

Short abstract
Expression of TNFα and IL‐10 is regulated differentially during SR‐A‐mediated macrophage adhesion via the production of PGE2.
Keywords: cyclooxygenase, inflammation, PGE2, phospholipase A2
Abstract
Inflammation is associated with modification of the extracellular environment, changes in cytokine expression, and the accumulation of immune cells. Such modifications create ligands that support SR‐A‐mediated macrophage adhesion and retention. This may be particularly important in settings, such as atherosclerosis and diabetes, as modified lipoproteins and gluc‐collagen are ligands for SR‐A. SR‐A‐mediated adhesion requires the PLA2‐dependent generation of AA and its metabolism by 12/15 LOX. In contrast, the inhibition of the COX‐dependent conversion of AA to PG had no effect on SR‐A‐mediated adhesion. In this study, macrophages were isolated from SR‐A+/+ and SR‐A−/− mice and plated on gluc‐collagen to test the hypothesis that COX‐derived PGs are produced during SR‐A‐mediated adhesion and regulate macrophage function. SR‐A‐mediated binding to gluc‐collagen induced a rapid but transient increase in PG production, which required the activation of PLA2 and Src kinase but not PI3K. SR‐A+/+ macrophages cultured on gluc‐collagen for 24 h secreted a similar amount of TNF‐α and 2.5‐fold more IL‐10 than SR‐A−/− macrophages. The inhibition of COX substantially increased TNF‐α production but reduced IL‐10 levels in SR‐A+/+ macrophages. These effects of COX inhibition were reversed by exogenous PGE2 and mimicked by specific antagonism of the EP4 receptor. Thus, in addition to the enhancement of macrophage adhesion, SR‐A binding to gluc‐collagen stimulates PG production, which in turn, differentially regulates the expression of inflammatory cytokines.
Abbreviations
- 12/15‐LOX
12/15‐lipoxygenase
- AA
arachidonic acid
- AACOCF3
arachidonyltrifluoro‐methyl ketone
- COX
cyclooxygenase
- ECM
extracellular matrix
- EP
prostaglandin E receptor
- gluc‐collagen
glucose‐modified collagen
- Gs
stimulatory G protein
- indomethacin
1‐(p‐chlorobenzoyl)‐5‐methoxy‐2‐methyl‐1H‐indole‐3‐acetic acid
- IP
prostacyclin receptor
- LOX
lipoxygenase
- MPM
mouse peritoneal macrophage
- NDGA
nordihydroguaiaretic acid
- PLA2
phospholipase A2
- PP2
4‐amino‐5‐(4‐chlorophenyl)‐7‐(t‐butyl)pyrazolo[3,4‐d]pyrimidine
- PP3
4‐amino‐7‐phenylpyrazol[3,4‐d]pyrimidine
- SR‐A
class A scavenger receptor
- TXA2/B2
thromboxane A2/B2
- WM
wortmannin
Introduction
Modifications of the ECM are characteristic of chronic inflammatory diseases, such as diabetes, atherosclerosis, and Alzheimer's disease. Such modifications create ligands that are recognized by a variety of cell‐surface pattern‐recognition receptors that modulate inflammation. SR‐As (CD204) mediate macrophage adhesion to a variety of modified ECM components, including modified and degraded collagens, β‐amyloid fibrils, and proteoglycans that are up‐regulated during inflammation [1, 2, 3, 4, 5–6]. SR‐A‐mediated adhesion is thought to promote granuloma formation and glomerular macrophage accumulation in diabetic animals [7, 8]. The production of SR‐A ligands, resulting from modification of ECM at sites of inflammation, and the subsequent accumulation of macrophages at these sites suggest a role for SR‐A‐mediated adhesion in chronic inflammatory disease.
The possibility that SR‐A may modulate macrophage function is supported by studies showing that the expression of inflammatory cytokines is altered in SR‐A−/− mice [7, 9, 10, 11–12]. For example, SR‐A−/− mice express more TNF‐α and less IL‐10 in a sterile peritonitis model [9]. Likewise, in a mouse model of myocardial infarction, SR‐A−/− mice displayed higher TNF‐α and less IL‐10 than the wild‐type control animals [10]. Such results suggest that SR‐A may suppress an inflammatory response. However, the signaling pathways that couple SR‐A to cytokine expression are not clear, and whether SR‐A‐mediated adhesion is linked specifically to cytokine regulation is not known.
Production of COX‐derived AA metabolites (i.e., PGs and TXA2) is an important mechanism for modulating inflammatory responses. In macrophages, PGE2 regulates the secretion of several cytokines, including TNF‐α, IL‐12, and IL‐10 [13, 14–15]. The possibility that SR‐A may regulate PG production is supported by results demonstrating that SR‐A‐dependent adhesion involves activation of a PLA2‐12/15‐LOX signaling pathway [16]. Although COX activity was not required for SR‐A‐mediated adhesion, the possibility that COX‐derived products were generated and coupled to other macrophage functions, such as cytokine secretion, was not assessed.
The aim of this study was to determine whether SR‐A binding to gluc‐collagen promotes PG production and regulates cytokine expression. We studied resident peritoneal macrophages isolated from SR‐A+/+ or SR‐A−/− mice that were cultured on gluc‐collagen. Our results show that SR‐A binding to gluc‐collagen results in the PLA2‐catalyzed release of AA and the production of COX‐derived PGs. We further show that the PGE2 produced acts via the EP4 receptor to differentially modulate TNF‐α and IL‐10 expression.
