

SUMMARY
Ependymoma is the third most common pediatric tumor with posterior fossa group A (PFA) being its most aggressive subtype. Ependymomas are generally refractory to chemotherapies and thus lack any effective treatment. Here, we report that elevated expression of CXorf67 (chromosome X open reading frame 67), which frequently occurs in PFA ependymomas, suppresses homologous recombination (HR)-mediated DNA repair. Mechanistically, CXorf67 interacts with PALB2 and inhibits PALB2-BRCA2 interaction, thereby inhibiting HR repair. Concordantly, tumor cells with high CXorf67 expression levels show increased sensitivity to poly(ADP-ribose) polymerase (PARP) inhibitors, especially when combined with radiotherapy. Thus, our findings have revealed a role of CXorf67 in HR repair and suggest that combination of PARP inhibitors with radiotherapy could be an effective treatment option for PFA ependymomas.
Graphical Abstract
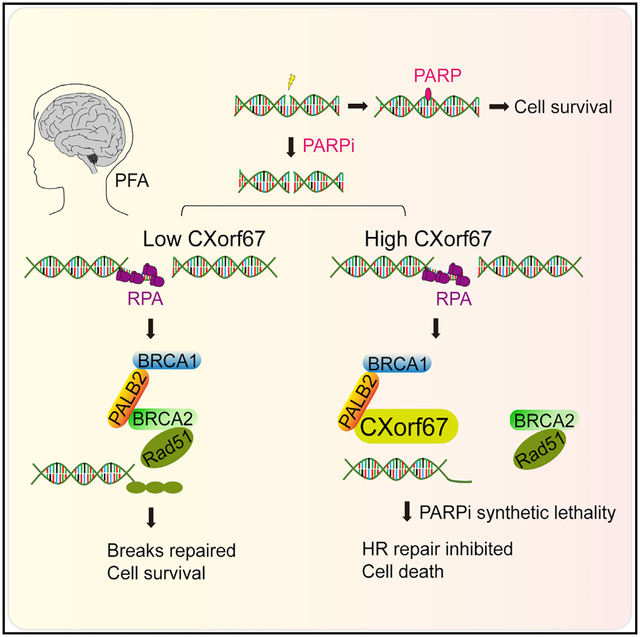
In Brief
Han et al. show that CXorf67 interacts with PALB2 and obstructs the PALB2-BRCA2 interaction, thereby inhibiting homologous recombination repair. A combination of PARP inhibitors with radiotherapy could be an effective treatment option for PFA ependymomas with elevated CXorf67 expression.
INTRODUCTION
Ependymoma develops throughout the neuraxis, which consists of three anatomic compartments of the central nervous system: supratentorial (ST), posterior fossa (PF), and spinal cord. In children, 90% of ependymomas arise intracranially, with two-thirds occurring in the PF and one-third being located in the ST (Pajtler et al., 2015, 2017). Based on DNA methylation status and gene expression profiles, PF ependymomas can be classified into three subgroups—PFA, PFB, and PF-SE. Among the three subgroups, PFA is the most common and aggressive one, and is mainly found in young children with a median age of ~3 years (Pajtler et al., 2015; Wani et al., 2012; Witt et al., 2011). Ependymomas respond inconsistently to conventional chemotherapies and, in most cases, remarkable resistance has been reported (Bouffet and Foreman, 1999; Garvin et al., 2012; Mack et al., 2014). So far, the standard therapy for intracranial ependymomas is surgical resection combined with radiotherapy. However, PFA tumors are often difficult to be completely resected due to their lateral localization. Together with the lack of chemotherapy options, PFA patients and especially those with recurrent tumors generally have very poor clinical outcomes.
Chromosome X open reading frame 67 (CXorf67) is an uncharacterized protein, whose function is poorly understood. Previous studies imply a possible role of CXorf67 in low-grade endometrial stromal sarcoma and prostate cancer in human (Dewaele et al., 2014). CXorf67 is normally expressed at a low level in all human tissues. It was recently reported that CXorf67 is specifically unregulated in PFA ependymoma (Pajtler et al., 2018). Furthermore, several studies suggest an involvement of CXorf67 in chromatin modification through affecting polycomb repressive complex 2 (PRC2) activity in PFA ependymoma (Hubner et al., 2019; Jain et al., 2019; Pajtler et al., 2018; Piunti et al., 2019). However, the potential clinical value of CXorf67 in the treatment of PFA ependymoma or other tumors remains unclear. In this study, we found that CXorf67 inhibits HR repair through blocking the PALB2-BRCA2 interaction. More importantly, we found that PFA tumor cells with high CXorf67 expression could be selectively killed by PARP inhibitors, especially when combined with radiotherapy. Therefore, our findings provide a strategy for the treatment of PFA ependymoma with immediate clinical applicability.
RESULTS
CXorf67 Is a DNA Damage Response Protein that Suppresses DNA Repair
CXorf67 is reported to be specifically highly expressed in PFA ependymoma, but not in other ependymoma subgroups (Pajtler et al., 2018), which we confirmed by analysis of the data from the GEO databases (Donson et al., 2017; Pajtler et al., 2015, 2018; Vladoiu et al., 2019) (Figure S1A). In addition, we examined CXorf67 protein levels in several human cell lines and found that CXorf67 was only detected in U2OS and Daoy cells (Figure S1B). To identify interaction partners of CXorf67, we immunoprecipitated CXorf67 from Daoy cells, followed by mass spectrometric analysis. The analysis identified several known CXorf67-interacting proteins, including EZH2, SUZ12, and EED (Hubner et al., 2019; Jain et al., 2019; Pajtler et al., 2018), and a number of DNA repair proteins, including PALB2 and BRCA1 (Table S1). Meanwhile, our analysis of the CellMiner Cross Database (CellMinerCDB), a web-based resource for interactive exploration of cancer cell line genomic and pharmacogenomics (Rajapakse et al., 2018), revealed a correlation of CXorf67 expression with cancer cell sensitivity to DNA-damaging agents, including camptothecin (CPT), etoposide, and doxorubicin (Figure S1C). Thus, we suspected that CXorf67 might play a role in the DNA damage response. So, we transfected EGFP-CXorf67 into U2OS and Daoy cells and monitored the localization kinetics of CXorf67 in response to laser-induced DNA damage. Indeed, CXorf67 was efficiently recruited to sites of DNA damage induced by laser micro-irradiation (Figure 1A). In addition, CXorf67 was enriched to the chromatin fraction in response to CPT (Figure 1B). Moreover, nuclear foci of endogenous CXorf67 were also readily detected in U2OS cells after CPT or ionizing radiation (IR) treatment (Figure S1D). Similarly, discrete foci of EGFP-tagged CXorf67, which were co-localized with γ-H2AX, were readily detected in U2OS cells after IR or CPT treatment (Figure 1C). Overall, these results are consistent with a possible function of CXorf67 in DNA damage repair.
Figure 1. CXorf67 Is a DNA Damage Response Protein and Suppresses DNA Repair.
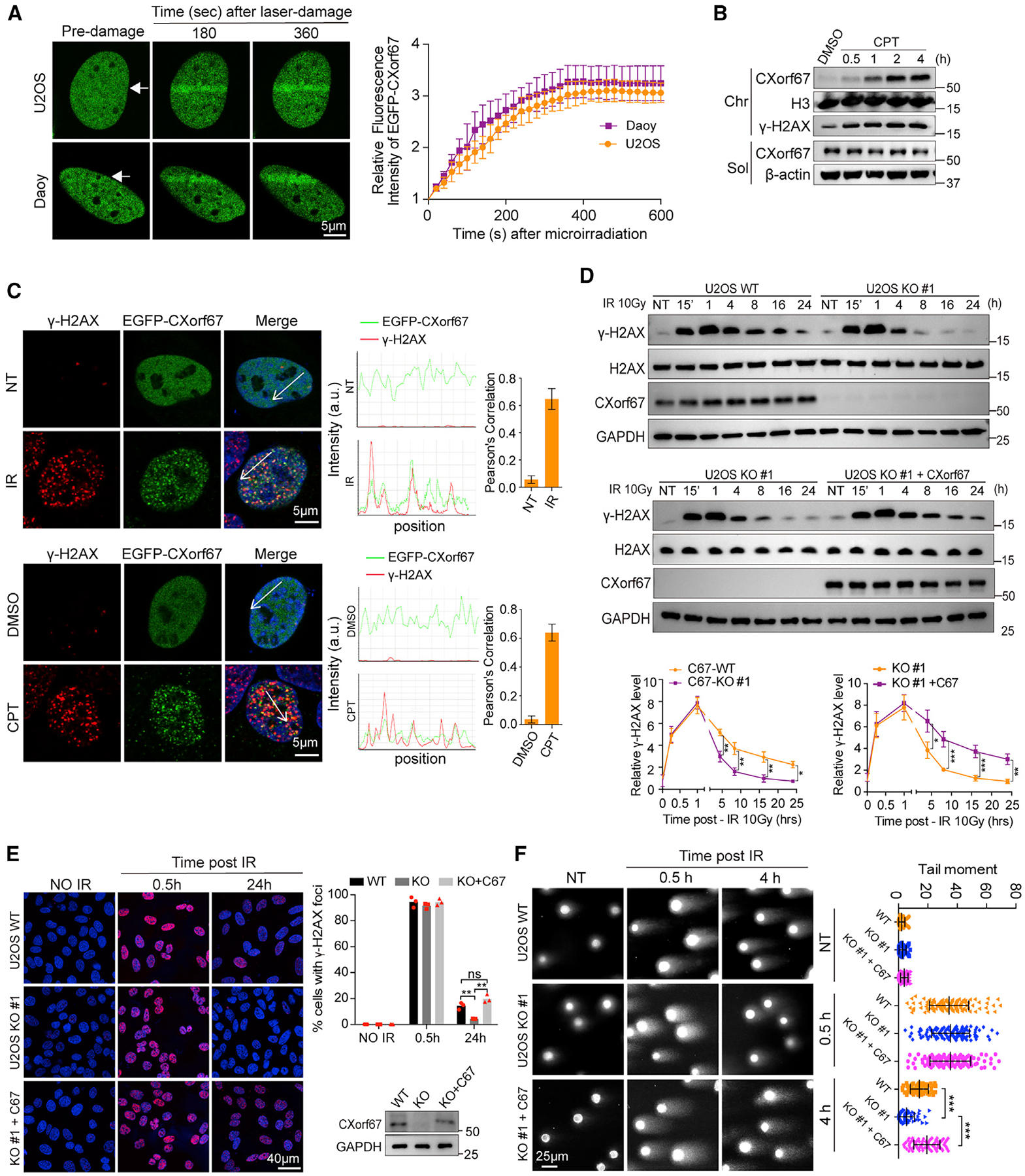
(A) Live-cell imaging of the recruitment of EGFP-CXorf67 proteins to laser damage tracks generated by laser micro-irradiation in U2OS and Daoy cells. Representative confocal images are shown. White arrows indicate irradiated regions. Accumulation of EGFP-CXorf67 on the DNA damage tracks was quantified. Data represented the mean values (±SD) from three independent experiments.
(B) U2OS cells were treated with CPT (0.1 μM) for 0.5, 1, 2, or 4 h. The chromatin fraction (Chr) and soluble fraction (Sol) were isolated for western blotting analysis.
(C) Representative confocal images for CXorf67 and γ-H2AX in U2OS cells transiently transfected with EGFP-CXorf67 and treated with IR (10 Gy) or CPT (0.1 μM) and immunostained after 4 h (left). Co-localization was confirmed with both fluorescent intensity profiles and Pearson’s correlation coefficient determination. Fluorescence-intensity profiles of CXorf67 and γ-H2AX were obtained using the Leica LAS AF software, along a straight line (white) crossing the nucleus of a representative cell (middle). Histograms represent mean ± SD of Pearson’s correlation coefficient between CXorf67 and γ-H2AX from 20 randomly selected cells in each group (right).
(D) Comparison of IR-induced γ-H2AX levels between CXorf67 WT and KO U2OS cells (upper panel); and between CXorf67 KO U2OS cells with and without CXorf67 restored (middle panel). Immunoblot detection of γ-H2AX was collected at the indicated time points after irradiation (10 Gy). NT, no treatment. Lower two panels: quantitative analysis of the relative levels of γ-H2AX normalized to H2AX loading control in the upper and middle panel by ImageJ software. Data are presented as mean ± SD from three independent experiments (two-way ANOVA; *p < 0.05, **p < 0.01, ***p < 0.001).
(E) γ-H2AX foci formation in C67-WT, C67-KO, and C67-KO with C67 re-expressed U2OS cells. Representative immunofluorescence (IF) images of cells at 0.5 h, 24 h after irradiation with 10 Gy are shown. Right: the percentage of positive cells with γ-H2AX foci (>10). At least 80 cells were analyzed for each datum point. Data are presented as mean ± SD (n = 3 independent experiments; paired t test; **p < 0.01). Western blot showing expression levels of CXorf67.
