

Abstract
INTRODUCTION:
Enzymes possess extraordinary catalytic proficiency and specificity. These properties ultimately derive from interactions not just between the active-site residues and the substrate but from functional interactions throughout a folded enzyme. Therefore, understanding the origins of catalytic proficiency and specificity will require the ability to make mutations throughout the protein. Traditionally, enzyme active sites have been characterized by means of site-directed mutagenesis (SDM), revealing much about the catalytic functions of these residues; nevertheless, SDM is low-throughput, costly, and labor intensive. By contrast, recently developed high-throughput mutational scanning techniques assay large numbers of sequences but provide only coarse estimates of function, such as the amount of product generated at a particular time under a particular set of conditions or overall organismal fitness.
RATIONALE:
New technologies are needed to overcome the limitations of current approaches and allow deep characterization of many enzyme variants in a cost- and time-efficient manner. To meet this challenge, we developed a high-throughput microfluidic platform that allows the simultaneous expression and purification of more than 1500 rationally chosen enzyme mutants in hours and allows their quantitative functional characterization in days. HT-MEK (High-Throughput Microfluidic Enzyme Kinetics) can be used with any enzyme system that can be tagged and expressed in vitro and has a direct or coupled fluorogenic assay.
RESULTS:
As a first application of HT-MEK, we functionally characterized 1036 single-site mutants that contain either a glycine or valine substitution at each position within PafA (phosphate-irrepressible alkaline phosphatase of Flavobacterium), a well-studied enzyme from the alkaline phosphatase superfamily. For each mutant, we measured Michaelis-Menten kinetics [apparent unimolecular rate constant (kcat), Michaelis constant (Km), and kcat/Km] for multiple substrates, inhibition constants, and effects on folding, obtaining more than 5000 kinetic and thermodynamic constants from more than 670,000 total reactions. We found that most mutations (702 of 1036) yielded statistically significant effects on some aspect of catalysis. By systematically and independently varying expression and assay conditions, we determined that 232 of these mutations reduced catalysis by promoting the formation of a long-lived, catalytically inactive misfolded state, whereas none did so through equilibrium unfolding under our assay conditions. Combining these functional measurements with prior mechanistic knowledge allowed us to systematically assess the effect of each mutation. Different groups of residues affected different aspects of function, with residues that affect a particular function forming large, spatially contiguous regions that spanned from the active site up to 20 Å from the active site and to the enzyme surface.
CONCLUSION:
HT-MEK has allowed us to uncover functional effects throughout PafA and to identify the catalytic features affected by different groups of residues. Some of these effects are readily rationalized through inspection of structural interconnections to the active site, whereas others were nonobvious, including large distal and surface effects and the discovery of a long-lived misfolded state. These results underscore the need to measure the effects of mutations on multiple kinetic and thermodynamic parameters across multiple reaction conditions and thus the need for this new technology. Because HT-MEK is applicable to any enzyme with a direct or coupled fluorescent readout and provides an in-depth and quantitative analysis of mutant space rapidly and at modest cost, it may be the method of choice to characterize new enzymes. In future applications, HT-MEK can be used to dissect potential evolutionary trajectories, determine the functional consequences of human disease-associated allelic variants, identify surfaces with nascent allosteric potential for rational control of catalysis, and direct the adaptation of natural and designed enzymes for new functions and roles.
Graphical Abstract
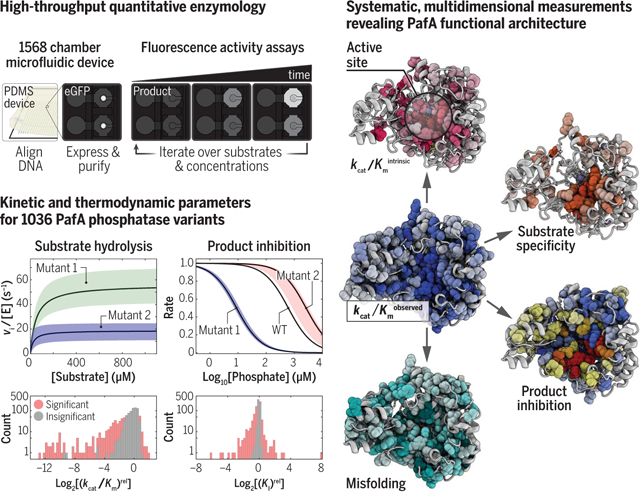
Simultaneous expression, purification, and biochemical characterization of enzyme variants in a microfluidic device makes it possible to measure Michaelis-Menten parameters and inhibition constants for more than 1500 variants in days. Mutational effects across multiple assays reveal an extensive functional architecture in which physically contiguous residue regions extending to the enzyme surface control or alter particular aspects of catalysis.
Systematic and extensive investigation of enzymes is needed to understand their extraordinary efficiency and meet current challenges in medicine and engineering. We present HT-MEK (High-Throughput Microfluidic Enzyme Kinetics), a microfluidic platform for high-throughput expression, purification, and characterization of more than 1500 enzyme variants per experiment. For 1036 mutants of the alkaline phosphatase PafA (phosphate-irrepressible alkaline phosphatase of Flavobacterium), we performed more than 670,000 reactions and determined more than 5000 kinetic and physical constants for multiple substrates and inhibitors. We uncovered extensive kinetic partitioning to a misfolded state and isolated catalytic effects, revealing spatially contiguous regions of residues linked to particular aspects of function. Regions included active-site proximal residues but extended to the enzyme surface, providing a map of underlying architecture not possible to derive from existing approaches. HT-MEK has applications that range from understanding molecular mechanisms to medicine, engineering, and design.
Understanding how sequence encodes function remains a fundamental challenge in biology. Linear chains of amino acids fold into three-dimensional protein structures that carry out the physical and chemical tasks needed for life, such as highly efficient and specific catalysis. Sequence variations across organisms and individuals confer beneficial and deleterious effects: Variation throughout evolution creates proteins with improved or new functions, but variation among individuals can also compromise function and cause disease (1–3). An enhanced predictive understanding of the sequence-function landscape could have profound impacts across biology, from enabling efficient protein design to improving detection of rare allelic variants that drive disease (4–7), but new approaches and data are needed to attain this goal.
Understanding sequence-function relationships within enzymes poses a particular challenge. Structural and biochemical studies of enzymes have revealed the sites of substrate binding and catalytic transformation, the residues directly involved in catalysis, and roles for these residues. Nevertheless, residues outside the active site are needed for the active site to assemble and function and for control of function by allosteric ligands and covalent modifications (8–10). Despite their importance, the roles played by residues outside the active site, which comprise the majority of amino acids in an enzyme, remain largely unexplored.
This dearth of knowledge stems from the nature of experimental approaches currently available. Site-directed mutagenesis (SDM) has traditionally been used to assess function by means of in-depth biochemical assays that yield kinetic and thermodynamic constants. However, SDM is time-, resource-, and labor-intensive, limiting investigation to a small number of residues. By contrast, deep mutational scanning (DMS) provides the ability to assay the effects of all 20 amino acids at every position within an enzyme (5, 11, 12). However, DMS lacks the depth and dimensionality of traditional SDM studies, typically providing a scalar readout with an uncertain relationship to the multiple fundamental physical constants needed to describe an enzyme’s function.
Marrying the strengths of traditional SDM and emerging DMS is needed to usher in a new era of mechanistic enzymology. Here, we present HT-MEK (High-Throughput Microfluidic Enzyme Kinetics), a platform capable of simultaneously expressing, purifying, and characterizing more than 1500 rationally chosen enzyme mutants in parallel with the depth and precision of traditional SDM. Each HT-MEK experiment provides 1000s of measurements and multiple kinetic and thermodynamic constants [such as the apparent unimolecular rate constant (kcat), Michaelis constant (Km), kcat/Km, and inhibition constant (Ki)] in days and at low cost.
To guide HT-MEK development and demonstrate its capabilities, we carried out a comprehensive mechanistic investigation of the effects of mutations to every residue within the alkaline phosphatase superfamily member PafA (phosphate-irrepressible alkaline phosphatase of Flavobacterium) (Fig. 1A and fig. S1). PafA and related phosphomonoesterases are among the most prodigious catalysts known, with rate enhancements of up to ~1027-fold, providing a large dynamic range to explore (13). We also anticipated that PafA, a secreted enzyme, would be highly stable, potentially allowing us to more deeply probe catalysis without obfuscation from global unfolding. We found that 702 of the 1036 mutants investigated have substantial functional consequences, with none arising from equilibrium unfolding. Additional experiments revealed that many mutations promote the formation of a long-lived, catalytically incompetent misfolded state both in vitro and in cells. The multidimensional measurements provided by HT-MEK allowed us to decouple this misfolding from catalytic effects and quantify mutational effects on particular aspects of catalysis and mechanism using an approach we call Functional Component Analysis. This approach revealed spatially contiguous regions of residues that extend from the active site to the enzyme surface responsible for optimizing particular catalytic strategies. Surface residues with functional effects upon mutation may compose candidate allosteric regions useful for engineering rational control of catalytic activity. The HT-MEK platform and the quantitative multidimensional datasets it can provide will have broad utility for future efforts to understand catalytic mechanisms, natural variation, and evolutionary trajectories and to design enzymes with new functions.
Fig. 1. Overview of HT-MEK and PafA.
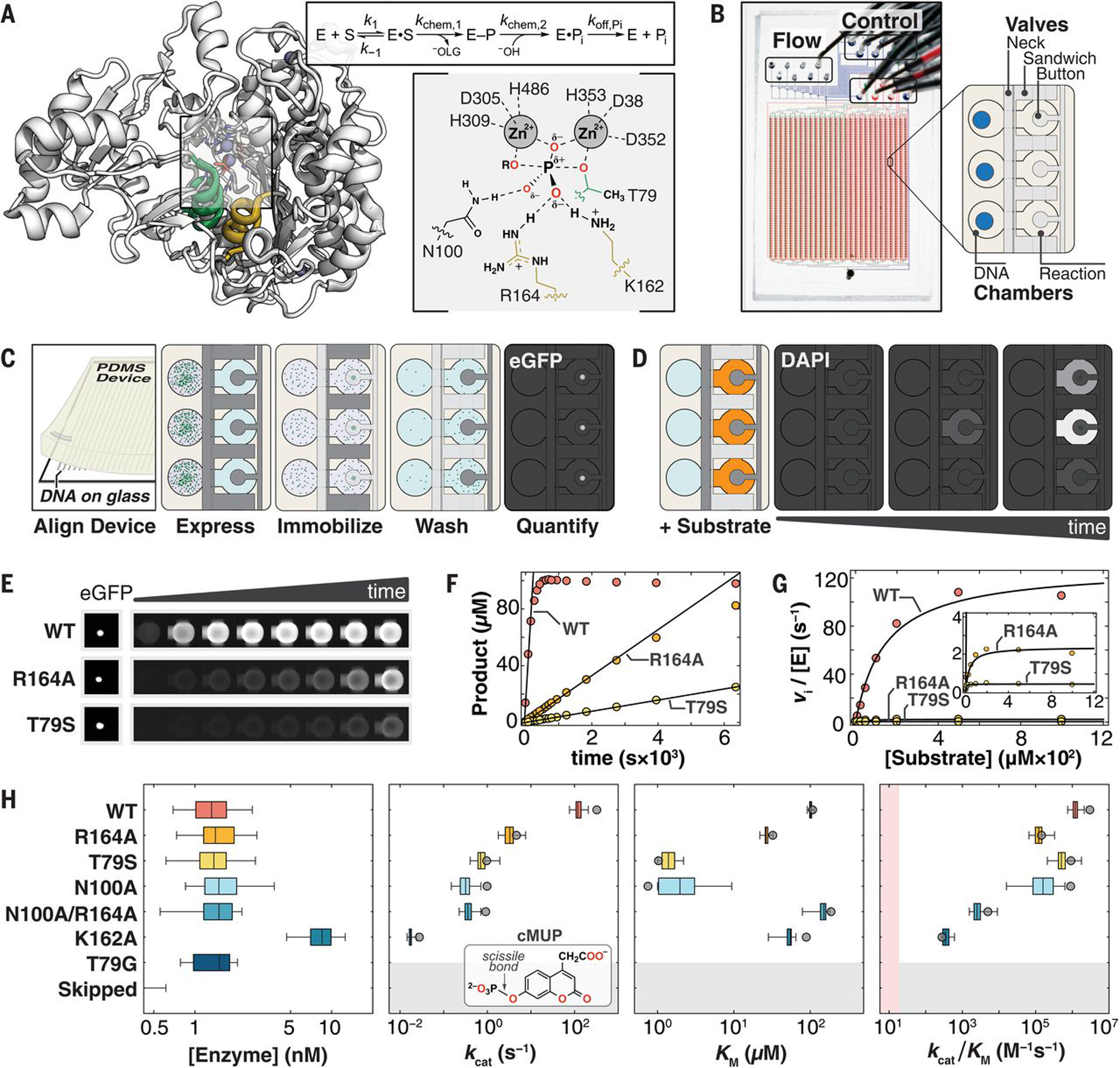
(A) (Left) Crystal structure of WT PafA [Protein Data Bank (PDB) ID: 5TJ3] highlighting “nucleophile” (residues 77 to 89, green) and “monoesterase” (residues 161 to 171, yellow) helices. (Right) PafA catalytic cycle (S, phosphate monoester substrate; E–P, covalent phospho-threonine intermediate; −OLG, alkoxide leaving group) and active site in the transition state of monoester hydrolysis. (Single-letter abbreviations for the amino acid residues are as follows: A, Ala; C, Cys; D, Asp; E, Glu; F, Phe; G, Gly; H, His; I, Ile; K, Lys; L, Leu; M, Met; N, Asn; P, Pro; Q, Gln; R, Arg; S, Ser; T, Thr; V, Val; W, Trp; and Y, Tyr. In the mutants, other amino acids were substituted at certain locations; for example, T79S indicates that threonine at position 79 was replaced by serine.) (B) HT-MEK microfluidic device image and schematic showing solution (Flow) and pneumatic manifold (Control) input ports, device valves, and chambers. (C) Schematic of on-chip enzyme expression pipeline. Dark and light gray valves are pressurized (closed) and depressurized (open), respectively. (D) Schematic of on-chip activity assays by using fluorogenic substrate in reaction chambers (orange). (E) Sample images of (left) immobilized enzyme and (right) fluorogenic product over time for WT PafA and two active-site mutants (R164A and T79S) with the substrate cMUP. (F) Example cMUP progress curves for chambers containing wild type and two active-site mutants and initial rate fits to these data. (G) Michaelis-Menten fits to initial rates yield kcat, Km, and kcat/Km for cMUP. (H) (Left) On-chip expressed concentrations for WT PafA and six active-site mutants calculated by using eGFP calibration curves. (Middle left to right) Comparisons of on-chip (box plots) and off-chip (gray circle) values of kcat, Km, and kcat/Km values (log10 axis) for cMUP for seven PafA variants (table S1) and (inset) cMUP structure. K162A was expressed at higher concentration in a later experimental tier (with higher [E]); “skipped” refers to chambers without plasmid DNA; pink shaded region denotes 10-fold above the median apparent second-order rate constant for T79G control chambers (supplementary materials, materials and methods).