MATERIALS AND METHODS
Chemicals
AACOCF3, PP2, and PP3 were purchased from Calbiochem (La Jolla, CA, USA). Indomethacin and AA were from Sigma Chemical (St. Louis, MO, USA). WM was from Enzo Life Sciences (Farmingdale, NY, USA). NDGA, PGE2, cicaprost, U46619, GW627368X, AH6809, and CAY10441 were purchased from Cayman Chemical (Ann Arbor, MI, USA).
Glucose modification of collagen
Collagen Bornstein and Traub Type I from calf skin, supplied as a 1 mg/ml solution in 0.1 M acetic acid (Sigma Chemical), were mixed with 200 mM glucose and incubated for 6 weeks at 4°C. The solution was then dialyzed against PBS and stored at 4°C until used. The conditions used for glucose modification of collagen were optimized to support SR‐A‐dependent macrophage adhesion. The ability to increase SR‐A‐dependent adhesion was used as a bioassay to confirm collagen modification in each preparation of gluc‐collagen.
Cell isolation and culture
Resident MPMs were harvested from wild‐type C57Bl/6 mice (The Jackson Laboratory, Bar Harbor, ME, USA) or SR‐A−/− on a C57Bl/6 background (The Jackson Laboratory) via peritoneal lavage with ice‐cold sterile saline and cultured in DMEM GlutaMax (Gibco‐BRL, Grand Island, NY, USA) containing FBS (10% v/v), penicillin, and streptomycin, as described previously [17]. Animal care and use for all procedures were done according to protocols reviewed and approved by the Institutional Animal Care and Use Committee at the University of Arkansas for Medical Sciences.
Cell adhesion assays
Cell adhesion assays were performed as described previously [16, 18]. In brief, MPMs were treated as described in individual figure legends and then plated for 2 h in Lab‐Tek slides (Nalge Nunc International, Naperville, IL, USA), precoated at 4°C for 16 h with native (unmodified) collagen type I or gluc‐collagen type I (30 μg/cm2). Trypan blue exclusion was used to confirm that treatments did not affect cell viability before plating. Nonadherent cells were removed by washing with PBS, and then adhered macrophages were fixed, permeabilized, and stained with Alexa Fluor568‐conjugated phalloidin and DAPI (Molecular Probes, Eugene, OR, USA). Digital images were captured and cell‐surface area quantified by use of AxioVision software (Carl Zeiss GmbH, Jena, Germany).
PG and cytokine ELISA
Freshly isolated MPMs were treated as described in individual figure legends and then cultured on tissue‐culture plates (Corning, Corning, NY, USA), precoated with collagen type I or gluc‐collagen type I (40 μg/cm2). Cells (∼106) were cultured in 1 ml complete medium for PG experiments and in serum‐free medium for cytokine production. Unless otherwise indicated in figure legends, the culture media were removed after 1 h and assayed by ELISA for different COX‐derived AA metabolites (Cayman Chemical) or after 24 h for cytokine ELISA (R&D Systems, Minneapolis, MN, USA, and Invitrogen, Grand Island, NY, USA), according to the manufacturersˈ protocols. Cell protein was quantified by bicinchoninic acid assay (Pierce, Rockford, IL, USA) and the amount of PG/cytokine normalized to total cell protein. Protein yield (∼0.075 mg/106 cells) was not altered by any of the culture condition or treatments. Trypan blue exclusion was used to confirm that macrophage viability was not affected by any of the treatments or incubation conditions.
Statistical analysis
Experiments were repeated at least 3 times and significance among treatment groups determined by use of GraphPad Prism. Values with P < 0.05 were considered to be statistically significant.
RESULTS
SR‐A mediates macrophage adhesion to gluc‐collagen and requires activation of Src kinase, PI3K, and PLA2‐mediated AA metabolism
SR‐A‐mediated adhesion progresses from an attached, rounded state to a state in which the cells are firmly adhered and spread [16, 18, 19]. Thus, the extent to which SR‐A enhances macrophage spreading can be used as an index of SR‐A‐mediated macrophage adhesion. We reported previously that SR‐A‐dependent macrophage adhesion to malondialdehyde‐modified BSA requires activation of a Src (Lyn)‐PI3K pathway [18], the PLA2‐catalyzed hydrolysis of AA from membrane phospholipids, and the metabolism of AA by 12/15‐LOX but was independent of COX activity [16]. To assess SR‐A‐mediated adhesion to gluc‐collagen, resident MPMs were isolated from SR‐A+/+ or SR‐A−/− mice and plated for 2 h in wells coated with gluc‐collagen. Polymerized actin (F‐actin) was stained with phalloidin and cell‐surface area quantified as an index of cell adhesion. SR‐A+/+ macrophages exhibited enhanced spreading on gluc‐collagen compared with SR‐A−/− macrophages, which remained mostly rounded (Fig. 1A). Neither SR‐A+/+ nor SR‐A−/− macrophages spread well when allowed to adhere to native collagen but spread equally well on fibronectin (data not shown). To examine the signaling pathways involved in SR‐A‐mediated adhesion to gluc‐collagen, macrophages were preincubated with specific inhibitors before adhesion (Fig. 1B). The preincubation of macrophages with the Src kinase inhibitor PP2 (50 μM) but not its inactive homolog PP3 abolished the increased macrophage spreading on gluc‐collagen. Likewise, the pretreatment of macrophages with inhibitors of PI3K (WM, 200 nM), PLA2 (AACOCF3, 30 µM), or LOX (NDGA, 50 µM) inhibited SR‐A‐mediated macrophage spreading on gluc‐collagen. In contrast, macrophages spreading on gluc‐collagen were not inhibited by the COX inhibitor indomethacin (10 µM). These data confirm that SR‐A‐mediated macrophage adhesion to gluc‐collagen requires the activation of a Src‐PI3K signaling pathway and LOX‐derived but not COX‐derived products of AA metabolism.