(F) C67-WT, C67-KO, and C67-KO with C67 re-expressed U2OS cells were harvested for the neutral comet assay at the indicated time after IR (10 Gy) treatment. The tail moment was analyzed using the CometScore software. Data are presented as mean ± SD (n = 60–117; unpaired t test; ***p < 0.001). The experiments were repeated three times.
To further investigate the potential role of CXorf67 in DNA damage repair, we generated CXorf67 knockout (KO) U2OS and Daoy cell lines (hereafter referred to as C67-KO U2OS or Daoy, whereas the parental cells are referred to as C67-wild type [WT] U2OS or Daoy) using the CRISPR-Cas9 technique. CXorf67 deficiency was confirmed by western blot and immunofluorescent analysis (Figures S1E and S1F). Both IR and CPT can induce DNA double-strand breaks (DSBs), which cause rapid increases in histone H2AX phosphorylation at serine 139 (referred to as γ-H2AX) at the DNA damage sites. The level of γ-H2AX subsequently declines as DNA damage is repaired (Rogakou et al., 1998). We compared the kinetics of γ-H2AX induced by IR between C67-WT and C67-KO cells in both U2OS and Daoy cells. Data from these different cell lines consistently showed that, while C67-deficiency did not alter the initial increases of γ-H2AX, the γ-H2AX levels in C67-KO cells were significantly lower than C67-WT cells 4 h after IR treatment (Figures 1D, S1G, and S1H). These results suggest that CXof67 may play a negative role in DNA damage repair. To confirm this, we carried out rescue experiments using C67-KO U2OS and C67-KO Daoy cells. Our result showed that re-expression of CXorf67 in C67-KO U2OS or Daoy cells increased the γ-H2AX levels in the post-IR recovery phase (Figures 1D and S1I). Consistently, immunofluorescent analysis showed that the number of γ-H2AX foci were nearly the same in C67-WT as that in C67-KO U2OS cells at 0.5 h after DNA damage; whereas the number of γ-H2AX foci in C67-KO cells was significantly lower than that in the C67-WT U2OS cells at 24 h. This difference at 24 h could be abrogated by re-expression of CXorf67 in C67-KO cells (Figure 1E). Finally, we measured the effect of CXorf67 depletion on DNA damage by using the neutral comet DNA repair assay in U2OS cells. As shown in Figure 1F, the tail moments of C67-KO cells were markedly shorter than those of C67-WT cells at 4 h after IR treatment, although no significant differences were observed at 0.5 h after IR treatment; meanwhile, re-expression of CXorf67 suppressed DNA repair and made the tail moments longer. Together, these results demonstrate that CXorf67 deficiency leads to enhanced DNA damage repair, suggesting that CXorf67 is a negative regulator of DNA damage repair.
CXorf67 Inhibits HR-Mediated DSB Repair
Organisms have two major DSB repair pathways: non-homologous end-joining (NHEJ) and homologous recombination (HR). NHEJ is an error-prone end-to-end ligation process that can occur at any time in the cell cycle. By contrast, HR is an error-free process that occurs primarily in the S and G2 phases of the cell cycle (Ciccia and Elledge, 2010). We next investigated which of these two DNA repair pathways might be regulated by CXorf67. For this purpose, we used U2OS DR-GFP and Daoy EJ5-GFP reporter assay. We found that knockdown of CXorf67 significantly increased HR DSB repair efficiency; reversely, CXorf67 overexpression significantly inhibited it (Figure 2A). However, CXorf67 overexpression and knockdown had no apparent effect on NHEJ efficiency (Figure S2A). These results suggest that CXorf67 may function in HR-mediated DNA repair. In HR-mediated DSB repair, formation of Rad51 foci is an important step. Therefore, we next examined whether CXorf67 may affect Rad51 foci formation. Our results showed that CXorf67 depletion significantly increased the number of Rad51 foci upon DNA damage induced by CPT in both Daoy and U2OS cells (Figures 2B, 2C, S2B, and S2C), and the increases could be reversed by expression of exogenous CXorf67 (Figures 2D and S2D). These results further indicate that CXorf67 has a role in DNA repair through the HR pathway. We then examined the role of CXorf67 in focus formation of RPA2 and BRCA1, which act upstream of Rad51 in the HR pathway, in Daoy and U2OS cells. Our result showed that both RPA2 and BRCA1 foci were not altered by CXorf67 deficiency (Figures 2E, 2F, S2E, and S2F). Meanwhile, the lack of CXorf67 did not affect Rad51, RPA2, and BRCA1 expression, or cell-cycle progression in these cells (Figures 2G, S2G, and S2H). Together, these results suggest that CXorf67 suppresses HR-mediated DSB repair probably through acting upstream of Rad51 and downstream of BRCA1.
Figure 2. CXorf67 Regulates HR- but Not NHEJ-Mediated DSB Repair.
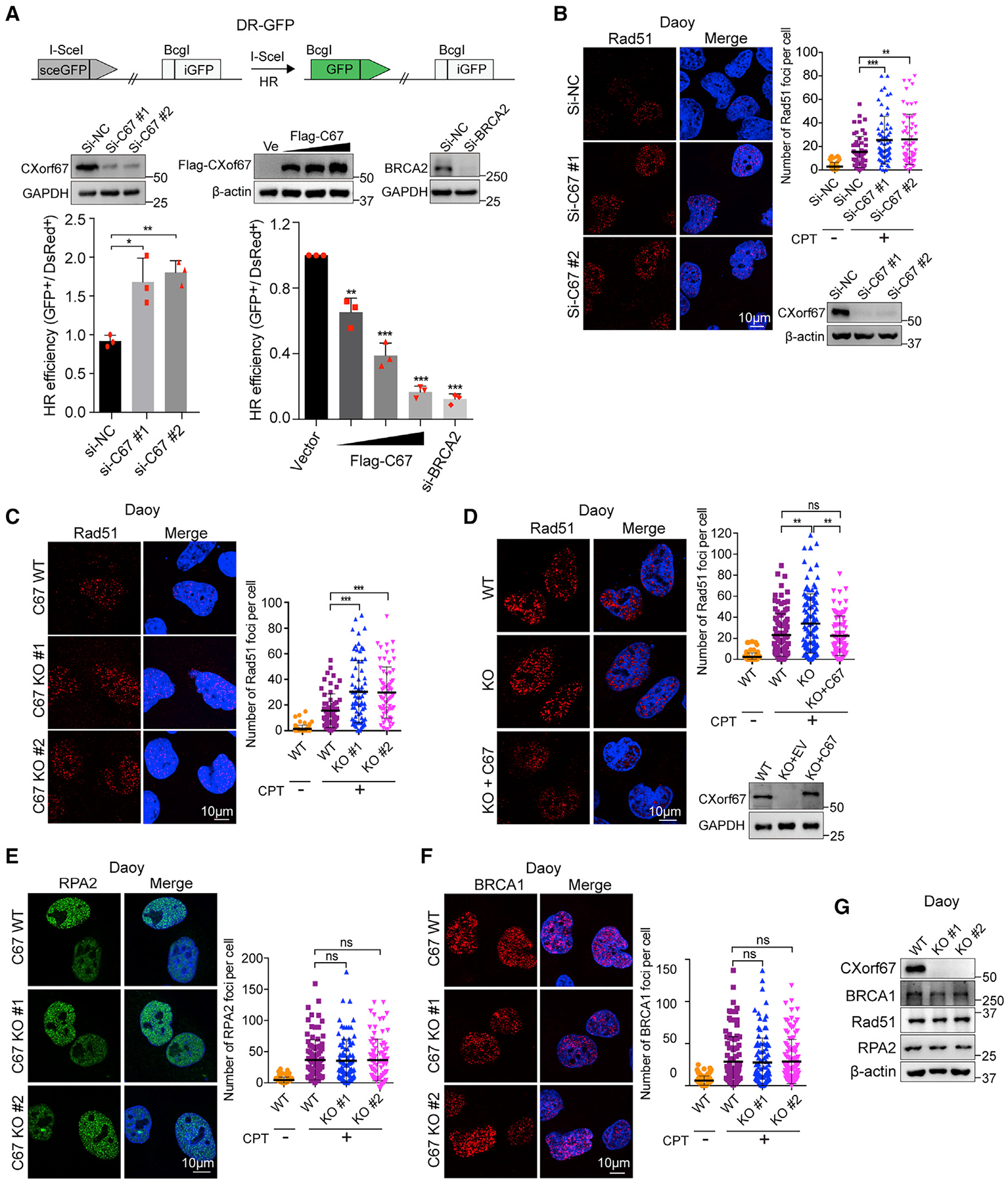
(A) Schematic illustration of the GFP-based HR reporter assay (DR-GFP). Effect of CXorf67 knockdown and overexpression on the efficiency of homologous recombination (HR) repair. U2OS DR-GFP cells were transfected with CXorf67-specific siRNA or Flag-tagged CXorf67 and control vector together with I-SceI plasmids, and the percentage of GFP-positive cells was analyzed by fluorescence-activated cell sorting 48 h after transfection. Knockdown of BRCA2 act as a positive control for inhibiting HR repair. Data are presented as mean ± SD (n = 3 independent experiments; paired t test; *p < 0.05, **p < 0.01, ***p < 0.001).
(B and C) Knockdown or knockout of CXorf67 leads to increased Rad51 foci formation. Daoy cells were transfected with CXorf67 siRNA or control siRNA for 48 h and then treated with CPT (0.1 μM) for 4 h and immunostained with an RAD51 antibody followed by Cy3-conjugated secondary antibody. Scatter dot plot represents the Rad51 foci per nuclei (right). Data are presented as mean ± SD (n = 56–64; unpaired t test; **p < 0.01, ***p < 0.001). (C) Same as above, but performed in the C67-WT and C67-KO Daoy cells. Scatter dot plot represents the Rad51 foci per nuclei (right). Data are presented as mean ± SD (n = 62–71; unpaired t test; ***p < 0.001).
(D) C67-KO 1# Daoy cells showed increased Rad51 foci formation, which is rescued by re-expression of CXorf67. Cells were treated with CPT (0.1 μM) for 4 h and immunostained with an RAD51 antibody followed by Cy3-conjugated secondary antibody. Scatter dot plot represents the Rad51 foci per nuclei (right). Data are presented as mean ± SD (n = 83–106; unpaired t test; **p < 0.01).
(E and F) Representative images of RPA2 foci and BRCA1 foci in Daoy C67-WT and C67-KO cells that were treated with CPT (0.1 μM) for 4 h. Quantification of RPA2 and BRCA1 foci per nuclei using the Image-Pro Plus software. Scatter dot plot represents the RPA2 foci per nuclei. Data are presented as mean ± SD (n = 88–96; unpaired t test; ns, not significant, p > 0.05) (E). Data are presented as mean ± SD (n = 98–110; unpaired t test; ns, not significant) (F).
(G) CXorf67, Rad51, RPA2, and BRCA1 gene expressions at the protein level in C67-WT and C67-KO Daoy cells were validated by western blotting.
See also Figure S2.