Results
HT-MEK device and experimental pipeline
HT-MEK is built around a two-layer poly-dimethylsiloxane (PDMS) microfluidic device with 1568 chambers and integrated pneumatic valves (Fig. 1B and fig. S2) (14, 15). Each chamber is composed of two compartments (DNA and Reaction) separated by a valve (Neck), with adjacent chambers isolated from one another by a second valve (Sandwich). A third valve (Button) reversibly excludes or exposes a circular patch of the reaction compartment surface, enabling surface patterning for on-chip protein immobilization and purification (Fig. 1C) and subsequent simultaneous initiation of successive on-chip reactions across the device (Fig. 1D). Each DNA compartment of each chamber is programmed with a specified enzyme variant by aligning the device to a spotted array of DNA plasmids that encode for the expression of C-terminally enhanced green fluorescent protein (eGFP)–tagged variants (fig. S3). After alignment, device surfaces are patterned with antibodies to eGFP beneath the Button valve and passivated with bovine serum albumin elsewhere. All enzymes are then expressed in parallel through the introduction of an Escherichia coli in vitro transcription-translation system and purified by means of capture with surface-patterned immobilized antibody and washing (fig. S4). Production of up to 1568 different purified enzymes takes ~10 hours, with most steps automated. Enzymes are immobilized under the Button valves that protect against flow-induced loss of enzyme during solution exchange and allow repeated synchronous initiation of reactions.
To obtain catalytic rate parameters, we quantify (i) the concentration of immobilized enzyme in each chamber, using an eGFP calibration curve (fig. S5), and (ii) the amount of product formed as a function of reaction time, using a chamber-specific product calibration curve (fig. S6). We then fit reaction progress curves in each chamber to obtain initial rates (υi) for each substrate concentration using a custom image-processing pipeline and convert observed rates (υi) to enzyme-normalized rate constants according to the eGFP intensity in each chamber and calibration curve (Fig. 1, E and F, and figs. S7 and S8). This process—repeated on a single device for multiple substrate concentrations, multiple substrates, and multiple inhibitors—provides the data necessary to obtain Michaelis-Menten parameters and other kinetic and thermodynamic constants (Fig. 1G and fig. S7).
HT-MEK reproduces kinetic constants previously measured with traditional assays
To demonstrate the technical capabilities of HT-MEK, we applied it to study seven previously characterized PafA variants: wild type, five active site mutants (T79S, N100A, R164A, K162A, and N100A/R164A), and one mutant lacking detectable activity (T79G, negative control) (16). Activities of wild-type (WT) PafA and the six mutants span a broad range in kcat (>104-fold), kcat/Km (>104-fold), and Km (>102-fold) for aryl phosphate monoester hydrolysis, providing a stringent initial test of HT-MEK dynamic range (table S1). Nearly all DNA-containing chambers expressed enzyme (>90%), and all mutants expressed at similar levels as determined from eGFP fluorescence (K162A was deliberately expressed at higher concentrations in a later experimental tier, described below) (Fig. 1H and fig. S9).
Although fluorogenic phosphate ester substrates permit kinetic assays of phosphatase activity with a high dynamic range, microfluidic assays that use the commercial 4-methylumbelliferyl phosphate ester (MUP) were complicated by partitioning of the hydrophobic fluorescent product into the hydrophobic PDMS, which increases background and distorts kinetic measurements. To address this limitation, we synthesized MUP derivatives of similar reactivity (cMUP and a corresponding methyl phosphodiester, MecMUP) that bear a charged moiety on the leaving group to eliminate PDMS absorption (figs. S10 to S12 and table S1).
Accurately resolving enzymatic rates spanning many orders of magnitude poses technical challenges because different acquisition times are needed at catalytic extremes (fig. S13), and even a small concentration of contaminating fast enzyme introduced into nearby chambers during fluid exchanges can obscure the true rates for the most catalytically compromised mutants. To address the first challenge, we expressed enzymes at two concentrations: ~1.5 nM (for accurate measurement of fast enzymes) and ~15 nM (to speed reactions for efficient detection of slow enzymes) (Fig. 1H and fig. S14). To identify regions of the device with contaminating enzymes from other chambers, we interspersed chambers that were empty or contained the inactive T79G mutant and also measured their apparent activity (figs. S15 and S16 and table S2). Per-device normalizations (0.4 and 1.4-fold) were used to account for small variations in apparent activity owing to nonspecific adsorption of mutant enzymes to chamber walls (supplementary materials, materials and methods). This normalization increased precision across replicates but did not affect conclusions (fig. S17). HT-MEK assays recapitulated cMUP kinetic parameters (kcat, Km, and kcat/Km) accurately and over a wide dynamic range (>104-fold in kcat/Km), indicating that the device and added eGFP tag did not alter activity (Fig. 1H and table S3).
Many mutations throughout PafA affect phosphate monoester hydrolysis
To explore functional effects of mutations throughout PafA, we created mutant libraries in which we introduced two residues with widely differing side-chain properties at each position: (i) glycine, to ablate side chain interactions and increase backbone flexibility, and (ii) valine, to introduce a branched chain hydrophobe of average volume. Native valine and glycine residues were mutated to alanine. Nearly all of these 1052 possible mutants (1036; 98%) were successfully cloned, sequenced, expressed, and assayed with HT-MEK (fig. S18).
We first measured the catalytic effect of each substitution on the steady-state kinetic parameters for cMUP hydrolysis (kcat, Km, and kcat/Km) (Fig. 2A). To facilitate efficient measurement, we performed experiments in three tiers based on reaction rates (table S2). In tier 1, we assayed devices that contained all variants at low enzyme concentration ([E] ~ 1.5 nM), with enzymes printed in duplicate and the above-noted active site mutants distributed throughout as fiducial controls. Tier 2 and 3 measurements, using higher enzyme concentration ([E] ≈ 15 nM), focused successively on the slowest variants with increasing assay times and increasing numbers of replicates to provide high-precision measurements of these slower mutants. Each device was used to measure tens of cMUP progress curves, and all expressed variants were stable over days, facilitating high-throughput data collection. Per-experiment data reports contain all data collected for each chamber, including initial rate plots and fit Michaelis-Menten curves (an example is provided in fig. S19, and full data sets are available as data files S1 and S2); per-mutant summaries combine data from all experiments and include estimates of statistical significance (Fig. 2, B and C, and fig. S20). In total, we acquired a median of nine and seven replicates for valine and glycine mutants, respectively, over 16 experiments (figs. S21 to 24 and table S2). The wealth and precision of these data allowed us to resolve differences across ranges of 104, 102, and 105-fold for kcat, Km, and kcat/Km, respectively.
Fig. 2. HT-MEK measurements of aryl phosphate monoester (cMUP) hydrolysis for valine and glycine scans of PafA.
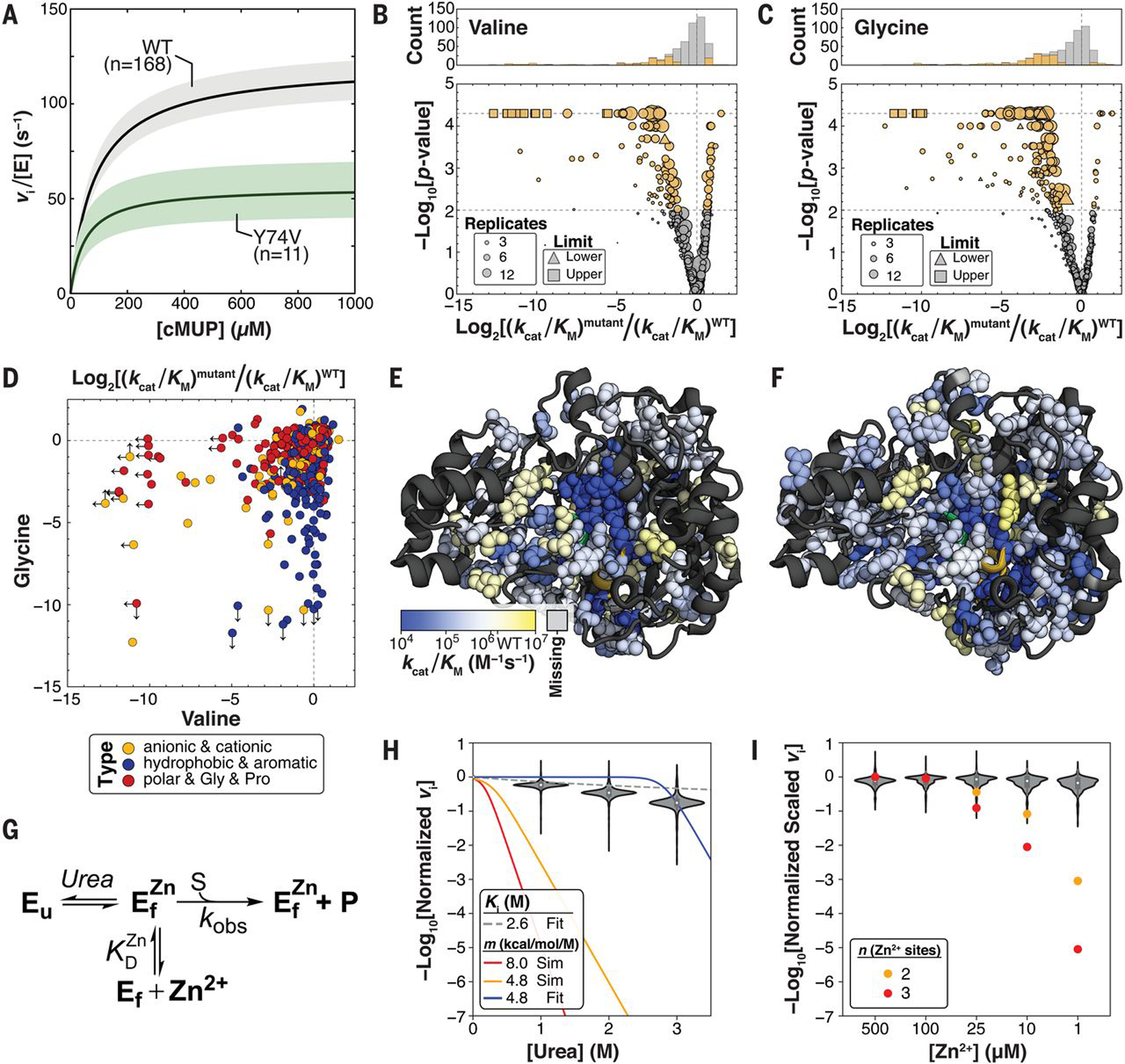
(A) Median Michaelis-Menten curves for WT PafA and an example mutant with kcat and Km effects. Colored regions denote 99% confidence intervals (CIs) on medians of parameters from replicates. (B) Valine substitutions at 126 positions alter cMUP kcat/Km at P < 0.01 (105 slower, 21 faster, gold markers; gray, P ≥ 0.01). (C) Glycine substitutions at 176 positions alter cMUP kcat/Km at P < 0.01 (162 slower, 14 faster, gold; gray, P ≥ 0.01). (D) Effect of glycine versus valine substitution on cMUP kcat/Km at each position, colored by identity type of the native residue. Arrows denote the presence and direction of measurement limits. (E and F) Valine (E) and glycine (F) substitution effects on cMUP hydrolysis on the PafA structure. P < 0.01 sites shown as spheres, P ≥ 0.01 and missing sites shown as ribbons. Alternate views are provided in fig. S21. (G) Model of equilibrium unfolding with varying urea and Zn2+. (H) Activity of mutants relative to wild type at 50 μM cMUP as a function of urea. Red and yellow lines show predicted dependencies if mutants were 10% unfolded in the absence of urea, assuming m values of 4.8 and 8 kcal/mol/M (supplementary text S1). The blue curve is the best fit of the data for m = 4.8 kcal/mol/M with fraction unfolded (fu) at 0 M [urea] as a free parameter (fit fu = 1 × 10−10), and the dashed gray curve is the fit competitive inhibition of WT PafA by urea (fig. S26). (I) Normalized activity of mutants relative to wild type as a function of [Zn2+]. Orange and red points show expected activities for Zn2+-dependent unfolding events if mutants were 10% unfolded at 100 μM Zn2+ assuming two or three Zn2+ binding events, respectively.