Figure 1.
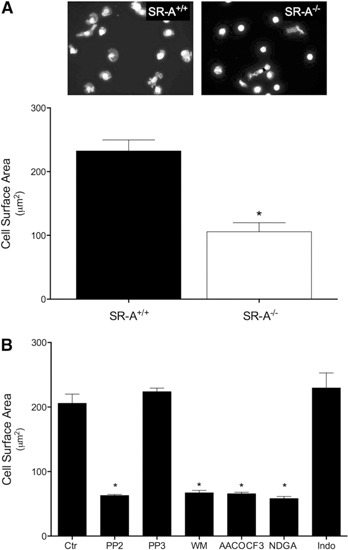
SR‐A‐mediated macrophage adhesion to gluc‐collagen requires activation of Src kinase, PI3K, and PLA2. (A) Resident MPMs isolated from SR‐A+/+ or SR‐A−/− mice were plated on gluc‐collagen‐coated slides. Cells were adhered for 2 h at 37°C, fixed, and stained with Alexa Fluor568‐conjugated phalloidin. Nuclei were stained with DAPI. Images were digitally captured, and the surface area of individual cells quantified. (B) MPMs isolated from SR‐A+/+ were pretreated as indicated with PP2 (50 µM), PP3 (50 µM), WM (200 nM), AACOCF3 (30 µM), NDGA (50 µM), or indomethacin (Indo; 10 µM) for 30 min in suspension and then plated on gluc‐collagen‐coated slides. Ctr, Control. Cells were stained and cell surface area quantified as described for A. The graphs depict the mean ± sem cell surface area determined in 3 separate experiments. Significant difference (*P < 0.05) from untreated SR‐A+/+ (control) cells.
SR‐A‐mediated macrophage adhesion is coupled to PG synthesis
Previous studies have shown that SR‐A‐dependent macrophage adhesion requires the activation of a Src (Lyn)‐PI3K pathway, the PLA2‐catalyzed hydrolysis of AA from membrane phospholipids, and the metabolism of AA by 12/15‐LOX [16, 18]. Although SR‐A‐mediated adhesion was shown to be independent of COX activity, these previous studies did not address whether COX‐derived products are produced and secreted during SR‐A‐mediated adhesion. To test this possibility, macrophages were isolated from wild‐type (SR‐A+/+) or SR‐A−/− mice and then cultured on gluc‐collagen‐coated plates. At various time‐points, the culture medium was collected and assayed for PG. As shown in Fig. 2A, PG production by SR‐A+/+ macrophages increased substantially during the first 2 h of adhesion. In contrast, PG production in SR‐A−/− macrophages was only elevated slightly. PG synthesis was fastest during the first hour of adhesion (Fig. 2B) and was abolished by indomethacin (Fig. 2B and C). As expected, pretreating macrophages with a PLA2 inhibitor, AACOCF3, abolished SR‐A‐mediated PG production (Fig. 2C). As SR‐A‐mediated macrophage adhesion involves the activation of Src kinases and PI3K [18], we investigated whether these kinases might also regulate PG production. As shown in Fig. 2C, PP2, a Src kinase inhibitor, but not WM, a PI3K inhibitor, blocked SR‐A‐mediated PG production (Fig. 2C). However, PP2 had no effect on AA‐stimulated PG production (Fig. 2D), indicating that inhibition of Src kinase prevents PG production by inhibiting AA production.
Figure 2.
