

Abstract
The transport of Ca2+ across membranes precedes the fusion and fission of various lipid bilayers. Yeast vacuoles under hyperosmotic stress become fragmented through fission events that requires the release of Ca2+ stores through the TRP channel Yvc1. This requires the phosphorylation of phosphatidylinositol-3-phosphate (PI3P) by the PI3P-5-kinase Fab1 to produce transient PI(3,5)P2 pools. Ca2+ is also released during vacuole fusion upon trans-SNARE complex assembly, however, its role remains unclear. The effect of PI(3,5)P2 on Ca2+ flux during fusion was independent of Yvc1. Here, we show that while low levels of PI(3,5)P2 were required for Ca2+ uptake into the vacuole, increased concentrations abolished Ca2+ efflux. This was as shown by the addition of exogenous dioctanoyl PI(3,5)P2 or increased endogenous production of by the hyperactive fab1T2250A mutant. In contrast, the lack of PI(3,5)P2 on vacuoles from the kinase dead fab1EEE mutant showed delayed and decreased Ca2+ uptake. The effects of PI(3,5)P2 were linked to the Ca2+ pump Pmc1, as its deletion rendered vacuoles resistant to the effects of excess PI(3,5)P2. Experiments with Verapamil inhibited Ca2+ uptake when added at the start of the assay, while adding it after Ca2+ had been taken up resulted in the rapid expulsion of Ca2+. Vacuoles lacking both Pmc1 and the H+/Ca2+ exchanger Vcx1 lacked the ability to take up Ca2+ and instead expelled it upon the addition of ATP. Together these data suggest that a balance of efflux and uptake compete during the fusion pathway and that the levels of PI(3,5)P2 can modulate which path predominates.
Keywords: Fab1, Fig4, PIKfyve, Pmc1, Vcx1, Yvc1
1 ∣. INTRODUCTION
The role of Ca2+ as a regulator of membrane fusion is best understood in the release of neurotransmitters through the fusion of synaptic vesicles with the presynaptic plasma membrane.1 Upon plasma membrane depolarization an influx of Ca2+ into the cytoplasm occurs which subsequently binds to the C2 domains of synaptotagmin-1. Once bound to Ca2+, synaptotagmin-1 changes conformation and aids in triggering SNARE-mediated fusion.2 Ca2+ transport across membranes is also a part of endolysosomal trafficking. In Saccharomyces cerevisiae vacuolar lysosomes serve as the major Ca2+ reservoir where it is complexed with inorganic polyphosphate.3 The transport of Ca2+ into the vacuole lumen is mediated through the Ca2+/H+ exchanger Vcx1, and the Ca2+ ATPase pump Pmc1, whereas the movement out of the lumen into the cytoplasm is performed by the Ca2+ channel Yvc1, a mucolipin transient receptor potential cation channel (TRPML) family orthologue.4
To date, we have a partial understanding of the mechanisms driving the release of Ca2+ during intracellular membrane trafficking. During osmotic shock or mechanical stress Yvc1 transports Ca2+ from the vacuole lumen to the cytosol in a manner that is controlled in part by the formation of PI(3,5)P2 by the PI3P 5-kinase Fab1/PIKfyve.5-8 Through uncertain mechanisms this cascade leads to the fragmentation of the vacuole into small vesicles through fission events.
During membrane fusion the release of Ca2+ is triggered by trans-SNARE pairing.9 However, a role for PI(3,5)P2 for fusion related Ca2+ efflux has not been tested. Previously, we showed that vacuole fusion was inhibited in the presence of elevated PI(3,5)P2.10 Fusion was blocked by either the addition of an exogenous dioctanoyl form of the lipid (C8-PI(3,5)P2) or through endogenous overproduction by the hyperactive fab1T2250A mutant. Fusion was arrested after the formation of trans-SNARE pairs but before mixing of the outer leaflets, that is, hemifusion. The possible role for the TRP Ca2+ channel Yvc1 in the inhibition of fusion by PI(3,5)P2 was eliminated, as vacuoles lacking the transporter fused well and were equally sensitive to PI(3,5)P2 relative to wild type vacuoles. Although Yvc1 was not involved in the arrest of fusion, others have shown that Ca2+ efflux still occurs in the absence of the channel.9 Thus, we postulated that PI(3,5)P2 could affect Ca2+ transport through a separate mechanism.
Various mechanisms control the involvement of Ca2+ transport in membrane fusion including Ca2+-binding protein calmodulin and a specific ABC transporter. Calmodulin acts as a sensor and participates in triggering late fusion events. Calmodulin tightly binding vacuoles upon Ca2+ release signals the completion of docking, which stimulates bilayer mixing.11,12 Another mechanism that regulates fusion events through modulating Ca2+ transport involves the vacuolar Class-C ABC transporters Ybt1.13,14
Here we continued our study of PI(3,5)P2 during fusion and found that elevated concentrations of the lipid blocked the release of Ca2+. Both exogenous C8-PI(3,5)P2 and overproduction by fab1T2250A inhibited Ca2+ efflux. In contrast, Ca2+ efflux was enhanced when Fab1 was inhibited by Apilimod, or when PI(3,5)P2 was sequestered by ML1-N. This effect was reproduced by the kinase dead fab1EEE mutation. Finally, we found that the effect of PI(3,5)P2 on Ca2+ transport depends on the Ca2+ uptake pump Pmc1 and not Yvc1.
2 ∣. RESULTS
2.1 ∣. Ca2+ efflux is inhibited by PI(3,5)P2
The release of vacuole luminal Ca2+ stores occurs upon trans-SNARE complex formation in the path towards fusion.9 The formation of vacuolar SNARE complexes also relies on the assembly of membrane microdomains enriched in regulatory lipids that include phosphoinositides such as the Fab1 substrate PI3P.15 However, the link between phosphoinositides and Ca2+ transport during fusion has remained unclear. By contrast, the dependence of Ca2+ efflux on PI(3,5)P2 during vacuole fission under hyperosmotic conditions has been established.7 Together, this served as the impetus to explore the role of PI(3,5)P2 in fusion-linked Ca2+ transport.
To address this gap in knowledge, we used isolated vacuoles for in vitro assays of Ca2+ flux. Ca2+ transport was monitored through changes in Fluo-4 (or Cal-520) fluorescence as previously demonstrated.13,16-18 Vacuoles were incubated under fusion conditions and reactions were started by the addition of ATP. As seen before, the addition of ATP triggers the rapid uptake of extraluminal Ca2+ from the buffer into the vacuole lumen as shown by the decrease in fluorescence (Figure 1A, black line). Omission of ATP prevented Ca2+ uptake and fluorescence remained flat (not shown). Shortly after, and in a SNARE-dependent manner, Ca2+ was released from vacuoles as shown by an increase in fluorescence. As a negative control for SNARE-dependent efflux, select reactions were incubated with antibody against Sec17 (α-SNAP), a co-chaperone of Sec18 (NSF) that aids in disrupting inactive cis-SNARE complexes to initiate the fusion pathway (Figure 1A, dotted line).19
FIGURE 1.