As expected, mutations of active-site residues and catalytic Zn2+ ligands were highly deleterious, and positional effects varied for valine and glycine (Fig. 2, D to F). Nevertheless, a surprisingly large number of mutations throughout the enzyme were deleterious, with decreases in kcat/Km observed for 267 of the 1036 mutants (P < 0.01). We also observed 35 mutants with increased activity (fig. S21). These measurements provide a quantitative survey of Michaelis-Menten kinetic constants for mutations throughout a large enzyme but do not tell us why so many mutations alter activity.
The most obvious explanation for these widespread effects would be destabilization leading to a significant fraction of unfolded enzyme. Beyond destabilization, mutations can have other repercussions, altering the catalytic effectiveness of particular active-site residues, reducing Zn2+ affinity at the bimetallo active site or altering enzymatic protonation states. The ability to efficiently measure catalytic activity for all mutants under different assay and expression conditions as afforded by HT-MEK allowed us to test each of these possibilities.
Widespread mutational effects do not arise from equilibrium unfolding
Reflecting its role as a secreted phosphatase designed to function in harsh and variable environments, WT PafA is highly stable and remains folded, as inferred from circular dichroism (CD) spectra, even after exposure to 4 M urea for 14 days (fig. S25). This stability suggests that any individual mutation is unlikely to substantially unfold the enzyme. To directly test this expectation, we measured cMUP activities in the presence of increasing concentrations of urea. If a variant were already partially unfolded in the absence of urea, then even low concentrations of added urea would cause substantial additional unfolding (17), proportionally lowering activity (Fig. 2G and supplementary text S1). By contrast, we observed only minor rate effects for all mutants (six ± onefold decrease at 3 M compared with 0 M urea, ±SEM), which is considerably less than the >109-fold decrease predicted for an unfolding effect and consistent with inhibition by urea, with Ki = 2.6 M for all variants (Fig. 2H and fig. S26).
Equilibrium unfolding would also predict a Zn2+ concentration dependence for the observed rate because added or removed Zn2+ would pull the equilibrium between folded (and Zn2+-bound) and unfolded states (Fig. 2G). Consistent with an absence of unfolding effects, we observed no dependence on Zn2+ concentration over a ~103-fold range (Fig. 2I and supplementary text S1). This observation also establishes that observed rate effects for mutants with measurable rates do not arise from loss of bound Zn2+ owing to lowered Zn2+ affinity. Last, mutants were unaltered in their pH dependencies (fig. S27 and supplementary text S1), ruling out altered protonation states as responsible for observed kinetic effects.
A general high-throughput assay for phosphate release and additional mutational effects
Although fluorogenic probes provide a sensitive and convenient method for directly visualizing enzyme activity in kinetic assays, many reactions lack a direct fluorogenic readout. To allow future application of HT-MEK to a much broader range of enzymes, we developed an on-chip coupled assay in which inorganic phosphate (Pi) is detected through fluorescence emitted upon binding to a modified phosphate binding protein (PBP) (Fig. 3A) (18). Calibration curves for Pi and PBP and control measurements using the PafA substrate methyl phosphate (MeP) established that this coupled on-chip assay can detect submicromolar Pi formation and accurately reproduce off-chip kinetic constants (figs. S28 to S30).
Fig. 3. Substrates with different intrinsic reactivity reveal kinetic folding traps.
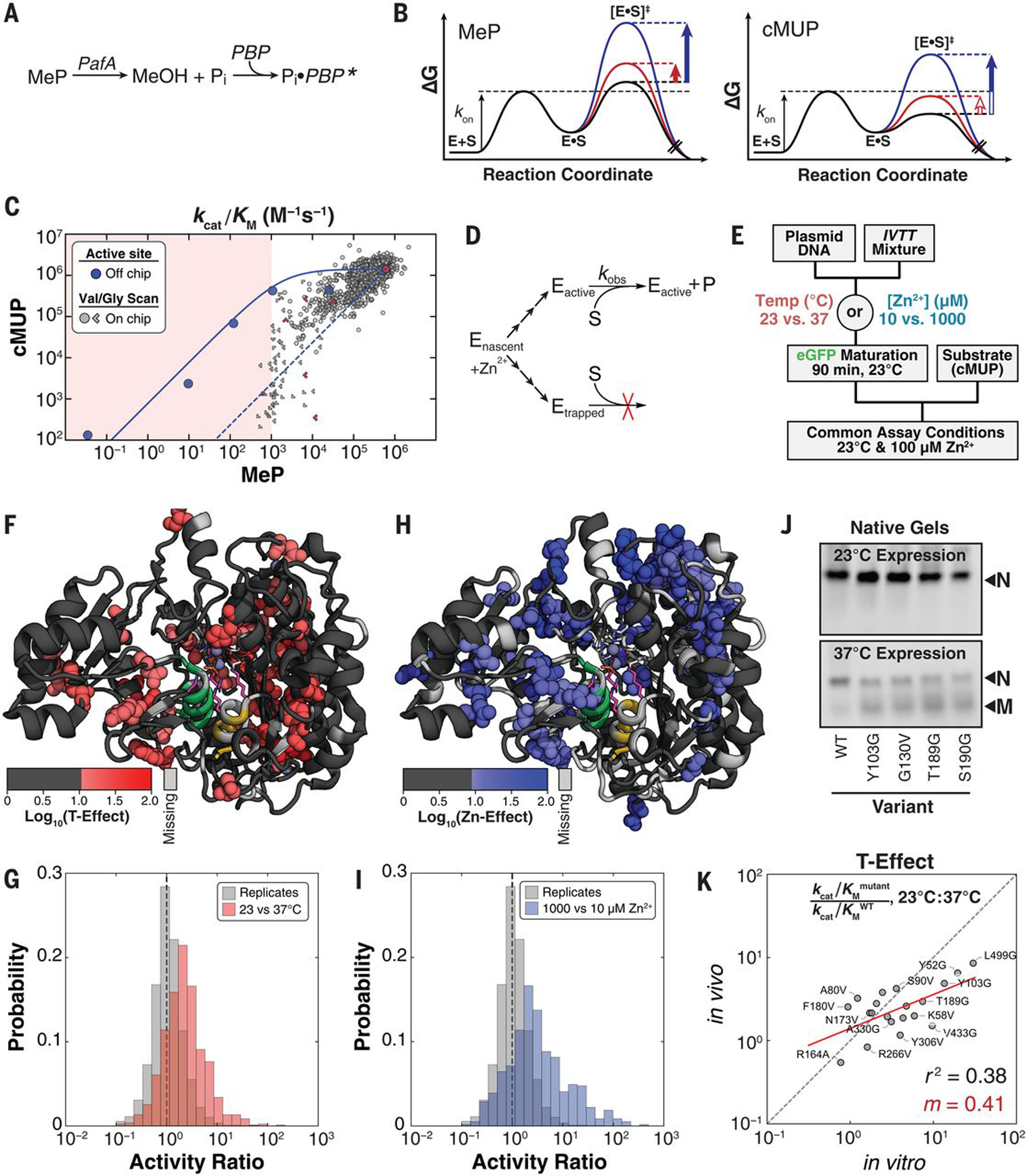
(A) Schematic illustrating real-time detection of Pi produced by MeP hydrolysis on-chip using a fluorophore-conjugated PBP (18). (B) Reaction coordinate diagrams schematically illustrating substrates with either chemistry (MeP) or binding (cMUP) as the rate-limiting step. Solid arrows indicate observable mutational effects on the chemical step, and open arrows indicate effects that are not observable. (C) Measured kcat/Km for MeP versus kcat/Km for cMUP for active-site mutants off-chip (blue points) and for valine and glycine mutants. Limits in one or both directions are indicated with chevrons pointing in the direction or quadrant of the limit. The red shaded region denotes lower limit of detection for MeP, the solid blue line indicates predicted relationship between kcat/Km for cMUP and MeP hydrolysis, and the dashed blue line shows predicted relationship for enzymes with WT activity but a varying fraction of inactive enzyme (supplementary text S2). (D) Scheme showing kinetic partitioning between folded (active) and misfolded (kinetically trapped) enzyme during expression. (E) Pulse-chase experiment to test for expression-dependent misfolding by varying expression conditions while keeping reaction conditions constant. (F) Residues with temperature-dependent activity changes shown as spheres on the PafA structure, with active-site residues N100, K162, R164, and T79 shown as purple sticks. (G) Histograms of the ratio of measured activities after expression at 23° and 37°C [cMUP (kcat/Kmmutant)/(kcat/KmWT), 23°C/37°C] (red) compared with the ratios of randomly paired replicates expressed under standard conditions (37°C and 100 μM Zn2+; gray). (H) Residues with expression [Zn2+]–dependent activity changes shown as spheres on the PafA structure. (I) Histograms of the ratio of measured activities across Zn2+ concentrations [cMUP (kcat/Kmmutant)/(kcat/KmWT), 1000 μM/10 μM Zn2+] (blue) compared with the ratios of randomly paired replicates expressed under standard conditions (37°C and 100 μM Zn2+; gray). (J) (Top) Native gels for enzymes expressed by means of in vitro transcription-translation off-chip at 23°C. All constructs appear as a single band corresponding to the natively folded species (“N”). (Bottom) At 37°C, non-WT variants have an additional band corresponding to the putatively misfolded (“M”) species. (K) Expression temperature effects on-chip (in vitro) versus in E. coli (in vivo). The red line indicates linear regression of log-transformed data with the indicated slope (values provided in table S5). In vivo protein levels were determined from eGFP fluorescence to estimate total expressed protein concentration.
Beyond their limited availability, fluorogenic substrates are often more reactive than naturally occurring substrates, potentially rendering binding rate–limiting and obscuring mutational effects on the chemical step of catalysis, and there is evidence for this behavior with PafA (16). On-chip MeP hydrolysis assays, therefore, complement the wide dynamic range of cMUP assays by identifying modest effects on the chemical step. Off-chip measurements of several active-site mutants revealed a decrease in observed MeP activity (owing to effects on the chemical step) without a concomitant change in cMUP activity, as expected for rate-limiting cMUP binding (Fig. 3B, red vertical arrows). Once transition-state destabilization was sufficiently large, MeP and cMUP reactions both slowed (Fig. 3B, blue vertical arrows). The solid blue line in Fig. 3C is a fit to the rate model derived from the free energy–reaction profiles in Fig. 3B for a series of active-site mutants (Fig. 3C, blue points; figs. S31; and supplementary text S2) (19) and predicts the kinetic behavior expected for the PafA glycine- and valine-scanning library mutants.
HT-MEK kinetic measurements revealed kcat/Km effects for almost half of the mutants (498 of 1035 with P < 0.01) (fig. S32), but few exhibited the predicted behavior (Fig. 3C, solid blue line versus gray symbols). Instead, the mutants tended to fall between the predicted line and a diagonal line representing equally deleterious effects on the reactions of both substrates (Fig. 3C, blue solid and dashed lines, respectively). Equally deleterious effects are expected for enzymes with WT activity but only a fraction of the enzyme in the active configuration, with less correctly folded variants further down the diagonal. Thus, observed intermediate effects could represent combinations of effects on the chemical step and on the fraction of the mutant population that is active.
