

Abstract
Sodium-glucose cotransporter-2 (SGLT2) inhibitors are drugs designed to lower plasma glucose concentration by inhibiting Na+-glucose–coupled transport in the proximal tubule. Clinical trials demonstrate these drugs have favorable effects on cardiovascular outcomes to include slowing the progression of CKD. Although most patients tolerate these drugs, a potential complication is development of ketoacidosis, often with a normal or only a minimally elevated plasma glucose concentration. Inhibition of sodium-glucose cotransporter-2 in the proximal tubule alters kidney ATP turnover so that filtered ketoacids are preferentially excreted as Na+ or K+ salts, leading to indirect loss of bicarbonate from the body and systemic acidosis under conditions of increased ketogenesis. Risk factors include reductions in insulin dose, increased insulin demand, metabolic stress, low carbohydrate intake, women, and latent autoimmune diabetes of adulthood. The lack of hyperglycemia and nonspecific symptoms of ketoacidosis can lead to delays in diagnosis. Treatment strategies and various precautions are discussed that can decrease the likelihood of this complication.
Keywords: euglycemic ketoacidosis, sodium glucose co-transporter inhibitor
Introduction
Underappreciated is the role the kidney plays in ketoacidosis defined as a primary decrease in plasma bicarbonate concentration resulting from increased ketoacids. Diabetic ketoacidosis (DKA) is a common cause of this acid-base disturbance and is accompanied by plasma glucose concentration >250 mg/dl, which is a criterion for diagnosis. There are patients with diabetes mellitus who develop ketoacidosis with less severe hyperglycemia, often <200 mg/dl. This form of ketoacidosis is euglycemic ketoacidosis and is a recognized complication of sodium-glucose cotransporter-2 inhibitors (SGLT2is). There are new data about the benefits of SGLT2i in individuals with kidney disorders; therefore, here we remind the clinician of alterations in kidney function induced by these drugs that increase the risk of ketoacidosis. We highlight unique risk factors and treatments of this disorder in individuals with kidney disease.
Risk of Ketoacidosis with Sodium-Glucose Cotransporter-2 Inhibitor
SGLT2is are drugs developed to reduce blood glucose levels in patients with diabetes mellitus. Randomized controlled trials demonstrate risk reductions for myocardial infarction, heart failure, kidney disease progression, and cardiovascular mortality, which likely will increase their use (1 –5). In 2015, the Food and Drug Administration (FDA) issued a safety warning on the risk of DKA (6). The frequency of ketoacidosis in clinical trials was small in patients with type 2 diabetes mellitus, but occurred in 4%–6% of patients when inhibitors were used in combination with insulin in patients with type 1 diabetes mellitus (7 –9). In the Kidney Disease Improving Global Outcomes 2020 clinical practice guideline for management of patients with diabetes and CKD, the risk of this disorder is described as generally a rare event in patients with type 2 diabetes mellitus (10). Reports examining postmarketing use suggest that the frequency of this disorder is much greater in real-world practice. An analysis of the FDA Adverse Event Reporting System found a seven‐fold higher risk of acidosis in treated patients with type 2 diabetes mellitus when compared with dipeptidyl peptidase-4 inhibitor therapy (11). This risk is approximately two- to four-fold greater than the numerical imbalance reported in clinical trials of canagliflozin and empagliflozin. Of those with ketoacidosis, euglycemia was present in 71% of the patients. More recently, a three-fold increase in the risk of DKA was found in a population cohort study of over 350,000 adults with type 2 diabetes mellitus compared with those treated with dipeptidyl peptidase-4 inhibitor (12). Importantly, fatality rates of this complication with SGLT2i therapy are greater when compared with all patients with DKA (13).
Increased Ketogenesis with Sodium-Glucose Cotransporter-2 Inhibitor
SGLT2i lowers plasma glucose by pharmacologically forcing excretion of glucose into the urine. The sodium-glucose cotransporter-2 (SGLT2) is located in the early (S1) proximal tubule and is responsible for reabsorption of 80%–90% of filtered glucose (14). SGLT1 is located in the more distal part of the proximal tubule (S2/S3) and is responsible for the reabsorption of the remaining 10%–20% of filtered glucose. Inhibition of these transporters reduces the transport maximum for glucose reabsorption, leading to glycosuria and reductions in plasma glucose concentrations. Decreased plasma glucose concentration signals a fall in insulin levels and causes plasma glucagon levels to increase due to a paracrine effect induced by diminished inhibition of insulin on glucagon release in the pancreas. In addition, inhibition of SGLT2 on pancreatic α-cells triggers release of glucagon. Glucagon further suppresses insulin by promoting hepatic release of kisspeptin-1 that, in turn, suppresses glucose-stimulated insulin secretion (15). The reduction in plasma insulin and glucose levels decreases carbohydrate oxidation and shifts metabolism toward lipid oxidation fueled by increased circulating fatty acids and lipolysis. Changes in fuel preference are detected by measurable drops in the respiratory quotient following initiation of SGLT2i therapy (16). By way of background, the respiratory quotient is a dimensionless number used in calculations of basal metabolic rate when estimated from carbon dioxide production. Greater amounts of fatty acid metabolism lower the quotient toward 0.7, whereas a shift to carbohydrate metabolism raises the quotient to values more than one.
SGLT2i causes daily glucose losses of 50–100 g corresponding to 200–400 kcal/d, which represent up to half of estimated carbohydrate intake in men and even higher amounts in women (17). SGLT2i therapy leads to weight loss in both rodents and humans in association with reductions in visceral and subcutaneous fat mass as measured by dual-energy x-ray absorptiometry (18). In rodent models, SGLT2is increase energy expenditure with activation and recruitment of brown and beige adipocytes, respectively (19). Caloric deficits due to glucose loss in the urine combined with enhanced lipid oxidation and increases in energy expenditure account for reductions in fat mass (19,20).
The combination of increased lipolysis and a reduced insulin-glucagon ratio leads to increased hepatic production of ketoacids. Increased production of acetyl-CoA from fatty acid β-oxidation is preferentially directed toward ketoacid production because glucagon stimulates and insulin inhibits the rate-limiting enzyme for this pathway (hydroxylmethylglutaryl-CoA). This pattern is similar to what occurs with fasting, except changes following SGLT2i are more rapid. In one study, fasting plasma ketone concentrations increased four-fold over baseline in the first 2 weeks of therapy with dapaglifozin (16). This shift to increased fat oxidation and ketogenesis also occurs in SGLT2i-treated individuals without diabetes mellitus, but to a lesser extent (21). Despite measurable increases in plasma ketone levels in treated patients, development of ketoacidosis is unusual. Those with the greatest increase in β-hydroxybutyrate levels have lower fasting and postmeal insulin levels and impaired β-cell insulin sensitivity when compared with subjects in the lower quartiles, suggesting the amount of endogenous insulin secretion reserve may factor into the magnitude of ketogenic response (22). As discussed below, factors leading to either a brisk reduction in exogenous insulin dose or inadequate endogenous insulin secretion predispose to overt ketoacidosis.