CXorf67 Interacts Directly with PALB2
HR-mediated DNA repair in eukaryotic cells is a complex, multi-step process that requires the coordination of a cohort of proteins. In brief, after the formation of γ-H2AX, BRCA1 recruits BRCA2 and RAD51 to the DSB sites through the interaction with PALB2. The BRCA1-PALB2-BRCA2 complex mediates the replication protein A (RPA) complex replacement by Rad51 and stimulates RAD51-dependent D loop formation and strand invasion (Sy et al., 2009; Xia et al., 2006; Zhang et al., 2009). Our results above suggest that CXorf67 functions between BRCA1 and Rad51. Moreover, our mass spectrometric data showed PALB2 and BRCA1 as potential binding partners of CXorf67 (Table S1). We thus wanted to know whether CXorf67 may function through interacting with PALB2 and/or BRCA1 to affect the BRCA1-PALB2-BRCA2 complex. We performed immunoprecipitation using Daoy cells and detected co-immuno-precipitation (co-IP) of PALB2 and BRCA1, but not RPA2 or Rad51, with endogenous CXorf67 (Figure 3A). Next, we overexpressed HA-tagged CXorf67 with Flag-tagged PALB2 or BRCA1 in HEK293T cells and conducted co-IP. We found that CXorf67 showed stronger interaction with PALB2 than BRCA1. Moreover, the CXorf67-BRCA1 interaction was markedly increased by PALB2 expression (Figure S3A). This result indicates that PALB2 might be the primary binding partner for CXorf67, whereas BRCA1 might interact with CXorf67 through PALB2. To identify the region of PALB2 that is required for its interaction with CXorf67, we generated a series of PALB2 deletion mutants and performed co-IP. As shown in Figure S3B, PALB2 interacted with CXorf67 largely via its C-terminal WD40 domain. To examine whether the interaction between CXorf67 and PALB2 WD40 domain is direct, we expressed and purified recombinant His-tagged CXorf67 and GST-tagged WD40 in Spodoptera frugiperda, Sf9 (Oliver et al., 2009), and then carried out an in vitro pull-down assay. Our result showed that CXorf67 and PALB2 WD40 domain interacted directly (Figure 3B). Of note, the WD40 domain of PALB2 is required for its interaction with BRCA2 in transduction of DNA repair signals (Ducy et al., 2019; Nepomuceno et al., 2017; Oliver et al., 2009). We noticed that a 13-residue sequence near the C-terminal end of CXorf67 shares amino acid sequence homology with the PALB2-binding motif of BRCA2 (Figure 3C). We next sought to determine if these 13 residues of CXorf67 (420–432) were sufficient to interact with PALB2 WD40 domain in vitro. To this end, we synthesized the biotin-labeled 13-residue peptide. Meanwhile, since Trp-31 of BRCA2, corresponding to Trp-425 of CXorf67, is reported to be crucial for the BRCA2-PALB2 interaction (Oliver et al., 2009; Xia et al., 2006), we also made a mutant peptide with Trp-425 being mutated to Cys. We then performed streptavidin pull-down using the WT or mutant peptide with purified recombinant GST-WD40. We found that the WT synthetic peptide, but not the mutant peptide, could readily pull down WD40 (Figure 3C). Consistent with this result, the full-length CXorf67 W425C mutant exhibited reduced binding to PALB2 compared with its WT counterpart (Figure 3D). Meanwhile, live-cell imaging revealed that the W425C mutation caused reduced recruitment of CXorf67 to sites of DNA damage (Figure 3E). Importantly, this W425C mutation markedly impaired CXorf67’s ability to inhibit Rad51 focus formation in Daoy cells and HR repair in DR-GFP HR reporter cells compared with WT CXorf67 (Figures 3F and 3G). In addition, the co-IP experiments revealed that CXorf67 could only interact with PALB2, but not with BRCA2 (Figures S3C–S3E). These results demonstrate that CXorf67 functions by interacting directly with PALB2 in a way similar to BRCA2 interacting with PALB2.
Figure 3. CXorf67 Interacts with the PALB2 WD40 Domain through the PALB2-Binding Motif.

(A) Immunoblot detection of anti-CXorf67 immunoprecipitates. Daoy CXorf67 WT and KO cells were treated with CPT (0.1 μM) for 1 h and lysates treated with Benzonase were precipitated with anti-CXorf67 and analyzed by immunoblotting with the indicated antibodies.
(B) Direct binding between recombinant GST-WD40 and His-CXorf67 in vitro. GST-agarose beads-bound GST-WD40 or GST proteins were incubated with purified His-CXorf67. After wash, the beads-bound proteins were subjected to western blot analysis using anti-GST and anti-His antibodies.
(C) Sequence alignment showing the similarity between CXorf67 and BRCA2 in a minimal PALB2-binging motif (top). Pull-down (PD) assays were performed by mixing purified recombinant GST-WD40 protein and biotinylated WT C67 (420–432) peptide or W425C mutant peptide for 2 h. The peptides were pulled down using streptavidin agarose beads followed by immunoblotting analyses.
(D) PALB2 co-immunoprecipitation with CXorf67 is lost in CXorf67 W425C mutant. Flag-tagged CXorf67 WT or W425C mutant were co-expressed with Myc-tagged PALB2 in HEK293T cells. CXorf67 was immunoprecipitated with an anti-Flag antibody.
(E) U2OS cells expressing EGFP-PALB2, EGFP-C67-WT, or W425C mutant were laser micro-irradiated and monitored using a live-cell imaging microscope. Representative confocal images are shown. White arrows indicate irradiated regions. Accumulation of EGFP-PALB2, EGFP-C67-WT, and W425C mutant on the DNA damage tracks was quantified. Data represent the mean values (±SD) from three independent experiments.
(F) CXorf67 W425C cannot inhibit Rad51 foci formation. Daoy C67-KO cells were transfected with HA-tagged WT CXorf67 or the W425C mutant. The cells were then treated with CPT (0.1 μM) for 4 h and stained with anti-Rad51 antibody followed by Cy3-conjugated secondary antibody. Scatter dot plot represents the Rad51 foci per nuclei. Data are presented as mean ± SEM (n = 104–113; unpaired t test; ns, not significant; *p < 0.05, **p < 0.01).
(G) CXorf67 W425C cannot inhibit HR repair in reporter cells. U2OS DR-GFP reporter cells were transfected with I-SceI and Flag-tagged WT CXorf67 or the W425C mutant. The HR efficiency was assessed by measuring the GFP-positive cells 48 h later.
Data are presented as mean ± SD (n = 3 independent experiments; paired t test; ns, not significant; *p < 0.05, **p < 0.01). See also Figure S3 and Table S1.
CXorf67 was recently also shown to inhibit the PRC2 function by interacting with EZH2, in which M406 of CXorf67 plays a key role because the CXorf67 M406E/K/R mutant no longer binds to EZH2 (Hubner et al., 2019; Jain et al., 2019; Piunti et al., 2019). To investigate whether the function of CXorf67 in HR is related to its inhibitory activity against EZH2, we tested the effect of CXorf67 M406 mutants on HR using the DR-GFP HR reporter and Rad51 focus formation assay in U2OS cells. We found that CXorf67 M406 mutants showed similar activity as its WT counterpart in inhibiting HR (Figures S3F and S3G). These results indicated that the function of CXorf67 in HR is unrelated to its inhibitory activity in PRC2 function.
CXorf67 Competes with BRCA2 for PALB2 Interaction
Since CXorf67 shares a similar PALB2-binding motif with BRCA2, we then asked whether CXorf67 may compete with BRCA2 for interaction with PALB2. For this, we carried out co-IP assays with increasing expression of CXorf67. Our results showed a dose-dependent inhibitory effect of CXorf67 on the PALB2-BRCA2 interaction (Figure 4A). Concordantly, expression of CXorf67 abrogated localization of BRAC2 to the PALB2 foci in U2OS cells (Figure 4B). To further confirm this, we used a single-cell assay assessing the co-localization of proteins at an integrated LacO array (Orthwein et al., 2015). By using this LacR/LacO system, we observed the PALB2-CXorf67 interaction recapitulated by a Myc-tagged LacR-PALB2 fusion protein with GFP-tagged CXorf67 (Figure 4C). Moreover, this system also enabled us to monitor the effect of CXorf67 on the PALB2-BRCA2 interaction. As shown in Figure 4D, expression of CXorf67 impaired the PALB2-BRCA2 interaction. Finally, we examined the endogenous effects of CXorf67 on DNA damage-induced recruitment of BRCA2 and PALB2 to chromatin. Analysis of chromatin-enriched fractions showed that DNA damage-induced accumulations of BRAC2 and Rad51 at the chromatin were significantly increased in Daoy C67-KO cells as compared with C67-WT cells, which could be reversed by restoration of CXorf67 expression in these KO cells (Figure 4E). By contrast, recruitments of RPA2, BRCA1, and PALB2 to chromatin were not affected by depletion of CXorf67 (Figure 4E). Together, these results demonstrate that CXorf67 disrupts the PALB2-BRCA2 interaction through competing with BRAC2 for PALB2 binding.
Figure 4. CXorf67 Competes with BRCA2 for PALB2 Interaction.
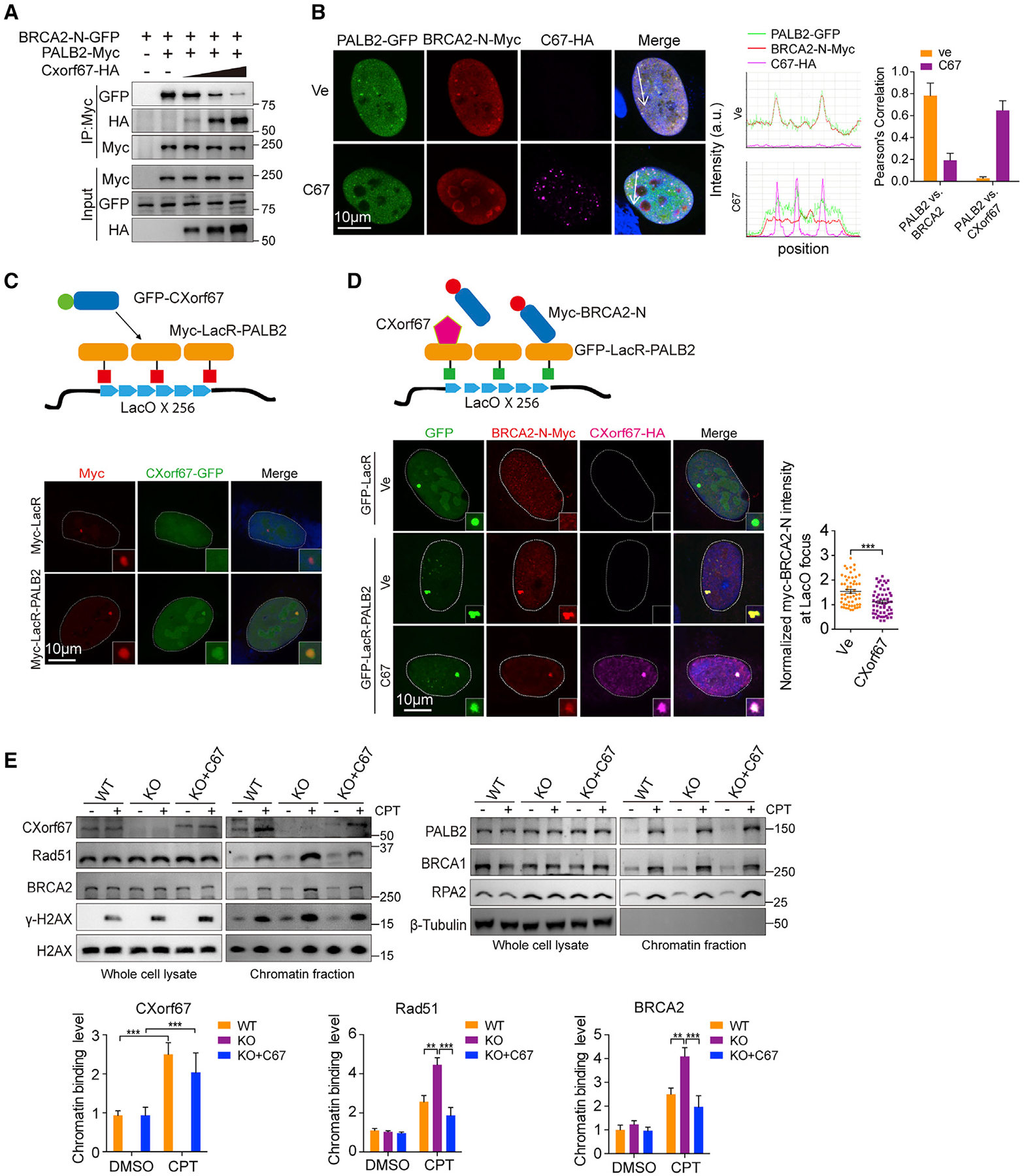
(A) Competition of CXorf67 with BRCA2 for binding with PALB2. HEK293T cells were transfected with plasmids expressing both Flag-tagged PALB2 and GFP-tagged BRCA2-N (1–200 amino acids [aa]) in the presence of increasing amounts of CXorf67 expression plasmid.
(B) CXorf67 overexpression can disrupt the interaction between PALB2 and BRCA2 in U2OS cells with CPT (0.1 μM) for 4 h (left). GFP-tagged PALB2 and Myc-tagged BRCA2-N (1–200 aa) were co-transfected with Vector (Ve) or HA-tagged CXorf67 and stained with anti-Myc (red, Cy3-conjugated secondary antibody), anti-HA (magenta, Alexa Fluor 647 conjugated secondary antibody). Co-localization was confirmed with both fluorescent intensity profiles and Pearson’s correlation coefficient determination. Fluorescence-intensity profiles of PALB2, BRCA2, and CXorf67 were obtained using Leica LAS AF software, along a straight line (white) crossing the nucleus of a representative cell (middle). Histograms represent mean ± SD of Pearson’s correlation coefficient between PALB2 and BRCA2 or PALB2 and CXorf67 from 20 randomly selected cells in each group (right).
(C) Schematic representation of the lacO/LacR chromatin-targeting protein interaction assay system. U2OS-lacO cells expressing Myc-LacR-PALB2 or Myc-LacR together with GFP-CXorf67 were immunostained with anti-Myc antibody followed by Cy3-conjugated secondary antibody.
