and Ni layers has been investigated by applying tandem hydrogenation of nitrobenzene to aniline using N2H4·H2O as a hydrogen source in water as a perfect model reaction, since the Ni layer can catalyze the decomposition of N2H4·H2O to produce hydrogen,36 while the Pt layer can catalyze nitrobenzene hydrogenation efficiently. The different functions of the Ni and Pt layers have been clearly confirmed for the two single reactions by control experiments (Figure S4). In the tandem reaction, the TiO2/Pt/10TiO2/Ni/TiO2 catalyst exhibits remarkably higher activity (24.8 mmol gCat.−1 h−1) than other catalysts with different structures, including TiO2/Pt/TiO2 (2.4 mmol gCat.−1 h−1) and TiO2/Ni/TiO2 (2.5 mmol gCat.−1 h−1) with one metal layer, TiO2/Pt/Ni/TiO2 without an interlayer (16.3 mmol gCat.−1 h−1), a physical mixture of TiO2/Pt/TiO2 and TiO2/Ni/TiO2 (5.8 mmol gCat.−1 h−1), and TiO2/Pt|Ni/TiO2 with a tube-in-tube structure (9.3 mmol gCat.−1 h−1) (Figures 3A and S5). This indicates the advantages of the spatially isolated Pt and Ni layers with an ultrathin TiO2 interlayer. We calculated the H2 consumption rate (74.3 mmol gCat.−1 h−1) in the tandem catalysis reaction, which is more than 100 times higher than the H2 generation rate from the single decomposition reaction of N2H4·H2O (0.62 mmol gCat.−1 h−1) over the same TiO2/Pt/10TiO2/Ni/TiO2 catalyst (Figures 3A and S4). Thus, hydrogen generation is accelerated during the tandem reaction. The influence of the distance between the Pt and Ni layers is conveniently investigated by precisely changing the thickness of the TiO2 interlayer. As shown in Figure 3B, the tandem catalytic activity increases as the ALD cycle number of the TiO2 interlayer decreases from 100 to 10, and TiO2/Pt/10TiO2/Ni/TiO2 exhibits the highest activity, indicating that a short distance is favorable for the high catalytic activity in this tandem reaction, while the formation of a Ni-Pt alloy must be avoided. Since the selectivity of aniline is 99% for all the tested catalyst, the distance only influences the catalytic activity in the tandem reaction. Furthermore, the effects of the size of the Ni component and interlayer materials on the catalytic performance have also been investigated. The same tendency is also observed when decreasing the ALD cycle number of the Ni layer from 300 to 100 for the five-layer catalysts. When porous Al2O3 or ZnO with different thicknesses were deposited as the interlayer (Figures 3C and S6), the activity in the tandem reaction was very low. In addition, the reusability of TiO2/Pt/10TiO2/Ni/TiO2 was tested. It exhibited nearly similar activity for five consecutive recycling runs, indicating its outstanding stability (Figure 3D). These results indicate that highly efficient synergy between the Ni and Pt sites at the subnanometer scale is generated by separating the two metal sites with an ultrathin reducible TiO2 interlayer.
Figure 3.

Catalytic Performance.
(A) Tandem catalytic performance for nitrobenzene hydrogenation using N2H4·H2O as the hydrogen source over TiO2/Ni/TiO2, TiO2/Pt/TiO2, a physical mixture of TiO2/Ni/TiO2 and TiO2/Pt/TiO2, TiO2/Pt|Ni/TiO2, TiO2/Pt/Ni/TiO2, and TiO2/Pt/10TiO2/Ni/TiO2.
(B) The influence of the interlayer thickness and Ni content of TiO2/Pt/yTiO2/xNi/TiO2 on the catalytic activity.
(C) Comparison of the catalytic performance of TiO2/Pt/10TiO2/Ni/TiO2, TiO2/Pt/10ZnO/Ni/TiO2, and TiO2/Pt/10Al2O3/Ni/TiO2 with different interlayers for the tandem reaction.
(D) Recycling tests of TiO2/Pt/10TiO2/Ni/TiO2 in the tandem reaction.