Sodium-Glucose Cotransporter-2 Inhibitor Effects on Kidney Function Predisposing to Ketoacidosis
Although changes in insulin and glucagon levels are central to increased production of ketoacids, SGLT2is further increase the risk of overt ketoacidosis by altering ATP turnover in the proximal tubule (Figure 1). Na+-coupled glucose reabsorption is an example of secondary active transport where ATP hydrolysis fuels activity of the basolateral Na+-K+-ATPase to maintain a low intracellular Na+ concentration and secondarily provide a favorable inward gradient for Na+-coupled glucose transport across the apical membrane. This pathway normally reabsorbs approximately 4%–6% of total filtered Na+ (22,23). In patients with diabetes mellitus, the threshold for glucose excretion is increased from 150 to 250 mg/dl due to glucose-mediated hypertrophy of the proximal tubule and increased expression of SGLT2 (24,25). It is estimated these changes lead to a doubling in percentage of filtered Na+ reabsorbed by this segment and a 50% increase in daily kidney ATP turnover (22,26).
Figure 1.
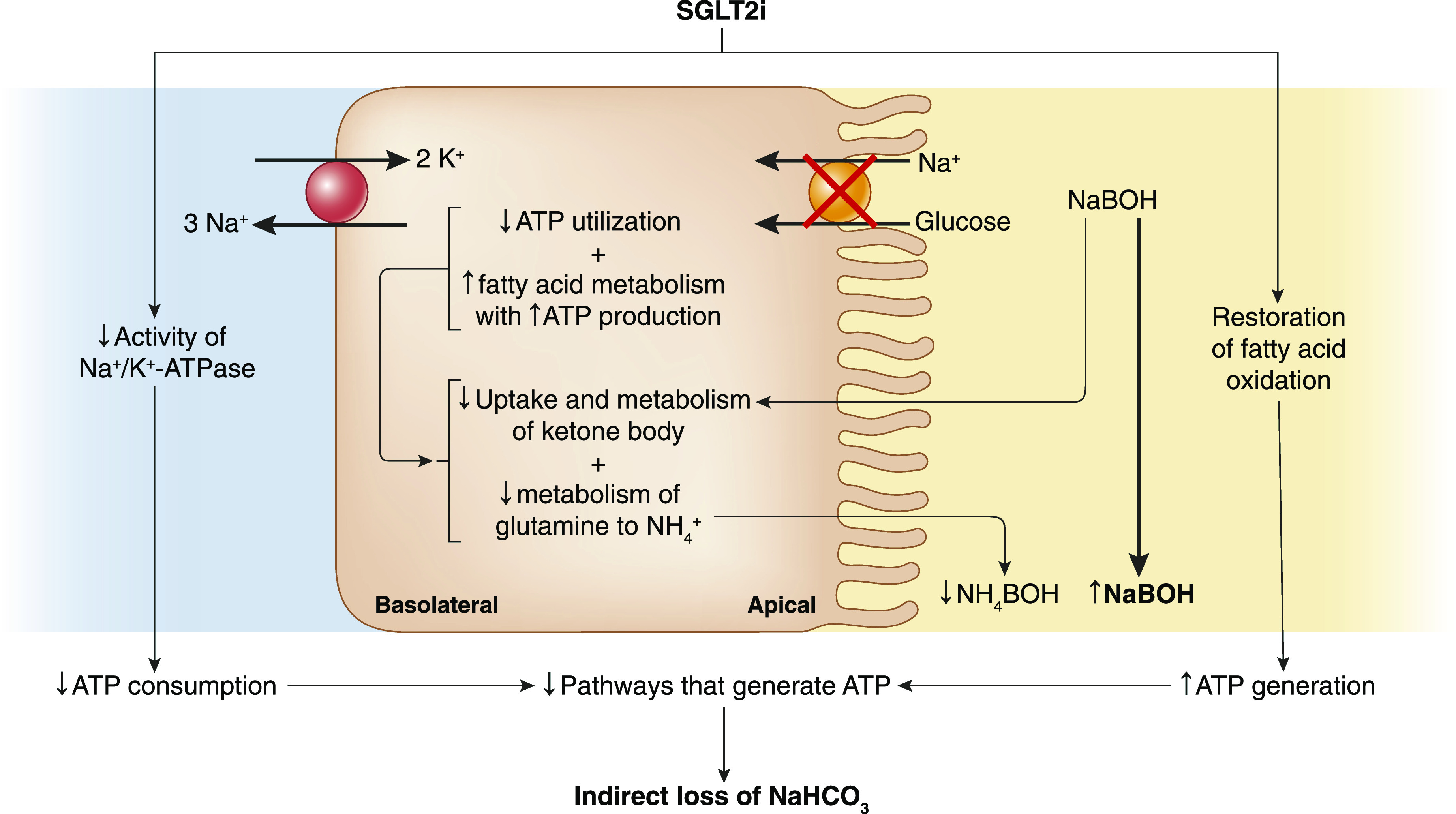
Alterations in ATP turnover predispose to systemic acidosis following sodium-glucose cotransporter-2 inhibitor (SGLT2i) administration. Decreased Na+ entry into the proximal tubular cell following inhibition of sodium-glucose cotransporter-2 decreases consumption of ATP by the Na+-K+-ATPase pump. SGLT2i also restores fatty acid oxidation, which delivers more energy per gram than glucose. The combined effect of reduced consumption along with increased generation of ATP decreases activity of other pathways that normally generate ATP to include ammoniagenesis and uptake and oxidation of filtered ketone bodies. These changes predispose to urinary loss of filtered ketone bodies as Na+ or K+ salts, causing indirect loss of bicarbonate from the body and development of metabolic acidosis under conditions of increased ketogenesis. NaBOH, sodium β-hydroxybutyrate; NaHCO3, sodium bicarbonate; NH4 −, ammonium; NH4BOH, ammonium betahydroxybutyrate.