(D) Representative micrograph and quantitative assessment of the PALB2-BRCA2-N (1–200 aa) interaction in U2OS-lacO cells transfected with CXorf67 or Ve (Vector). Right: the relative enrichment of Myc-BRCA2-N (1–200 aa) was determined as the ratio of its fluorescence intensity at lacO to the fluorescence intensity of LacR-PALB2 at lacO in U2OS-lacO cell line with CXorf67 or Ve. Data are presented as mean ± SEM (n = 56; unpaired t test; ***p < 0.001).
(E) Western blots with total or chromatin fractions from Daoy C67-WT cells, C67-KO cells, or C67-KO cells re-expressing C67 at 4 h after CPT (0.1 μM) treatment using the indicated antibodies. The protein levels of CXorf67, Rad51, and BRCA2 in chromatin-enriched fractions were measured by ImageJ software.
Data are presented as mean ± SD (paired t test; *p < 0.05, **p < 0.01, ***p < 0.001).
CXorf67 High Expression Sensitizes Tumor Cells to PARP1/2 Inhibition
Poly(ADP-ribose) polymerase (PARP) inhibition was initially identified as a synthetic lethal therapy for patients with BRCA-mutant cancers (Bryant et al., 2005; Farmer et al., 2005). Subsequently, PARP inhibitors were found to selectively kill tumors with defects in HR repair caused by mechanisms other than BRCA mutations (Lord and Ashworth, 2016, 2017). Thus, we asked whether tumor cells with high CXorf67 expression might have increased sensitivity to PARP inhibitors. To test this possibility, we added the PARP inhibitor talazoparib to Daoy C67-WT and -KO cells. Our results showed that knockout of CXorf67 resulted in decreased sensitivity to the PARP inhibitor (Figure 5A). Next, we added PARP inhibitor talazoparib or olaparib to C67-KO Daoy cells with or without exogenous expression of CXorf67. Our results showed that re-expression of CXorf67 in C67-KO Daoy cells led to significantly enhanced sensitivity to both talazoparib and olaparib (Figure 5B). Moreover, we included two human glioblastoma cell lines, U251 and U87, which showed no detectable CXorf67 protein. Expression of CXorf67 in these two cell lines also led to an increased cell sensitivity to the PARP inhibitor (Figure S4A).
Figure 5. High CXorf67 Expression is Correlated with Increased Sensitivity to Poly(ADP-Ribose) Polymerase Inhibitors.
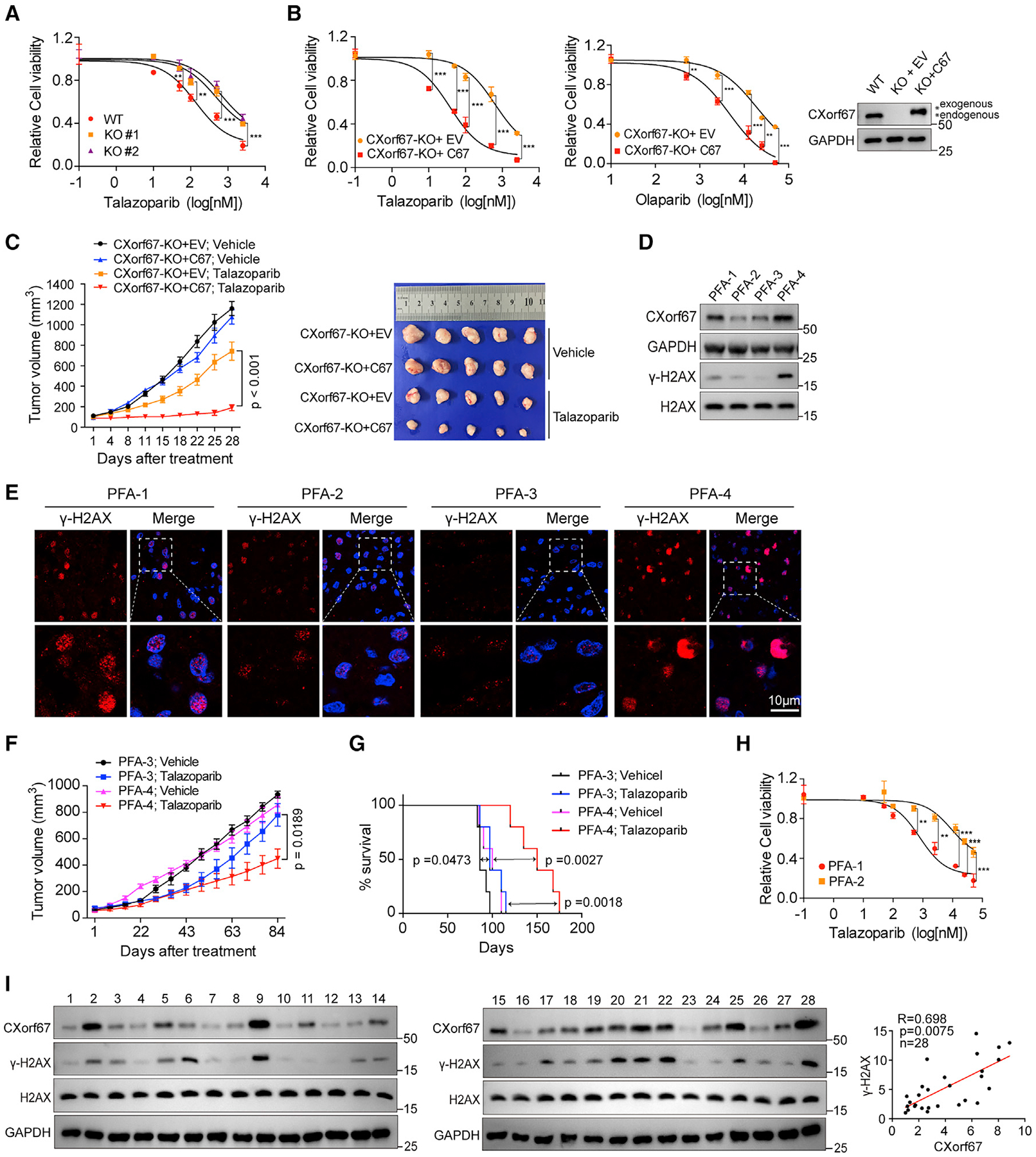
(A) CXorf67 deficiency in Daoy cells decreases sensitivity to the PARP inhibitor talazoparib. Cell viability was assessed by the CellTiter-Glo assay. Data are presented as mean ± SD (n = 3, two-way ANOVA; **p < 0.01, ***p < 0.001).
(B) Re-expression of CXorf67 in Daoy C67-KO cells increases sensitivity to PARPi. Cell viability was assessed by the CellTiter-Glo assay. Data are presented as mean ± SD (n = 3, two-way ANOVA; **p < 0.01, ***p < 0.001).
(C) Daoy xenograft tumor model. 3 ×106 C67-KO cells and C67 re-expressing Daoy cells were injected subcutaneously. When tumors reached approximately 100 mm3, mice were randomly assigned to treatment with vehicle or talazoparib (0.33 mg/kg) (n = 5 for each group). Average tumor volumes ± SEM are shown. Statistical significance differences between groups were calculated using two-way ANOVA. Bottom: the tumors were collected and photographed at 28 days of treatment.
(D) Western blots showing CXorf67 and γ-H2AX levels in untreated patient-derived posterior fossa brain tumors (four ependymoma tumors: PFA1–4).
(E) Representative IF micrographs showing γ-H2AX expression in PFA1–4 tumor specimens.
(F) Tumor volume (mm3) measurements in PFA-3 and PFA-4 PDX models treated with vehicle and talazoparib. Significant difference in tumor volume from PFA-3 and PFA-4 is calculated (two-way ANOVA).
(G) Kaplan-Meier survival plots for PDX xenografts (PFA-3 and PFA-4) treated with vehicle or talazoparib (0.33 mg/kg) (n = 5 for each group). Mice were euthanized when tumor volume reached 1,000 mm3, p values from log rank (Mantel-Cox) test are shown.
(H) Sensitivity of two primary PFA cells to talazoparib. Survival analysis of PFA-1 and PFA-2 cells treated for 5 days with talazoparib. Data are presented as mean ± SD (two-way ANOVA; **p < 0.01, ***p < 0.001).
(I) Western blot analyses of 28 untreated specimens from posterior fossa ependymoma patients were performed using anti-CXorf67 and anti-γ-H2AX antibodies. ImageJ software was used for quantification. The levels of CXorf67 were normalized to GAPDH loading control. The levels of γ-H2AX were normalized to H2AX loading control. Pearson correlation coefficient analysis between normalized CXorf67 and γ-H2AX was performed.
See also Figure S4.
To investigate the potential role of CXorf67 in tumor cell sensitivity to PARP inhibition in vivo, we tested talazoparib sensitivity using xenograft tumor models inoculated with Daoy C67-KO cells or Daoy C67-KO cells re-expressing CXorf67. We found that Daoy tumors expressing CXorf67 were more sensitive to talazoparib treatment than those lacking CXorf67 (Figures 5C and S4B). Of note, the tumors with and without CXorf67 expression showed similar growth rates without the treatment (Figure 5C). Knowing that CXorf67 is upregulated in PFA, we then asked whether PFA tumors could respond to PARP inhibitors. For this purpose, we collected four fresh brain PFA ependymoma tumors (PFA1–4) with varying levels of CXorf67 expression (Figures 5D and S4C). We also noticed that there seemed to be a positive correlation between the levels of CXorf67 and γ-H2AX in these four PFA ependymoma tumors (Figures 5D and 5E).
We were able to successfully establish patient-derived xenograft (PDX) models from PFA-3 and PFA-4. The PDX tumors derived from PFA-4, which had higher CXorf67 expression than PFA-3, showed significant greater sensitivity to talazoparib than those from PFA-3 (Figures 5F, 5G, and S4D). Although we were only able to establish short-term cultures from the PFA-1 and PFA-2 tumors, they were tested for their sensitivity to talazoparib. The PFA-1 culture, which had higher CXorf67 expression than the PFA-2 culture, also exhibited significantly higher sensitivity to talazoparib than did PFA-2 (Figure 5H). Together, these results further support a positive correlation between the CXorf67 levels and tumor cell sensitivity to PARP inhibitors in PFA tumors.
To further confirm the positive correlation between the levels of CXorf67 and γ-H2AX, we collected 28 human PFA ependymoma specimens and examined the CXorf67 and γ-H2AX levels by western blot analysis. Significant positive correlation between the CXorf67 protein and γ-H2AX levels was observed (Figure 5I). The increases in the steady-state levels of γ-H2AX that are associated with elevated CXorf67 protein level are likely due to impaired DNA damage repair caused by high CXorf67 expression. We also examined the interaction of CXorf67 with PALB2-and CPT-induced Rad51 focus formation in cultured PFA-3 and PFA-4 cells. We found that CXorf67 and PALB2 also coimmunoprecipitated in these primary PFA cells (Figure S4E). Importantly, high expression of CXorf67 in PFA-4 appeared to be correlated with reduced co-IP of BARC2 (Figure S4E) and number of Rad51 foci (Figure S4F) in these cells in comparison with PFA-3 cells, in which CXorf67 expression is lower than that in PFA-4. These results from primary PFA cells further confirm the conclusion that CXorf67 inhibits the PALB2-BRCA2 interaction and Rad51 foci formation.
PARPi and IR in Combination Effectively Kill Tumor Cells Expressing CXorf67
Radiotherapy is a standard therapy for PFA patients. Knowing that CXorf67 inhibits HR, we thus postulated that radiotherapy might facilitate PFA tumor cell sensitivity to PARP inhibitors. To investigate this possibility, we exposed C67-WT and -KO Daoy cells to different does of IR followed by treatment of talazoparib. We found that C67-WT Daoy cells that are sensitive to talazoparib were also more sensitive to IR than C67-KO cells. Importantly, talazoparib and IR treatment in combination showed additional effects in suppressing tumor cell growth of both C67-WT and -KO Daoy cells (Figure S5A). More importantly, the half maximal inhibitory concentration (IC50) of talazoparib was reduced from 140.4 to 7.8 nM in C67-WT Daoy cells when irradiation was used, indicating a strong combination effect between PARPi and IR (Figure 6A). The combination effects were also observed in C67-KO Daoy cells restored with CXorf67 expression, when cells were irradiated in the presence of three different PARP inhibitors (talazoparib, niraparib, and olaparib) (Figures 6B and S5B). We also examined the combination effect of PARPi and IR on the induction and persistence of DSBs and apoptosis in C67-KO Daoy cells with or without CXorf67 expression restored. We found that the combination of talazoparib and IR resulted in further increases in γ-H2AX foci at 24 h after treatment (Figure 6C) and apoptosis (Figure S5C) compared with single treatment. In addition, the effects of these treatments are stronger in cells with CXorf67 expression restored than the KO cells.
Figure 6. PARPi and IR in Combination Effectively Kill Tumor Cell-Expressing CXorf67.