Transfer Mechanism of Active Hydrogen from Ni to Pt Sites
In the tandem reaction, the active hydrogen generated from the decomposition of N2H4·H2O on the Ni layer needs to be transferred to the Pt layer for hydrogenation since the Ni and Pt sites of TiO2/Pt/yTiO2/Ni/TiO2 are separated by the TiO2 interlayer. The distance effect may be due to a change in the transfer mechanism of active hydrogen during the tandem reaction. To unveil the transfer mechanism of active hydrogen, isotopic experiments were then conducted to investigate the kinetic isotope effect using D2O/ethanol as the solvent over the TiO2/Pt/10TiO2/Ni/TiO2 catalyst. At first, for single reactions, D2O has nearly no isotope effect on either N2H4·H2O decomposition or nitrobenzene hydrogenation in H2 due to the low of approximately 1 (Figures 4A and 4B). However, there is an obvious activity decrease in D2O/ethanol in the tandem reaction and a high of 2.8 (Figure 4C). Further gas chromatography-mass spectrometry analysis shows that C6H5NDH (m/z = 94, 41.8%) and C6H5ND2 (m/z = 95, 41.0%) are the main products of the tandem reaction in D2O/ethanol over the TiO2/Pt/10TiO2/Ni/TiO2 catalyst (Figures S7 and S8;Table S4). It can be seen that 62% of hydrogen atoms for the hydrogenation step are from D2O instead of N2H4·H2O. Therefore, it can be inferred that the active hydrogen transfer involving cleavage of the O-H bond in the H2O solvent is the rate-determining step in the tandem reaction over the TiO2/Pt/10TiO2/Ni/TiO2 catalyst.
Figure 4.

Isotope Experiments.
(A) Isotope effect in the decomposition of N2H4·H2O to hydrogen over TiO2/Pt/10TiO2/Ni/TiO2.
(B) Isotope effect in nitrobenzene hydrogenation using H2 (10 bar) as a hydrogen source over TiO2/Pt/10TiO2/Ni/TiO2.
(C) Isotope effect in the tandem decomposition of N2H4·H2O and nitrobenzene hydrogenation over TiO2/Pt/10TiO2/Ni/TiO2.
(D) The correlation between the ALD cycle number of the TiO2 interlayer and the in the tandem reaction over TiO2/Pt/yTiO2/Ni/TiO2.
(E) The correlation between and reaction rate over the TiO2/Pt/yTiO2/Ni/TiO2 five-layer catalysts in the tandem reaction (the number near the solid square is the thickness value of the TiO2 interlayer).
Isotopic experiments for the tandem reaction were also conducted for different five-layer catalysts to reveal the influence of the thickness of the TiO2 interlayer on hydrogen transfer. The value decreased from 2.8 to 1.5 as the ALD cycle number of the TiO2 interlayer increased from 10 to 300 (Figure 4D). This indicates that the active hydrogen transfer involving cleavage of the O-H bond is dramatically changed with the thickness of the TiO2 interlayer. In principle, the decrease in the isotope effect is related to the decrease in activation energy and/or active site number on the interlayer for hydrogen transfer.37 However, the dynamic experiments show that the calculated apparent activation energy is almost unchanged over different five-layer catalysts (Figure S9). This implies that the extent of water participation involving cleavage of the O-H bond during active hydrogen transfer is dramatically changed with the thickness of the TiO2 interlayer. Thus, the decrease in the isotope effect indicates the decrease in active sites for hydrogen transfer on a thick TiO2 interlayer. The corresponding perfect linear relationship between and the reaction rate shown in Figure 4E further confirms this conclusion.
Discussion
Although many technologies have been developed for the distance control and spatial isolation of different functional sites, there is still no report on the precise control of the distance on the subnanometer scale to reveal the distance effect. In this work, the distance between the Ni and Pt layers was regulated precisely on the subnanometer scale by changing the thickness of the interlayer. The distance effect was revealed using the tandem decomposition of N2H4·H2O and hydrogenation of nitrobenzene as a model reaction. In this system, the Ni layer catalyzes only the decomposition of N2H4·H2O, while the hydrogenation of nitrobenzene proceeds on the Pt layer. The synergy of the two metal layers dramatically increases the activity in the tandem reaction and is enhanced with a short distance through a more efficient path. In this path, the active hydrogen atoms generated from the decomposition of N2H4·H2O on the Ni layer are instantly transferred to the Pt layer for hydrogenation instead of desorption to produce H2 molecules for the hydrogenation step. The strong isotope effect of D2O indicates that the active hydrogen transfer step via the dissociation and formation of the O-H bond on the surface of the TiO2 interlayer predominantly determines the efficiency of the overall tandem reaction. A linear relationship between the value and the reaction rate and unchanged apparent activation energy over the five-layer catalysts with different thicknesses of the TiO2 interlayer indicates that the number of active sites for hydrogen transfer increases with decreasing thickness of the TiO2 interlayer.