PI(3,5)P2 blocks Ca2+ efflux from vacuoles during fusion. Vacuoles were harvested from BJ3505 and 2X fusion reactions containing 150 μM Fluo-4 dextran. After 10 minutes of incubation with ATP (arrow) at 27°C to allow for the uptake of Ca2+, reactions were incubated with C8-PI(3,5)P2 (A), C8-PI3P (B) or C8-PA (C) at the indicated concentrations. Reactions were further incubated at 27°C and fluorescence was measured every 30 seconds for 80 minutes. Values were normalized to 1.0 representing the extraluminal Ca2+ at the beginning of the reaction and expressed as relative (Rel.) values compared to the untreated control. Separate reactions were incubated with 140 μg/mL anti-Sec17 IgG to block SNARE-dependent Ca2+ efflux. D, Average Ca2+ efflux from A to C. The maximum efflux for the untreated control was normalized to 1 and the results from the treatments were calculated in relation to the untreated control. Error bars are SEM (n = 3). Significant differences were in comparison the untreated control (buffer) *P < .05, **P < .01, ***P < .001 (Unpaired t test)
Here, we tested the effects of PI(3,5)P2 on Ca2+ transport. We incubated reactions with dioctanoyl (C8) derivatives of PI(3,5)P2, PI3P and phosphatidic acid (PA). PA was included due to its inhibitory effect on vacuole fusion through the inhibition of cis-SNARE priming,20 while PI3P was included as a precursor to PI(3,5)P2 and as a pro-fusion lipid.16,21-24 Short chain lipids were used to avoid solubility issues. As a result, seemingly high concentrations of C8 lipids were needed to see an effect. This, however, does not reflect the mol% of the lipids that partition into the membrane bilayer. Collins and Gordon showed that only a small fraction of C8-lipids incorporate into the membrane.25 Based on their work we estimated that 100 μM C8-PI(3,5)P2 correlated to ~1 mol% partitioned to the membrane fraction, which was in keeping with what reconstituted proteoliposome studies use in fusion models.26 Lipids were added after Ca2+ uptake plateaus (arrows). We found that C8-PI(3,5)P2 blocked Ca2+ efflux in a dose dependent manner and as effectively as the anti-Sec17 IgG negative control (Figure 1A,D, red lines). In parallel we found that C8-PI3P only showed a modest inhibition at the highest concentration tested when compared to C8-PI(3,5)P2 (Figure 1B,D). When C8-PA was tested we found that it had no effect on Ca2+ efflux (Figure 1C,D). These data indicated that the Ca2+ efflux associated with vacuole fusion was blocked by PI(3,5)P2. This is in contrast to the stimulatory effect of PI(3,5)P2 on Ca2+ efflux associated with vacuole fission. Importantly the concentrations used do not fully inhibit fusion, suggesting that Ca2+ transport can be uncoupled from fusion by altering the lipid composition of vacuole.10
2.2 ∣. Inhibition of the PI3P 5-kinase Fab1 leads to enhanced Ca2+ efflux
To verify the effect of PI(3,5)P2 on Ca2+ efflux we sought to counter its effects through inhibiting its production. The small molecule Apilimod had been shown to inhibit mammalian PIKfyve.27,28 Thus, we first tested if Apilimod could inhibit yeast Fab1 activity. For this we measured the conversion of BODIPY-TMR C6-PI3P to BODIPY-TMR C6-PI(3,5)P2 by isolated vacuoles. Before we tested the effects of Apilimod, we verified our detection system by using fig4Δ and fab1Δ vacuoles that reduce or abolish PI(3,5)P2 production, respectively. Using thin layer chromatography, we found that wild type vacuoles phosphorylated BODIPY-TMR C6-PI3P to make BODIPY-TMR C6-PI(3,5)P2 (Figure 2A,B). The half-life of the product was observed to be short as the levels of BODIPY-TMR C6-PI(3,5)P2 was significantly reduced after 5 minutes of incubation. This was in keeping reports by others showing that PI(3,5)P2 has a short half-life during vacuole fission.29-31 Next, vacuoles were incubated in the presence of BODIPY-TMR C6-PI3P and incubated with either vehicle or Apilimod to inhibit Fab1 activity. Indeed, Apilimod treatment abolished BODIPY-TMR C6-PI(3,5)P2 generation (Figure 2C,D). It should be noted that high concentrations of Apilimod were needed to inhibit PI(3,5)P2 production relative to the nM IC50 of Apilimod for mammalian PIKfyve.27 The high concentrations needed here are probably due to the high dissociation rates of the molecule to the active site of yeast Fab1. This is often seen when inhibitors of mammalian orthologs are used with yeast where the effective dose can differ by orders of magnitude. This is exemplified by the effects of propranolol on inhibiting PA phosphatase activity on mammalian Lipin1 (μM) vs yeast Pah1 (mM),32,33 Latrunculin-B on actin polymerization (nM vs μM),34,35 and the PI 3-kinase inhibitor wortmannin (nM vs μM).21,36
FIGURE 2.

Apilimod inhibits Fab1 activity. A, Vacuoles were incubated with BODIPY-TMR C6-PI3P at 27°C after which reactions were quenched with acetone and lipids were extracted and resolved by TLC to detect the production of BODIPY-TMR C6-PI(3,5)P2 by Fab1 activity. Wild type vacuoles were tested in parallel to those from fig4Δ and fab1Δ strains. Pure BODIPY-TMR C6-PI3P and C6-PI(3,5)P2 were used as standards in lanes 1 and 2. B, Average of three experiments in panel A. C, Vacuoles were incubated with DMSO (carrier) or 500 μM Apilimod. D, Average of three experiments in panel C. E, Vacuoles were incubated under docking conditions for 20 minutes. Endogenous PI(3,5)P2 was labeled with 2.5 μM Cy3-ML1-N and the total membranes were labeled with 1 μM MDY-64. Vacuoles were incubated with Apilimod or DMSO. Arrows point at examples of PI(3,5)P2 puncta. Error bars are SEM (n = 3). Significant differences were in comparison wild type or DMSO at 1 minute **P < .01 (unpaired t test). Scale bar: 2 μm
To visualize the effect of Apilimod on intact vacuoles we performed docking assays where endogenously produced PI(3,5)P2 was labeled with Cy3-ML1-N as described previously.10 ML1-N is the N-terminal polypeptide from the endolysosomal mucolipin transient receptor potential (TRPML) Ca2+ channel.7 ML1-N preferentially binds to PI(3,5)P2 on isolated yeast vacuoles as no labeling is observed with vacuoles from the kinase inactive fab1EEE mutant.10 In Figure 2E, we show that vacuoles incubated with carrier (DMSO) showed distinct PI(3,5)P2 punctate staining as previously reported. When vacuoles were incubated with Apilimod, we found that Cy3-ML1-N puncta were eliminated, further illustrating that Fab1 activity was inhibited during the experiment. Some free dye accumulated in the vacuole lumen to give the background fluorescence.
2.3 ∣. Apilimod alters Ca2+ transport
The effects of Apilimod were next tested on Ca2+ efflux. First, we added a concentration range of Apilimod, or DMSO (carrier) at the beginning of the assay. This showed that Ca2+ uptake was inhibited by Apilimod in a dose dependent manner (Figure 3A,B). This suggested that some Fab1 activity was needed at the beginning of the reaction to allow Ca2+ transport into the vacuole lumen. In parallel, we tested whether acute inhibition of PI(3,5)P2 production would have an effect on vacuole fusion. In Figure 3B, we show that vacuole fusion was not significantly affected by Apilimod, indicating that newly synthesized PI(3,5)P2 was not necessary for fusion to occur. Importantly, it also indicated that Apilimod did not have broad non-specific effects on the fusion machinery. We tested Apilimod after Ca2+ was taken up by vacuoles to examine its effects on efflux. The addition of Apilimod enhanced the release of Ca2+ in a dose dependent manner (Figure 3C,D). This suggested that blocking PI(3,5)P2 production promoted Ca2+ efflux, which complimented the inhibition of Ca2+ release through the addition of C8-PI(3,5)P2 shown in Figure 1. The DMSO carrier control had no effect as shown by the overlapping curve with buffer control. It is important to note that pre-existing PI(3,5)P2 remained on the vacuole upon Apilimod treatment. To compare the effects of new lipid production with the starting pool of PI(3,5)P2 we tested whether physically blocking PI(3,5)P2 with ML1-N would reproduce the effects seen with Apilimod. When ML1-N was added we found that efflux was increased, which partially reproduced the Apilimod affect (Figure 3E,F). However, higher concentrations (≥1 μM) started to reduce efflux. Unlike Apilimod, high levels of ML1-N was expected to sequester any unbound PI(3,5)P2. This was in agreement with the notion that low levels of PI(3,5)P2 are needed for the fusion pathway to progress, while its absence or excess have deleterious effects on the system.10
FIGURE 3.

Apilimod modulates Ca2+ flux during vacuole fusion. A, Vacuoles were incubated with buffer, 140 μg/mL anti-Sec17 IgG, Apilimod or DMSO (Carrier) at the beginning of the reaction and incubated for 30 minutes to monitor Ca2+ flux. B, Quantitation of Ca2+ efflux at 30 minutes normalized to the untreated control. In parallel, vacuole fusion was measured in the presence of Apilimod. The maximum Ca2+ efflux for the untreated control was normalized to 1 and the results from the treatments were calculated in relation to the untreated control. C, Vacuoles were incubated with buffer or 140 μg/mL anti-Sec17 IgG and incubated for 8 to 10 minutes to allow Ca2+ uptake. Next, vacuoles were incubated with a concentration range of Apilimod or DMSO (Carrier). Reactions were further incubated for a total 40 minutes. D, Quantitation of Ca2+ efflux at 30 minutes normalized to the untreated control. Results were normalized as in described in B. E, Vacuoles were incubated with buffer or anti-Sec17 IgG and incubated for 10 minutes to allow Ca2+ uptake. Next, vacuoles were incubated with a concentration range of GST-ML1-N and reactions were further incubated for a total of 30 minutes. F, Ca2+ release at 30 minutes. Results were normalized as in described in B. Error bars are SEM (n = 3). *P < .05, **P < .01, ***P < .001 (unpaired t test). Significant differences were in comparison to untreated wild type efflux
2.4 ∣. Fab1 kinase activity mutants differentially affect Ca2+ transport
Thus far we have used exogenous C8-PI(3,5)P2, chemical inhibition of Fab1, and lipid sequestration to show that Ca2+ efflux is affected by this lipid. To further confirm these findings, we next used vacuoles from yeast that expressed Fab1 mutations that altered kinase activity. First, we used vacuoles that contained the hyperactive Fab1 kinase mutation T2250A.37 We found that fab1T2250A vacuoles showed blocked Ca2+ release early in the assay when wild type vacuoles showed peak release at ~15 minutes (Figure 4A, red). This was followed by delayed release by fab1T2250A vacuoles that eventually reached wild type levels only after 60 minutes of incubation. We propose that the delay in Ca2+ release by fab1T2250A vacuoles could be due to an increased time required for phosphatase Fig4 to reduce the levels of PI(3,5)P2 present on these organelles. This was in keeping with the effects of adding exogenous C8-PI(3,5)P2 in Figure 1A and supports the idea that elevated PI(3,5)P2 concentrations suppress Ca2+ efflux. The delay in efflux seen with low concentrations of C8-PI(3,5)P2 was close to what we observed with fab1T2250A vacuoles and suggests that the prolonged inhibition by higher lipid concentrations was due to overwhelming the capacity of Fig4 to act on PI(3,5)P2 in the duration of the experiment. To verify that the decreased Ca2+ efflux seen with fab1T2250A vacuoles was due to directly to changes in PI(3,5)P2 concentrations, we added Apilimod to block PI(3,5)P2 synthesis. We found that blocking Fab1 activity restored Ca2+ release from fab1T2250A vacuoles at low concentrations, while at higher concentrations led to enhanced efflux relative to wild type vacuoles (Figure 4B). The data points reflect the normalized levels of fluorescence after 30 minutes of incubation. In parallel we found that sequestering “excess” lipid with ML1-N also restored Ca2+ efflux fab1T2250A vacuoles to wild type levels (Figure 4C). ML1-N at concentrations at or above 500 nM reduced Ca2+ as seen in Figure 3. Although the rescue was not as pronounced as the effect of Apilimod, the trend is consistent with the notion that elevated PI(3,5)P2 reduces Ca2+ efflux.