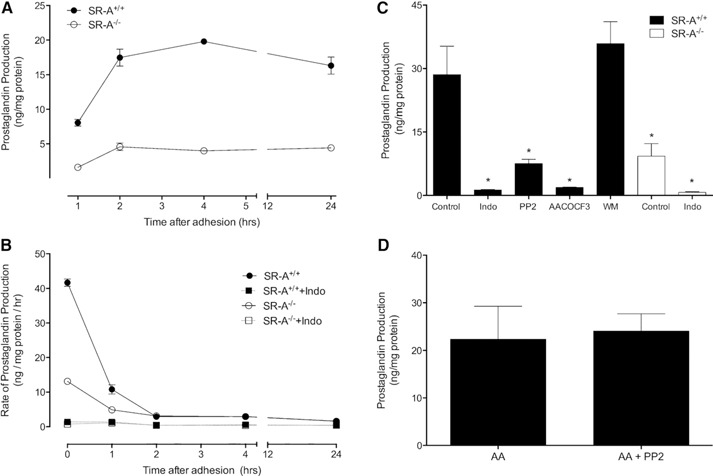
SR‐A‐mediated adhesion induces PG production by macrophages. MPMs isolated from SR‐A+/+ or SR‐A−/− mice were cultured on gluc‐collagen‐coated plates as indicated, and PG released into culture medium was measured by ELISA. (A) PG production was assessed at the indicated times. Data are from a representative experiment that was repeated 3 times with similar results. (B) MPMs were pretreated in the presence or absence of indomethacin (10 µM, 30 min) before plating on gluc‐collagen‐coated plates. At the indicated times, culture medium was exchanged for fresh medium and PG release assessed after 1 h. Data are from a representative experiment that was repeated 3 times with similar results. (C) MPMs were pretreated in the absence (Control) or presence of indomethacin (10 µM), PP2 (50 μM), AACOCF3 (30 µM), or WM (200 nM) for 30 min before plating. After 1 h, the culture medium was collected and assayed for PG. The graph depicts the mean ± sem of multiple experiments. Data were analyzed by 1‐way ANOVA and significant (P < 0.05) differences from control determined by use of a Dunnett's post hoc test. ∗Significant difference from untreated SR‐A+/+ (control) macrophages. (D) MPMs isolated from SR‐A+/+ mice were pretreated in the absence or presence of PP2 (50 μM) before adhesion. AA (30 μM) was added and cells adhered to gluc‐collagen plates for 1 h, and then PG production was assessed. The graph depicts the mean ± sem of 5 separate experiments. A significant difference (P < 0.05) was assessed by use of an unpaired t‐test.
SR‐A‐mediated binding to gluc‐collagen differentially regulates TNF‐α and IL‐10 release
Several studies associate SR‐A with altered cytokine production, particularly for TNF‐α and IL‐10 [7, 9, 10, 11–12]. Therefore, we examined whether SR‐A binding to gluc‐collagen would alter production of these cytokines. Resident peritoneal macrophages were isolated from SR‐A+/+ or SR‐A−/− mice and adhered to gluc‐collagen‐coated plates. After 24 h, the culture medium was collected and TNF‐α and IL‐10 measured by specific ELISA.
As shown in Fig. 3, a similar amount of TNF‐α (Fig. 3A) was detected in the culture medium of SR‐A+/+ and SR‐A−/− macrophages that were bound to gluc‐collagen for 24 h. In contrast, SR‐A+/+ macrophages produced a significantly greater amount of IL‐10 (Fig. 3B) than SR‐A−/− macrophages when bound to gluc‐collagen for 24 h. To determine if cytokine expression might be modulated by PG production, macrophages were pretreated with indomethacin to block PG synthesis before binding to gluc‐collagen. Indomethacin pretreatment significantly increased TNF‐α production by SR‐A+/+ and SR‐A−/− macrophages; however, the increase in TNF‐α secretion by indomethacin‐pretreated SR‐A+/+ macrophages was substantially higher than that of SR‐A−/− macrophages. The pretreatment of SR‐A+/+ macrophages with indomethacin decreased SR‐A‐dependent IL‐10 secretion to a level that was similar to that secreted by SR‐A−/− macrophages, which was not altered significantly by indomethacin. A similar pattern of regulation was observed by use of quantitative RT‐PCR to quantify cytokine gene expression (data not shown). These results suggest that PGs produced as a consequence of SR‐A‐mediated binding suppress TNF‐α expression but enhance that of IL‐10.
Figure 3.
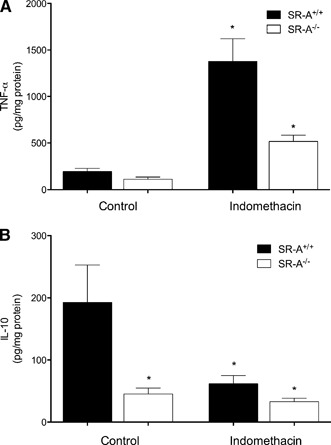
PGs produced during SR‐A‐mediated adhesion modulate TNF‐α and IL‐10 production. MPMs from SR‐A+/+ or SR‐A−/− mice were isolated and pretreated in the absence (Control) or presence of indomethacin (10 μM) for 30 min before plating on gluc‐collagen‐coated plates. After 24 h, the culture medium was removed and assayed for TNF‐α (A) or IL‐10 (B) by ELISA. The graph depicts the mean ± sem of at least 3 separate experiments. Log‐transformed data were analyzed by 1‐way ANOVA. Significant differences (*P < 0.05) from untreated SR‐A+/+ control macrophages by use of a Dunnett's post hoc test.
SR‐A‐mediated adhesion promotes production of COX‐derived AA metabolites
Previous studies indicate that macrophages secrete a variety of COX‐derived PGs [20, 21]. Thus, we used ELISAs to identify specific COX‐dependent AA products produced during SR‐A‐mediated adhesion to gluc‐collagen. As shown in Fig. 4, when adhered to gluc‐collagen for 1 h, SR‐A+/+ macrophages produced significantly more PGE2, PGI2 (measured as its stable metabolite 6‐keto‐PGF1 α), and TXA2 (measured as its stable metabolite TXB2) than SR‐A−/− macrophages. We could not detect any PGD2 or PGF2 α production (data not shown). As expected, production of the COX‐derived products was abolished by indomethacin.
Figure 4.