Many mutations reduce catalysis by altering folding
The urea, Zn2+, and pH data presented above provided strong evidence against equilibrium unfolding for any of the variants (Fig. 2, H and I). We therefore considered and tested an alternate model in which inactive enzyme resulted from a nonequilibrium process: the formation of long-lived misfolded proteins during expression (Fig. 3D) (20). Because temperature is known to affect folding efficiency (21, 22), and because PafA binds multiple Zn2+ ions during folding, we varied the expression temperature (23° and 37°C) and Zn2+ concentration (10 and 1000 μM) from our standard expression conditions (37°C and 100 μM Zn2+). We then measured reaction rates under identical assay conditions (23°C and 100 μM Zn2+, our standard assay conditions), so that any observed rate changes must arise from differences during folding that persisted over time (Fig. 3E). Many mutations had differential effects on observed catalytic activity when expressed at 23° versus 37°C (“T-Effect”) or with different concentrations of Zn2+ (“Zn-Effect”). Whereas T-Effects were found predominantly in the enzyme core, Zn-Effects were concentrated around the distal Zn2+ (Fig. 3, F to I, and figs. S33 and S34). These results strongly support the presence of persistent nonequilibrium folding effects, which have been anecdotally observed in other systems (20, 21, 23–25). A second prediction of the misfolding model was also met: Variants with T- and Zn-Effects were not systematically altered in Km under varied expression conditions (fig. S35), as expected under a model of nonequilibrating states. PafA folding thus apparently involves one or more branchpoints sensitive to temperature and Zn2+ that lead to active PafA or one or more long-lived inactive states (Fig. 3D).
Altered folding pathways promote a long-lived inactive state in vitro and in vivo
Misfolding could be an artifact of high-throughput on-chip expression or could also arise during standard expression in vitro and possibly in vivo. To test for chip-induced misfolding effects, we selected 19 variants with varying predicted amounts of misfolding for off-chip expression by means of in vitro transcription-translation and kinetic characterization (fig. S36 and table S4). Activities were similar off- and on-chip (fig. S37), suggesting that the chip is not responsible for the observed misfolding.
Native gels and kinetic assays provided additional support for the misfolded state. Mutants predicted to misfold had an additional band of distinct mobility when expressed at high temperature that was not present or diminished when expressed at lower temperature (Fig. 3J, misfolded state “M”). Transient treatment with thermolysin, a protease that cleaves within exposed hydrophobic regions that occur in unfolded or misfolded proteins, resulted in loss of M but not the native state (native state “N”) (fig. S38A) (26). Nevertheless, despite degradation of the majority of the protein (present as M), the total observed enzyme activity for each mutant was unchanged before and after degradation (fig. S38B), indicating that M lacked substantial activity and that N and M did not equilibrate over the hours taken to carry out these experiments. The observation that different temperature-sensitive mutants gave the same mobility on a native gel and similar protease sensitivity suggests the formation of a single misfolded state or set of states with similar properties.
Our observation of a long-lived inactive state raised the question of whether analogous misfolding occurs in cells, where cellular machinery can assist folding. We recombinantly expressed 21 variants in E. coli that did and did not undergo temperature-dependent misfolding in vitro (fig. S36 and table S5). Expression in vivo was also temperature dependent, with changes in apparent kcat/Km values for the in vivo–expressed PafA mutants that correlated with the change observed in vitro [coefficient of determination (R2) = 0.38] (Fig. 3K). Temperature effects were generally smaller for in vivo–expressed mutants, suggesting that cellular factors may partially rescue or preferentially degrade the misfolded state, analogous to cellular degradation of thermodynamically destabilized mutants (27).
To test whether misfolding was a consequence of the eGFP tag, we overexpressed a non–eGFP-tagged variant (Y103G). We found wild type–like activity when it was expressed at 23°C but 260-fold less activity when it was expressed at 37°C (table S6 and fig. S39), suggesting that >99% of the 37°C-expressed enzyme was in the M state. CD spectra of the purified Y103G mutant provided independent evidence for a structural alteration that accompanied misfolding in vivo. WT PafA exhibits identical CD spectra when expressed at 37° or 23°C; by contrast, the CD spectrum for Y103G matches that of wild type when the mutant is expressed at low temperature (23°C) but not at higher temperature (37°C) (fig. S40), with the observed difference at 37°C suggesting loss of about one-third of PafA’s α-helical character in M (table S7).
Together, these results suggest that cellular folding conditions and chaperones are insufficient to prevent mutations from causing PafA to misfold and form long-lived inactive states in an E. coli expression strain [BL21 (DE3)]. However, the degree to which misfolding is rescued likely varies between strains and is sensitive to growth conditions and expression levels (28). A tendency to form kinetically stable misfolded states may therefore exert a selective pressure and influence the fitness landscape of proteins in cells (29–33).
Dissecting the origins of observed catalytic effects
HT-MEK assays allow us to quantify and dissect the degree to which observed changes in activity arise from changes in the amount of expressed protein, the amount that is correctly folded, and the catalytic efficiency of the correctly folded enzyme. Below, we isolate the catalytic effects for our PafA variants. We then take advantage of HT-MEK’s ability to provide quantitative kinetic and thermodynamic constants for multiple substrates and inhibitors and use these data to probe PafA’s functional architecture at a global level.
To remove folding effects, we compared kcat/Km values measured for substrates with different rate-limiting steps (cMUP and MeP) (Fig. 3C and supplementary text S3 and S4), represented the datapoint for each PafA variant in Fig. 3C as a superposition of a catalytic effect (Fig. 3C, blue solid line, defined by PafA active site variants that do not have misfolding effects; fig. S41) and a misfolding effect (Fig. 3C, diagonal blue dashed line), and solved for both (Fig. 4A and fig. S42). With this approach, we were able to quantify catalytic effects (kcat/Kmchem) for 946 of the 1036 variants and obtain upper limits for an additional 65 (fig. S43). Deleterious catalytic effects were found for mutations at 161 of PafA’s 526 positions (Fig. 4B, figs. S44 and S45, and table S8). Mutations at an even larger number of positions, 232, gave folding effects. The magnitude of folding effects did not correlate with observed eGFP fluorescence intensities, establishing that enzyme misfolding does not influence eGFP folding (fig. S46).
Fig. 4. Separating mutational effects on catalysis from changes in the fraction of active enzyme (fa).
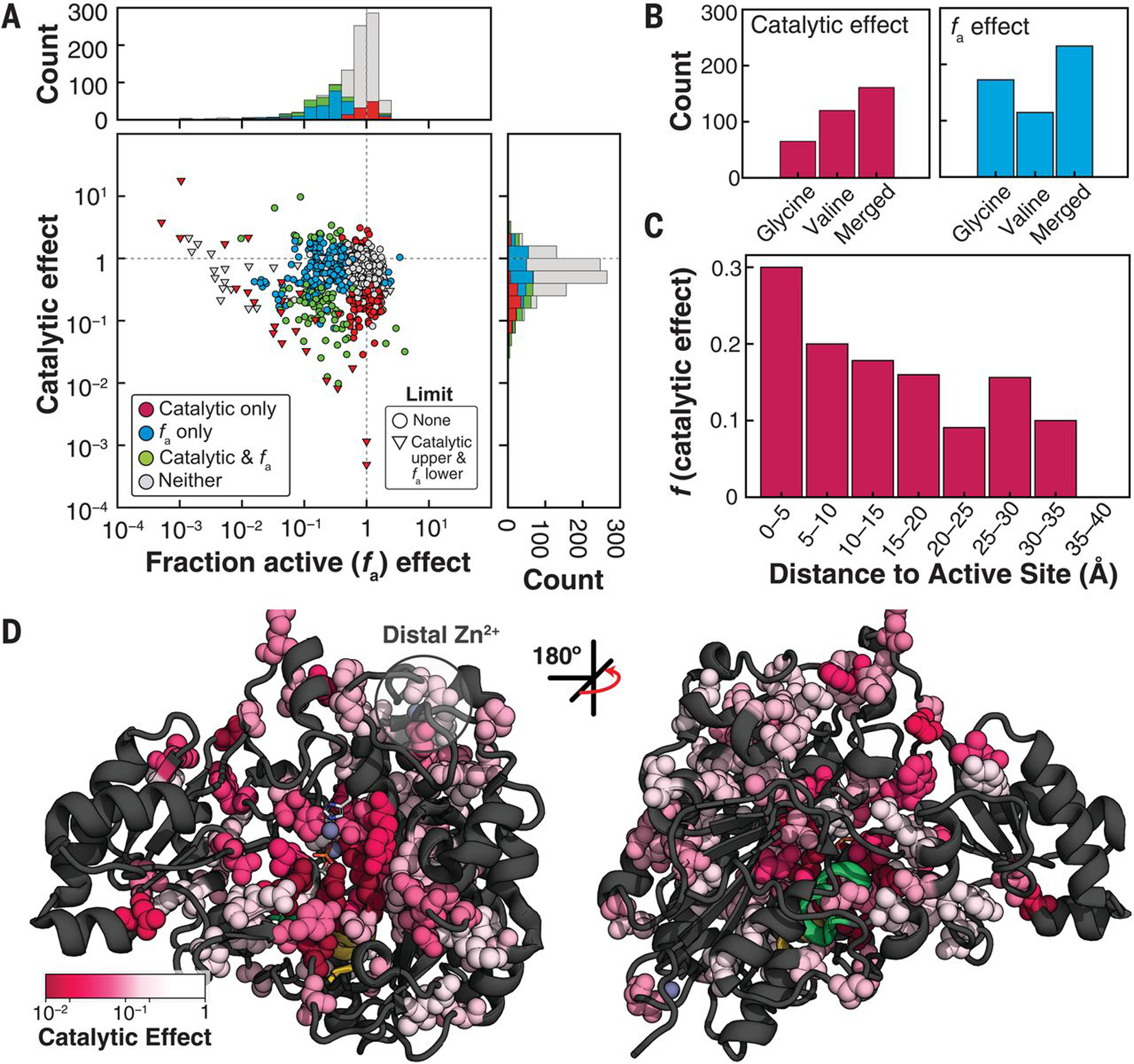
(A) Catalytic and fa effects (relative to wild type) for all variants colored by effect. Distributions of effects are projected along axes (stacked). (B) Counts of mutants with catalytic (P < 0.05) and fa effects (P < 0.01) for the valine and glycine libraries, and total number of positions with effects (out of 525, “Merged”) (fig. S45). (C) Fraction of non–active-site mutants within each distance bin with deleterious catalytic effects. P < 0.05; 5-Å bins (table S8). (D) Positions with deleterious catalytic effects on the PafA structure [161; “Merged” set in (B)]. Residues with deleterious effects (greater than fivefold down from wild type) are colored according to the most deleterious effect and shown as spheres.
The largest catalytic effects cluster in and around the active site, with the fraction of residues exhibiting effects diminishing with distance from the active site (Fig. 4C and table S8). Although positions that give catalytic effects tend to cluster, the pattern is asymmetric and complex (Fig. 4D). Many large distal effects are found surrounding the bound Zn2+ that is >15 Å from the active site (“distal Zn2+”), suggesting that its coordination may restrain the conformational mobility of surrounding residues and propagate to the active site. Nevertheless, it is difficult to rationalize catalytic effects, especially more distal effects. To better understand these patterns and to relate effects to the specific mechanisms used in catalysis, we defined functional components (FCs) from prior mechanistic studies of PafA and other alkaline phosphatase (AP) superfamily members (16, 34, 35) and systematically assessed the effect of each mutation on each FC.
FC1: Mutations disrupting O2 phosphoryl oxygen atom interactions
Our first FC (FC1) is derived from the observation that removal of two active-site side chains, K162 and R164, reduces monoester hydrolysis by 105-fold but has no effect on diester hydrolysis, rendering PafA an equally potent phosphate mono- and diesterase (fig. S47) (16). Because K162 and R164 interact with the phosphoryl oxygen that is anionic in monoesters but esterified in diesters (Fig. 5A, O2) (34), we expect other residues that support the formation of these interactions to similarly disrupt monoester but not diester hydrolysis. We define these effects as FC1 = Δdiester/Δmonoester, where Δ = (kcat/Km)mutant/(kcat/Km)WT. Although the simplest expectation is that mutations to residues neighboring K162 and R164 will have FC1 effects, we cannot predict how large and how varied these effects are, how far they extend, or whether there are remote regions or surface sites that have large effects. In addition, we cannot predict whether residues that affect FC1 also contribute to other catalytic mechanisms, represented as other FCs below.