FIGURE 4.

FAB1 kinase mutations differentially affect Ca2+ transport. Vacuoles from wild type yeast were tested for Ca2+ transport in parallel with those from fab1T2250A (A), fab1EEE (D) or fig4Δ (F) yeast. B, Fab1T2250A vacuoles were incubated with a concentration range of Apilimod after 10 minutes of incubation. Reactions were then incubated for a total of 60 minutes. Average Ca2+ efflux values at 30 minutes of incubation were normalized relative to wild type given a value of 1 (dotted line). C, Fab1T2250A vacuoles were incubated with a concentration range of GST-ML1-N as done with Apilimod. Values were normalized to wild type efflux at 30 minutes. D, fab1EEE vacuoles were incubated with a dose curve of C8-PI(3,5)P2 and incubated in parallel with wild type vacuoles. Values were taken at 30 minutes and normalized wild type efflux without treatment at 30 minutes. E, Quantitation of fab1EEE vacuole flux in the presence of C8-PI(3,5)P2. Values were normalized to wild type efflux, which was set to 1. F, Wild type vacuoles were incubated in parallel with and fig4Δ vacuoles incubated with buffer or C8-PI(3,5)P2. Efflux values were taken at 30 minutes with or without treatment for wild type and fig4Δ vacuoles. G, Comparison of the maximal effects of C8-PI(3,5)P2 on Ca2+ flux between wild type and fig4Δ vacuoles. Values were normalized to the no treatment condition for each vacuole type. Error bars are SEM (n = 3). Significant differences were in comparison wild type. *P < .05, **P < .01, ***P < .001, ****P < .0001 (unpaired t test)
To better our sense of how PI(3,5)P2 concentrations serve as a switch to affect Ca2+ transport, we next used vacuoles from yeast that expressed the kinase-dead fab1EEE mutation.38 In Figure 4D, we show that fab1EEE showed a delay and overall reduction in Ca2+ uptake compared to wild type. The eventual release of Ca2+ was also delayed, yet sizeable. Together the altered uptake and release of Ca2+ by fab1EEE vacuoles reproduced the effects of adding Apilimod alone. The delay itself could be attributed to the reduced level of Sec17 present on fab1EEE vacuoles.10 While priming was not tested it remains possible that a reduction in Sec17 would delay the critical amount of trans-SNARE paring to trigger Ca2+ release earlier during the incubation to match the other assays.
To see if the effects of fab1EEE were due to the lack of PI(3,5)P2, we added exogenous C8-PI(3,5)P2 and found that the Ca2+ efflux of fab1EEE vacuoles was shifted down to wild type levels (Figure 4E). In contrast, our previous work showed PI(3,5)P2 was unable to restore the fusion defect of fab1EEE, which we attributed to separate vacuolar sorting defects.10 This suggests that the regulation of Ca2+ efflux by PI(3,5)P2 is distinct from its effects on protein sorting to the vacuole. Collectively with the effect of overproducing PI(3,5)P2, these data demonstrate that PI(3,5)P2 can serve as a rheostat to either inhibit or enhance Ca2+ release from vacuoles. Furthermore, the effects of PI(3,5)P2 on Ca2+ release appear to be oppositely regulated during osmotic shock vs isotonic conditions, as elevated PI(3,5)P2 induces Ca2+ efflux under hyperosmotic conditions.7
In parallel, we tested the effect of deleting the PI(3,5)P2 5-phosphatase FIG4 on Ca2+ transport. Cells lacking Fig4 have reduced levels of PI(3,5)P2 produced during osmotic shock,29,30 and, thus, we predicted that it would have an intermediate effect compared to vacuoles with the fab1EEE mutation. As expected fig4Δ vacuoles showed attenuated Ca2+ uptake similar to fab1EEE yet did not show the same delays in Ca2+ uptake or efflux (Figure 4E). Due to the reduced level of PI(3,5)P2 present on fig4Δ vacuoles we expected that Ca2+ efflux would be resistant to added C8-PI(3,5)P2. In accord with the other data, fig4Δ vacuoles were indeed resistant to C8-PI(3,5)P2 compared to wild type vacuoles (Figure 4F). This is consistent with our previous finding with fig4Δ vacuoles and lipid mixing.10
2.5 ∣. PI(3,5)P2 affects Ca2+ transport in a Pmc1 dependent manner
To hone in on the mechanism by which PI(3,5)P2 regulates Ca2+ flux during fusion we generated knockouts of the known vacuolar Ca2+ transporters: Pmc1, Vcx1, and Yvc1. As reported by Merz and Wickner, deletion of these transporters was unable to abolish the release of Ca2+ during fusion.9 In keeping with their findings, we found that Ca2+ efflux was close to wild type with each of the deletion strains (Figure 5A,B). We next asked if treating these deletion strains with C8-PI(3,5)P2 would inhibit Ca2+ flux to the same extent as seen with wild type vacuoles. We found that C8-PI(3,5)P2 inhibited Ca2+ efflux in vacuoles from yvc1Δ and vcx1Δ strains, but not those from pmc1Δ yeast (Figure 5A,B). Thus, it was probably that PI(3,5)P2 affected Ca2+ transport in a Pmc1-dependent manner. Because Pmc1 transports Ca2+ into the vacuole, the effect on the observed efflux could be due to the hyper-stimulation of Pmc1. It is important to emphasize that deleting YVC1 had no effect on the inhibition of Ca2+ flux by PI(3,5)P2. While Yvc1 is essential for the PI(3,5)P2 activated Ca2+ transport during vacuole fission, this channel does not appear to play a role during vacuole fusion, which is in keeping with our previous report.10 Others have suggested that leakage can occur during vacuole fusion when SNAREs are overexpressed.39 Thus, it could be that a similar leakage could serve as a transient Ca2+ pore. However, the ability of PI(3,5)P2 to inhibit net Ca2+ efflux at concentrations that do not inhibit fusion argues that the effects seen here are not due to non-specific leakage.
FIGURE 5.

PI(3,5)P2 regulates Ca2+ transport through Pmc1 during fusion. A, Vacuoles isolated from wild type as well as pmc1Δ, yvc1Δ and vcx1Δ strains were used in Ca2+ transport assays as described. Reactions were incubated with 140 μg/mL anti-Sec17, buffer or 116 μM C8-PI(3,5)P2 added after 5 minutes of incubation. Reactions were incubated for a total of 40 minutes and fluorescence was measured every 60 seconds. B, Average Ca2+ transport levels for multiple experiments with wild type, pmc1Δ, yvc1Δ or vcx1Δ vacuoles. After 10 minutes of incubation reactions were incubated with buffer, DMSO, 116 μM C8-PI(3,5)P2, 500 μM Verapamil or C8-PI(3,5)P2 and Verapamil together and incubated for a total of 40 minutes. The extraluminal Ca2+ at the end of 40 minutes was normalized to 1 for the buffer and DMSO controls. The amount of extraluminal Ca2+ for each treatment was then normalized to the controls. Error bars are SEM (n = 3). Significant differences were in comparison the untreated control (buffer) *P < .05, ***P < .001 (unpaired t test)
If PI(3,5)P2 was truly acting to trigger Pmc1 activity we hypothesized that the effect could be reversible by the Ca2+ pump inhibitor Verapamil.40,41 While there are no report of Verapamil inhibiting Pmc1, the sarcoendoplasmic reticulum Ca2+ ATPase pump (SERCA) has been shown to be inhibited by Verapamil at 290 μM.42 In our experiments we found that Verapamil blocked the effect of C8-PI(3,5)P2 on Ca2+ efflux (Figure 5B). This further indicates that the observed net Ca2+ efflux could instead be the inhibition of further Ca2+ uptake while the cation is released at a constant rate by another mechanism.