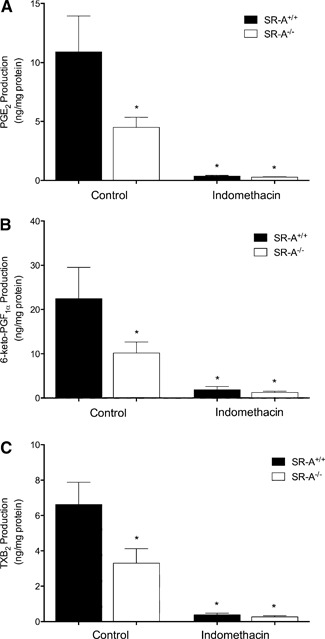
Multiple COX‐derived AA products are produced during SR‐A‐mediated macrophage adhesion. MPMs from SR‐A+/+ or SR‐A−/− mice were isolated and pretreated in the absence (Control) or presence of indomethacin (10 μM) for 30 min before plating on gluc‐collagen‐coated plates. After 1 h, the culture medium was removed and assayed by ELISA for PGE2 (A), the stable PGI2 metabolite 6‐keto‐PGF1 α (B), and the stable TXA2 metabolite TXB2 (C). The graph depicts the mean ± sem of at least 3 separate experiments. Log‐transformed data were analyzed by 1‐way ANOVA. Significant differences (*P < 0.05) from untreated SR‐A+/+ control macrophages by use of a Dunnett's post hoc test.
SR‐A‐dependent cytokine regulation is mediated by the EP4 receptor
Macrophages isolated from SR‐A+/+ mice were pretreated with indomethacin and then adhered to gluc‐collagen for 24 h in the presence or absence of exogenous PGE2, cicaprost (a stable PGI2 analog), or U46619 (a stable TXA2 analog). As shown in Fig. 5A, the addition of PGE2 or cicaprost completely reversed the enhancing effect of indomethacin on TNF‐α production. In contrast, augmented TNF‐α production, in the presence of indomethacin, was not altered by the addition of U46619. Likewise, when added to indomethacin‐pretreated macrophages, PGE2 and cicaprost restored IL‐10 production to levels obtained in untreated cells (Fig. 5B). U46619 did not alter the inhibitory effect of indomethacin on IL‐10 production.
Figure 5.
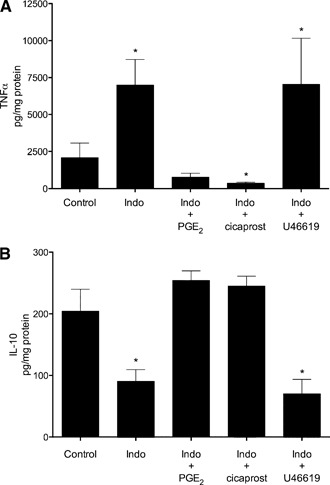
Exogenous PG agonists differentially regulate cytokine production during SR‐A‐mediated macrophage adhesion. MPMs from SR‐A+/+ mice were isolated and pretreated in the absence (Control) or presence of indomethacin (10 μM) for 30 min. As indicated, exogenous PGE2 (0.1 μM), the stable PGI2 analog cicaprost (0.1 μM), or the stable TXA2 analog U46619 (0.1 μM) was added and cells plated on gluc‐collagen‐coated plates. After 24 h, the culture medium was removed and assayed for TNF‐α (A) or IL‐10 (B). The graph depicts the mean ± sem of at least 3 separate experiments. Data were analyzed by 1‐way ANOVA. Significant differences (*P < 0.05) from untreated SR‐A+/+ control macrophages by use of a Dunnett's post hoc test.
The results presented in Fig. 4 indicate that PGE2 and/or PGI2 but not TXA2 can modulate TNF‐α and IL‐10 expression, resulting from SR‐A binding to gluc‐collagen. The cellular effects of PGE2 are mediated by binding to G protein‐coupled EP receptors, of which there are 4 isoforms (EP1, EP2, EP3, and EP4). PGI2 mediates its effects via the G protein‐coupled IP receptor. To examine the coupling of PGE2 and PGI2 with modulating cytokine production during SR‐A‐mediated adhesion, macrophages were pretreated with specific antagonists to EP4 receptors (GW627368X), EP1–3 receptors (AH6809), or IP receptors (CAY10441) before adhering to gluc‐collagen. As shown in Fig. 6, the preincubation of macrophages with GW627368X augmented TNF‐α production (Fig. 6A) and inhibited IL‐10 production (Fig. 6B) to the same extent as indomethacin. In contrast, preincubation with AH6809 or CAY10441 had no effect on TNF‐α or IL‐10 production. To confirm that CAY10441 was blocking IP receptor activation, we demonstrated that the effects of cicaprost were inhibited by CAY10441 (data not shown). These results indicate that the PG‐mediated modulation of cytokine release during SR‐A‐dependent macrophage binding to gluc‐collagen is mediated by PGE2 acting on EP4 receptors.
Figure 6.
