

Significance Statement
The complement pathway represents one of the critical arms of the innate immune system. We combined genome-wide and phenome-wide association studies using medical records data for C3 and C4 levels to discover common genetic variants controlling systemic complement activation. Three genome-wide significant loci had large effects on complement levels. These loci encode three critical complement genes: CFH, C3, and C4. We performed detailed functional annotations of the significant loci, including multiallelic copy number variant analysis of the C4 locus to define two structural genomic variants with large effects on C4 levels. Blood C4 levels were strongly correlated with the copy number of C4A and C4B genes. Lastly, using genome-wide genetic correlations and electronic health records–based phenome-wide association studies in 102,138 participants, we catalogued a spectrum of human diseases genetically related to systemic complement activation, including inflammatory, autoimmune, cardiometabolic, and kidney diseases.
Key words: electronic health records, genome wide association study, phenome wide association study, complement system, autoimmunity
Abstract
Background
Genetic variants in complement genes have been associated with a wide range of human disease states, but well-powered genetic association studies of complement activation have not been performed in large multiethnic cohorts.
Methods
We performed medical records–based genome-wide and phenome-wide association studies for plasma C3 and C4 levels among participants of the Electronic Medical Records and Genomics (eMERGE) network.
Results
In a GWAS for C3 levels in 3949 individuals, we detected two genome-wide significant loci: chr.1q31.3 (CFH locus; rs3753396-A; β=0.20; 95% CI, 0.14 to 0.25; P=1.52x10-11) and chr.19p13.3 (C3 locus; rs11569470-G; β=0.19; 95% CI, 0.13 to 0.24; P=1.29x10-8). These two loci explained approximately 2% of variance in C3 levels. GWAS for C4 levels involved 3998 individuals and revealed a genome-wide significant locus at chr.6p21.32 (C4 locus; rs3135353-C; β=0.40; 95% CI, 0.34 to 0.45; P=4.58x10-35). This locus explained approximately 13% of variance in C4 levels. The multiallelic copy number variant analysis defined two structural genomic C4 variants with large effect on blood C4 levels: C4-BS (β=−0.36; 95% CI, −0.42 to −0.30; P=2.98x10-22) and C4-AL-BS (β=0.25; 95% CI, 0.21 to 0.29; P=8.11x10-23). Overall, C4 levels were strongly correlated with copy numbers of C4A and C4B genes. In comprehensive phenome-wide association studies involving 102,138 eMERGE participants, we cataloged a full spectrum of autoimmune, cardiometabolic, and kidney diseases genetically related to systemic complement activation.
Conclusions
We discovered genetic determinants of plasma C3 and C4 levels using eMERGE genomic data linked to electronic medical records. Genetic variants regulating C3 and C4 levels have large effects and multiple clinical correlations across the spectrum of complement-related diseases in humans.
The complement system provides a primary defense mechanism against various pathogens, especially encapsulated bacterial organisms, such as Neisseria species.1 At the same time, uncontrolled activation of the complement system may lead to increased inflammation and tissue injury with multiorgan manifestations.2 For example, systemic complement activation plays an important role in the pathogenesis of septic shock and vascular thrombosis. It may be responsible for multiorgan complications of systemic infections, such as microangiopathy, coagulopathy, and lung and kidney injury in the setting of severe acute respiratory syndrome coronavirus 2 infection.3–5 Activation of the complement system in the kidney often leads to microvascular injury and GN, including membranoproliferative GN, poststreptococcal GN, lupus nephritis, and IgA nephropathy.6 Activation of complement in the retina is involved in the pathogenesis of age-related macular degeneration.7 There are also rare genetic disorders due to mutations in specific complement regulatory genes; for example, mutations in complement factor H (CFH) and CFHR genes cause complement component 3 (C3) GN, dense deposit disease, or atypical hemolytic uremic syndrome,8,9 and some of them overlap with those that cause age-related macular degeneration.10,11 Additionally, recent genome-wide association studies (GWAS) have identified a number of common functional variants in complement genes that have specific disease associations, including for age-related macular degeneration, SLE, IgA nephropathy, and Neisseria infections.12–15 The full spectrum of disease associations for these complement-related loci has not been comprehensively assessed in large datasets.