Fig. 5. FC1: Catalytic effects through the O2 phosphoryl oxygen atom.
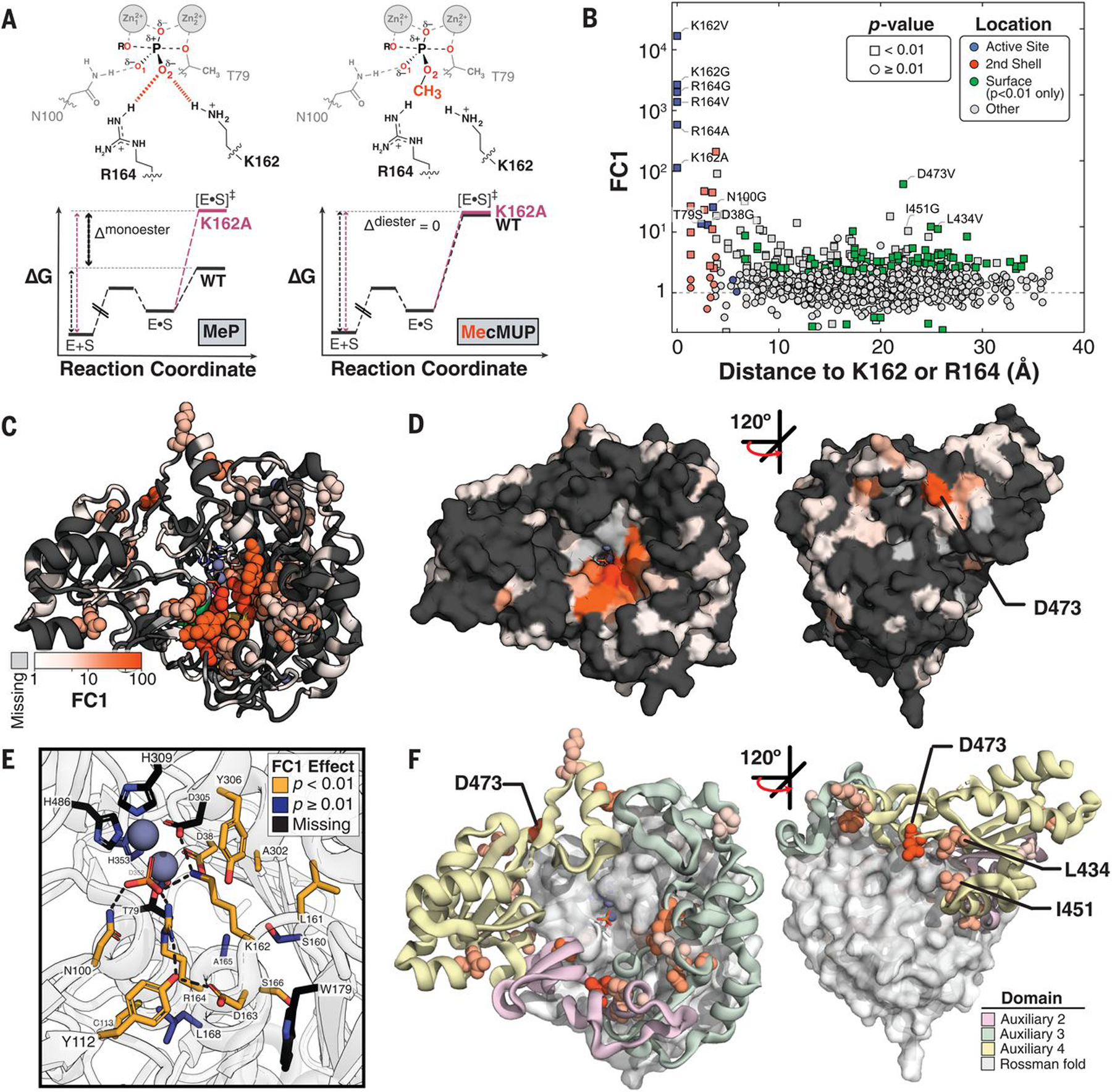
(A) Schematic of the transition states for reaction of (left) MeP and (right) MecMUP highlighting the methyl group on the O2 phosphoryl atom of MecMUP (orange) and the O2 interactions with K162 and R164. Thin black and pink arrows indicate the reaction barrier for wild type and a mutant PafA, respectively, for each reaction; the thick black arrow denotes their difference for the two reactions (Δmonoester and Δdiester). The ratio of kcat/Km effects (the difference between Δmonoester and Δdiester in energy space) gives FC1. (B) FC1 values as a function of the minimum distance to K162 or R164, with active site, second shell, and significantly affected surface residues colored. (C) PafA positions with FC1 effects (P < 0.01) when mutated to valine or glycine, colored by FC1 magnitude. Effects greater than fivefold are shown as spheres, with ribbon coloring for positions with effects less than or equal to fivefold. (D) PafA surface representation with FC1 effects colored as in (C). (E) FC1 effects of active site and second-shell residues indicating residues with significant FC1 effects (yellow), without FC1 effects (blue), and without measurements (black) (table S9). (F) Distribution of FC1 effects within the Rossmann core and Auxiliary Domains. Distal (greater than or equal to third shell) positions with effects greater than fivefold shown as spheres colored corresponding to their auxiliary domain. Only effects in auxiliary domains are shown.
To measure diester activity on-chip and determine FC1 effects, we synthesized a fluorogenic diester substrate suitable for HT-MEK (MecMUP) and measured kcat/Km for the PafA mutant libraries (high Km values for the non-cognate diesterase activities preclude estimation of kcat and Km separately) (fig. S48) (16). We obtained kcat/Km values for 857 of the 1036 mutants and upper limits for an additional 178 (fig. S49, A and B), and on-chip diester rate constants matched off-chip measurements (fig. S49C). Because folding affects hydrolysis of both substrates equally, we compared measured rates directly, without correcting for folding effects (fig. S50A), allowing stronger statistical inference (fig. S50B).
Many mutants had FC1 effects: 88 Val and 93 Gly mutations (fig. S50C), corresponding to 156 of the 494 measurable positions (of a total of 526 positions) (Fig. 5, B to D). Seven of the 10 measurable non–active-site residues directly contacting K162 or R164 (second-shell residues) exhibited FC1 effects (Fig. 5E; fig. S51, A and B; and table S9A), which is consistent with frequent second-shell effects observed in directed evolution experiments (36–38). Of the three active-site Zn2+ ligands with measurable effects upon mutation, we observed an FC1 effect for D38G (which accepts a hydrogen bond from K162) but no FC1 effect for D352G or H353V (which do not interact with K162 or R164). The active-site variants T79S and N100G also had FC1 effects (tables S9B and S10), which is consistent with coupling between active-site residues attributable to shared contacts with K162 and R164 (Fig. 5E).
Although the largest effects were observed for active-site residues and next-largest for the second-shell residues, there was no additional drop in effect size after the third shell; the majority of residues with >10-fold FC1 effects were found in the third shell and beyond (15 of 23) (fig. S51 and table S11). Four of these lie at the enzyme surface (Fig. 5D and fig. S51), which is consistent with the hypothesis that enzymes possess a reservoir of allosteric potential and suggests that HT-MEK can be used to identify regions that are potential sites for allosteric inhibitors and drugs (39–43).
PafA has three of four nonterminal auxiliary domains (ADs) found within the AP superfamily, referred to as ADs 2 to 4 (supplementary text S5), that sit around the universally conserved Rossmann fold (Fig. 5F and fig. S52). ADs 2 and 4 are present in both AP superfamily phosphate mono- and diesterases, whereas AD 3 contains K162 and R164 and is considerably more extensive in the monoesterases (figs. S52 and S53 and table S12). Despite these apparent functional and evolutionary differences, FC1 effects are found in all three ADs to a similar extent and with similar magnitudes (fig. S54 and table S13). The largest FC1 effect outside of the active site or second shell comes from a solvent-exposed surface residue, D473, within AD 4 (Fig. 5F, yellow); a dramatically larger effect for the valine substitution (>60-fold versus less than twofold for D473G) suggests that a change in local folding may allosterically disrupt the O2 site 20 Å away. Two nearby residues (L434 and I451) also exhibit >10-fold FC1 effects, and all three lie along a three-way interface between ADs 3 and 4 and the Rossmann core domain (Fig. 5F). Future multimutant cycle experiments (44–46) using HT-MEK may allow testing of whether mutations at interfaces result in more extensive conformational rearrangements than mutations within a single domain and dissect the underlying structural properties accounting for long-range effects.
FC2 and FC3: Effects on phosphate affinity
To provide catalysis, enzymes must bind their transition states more strongly than their substrates; otherwise, the energetic barrier for the reaction and reaction rate would remain the same as in solution (47, 48). Enzymes must also limit the binding of substrates and products to allow sufficient turnover in the presence of higher substrate and product concentrations (49, 50). Ground-state destabilization has been considered a possible mechanism for achieving this, and there is evidence for ground-state destabilization by PafA and other AP superfamily members through electrostatic repulsion between the anionic nucleophile, T79 in PafA, and the negatively charged phosphoryl oxygens (Fig. 6A) (16, 35, 51). For PafA, mutating T79 to serine increases the affinity for Pi, the reaction’s product and a ground-state analog, by 100-fold, whereas in E. coli AP, the nucleophile S102G mutation increases affinity more than 1000-fold (16, 35); the S102G mutation ablates the nucleophile entirely, and the chemically conservative T79S substitution presumably allows greater mobility, reducing electrostatic repulsion (16). We therefore expect that mutations that perturb this mechanism will increase Pi affinity and define the second FC, FC2, as strengthened Pi binding (Kimutant/KiWT < 1).
Fig. 6. FC2 and FC3: Mutational effects on affinity for Pi.
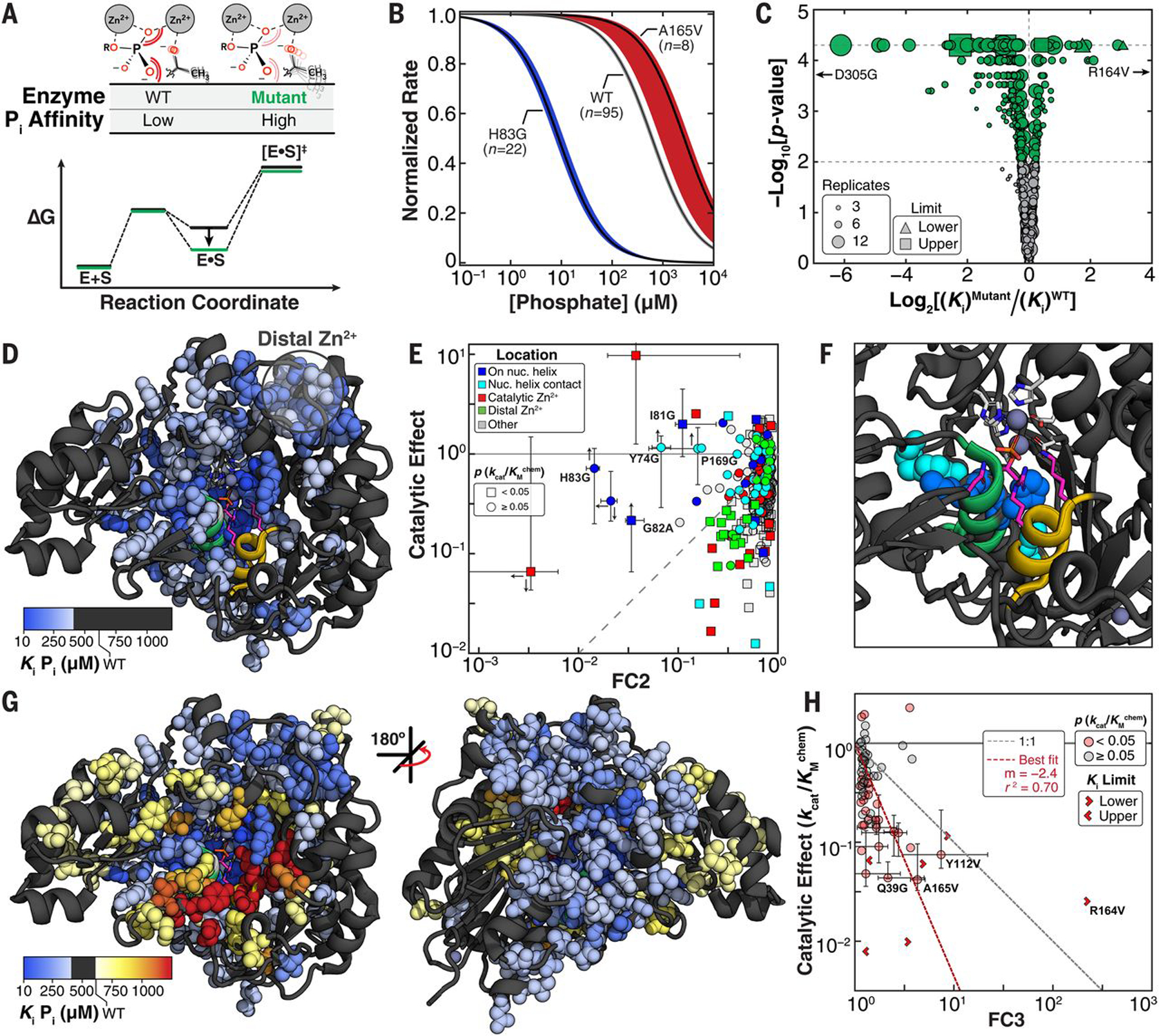
(A) Reaction coordinate diagram for hypothetical mutant (green) with diminished destabilization relative to wild type (black) and schematic illustrating a model for an FC2 effect through increased flexibility of the T79 nucleophile. (B) Median Pi inhibition curves for wild type and two mutants with FC2 (H83G) and FC3 (A165V) effects. Colored regions denote 99% CIs on the medians of replicate measurements. (C) Volcano plot of Ki Pi effects for glycine and valine scan mutants. P < 0.01, green markers; P ≥ 0.01, gray markers. (D) PafA structure with positions for which Ki Pi is >1.5-fold tighter than that of wild type (at P < 0.01) shown as spheres colored by the magnitude of the larger effect. The “nucleophile” helix (green) is largely obscured. (E) Scatter plot of catalytic and FC2 effects (P < 0.01) colored by location. Error bars correspond to 95% CIs determined from bootstrapping; up and down arrows indicate lower and upper kcat/Kmchem limits, respectively; left and right arrows indicate upper and lower Ki Pi limits. (F) PafA active-site (pink residues) and the five mutants possessing large FC2 effects without kcat/Kmchem effects shown as spheres, corresponding to the labeled mutants in (E). (G) Front and back views of FC2 (blue spheres) and FC3 effects (red spheres). FC2 effects are those shown in (D), and FC3 effects are shown for P < 0.01. (H) Scatter plot of catalytic (kcat/Kmchem) and FC3 effects for mutants with FC3 effects (P < 0.01). Gray and red points indicate significant and insignificant kcat/Kmchem effects, respectively. Error bars correspond to 95% CIs, as in (E); red dashed line indicates the best fit line to mutants with significant FC3 and catalytic (kcat/Kmchem) effects, excluding active-site mutants.