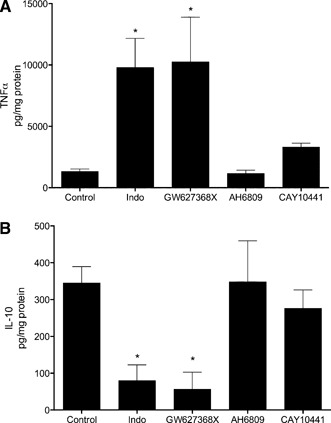
PG antagonists differentially regulate cytokine production during SR‐A‐mediated macrophage adhesion. MPMs from SR‐A+/+ mice were isolated and left untreated (Control) or pretreated in the presence of indomethacin (10 μM) for 30 min or for 5 min with GW627368X (10 μM), AH6809 (10 μM), or CAY10441 (10 μM). Cells were then plated on gluc‐collagen‐coated plates, and after 24 h, the culture medium was removed and assayed for TNF‐α (A) or IL‐10 (B). The graph depicts the mean ± sem of at least 3 separate experiments. Data were analyzed by 1‐way ANOVA. Significant differences (*P < 0.05) from untreated control macrophages, determined by use of a Dunnett's post hoc test.
DISCUSSION
SR‐A is a pattern recognition receptor that recognizes a variety of ligands, and several studies have documented the involvement of intracellular signaling cascades in regulating SR‐A function [16, 17, 18–19, 22, 23, 24, 25–26]. For example, SR‐A‐mediated adhesion involves the activation of a Src kinase (Lyn)‐PI3K‐paxillin and a PLA2‐12/15‐LOX‐Cdc42/Rac signaling pathway [16, 18]. Although increased macrophage adhesion to an SR‐A ligand is independent of COX activation, the results shown in Fig. 1 demonstrate that the COX‐derived products are formed as a consequence of SR‐A binding to gluc‐collagen. In addition, the finding that inhibition of Src kinases (e.g., Lyn) abolished SR‐A‐mediated but not exogenous AA‐mediated PG production indicates that Src kinase is activated upstream of PLA2. Together with our previous finding that Lyn activation is required for PI3K activation and the current finding that PI3K inhibition had no effect on PG production, our results indicate that the 2 signaling pathways diverge downstream of the Src kinase (e.g., Lyn).
Studies that compare SR‐A+/+ and SR‐A−/− mice indicate an important role for SR‐A in regulating cytokine expression in settings of diabetic nephropathy [7], sterile peritonitis [9], myocardial infarction [10], and microbial infections [11, 12]. The mechanism(s) that couple SR‐A to cytokine expression and macrophage activation have not been defined. Our results indicate that SR‐A differentially modulates TNF‐α and IL‐10 expression by promoting the release of PGE2, which in turn, activates EP4 receptors in an autocrine/paracrine manner. Details of how EP4 receptors, which are coupled to Gs and adenylyl cyclase activation, regulate TNF‐α and IL‐10 expression are not defined, and it is possible that these cytokines regulate each others’ expression. The ability of SR‐A to regulate cytokine expression by increasing PGE2 production has important implications, as it was recently shown that genetic deletion of EP4, specifically in hematopoietic cells, enhances inflammation and increases atherosclerosis and abdominal aortic aneurysm formation in mice [27, 28]. Thus, the stimulation of PGE2 production represents a distinct mechanism by which SR‐A may modulate macrophage function and an inflammatory response.
In addition to PGE2, we detected PGI2 and TXA2 production during SR‐A‐mediated adhesion. The stable IP receptor agonist cicaprost completely reversed the effects of indomethacin on macrophage cytokine production during SR‐A‐mediated adhesion, indicating the presence of functional IP receptors. However, the high‐affinity IP receptor‐specific antagonist CAY10441 did not alter cytokine production, indicating that the concentration of endogenously produced PGI2 is not sufficient to elicit this effect, perhaps as a result of the instability of this PG. In contrast to PGE2 and PGI2, the addition of a stable TXA2 analog U46619 did not alter cytokine production. This may reflect a lack of TXA2 receptor expression and/or that TXA2 receptors transduce signals that do not regulate cytokine expression in macrophages. However, PGI2 and TXA2 have potent effects in vascular and blood cells (e.g., platelets), and their release during SR‐A‐mediated macrophage adhesion may have pathophysiological effects via their action on other cell types.
Overall, our results identify a novel role for PG production and autocrine/ paracrine signaling in differentially coupling SR‐A to cytokine expression during macrophage adhesion. Together with previous studies, our results define a model (Fig. 7) by which SR‐A enhances macrophage adhesion and modulates the expression of inflammatory cytokines. Specifically, SR‐A binding to a modified matrix promotes activation of a Src kinase (Lyn) and the subsequent activation of PI3K [18]. Activation of Src kinase (Lyn) is also coupled to PLA2‐mediated AA release. AA is metabolized by 12/15‐LOX to enhance macrophage adhesion [16] and by COX to promote PG production and regulate cytokine expression. An important implication of our results is that in diseases, such as diabetes and atherosclerosis, SR‐A‐mediated adhesion might modulate local inflammation by increasing macrophage retention, stimulating the production of COX‐derived PGs, and differentially regulating the expression of inflammatory cytokines.
Figure 7.