The energy required to fuel Na+-K+-ATPase pump activity to maintain a low intracellular Na+ concentration decreases following pharmacologic inhibition of SGLT2. In addition to decreasing Na+ entry into the cell coupled to glucose, SGLT2is also decrease Na+ entry by inhibiting Na+-H+ antiporter (NHE3) activity. Studies in rodents show SGLT2 and NHE3 colocalize in the proximal tubule brush border membrane (27). Inhibition of SGLT2 decreases activity of NHE3, and the natriuretic effect of SGLT2i is diminished in models where NHE3 is knocked down (28). The estimated daily kidney ATP turnover decreases by approximately 50% in patients with diabetes mellitus treated with SGLT2i (26). Because ATP is not stored, reduced consumption will secondarily decrease pathways that normally generate ATP (29). Because metabolism of glutamine to NH4 + and HCO3 − in the proximal tubule results in an obligatory production of ATP, ammoniagenesis decreases following SGLT2i therapy. A similar reduction in ammoniagenesis occurs when cellular ATP utilization is lowered by decreasing the filtered Na+ or directly inhibiting Na+-K+-ATPase pump activity in the proximal tubule, supporting the notion that the rate of ATP utilization places an upper limit on ammonia production in the kidney (30).
Inhibition of ammonia synthesis indirectly predisposes to HCO3 − loss from the body and development of metabolic acidosis. The production of β-hydroxybutyric acid consumes HCO3 − and generates Na-β-hydroxybutyrate and H2CO3, the latter of which quickly disassociates into CO2 (exhaled by the lungs) and water. When β-hydroxybutyrate is excreted in the urine coupled to NH4 +, there is no net change in acid-base balance because production of NH4 + from glutamine generates HCO3 −. By contrast, urinary excretion of β-hydroxybutyrate as either an Na+ or K+ salt is the equivalent to losing HCO3 − from the body and will generate metabolic acidosis (31,32). The inhibitory effect on ammonia synthesis by SGLT2i favors excretion of Na+- and/or K+-coupled β-hydroxybutyrate in the urine.
SGLT2i also suppresses ammoniagenesis by causing a shift toward greater fatty acid oxidation by the proximal tubule. Under normal circumstances, this nephron segment preferentially utilizes fatty acids as a fuel source to generate ATP to meet the high metabolic demand of the tubule because fatty acids provide more energy per gram as compared with glucose. Studies in humans and rodent models of diabetes mellitus show metabolism in the kidney is shifted toward utilization of glucose for anaerobic glycolysis and away from oxidative phosphorylation of fatty acids (33,34). Rather than being utilized as fuel, lipid accumulates in the cell, where it has been linked to tubular injury through lipotoxicity. Upregulation of hypoxia-inducible factor is etiologically linked to this change in fuel preference (35). Administration of dapagliflozin prevents stabilization of HIF and attenuates the metabolic shift, suggesting that reabsorption of excess glucose by the diabetic kidney is responsible for the switch from fatty acid oxidation to glycolysis. The increase in ATP production following restoration of fatty acid oxidation in combination with reduced utilization accounts for the decrease in ammoniagenesis that occurs with SGLT2i (36).
The increase in fat oxidation following SGLT2i can further exacerbate indirect loss of HCO3 − from the body by impairing uptake and oxidation of filtered β-hydroxybutyrate. The kidney normally has a large capacity to reabsorb filtered ketone bodies. With prolonged fasting, blood concentrations and the filtered load of acetoacetate and β-hydroxybutyrate progressively increase. At the same time, the absolute reabsorption rate of ketone bodies increases linearly with no evidence of a tubular maximum (37,38). Reabsorption and subsequent oxidation in the kidney regenerate consumed HCO3 −, thereby lessening the degree of acidosis that would otherwise occur if the ketone was excreted as an Na+ or K+ salt. The complete oxidation of ketones by the tubule also generates ATP, adding to the pool derived from fatty acid oxidation. This additional amount of ATP adds to the inhibitory effect on ammonia production. In starvation, the decrease in ammonia availability does not adversely affect acid-base balance because oxidation of the ketone body in the proximal tubule regenerates an amount of HCO3 − equal to what was consumed in their production. This suppressive effect on ammoniagenesis by way of ATP turnover is advantageous during starvation because the requirement for glutamine uptake by the kidney will be reduced, resulting in less proteolysis and providing a protein-sparing effect. In this regard, infusion of β-hydroxybutyrate has been shown to reduce kidney NH4 + production in dogs and humans with chronic metabolic acidosis (39 –41). Following SGLT2i, the reduction in ATP utilization in combination with the large quantity of ATP produced from fat oxidation will not only suppress ammoniagenesis but also decrease uptake and oxidation of ketone bodies. These changes cause increased urinary excretion of β-hydroxybutyrate coupled to Na+ or K+, predisposing to acidosis through the indirect loss of HCO3 − from the body (42,43). In a study of 66 patients treated with empagliflozin, kidney β-hydroxybutyrate excretion rose three-fold, and fractional excretion increased by approximately 70% after 28 days of therapy (42).
Risk Factors for Development of Ketoacidosis
Changes in systemic metabolism and kidney ATP turnover following SGLT2i do not cause overt metabolic acidosis in most treated patients. Rather, these changes create a tenuous state whereby metabolic acidosis can quickly develop following some precipitating factor that leads to accelerated ketogenesis (Table 1). This situation occurs when insulin deficiency becomes more pronounced, explaining reports of ketoacidosis when SGLT2is are used off label in patients with type 1 diabetes mellitus or when the insulin dose is reduced by either patients or health care workers due to reductions in plasma glucose levels.
Table 1.