The development of large-scale biobanks linking genomic data to electronic health records (EHRs) enables powerful approaches to explore pleiotropic associations of genetic variants through phenome-wide association studies (PheWAS).16 In this study, we leveraged the resources of the Electronic Medical Records and Genomics (eMERGE) network and the laboratory test results captured in medical records to perform a combined GWAS-PheWAS study of circulating levels of C3 and C4. These markers are commonly measured as a clinical screening test for several suspected autoimmune conditions. Depressed levels of C4 provide an indirect measure of the classic complement pathway activation, whereas depressed plasma C3 levels may reflect either the classical or alternative pathway activity.1,17–19 Previous studies have demonstrated that both C3 and C4 levels are highly heritable.20,21 To date, there have only been two GWAS studies of complement levels, a Chinese study of C3 and C4 levels,22 and a Swedish study of only C3 levels.23 The relevance of these studies to more diverse populations is unknown, and specific disease associations of the significant loci have not been examined.
Motivating our study is the fact that the complement system plays a pathogenic role in a wide spectrum of kidney disorders, yet the effect of common germline genetic variation on the activity of the complement system in diverse human populations remains poorly understood. Common genetic variation affecting complement activation could be clinically relevant, as it may potentially contribute to the severity of complement-mediated tissue injury and may also be important in pharmacologic response to complement-targeting drugs. Considering a growing number of complement inhibitors currently in clinical trials for kidney disorders, this hypothesis is especially relevant to the field of nephrology. In this study, we aim to demonstrate that medical record–contained laboratory data can be used effectively to map common inherited variants with regulatory effects on the complement pathway. Our results provide evidence for genome-wide significant loci with large effects on the complement levels and show a significant contribution of common inherited variation in the determination of circulating levels of C3 and C4.
Methods
The study design and the overall workflow are shown in Figure 1. The primary objective of this study is to gain new insights into the genetic regulation of the complement system through quantitative GWAS of C3 and C4 endophenotypes. The secondary objective is to perform comprehensive in silico annotations of the newly discovered loci in order to better understand genetic mechanisms underlying these associations. We also undertake a number of exploratory analyses on the basis of GWAS results in order to comprehensively assess the pleiotropy of the newly discovered loci and to test for a shared polygenic architecture of complement levels with a broad range of relevant complex traits previously studied by GWAS. These analyses include genome-wide genetic correlation (single-nucleotide polymorphism [SNP]–based co-heritability) analyses of C3 and C4 levels with a range of autoimmune, infectious, cardiometabolic, and renal traits, as well as phenome-wide association analyses against all common EHR-contained diagnoses.
Figure 1.

Study design and workflow summary. Analytical workflow for GWAS for C3 and C4 levels in the eMERGE network (primary analysis; top); refinement of genome-wide significant loci by conditional analysis, imputation, annotations, and single-locus PheWAS (secondary analyses; bottom right); and derivation of polygenic scores for C3 and C4 followed by a polygenic score–based PheWAS (exploratory analyses; bottom left). QC, quality control.
Ethics Statement
The study was approved by the Columbia University Institutional Review Board and individual institutional review boards at all Electronic Medical Records and Genomics phase 3 (eMERGE-III) network sites contributing human genetic and clinical data. BioVU operated on an opt-out basis until January 2015 and on an opt-in basis since. The phenotypic data in BioVU are all deidentified, and the study was designated “nonhuman subjects” research by the Vanderbilt Institutional Review Board. All other participants provided informed consent to participate in genetic studies.