Conversely, active-site residues typically make both ground-state and transition-state interactions, so their removal weakens binding and diminishes catalysis, in some instances to a similar extent (“uniform binding”) and in others preferentially destabilizing the transition state (52–55). As expected, mutations to the PafA active-site residues that interact with the phosphoryl O1 and O2 oxygen atoms (N100, K162, and R164) weaken Pi binding, but N100 and K162 mutations have much larger effects on catalysis, indicating preferential transition-state stabilization (fig. S55) (16). We therefore define the third FC (FC3) as weakened Pi binding (Kimutant/KiWT > 1).
To measure inhibition constants, we quantified rates of cMUP hydrolysis as a function of Pi concentration and fit observed initial rates to a competitive inhibition model (Fig. 6B and fig. S56). HT-MEK–determined inhibition constants agreed with previous off-chip measurements (fig. S57) and are of higher precision than kinetic constants because they are not sensitive to uncertainties in total enzyme concentration or active fraction (Fig. 6C; reports of inhibition measurements are available in data files S1 and S2).
We uncovered 331 mutants that increased Pi affinity (FC2) and 73 that decreased Pi affinity (FC3) (Fig. 6C and fig. S58). Thus, about one-third of all mutants measurably altered affinity, and four times as many mutations enhanced binding as weakened it. Because it is highly unusual to enhance function through random variation, this observation suggests that residues at many positions are evolutionarily selected to prevent tight Pi binding. Mutations with ground-state destabilization effects (FC2) were located in an extended yet spatially contiguous region that included the helix containing the T79 oxyanion (“nucleophile helix”), the catalytic Zn2+ ions, and the distal Zn2+ site (Fig. 6D and tables S14 and S15).
Catalytic effects (kcat/Kmchem) for mutants with FC2 effects ranged from insignificant to 100-fold reductions (Fig. 6E and fig. S59). For most, the catalytic effect was greater than the FC2 effect, which is consistent with functional roles beyond the prevention of too-tight binding. Nevertheless, five mutants of residues that form a spatially contiguous subregion on and adjacent to the nucleophile helix have the largest FC2 effects but little or no catalytic effect (Fig. 6E and F), suggesting that these mutations allow rearrangements to reduce electrostatic repulsion in the presence of bound substrate.
Mutants that weakened Pi binding (FC3 effects) were also located in a contiguous region (Fig. 6G). The largest FC3 effects were near active-site residues K162 and R164 and had larger effects on catalysis (kcat/Kmchem) than Pi binding (Fig. 6H, red, and figs. S60 to S62), presumably reflecting the greater constraints present in the transition state than in the ground state (56, 57).
Comparisons of the mutational effects across FC1 to FC3 reveal that many mutations outside the active site preferentially affect either FC1 or FC2. Further, several preferentially alter FC2 without dramatically altering FC1, reducing the fraction of active enzyme, or reducing overall catalysis (fig. S63). A small number of residues at the interface of FC2 and FC3 effects yield either an FC2 or FC3 effect depending on the substitution (fig. S64). The ability to selectively alter particular properties through specific mutations provides a potential starting point for attempts to engineer enzymes with desired kinetic and thermodynamic constants and behaviors.
FC4: Rates of phosphoenzyme hydrolysis
Linking observed mutational rate effects to their physical and chemical origins requires knowledge of the step that is being observed: the rate-limiting step. Pre–steady-state approaches (such as stopped flow and rapid quench) are the gold standard for determining rates of individual reaction steps but do not readily scale to large libraries. For PafA, extensive prior mechanistic knowledge allowed us to determine mutational effects on individual PafA reaction steps for 992 PafA variants without requiring pre–steady-state measurements.
The steady-state kinetic constant kcat/Km can be limited by substrate binding or chemical cleavage of the substrate to form the covalent enzyme-phosphate (E–P) species (Fig. 7A, k1 and kchem,1 steps), and kcat can be limited by hydrolysis of E–P (kchem,2) or dissociation of product Pi (koff,Pi) (Fig. 7A). We deconvoluted mutational effects on kchem,2 using measurements of kcat for cMUP and Ki for Pi, and these kchem,2 effects define FC4 (supplementary text S6). Seven mutants changed the rate-limiting step from E–P hydrolysis to Pi release (koff,Pi < kchem,2) (fig. S65), which is consistent with the observation of naturally occurring alkaline phosphatases of the AP superfamily with either of these steps rate limiting (58).
Fig. 7. FC4: Mutational effects on phosphoenzyme intermediate hydrolysis.
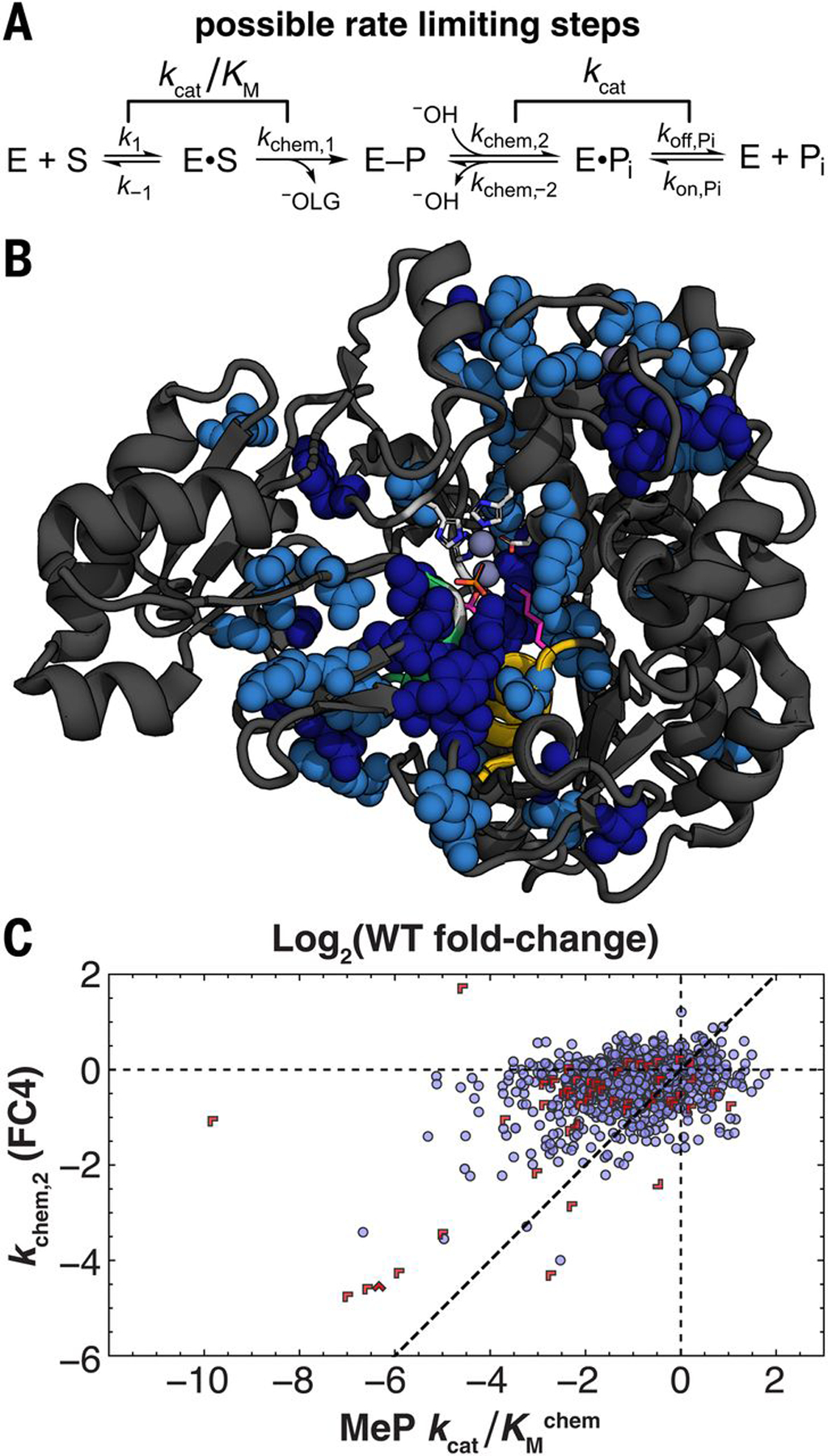
(A) Schematic of PafA catalytic cycle with possible rate-limiting steps under saturating (kcat) and subsaturating (kcat/Km) conditions. (B) Structure showing positions with decreased kchem,2 (P < 0.05 and P < 0.1 in dark and light blue, respectively) upon mutation (spheres). Positions lacking an estimate of kchem,2 for both substitutions are light gray ribbons, and positions without significant effects are black ribbons. (C) Scatter plot comparing mutational effects on phosphoenzyme hydrolysis (kchem,2, or FC4) and kcat/Kmchem. Limits are indicated with red chevrons pointing in limit directions (supplementary text S6).
Overall, we found 18 Val and 36 Gly mutants that reduce kchem,2 (Fig. 7B and fig. S66), and these overlap substantially with those affecting kcat/Kmchem (table S16). In the simplest scenario, mutations reducing kchem,2 would be a subset of those reducing kcat/Kmchem because kcat/Km includes both a phosphoryl transfer chemical step and an additional step (binding). Consistent with this expectation, mutations affect either kcat/Kmchem alone or kcat/Kmchem and kchem,2 to a similar or smaller extent (Fig. 7C).
Discussion
HT-MEK uses automatic valved microfluidics to carry out high-throughput expression, purification, and comprehensive biochemical characterization of enzymes at unprecedented scale and mechanistic depth. In the future, HT-MEK can be applied to the vast number of enzymes whose activity can be monitored through fluorescence directly or with a coupled assay. In particular, the ability to sensitively detect Pi (18) renders HT-MEK immediately applicable to adenosine triphosphatases, guanosine triphosphatases, helicases, protein chaperones, polymerases (with pyrophosphatase present), and many others.
Deleterious effects throughout the enzyme and at the surface, with many of the largest effects distant from the active site, mirror results obtained from DMS studies of other enzymes that often reveal distal effects on product formation or organismal fitness (12, 59–61). However, the ability of HT-MEK to provide quantitative measurements of a battery of kinetic and thermodynamic parameters yields an atlas of PafA functional “architecture” with unprecedented detail (table S17). Regions of residues with similar catalytic signatures extending throughout the enzyme affirm that the enzyme beyond the active site is not a passive, monolithic scaffold but rather contributes to function in multiple ways.