The effects of PI(3,5)P2 on the different strains suggested that the net Ca2+ efflux observed during fusion is in fact due to a change in the balance between uptake and efflux. This would consistent with the effect of PI(3,5)P2 Ca2+ flux in a Pmc1-dependent manner. To further examine this, we used vacuoles from vcx1Δ pmc1Δ double deletion strains. The double deletion of these importers was expected to abolish Ca2+ uptake in the presence of ATP. In Figure 6A we show that in the absence of ATP, both wild type and vcx1Δ pmc1Δ vacuoles were unable to take up Ca2+. Unexpectedly, upon the addition of ATP, vcx1Δ pmc1Δ vacuoles immediately expelled Ca2+, the opposite of the uptake observed with wild type vacuoles. The levels of Ca2+ release by vcx1Δ pmc1Δ vacuoles reached the maximum detection levels of the plate reader when set to detect to Ca2+ flux from both mutant and wild type vacuoles. Lowering the gain for vcx1Δ pmc1Δ vacuoles would render wild type flux undetectable. The expulsion of Ca2+ by vcx1Δ pmc1Δ vacuoles was resistant to anti-Sec17 IgG, indicating that the release was SNARE independent. We next tested if the efflux of Ca2+ by vcx1Δ pmc1Δ vacuoles was sensitive to PI(3,5)P2 and found that the addition of this lipid had no effect on Ca2+ release (Figure 6B,C). This suggests that the effects of PI(3,5)P2 occur during the Ca2+ uptake phase. Based on these data we hypothesized that in the presence of ATP, vacuoles constitutively release Ca2+, and that its detection is normally overcome by the dual action of Vcx1 and Pmc1. The production of PI(3,5)P2 early in the reaction promotes Ca2+ uptake in a Pmc1-dependent manner. The short half-life of the lipid could then reduce uptake, resulting in the in the observed Ca2+ efflux.
FIGURE 6.

Vacuoles lacking both Pmc1 and Vcx1 expel Ca2+ in an ATP-dependent manner. A, Compares the Ca2+ transport profiles of wild type vs the pmc1Δ vcx1Δ double deletion vacuoles. Individual reactions were in the presence of reaction buffer alone, buffer with ATP, or buffer with ATP and 140 μg/mL anti-Sec17 IgG. B, Effect of C8-PI(3,5)P2 on Ca2+ flux by pmc1Δ vcx1Δ double deletion vacuoles. C, Quantitation of relative Ca2+ flux into or out of vacuole from wild type and pmc1Δ vcx1Δ double deletion yeast after 5 minutes of incubation. The extraluminal Ca2+ in the absence of ATP was set to 1 and the results in the presence of ATP were normalized to the no ATP control. D, Vacuoles from vcx1Δ yeast were incubated with 140 μg/mL anti-Sec17 IgG, 500 μM Verapamil or 125 μM Apilimod added at T = 0 min. E, Quantitation of relative Ca2+ uptake wild type and vcx1Δ vacuoles after 10 minutes of incubation. The extraluminal Ca2+ in the untreated control was set to 1 and the results with Apilimod and Verapamil was normalized to the untreated control. Error bars are SEM (n = 3). Significant differences were in comparison the untreated control (buffer) **P < .01, ***P < .001 (unpaired t test)
The yeast vacuole is predicted to only have Pmc1 and Vcx1 to import Ca2+. This was supported by the data above as deletion of both abolished vacuolar Ca2+ uptake. Thus, deleting VCX1 alone would leave only Pmc1 on the vacuole, and results seen with vcx1Δ vacuoles would serve as a reporter for Pmc1 activity. Based on this, we next tested the effect of adding Verapamil and Apilimod at the beginning of the assay to measure Ca2+ uptake with vcx1Δ vacuoles. We observed that Ca2+ uptake was inhibited by both reagents, with Verapamil having a greater effect vs Apilimod (Figure 6D,E). These data were consistent with the hypothesis that Pmc1 mediated Ca2+ uptake is sensitive to PI(3,5)P2 levels.
To additionally test the balance of Ca2+ uptake and release during the pathway, we further examined the effects of Verapamil. First, we showed that verapamil blocks Ca2+ uptake in a dose dependent manner and in the range seen to inhibit mammalian SERCA. Vacuoles incubated with a concentration range of Verapamil at the beginning of the assay. Using wild type vacuoles, we found that Verapamil inhibited Ca2+ uptake in a dose dependent manner when added at the beginning of the assay (Figure 7A,B). This had little effect on vacuole fusion. Next, we tested if Ca2+ uptake continues late in the reaction by adding Verapamil after the initial uptake of Ca2+. As seen in Figure 7C, D, adding Verapamil after Ca2+ uptake resulted in a rapid accumulation of extraluminal Ca2+. This suggests that under control conditions, Ca2+ uptake continues through the docking stage, 10 to 15 minutes into the pathway, after which uptake is either inactivated or simply overcome by Ca2+ efflux.
FIGURE 7.

Verapamil inhibits Ca2+ uptake. A, Wild type vacuoles were incubated with a dose curve of Verapamil at T = 0 minute to block Ca2+ uptake. Separate reactions were incubated with buffer, DMSO or anti-Sec17 as controls. Reactions were incubated for a total of 30 minutes and fluorescence was measured every 30 seconds. B, Average Ca2+ transport levels for multiple experiments shown in panel A (green). The extraluminal Ca2+ for the untreated control after 5 minutes of incubation was normalized to 1. The result for each concentration of Verapamil was normalized as a fraction of the untreated control. In parallel, the effects of Verapamil on vacuole fusion (black). C, Wild type vacuoles were incubated with 500 μM Verapamil or DMSO after 8 minutes of incubation to allow initial Ca2+ uptake. Reactions were analyzed as described in panel A. D, Average Ca2+ transport levels for multiple experiments shown in panel C. The changes were determined by taking the difference between maximum uptake and efflux signals for each condition and normalized relative to control (buffer) conditions set to 1
Given the surprising result that Ca2+ efflux appeared to be triggered by a reduction in Pmc1 activity we sought to clarify whether the main cause of Ca2+ efflux during membrane fusion was through the generation of transient pores as has been proposed. If transient pore formation serves as the primary efflux pathway, then we would only expect Verapamil to trigger Ca2+ efflux if fusion was allowed to proceed. We found that extraluminal Ca2+ increased over the baseline in the presence of Verapamil and anti-Sec17 IgG, however, the amount detected was half of what was seen with Verapamil alone. This suggests that the increase in extraluminal Ca2+ seen with Verapamil was not due to pore formation.
2.6 ∣. PI(3,5)P2 and Pmc1 interactions with Nyv1 and the V-ATPase
Others have previously shown that Pmc1 activity is inhibited in part through its binding to the vacuolar R-SNARE Nyv1.43 Separately, PI(3,5)P2 has been shown to stabilize Vo – V1 holoenzyme assembly.38 Further, Pmc1 physically interacts with the V-ATPase. Thus, we thought it important to examine if this lipid affected the interactions between these proteins to shed further light onto a novel mechanism that controls Ca2+ transport. To accomplish this, we used vacuoles that harbored Pmc1 tagged with the triple hemagglutinin (HA) epitope in order to perform co-immunoprecipitation (IP) assays.44 Figure 8A shows that IP of wild type vacuoles anti-HA antibody did not pull down anything, ensuring that complexes pulled down from the Pcm1-HA3 strain would not be spurious. When Pmc1-HA3 vacuoles were used we observed a clear band corresponding to the tagged pump. IP of Pmc1-HA3 with anti-HA IgG isolated Pmc1-HA3 in addition to Nyv1 and Vph1, a component of the membrane integrated VO complex of the V-ATPase. Vph1 has been shown to bind Pmc1 in a high throughput screen of the yeast protein interactome.45
FIGURE 8.
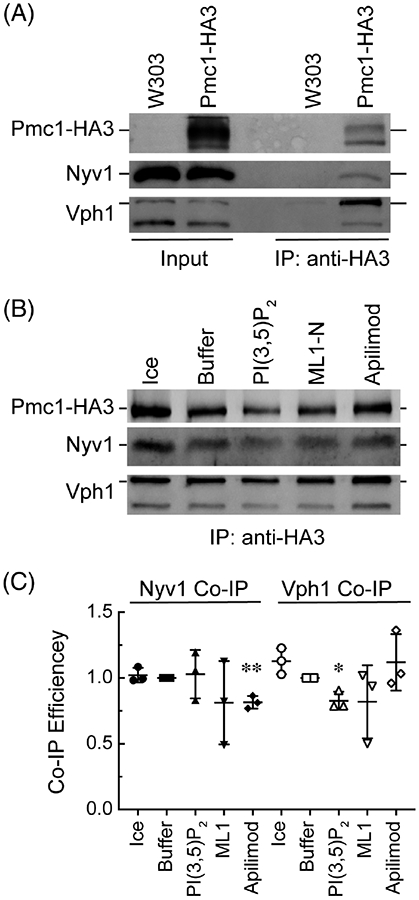
The effects of PI(3,5)P2 levels on Pmc1 interactions. A, Vacuoles from wild type yeast and a strain expressing HA-tagged Pmc1 were compared by immunoprecipitation with anti-HA beads and immunoblotting for isolated complexes. B, Vacuoles expressing Pmc1-HA3 were incubated in large scale reactions (20X) in the presence of 150 μM C8-PI(3,5)P2, 500 nM ML1-N, 500 μM Apilimod or buffer alone. Reactions were incubated for 30 minutes at 27°C. After incubating, the reactions were place on ice and solubilized and processed for immunoprecipitation with anti-HA agarose. Protein complexes were eluted with sodium thiocyanate and resolved by SDS-PAGE and probed by Western blotting for the presence of Pmc1-HA3 and Nyv1. C, Average quantitation of Nyv1 and Vph1 bound to Pmc1-HA. Western blot band intensities were measured using ImageJ (NIH). Values for the buffer control at 27°C were set to 1 and the other values were normalized relative to this control. Values represent concentrations relative to Pmc1-HA3 at T = 30 minutes. Error bars represent SD (n = 3). Significant differences were in comparison to the buffer control at 27°C *P < .05, **P < .01 (unpaired t test)
Next, we examined Pmc1 interactions in the presence of buffer, C8-PI(3,5)P2, ML1-N and Apilimod. We found that C8-PI(3,5)P2 did not alter the interactions between Pmc1 and Nyv1. However, blocking PI(3,5)P2 production with Apilimod partially reduced the amount of Nyv1 that co-immunoprecipitated with Pmc1 (Figure 8B,C). Although, PI(3,5)P2 is not known to bind either Nyv1 or Pmc1 directly, it has been shown to bind to the VO component Vph1 where it stabilizes VO – V1 holoenzyme assembly.38 Because the V-ATPase is known to bind Pmc1 we tested if C8-PI(3,5)P2 would affect Pmc1-Vph1 interactions.45 We found that PI(3,5)P2 led to a partial reduction in Vph1 interactions with Pmc1. Although, these interactions were not completely abolished, it is still possible that function was altered with minimal changes to the stoichiometry of the complex. Such a scenario has been documented with SNARE complexes on yeast vacuoles, where trans-SNARE complexes form, yet fail to induce vacuole fusion for various reasons.10,46 It is also possible that complex sub-units that remain to be identified could be affected more drastically by changes in PI(3,5)P2. While it is not possible to conclusively say that PI(3,5)P2 affects the Pmc1 protein complex, we can conclude that changes in PI(3,5)P2 concentrations act as a controller affecting Ca2+ transport. Future studies will elucidate the relationship between the lipid and Ca2+ flux as well as how these effects could alter V-ATPase activity.