Risk factors for sodium-glucose cotransporter-2 inhibitor–associated ketoacidosis
| Risk Factor | Mechanism | Precautions to Reduce Risk |
|---|---|---|
| Reduction in insulin dose | Lowering of plasma glucose concentration following SGLT2i may prompt lowering of insulin dose by patient or prescriber | Avoid >20% reductions in insulin dose; implement frequent pre- and postprandial glucose monitoring following changes in insulin dose |
| Decreased insulin effect stimulates lipolysis and increases glucagon-insulin ratio, predisposing to ketogenesis in liver | Frequent monitoring of urine ketones following insulin dose adjustments | |
| Off-label use in type 1 diabetes mellitus | Increased risk with pump therapy due to operational failures causing sudden decrease in insulin dose; lower risk with subcutaneous regimens utilizing longer-acting formulations compared with rapid-acting analogs in pump regimens | Reduce insulin up to 20% following first dose of SGLT2i, then up titrate to original baseline or maintain stable insulin regimen for first 7 treatment d, and adjust thereafter according to glucose levels.a Assess ketones to detect early DKA in event of nonspecific symptoms regardless of glucose levels; adhere to optimized sick-day protocols |
| Restriction in dietary carbohydrate availability as seen with the use of ketogenic diets | Glycosuric effect of SGLT2i combined with decreased dietary carbohydrate intake leads to decreased insulin levels | Avoid very low–carbohydrate diets |
| Increased lipolysis due to reductions in carbohydrate intake leads to an increased glucagon-insulin ratio, which predisposes to ketogenesis | ||
| Surgical stress, trauma, intercurrent illness such as gastroenteritis where there is an inability to eat or drink | Decreased food intake leads to reduced insulin levels, thereby increasing likelihood of ketogenesis | Discontinue SGLT2i 3 d prior to elective surgery |
| Nonspecific symptoms, such as malaise and nausea, along with mild or absent glycemia may cause patient to decrease insulin dose, thus increasing risk of ketosis | Discontinue SGLT2i with acute illness | |
| Increased levels of counter-regulatory hormones (catecholamines and cortisol) accelerate lipolysis and increase insulin demand | Educate patient to monitor for ketonuria, in addition to glucose, and contact provider with any symptoms or detectable ketonuria | |
| Supplemental doses of rapid-acting insulin, along with fluids and ingestion of carbohydrates | ||
| Lean body habitus | Carbohydrate deficit is greater in lean versus obese individual because the degree of glycosuria relative to filtered load is independent of body size following SGLT2i | Educate lean/muscle-bound patients as to their increased risk and symptoms |
| Alcohol use disorder and salicylate toxicity | Alcohol withdrawal and salicylate toxicity are both associated with increased lipolysis and decreased insulin-glucagon ratio, thereby increasing risk of ketogenesis, conditions made worse following SGLT2i | Discontinue SGLT2i |
| Be aware of patients with chronic salicylate toxicity | ||
| Women | Women preferentially oxidize fatty acids and exhibit greater increase in ketogenesis with metabolic stress | Patient education that increased risk is confined to premenopausal women or postmenopausal women taking hormonal replacement therapy to include estrogens |
| Estrogen enhances hormone-sensitive lipase in adipocytes | ||
| LADA | LADA is phenotypically similar to type 2 diabetes mellitus, but insulin dependency develops rapidly (5–6 yr) due to immune-mediated β-cell injury | Increased awareness of disorder |
| Unrecognized rapid loss of β-cell secretory reserve combined with metabolic stress increases risk in the setting of SGLT2i | Prevalence is about 10% among patients diagnosed with type 2 diabetes mellitus with islet antibodies | |
| Presence of impaired β-cell function at diagnosis warrants early insulin therapy | ||
| Long-standing type 2 diabetes mellitus | Longer duration of disease increases likelihood of marked β-cell insufficiency | Patient education and more frequent monitoring when SGLT2i is given to patients with long duration of disease |
| Imposition of metabolic stress readily unmasks limited insulin secretory reserve |
Adjunctive therapy with SGLT2i to intensive insulin regimens improves glycemic control in type 1 diabetes mellitus but is associated with a dose-dependent increase in the absolute risk of DKA (44 –47). The higher risk is attributable to the failure to recognize early manifestations of metabolic decompensation secondary to a blunted rise in plasma glucose levels due to increased urinary glucose excretion (48). Use of SGLT2i in type 1 diabetes mellitus should be restricted to specialists with the resources necessary to train, educate, and support carefully selected patients.
A sudden restriction in carbohydrate availability will increase lipolysis and lipid oxidation, explaining reports of ketoacidosis under conditions of surgical stress, intercurrent illness, or prolonged fasting (49). A weight loss strategy centered on carbohydrate restriction is a risk factor and should be discouraged or implemented with close monitoring. The glycosuric response of SGLT2i relative to the filtered glucose load is independent of body size. This finding may explain the higher risk of acidosis in lean individuals on a low-carbohydrate diet because they are exposed to a higher deficit of carbohydrate when compared with obese subjects (50). Use of SGLT2i in the setting of alcohol use or salicylate toxicity is a risk factor because these conditions are characterized by increased ketogenesis (32,51).
Sex-based differences in substrate metabolism may explain the higher risk of ketoacidosis reported in women (11,45,52). During fasting, women demonstrate a greater increase in circulating nonesterified fatty acids and ketone bodies and a greater fall in the rate of endogenous glucose production and utilization when compared with men (53). Women also demonstrate increased lipid and reduced carbohydrate utilization as a fuel during exercise, whereas the opposite is true in men (54). This proclivity to utilize lipids as a fuel under conditions of metabolic stress is due to the permissive effect of estrogen on lipolysis through enhancement of hormone-sensitive lipase in adipocytes. In addition, a sexually dimorphic response of autonomic nerve activity may play a role (55). Circulating catecholamines and skeletal muscle sympathetic nerve activity with exercise are lower in women as compared with men. Despite the blunted response, women demonstrate a greater degree of lipolysis but lower rates of glucose production and utilization. Lipolysis in women is solely mediated by β-adrenergic receptors, whereas in men, there is also activation of α-adrenergic receptors, which exert an inhibitory effect on lipolysis (56). Given the divergent effects of α- and β-receptor activation, stimulation of both receptors in men will blunt the degree of lipolysis when compared with women, in whom only lipolytic β-receptors are stimulated. The shift to greater fat oxidation in women during metabolic stress increases the risk of ketoacidosis with SGLT2i.
Patients with a longer duration of disease or the requirement for insulin have more impaired insulin secretory reserve and are at higher risk. In addition to type 1 diabetes mellitus, an analysis of canagliflozin trial data found DKA events higher in patients with latent autoimmune diabetes of adulthood or those testing positive for glutamic acid decarboxylase 65-kD isoform antibodies (57). In one series of SGLT2i-associated ketoacidosis, approximately one third of patients had this form of the disease (49). These patients have circulating islet cell antibodies and are diagnosed with diabetes mellitus later in life. Upon presentation, insulin may not be required due to the presence of residual β-cell function, leading to misclassification as type 2 diabetes mellitus. Soon after diagnosis, there is a relatively rapid decline in insulin release due to autoantibody-mediated injury. SGLT2i can acutely accelerate this decline due to increased release of glucagon, rendering the patient at risk for ketoacidosis following a minimal metabolic stress.
Because urinary excretion of ketoacid salts coupled to NH4 + is neutral with regard to acid-base balance, it will be important to determine if people with diabetes and reduced GFR or those on renin angiotensin system blockers are at increased risk for ketoacidosis because these conditions can limit ammoniagenesis in the kidney. There may also be additional concerns in individuals with type 4 renal tubular acidosis because hyperkalemia further suppresses ammoniagenesis.