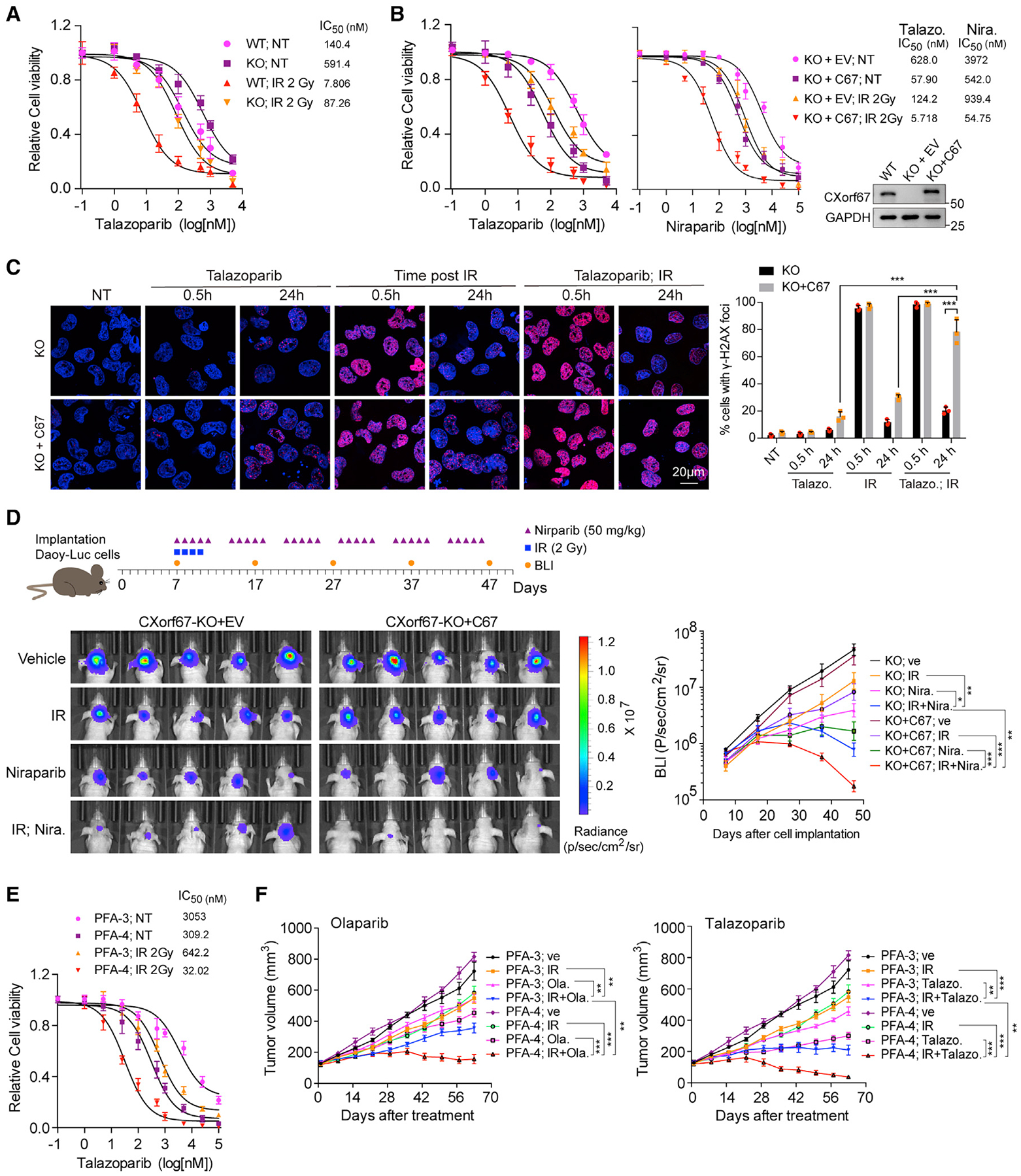
(A) PARP inhibitor and IR show more effective in killing Daoy cells expressing CXorf67. C67-WT and -KO Daoy cells were treated with different doses of talazoparib immediately after irradiation (2 Gy). Cell viability was assessed by the CellTiter-Glo assay after a 5-day treatment. Data are presented as mean ± SD from three independent experiments. The IC50 value was calculated using GraphPad software.
(B) Re-expression of CXorf67 in C67-KO Daoy cells increases sensitivity to PARPi and IR combination. Cell viability was assessed by the CellTiter-Glo assay. Data are presented as mean ± SD. Western blot showing expression levels of CXorf67.
(C) γ-H2AX foci formation in C67-KO and C67-KO with C67 re-expressed Daoy cells. Representative IF images of cells at 0.5 h, 24 h after talazoparib (5 nM), IR (2 Gy), or combination treatment are shown. Right: the percentage of positive cells with γ-H2AX foci (>10). At least 50 cells were analyzed for each datum point. Data are presented as mean ± SD (n = 3 independent experiments; paired t test; *p < 0.05, **p < 0.01).
(D) Representative scheme and images for orthotopic xenograft experiment. Luciferase-expressing stable cell lines in C67-KO and C67-KO with C67 re-expressed Daoy cells were established and implanted in the cerebellum of nude mice (2 ×105 cells per mouse). The mice were randomized into four treatment groups (n = 5 per group) after 7 days from the implantation: vehicle (0.5% methylcellulose in PBS), niraparib (50 mg/kg in 0.5% methylcellulose, orally once daily for 5 days per week), 8 Gy fractionated radiotherapy (2 Gy daily for 4 days), and niraparib combined with 8 Gy fractionated radiotherapy. The bioluminescence imaging levels were acquired every 10 days using an IVIS SpectrumCT imaging system. The radiance (photons) within each area of interest was determined using Living Image Software 4.5.4 (PerkinElmer). Data are presented as mean ± SD (two-way ANOVA; *p < 0.05, **p < 0.01, ***p < 0.001).
(E) Combined treatment with talazoparib and ionizing radiation (IR) enhanced killing of ependymoma cell cultures. PFA-3 and PFA-4 cells were treated with different dose of talazoparib after exposed to IR (2 Gy). Cell viability was assessed by the CellTiter-Glo assay. Data are presented as mean ± SD.
(F) Tumor volume (mm3) measurements in PFA-3 and PFA-4 PDX models treated with vehicle (10% β-cyclodextrin in PBS), olaparib (50 mg/kg in 10% β-cyclodextrin, orally once daily for 5 days per week), talazoparib (0.33 mg/kg in 10% DMAC and 6% Solutol, orally once daily for 5 days per week), IR (2 Gy daily for 4 days), olaparib combined with IR, and talazoparib combined with IR. Data are presented as mean ± SD (two-way ANOVA; *p < 0.05, **p < 0.01, ***p < 0.001). The experiments in the left and right panels were performed simultaneously with a single control experiment (P3; Ve, P4; Ve, P3; IR, and P4; IR).
See also Figure S5.
To investigate the effect of PARPi on tumors located in the brain, we generated stable luciferase-expressing cell lines in Daoy cells that express CXorf67. After implanting corresponding cells into the cerebellum of immune compromised nude mice, we treated the mice with niraparib (a PARP inhibitor that can pass the blood-brain barrier), IR, and their combination. As shown in Figure 6D, the combination of niraparib and IR showed strongest effect on suppression of tumor growth. We also tested the effect of PARPi and IR combination on PFA-3 and PFA-4 cell cultures and found that the PFA-4 culture, which had higher CXorf67 expression than the PFA-3 culture, exhibited higher sensitivity than PFA-3 did (Figure 6E). Meanwhile, we knocked down CXorf67 using short hairpin RNA (shRNA) in PFA-3/4 cells and found that CXorf67 knockdown reduced tumor cell sensitivity to talazoparib compared with control (Figure S5D). In addition, we performed combination treatment of PARPi with IR in PFA-3 and PFA-4 PDX models. Similar to the in vitro results observed in patient-derived primary cells (Figure 6E), talazoparib or olaparib in combination with radiotherapy showed stronger effect on tumor growth suppression in PFA-4 than in PFA-3 PDX (Figure 6F). In fact, the combinations resulted in almost complete tumor regression in most mice of the PFA-4 group (Figure S5E). Taken together, these results consistently showed that, when combined with IR, the efficiency of PARPi treatment is substantially enhanced in tumor cells with high CXorf67 expression.
DISCUSSION
PFA ependymoma has a very poor prognosis; there are currently no effective chemotherapies for PFA patients and up to 40% of cases are incurable. In this study, we uncovered that CXorf67, which is overexpressed in PFA ependymomas, suppressed HR repair by interacting directly with PALB2 to block its interaction with BRCA2. More importantly, our results show that tumor cells with higher CXorf67 expression are more sensitive to PARP inhibitors. Therefore, our findings identify CXorf67 as a potential biomarker of response to PARP inhibitors. In addition to PFA ependymoma, we found upregulation of CXorf67 in several other cancer types. Our analysis reveals that high-level CXorf67 expression is correlated with reduced overall survival in kidney renal clear cell carcinoma and kidney renal papillary cell carcinoma (data not shown). Whether CXorf67 could be a biomarker for PARP inhibitor treatment in other tumors certainly warrants further investigations.
So far, the molecular basis for CXorf67’s upregulation in PFA ependymoma remains unknown. During the preparation of this manuscript, we noted a newly released work that links upregulation of CXorf67 in PFA with hypoxia (Michealraj et al., 2020). This study shows that PFA ependymomas are maintained in hypoxic condition, which may induce CXorf67 expression. CXorf67 upregulation inhibits PRC2 function, which leads to epigenetic changes and consequently contributes to PFA tumor growth. To understand the mechanisms behind CXorf67 upregulation in PFA ependymoma, more work is required to dissect the relationships among hypoxia, expression of CXorf67, and epigenetic changes of CXorf67 promoter.
PFA ependymomas are known as epigenetically deregulated tumors, and current therapeutic efforts mainly focus on targeting epigenetics-related mechanisms (Mack et al., 2014; Michealraj et al., 2020). Previous studies revealed CXorf67 as a regulator of PRC2 function (Hubner et al., 2019; Jain et al., 2019; Piunti et al., 2019). In this work, we reveal another function of CXorf67 in inhibiting DNA HR repair. Despite a modest effect of CXorf67 on sensitizing to PARPi, when combined with radiotherapy, the IC50 of PARPi is dramatically decreased from a micromolar level to a nanomolar level for PFA tumors with high CXorf67 expression. Therefore, our work here provides another strategy for the treatment of PFA ependymoma with immediate clinical applicability.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Lin Li (lli@sibs.ac.cn).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
This study did not generate datasets and code. Previously published expression datasets used in this study are available through GEO: GSE64415, GSE94349 as well as The Cancer Genome Atlas (TCGA). The raw data of this study have been deposited in Mendeley Data and available at https://doi.org/10.17632/rjxfxzsh5m.1.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patients and Tumor Samples
Ependymoma tumor samples were collected from Children’s Hospital of Fudan University using protocols approved by the Institutional Review Board. Informed consent was obtained from all patients. Tissue samples were snap-frozen at the time of resection. No patient underwent chemotherapy or radiotherapy before the surgical removal of the primary tumor. At least 80% of tumor cell content was estimated by staining cryosections of each sample with haematoxylin and eosin. Diagnoses were confirmed by histopathologic assessment by at least two neuropathologists, including a central pathology review that use the 2007 World Health Organization classification for Central Nervous System tumors.
Animals
All the animal experiments and protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of Shanghai Institute of Biochemistry and Cell Biology (SIBCB), Chinese Academy of Sciences. The approval ID for the use of animals was SIBCB1812030 issued by the Animal Core Facility of SIBCB.
BALB/cASlac-nu (BALB/c nude) mice were purchased from Shanghai SLAC laboratory and NOD.CB17-Prkdcscid/NcrCrl (NOD SCID) mice from Vital River Laboratories (Beijing, China). Mouse models of Daoy cell line-dervied subcutaneous xenografts and orthotopic xenografts for bioluminescence imaging were established by transplanting Daoy C67-KO cells or Daoy C67-KO cells re-expressing CXorf67 into 6-week-old female BALB/c nude mice. The subcutaneous ependymoma PDX model was established by transplanting minced fresh tumor tissue into 6-week-old male NOD SCID mice. After establishment of PFA-3 and PFA-4 PDX models, tumor tissue was minced and dissociated into single cells, which were then injected s.c. into right flanks of nude mice (female, six weeks). Mice were randomly assigned into control or treatment groups. Prior to experiments, mice were allowed one week to acclimate to housing conditions at the SIBCB Animal Facility. All experimental mice were housed in a pathogen-free environment and treated in strictly accordance with approved protocols.