3 ∣. DISCUSSION
The yeast vacuole serves as the primary reservoir for Ca2+ ions, which are translocated across the membrane through multiple transporters including Pmc1, Yvc1, and Vxc1. Although the regulatory mechanisms for each of these transporters are not fully understood, it has been shown that various lipids can affect their function in an organelle specific manner. For instance, PI(3,5)P2 regulates Ca2+ release through Yvc1 during the osmotically driven vacuole fission.7 Ca2+ is also released from the vacuole during the fusion process upon the formation of trans-SNARE complexes.9,12,47 More recently, we found that elevated levels of PI(3,5)P2 inhibits vacuole fusion at a stage between trans-SNARE pairing and outer-leaflet lipid mixing, that is, hemifusion and full bilayer fusion.10 This was observed through adding exogenous C8-PI(3,5)P2 or through using FAB1 mutations that were either kinase dead or overproduced the lipid. That study established that low levels of PI(3,5)P2 are needed for fusion to occur, but elevated concentrations are inhibitory. At this point, it is unclear whether the effects of PI(3,5)P2 are direct or if the ratio with its precursor PI3P and downstream degradation product(s) also play a role. In either circumstance, it is clear that PI(3,5)P2 has a biphasic effect on vacuole fusion.
The biphasic nature of PI(3,5)P2 in vacuole fusion is not limited to this lipid. For instance, PA is needed for reconstituted proteoliposome fusion to occur and for Vam7 binding to vacuoles.16,26 Yet, inhibiting PA phosphatase activity to produce diacylglycerol (DAG), resulting in elevating PA concentrations, blocks vacuole fusion at the priming stage through sequestering Sec18 from cis-SNARE complexes.20,48 Likewise, low levels of phospholipase C (PLC) stimulates vacuole fusion through converting PI(4,5)P2 to DAG, while high levels of PLC potently inhibits fusion.49 Finally, while PI(4,5)P2 is well known to stimulate vacuole fusion, an excess of the lipid can also inhibit the pathway.10,15,23,50 These examples show that the balance of substrate and product lipids can serve to move a pathway forward or arrest its progress.
In this study, we continued our investigation on the inhibitory nature of PI(3,5)P2 on vacuole fusion. Our work has identified that elevated levels of PI(3,5)P2 block the net Ca2+ efflux observed during fusion vacuole fusion. The effect of PI(3,5)P2 on Ca2+ transport was shown by various means including the use of the kinase dead fab1EEE and hyperactive fab1T2250A mutants. While both mutants were previously shown to inhibit hemifusion,10 here we see that only fab1T2250A inhibited Ca2+ efflux, which is in accord with a scenario in which excessive PI(3,5)P2 is a negative regulator of the fusion pathway. In contrast, kinase dead fab1EEE vacuoles attenuated Ca2+ uptake followed by a delayed release of the cation. In both instances, Ca2+ flux was restored to near wild type levels when the amount of free PI(3,5)P2 was increased on fab1EEE vacuoles or reduced on fab1T2250A vacuoles. The Ca2+ efflux seen with fab1EEE vacuoles observed here, albeit delayed, combined with our previous report showing that hemifusion was inhibited by this mutant indicates that Ca2+ flux precedes hemifusion.
Another revelation that comes from these experiments is that the effect of PI(3,5)P2 on Ca2+ transport was independent of the TRP channel Yvc1. This is contrary to what occurs during vacuole fission induced by osmotic stress, when PI(3,5)P2 interacts with Yvc1 leading to the release of Ca2+ from the vacuole lumen.7 Our previous work with PI(3,5)P2 suggested that this lipid inversely regulates fission and fusion. The current work further supports this model and specifies that the effects occur through differentially modulating Ca2+ transport across the vacuole bilayer. Surprisingly, we found that the inhibition of Ca2+ transport by PI(3,5)P2 was dependent on the Ca2+ ATPase Pmc1, which utilizes ATP hydrolysis to move Ca2+ into the vacuole lumen against the concentration gradient. Furthermore, it appears that the effect on Ca2+ uptake is immediate, as treatment with the Fab1/PIKfyve inhibitor Apilimod led to a rapid release of Ca2+, suggesting that PI(3,5)P2 needs to be newly generated at low levels to regulate Ca2+ transport. Under unencumbered conditions it is probably that PI(3,5)P2 levels are kept in check through the phosphatase activity of Fig4. One of the most striking findings from this study is that the Ca2+ efflux associated with fusion appears to be in part through the inactivation of Ca2+ influx. Additionally, our data suggests that the primary route of efflux can be uncoupled from membrane fusion as inhibition of SNARE priming was unable to prevent the trigger of Ca2+ efflux following treatment with Verapamil. We therefore conclude that there is a Ca2+ efflux route is opposed by Pmc1.
The regulation of Pmc1 on yeast vacuoles is unclear relative to other P-type Ca2+ ATPases. Notwithstanding, others have shown that that the R-SNARE Nyv1 directly interacts with Pmc1 to reduce Ca2+ transport.43 Although not directly shown, their model posits that increased Nyv1 would proportionally have increased interactions with Pmc1 to further reduce Ca2+ transport. We sought to learn if elevated PI(3,5)P2 levels would dissociate the Nyv1-Pmc1 complex to derepress Ca2+ transport. In our hands, we found that the Nyv1-Pmc1 interaction was only reduced by ~25% when PI(3,5)P2 production was inhibited by Apilimod. Alone, this makes it difficult to concluded whether the change was sufficient to directly cause the observed results. In parallel, adding C8-PI(3,5)P2 reduced the amount of Vph1 that co-isolated with Pmc1. Together, these data are supportive of the idea that changes in PI(3,5)P2 levels could affect complex assembly. This also suggests that changes in PI(3,5)P2 levels could affect V-ATPase function. This is in accord with work from the Kane lab showing that PI(3,5)P2 can alter VO – V1 complex stability and the translocation of protons into the vacuole lumen.38,51
Neither Pmc1, nor Nyv1 have been shown to bind PI(3,5)P2 directly, which raises the question as to how this lipid could affect the complex? While a direct interaction has not been shown it is still possible that an indirect path exists. One of the few bona fide PI(3,5)P2 binding proteins on the vacuole is the VO component Vph1.38 Their work showed that PI(3,5)P2 stabilized the assembly of the V1 – VO holoenzyme. Because Vph1 binds Pmc145 and R-SNAREs have been shown to directly bind VO,52 we hypothesized that PI(3,5)P2 could affect the results of Vph1-Pmc1 and Pmc1-Nyv1 interactions. Our Pmc1 immunoprecipitations showed that Vph1 indeed interacted with Pmc1 and that adding C8-PI(3,5)P2 reduced the Pmc1 interaction with Vph1, though the reduction was moderate. When we consider that blocking V-ATPase activity results in a decrease in Ca2+ uptake by vacuoles,53 we can venture to think that promoting V-ATPase activity through PI(3,5)P2-Vph1 interactions and promotion of V1 – VO complex could inversely promote Ca2+ uptake by reducing Vph1-Pmc1 interactions. Our data taken together with the finding that blocking Vph1 activity anti-Vph1 antibody inhibits Ca2+ efflux47 suggests Vph1 could bind and affect Pmc1, and that the addition of C8-PI(3,5)P2 or anti-Vph1 might attenuate productive interactions. Thus, it is possible that PI(3,5)P2 regulates Pmc1 in part through binding Vph1 and induce conformational change that alter the downstream outcomes of Pmc1-Vph1 interactions. Although this model is incomplete, we believe these data gives further insight into the mechanism behind Ca2+ efflux during the fusion process as well as the ability of PI(3,5)P2 to act as a potent fusion-fission switch.