Clinical Features and Treatment of Sodium-Glucose Cotransporter-2 Inhibitor–Induced Ketoacidosis
A normal anion gap hyperchloremic metabolic acidosis is present in the initial stages of ketoacidosis (31). Loss of Na+-β-hydroxybutyrate in the urine secondarily causes kidney retention of NaCl in an attempt to maintain extracellular fluid volume near normal. Volume depletion or a reduction in GFR will limit filtration of ketoacid salts, causing Na+-β-hydroxybutyrate to accumulate in the plasma resulting in an increased anion gap metabolic acidosis. At presentation, most patients exhibit some combination of increased anion gap and a normal anion gap acidosis (58). Because insulin deficiency induced by SGLT2i is less severe compared with DKA, glucose overproduction and underutilization are quantitatively less pronounced (Table 2). This effect, combined with pharmacologic inhibition of glucose reabsorption in the kidney, likely accounts for why the plasma glucose is only minimally elevated in patients with SGLT2i-induced ketoacidosis. The lack of significant hyperglycemia despite development of ketoacidosis can delay the diagnosis because plasma glucose is what is typically monitored by the patients on a day-to-day basis. Most patients present with abdominal pain, nausea, anorexia, vomiting, or other nonspecific symptoms (49) (Table 3).
Table 2.
Selected clinical and laboratory findings distinguishing ketoacidosis due to sodium-glucose cotransporter-2 inhibitor from typical diabetic ketoacidosis
| Factor | Sodium-Glucose Cotransporter-2 Inhibitor–Induced Diabetic Ketoacidosis | Diabetic Ketoacidosis |
|---|---|---|
| Endogenous glucose production | ↑ | ↑↑ |
| Insulin release | ↓ | ↓↓ |
| Insulin resistance | ↑ | ↑↑ |
| Tissue glucose disposal | ↓ | ↓↓ |
| Kidney glucose clearance | ↑↑ | ↑ |
| Plasma glucose levels | Normal or increased, often <250 mg/dla | Typically 350–800 mg/dl |
| Extracellular fluid volume | ↓ | ↓↓ |
| Presenting symptoms | More nonspecific to include malaise, nausea, anorexia, abdominal pain | Polyuria and polydipsia due to osmotic diuresis, nausea, vomiting, shortness of breath |
Because the plasma glucose is either normal or only modestly increased in most patients, transcellular shift in water and K+ is minimal, making disturbances in plasma Na+ and K+ concentration less severe when compared with typical diabetic ketoacidosis (mechanisms are reviewed in ref. 31).
Table 3.
Recommendations to patients when prescribed sodium-glucose cotransporter-2 inhibitor
| Recommendations |
|---|
| Maintain appropriate fluid intake |
| Ensure adequate carbohydrate intake and avoid low-carbohydrate diets |
| Avoid skipping insulin doses and skipping meals |
| In situations of acute illness, vomiting, diarrhea, or inability to eat or drink |
| Discontinue SGLT2i |
| Contact provider even if blood glucose levels are not elevated |
| Continue to monitor glucose levels and monitor for presence of urinary ketones |
| If on insulin therapy, contact provider for dose adjustments; do not stop insulin |
| Ketoacidosis symptoms are nonspecific (malaise, nausea, anorexia, vomiting) and can occur despite normal or minimally elevated blood glucose level |
| If ketonuria detected, patient may be advised to administer dose of rapid-acting insulin and consume 30 g carbohydrate |
| Restart SGLT2i when eating and drinking normally, usually after 24–48 h as directed by medical provider |
Recommendations to patients when prescribed sodium-glucose cotransporter-2 inhibitor (SGLT2i) are especially important for lean women who are premenopausal or postmenopausal on hormone replacement therapy.
Given the benefits demonstrated in nondiabetic kidney disease in the Dapagliflozin and Prevention of Adverse Outcomes in Chronic Kidney Disease study, use of SGLT2i by nephrologists is likely to increase. Little is known about development of ketoacidosis in such individuals, but certain risk factors may increase the risk of this complication. Insulin resistance is a feature of uremia and increases in severity with advancing stages of CKD (59). Concomitant use of immunosuppressive drugs, particularly corticosteroids, can worsen insulin resistance and predispose to ketoacidosis. In nondiabetic rodents, euglycemic ketoacidosis developed with SGLT2i when animals were stressed with volume depletion (60). This observation suggests clinical conditions predisposing to insulin resistance, such as activation of sympathetic nerve activity or increased circulating glucocorticoids, may be sufficient to cause this complication in susceptible individuals without known diabetes mellitus.
The risk of SGLT2i-induced ketoacidosis can be minimized by close monitoring and making appropriate adjustments in medications upon changing clinical conditions. For example, major surgery or trauma is a common precipitating risk factor. These conditions lead to stress-induced increases in circulating catecholamines and cortisol, which, in combination, can reduce insulin sensitivity. The glycosuric effect of SGLT2i may prevent significant increases in plasma glucose concentration, masking the true requirement for insulin and increasing the risk of ketoacidosis. Current recommendations suggest SGLT2i should be discontinued at least 3 days prior to an elective surgical or invasive procedure (61). Insulin can be used as a bridge for glycemic control during this period. Although this time period provides approximately five t 1/2 of elimination, it should be noted that pharmacologic effects of the drugs, such as glycosuria and ketonemia, can be detected up to 10 days after discontinuation (62). Patient reports have described the onset of DKA up to 14 days after drug discontinuation (63). Patients should be educated about possible symptoms of ketoacidosis and, if present, instructed to contact their physician even if glucose monitoring does not indicate hyperglycemia (Table 3). When SGLT2is are prescribed to patients treated with insulin, the dose of insulin may need to be reduced to avoid hypoglycemia. Such a change requires close monitoring as the reduction in inulin dose increases the risk for ketoacidosis.
There are no specific guidelines for treatment of SGLT2i-induced ketoacidosis; however, the treatment approach is like that of typical DKA (31). In addition to drug discontinuation, restoration of extracellular fluid volume with normal saline should be the initial approach. The degree of volume depletion may be less severe because hyperglycemia is less pronounced, minimizing the severity of osmotic diuresis. Early intravenous insulin is required to facilitate utilization of glucose and diminish ketogenesis. In typical DKA, a 5% dextrose solution is recommended after the plasma glucose falls to 250 mg/dl. This solution along with insulin may be appropriate earlier in the treatment of SGLT2i-induced ketoacidosis because the plasma glucose concentration may only be minimally increased. In the absence of life-threatening acidosis or a pH<7.1, alkali therapy is generally not required because insulin administration slows the rate of ketoacid production, and oxidation of ketoanions regenerates HCO3 −. The presence of a predominately normal gap acidosis indicates that only a small amount of organic anions is available for regeneration of HCO3 −, and the indirect loss of HCO3 − from the body has been considerable. Regeneration of HCO3 − and establishment of normal acid-base balance can take up to 72 hours or even longer in the presence of CKD in this setting. Alkali therapy may be useful to shorten the total duration of acidemia in such patients.