Cell Line
All cell lines were grown at 37°C and 5% CO2. All culture media were supplemented with 10% fetal bovine serum (FBS). U-2-OS (U2OS) cells were purchased from ATCC and cultured in 1640 medium (Gibco). Daoy cells were kindly provided by Stem Cell Bank, Chinese Academy of Sciences and grown in Minimum Essential Media (MEM; Gibco). U2OS DR-GFP cell line were kindly provided by Dr. Jun Huang (Zhejiang University). U2OS-LacO cell line was kindly provided by Dr. Fangwei Wang (Zhejiang University). They were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Gibco). HEK293T cells were also maintained in DMEM. All these cell lines were confirmed to be free of mycoplasma contamination and authenticated by STR DNA profiling. Ependymoma short-term primary cell culture (PFA-1 and PFA-2) and ependymoma cell culture from xenografts (PFA-3 and PFA-4) were cultured in Neurobasal meida (Gibco) consisting of B27 (Gibco), N2 (Gibco), GlutaMAX (Gibco), EGF (Gibco), FGF (Gibco) and heparin (Sigma).
METHOD DETAILS
Plasmids
Full-length CXorf67 and PALB2 were amplified by PCR from cDNA library and cloned into the pCMV-Flag, pCMV-HA, pEGFP-C1, GFP-LacR and Myc-LacR vectors using ClonExpress II (Vazyme). The GFP-LacR and Myc-LacR vectors were gifts from Fangwei Wang at Zhejiang University. The WD40 domain of PALB2 (residues 853–1186) was amplified by PCR and cloned into the GST expression vector pFastBacI. Full-length CXorf67 was amplified by PCR and cloned into vector pET28a carrying an N-terminal His tag. Truncated forms of PALB2 were generated by PCR and cloned into pCMV-Flag vector. The N-terminal domain of BRCA2 (residues 1–200) was amplified by PCR and induced into GFP-LacR and Myc-LacR vectors.
Cell Culture and Transfection
Plasmid transfection was performed using Lipofectamine 3000 Transfection Reagent (Invitrogen) according to the manufacture’s protocol. RNA interference (RNAi) transfection was carried out using Lipofectamine RNAiMax (Invitrogen) in a forward transfection mode. Small-interfering RNA (siRNA) duplexes targeting CXorf67 (#1: GGAGGGCUGAACAACGAAAUU; #2: GCUGCUGUGCUGACGGAGAUU); targeting BRCA2 (GAAGAACAAUAUCCUACUA); targeting Ku80 (GCGAGUAACCAGCUCAUAA), and a non-targeting control siRNA (UUCUCCGAACGUGUCACGUUU) were synthesized by GenePharma (Shanghai, China).
Neutral Comet Assays
Single-cell gel electrophoretic comet assays were performed as previously described (Xie et al., 2018). Briefly, the cells were irradiated with 10 Gy of IR and collected at the indicated time. After cells were harvested and rinsed twice with ice-cold PBS, 5 ×105 cells/ml were mixed with OxiSelect™ Comet Agarose (235002, Cell Biolabs) at a ratio of 1:3(V/V) and immediately pipetted onto OxiSelect™ 3-Well Comet Slides (STA-352). The slides were put in the 4°C refrigerator for 30min and then treated with neutral lysis buffer (2.5 M NaCl, 100 mM Na2EDTA, 1% sodium lauroyl sarcosine, 10 mM Tris, pH 7.5, 1% Triton-X100 and 10% DMSO) overnight. Next, the slides were subjected to electrophoresis at 25 V for 30 minutes and stained in 50 μg/ml DAPI (D8417, Sigma) for 10 minutes. Images were captured using a fluorescence microscope (Olympus BX51) and analyzed with the CometScore software.
Immunofluorescence and Quantification
Cells were grown on glass coverslips (12-545-80, Fisherbrand), fixed with 4% (w/v) paraformaldehyde (P6148, Sigma) in PBS for 15 minutes at room temperature, washed with 1 x PBS, permeabilized with 0.2% (v/v) Triton X-100 for 15 minutes and blocked with 5% BSA in PBS for 1 h. Subsequently, cells were washed three times for 5 minutes with PBS and then incubated with the primary antibody diluted in PBS containing 0.25% BSA at 4°C overnight. Primary antibodies used were Rad51 (ab88572, Abcam), pS139 H2A.X (05-636, Millipore), BRCA1 (D-9, Santa Cruz), RPA2 (A2189, ABclonal), LOC340602 (sc-515296, Santa Cruz), HA (3724, Cell Signaling), Flag (F1804, Sigma), Myc (2276, Cell Signaling). Cells were next washed three times with PBS and then incubated with the appropriated secondary antibody supplemented with 0.5 μg/ml of DAPI (D8417, Sigma) for 1 h at room temperature. Corresponding Alexa Fluor 488 (A21206, Invitrogen), Cyanine Cy3 (115-165-062, Jackson ImmunoResearch) and Alexa Fluor 647 (A21245, Invitrogen) were used as secondary antibody. Coverslips were mounted onto slides with Vectashield antifade mounting medium (H-1400, Vector). Confocal images were acquired using a Lecia TCS SP8 laser scanning microscope. Rad51, BRCA1 and RPA2 foci were quantified using the Image-Pro Plus software. Foci pictures of each individual experiment were obtained with the same exposure parameters.
Immunofluorescence for PALB2 and BRCA2 interactions were performed as follows. U2OS-LacO cell line was a generous gift from Dr. Fangwei Wang at Zhejiang University. The cells were transiently transfected with GFP-LacR-PALB2 and Myc-BRCA2-N with or without CXorf67-HA co-transfected. The process of immunofluorescence staining and images capture were indicated above. Quantification of fluorescent intensity was performed with ImageJ software using images acquired at identical illumination settings following previously described protocols (Zhou et al., 2017).
Chromatin Isolation
Chromatin isolation was carried out by previously described. Briefly, U2OS cells were treat with CPT for indicated time, and 4 × 106 cells were harvested and washed with PBS. Cells were resuspended in 200 μl of buffer A (10 mM HEPES, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.34 M Sucrose, 10% glycerol, 1 mM DTT, 10 mM NaF, 1 mM Na2VO3, 1 mM PPi and protease inhibitor) containing 0.1% Triton X-100, and incubated for 10 min on ice. After centrifugation (1300 g at 4°C, 5 min), cytoplasmic proteins were separated from nuclei. The nuclei were then washed once with buffer A and resuspened with 200 μl of buffer B (3 mM EDTA, 0.2 mM EGTA, 1 mM DTT, 10 mM NaF, 1 mM Na3VO4, 1 mM PPi and protease inhibitor), and incubated for 40 min at 4°C. Insoluble chromatin was collected by centrifugation (1700 g at 4°C, 5 min) and washed once with buffer B. The chromatin pellet was resuspended in Laemmli buffer and sonicated for 60 s.
Generation of Cxorf67 Knockout Cells by CRISPR/Cas9
To delete Cxorf67 gene in U2OS or Daoy cell line, we designed two independent guide RNAs (gRNAs) using the CRISPR design tool (http://crispr.mit.edu). The gRNA sequences were as follows: Cxorf67 #1: ACGGCTCAGGCGGTGTTGCG and Cxorf67 #2: ATCTTGATTCCCGGTCCCGC. The gRNA sequences were cloned into pSpCas9(BB)-2A-GFP (PX458) all-in-one plasmid (48138, Addgene) according to the standard protocol. The gRNA/Cas9 expression construct #1 and #2 were co-transfected into U2OS or Daoy cells. 24 h after transfection, the cells were enriched by fluorescent-based sorting using a FACS Aria Sorp (BD Biosciences) and transferred into 96-well plates at ~ 1 cell per well. After PCR screening, the candidate clones were analyzed by western blotting and sequencing.
HR and NHEJ Reporter Assays
U2OS DR-GFP was a generous gift from Dr. Jun Huang at Zhejiang University. Daoy EJ5-GFP cell line was generated by our lab. To monitor transfection efficiency, we developed a new I-SceI-T2A-dsRed construct on the basis of I-SceI (pCBASceI). The efficiency of homologous recombination was determined by the ratio of cells exhibiting both GFP and dsRed signals to all dsRed cells as previously described(Lou et al., 2017). Briefly, 3 × 105 U2OS DR-GFP or EJ5-GFP cells were seeded in 6-well plates and were cultured for 24 h. The cells then were transfected with 3 μg of I-SceI and different concentrations of CXorf67-HA using Lipofectamine 3000 transfection kit (L3000015, Invitrogen). After 48 h, the cells were harvested and analyzed by fluorescence-activated cell sorting (FACS) using LSR II (BD Biosciences) or CytoFLEX LX (Beckman Coulter). At least 50,000 cells were counted. Three independent experiments were performed.
Immunoprecipitation (IP) and Mass Spectrometry
For co-immunoprecipitation experiments, U2OS or HEK293T cells were transfected with indicated plasmids for 24 h. Cells were collected and washed once with PBS, and then resuspended in lysis buffer (0.5% NP40, 2 mM EDTA, 50 mM Tris-HCl and 150 mM NaCl) supplemented with protease inhibitor cocktail (Roche), 10 mM NaF, 2 mM Na3VO4 and 1 mM PPi. Cells were sonicated three times for 10s each using lower power (Bioruptor UCD-200, Diagenode) and treated with 50 U Benzonase (70746, Novagen) for 30 min at 4°C. After centrifugation (13500 rpm, 15 min, 4°C), the supernatants were incubated with corresponding antibodies for 2 h or overnight at 4°C. The antibodies used were Flag (2368, CST), HA (901515, Biolegend), Myc (2276, CST), GFP (11814460001, Roche), PALB2 (ab220861, Abcam) and CXorf67 (sc-515296, Santa Cruz). Protein A/G Plus-Agarose beads (sc-2003, Santa Cruz) were added for an additional hour and then washed three times. The beads were resuspended in SDS loading buffer and boiled at 95°C for 5 min.
For mass spectrometry, whole cell lysates from Daoy cells were prepared for IP using the CXorf67 antibody (sc-515296, Santa Cruz) and normal mouse IgG (sc-2025, Santa Cruz). The process of IP was carried out as described above. The IP samples were analyzed by mass spectrometry.
Laser Micro-Irradiation and Live-Cell Imaging
U2OS or Daoy cells were grown overnight on 35-mm glass bottom dish (D35-20-1-N, Cellvis) and then transfected with pEGFP-CXorf67 WT or W425C mutant construct, which were maintained with 10 mM BrdU (B9285, Sigma) for 24 h. Cells were stained with Hoechst 33342 (6249, ThermoFisher scientific) for 15 min and washed three times with new medium prior to micro-irradiation. Next, the cells were placed into a cell culture chamber (37°C, 5% CO2) on an inverted microscope (Eclipse Ti, Nikon). Micro-irradiation was carried out by scanning the regions of interest 20 times using 20% of the 405-nm laser power controlled by NIS-Elements software. Images were acquired every 30 s for 10 min and exported as TIFF files.
Protein Purification and GST Pull-down Assay
CXorf67 was cloned into pET-28a vector with an N-terminal His tag and transformed into E. Coli BL21 cells grown at 37°C in Terrific Broth (TB) medium supplemented with 25 mg/L kanamycin and 25 mg/L chloramphenicol. At absorbance at 600nm (A600) =0.6, expression was induced by adding 0.2 mM IPTG for 16 h at 16°C. Cells were then harvested, resuspended in lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM DTT and 5% glycerol) containing protease inhibitor cocktail (Roche) and disrupted by a low-temperature homogenizers FB-110X (LiTu, China) at 800 MPa. After centrifugation at 20,000 g for 60 min at 4°C, the soluble fraction was incubated with Ni Sepharose (17-5318-02, GE Healthcare) for 2 h at 4°C and washed three times with washing buffer (50mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM DTT, 5% glycerol and 20 mM imidazole). The protein was then eluted with 500 mM imidazole and desalted using a HiPrep Desalting Column (17-5087-01, GE Healthcare) according to the manufactures’ protocols. The samples were stored in aliquots at −80°C.
PALB2-WD40 (853–1186 aa) was cloned into pFastBacI vector with an C-terminal GST tag. The GST-WD40 protein was expressed in Spodoptera frugiperda (Sf9) cells (Expression Systems) using Bac-to-Bac Baculovirus Expression System (A11101, Invitrogen). Briefly, cells were harvested by centrifugation and the pellet was resuspended on ice in Buffer A: 50 mM HEPES, pH 7.5, 250 mM NaCl, 10 mM imidazole, 1mM DTT, supplemented with protease inhibitors (Roche). Cells were lysed through hand-homogenization combined with thawing process. After centrifugation, the supernatant was incubated with Glutathione Sepharose beads (17-5132-01, GE Healthcare) for 2 h at 4°C and washed three times with GST buffer. GST-tagged PALB2-WD40 was eluted with elution buffer (50 mM Tris-HCl, 20 mM reduced glutathione, pH 8.0) followed by desalting. The purified protein samples were stored at −80°C.