Our finding that PI(3,5)P2 affects Pmc1 and not Yvc1 during fusion presents an apparent paradox with what is known about this lipid during fission. The dual role of PI(3,5)P2 in fission and fusion is indeed complex and this study goes to further our understanding of the difference. During fission, Fab1 is activated leading to a spike of PI(3,5)P2 on the vacuole. This activates the mechanosensitive TRP channel Yvc1 and efflux of luminal Ca2+ stores into the cytosol.7 During fusion, Fab1 is active and elevated levels of PI(3,5)P2 inhibits fusion. In this paper, we show that one of the effects of elevated PI(3,5)P2 is that the Ca2+ efflux associated with trans-SNARE complex formation is blocked. In order to tease these mechanisms apart we have to take into consideration the post-translational modification of Yvc1. During osmotic shock, 6 of the 9 surface exposed Cys on Yvc1 are modified with glutathione by Gtt1.54,55 After recovering from shock the attached glutathiones are removed by the thioreductase Trx2. Mutation of key Cys residues to Ser, or deletion of GTT1 to prevent the addition of glutathione blocks Yvc1 from transporting Ca2+. Another consideration is that Yvc1 exists as a multimer in isotonic conditions as shown in large-scale proteomic studies.45,56 We propose that Gtt1 activity interferes with Yvc1 dimers to allow activation by PI(3,5)P2 and promote Ca2+ flux. Taken together, we theorize that under isotonic conditions Yvc1 is not modified and the dimer is inactive. Further, Yvc1 is probably unable to interact with PI(3,5)P2 in a manner that promotes Ca2+ transport. This is probably due to changes in affinity or steric hindrance by the unmodified Ycv1 dimer.
The spike in PI(3,5)P2 production during osmotic shock not only activates Yvc1, but also stimulates V-ATPase activity.57 The latter is probably due to the stabilization of the V1 – VO complexes.38 As a result, the vacuole becomes more acidic and Ca2+ is released from the vacuole during fission. Under isotonic conditions, the lower levels of PI(3,5)P2 present may be enough to activate V-ATPase activity, but not to overly acidify the vacuole. Thus, another important facet of PI(3,5)P2 activity is the regulation of vacuolar acidification. In conclusion, this work adds to the body of evidence showing that PI(3,5)P2 has multiple roles in vacuole dynamics including the switch between fusion and fission and the transport of Ca2+ and H+ across the membrane.
4 ∣. MATERIALS AND METHODS
4.1 ∣. Reagents
Soluble reagents were dissolved in PIPES-Sorbitol (PS) buffer (20 mM PIPES-KOH, pH 6.8, 200 mM sorbitol) with 125 mM KCl unless indicated otherwise. Anti-Sec17 IgG,19 and Pbi258 were prepared as described previously. C8-PA (1,2-dioctanoyl-sn-glycero-3-phosphate), C8-PI3P (1,2-dioctanoyl-phosphatidylinositol 3-phosphate), and C8-PI (3,5)P2 (1,2-dioctanoyl-phosphatidylinositol 3,5-bisphosphate), BODIPY-TMR C6-PI3P and BODIPY-TMR C6-PI(3,5)P2 were purchased from Echelon Inc. Apilimod was from MedKoo Biosciences and Verapamil was from Cayman Chemical. Both were dissolved in DMSO as stock solutions. Anti-HA antibody was from Thermo-Fisher. GST-ML1-N was produced as described.7,10 Cy3-maleimide (GE Healthcare) was used to label GST-ML1-N according to the manufacturer's instructions.
4.2 ∣. Strains
Vacuoles from BJ3505 genetic backgrounds were used for Ca2+ flux assays (Table 1). PMC1 was deleted by homologous recombination using PCR products amplified using 5′-PMC1-KO (5′-TTCTAAAAAAAAAAAAACTGTGTGCGTAACAAAAAAAATAGACATGGAGGCCCAGAATAC-3′) and 3′-PMC1-KO (5′-TTGGTCACTTACATTTGTATAAACATATAGAGCGCGTCTACAGTATAGCGACCAGCATTC-3′) primers with homology flanking the PMC1 coding sequence. The PCR product was transformed into yeast by standard lithium acetate methods and plated on YPD media containing G418 (250 μg/μL) to generate BJ3505 pmc1::kanMX6 (RFY84). Similarly, VCX1 was deleted from BJ3505 by recombination using 5′-VCX1-KO (5′-TTCATCGGCTGCTGATAGCAAATAAAACAACATAGATACAGACATGGAGGCCCAGAATAC-3′) and 3′-VCX1-KO (5′-ATATAAAAATTAGTTGCGTAAACATAATATGTATAATATACAGTATAGCGACCAGCATTC-3′) primers that flanked the VCX1 open reading frame to make RFY86. The vcx1Δ pmc1Δ strain was from Dr. William Wickner (Dartmouth Medical School).59 The yeast strain expressing HA-tagged Pmc1-HA was from Dr. Kyle Cunningham (Johns Hopkins University)44 and W303-A1 was from Dr. Rodney Rothstein (Columbia University).
TABLE 1.
Yeast strains used in this study
| Strain | Genotype | Source |
|---|---|---|
| BJ3505 | MATa ura3–52 trp1-Δ101 his3-200 lys2-801 gal2 (gal3) can1 prb1-Δ1.6R pep4::HIS3 | 63 |
| DKY6281 | MATα pho8::TRP1 leu2–3 leu 2-112 ura3-52 his3-200 trp1-Δ901 lys2-801 | 60 |
| W303-1A | MATa leu2-3112 trp1-1 ura3-1 his3-11 can1-100 | 64 |
| K699 | W303-1A, PMC1::3XHA | 44 |
| vcx1Δ pmc1Δ | BJ3505, vcx1::URA3 pmc1::TRP1 | 59 |
| RFY74 | BJ3505, yvc1::kanMX6 | 10 |
| RFY76 | BJ3505, fab1::kanMX6 | 10 |
| RFY78 | RFY76, pRS416-FAB1T2250A | 10 |
| RFY80 | RFY76, pRS416-FAB1EEE | 10 |
| RFY82 | BJ3505, fig4::kanMX6 | 10 |
| RFY84 | BJ3505, pmc1::kanMX6 | This study |
| RFY86 | BJ3505, vcx1::kanMX6 | This study |
4.3 ∣. Vacuole Isolation and in vitro fusion
Vacuoles were isolated as described.60 in vitro fusion reactions (30 μL) contained 3 μg each of vacuoles from BJ3505 (PHO8 pep4Δ) and DKY6281 (pho8Δ PEP4) backgrounds, reaction buffer 20 mM PIPES-KOH pH 6.8, 200 mM sorbitol, 125 mM KCl, 5 mM MgCl2), ATP regenerating system (1 mM ATP, 0.1 mg/mL creatine kinase, 29 mM creatine phosphate), 10 μM CoA, and 283 nM Pbi2 (Protease B inhibitor). Fusion was determined by the processing of pro-Pho8 (alkaline phosphatase) from BJ3505 by the Pep4 protease from DK6281. Fusion reactions were incubated at 27°C for 90 minutes and Pho8 activity was measured in 250 mM Tris-HCl pH 8.5, 0.4% Triton X-100, 10 mM MgCl2, and 1 mM p-nitrophenyl phosphate. Pho8 activity was inhibited after 5 minutes by addition of 1 M glycine pH 11 and fusion units were measured by determining the p-nitrophenolate produced by detecting absorbance at 400 nm.
4.4 ∣. In vitro Ca2+ flux assay
Vacuolar Ca2+ flux was measured as described.13,16,18 in vitro Ca2+ transport reactions (60 μL) contained 20 μg vacuoles from BJ3505 backgrounds, fusion reaction buffer, 10 μM CoA, 283 nM Pbi2 (inhibitor of Proteinase B), and 150 nM of the Ca2+ probe Fluo-4 dextran conjugate MW 10000 (Invitrogen) or Cal-520 dextran conjugate MW 10000 (AAT Bioquest). Reaction mixtures were loaded into a black, half-volume 96-well flat-bottom plate with nonbinding surface (Corning). ATP regenerating system or buffer was added, and reactions were incubated at 27°C while Fluo-4 or Cal-520 fluorescence was monitored. Samples were analyzed using a POLARstar Omega fluorescence plate reader (BMG Labtech) with the excitation filter at 485 nm and emission filter at 520 nm for Fluo-4 or Cal-520. Reactions were initiated with the addition of ATP regenerating system following the initial measurement. The effects of inhibitors on efflux were determined by the addition of buffer, inhibitors, or C8-lipids immediately following Ca2+ influx. Calibration was done using buffered Ca2+ standards (Invitrogen).
4.5 ∣. PI3P 5-kinase assay and thin-layer chromatography
Fab1 kinase activity was measured with an assay adapted from the detection of Fig4 and Plc1 activity49,61 with some modifications. Kinase reactions (30 μL) contained 6 μg vacuoles from BJ3505 backgrounds, fusion reaction buffer, ATP regenerating system, 10 μM CoA, 283 nM Pbi2, 2 μM BODIPY-TMR C6-PI3P, and 1 mM sodium orthovanadate. Reaction mixtures were incubated for 1 or 5 minutes at 27°C and the placed on. The reactions were immediately quenched with acetone (100 μL). Following incubation acidic phospholipids were extracted from all reactions.62 TLC plates (Partisil LK6D Silica Gel Plates [60 Å], Whatman) were pretreated with 1.2 mM EGTA and 1% potassium oxalate (wt/vol) in MeOH/Water (3:2) and then dried at 100°C for 30 minutes prior to use. Dried lipids were resuspended in CHCl3/MeOH (1:1) (40 μL) and 5 μL was spotted on the plate. Plates were run in CHCl3/acetone/MeOH/AcOH/Water (46:17:15:14:8). Individual channels were loaded with PI3P and PI(3,5)P2 standards (Echelon). Imaging of plates was performed using a ChimiDoc MP System (BioRad) and densitometry was determined with Image Lab 4.0.1 software.