Comparing patterns of mutational effects between FCs reveals that these architectural solutions are idiosyncratic. For example, K162/R164, and the T79 nucleophile, sit at the N-termini of their respective helices, but the largest FC2 effects were on and around the nucleophile helix, whereas FC1 effects were not similarly distributed around the monoesterase helix. This difference perhaps reflects a need for more interactions to secure the nucleophile helix against ground-state electrostatic repulsive forces. Twice as many glycine as valine mutations increased Pi binding (fig. S58), potentially because side-chain ablation allows structural rearrangements to reduce electrostatic repulsion without disrupting favorable binding interactions.
The need for interactions throughout the enzyme for optimal catalysis highlights the challenges inherent in de novo enzyme design because it suggests that computational efforts must consider in detail the interactions of a prohibitively large number of residues. The detailed architectural maps provided by HT-MEK can focus computational and experimental mutagenesis by distinguishing catalytic from folding effects and by identifying residues and regions that affect particular aspects of catalysis.
The presence of a long-lived inactive population of many PafA variants emphasizes how different underlying (and unexpected) biophysical mechanisms can compromise an enzyme’s activity and thus the need to explicitly decouple effects on folding and catalysis to understand function. Analogous behavior observed for these mutants in E. coli is consistent with selective pressure to avoid misfolding in vivo and adds to growing evidence that kinetic factors affect stable protein expression in cells (20, 30, 62–64). We speculate that highly stable proteins such as PafA and other secreted enzymes may be more prone to forming kinetically trapped states.
Although PafA functional regions have superficial similarities to sectors and other measures of evolutionary covariation and coconservation (65–69), our data report on sequence-function relationships in ways that sequence analyses alone cannot. On the basis of a metagenomic alignment of 14,505 PafA-like AP superfamily sequences (fig. S67 and supplementary text S7), we found that observed kcat/Km values correlate only modestly with conservation at a given position (figs. S68 and S69). Positions at which mutations promote misfolding tend to be most conserved, suggesting that the largest selective pressure at the residue level may be related to ensuring correct folding, whereas correlations between individual FCs and conservation are weak, with many large effects at nonconserved residues (figs. S69 and S70). For PafA, selective pressures will differ with the available Pi at the organism’s physical location, whether available Pi varies temporally, and whether there is competition for Pi from other organisms in the same ecological niche. Residues with critical roles in responding to these adaptive pressures would be poorly conserved, changing frequently despite being tightly linked to survival. These results underscore the need for in-depth functional studies to uncover the relationships between the evolution of particular functions and conservation.
Enzymes are the targets of many therapeutics, are altered in genetic diseases, serve as tools for molecular biology, and play critical roles in industrial processes. Its rapidity and low cost make HT-MEK a powerful tool for future applications across all these areas. In basic research, the large, highly quantitative datasets provided by HT-MEK can greatly extend and even supplant traditional SDM approaches for the initial characterization of new enzymes and for in-depth mechanistic investigation. Combined with recent advances in gene synthesis, HT-MEK can rapidly functionally characterize metagenomic variants, providing a critically needed dimension to phylogenetic analyses. In medicine, we anticipate that HT-MEK will rapidly determine the functional effects of human enzyme allelic variants of unknown relevance identified from sequence data and systematically identify candidate allosteric surfaces within currently “undruggable” therapeutic target enzymes. We anticipate HT-MEK contributing to these and still more areas of basic and applied biology, medicine, and engineering.
Materials and methods summary
A full description of materials and methods is provided in the supplementary materials. Briefly, we fabricated microfluidic devices and aligned them to plasmid DNA arrays as previously described (70–72), then connected devices to a custom pneumatic manifold (73) and imaged using a fully automated fluorescence microscope. The plasmid DNAs coded for a set of specified PafA variants, each with a C-terminal eGFP tag. The PafA-eGFP variants were expressed in vitro on-chip and immobilized to Button valves by means of antibodies to eGFP (72) in parallel, before quantification of the enzyme concentration within each chamber by using an eGFP standard curve. This entire process takes ~10 hours to complete. Before measuring kinetics of substrate hydrolysis for cMUP, MeP, and MecMUP, we determined product standard curves for each chamber by introducing a series of product concentrations [cMU for cMUP and MecMUP hydrolysis and Pi in the presence of 30 μM of modified PBP (18) for MeP hydrolysis], imaging the device after each addition. To measure rates of hydrolysis, we flowed a single concentration of substrate into the device with the Button valves closed and then started reactions by (i) closing the Sandwich valves to isolate individual reaction chambers and (ii) opening the Button valves to expose the immobilized enzyme within each chamber to substrate. We quantified initial rates of product formation for each chamber by imaging over time across the entire device and converting measured intensities to product concentrations using standard curves. To determine kcat, Km, and kcat/Km for cMUP hydrolysis and kcat/Km for MeP and MecMUP hydrolysis, we quantified initial rates of product formation for a series of increasing substrate concentrations, scaled rates by measured enzyme concentration, and fit to a simple Michaelis-Menten model. For the nonfluorogenic MeP substrate, we detected formation of Pi by performing reactions in the presence of 30 μM PBP. To determine the Ki for Pi inhibition, we assayed rates of cMUP hydrolysis at 10 or 50 μM cMUP in the presence of increasing Pi concentration, fit initial rates as described above, and then fit these rates to a competitive inhibition model. For each assay type, we carried out multiple replicate experiments for each parameter for each mutant, allowing us to perform extensive quality control measurements and determine the statistical significance of effects.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Herschlag and Fordyce laboratories for discussions and review of the manuscript, M. Madsen for technical assistance, K. Brower for microfluidic mold fabrication and optics assistance, and S. Costello and S. Marqusee for helpful discussions.
Funding:
This research was supported by the NIH grant R01 (GM064798) awarded to D.H. and P.M.F., a Joint Initiative for Metrology in Biology (JIMB) seed grant, a Stanford Bio-X Interdisciplinary Initiative Seed Grant, an Ono Pharma Foundation Breakthrough Innovation Prize, and the Gordon and Betty Moore Foundation (grant 8415). P.M.F. acknowledges the support of an Alfred P. Sloan Foundation fellowship and is a Chan Zuckerberg Biohub Investigator. C.J.M. acknowledges the support of a Canadian Institutes of Health Research (CIHR) Postdoctoral Fellowship. D.A.M. acknowledges support from the Stanford Medical Scientist Training Program and a Stanford Interdisciplinary Graduate Fellowship (Anonymous Donor) affiliated with Stanford ChEM-H. E.A. acknowledges support from the NIH (GM0595). This research used resources of the National Energy Research Scientific Computing Center (NERSC), a US Department of Energy (DOE) Office of Science User Facility operated under contract DE-AC02-05CH11231, as well as resources obtained from the Facilities Integrating Collaborations for User Science (FICUS) program associated with the Joint Genome Institute of the DOE, proposal 503369.
Footnotes
Competing interests: The authors declare no competing financial interests.
SUPPLEMENTARY MATERIALS
science.sciencemag.org/content/373/6553/eabf8761/suppl/DC1
MDAR Reproducibility Checklist
Data files S1 and S2
Data and materials availability:
Summary tables of all kinetic and thermodynamic parameters measured for each mutant are included in the supplementary materials. All data acquired in this study, all code used to obtain and process images and fit kinetic and thermodynamic parameters, and PyMOL files corresponding to all figures containing structural information are available in a registered Open Science Foundation Repository (DOI: 10.17605/OSF.IO/QRN3C) (74). Additional per-experiment and per-mutant summary PDFs are available on the Fordyce Lab website (www.fordycelab.com/publication-data).
REFERENCES AND NOTES
- 1.Wang Q et al. , Landscape of multi-nucleotide variants in 125,748 human exomes and 15,708 genomes. Nat. Commun 11, 2539 (2020). doi: 10.1038/s41467-019-12438-5; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Claussnitzer M et al. , A brief history of human disease genetics. Nature 577, 179–189 (2020). doi: 10.1038/s41586-019-1879-7; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lek M et al. , Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). doi: 10.1038/nature19057; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stein A, Fowler DM, Hartmann-Petersen R, Lindorff-Larsen K, Biophysical and mechanistic models for disease-causing protein variants. Trends Biochem. Sci 44, 575–588 (2019). doi: 10.1016/j.tibs.2019.01.003; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kinney JB, McCandlish DM, Massively parallel assays and quantitative sequence-function relationships. Annu. Rev. Genomics Hum. Genet 20, 99–127 (2019). doi: 10.1146/annurev-genom-083118-014845; [DOI] [PubMed] [Google Scholar]
- 6.Blanco C, Janzen E, Pressman A, Saha R, Chen IA, Molecular fitness landscapes from high-coverage sequence profiling. Annu. Rev. Biophys 48, 1–18 (2019). doi: 10.1146/annurev-biophys-052118-115333; [DOI] [PubMed] [Google Scholar]
- 7.Weile J, Roth FP, Multiplexed assays of variant effects contribute to a growing genotype-phenotype atlas. Hum. Genet 137, 665–678 (2018). doi: 10.1007/s00439-018-1916-x; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lisi GP, Loria JP, Allostery in enzyme catalysis. Curr. Opin. Struct. Biol 47, 123–130 (2017). doi: 10.1016/j.sbi.2017.08.002; [DOI] [PubMed] [Google Scholar]
- 9.Reynolds KA, McLaughlin RN, Ranganathan R, Hot spots for allosteric regulation on protein surfaces. Cell 147, 1564–1575 (2011). doi: 10.1016/j.cell.2011.10.049; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keedy DA et al. , An expanded allosteric network in PTP1B by multitemperature crystallography, fragment screening, and covalent tethering. eLife 7, e36307 (2018). doi: 10.7554/eLife.36307; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fowler DM et al. , High-resolution mapping of protein sequence-function relationships. Nat. Methods 7, 741–746 (2010). doi: 10.1038/nmeth.1492; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Araya CL, Fowler DM, Deep mutational scanning: Assessing protein function on a massive scale. Trends Biotechnol. 29, 435–442 (2011). doi: 10.1016/j.tibtech.2011.04.003; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lassila JK, Zalatan JG, Herschlag D, Biological phosphoryl-transfer reactions: Understanding mechanism and catalysis. Annu. Rev. Biochem 80, 669–702 (2011). doi: 10.1146/annurev-biochem-060409-092741; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fordyce PM et al. , De novo identification and biophysical characterization of transcription-factor binding sites with microfluidic affinity analysis. Nat. Biotechnol 28, 970–975 (2010). doi: 10.1038/nbt.1675; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maerkl SJ, Quake SR, A systems approach to measuring the binding energy landscapes of transcription factors. Science 315, 233–237 (2007). doi: 10.1126/science.1131007; [DOI] [PubMed] [Google Scholar]
- 16.Sunden F et al. , Mechanistic and evolutionary insights from comparative enzymology of phosphomonoesterases and phosphodiesterases across the alkaline phosphatase superfamily. J. Am. Chem. Soc 138, 14273–14287 (2016). doi: 10.1021/jacs.6b06186; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Santoro MM, Bolen DW, Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl α-chymotrypsin using different denaturants. Biochemistry 27, 8063–8068 (1988). doi: 10.1021/bi00421a014; [DOI] [PubMed] [Google Scholar]