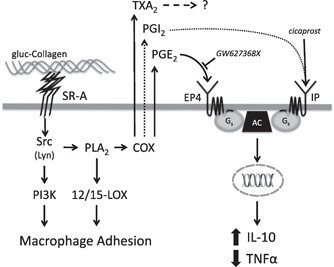
The role of PG in SR‐A‐mediated signaling during macrophage adhesion. SR‐A‐mediated macrophage adhesion involves the activation of a Src kinase, PI3K, PLA2, and 12/15‐LOX (Fig. 1 and refs. [16, 18]). Previous studies show that the Src kinase Lyn mediates the activation of PI3K during SR‐A‐mediated adhesion [18]. The current results (Fig. 2) indicate that PLA2 is also activated by a Src kinase (e.g., Lyn) and that COX‐derived products (PGE2, PGI2, and TXA2) are produced following the PLA2‐dependent generation of AA (Fig. 4). PGE2 suppresses TNF‐α and promotes IL‐10 production via binding to the EP4 receptor (Figs. 5 and 6), which elicits responses via Gs and adenylyl cyclase (AC). Although activation of the Gs‐coupled IP receptor can mediate similar effects on cytokine production (dotted lined), the amount of PGI2 produced during SR‐A‐mediated macrophage adhesion is not sufficient to elicit these responses. TXA2, which is also produced, has no effect on cytokine production during SR‐A‐mediated macrophage adhesion.
AUTHORSHIP
D.M.N., S.V., and B.H. contributed to performing and analyzing experiments and preparing the manuscript. J.W., T.K., and S.R.P. participated in designing experiments, interpreting results, and preparing the manuscript.
DISCLOSURES
The authors have no conflicts of interest to disclose.
ACKNOWLEDGMENTS
This work was supported by grants from the National Heart, Lung, and Blood Institute U.S. National Institutes of Health (R01HL089588) and the Sturgis Charitable Trust (to S.R.P.) and an American Heart Association predoctoral fellowship (to S.V.). The authors thank Maria Mercado, Jessica Webber, Amir Mortazavi, and Jacqueline Post for technical assistance. The authors also thank Drs. Ginell Post and Anna Mazur for critical reading of the manuscript.
REFERENCES
- 1.Gowen, B.B., Borg, T.K., Ghaffar, A., Mayer, E.P. (2000) Selective adhesion of macrophages to denatured forms of type I collagen is mediated by scavenger receptors. Matrix Biol. 19, 61–71. [DOI] [PubMed] [Google Scholar]
- 2.El Khoury, J., Hickman, S.E., Thomas, C.A., Cao, L., Silverstein, S.C., Loike, J.D. (1996) Scavenger receptor‐mediated adhesion of microglia to beta‐amyloid fibrils. Nature 382, 716–719. [DOI] [PubMed] [Google Scholar]
- 3.El Khoury, J., Thomas, C.A., Loike, J.D., Hickman, S.E., Cao, L., Silverstein, S.C. (1994) Macrophages adhere to glucose‐modified basement membrane collagen IV via their scavenger receptors. J. Biol. Chem. 269, 10197–10200. [PubMed] [Google Scholar]
- 4.Kirkham, P.A., Spooner, G., Ffoulkes‐Jones, C., Calvez, R. (2003) Cigarette smoke triggers macrophage adhesion and activation: role of lipid peroxidation products and scavenger receptor. Free Radic. Biol. Med. 35, 697–710. [DOI] [PubMed] [Google Scholar]
- 5.Santiago‐García, J., Mas‐Oliva, J., Innerarity, T.L., Pitas, R.E. (2001) Secreted forms of the amyloid‐beta precursor protein are ligands for the class A scavenger receptor. J. Biol. Chem. 276, 30655–30661. [DOI] [PubMed] [Google Scholar]
- 6.Santiago‐García, J., Kodama, T., Pitas, R.E. (2003) The class A scavenger receptor binds to proteoglycans and mediates adhesion of macrophages to the extracellular matrix. J. Biol. Chem. 278, 6942–6946. [DOI] [PubMed] [Google Scholar]
- 7.Horiuchi, S., Unno, Y., Usui, H., Shikata, K., Takaki, K., Koito, W., Sakamoto, Y., Nagai, R., Makino, K., Sasao, A., Wada, J., Makino, H. (2005) Pathological roles of advanced glycation end product receptors SR‐A and CD36. Ann. N. Y. Acad. Sci. 1043, 671–675. [DOI] [PubMed] [Google Scholar]
- 8.Daugherty, A., Kosswig, N., Cornicelli, J.A., Whitman, S.C., Wolle, S., Rateri, D.L. (2001) Macrophage‐specific expression of class A scavenger receptors enhances granuloma formation in the absence of increased lipid deposition. J. Lipid Res. 42, 1049–1055. [PubMed] [Google Scholar]
- 9.Cotena, A., Gordon, S., Platt, N. (2004) The class A macrophage scavenger receptor attenuates CXC chemokine production and the early infiltration of neutrophils in sterile peritonitis. J. Immunol. 173, 6427–6432. [DOI] [PubMed] [Google Scholar]
- 10.Tsujita, K., Kaikita, K., Hayasaki, T., Honda, T., Kobayashi, H., Sakashita, N., Suzuki, H., Kodama, T., Ogawa, H., Takeya, M. (2007) Targeted deletion of class A macrophage scavenger receptor increases the risk of cardiac rupture after experimental myocardial infarction. Circulation 115, 1904–1911. [DOI] [PubMed] [Google Scholar]