4.6 ∣. Vacuole docking
Docking reactions (30 μL) contained 6 μg of wild type vacuoles were incubated in docking buffer (20 mM PIPES-KOH pH 6.8, 200 mM sorbitol, 100 mM KCl, 0.5 mM MgCl2), ATP regenerating system (0.3 mM ATP, 0.7 mg/mL creatine kinase, 6 mM creatine phosphate), 20 μM CoA, and 283 nM Pbi2.15 PI(3,5)P2 was labeled with 2 μM Cy3-GST-ML1-N. Reactions were incubated at 27°C for 20 minutes. After incubating, reaction tubes were placed on ice and vacuoles were stained with 1 μM MDY-64. Reactions were next mixed with 50 μL of 0.6% low-melt agarose (in PS buffer), vortexed to disrupt non-specific clustering, and mounted on slides for observation by fluorescence microscopy. Images were acquired using a Zeiss Axio Observer Z1 inverted microscope equipped with an X-Cite 120XL light source, Plan Apochromat 63X oil objective (NA 1.4), and an AxioCam CCD camera.
4.7 ∣. Pmc1-HA immunoprecipitation
Pmc1-HA complex isolation was performed using 20X fusion reactions. Individual reactions were incubated with 150 μM C8-PI(3,5)P2, 500 nM GST-ML1-N, 500 μM Apilimod or buffer alone. After 30 minutes, reactions were placed on ice. Next, reactions were sedimented (11 000g, 10 minutes, 4°C), and the supernatants were discarded before extracting vacuoles with solubilization buffer (SB: 50 mM Tris-HCl, pH 8.0, 2 mM EDTA, 150 mM NaCl, 0.5% Tween-20, 1 mM PMSF). Reactions were then nutated for 1 hour at 4°C. Insoluble debris was sedimented (16 000 g, 10 minutes, 4°C) and 350 μL of supernatants were removed and placed in chilled tubes. Next, 35 μL was removed from each reaction as 10% total samples, mixed with 17.5 μL of 5X SDS loading buffer and 17.5 μL PS buffer. Equilibrated anti-HA agarose beads (60 μL) were incubated with the extracts (15 hours, 4°C, nutation). Beads were sedimented and washed 5X with 1 mL SB (800g, 2 minutes, 4°C), and bound material was eluted with 0.1 M Glycine, pH 2.2 and eluates were mixed with SDS-loading buffer. Protein complexes were resolved by SDS-PAGE and examined by Western blotting.
4.8 ∣. Data analysis and statistics
Results are expressed as the mean ± SEM. Experimental replicates (n) are defined as the number of separate experiments with different vacuole preparations. Significant differences were calculated using unpaired t tests. P values ≤.05 were considered as significant.
ACKNOWLEDGMENTS
We thank Drs. Kyle Cunningham (Johns Hopkins University), Rodney Rothstein (Columbia University), Lois Weisman (University of Michigan), William Wickner (Dartmouth Medical School) and Haoxing Xu (University of Michigan) for plasmids and yeast strains. This research was supported by grants from the National Institutes of Health (R01-GM101132) and National Science Foundation (MCB 1818310) to RAF.
Funding information
National Institute of General Medical Sciences, Grant/Award Number: GM101132; National Science Foundation, Grant/Award Number: MCB 1818310
Footnotes
CONFLICT OF INTEREST
The authors declare that they do not have conflicts of interest with the contents of this article.
REFERENCES
- 1.Südhof TC. Calcium control of neurotransmitter release. Cold Spring Harb Perspect Biol. 2012;4(1):a011353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chapman ER. How does synaptotagmin trigger neurotransmitter release. Annu Rev Biochem. 2008;77:615–641. [DOI] [PubMed] [Google Scholar]
- 3.Dunn T, Gable K, Beeler T. Regulation of cellular Ca2+ by yeast vacuoles. J Biol Chem. 1994;269(10):7273–7278. [PubMed] [Google Scholar]
- 4.Cunningham KW. Acidic calcium stores of Saccharomyces cerevisiae. Cell Calcium. 2011;50(2):129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonangelino CJ, Nau JJ, Duex JE, et al. Osmotic stress-induced increase of phosphatidylinositol 3,5-bisphosphate requires Vac14p, an activator of the lipid kinase Fab1p. J Cell Biol. 2002;156(6):1015–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Denis V, Cyert MS. Internal Ca(2+) release in yeast is triggered by hypertonic shock and mediated by a TRP channel homologue. J Cell Biol. 2002;156(1):29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dong XP, Shen D, Wang X, et al. PI(3,5)P2 controls membrane trafficking by direct activation of mucolipin Ca(2+) release channels in the endolysosome. Nat Commun. 2010;1:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Su Z, Anishkin A, Kung C, Saimi Y. The core domain as the force sensor of the yeast mechanosensitive TRP channel. J Gen Physiol. 2011; 138(6):627–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Merz AJ, Wickner W. Trans-SNARE interactions elicit Ca2+ efflux from the yeast vacuole lumen. J Cell Biol. 2004;164:195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miner GE, Sullivan KD, Guo A, et al. Phosphatidylinositol 3,5-Bisphosphate regulates the transition between trans-SNARE complex formation and vacuole membrane fusion. Mol Biol Cell. 2019;30:201–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ostrowicz CW, Meiringer CT, Ungermann C. Yeast vacuole fusion: a model system for eukaryotic endomembrane dynamics. Autophagy. 2008;4(1):5–19. [DOI] [PubMed] [Google Scholar]
- 12.Peters C, Mayer A. Ca2+/calmodulin signals the completion of docking and triggers a late step of vacuole fusion. Nature. 1998;396(6711):575–580. [DOI] [PubMed] [Google Scholar]
- 13.Sasser TL, Padolina M, Fratti RA. The yeast vacuolar ABC transporter Ybt1p regulates membrane fusion through Ca2+ transport modulation. Biochem J. 2012;448(3):365–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sasser TL, Fratti RA. Class C ABC transporters and vacuole fusion. Cell Logist. 2014;4(3):e943588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fratti RA, Jun Y, Merz AJ, Margolis N, Wickner W. Interdependent assembly of specific regulatory lipids and membrane fusion proteins into the vertex ring domain of docked vacuoles. J Cell Biol. 2004;167(6):1087–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miner GE, Starr ML, Hurst LR, Sparks RP, Padolina M, Fratti RA. The central polybasic region of the soluble SNARE (soluble N-Ethylmaleimide-sensitive factor attachment protein receptor) Vam7 affects binding to phosphatidylinositol 3-phosphate by the PX (Phox homology) domain. J Biol Chem. 2016;291(34):17651–17663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miner GE, Starr ML, Hurst LR, Fratti RA. Deleting the DAG kinase Dgk1 augments yeast vacuole fusion through increased Ypt7 activity and altered membrane fluidity. Traffic. 2017;18(5):315–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miner GE, Fratti R. Real-time fluorescence detection of calcium efflux during vacuolar membrane fusion. Methods Mol Biol. 1860;2019: 323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mayer A, Wickner W, Haas A. Sec18p (NSF)-driven release of Sec17p (alpha-SNAP) can precede docking and fusion of yeast vacuoles. Cell. 1996;85(1):83–94. [DOI] [PubMed] [Google Scholar]
- 20.Starr ML, Hurst LR, Fratti RA. Phosphatidic acid sequesters Sec18p from cis-SNARE complexes to inhibit priming. Traffic. 2016;17(10):1091–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boeddinghaus C, Merz AJ, Laage R, Ungermann C. A cycle of Vam7p release from and PtdIns 3-P-dependent rebinding to the yeast vacuole is required for homotypic vacuole fusion. J Cell Biol. 2002;157(1):79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karunakaran S, Sasser T, Rajalekshmi S, Fratti RA. Snares, HOPS, and regulatory lipids control the dynamics of vacuolar actin during homotypic fusion. J Cell Sci. 2012;14:1683–1692. [DOI] [PubMed] [Google Scholar]
- 23.Karunakaran S, Fratti R. The lipid composition and physical properties of the yeast vacuole affect the hemifusion-fusion transition. Traffic. 2013;14(6):650–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lawrence G, Brown CC, Flood BA, et al. Dynamic association of the PI3P-interacting Mon1-Ccz1 GEF with vacuoles is controlled through its phosphorylation by the type-1 casein kinase Yck3. Mol Biol Cell. 2014;25(10):1608–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Collins MD, Gordon SE. Short-chain phosphoinositide partitioning into plasma membrane models. Biophys J. 2013;105(11):2485–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mima J, Wickner W. Complex lipid requirements for SNARE-and SNARE chaperone dependent membrane fusion. J Biol Chem. 2009;284:27114–27122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cai X, Xu Y, Cheung AK, et al. PIKfyve, a class III PI kinase, is the target of the small molecular IL-12/IL-23 inhibitor apilimod and a player in toll-like receptor signaling. Chem Biol. 2013;20(7):912–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dayam RM, Saric A, Shilliday RE, Botelho RJ. The Phosphoinositidegated Lysosomal Ca(2+) channel, TRPML1: is required for phagosome maturation. Traffic. 2015;16(9):1010–1026. [DOI] [PubMed] [Google Scholar]