According to the EELS characterization, more reduced Ti oxide species (≤3+) with oxygen vacancy defect sites ( sites) are generated on the thinner TiO2 interlayer. Furthermore, H2 temperature-programmed reduction (H2-TPR, Figure S10) results show that the reduction of TiO2 shifts to lower temperatures for the five-layer catalyst with a thinner interlayer, indicating the formation of more reduced Ti oxide species with a thinner layer. These sites on anatase TiO2 can interact and coexist with water molecules.38 With the assistance of the sites and reduced Ti oxide species, the water molecules will easily interact with the surface O atoms of the TiO2 interlayer to form surface O-H species (H2O + -Ti-VO- + -Ti-O- → -Ti-OH)38,39 and are expected to continuously adsorb/desorb on the water/TiO2 interface facilitating hydrogen transfer. The hydrogen transfer can be well coupled with the spillover of active hydrogen at the metal-oxide interface with the -Ti-OH surface intermediate.40 Therefore, we propose a plausible mechanism for active hydrogen transfer on the five-layer catalyst as shown in Figure 5. The active hydrogen atoms from the decomposition of N2H4·H2O (H-Ni) transfer from the Ni layer to the TiO2 interlayer and further to the Pt interlayer for hydrogenation. Considering that the spillover of hydrogen is easy at the metal-oxide interface, the transfer of hydrogen on the interlayer with the aid of water predominantly determines the overall reaction rate of the tandem reaction. Since the reducibility of Al2O3 and ZnO is not comparable with that of TiO2, the synergistic effect is considerably decreased due to less available surface O-H species when they are used as the interlayers. Therefore, both proper distance and medium for the transfer of active intermediates are critically important for the efficient synergy of dual-site tandem catalysts.
Figure 5.

Transfer Mechanism of Active Hydrogen on the TiO2 Interlayer by Water Participation in the Tandem Reaction
Conclusions
In summary, we successfully revealed the distance effect of Ni and Pt sites in the tandem reaction by developing a TiO2/Pt/TiO2/Ni/TiO2 five-layer catalyst with a precise thickness-controlled TiO2 interlayer. Higher efficiency on the tandem reaction is observed with a small distance on the subnanometer scale. The distance effect controls the transfer of active hydrogen, which is the rate-determining step of the tandem reaction in water. The reduced Ti species with oxygen vacancies on the TiO2 interlayer provide the active sites for hydrogen transfer with the -Ti-OH surface intermediates via the continuous chemisorption/desorption of water. A smaller distance induces more active sites for hydrogen transfer and higher efficiency in the synergy of Ni and Pt sites.
Materials and Methods
ALD
The ALD processes of TiO2, Pt nanoparticles, NiO, polyimide, Al hybrid film, and ZnO were conducted at a hot-wall closed chamber-type ALD reactor with different ALD processes as we have described.31 The Al-hybrid film was deposited at 180°C with trimethylaluminum (TMA) and ethylene glycol (EG) (80°C) as precursors. The pulse, exposure, and purge time were 0.02, 8, and 25 s for TMA and 1, 10, and 25 s for EG, respectively. Porous Al2O3 film is obtained after the calcination of the Al-hybrid film. The deposition of ZnO was conducted at 180°C with diethylzinc and deionized water as precursors. The pulse, exposure, and purge time were 0.02, 8, and 25 s for diethyl zinc and 1, 10, and 25 s for H2O, respectively.