Study Cohort and Electronic Phenotypes
The eMERGE network consortium consists of 12 medical centers with EHRs linked to genome-wide genotype data for 102,138 individuals (Supplemental Table 1). More detailed characterization of the eMERGE-III cohorts and related datasets has been published recently.24 The C3 and C4 levels were extracted by a laboratory value query performed by the eight active sites of the eMERGE-III. Individual site–level laboratory data were provided for further phenotype quality control analyses. For individuals with multiple C3 and C4 measurements, we used the nadir level (lowest value recorded) to capture the maximum level of complement depletion across all available tests for a given participant. Supplemental Table 2 provides the distribution of eMERGE-III participants by the number of C3/C4 tests performed. After quality control, C3 levels were available for 3949 individuals (3210 European, 589 African, and 150 East Asian ancestry). C4 levels were available for 3998 individuals (3247 European, 600 African, and 151 East Asian ancestry). There was a near-complete (96%) overlap between the individuals in C3 and C4 studies because these tests are usually bundled together in a single clinical order set.
The distributions of C3 and C4 levels were positively skewed and required a logarithmic transformation. We additionally adjusted for age, sex, site, and known diagnoses of autoimmune and inflammatory diseases, including SLE, Sjogren syndrome, mixed connective tissue disease, rheumatoid arthritis, polymyalgia rheumatica, Crohn's disease, psoriasis, vasculitis, and arteritis. The overall distribution of autoimmune disorders among GWAS participants is provided in Supplemental Table 3. These phenotypes were defined electronically using a validated eMERGE electronic autoimmunity phenotype (https://phekb.org/phenotype/autoimmune-disease-phenotype) from the Phenotype Knowledge Base.25 After regressing these covariates against log-transformed C3 and C4 levels, we derived standard normal residuals, which were used as quantitative traits in genome-wide association analyses.
Genotyping, Imputation, and Quality Control
The genotyping and imputation of the eMERGE cohort have been recently described in detail.26 Briefly, we implemented the mimimac3 missing variant imputation model with genome-wide imputation using the HRC1.1 reference (Michigan Imputation Server) in genome build 37 (hg19) for each genotyping platform in a separate batch. After imputation, we merged all of the 81 imputed batches on the basis of position using bcftools (http://researchcomputing.syr.edu/bcftools/). We filtered out markers with minor allele frequency <1%. In addition, our quality control filters required imputation quality R2>0.8 in at least 75% of 81 imputed batches. We applied a principal component (PC) analysis of each cohort using FlashPCA.27 We used KING software to identify cryptically related subjects, and we removed one individual per related pair with a second degree or higher relatedness.28 We removed ancestry outliers, reran PC analysis, and adjusted GWAS analysis for significant PCs to reduce any potential bias from population stratification (Supplemental Figure 1).
GWAS Meta-Analysis and Conditional Analysis
For both C3 and C4 levels, association tests were performed using linear regression of imputed dosage data with adjustment for significant PCs of ancestry. The association testing was performed for each major ancestral population (European, African, and East Asian) separately, and then, the results were combined using an inverse variance–weighted fixed effects meta-analysis with METAL.29 The analyses were performed using a combination of VCFtools, PLINK, and custom scripts in PYTHON and R.30,31
C4 Copy Number Variations Imputation
For C4 copy number imputation, we used the C4 copy number reference panel from Sekar et al.32 We used Eagle.v2.4 software to phase the haplotypes followed by imputation using mimimac3.33–35 We imputed 14 C4 alleles and 2709 biallelic SNPs in the region. We filtered the high-quality common variants on the basis of R2 and MAF before association analysis. Postimputation, we analyzed the top six copy number alleles that were common (MAF>1%) and imputed with adequate quality (R2≥0.3). We performed both biallelic association tests and multiallelic tests of copy number variants using linear regression with dosage-coded imputed genotypes as predictors and C4 levels as an outcome. The analyses were adjusted for significant PCs of ancestry.