- 29.Duex JE, Nau JJ, Kauffman EJ, Weisman LS. Phosphoinositide 5-phosphatase fig 4p is required for both acute rise and subsequent fall in stress-induced phosphatidylinositol 3,5-bisphosphate levels. Eukaryot Cell. 2006;5(4):723–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duex JE, Tang F, Weisman LS. The Vac14p-Fig4p complex acts independently of Vac7p and couples PI3,5P2 synthesis and turnover. J Cell Biol. 2006;172(5):693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jin N, Chow CY, Liu L, et al. VAC14 nucleates a protein complex essential for the acute interconversion of PI3P and PI(3,5)P(2) in yeast and mouse. EMBO J. 2008;27(24):3221–3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morlock KR, McLaughlin JJ, Lin YP, Carman GM. Phosphatidate phosphatase from Saccharomyces cerevisiae. Isolation of 45- and 104-kDa forms of the enzyme that are differentially regulated by inositol. J Biol Chem. 1991;266(6):3586–3593. [PubMed] [Google Scholar]
- 33.Pappu AS, Hauser G. Propranolol-induced inhibition of rat brain cytoplasmic phosphatidate phosphohydrolase. Neurochem Res. 1983;8(12):1565–1575. [DOI] [PubMed] [Google Scholar]
- 34.Eitzen G, Wang L, Thorngren N, Wickner W. Remodeling of organelle-bound Actin is required for yeast vacuole fusion. J Cell Biol. 2002;158(4):669–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spector I, Shochet NR, Blasberger D, Kashman Y. Latrunculins–novel marine macrolides that disrupt microfilament organization and affect cell growth: I. comparison with cytochalasin D. Cell Motil Cytoskeleton. 1989;13(3):127–144. [DOI] [PubMed] [Google Scholar]
- 36.Futter CE, Collinson LM, Backer JM, Hopkins CR. Human VPS34 is required for internal vesicle formation within multivesicular endosomes. J Cell Biol. 2001;155(7):1251–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lang MJ, Strunk BS, Azad N, Petersen JL, Weisman LS. An intramolecular interaction within the lipid kinase Fab1 regulates cellular phosphatidylinositol 3,5-bisphosphate lipid levels. Mol Biol Cell. 2017;28(7):858–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li SC, Diakov TT, Xu T, et al. The signaling lipid PI(3,5)P2 stabilizes V1-V(o) sector interactions and activates the V-ATPase. Mol Biol Cell. 2014;25(8):1251–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Starai VJ, Jun Y, Wickner W. Excess vacuolar SNAREs drive lysis and Rab bypass fusion. Proc Natl Acad Sci U S A. 2007;104(34):13551–13558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Calvert CM, Sanders D. Inositol trisphosphate-dependent and -independent Ca2+ mobilization pathways at the vacuolar membrane of Candida albicans. J Biol Chem. 1995;270(13):7272–7280. [DOI] [PubMed] [Google Scholar]
- 41.Teng J, Goto R, Iida K, Kojima I, Iida H. Ion-channel blocker sensitivity of voltage-gated calcium-channel homologue Cch1 in Saccharomyces cerevisiae. Microbiology. 2008;154(Pt 12):3775–3781. [DOI] [PubMed] [Google Scholar]
- 42.Paydar MJ, Pousti A, Farsam H, Amanlou M, Mehr SE, Dehpour AR. Effects of diltiazem or verapamil on calcium uptake and release from chicken skeletal muscle sarcoplasmic reticulum. Can J Physiol Pharmacol. 2005;83(11):967–975. [DOI] [PubMed] [Google Scholar]
- 43.Takita Y, Engstrom L, Ungermann C, Cunningham KW. Inhibition of the Ca(2+)-ATPase Pmc1p by the v-SNARE protein Nyv1p. J Biol Chem. 2001;276(9):6200–6206. [DOI] [PubMed] [Google Scholar]
- 44.Cunningham KW, Fink GR. Calcineurin inhibits VCX1-dependent H+/Ca2+ exchange and induces Ca2+ ATPases in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16(5):2226–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tarassov K, Messier V, Landry CR, et al. An in vivo map of the yeast protein interactome. Science. 2008;320(5882):1465–1470. [DOI] [PubMed] [Google Scholar]
- 46.Fratti RA, Collins KM, Hickey CM, Wickner W. Stringent 3Q: 1R composition of the SNARE 0-layer can be bypassed for fusion by compensatory SNARE mutation or by lipid bilayer modification. J Biol Chem. 2007;282(20):14861–14867. [DOI] [PubMed] [Google Scholar]
- 47.Bayer MJ, Reese C, Buhler S, Peters C, Mayer A. Vacuole membrane fusion: V0 functions after trans-SNARE pairing and is coupled to the Ca2+–releasing channel. J Cell Biol. 2003;162(2):211–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sasser T, Qiu QS, Karunakaran S, et al. Yeast lipin 1 orthologue pah1p regulates vacuole homeostasis and membrane fusion. J Biol Chem. 2012;287(3):2221–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jun Y, Fratti RA, Wickner W. Diacylglycerol and its formation by phospholipase C regulate Rab- and SNARE-dependent yeast vacuole fusion. J Biol Chem. 2004;279:53186–53195. [DOI] [PubMed] [Google Scholar]
- 50.Mayer A, Scheglmann D, Dove S, Glatz A, Wickner W, Haas A. Phosphatidylinositol 4,5-bisphosphate regulates two steps of homotypic vacuole fusion. Mol Biol Cell. 2000;11(3):807–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Banerjee S, Clapp K, Tarsio M, Kane PM. Interaction of the late endolysosomal lipid PI(3,5)P2 with the Vph1 isoform of yeast V-ATPase increases its activity and cellular stress tolerance. J Biol Chem. 2019;294:9161–9171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Di Giovanni J, Boudkkazi S, Mochida S, et al. V-ATPase membrane sector associates with synaptobrevin to modulate neurotransmitter release. Neuron. 2010;67(2):268–279. [DOI] [PubMed] [Google Scholar]
- 53.Miseta A, Kellermayer R, Aiello DP, Fu L, Bedwell DM. The vacuolar Ca2+/H+ exchanger Vcx1p/Hum1p tightly controls cytosolic Ca2+ levels in S. cerevisiae. FEBS Lett. 1999;451(2):132–136. [DOI] [PubMed] [Google Scholar]
- 54.Chandel A, Das KK, Bachhawat AK. Glutathione depletion activates the yeast vacuolar transient receptor potential channel, Yvc1p, by reversible glutathionylation of specific cysteines. Mol Biol Cell. 2016;27(24):3913–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hamamoto S, Mori Y, Yabe I, Uozumi N. In vitro and in vivo characterization of modulation of the vacuolar cation channel TRPY1 from Saccharomyces cerevisiae. FEBS J. 2018;285(6):1146–1161. [DOI] [PubMed] [Google Scholar]
- 56.Schlecht U, Miranda M, Suresh S, Davis RW, St Onge RP. Multiplex assay for condition-dependent changes in protein-protein interactions. Proc Natl Acad Sci U S A. 2012;109(23):9213–9218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li SC, Diakov TT, Rizzo JM, Kane PM. Vacuolar H+-ATPase works in parallel with the HOG pathway to adapt Saccharomyces cerevisiae cells to osmotic stress. Eukaryot Cell. 2012;11(3):282–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Slusarewicz P, Xu Z, Seefeld K, Haas A, Wickner WT. I2B is a small cytosolic protein that participates in vacuole fusion. Proc Natl Acad Sci U S A. 1997;94(11):5582–5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ungermann C, Wickner W, Xu Z. Vacuole acidification is required for trans-SNARE pairing, LMA1 release, and homotypic fusion. Proc Natl Acad Sci U S A. 1999;96(20):11194–11199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Haas A, Conradt B, Wickner W. G-protein ligands inhibit in vitro reactions of vacuole inheritance. J Cell Biol. 1994;126(1):87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rudge SA, Anderson DM, Emr SD. Vacuole size control: regulation of PtdIns(3,5)P2 levels by the vacuole-associated Vac14-Fig4 complex, a PtdIns(3,5)P2-specific phosphatase. Mol Biol Cell. 2004;15(1):24–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sasser TL, Lawrence G, Karunakaran S, Brown C, Fratti RA. The yeast ABC transporter Ycf1p enhances the recruitment of the soluble SNARE Vam7p to vacuoles for efficient membrane fusion. J Biol Chem. 2013;288:18300–18310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jones EW, Zubenko GS, Parker RR. PEP4 gene function is required for expression of several vacuolar hydrolases in Saccharomyces cerevisiae. Genetics. 1982;102(4):665–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wallis JW, Chrebet G, Brodsky G, Rolfe M, Rothstein R. A hyper-recombination mutation in S. cerevisiae identifies a novel eukaryotic topoisomerase. Cell. 1989;58(2):409–419. [DOI] [PubMed] [Google Scholar]