For pulldown assay, purified His-CXorf67 (0.25 μg) was incubated with 0.25 μg of purified GST-PALB2-WD40 or purified GST for 20 min at 4°C in 100 μl NETN buffer (100 mM NaCl, 20 mM Tris-HCl, 0.5 mM EDTA and 0.5% NP-40). Then, 25 μl of GST beads and 400 μl of NETN buffer were added for 30 min. Beads were washed four times with NETN buffer. Proteins were detected by western blotting using the indicated antibodies.
Biotin Pull-down Assay
Pull-down assay was carried out with biotinylated peptides corresponding to either residues 420–432 of CXorf67 (Biotin-GGPIPQQWDESSSSS) or to the same sequence containing a single point mutant W425C (Biotin-GG-PIPQQCDESSSSS). Excess of peptides were incubated with 30 μl of Streptavidin Agarose beads (SA100–04, Invitrogen) pre-equilibrated with binding buffer (20 mM HEPES, pH 7.5, 100 mM NaCl, 1 mM DTT, 1 mM EDTA, 0.01% IGEPAL CA-630) for 1 h at 4°C. Beads were washed three times and then incubated with purified recombinant GST-WD40 (0.5 μg) for 2 h at 4°C. The beads were washed three times and resuspended in 30 μl of 2 x SDS loading buffer followed by immunoblotting analyses.
Primary Cell Culture and Cell Viability Assays
Following patient’s consent to the Institutional Review Board-approved tissue bank protocol at Children’s Hospital of Fudan University, fresh ependymoma samples (PFA-1: male, 29 months; PFA-2: male, 62 months) were collected during surgery and quickly transferred to the tissue culture room in Neurobasal medium on ice. Tumor tissues were washed with sterile phosphate-buffered saline (PBS) and minced into small fragments, followed by dissociation with 0.2% collagenase type 1 (LS004196, Worthington) and Accutase (A6964, Sigma). Single cells were collected with a 70 μm cell strainer, and then resuspended and cultured on Laminin (L2020, Sigma) coated 60 mm dishes in Neurobasal media (21103049, Gibco) consisting of B27 (17504044, Gibco), N2 (17502048, Gibco), GlutaMAX (35050061, Gibco), EGF (PHG0311, Gibco), FGF (PHG0261, Gibco) and heparin (H3149, Sigma). The culture media was replenished every other day while leaving half of the conditioned media to encourage cell proliferation. Cell viability assays: the cells were seeded in white-walled clear-bottom 96-well plates (3160, Costar) and treated with DMSO or various concentrations of drug in the following day. After incubation for 5 days, cell viability was measured using the CellTiter-Glo Kit (G7572, Promega) according to manufacturer’s protocol. Talazoparib (HY-16106), Niraparib (HY-10619) and Olaparib (HY-10162) were obtained from MedChem Express.
For PFA-3 and PFA-4 cell viability assays, the cells were isolated from corresponding PDXs, and cultured as described above. The shRNAs using to knockdown CXorf67 in PFA-3 and PFA-4 cells were cloned into pLKO.1-puro lenti-viral vector (Addgene). The sequence targeting CXorf67 are: (#1: GGAGGGCTGAACAACGAAACC; #2: GCTGCT- GTGCTGACGGAGAGG), and a non-targeting control shRNA (ACAGTTAACCACTTTTTGA AT) were synthesized by GenePharma (Shanghai, China). Viral packaging and infection of cells following the manufacture’s recommended protocol. Briefly, lentiviral particles were generated by co-transfecting pLKO.1 transfer vector, psPAX2, and pVSV-G into HEK293T cells. 48 h later, the viruses were collected and added to PFA-3/4 cells in the presence of polybrene, and replaced with fresh medium after 12 h. After 60 h later, the infected cells were used for cell viability assays.
Daoy Subcutaneous and Orthotopic Xenografts
3 × 106 Daoy C67-KO cells and C67-stable expressing Daoy cells were injected s.c. into right flanks of nude mice (female, six weeks) in a 1:1 mix of PBS and Matrigel (354234, Corning). One week after injection, tumors were measured using calipers. Tumors volumes were calculated using the formula: 0.5 × length × width2. When tumors reached approximately 100 mm3, mice were randomly assigned to treatment with vehicle or talzaoparib (0.33 mg/kg). Talazoparib was diluted in diluent (10% DMAC and 6% Solutol in PBS) and administered by oral gavage, 5 days per week. For the vehicle, an equivalent volume of DMSO was diluted as above. Mice weight and tumor volume were recorded twice weekly. After nearly 4 weeks of treatment, mice were euthanized and tumor weight was measured.
For in vivo experiments, we generated stable luciferase expressing cell lines in CXorf67-KO Daoy cells with or without CXorf67 re-expressed. Sixty nude mice (female, six weeks old) were injected with 2 × 105 Daoy cells, to establish cerebellar tumors. Cerebellar coordinates were −2 from lambda, + 1 laterally, and 3 mm deep from the surface of the skull. The tumor formation in brain were confirmed by bioluminescence, and the mice were randomized into 4 groups (n=5 per group) after 7 days from the implantation for different treatments: vehicle (0.5% methylcellulose in PBS), niraparib (50 mg/kg in 0.5% methylcellulose, orally once a day for 5 days per week), 8 Gy fractionated radiotherapy (2 Gy daily for 4 days), and niraparib combined with 8 Gy fractionated radiotherapy. The bioluminescence imaging (BLI) levels were acquired every 10 days using IVIS SpectrumCT imaging system. The mice were intraperitoneally injected with 200 μl of 15 mg/ml D-luciferin (122799, Perkin Elmer) and subsequently anesthetized with isoflurane inhalation and photographed after 10 min injection. The Radiance (photons) within each area of interest was determined using the Living Image Software 4.5.4 (Perkin Elmer).
Patient Derived Xenografts (PDXs)
The collection of brain tumor surgical specimens was approved by the Institutional Review Board of Children’s Hospital of Fudan University. Informed consent of parent or legal guardian was obtained according to institutional regulatory standards before surgery. In total, two patient tumor samples (PFA-3: male, 34 months; PFA-4: female, 32 months) were collected and classified as ependymoma by at least two neuropathologists according to the 2016 WHO classification for central nervous system tumors. Minced fresh tumor tissue were transplanted subcutaneously to six weeks old male NOD SCID mice. After PDX model establishment, tumor tissue was minced and dissociated into single cell with Accutase (A6964, Sigma). 3×106 viable cells (per mouse) were injected s.c. into right flanks of nude mice (female, six weeks) in a 1:1 mix of Hank’s balanced salt solution and Matrigel. When tumors reached 50–150 mm3, mice were randomly assigned to treatment with vehicle or talzaoparib (0.33 mg/kg) as described above.
For combination treatment experiments, PFA-3 and PFA-4 PDX models were established as described above and treated with vehicle (10% β-cyclodextrin in PBS), olaparib (50 mg/kg in 10% β-cyclodextrin, orally once a day for 5 days per week), talazoparib (0.33 mg/kg in 10% DMAC and 6% Solutol, orally once a day for 5 days per week), IR (2 Gy daily for 4 days), olaparib combined with IR, and talazoparib combined with IR. Mice weight and tumor volume were recorded once weekly. Tumors volumes were calculated using the formula: 0.5 × length × width2.
Bioinformatics Analysis
Differentially expressed CXorf67 in PFA: gene expression data were collected from GEO database with the accession number GSE64415 and GSE94349. The processed data were used for differential expression analysis with t-test between samples of PFA and other samples. CXorf67 expression level across cancers and survival analysis: gene expression data and the corresponding clinical information were collected from The Cancer Genome Atlas (TCGA) project (https://portal.gdc.cancer.gov).
Drug activity data and mRNA expression level of CXorf67 in Brain or CNS cell lines of GDSC database were downloaded from CellMinerCDB (https://discover.nci.nih.gov/cellminercdb/). 93 drugs were filtered out with spearman correlation coefficient > 0.1 and p value < 0.05. Among them 59 filtered drugs with GDSC annotation were used for the barplot.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using GraphPad Prism 6 software (GraphPad). Statistical parameters are expressed as the mean ± s.e.m or SD and corresponding sample size and p-values are reported in the Figures and Figure legends. No samples or animals were excluded from data analyses. Student’s t test, unpaired, two-sided, two-tailed, one-way or two-way analysis of variance (ANOVA) were used to analyze data as indicated. Statistical analysis between two groups were done by paired or unpaired and two-tailed t-test. For comparisons among multiple groups, ANOVA was used. Survival analyses of data obtained from the TCGA was performed using median cut-off. Overall survival data were plotted as Kaplan-Meier curves and the Log-rank test was used for analyzing. p< 0.05 was defined to be statistically significant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse polyclonal anti-Rad51 (IF and WB) | Abcam | Cat# ab88572; RRID: AB_2042762 |
| Mouse monoclonal anti-BRCA1 (IF and WB) | Santa Cruz Bio | Cat# sc-6954; RRID: AB_626761 |
| Mouse monoclonal anti-phospho-Histone H2A.X (Ser139), clone JBW301, (IF and WB) | Millipore | Cat# 05-636; RRID: AB_309864 |
| Rabbit monoclonal anti-HA-Tag (C29F4) (IF) | Cell Signaling Technology | Cat# 3724; RRID: AB_1549585 |
| Mouse monoclonal anti-HA.11 Epitope Tag Antibody (WB) | Biolegend | Cat# 901515; RRID: AB_2565334 |
| Mouse monoclonal anti-Flag, Clone M2, (IF) | Sigma-Aldrich | Cat# F1804; RRID: AB_262044 |
| Rabbit polyclonal anti-Flag Tag (WB) | Cell Signaling Technology | Cat# 2368; RRID: AB_2217020 |
| Mouse monoclonal anti-Myc-Tag (9B11) (IF, WB) | Cell Signaling Technology | Cat# 2276; RRID: AB_331783 |
| Mouse monoclonal anti-LOC340602(CXorf67) (D-1) (IF, WB and IHC) | Santa Cruz Bio | Cat# sc-515296 |
| Normal mouse IgG | Santa Cruz Bio | Cat# sc-2025; RRID: AB_737182 |
| Mouse mixture of two monoclonal anti-Green Fluorescent Protein (7.1 and 13.1) (WB) | Roche | Cat# 11814460001; RRID: AB_390913 |
| Rabbit monoclonal anti-histone H3 (D2B12) (WB) | Cell Signaling Technology | Cat# 4620; RRID: AB_1904005 |
| Rabbit polyclonal anti-PALB2 (WB) | Abcam | Cat# ab220861 |
| Mouse monoclonal anti-BRCA2 (Clone 234403) (WB) | R&D Systems | Cat# MAB2476; RRID: AB_2259370 |
| Rabbit polyclonal anti-KU80 (WB) | Cell Signaling Technology | Cat# 2753; RRID: AB_2257526 |
| Rabbit polyclonal anti-H2A.X (WB) | Abcam | Cat# 11175; RRID: AB_297814 |
| Mouse monoclonal anti-β-Actin (WB) | Santa Cruz Bio | Cat# sc-47778; RRID: AB_626632 |
| Mouse monoclonal anti-GST (WB) | Sigma-Aldrich | Cat# G1166; RRID: AB_259846 |
| Mouse monoclonal anti-His (WB) | Sigma-Aldrich | Cat# H1029; RRID: AB_260015 |
| Rabbit polyclonal anti-GAPDH | Bioworld Technology | Cat# AP0063; RRID: AB_2651132 |
| Rabbit polyclonal anti-RPA2 | ABclonal | Cat# A2189; RRID: AB_2764207 |
| Mouse monoclonal anti-β-Tubilin | Sigma-Aldrich | Cat# T5293; RRID: AB_477580 |
| Bacterial and Virus Strains | ||
| DH10Bac chemically compotent cell | Shanghai Weidi Biotechnology | Cat# DL1071 |
| Escherichia coli DH5α | Tiangen Biotech | Cat# CB101 |
| Escherichia coli BL21 (DE3) | Tiangen Biotech | Cat# CB105 |
| Biological Samples | ||
| Ependymoma tumor samples | Children’s Hospital of Fudan University | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| D-Luciferin | Perkin Elmer | Cat# 122799 |
| Niraparib | MedChem Express | Cat# HY-10619 |
| CXorf67 (420-432aa) peptide | This paper | N/A |
| CXorf67 (420-432aa) W425C peptide | This paper | N/A |
| Olaparib | MedChem Express | Cat# HY-10162 |
| DAPI | Sigma-Aldrich | Cat# D8417 |
| EGF | Gibco | Cat# PHG0311 |
| FGF | Gibco | Cat# PHG0261 |
| DMAC | Sangon Biotech | Cat# A504006 |
| Solutol HS-15 | MedChem Express | Cat# HY-Y1893 |
| SBE-β-CD | MedChem Express | Cat# HY-17031 |
| Methylcellulose | MedChem Express | Cat# HY-125861 |
| Talazoparib | MedChem Express | Cat# HY-16106 |
| Critical Commercial Assays | ||
| CellTiter-Glo Kit | Promega | Cat# G7572 |
| H&E staining Kit | Beyotime | Cat# C0105 |
| ClonExpress II one step cloning Kit | Vazyme | Cat# C112 |
| Lipofectamine 3000 transfection Kit | Invitrogen | Cat# L3000015 |
| DAB Kit | Beyotime | Cat# P0203 |
| FITC Annexin V Apoptosis Detection Kit | BD Biosciences | Cat# 556547 |
| OxiSelect Comet Assay Kit | Cell Biolabs | Cat# STA-350 |
| Deposited Data | ||
| Microarray gene expression | (Donson et al., 2017; Pajtler et al., 2015) | GEO: GSE64415 and GSE94349 |
| TCGA mRNA data and clinical data | TCGA | https://portal.gdc.cancer.gov |
| The raw data of this study | This paper | https://doi.org/10.17632/rjxfxzsh5m.1 |
| CellMinerCDB | (Rajapakse et al., 2018) | https://discover.nci.nih.gov/cellminercdb/ |
| Experimental Models: Cell Lines | ||
| Human: U2OS DR-GFP | (Lou etal., 2017) | N/A |
| Human: U2OS-LacO | (Zhou etal., 2017) | N/A |
| Human: U2OS | ATCC | Cat# HTB-96; RRID: CVCL_0042 |
| Human: Daoy | Cell library, SIBCB | SCSP-509 |
| Human: 293T | Cell library, SIBCB | SCSP-502 |
| Ependymoma primary cell cultures: PFA-1 and PFA-2 | This paper | N/A |
| Ependymoma cell cultures from xenograft: PFA-3 and PFA-4 | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: NOD.CB17-Prkdcscid/NcrCrl (NOD-SCID) | Vital River Laboratories | Cat# 406 |
| Mouse: BALB/cASlac-nu | SLAC Laboratory Animal | N/A |
| Oligonucleotides | ||
| siRNA targeting sequence: BRCA2: GAAGAACAAUAUCCUACUA | This paper | N/A |
| siRNA targeting sequence: Ku80: GCGAGUAACCAGCUCAUAA | This paper | N/A |
| Non-targeting control siRNA sequence: UUCUCCGAACGUGUCACGUUU | This paper | N/A |
| siRNA targeting sequence: CXorf67 #1: GGAGGGCUGAACAACGAAAUU | This paper | N/A |
| siRNA targeting sequence: CXorf67 #2: GCUGCUGUGCUGACGGAGAUU | This paper | N/A |
| Recombinant DNA | ||
| GFP/Myc-LacR | (Zhou etal., 2017) | N/A |
| pEGFP-CXorf67 | This paper | N/A |
| pCMV-CXorf67-HA | This paper | N/A |
| pCMV-CXorf67-Flag | This paper | N/A |
| Lentiviral pLVX-CXorf67 | This paper | N/A |
| Lentiviral pLKO.1-puro | Addgene | Cat# 8453; RRID: Addgene_8453 |
| pSpCas9(BB)-2A-GFP (PX458) | Addgene | Cat# 48138; RRID: Addgene_48138 |
| pCBASceI | Addgene | Cat# 26477; RRID: Addgene_26477 |
| Software and Algorithms | ||
| FlowJo (v10.4) | FlowJo software | https://www.flowjo.com |
| Leica LAS AF software (LAS AF 2.4.1) | Leica | In house license |
| NIS-Elements software (v4.5) | Nikon | In house license |
| GraphPad Prism (v6.0) | GraphPad software lnc. | https://www.graphpad.com |
| ImageJ software (v1.8.0) | National Institutes of Health | https://imagej.nih.gov/ij/ |
| CometScore software (v2.0) | Rex Hoover | http://rexhoover.com/index.php?id=cometscore |
| Living Image Software (v4.5.4) | Perkin Elmer | In house license |
Highlights.