SNP-Based Heritability, Genetic Correlation, and Pathway Analyses
We estimated the fraction of additive genetic variance explained by GWAS for each phenotype using linkage disequilibrium (LD) score regression software.36 This method models polygenicity of the traits by accounting for the relationship between test statistics and LD. The same method was used to derive genome-wide genetic correlations with various GWAS traits. We performed both gene-based and pathway-based enrichment analyses that account for LD using VEGAS2.37
Functional Annotations of GWAS Loci
In order to capture all variants that could potentially underlie our genome-wide significant signals, we first cataloged all common variants in LD (r2>0.8) with the top SNP at each locus. We performed annotations of these SNPs using tissue-specific FUN-LDA, which provides a new method for functional scoring of noncoding genetic variants in a cell type– and tissue-specific manner.38 Additionally, we interrogated the Genotype-Tissue Expression (GTEx-V8) project data to identify significant expression QTLs and splice QTLs (sQTLs) across 47 human tissues.39 To increase power for eQTL detection, we also used the latest blood eQTL dataset on the basis of the meta-analysis of 31,684 individuals.40(preprint)
Polygenic Score Derivation and PheWAS
We used LDPred39 to derive genome-wide polygenic predictors of blood C3 and C4 levels on the basis of our GWAS summary statistics and 1000 Genomes reference panel for all ancestries41 and assuming maximum polygenicity with all common variants contributing to the traits. The derived polygenic predictors for C3 and C4 were then used to score all 102,138 eMERGE participants with available GWAS and EHR data. In order to test each polygenic predictor for disease associations phenome wide, we first harmonized the coded diagnoses data by converting all available ICD-10-CM codes to the ICD-9-CM system. This approach was motivated by the facts that the great majority of data for eMERGE-III participants is already coded using the ICD-9 system; ICD-10 codes are more granular and thus, a reverse conversion is more prone to mapping errors; and the current PheWAS R library supports only ICD-9 codes. After the conversion, the 102,138 genotyped eMERGE participants had a total of 20,783 unique ICD-9 codes that were then mapped to 1817 distinct phecodes (disease-specific groupings of ICD codes). Phenome-wide associations were performed using the PheWAS R package.16 The package uses predefined “control” groups for each phecode. The case definition requires a minimum of two ICD-9 codes from the “case” grouping of each phecode. In total, all 1817 phecodes were tested using logistic regression with each phecode patient-control status as an outcome and the polygenic score for C3 and C4 adjusted for age, sex, study site, and three PCs of ancestry as a predictor. To establish significant disease associations in PheWAS, we set the Bonferroni-corrected statistical significance threshold at 2.75×10−05 (0.05 divided by 1817), correcting for 1817 independent phecodes tested.
Results
GWAS for C3 Levels
We performed GWAS for the nadir level of C3 in 3949 individuals with available C3 measurements (Table 1). The QQ plot revealed no global deviation of the test statistic from the expected null distribution (Supplemental Figure 2A), and the corresponding genomic inflation (λ) was estimated at 1.0004, confirming negligible inflation of our test statistics genome wide. The Manhattan plot and regional plots of significant loci are displayed in Figure 2. We observed two independent genome-wide significant loci for C3 levels on chr.1q31.3 (CFH locus; rs3753396-A; β=0.20; 95% confidence interval [95% CI], 0.14 to 0.25; P=1.52x10-11) and chr.19p13.3 (C3 locus; rs11569470-G; β=0.19; 95% CI, 0.13 to 0.24; P=1.29x10-08) (Table 2). In conditional analysis, a single SNP explained the entire signal at each locus without evidence of significant independent association signals. As expected, both loci are predominantly driven by the largest (European) ancestral group (Supplemental Figures 3 and 4).
Table 1.