Catalysts Preparation
All catalysts were prepared by template-assisted ALD. Typically, TiO2/Pt/yTiO2/xNi/TiO2 was prepared by the sequential deposition of 300 cycles TiO2, x cycles Ni (x = 100 or 300), y cycles TiO2 interlayer (y = 10–100), 20 cycles Pt, 300 cycles TiO2 on CNFs, followed by calcination in the air to remove the CNFs at 450°C for 1.5 h and then reduced in 5% H2/Ar at 380°C for 2 h. Typically, the 20 mg of CNFs were dispersed into the alcohol and uniformly spread over a quartz wafer (8 × 8 cm). After the evaporation of ethanol, the quartz wafer with CNFs was transferred to the ALD chamber. For the five-layer catalyst, about 30 mg of catalysts are obtained after the calcination to remove the CNFs for every ALD process. TiO2/Ni/TiO2 and TiO2/Pt/TiO2 with one metal layer and TiO2/Pt/Ni/TiO2 with no interlayer were prepared by a similar method. TiO2/Pt/ZnO/Ni/TiO2 and TiO2/Pt/Al2O3/Ni/TiO2 were prepared by changing the interlayer to ZnO or Al2O3. The TiO2/Pt|Ni/TiO2 with a tube-in-tube structure were synthesized using polyimide as the sacrificial layer. The CNFs were synthesized at 280°C by chemical vapor deposition using copper nanoparticles as catalyst and acetylene as a feed gas.41 The CNFs were carbonized at 900°C in Ar and further treated by an HNO3 aqueous solution (25 wt%) at 100°C before the deposition.
Catalysts Characterization
TEM and high-resolution TEM were conducted using a JEOL-2100F field emission transmission microscope. HAADF-STEM images and EDX mapping profiles were collected on an atomic resolution analytical microscope (JEOL ARM-200F) operated at 200 kV. The XRD patterns were recorded using a Philips X'Pert Pro Super X-ray diffractometer with Cu Kα radiation (λ = 1.540 Å). N2 adsorption-desorption experiments were performed on a BELSORP-Mini system at 77 K. The specific surface area was determined using the Brunauer-Emmett-Teller method, and the pore size distributions were calculated by the Barrett-Joyner-Halenda method according to the desorption branches. The XANES and EXAFS spectra of the Pt L3-edge and Ni K-edge were measured on the BL14W1 beamline of the Shanghai Synchrotron Radiation Facility, Shanghai Institute of Applied Physics, China, operated at 3.5 GeV.42 A Si (111) double-crystal monochromator was used to reduce the harmonic component of the monochrome beam. Pt foil, PtO2, and Ni foil were used as reference samples. The XANES and EXAFS spectra of the Pt L3-edge were measured in fluorescence mode, while the XANES and EXAFS spectra of the Ni K-edge were measured in transmission mode. Hydrogen pulse adsorption experiments and H2-TPR were performed in a tubular quartz reactor (TP-5080 Tianjin Xianquan, China). Typically, 50 mg of sample was loaded and pre-treated at 380°C for 2 h in 10% H2/N2 flow, and then swept with N2 at a flow rate of 40 mL/min. H2 pulse adsorption was performed in N2 flow at 30°C to test the number of active metal sites. H2-TPR was performed by heating the sample from 30°C to 900°C at a ramp rate of 10°C/min in a 10% H2/N2 flow.
Catalytic Performance Tests
Tandem decomposition of N2H4·H2O and hydrogenation of nitrobenzene was conducted in a 25-mL stainless-steel autoclave. Typically, 240 mg nitrobenzene, 800 mg N2H4·H2O, 10 mg catalysts, 5 mL deionized water, and 5 mL ethanol were added into the autoclave. After flushing with ultrahigh-purity Ar for three cycles, the reaction proceeded under magnetic stirring with a rate of 700 rpm at 30°C for 2 h. For recycling tests of the TiO2/Pt/10TiO2/Ni/TiO2 catalyst in the tandem reaction, the reaction time increases to 4 h for each run. After catalytic reaction, the catalysts were collected by centrifugation, washing, drying, and reduction (380°C in 5% H2/Ar) for the next run during the recycling tests. The liquid reaction products were analyzed by gas chromatography (Zhejiang Full Chromatogram Analysis, China) equipped with a flame ionization detector. The hydrogenation of nitrobenzene in H2 followed a similar procedure of tandem reaction, except for changing the hydrogen source from N2H4·H2O to 1.0 MPa H2. The decomposition of N2H4·H2O was tested in a 25-mL three-necked flask; 800 mg N2H4·H2O, 10 mg catalysts, and 10 mL deionized water were co-added into the flask under magnetic stirring with a rate of 700 rpm at 30°C.
Acknowledgments
We acknowledge the characterization support from the Shanghai Synchrotron Radiation Facility, Shanghai Institute of Applied Physics, Shanghai, People's Republic of China. This work was supported by National Natural Science Foundation of China (21872160, 21673269, and U1832208), National Science Fund for Distinguished Young Scholars (21825204), the National Key R&D Program of China (2017YFA0700101 and 2018YFB1501602), the Youth Innovation Promotion Association CAS (2017204), and Natural Science Foundation of Shanxi Province (201901D211591).