In summary, use of SGLT2i is likely to increase dramatically given the evidence that these drugs confer cardiovascular benefit. These drugs alter ATP turnover in the kidney, predisposing to euglycemic ketoacidosis under conditions of accelerated ketogenesis. Clinicians should be familiar with risk factors so the likelihood of this disorder can be minimized.
Disclosures
All authors have nothing to disclose.
Funding
None.
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
References
- 1.Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ, Broedl UC, Inzucchi SE; EMPA-REG OUTCOME Investigators: Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med 373: 2117–2128, 2015 [DOI] [PubMed] [Google Scholar]
- 2.McMurray JJV, Solomon SD, Inzucchi SE, Køber L, Kosiborod MN, Martinez FA, Ponikowski P, Sabatine MS, Anand IS, Bělohlávek J, Böhm M, Chiang CE, Chopra VK, de Boer RA, Desai AS, Diez M, Drozdz J, Dukát A, Ge J, Howlett JG, Katova T, Kitakaze M, Ljungman CEA, Merkely B, Nicolau JC, O’Meara E, Petrie MC, Vinh PN, Schou M, Tereshchenko S, Verma S, Held C, DeMets DL, Docherty KF, Jhund PS, Bengtsson O, Sjöstrand M, Langkilde AM; DAPA-HF Trial Committees and Investigators: Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med 381: 1995–2008, 2019 [DOI] [PubMed] [Google Scholar]
- 3.Packer M, Anker SD, Butler J, Filippatos G, Pocock SJ, Carson P, Januzzi J, Verma S, Tsutsui H, Brueckmann M, Jamal W, Kimura K, Schnee J, Zeller C, Cotton D, Bocchi E, Böhm M, Choi DJ, Chopra V, Chuquiure E, Giannetti N, Janssens S, Zhang J, Gonzalez Juanatey JR, Kaul S, Brunner-La Rocca HP, Merkely B, Nicholls SJ, Perrone S, Pina I, Ponikowski P, Sattar N, Senni M, Seronde MF, Spinar J, Squire I, Taddei S, Wanner C, Zannad F; EMPEROR-Reduced Trial Investigators: Cardiovascular and renal outcomes with empagliflozin in heart failure. N Engl J Med 383: 1413–1424, 2020 [DOI] [PubMed] [Google Scholar]
- 4.Perkovic V, Jardine MJ, Neal B, Bompoint S, Heerspink HJL, Charytan DM, Edwards R, Agarwal R, Bakris G, Bull S, Cannon CP, Capuano G, Chu PL, de Zeeuw D, Greene T, Levin A, Pollock C, Wheeler DC, Yavin Y, Zhang H, Zinman B, Meininger G, Brenner BM, Mahaffey KW; CREDENCE Trial Investigators: Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N Engl J Med 380: 2295–2306, 2019 [DOI] [PubMed] [Google Scholar]
- 5.Heerspink HJL, Stefánsson BV, Correa-Rotter R, Chertow GM, Greene T, Hou FF, Mann JFE, McMurray JJV, Lindberg M, Rossing P, Sjöström CD, Toto RD, Langkilde AM, Wheeler DC; DAPA-CKD Trial Committees and Investigators: Dapagliflozin in patients with chronic kidney disease. N Engl J Med 383: 1436–1446, 2020 [DOI] [PubMed] [Google Scholar]
- 6.US Food and Drug Administration: Drug Safety Communication: FDA warns that SGLT2 inhibitors for diabetes may result in a serious condition of too much acid in the blood, 2015. Available at: www.fda.gov/downloads/Drugs/DrugSafety/UCM446954.pdf. Accessed June 22, 2015
- 7.Bonora BM, Avogaro A, Fadini GP: Sodium-glucose co-transporter-2 inhibitors and diabetic ketoacidosis: An updated review of the literature. Diabetes Obes Metab 20: 25–33, 2018 [DOI] [PubMed] [Google Scholar]
- 8.Peters AL, Henry RR, Thakkar P, Tong C, Alba M: Diabetic ketoacidosis with canagliflozin, a sodium‐glucose cotransporter 2 inhibitor, in patients with type 1 diabetes. Diabetes Care 39: 532–538, 2016 [DOI] [PubMed] [Google Scholar]
- 9.Dandona P, Mathieu C, Phillip M, Hansen L, Tschöpe D, Thorén F, Xu J, Langkilde AM; DEPICT-1 Investigators: Efficacy and safety of dapagliflozin in patients with inadequately controlled type 1 diabetes: The DEPICT‐1 52‐week study. Diabetes Care 41: 2552–2559, 2018 [DOI] [PubMed] [Google Scholar]
- 10.Kidney Disease: Improving Global Outcomes (KDIGO) Diabetes Work Group: KDIGO 2020. clinical practice guideline for diabetes management in chronic kidney disease (CKD). Available at https://kdigo.org/guidelines/diabetes-ckd/
- 11.Blau JE, Tella SH, Taylor SI, Rother KI: Ketoacidosis associated with SGLT2 inhibitor treatment: Analysis of FAERS data. Diabetes Metab Res Rev 33: e2924, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Douros A, Lix LM, Fralick M, Dell’Aniello S, Shah BR, Ronksley PE, Tremblay É, Hu N, Alessi-Severini S, Fisher A, Bugden SC, Ernst P, Filion KB: Sodium-glucose cotransporter-2 inhibitors and the risk for diabetic ketoacidosis: A multicenter cohort study. Ann Intern Med 173: 417–425, 2020 [DOI] [PubMed] [Google Scholar]
- 13.Diaz-Ramos A, Eilbert W, Marquez D: Euglycemic diabetic ketoacidosis associated with sodium-glucose cotransporter-2 inhibitor use: A case report and review of the literature. Int J Emerg Med 12: 27, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghezzi C, Loo DDF, Wright EM: Physiology of renal glucose handling via SGLT1, SGLT2 and GLUT2. Diabetologia 61: 2087–2097, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song WJ, Mondal P, Wolfe A, Alonso LC, Stamateris R, Ong BW, Lim OC, Yang KS, Radovick S, Novaira HJ, Farber EA, Farber CR, Turner SD, Hussain MA: Glucagon regulates hepatic kisspeptin to impair insulin secretion. Cell Metab 19: 667–681, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daniele G, Xiong J, Solis-Herrera C, Merovci A, Eldor R, Tripathy D, DeFronzo RA, Norton L, Abdul-Ghani M: Dapagliflozin enhances fat oxidation and ketone production in patients with type 2 diabetes. Diabetes Care 39: 2036–2041, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosenstock J, Ferrannini E: Euglycemic diabetic ketoacidosis: A predictable, detectable, and preventable safety concern with SGLT2 inhibitors. Diabetes Care 38: 1638–1642, 2015 [DOI] [PubMed] [Google Scholar]