Basic characteristic of study subjects with C3 and C4 levels
| Phenotype and Ancestry | Sample Size, N | Mean Age (SD), yr | Mean Nadir C3 Level (SD), mg/dl | Women, % |
|---|---|---|---|---|
| C3 levels | ||||
| European | 3210 | 55.3 (16.9) | 124.74 (35.09) | 63 |
| African | 589 | 47.9 (16.8) | 129.75 (37.22) | 75 |
| East Asian | 150 | 40.8 (18.8) | 110.75 (37.73) | 71 |
| Total | 3949 | 48.01 (17.5) | 121.74 (36.68) | 70 |
| C4 levelsa | ||||
| European | 3247 | 55.6 (16.7) | 26.84 (10.31) | 63 |
| African | 600 | 48.2 (16.7) | 33.98 (13.07) | 75 |
| East Asian | 151 | 40.8 (16.8) | 26.28 (11.88) | 71 |
| Total | 3998 | 48.2 (16.7) | 29.03 (11.75) | 70 |
Ninety-six percent of participants overlap between the two datasets.
Figure 2.
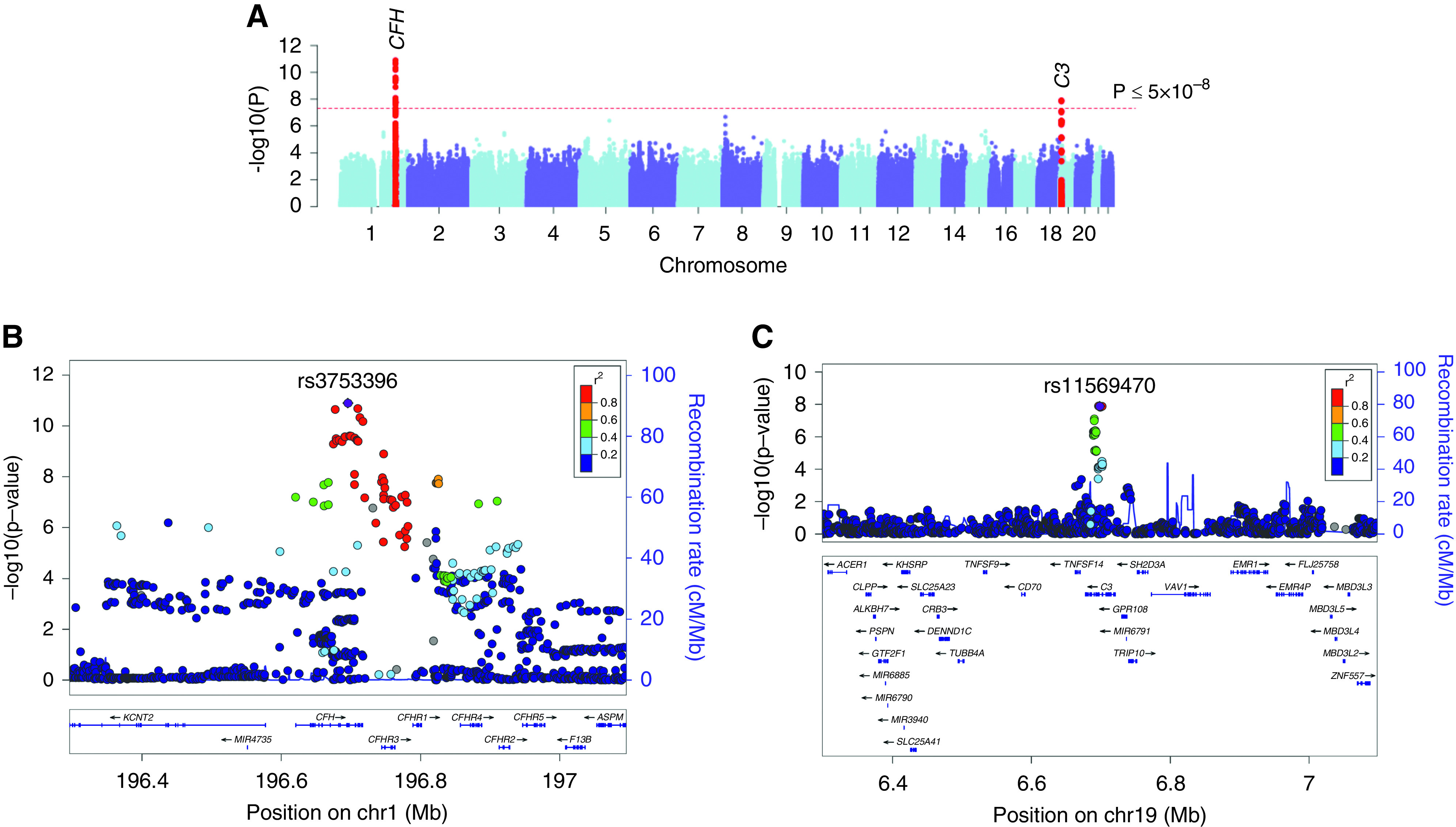
GWAS for C3 levels. (A) Manhattan plot, (B) regional plot for the CFH locus, and (C) regional plot for the C3 locus. The y axis represents −log-transformed P values, and the x axis represents genomic position. The 1000 Genomes Europeans reference panel was used for LD reference.