Declaration of Interests
The authors declare no competing interests.
Published: August 28, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.xinn.2020.100029.
Contributor Information
Bin Zhang, Email: zhangbin2009@sxicc.ac.cn.
Yong Qin, Email: qinyong@sxicc.ac.cn.
Supplemental Information
References
- 1.Lohr T.L., Marks T.J. Orthogonal tandem catalysis. Nat. Chem. 2015;7:477–482. doi: 10.1038/nchem.2262. [DOI] [PubMed] [Google Scholar]
- 2.Wasilke J.-C., Obrey S.J., Baker R.T., Bazan G.C. Concurrent tandem catalysis. Chem. Rev. 2005;105:1001–1020. doi: 10.1021/cr020018n. [DOI] [PubMed] [Google Scholar]
- 3.Yamada Y., Tsung C.-K., Huang W., Huo Z., Habas S.E., Soejima T., Aliaga C.E., Somorjai G.A., Yang P. Nanocrystal bilayer for tandem catalysis. Nat. Chem. 2011;3:372–376. doi: 10.1038/nchem.1018. [DOI] [PubMed] [Google Scholar]
- 4.Li Z., Assary R.S., Atesin A.C., Curtiss L.A., Marks T.J. Rapid ether and alcohol C–O bond hydrogenolysis catalyzed by tandem high-valent metal triflate + supported Pd catalysts. J. Am. Chem. Soc. 2014;136:104–107. doi: 10.1021/ja411546r. [DOI] [PubMed] [Google Scholar]
- 5.Zecevic J., Vanbutsele G., de Jong K.P., Martens J.A. Nanoscale intimacy in bifunctional catalysts for selective conversion of hydrocarbons. Nature. 2015;528:245–248. doi: 10.1038/nature16173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiao F., Li J., Pan X., Xiao J., Li H., Ma H., Wei M., Pan Y., Zhou Z., Li M., et al. Selective conversion of syngas to light olefins. Science. 2016;351:1065–1068. doi: 10.1126/science.aaf1835. [DOI] [PubMed] [Google Scholar]
- 7.Gao P., Li S., Bu X., Dang S., Liu Z., Wang H., Zhong L., Qiu M., Yang C., Cai J., et al. Direct conversion of CO2 into liquid fuels with high selectivity over a bifunctional catalyst. Nat. Chem. 2017;9:1019–1024. doi: 10.1038/nchem.2794. [DOI] [PubMed] [Google Scholar]
- 8.Gao P., Dang S., Li S., Bu X., Liu Z., Qiu M., Yang C., Wang H., Zhong L., Han Y., et al. Direct production of lower olefins from CO2 conversion via bifunctional catalysis. ACS Catal. 2018;8:571–578. [Google Scholar]
- 9.Bao J., He J., Zhang Y., Yoneyama Y., Tsubaki N. A core/shell catalyst produces a spatially confined effect and shape selectivity in a consecutive reaction. Angew. Chem. Int. Ed. 2008;47:353–356. doi: 10.1002/anie.200703335. [DOI] [PubMed] [Google Scholar]
- 10.Yang G., Tsubaki N., Shamoto J., Yoneyama Y., Zhang Y. Confinement effect and synergistic function of H-ZSM-5/Cu-ZnO-Al2O3 capsule catalyst for one-step controlled synthesis. J. Am. Chem. Soc. 2010;132:8129–8136. doi: 10.1021/ja101882a. [DOI] [PubMed] [Google Scholar]
- 11.Cheng K., Zhou W., Kang J., He S., Shi S., Zhang Q., Pan Y., Wen W., Wang Y. Bifunctional catalysts for one-step conversion of syngas into aromatics with excellent selectivity and stability. Chem. 2017;3:334–347. [Google Scholar]
- 12.Zhao B., Zhai P., Wang P., Li J., Li T., Peng M., Zhao M., Hu G., Yang Y., Li Y.-W., et al. Direct transformation of syngas to aromatics over Na-Zn-Fe5C2 and hierarchical HZSM-5 tandem catalysts. Chem. 2017;3:323–333. [Google Scholar]
- 13.George S.M. Atomic layer deposition: an overview. Chem. Rev. 2010;110:111–131. doi: 10.1021/cr900056b. [DOI] [PubMed] [Google Scholar]
- 14.Zhang B., Qin Y. Interface tailoring of heterogeneous catalysts by atomic layer deposition. ACS Catal. 2018;8:10064–10081. [Google Scholar]