- 18.Bolinder J, Ljunggren Ö, Kullberg J, Johansson L, Wilding J, Langkilde AM, Sugg J, Parikh S: Effects of dapagliflozin on body weight, total fat mass, and regional adipose tissue distribution in patients with type 2 diabetes mellitus with inadequate glycemic control on metformin. J Clin Endocrinol Metab 97: 1020–1031, 2012 [DOI] [PubMed] [Google Scholar]
- 19.Xu L, Nagata N, Nagashimada M, Zhuge F, Ni Y, Chen G, Mayoux E, Kaneko S, Ota T: SGLT2 inhibition by empagliflozin promotes fat utilization and browning and attenuates inflammation and insulin resistance by polarizing M2 macrophages in diet-induced obese mice. EBioMedicine 20: 137–149, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferrannini E, Muscelli E, Frascerra S, Baldi S, Mari A, Heise T, Broedl UC, Woerle HJ: Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J Clin Invest 124: 499–508, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferrannini E, Baldi S, Frascerra S, Astiarraga B, Heise T, Bizzotto R, Mari A, Pieber TR, Muscelli E: Shift to fatty substrate utilization in response to sodium-glucose cotransporter 2 inhibition in subjects without diabetes and patients with type 2 diabetes. Diabetes 65: 1190–1195, 2016 [DOI] [PubMed] [Google Scholar]
- 22.Brady JA, Hallow KM: Model-based evaluation of proximal sodium reabsorption through SGLT2 in health and diabetes and the effect of inhibition with canagliflozin. J Clin Pharmacol 58: 377–385, 2018 [DOI] [PubMed] [Google Scholar]
- 23.Thomas MC, Cherney DZI: The actions of SGLT2 inhibitors on metabolism, renal function and blood pressure. Diabetologia 61: 2098–2107, 2018 [DOI] [PubMed] [Google Scholar]
- 24.Wang XX, Levi J, Luo Y, Myakala K, Herman-Edelstein M, Qiu L, Wang D, Peng Y, Grenz A, Lucia S, Dobrinskikh E, D’Agati VD, Koepsell H, Kopp JB, Rosenberg AZ, Levi M: SGLT2 protein expression is increased in human diabetic nephropathy: SGLT2 protein inhibition decreases renal lipid accumulation, inflammation, and the development of nephropathy in diabetic mice. J Biol Chem 292: 5335–5348, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mather A, Pollock C: Glucose handling by the kidney. Kidney Int Suppl 120: S1–S6, 2011 [DOI] [PubMed] [Google Scholar]
- 26.Ferrannini E: Sodium-glucose co-transporters and their inhibition: Clinical physiology. Cell Metab 26: 27–38, 2017 [DOI] [PubMed] [Google Scholar]
- 27.Pessoa TD, Campos LC, Carraro-Lacroix L, Girardi AC, Malnic G: Functional role of glucose metabolism, osmotic stress, and sodium-glucose cotransporter isoform-mediated transport on Na+/H+ exchanger isoform 3 activity in the renal proximal tubule. J Am Soc Nephrol 25: 2028–2039, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Onishi A, Fu Y, Patel R, Darshi M, Crespo-Masip M, Huang W, Song P, Freeman B, Kim YC, Soleimani M, Sharma K, Thomson SC, Vallon V: A role for tubular Na+/H+ exchanger NHE3 in the natriuretic effect of the SGLT2 inhibitor empagliflozin. Am J Physiol Renal Physiol 319: F712–F728, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Halperin ML, Cheema-Dhadli S: Renal and hepatic aspects of ketoacidosis: A quantitative analysis based on energy turnover. Diabetes Metab Rev 5: 321–336, 1989 [DOI] [PubMed] [Google Scholar]
- 30.Halperin ML, Vinay P, Gougoux A, Pichette C, Jungas RL: Regulation of the maximum rate of renal ammoniagenesis in the acidotic dog. Am J Physiol 248: F607–F615, 1985 [DOI] [PubMed] [Google Scholar]
- 31.Palmer BF, Clegg DJ: Electrolyte and acid-base disturbances in patients with diabetes mellitus [published correction appears in N Engl J Med 381: 1598, 2019 10.1056/NEJMx190026]. N Engl J Med 373: 548–559, 2015 [DOI] [PubMed] [Google Scholar]
- 32.Palmer BF, Clegg DJ: Electrolyte disturbances in patients with chronic alcohol-use disorder. N Engl J Med 377: 1368–1377, 2017 [DOI] [PubMed] [Google Scholar]
- 33.Jang HS, Noh MR, Kim J, Padanilam BJ: Defective mitochondrial fatty acid oxidation and lipotoxicity in kidney diseases. Front Med (Lausanne) 7: 65, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kang HM, Ahn SH, Choi P, Ko YA, Han SH, Chinga F, Park AS, Tao J, Sharma K, Pullman J, Bottinger EP, Goldberg IJ, Susztak K: Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med 21: 37–46, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cai T, Ke Q, Fang Y, Wen P, Chen H, Yuan Q, Luo J, Zhang Y, Sun Q, Lv Y, Zen K, Jiang L, Zhou Y, Yang J: Sodium-glucose cotransporter 2 inhibition suppresses HIF-1α-mediated metabolic switch from lipid oxidation to glycolysis in kidney tubule cells of diabetic mice. Cell Death Dis 11: 390, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vinay P, Lemieux G, Cartier P, Ahmad M: Effect of fatty acids on renal ammoniagenesis in in vivo and in vitro studies. Am J Physiol 231: 880–887, 1976 [DOI] [PubMed] [Google Scholar]
- 37.Sapir DG, Owen OE: Renal conservation of ketone bodies during starvation. Metabolism 24: 23–33, 1975 [DOI] [PubMed] [Google Scholar]
- 38.Owen OE, Caprio S, Reichard GA Jr, Mozzoli MA, Boden G, Owen RS: Ketosis of starvation: A revisit and new perspectives. Clin Endocrinol Metab 12: 359–379, 1983 [DOI] [PubMed] [Google Scholar]
- 39.Desir G, Bratusch-Marrain P, DeFronzo RA: Effect of hyperketonemia on renal ammonia excretion in man. Metabolism 35: 736–743, 1986 [DOI] [PubMed] [Google Scholar]
- 40.Sherwin RS, Hendler RG, Felig P: Effect of ketone infusions on amino acid and nitrogen metabolism in man. J Clin Invest 55: 1382–1390, 1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lemieux G, Pichette C, Vinay P, Gougoux A: Cellular mechanisms of the antiammoniagenic effect of ketone bodies in the dog. Am J Physiol 239: F420–F426, 1980 [DOI] [PubMed] [Google Scholar]