CXorf67 suppresses HR-mediated DNA repair
CXorf67 interacts directly with PALB2, thereby blocking BRCA2-PALB2 interaction
CXorf67 expression sensitizes tumor cells to PARP1/2 inhibitors (PARPi)
PARPi combined with radiotherapy efficiently kills CXorf67-positive tumor cells
ACKNOWLEDGMENTS
We greatly appreciate the gift of U2OS DR-GFP and EJ5-GFP cells from Jun Huang (Zhejiang University), and U2OS-LacO cells and related plasmids from Fangwei Wang (Zhejiang University). We acknowledge the assistance of Yanhua Wang from R.Z.’s laboratory for the mass spectrometry experiments. This work was supported by the National Key Research and Development Program of China (2019YFA0802002, 2017YF0503600), CAS Strategic Priority Research Program (XDB19030000), and the National Natural Science Foundation of China (31530094; 31571456).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.ccell.2020.10.009.
DECLARATION OF INTERESTS
L.L. and J.H. have filed a patent (202010351558X) for the use of CXorf67 expression for prediction of PARP1/2 inhibitors efficacy in China. All other authors declare no competing interests.
REFERENCES
- Bouffet E, and Foreman N (1999). Chemotherapy for intracranial ependymomas. Childs Nerv. Syst 15, 563–570. [DOI] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, and Helleday T (2005). Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917. [DOI] [PubMed] [Google Scholar]
- Ciccia A, and Elledge SJ (2010). The DNA damage response: making it safe to play with knives. Mol. Cell 40, 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewaele B, Przybyl J, Quattrone A, Finalet Ferreiro J, Vanspauwen V, Geerdens E, Gianfelici V, Kalender Z, Wozniak A, Moerman P, et al. (2014). Identification of a novel, recurrent MBTD1-CXorf67 fusion in low-grade endometrial stromal sarcoma. Int. J. Cancer 134, 1112–1122. [DOI] [PubMed] [Google Scholar]
- Donson AM, Apps J, Griesinger AM, Amani V, Witt DA, Anderson RCE, Niazi TN, Grant G, Souweidane M, Johnston JM, et al. (2017). Molecular analyses reveal inflammatory mediators in the solid component and cyst fluid of human adamantinomatous craniopharyngioma. J. Neuropathol. Exp. Neurol 76, 779–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducy M, Sesma-Sanz L, Guitton-Sert L, Lashgari A, Gao Y, Brahiti N, Rodrigue A, Margaillan G, Caron MC, Cote J, et al. (2019). The tumor suppressor PALB2: inside out. Trends Biochem. Sci 44, 226–240. [DOI] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921. [DOI] [PubMed] [Google Scholar]
- Garvin JH Jr., Selch MT, Holmes E, Berger MS, Finlay JL, Flannery A, Goldwein JW, Packer RJ, Rorke-Adams LB, Shiminski-Maher T, et al. (2012). Phase II study of pre-irradiation chemotherapy for childhood intracranial ependymoma. Children’s Cancer Group protocol 9942: a report from the Children’s Oncology Group. Pediatr. Blood Cancer 59, 1183–1189. [DOI] [PubMed] [Google Scholar]
- Hubner JM, Muller T, Papageorgiou DN, Mauermann M, Krijgsveld J, Russell RB, Ellison DW, Pfister SM, Pajtler KW, and Kool M (2019). EZHIP/CXorf67 mimics K27M mutated oncohistones and functions as an intrinsic inhibitor of PRC2 function in aggressive posterior fossa ependymoma. Neuro Oncol. 21, 878–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain SU, Do TJ, Lund PJ, Rashoff AQ, Diehl KL, Cieslik M, Bajic A, Juretic N, Deshmukh S, Venneti S, et al. (2019). PFA ependymoma-associated protein EZHIP inhibits PRC2 activity through a H3 K27M-like mechanism. Nat. Commun 10, 2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord CJ, and Ashworth A (2016). BRCAness revisited. Nat. Rev. Cancer 16, 110–120. [DOI] [PubMed] [Google Scholar]
- Lord CJ, and Ashworth A (2017). PARP inhibitors: synthetic lethality in the clinic. Science 355, 1152–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou J, Chen H, Han J, He H, Huen MS, Feng X. h., Liu T, and Huang J (2017). AUNIP/C1orf135 directs DNA double-strand breaks towards the homologous recombination repair pathway. Nat. Commun 8, 985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack SC, Witt H, Piro RM, Gu L, Zuyderduyn S, Stütz AM, Wang X, Gallo M, Garzia L, Zayne K, et al. (2014). Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 506, 445–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michealraj KA, Kumar SA, Kim LJY, Cavalli FMG, Przelicki D, Wojcik JB, Delaidelli A, Bajic A, Saulnier O, MacLeod G, et al. (2020). Metabolic regulation of the epigenome drives lethal infantile ependymoma. Cell 181, 1329–1345.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nepomuceno TC, De Gregoriis G, de Oliveira FMB, Suarez-Kurtz G, Monteiro AN, and Carvalho MA (2017). The role of PALB2 in the DNA damage response and cancer predisposition. Int. J. Mol. Sci 18, 10.3390/ijms18091886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver AW, Swift S, Lord CJ, Ashworth A, and Pearl LH (2009). Structural basis for recruitment of BRCA2 by PALB2. EMBO Rep. 10, 990–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orthwein A, Noordermeer SM, Wilson MD, Landry S, Enchev RI, Sherker A, Munro M, Pinder J, Salsman J, Dellaire G, et al. (2015). A mechanism for the suppression of homologous recombination in G1 cells. Nature 528, 422–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajtler KW, Mack SC, Ramaswamy V, Smith CA, Witt H, Smith A, Hansford JR, von Hoff K, Wright KD, Hwang E, et al. (2017). The current consensus on the clinical management of intracranial ependymoma and its distinct molecular variants. Acta Neuropathol. 133, 5–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajtler KW, Wen J, Sill M, Lin T, Orisme W, Tang B, Hübner J-M, Ramaswamy V, Jia S, Dalton JD, et al. (2018). Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathol. 136, 211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajtler KW, Witt H, Sill M, Jones David T.W., Hovestadt V, Kratochwil F, Wani K, Tatevossian R, Punchihewa C, Johann P, et al. (2015). Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell 27, 728–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piunti A, Smith ER, Morgan MAJ, Ugarenko M, Khaltyan N, Helmin KA, Ryan CA, Murray DC, Rickels RA, Yilmaz BD, et al. (2019). CATACOMB: an endogenous inducible gene that antagonizes H3K27 methylation activity of Polycomb repressive complex 2 via an H3K27M-like mechanism. Sci. Adv 5, eaax2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajapakse VN, Luna A, Yamade M, Loman L, Varma S, Sunshine M, Iorio F, Sousa FG, Elloumi F, Aladjem MI, et al. (2018). CellMinerCDB for integrative cross-database genomics and pharmacogenomics analyses of cancer cell lines. iScience 10, 247–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, and Bonner WM (1998). DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem 273, 5858–5868. [DOI] [PubMed] [Google Scholar]
- Sy SM, Huen MS, and Chen J (2009). PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc. Natl. Acad. Sci. U S A 106, 7155–7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vladoiu MC, El-Hamamy I, Donovan LK, Farooq H, Holgado BL, Sundaravadanam Y, Ramaswamy V, Hendrikse LD, Kumar S, Mack SC, et al. (2019). Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature 572, 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wani K, Armstrong TS, Vera-Bolanos E, Raghunathan A, Ellison D, Gilbertson R, Vaillant B, Goldman S, Packer RJ, Fouladi M, et al. (2012). A prognostic gene expression signature in infratentorial ependymoma. Acta Neuropathol. 123, 727–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witt H, Mack SC, Ryzhova M, Bender S, Sill M, Isserlin R, Benner A, Hielscher T, Milde T, Remke M, et al. (2011). Delineation of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cell 20, 143–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, Liu X, Jasin M, Couch FJ, and Livingston DM (2006). Control of BRCA2 cellular and clinical functions by a nuclear partner, Palb2. Mol. Cell 22, 719–729. [DOI] [PubMed] [Google Scholar]
- Xie X, Hu H, Tong X, Li L, Liu X, Chen M, Yuan H, Xie X, Li Q, Zhang Y, et al. (2018). The mTOR-S6K pathway links growth signalling to DNA damage response by targeting RNF168. Nat. Cell Biol 20, 320–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Ma J, Wu J, Ye L, Cai H, Xia B, and Yu X (2009). PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr. Biol 19, 524–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Liang C, Chen Q, Zhang Z, Zhang B, Yan H, Qi F, Zhang M, Yi Q, Guan Y, et al. (2017). The N-terminal non-kinase-domain-mediated binding of haspin to Pds5B protects centromeric cohesion in mitosis. Curr. Biol 27, 992–1004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate datasets and code. Previously published expression datasets used in this study are available through GEO: GSE64415, GSE94349 as well as The Cancer Genome Atlas (TCGA). The raw data of this study have been deposited in Mendeley Data and available at https://doi.org/10.17632/rjxfxzsh5m.1.