- 15.Detavernier C., Dendooven J., Sree S.P., Ludwig K.F., Martens J.A. Tailoring nanoporous materials by atomic layer deposition. Chem. Soc. Rev. 2011;40:5242–5253. doi: 10.1039/c1cs15091j. [DOI] [PubMed] [Google Scholar]
- 16.Knez M., Nielsch K., Niinistö L. Synthesis and surface engineering of complex nanostructures by atomic layer deposition. Adv. Mater. 2007;19:3425–3438. [Google Scholar]
- 17.Lu J., Elam J.W., Stair P.C. Synthesis and stabilization of supported metal catalysts by atomic layer deposition. Acc. Chem. Res. 2013;46:1806–1815. doi: 10.1021/ar300229c. [DOI] [PubMed] [Google Scholar]
- 18.O'Neill B.J., Jackson D.H.K., Lee J., Canlas C., Stair P.C., Marshall C.L., Elam J.W., Kuech T.F., Dumesic J.A., Huber G.W. Catalyst design with atomic layer deposition. ACS Catal. 2015;5:1804–1825. [Google Scholar]
- 19.Zhang B., Guo X.-W., Liang H., Ge H., Gu X., Chen S., Yang H., Qin Y. Tailoring Pt–Fe2O3 interfaces for selective reductive coupling reaction to synthesize imine. ACS Catal. 2016;6:6560–6566. [Google Scholar]
- 20.Cao K., Cai J., Liu X., Chen R. Catalysts design and synthesis via selective atomic layer deposition. J. Vac. Sci. Technol. A. 2018;36:010801. [Google Scholar]
- 21.Zhang S., Zhang B., Liang H., Liu Y., Qiao Y., Qin Y. Encapsulation of homogeneous catalysts in mesoporous materials using diffusion-limited atomic layer deposition. Angew. Chem. Int. Ed. 2018;57:1091–1095. doi: 10.1002/anie.201712010. [DOI] [PubMed] [Google Scholar]
- 22.Cao L., Liu W., Luo Q., Yin R., Wang B., Weissenrieder J., Soldemo M., Yan H., Lin Y., Sun Z., et al. Atomically dispersed iron hydroxide anchored on Pt for preferential oxidation of CO in H2. Nature. 2019;565:631–635. doi: 10.1038/s41586-018-0869-5. [DOI] [PubMed] [Google Scholar]
- 23.Zhang H.B., Marshall C.L. Atomic layer deposition: catalytic preparation and modification technique for the next generation. Chin. J. Catal. 2019;40:1311–1323. [Google Scholar]
- 24.Yang H.M., Chen Y., Qin Y. Application of atomic layer deposition in fabricating high-efficiency electrocatalysts. Chin. J. Catal. 2020;41:227–241. [Google Scholar]
- 25.Gao Z., Dong M., Wang G.G., Sheng P., Wu Z., Yang H., Zhang B., Wang G.G., Wang J., Qin Y. Multiply confined nickel nanocatalysts produced by atomic layer deposition for hydrogenation reactions. Angew. Chem. Int. Ed. 2015;54:9006–9010. doi: 10.1002/anie.201503749. [DOI] [PubMed] [Google Scholar]
- 26.Gao Z., Qin Y. Design and properties of confined nanocatalysts by atomic layer deposition. Acc. Chem. Res. 2017;50:2309–2316. doi: 10.1021/acs.accounts.7b00266. [DOI] [PubMed] [Google Scholar]
- 27.Ge H., Zhang B., Liang H., Zhang M., Fang K., Chen Y., Qin Y. Photocatalytic conversion of CO2 into light olefins over TiO2 nanotube confined Cu clusters with high ratio of Cu+ Appl. Catal. B Environ. 2020;263:118133. [Google Scholar]
- 28.Liang H., Zhang B., Ge H., Gu X., Zhang S., Qin Y. Porous TiO2/Pt/TiO2 sandwich catalyst for highly selective semihydrogenation of alkyne to olefin. ACS Catal. 2017;7:6567–6572. [Google Scholar]
- 29.Zhang J., Yu Z., Gao Z., Ge H., Zhao S., Chen C., Chen S., Tong X., Wang M., Zheng Z., Qin Y. Porous TiO2 nanotubes with spatially separated platinum and CoOx cocatalysts produced by atomic layer deposition for photocatalytic hydrogen production. Angew. Chem. Int. Ed. 2017;56:816–820. doi: 10.1002/anie.201611137. [DOI] [PubMed] [Google Scholar]