- 11.Haworth, R., Platt, N., Keshav, S., Hughes, D., Darley, E., Suzuki, H., Kurihara, Y., Kodama, T., Gordon, S. (1997) The macrophage scavenger receptor type A is expressed by activated macrophages and protects the host against lethal endotoxic shock. J. Exp. Med. 186, 1431–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Plüddemann, A., Hoe, J.C., Makepeace, K., Moxon, E.R., Gordon, S. (2009) The macrophage scavenger receptor A is host‐protective in experimental meningococcal septicaemia. PLoS Pathog. 5, e1000297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van der Pouw Kraan, T.C., Boeije, L.C., Smeenk, R.J., Wijdenes, J., Aarden, L.A. (1995) Prostaglandin‐E2 is a potent inhibitor of human interleukin 12 production. J. Exp. Med. 181, 775–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takayama, K., García‐Cardena, G., Sukhova, G.K., Comander, J., Gimbrone, Jr., M.A., Libby, P. (2002) Prostaglandin E2 suppresses chemokine production in human macrophages through the EP4 receptor. J. Biol. Chem. 277, 44147–44154. [DOI] [PubMed] [Google Scholar]
- 15.Suram, S., Silveira, L.J., Mahaffey, S., Brown, G.D., Bonventre, J.V., Williams, D.L., Gow, N.A. R., Bratton, D.L., Murphy, R.C., Leslie, C.C. (2013) Cytosolic phospholipase A(2)α and eicosanoids regulate expression of genes in macrophages involved in host defense and inflammation. PLoS ONE 8, e69002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nikolic, D.M., Gong, M.C., Turk, J., Post, S.R. (2007) Class A scavenger receptor‐mediated macrophage adhesion requires coupling of calcium‐independent phospholipase A(2) and 12/15‐lipoxygenase to Rac and Cdc42 activation. J. Biol. Chem. 282, 33405–33411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whitman, S.C., Daugherty, A., Post, S.R. (2000) Regulation of acetylated low density lipoprotein uptake in macrophages by pertussis toxin‐sensitive G proteins. J. Lipid Res. 41, 807–813. [PubMed] [Google Scholar]
- 18.Nikolic, D.M., Cholewa, J., Gass, C., Gong, M.C., Post, S.R. (2007) Class A scavenger receptor‐mediated cell adhesion requires the sequential activation of Lyn and PI3‐kinase. Am. J. Physiol. Cell Physiol. 292, C1450‐C1458. [DOI] [PubMed] [Google Scholar]
- 19.Post, S.R., Gass, C., Rice, S., Nikolic, D., Crump, H., Post, G.R. (2002) Class A scavenger receptors mediate cell adhesion via activation of G(i/o) and formation of focal adhesion complexes. J. Lipid Res. 43, 1829–1836. [DOI] [PubMed] [Google Scholar]
- 20.Stenson, W.F., Parker, C.W. (1980) Prostaglandins, macrophages, and immunity. J. Immunol. 125, 1–5. [PubMed] [Google Scholar]
- 21.Dalli, J., Serhan, C.N. (2012) Specific lipid mediator signatures of human phagocytes: microparticles stimulate macrophage efferocytosis and pro‐resolving mediators. Blood 120, e60–e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miki, S., Tsukada, S., Nakamura, Y., Aimoto, S., Hojo, H., Sato, B., Yamamoto, M., Miki, Y. (1996) Functional and possible physical association of scavenger receptor with cytoplasmic tyrosine kinase Lyn in monocytic THP‐1‐derived macrophages. FEBS Lett. 399, 241–244. [DOI] [PubMed] [Google Scholar]
- 23.Coller, S.P., Paulnock, D.M. (2001) Signaling pathways initiated in macrophages after engagement of type A scavenger receptors. J. Leukoc. Biol. 70, 142–148. [PubMed] [Google Scholar]
- 24.Fong, L.G., Le, D. (1999) The processing of ligands by the class A scavenger receptor is dependent on signal information located in the cytoplasmic domain. J. Biol. Chem. 274, 36808–36816. [DOI] [PubMed] [Google Scholar]
- 25.Hsu, H.Y., Hajjar, D.P., Khan, K.M., Falcone, D.J. (1998) Ligand binding to macrophage scavenger receptor‐A induces urokinase‐type plasminogen activator expression by a protein kinase‐dependent signaling pathway. J. Biol. Chem. 273, 1240–1246. [DOI] [PubMed] [Google Scholar]
- 26.Pollaud‐Cherion, C., Vandaele, J., Quartulli, F., Séguélas, M.H., Decerprit, J., Pipy, B. (1998) Involvement of calcium and arachidonate metabolism in acetylated‐low‐density‐lipoprotein‐stimulated tumor‐necrosis‐factor‐alpha production by rat peritoneal macrophages. Eur. J. Biochem. 253, 345–353. [DOI] [PubMed] [Google Scholar]
- 27.Tang, E.H. C., Shimizu, K., Christen, T., Rocha, V.Z., Shvartz, E., Tesmenitsky, Y., Sukhova, G., Shi, G.‐P., Libby, P. (2011) Lack of EP4 receptors on bone marrow‐derived cells enhances inflammation in atherosclerotic lesions. Cardiovasc. Res. 89, 234–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tang, E.H., Shvartz, E., Shimizu, K., Rocha, V.Z., Zheng, C., Fukuda, D., Shi, G.P., Sukhova, G., Libby, P. (2011) Deletion of EP4 on bone marrow‐derived cells enhances inflammation and angiotensin II‐induced abdominal aortic aneurysm formation. Arterioscler. Thromb. Vasc. Biol. 31, 261–269. [DOI] [PMC free article] [PubMed] [Google Scholar]