- 42.Ferrannini E, Baldi S, Frascerra S, Astiarraga B, Barsotti E, Clerico A, Muscelli E: Renal handling of ketones in response to sodium-glucose cotransporter 2 inhibition in patients with type 2 diabetes. Diabetes Care 40: 771–776, 2017 [DOI] [PubMed] [Google Scholar]
- 43.Yokono M, Takasu T, Hayashizaki Y, Mitsuoka K, Kihara R, Muramatsu Y, Miyoshi S, Tahara A, Kurosaki E, Li Q, Tomiyama H, Sasamata M, Shibasaki M, Uchiyama Y: SGLT2 selective inhibitor ipragliflozin reduces body fat mass by increasing fatty acid oxidation in high-fat diet-induced obese rats. Eur J Pharmacol 727: 66–74, 2014 [DOI] [PubMed] [Google Scholar]
- 44.Mathieu C, Dandona P, Gillard P, Senior P, Hasslacher C, Araki E, Lind M, Bain SC, Jabbour S, Arya N, Hansen L, Thorén F, Langkilde AM; DEPICT-2 Investigators: Efficacy and safety of dapagliflozin in patients with inadequately controlled type 1 diabetes (the DEPICT-2 study): 24-week results from a randomized controlled trial. Diabetes Care 41: 1938–1946, 2018 [DOI] [PubMed] [Google Scholar]
- 45.Rosenstock J, Marquard J, Laffel LM, Neubacher D, Kaspers S, Cherney DZ, Zinman B, Skyler JS, George J, Soleymanlou N, Perkins BA: Empagliflozin as adjunctive to insulin therapy in type 1 diabetes: The EASE trials. Diabetes Care 41: 2560–2569, 2018 [DOI] [PubMed] [Google Scholar]
- 46.Garg SK, Henry RR, Banks P, Buse JB, Davies MJ, Fulcher GR, Pozzilli P, Gesty-Palmer D, Lapuerta P, Simó R, Danne T, McGuire DK, Kushner JA, Peters A, Strumph P: Effects of sotagliflozin added to insulin in patients with type 1 diabetes. N Engl J Med 377: 2337–2348, 2017 [DOI] [PubMed] [Google Scholar]
- 47.Wolfsdorf JI, Ratner RE: SGLT inhibitors for type 1 diabetes: Proceed with extreme caution. Diabetes Care 42: 991–993, 2019 [DOI] [PubMed] [Google Scholar]
- 48.Patel NS, Van Name MA, Cengiz E, Carria LR, Weinzimer SA, Tamborlane WV, Sherr JL: Altered patterns of early metabolic decompensation in type 1 diabetes during treatment with a SGLT2 inhibitor: An insulin pump suspension study. Diabetes Technol Ther 19: 618–622, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burke KR, Schumacher CA, Harpe SE: SGLT2 inhibitors: A systematic review of diabetic ketoacidosis and related risk factors in the primary literature. Pharmacotherapy 37: 187–194, 2017 [DOI] [PubMed] [Google Scholar]
- 50.Herring RA, Shojaee-Moradie F, Garesse R, Stevenage M, Jackson N, Fielding BA, Mendis A, Johnsen S, Umpleby AM, Davies M, Russell-Jones DL: Metabolic effects of an SGLT2 inhibitor (Dapagliflozin) during a period of acute insulin withdrawal and development of ketoacidosis in people with type 1 diabetes. Diabetes Care 43: 2128–2136, 2020 [DOI] [PubMed] [Google Scholar]
- 51.Palmer BF, Clegg DJ: Salicylate toxicity. N Engl J Med 382: 2544–2555, 2020 [DOI] [PubMed] [Google Scholar]
- 52.Fadini GP, Bonora BM, Avogaro A: SGLT2 inhibitors and diabetic ketoacidosis: Data from the FDA adverse event reporting system. Diabetologia 60: 1385–1389, 2017 [DOI] [PubMed] [Google Scholar]
- 53.Marinou K, Adiels M, Hodson L, Frayn KN, Karpe F, Fielding BA: Young women partition fatty acids towards ketone body production rather than VLDL-TAG synthesis, compared with young men. Br J Nutr 105: 857–865, 2011 [DOI] [PubMed] [Google Scholar]
- 54.Hedrington MS, Davis SN: Sexual dimorphism in glucose and lipid metabolism during fasting, hypoglycemia, and exercise. Front Endocrinol (Lausanne) 6: 61, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Davis SN, Galassetti P, Wasserman DH, Tate D: Effects of gender on neuroendocrine and metabolic counterregulatory responses to exercise in normal man. J Clin Endocrinol Metab 85: 224–230, 2000 [DOI] [PubMed] [Google Scholar]
- 56.Palmer BF, Clegg DJ: The sexual dimorphism of obesity. Mol Cell Endocrinol 402: 113–119, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Erondu N, Desai M, Ways K, Meininger G: Diabetic ketoacidosis and related events in the canagliflozin type 2 diabetes clinical program. Diabetes Care 38: 1680–1686, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sampani E, Sarafidis P, Papagianni A: Euglycaemic diabetic ketoacidosis as a complication of SGLT-2 inhibitors: Epidemiology, pathophysiology, and treatment. Expert Opin Drug Saf 19: 673–682, 2020 [DOI] [PubMed] [Google Scholar]
- 59.Kobayashi S, Maesato K, Moriya H, Ohtake T, Ikeda T: Insulin resistance in patients with chronic kidney disease. Am J Kidney Dis 45: 275–280, 2005 [DOI] [PubMed] [Google Scholar]
- 60.Perry RJ, Rabin-Court A, Song JD, Cardone RL, Wang Y, Kibbey RG, Shulman GI: Dehydration and insulinopenia are necessary and sufficient for euglycemic ketoacidosis in SGLT2 inhibitor-treated rats. Nat Commun 10: 548, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.US Food and Drug Administration : FDA approves label changes to SGLT2 inhibitors regarding temporary discontinuation of medication before scheduled surgery. Available at: https://www.fda.gov/drugs/drug-safety-and-availability/fda-revises-labels-sglt2-inhibitors-diabetes-include-warnings-about-too-much-acid-blood-and-serious. Accessed March 23, 2020
- 62.Yehya A, Sadhu A: Sodium-glucose cotransporter 2 inhibitor-associated prolonged euglycemic diabetic ketoacidosis in type 2 diabetes: A case report and literature review. Clin Diabetes 38: 112–116, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Iqbal I, Hamid M, Khan MAA, Kainat A, Tariq S: Dapagliflozin-induced late-onset euglycemic diabetic ketoacidosis. Cureus 11: e6089, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]