- 30.Zhang J., Gao Z., Wang S., Wang G., Gao X., Zhang B., Xing S., Zhao S., Qin Y. Origin of synergistic effects in bicomponent cobalt oxide-platinum catalysts for selective hydrogenation reaction. Nat. Commun. 2019;10:4166. doi: 10.1038/s41467-019-11970-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ge H., Zhang B., Gu X., Liang H., Yang H., Gao Z., Wang J., Qin Y. A tandem catalyst with multiple metal oxide interfaces produced by atomic layer deposition. Angew. Chem. Int. Ed. 2016;55:7081–7085. doi: 10.1002/anie.201600799. [DOI] [PubMed] [Google Scholar]
- 32.Lu X., Chen A., Luo Y., Lu P., Dai Y., Enriquez E., Dowden P., Xu H., Kotula P.G., Azad A.K., et al. Conducting interface in oxide homojunction: understanding of superior properties in black TiO2. Nano Lett. 2016;16:5751–5755. doi: 10.1021/acs.nanolett.6b02454. [DOI] [PubMed] [Google Scholar]
- 33.Bertoni G., Beyers E., Verbeeck J., Mertens M., Cool P., Vansant E.F., Van Tendeloo G. Quantification of crystalline and amorphous content in porous samples from electron energy loss spectroscopy. Ultramicroscopy. 2006;106:630–635. [Google Scholar]
- 34.Dinh C.-T., Jain A., de Arquer F.P.G., De Luna P., Li J., Wang N., Zheng X., Cai J., Gregory B.Z., Voznyy O., et al. Multi-site electrocatalysts for hydrogen evolution in neutral media by destabilization of water molecules. Nat. Energy. 2019;4:107–114. [Google Scholar]
- 35.Beale A.M., Weckhuysen B.M. EXAFS as a tool to interrogate the size and shape of mono and bimetallic catalyst nanoparticles. Phys. Chem. Chem. Phys. 2010;12:5562–5574. doi: 10.1039/b925206a. [DOI] [PubMed] [Google Scholar]
- 36.He L., Liang B., Huang Y., Zhang T. Design strategies of highly selective nickel catalysts for H2 production via hydrous hydrazine decomposition: a review. Natl. Sci. Rev. 2017;5:356–364. [Google Scholar]
- 37.Wiberg K.B. The deuterium isotope effect. Chem. Rev. 1955;55:713–743. [Google Scholar]
- 38.Fisicaro G., Filice S., Scalese S., Compagnini G., Reitano R., Genovese L., Goedecker S., Deretzis I., La Magna A. Wet environment effects for ethanol and water adsorption on anatase TiO2 (101) surfaces. J. Phy. Chem. C. 2020;124:2406–2419. [Google Scholar]
- 39.Ketteler G., Yamamoto S., Bluhm H., Andersson K., Starr D.E., Ogletree D.F., Ogasawara H., Nilsson A., Salmeron M. The nature of water nucleation sites on TiO2(110) surfaces revealed by ambient pressure X-ray photoelectron spectroscopy. J. Phy. Chem. C. 2007;111:8278–8282. [Google Scholar]
- 40.Wan W., Nie X., Janik M.J., Song C., Guo X. Adsorption, dissociation, and spillover of hydrogen over Au/TiO2 catalysts: the effects of cluster size and metal–support interaction from DFT. J. Phy. Chem. C. 2018;122:17895–17916. [Google Scholar]
- 41.Qin Y., Zhang Z., Cui Z. Helical carbon nanofibers prepared by pyrolysis of acetylene with a catalyst derived from the decomposition of copper tartrate. Carbon. 2003;41:3072–3074. [Google Scholar]
- 42.Yu H.-S., Wei X.-J., Li J., Gu S.-Q., Zhang S., Wang L.-H., Ma J., Li L.-N., Gao Q., Si R., et al. The XAFS beamline of SSRF. Nucl. Sci. Tech. 2015;26:050102. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.