- 18.Brune M, Hunter JL, Corrie JET, Webb MR, Direct, real-time measurement of rapid inorganic phosphate release using a novel fluorescent probe and its application to actomyosin subfragment 1 ATPase. Biochemistry 33, 8262–8271 (1994). doi: 10.1021/bi00193a013; [DOI] [PubMed] [Google Scholar]
- 19.O’Brien PJ, Herschlag D, Alkaline phosphatase revisited: Hydrolysis of alkyl phosphates. Biochemistry 41, 3207–3225 (2002). doi: 10.1021/bi012166y; [DOI] [PubMed] [Google Scholar]
- 20.Addabbo RM et al. , Complementary role of co- and post-translational events in de novo protein biogenesis. J. Phys. Chem. B 124, 6488–6507 (2020). doi: 10.1021/acs.jpcb.0c03039; [DOI] [PubMed] [Google Scholar]
- 21.Sturtevant JM, Yu MH, Haase-Pettingell C, King J, Thermostability of temperature-sensitive folding mutants of the P22 tailspike protein. J. Biol. Chem 264, 10693–10698 (1989). doi: 10.1016/S0021-9258(18)81678-7; [DOI] [PubMed] [Google Scholar]
- 22.Guo M, Xu Y, Gruebele M, Temperature dependence of protein folding kinetics in living cells. Proc. Natl. Acad. Sci. U.S.A 109, 17863–17867 (2012). doi: 10.1073/pnas.1201797109; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith DH, King J, Temperature-sensitive mutants blocked in the folding or subunit assembly of the bacteriophage P22 tail spike protein. III. Intensive polypeptide chains synthesized at 39°C. J. Mol. Biol 145, 653–676 (1981). doi: 10.1016/0022-2836(81)90308-9; [DOI] [PubMed] [Google Scholar]
- 24.Baker D, Sohl JL, Agard DA, A protein-folding reaction under kinetic control. Nature 356, 263–265 (1992). doi: 10.1038/356263a0; [DOI] [PubMed] [Google Scholar]
- 25.Im H, Woo M-S, Hwang KY, Yu M-H, Interactions causing the kinetic trap in serpin protein folding. J. Biol. Chem 277, 46347–46354 (2002). doi: 10.1074/jbc.M207682200; [DOI] [PubMed] [Google Scholar]
- 26.Park C, Marqusee S, Probing the high energy states in proteins by proteolysis. J. Mol. Biol 343, 1467–1476 (2004). doi: 10.1016/j.jmb.2004.08.085; [DOI] [PubMed] [Google Scholar]
- 27.Thompson S, Zhang Y, Ingle C, Reynolds KA, Kortemme T, Altered expression of a quality control protease in E. coli reshapes the in vivo mutational landscape of a model enzyme. eLife 9, e53476 (2020). doi: 10.7554/eLife.53476; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gidalevitz T, Prahlad V, Morimoto RI, The stress of protein misfolding: From single cells to multicellular organisms. Cold Spring Harb. Perspect. Biol 3, a009704–a009704 (2011). doi: 10.1101/cshperspect.a009704; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sohl JL, Jaswal SS, Agard DA, Unfolded conformations of α-lytic protease are more stable than its native state. Nature 395, 817–819 (1998). doi: 10.1038/27470; [DOI] [PubMed] [Google Scholar]
- 30.Jaswal SS, Sohl JL, Davis JH, Agard DA, Energetic landscape of α-lytic protease optimizes longevity through kinetic stability. Nature 415, 343–346 (2002). doi: 10.1038/415343a; [DOI] [PubMed] [Google Scholar]
- 31.Watters AL et al. , The highly cooperative folding of small naturally occurring proteins is likely the result of natural selection. Cell 128, 613–624 (2007). doi: 10.1016/j.cell.2006.12.042; [DOI] [PubMed] [Google Scholar]
- 32.Bemporad F et al. , Biological function in a non-native partially folded state of a protein. EMBO J. 27, 1525–1535 (2008). doi: 10.1038/emboj.2008.82; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jefferson RE, Blois TM, Bowie JU, Membrane proteins can have high kinetic stability. J. Am. Chem. Soc 135, 15183–15190 (2013). doi: 10.1021/ja407232b; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zalatan JG, Fenn TD, Herschlag D, Comparative enzymology in the alkaline phosphatase superfamily to determine the catalytic role of an active-site metal ion. J. Mol. Biol 384, 1174–1189 (2008). doi: 10.1016/j.jmb.2008.09.059; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andrews LD, Fenn TD, Herschlag D, Ground state destabilization by anionic nucleophiles contributes to the activity of phosphoryl transfer enzymes. PLOS Biol. 11, e1001599 (2013). doi: 10.1371/journal.pbio.1001599; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hilvert D, Design of protein catalysts. Annu. Rev. Biochem 82, 447–470 (2013). doi: 10.1146/annurev-biochem-072611-101825; [DOI] [PubMed] [Google Scholar]
- 37.Freiberger MI, Guzovsky AB, Wolynes PG, Parra RG, Ferreiro DU, Local frustration around enzyme active sites. Proc. Natl. Acad. Sci. U.S.A 116, 4037–4043 (2019). doi: 10.1073/pnas.1819859116; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morley KL, Kazlauskas RJ, Improving enzyme properties: When are closer mutations better? Trends Biotechnol. 23, 231–237 (2005). doi: 10.1016/j.tibtech.2005.03.005; [DOI] [PubMed] [Google Scholar]
- 39.Dagliyan O, Dokholyan NV, Hahn KM, Engineering proteins for allosteric control by light or ligands. Nat. Protoc 14, 1863–1883 (2019). doi: 10.1038/s41596-019-0165-3; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCormick JW, Pincus D, Resnekov O, Reynolds KA, Strategies for engineering and rewiring kinase regulation. Trends Biochem. Sci 45, 259–271 (2020). doi: 10.1016/j.tibs.2019.11.005; [DOI] [PubMed] [Google Scholar]
- 41.Gunasekaran K, Ma B, Nussinov R, Is allostery an intrinsic property of all dynamic proteins? Proteins 57, 433–443 (2004). doi: 10.1002/prot.20232; [DOI] [PubMed] [Google Scholar]
- 42.Hardy JA, Wells JA, Searching for new allosteric sites in enzymes. Curr. Opin. Struct. Biol 14, 706–715 (2004). doi: 10.1016/j.sbi.2004.10.009; [DOI] [PubMed] [Google Scholar]
- 43.Bowman GR, Bolin ER, Hart KM, Maguire BC, Marqusee S, Discovery of multiple hidden allosteric sites by combining Markov state models and experiments. Proc. Natl. Acad. Sci. U.S.A 112, 2734–2739 (2015). doi: 10.1073/pnas.1417811112; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sunden F, Peck A, Salzman J, Ressl S, Herschlag D, Extensive site-directed mutagenesis reveals interconnected functional units in the alkaline phosphatase active site. eLife 4, e06181 (2015). doi: 10.7554/eLife.06181; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carter PJ, Winter G, Wilkinson AJ, Fersht AR, The use of double mutants to detect structural changes in the active site of the tyrosyl-tRNA synthetase (Bacillus stearothermophilus). Cell 38, 835–840 (1984). doi: 10.1016/0092-8674(84)90278-2; [DOI] [PubMed] [Google Scholar]
- 46.Carter P, Wells JA, Dissecting the catalytic triad of a serine protease. Nature 332, 564–568 (1988). doi: 10.1038/332564a0; [DOI] [PubMed] [Google Scholar]
- 47.Herschlag D, Natarajan A, Fundamental challenges in mechanistic enzymology: Progress toward understanding the rate enhancements of enzymes. Biochemistry 52, 2050–2067 (2013). doi: 10.1021/bi4000113; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jencks WP, Mechanism of enzyme action. Annu. Rev. Biochem 32, 639–676 (1963). doi: 10.1146/annurev.bi.32.070163.003231; [DOI] [PubMed] [Google Scholar]
- 49.Fersht AR, Enzyme Structure and Mechanism (W.H. Freeman and Co., ed. 2, 1985). [Google Scholar]
- 50.Jencks WP, Binding energy, specificity, and enzymic catalysis: The Circe effect. Adv. Enzymol. Relat. Areas Mol. Biol 43, 219–410 (1975). doi: 10.1002/9780470122884.ch4; [DOI] [PubMed] [Google Scholar]
- 51.Andrews LD, Deng H, Herschlag D, Isotope-edited FTIR of alkaline phosphatase resolves paradoxical ligand binding properties and suggests a role for ground-state destabilization. J. Am. Chem. Soc 133, 11621–11631 (2011). doi: 10.1021/ja203370b; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Albery WJ, Knowles JR, Evolution of enzyme function and the development of catalytic efficiency. Biochemistry 15, 5631–5640 (1976). doi: 10.1021/bi00670a032; [DOI] [PubMed] [Google Scholar]
- 53.Menger FM, Analysis of ground-state and transition-state effects in enzyme catalysis. Biochemistry 31, 5368–5373 (1992). doi: 10.1021/bi00138a018; [DOI] [PubMed] [Google Scholar]
- 54.McCarter JD, Adam MJ, Withers SG, Binding energy and catalysis. Fluorinated and deoxygenated glycosides as mechanistic probes of Escherichia coli (lacZ) β-galactosidase. Biochem. J 286, 721–727 (1992). doi: 10.1042/bj2860721; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moore SA, Jencks WP, Formation of active site thiol esters of CoA transferase and the dependence of catalysis on specific binding interactions. J. Biol. Chem 257, 10893–10907 (1982). doi: 10.1016/S0021-9258(18)33908-5; [DOI] [PubMed] [Google Scholar]
- 56.Whitty A, Fierke CA, Jencks WP, Role of binding energy with coenzyme A in catalysis by 3-oxoacid coenzyme A transferase. Biochemistry 34, 11678–11689 (1995). doi: 10.1021/bi00037a005; [DOI] [PubMed] [Google Scholar]
- 57.Zhang Y-L et al. , Impaired transition state complementarity in the hydrolysis of O-arylphosphorothioates by protein-tyrosine phosphatases. Biochemistry 38, 12111–12123 (1999). doi: 10.1021/bi990836i; [DOI] [PubMed] [Google Scholar]
- 58.Hull WE, Halford SE, Gutfreund H, Sykes BD, Phosphorus-31 nuclear magnetic resonance study of alkaline phosphatase: The role of inorganic phosphate in limiting the enzyme turnover rate at alkaline pH. Biochemistry 15, 1547–1561 (1976). doi: 10.1021/bi00652a028; [DOI] [PubMed] [Google Scholar]
- 59.Wrenbeck EE, Azouz LR, Whitehead TA, Single-mutation fitness landscapes for an enzyme on multiple substrates reveal specificity is globally encoded. Nat. Commun 8, 15695 (2017). doi: 10.1038/ncomms15695; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Klesmith JR, Bacik J-P, Wrenbeck EE, Michalczyk R, Whitehead TA, Trade-offs between enzyme fitness and solubility illuminated by deep mutational scanning. Proc. Natl. Acad. Sci. U.S.A 114, 2265–2270 (2017). doi: 10.1073/pnas.1614437114; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bandaru P et al. , Deconstruction of the Ras switching cycle through saturation mutagenesis. eLife 6, e27810 (2017). doi: 10.7554/eLife.27810; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dobson CM, Protein folding and misfolding. Nature 426, 884–890 (2003). doi: 10.1038/nature02261; [DOI] [PubMed] [Google Scholar]
- 63.Faber MS, Wrenbeck EE, Azouz LR, Steiner PJ, Whitehead TA, Impact of in vivo protein folding probability on local fitness landscapes. Mol. Biol. Evol 36, 2764–2777 (2019). doi: 10.1093/molbev/msz184; [DOI] [PubMed] [Google Scholar]
- 64.Zhao V, Jacobs WM, Shakhnovich EI, Effect of protein structure on evolution of cotranslational folding. Biophys. J 119, 1123–1134 (2020). doi: 10.1016/j.bpj.2020.06.037; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Halabi N, Rivoire O, Leibler S, Ranganathan R, Protein sectors: Evolutionary units of three-dimensional structure. Cell 138, 774–786 (2009). doi: 10.1016/j.cell.2009.07.038; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rivoire O, Reynolds KA, Ranganathan R, Evolution-based functional decomposition of proteins. PLOS Comput. Biol 12, e1004817 (2016). doi: 10.1371/journal.pcbi.1004817; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Salinas VH, Ranganathan R, Coevolution-based inference of amino acid interactions underlying protein function. eLife 7, e34300 (2018). doi: 10.7554/eLife.34300; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Teşileanu T, Colwell LJ, Leibler S, Protein sectors: Statistical coupling analysis versus conservation. PLOS Comput. Biol 11, e1004091 (2015). doi: 10.1371/journal.pcbi.1004091; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Creixell P et al. , Hierarchical organization endows the kinase domain with regulatory plasticity. Cell Syst. 7, 371–383.e4 (2018). doi: 10.1016/j.cels.2018.08.008; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fordyce PM et al. , Basic leucine zipper transcription factor Hac1 binds DNA in two distinct modes as revealed by microfluidic analyses. Proc. Natl. Acad. Sci. U.S.A 109, E3084–E3093 (2012). doi: 10.1073/pnas.1212457109; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Le DD et al. , Comprehensive, high-resolution binding energy landscapes reveal context dependencies of transcription factor binding. Proc. Natl. Acad. Sci. U.S.A 115, E3702–E3711 (2018). doi: 10.1073/pnas.1715888115; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Aditham AK, Markin CJ, Mokhtari DA, DelRosso N, Fordyce PM, High-throughput affinity measurements of transcription factor and DNA mutations reveal affinity and specificity determinants. Cell Syst. 12, 112–127.e11 (2021). doi: 10.1016/j.cels.2020.11.012; [DOI] [PubMed] [Google Scholar]
- 73.Brower K et al. , An open-source, programmable pneumatic setup for operation and automated control of single- and multi-layer microfluidic devices. HardwareX 3, 117–134 (2018). doi: 10.1016/j.ohx.2017.10.001; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Markin CJ et al. , Revealing enzyme functional architecture via high-throughput microfluidic enzyme kinetics. Open Science Repository (2021); doi: 10.17605/OSF.IO/QRN3C [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.