Table 2.
Top genome-wide significant SNPs in GWAS for C3 and C4 levels, their allelic frequencies, effect estimates (expressed in standard normal units), and P values by ancestry
| Locus | Test Allele | European Ancestry | African Ancestry | East Asian Ancestry | Combined Meta-Analysis | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Frequency | β (95% CI) | P Value | Frequency | β (95% CI) | P Value | Frequency | β (95% CI) | P Value | Frequency | β (95% CI) | P Value | I2, % | ||
| C3 levels | ||||||||||||||
| CFH | rs3753396-A | 0.17 | 0.19 (0.13 to 0.25) | 1.52E-09 | 0.06 | 0.22 (−0.01 to 0.42) | 0.06 | 0.06 | 0.45 (0.15 to 0.74) | 0.004 | 0.16 | 0.20 (0.14 to 0.25) | 1.52E-11 | 26 |
| C3 | rs11569470-G | 0.14 | 0.18 (0.12 to 0.23) | 1.41E-07 | 0.05 | 0.26 (0.01 to 0.51) | 0.04 | 0.05 | 0.39 (−0.25 to 1.03) | 0.23 | 0.12 | 0.19 (0.13 to 0.24) | 1.29E-08 | 0 |
| C4 levels | ||||||||||||||
| C4 | rs3135353-C | 0.15 | 0.40 (0.34 to 0.45) | 2.00E-33 | 0.02 | 0.28 (−0.07 to 0.63) | 0.12 | 0.05 | 0.45 (−0.17 to 1.07) | 0.16 | 0.12 | 0.40 (0.34 to 0.45) | 4.58E-35 | 0 |
| C4 | rs6459-T | 0.10 | 0.45 (0.39 to 0.50) | 8.04E-30 | 0.19 | 0.13 (−0.01 to 0.26) | 0.06 | 0.14 | 0.48 (0.06 to 0.89) | 0.04 | 0.12 | 0.38 (0.32 to 0.43) | 7.87E-29 | 87 |
| C4 | rs16869834-A | 0.05 | 0.47 (0.37 to 0.56) | 2.29E-18 | 0.06 | 0.30 (0.01 to 0.50) | 0.009 | 0.04 | 0.46 (−0.20 to 1.12) | 0.17 | 0.05 | 0.44 (0.36 to 0.51) | 8.28E-20 | 2 |
| C4 | rs9267822-C | 0.01 | 0.61 (0.41 to 0.80) | 7.15E-09 | 0.05 | 0.64 (0.40 to 0.87) | 6.11E-07 | 0.05 | 1.08 (0.49 to 1.67) | 0.0005 | 0.02 | 0.65 (0.51 to 0.78) | 6.31E-17 | 6 |
| C4 | rs805285-C | 0.25 | 0.18 (0.14 to 0.22) | 1.69E-10 | 0.36 | 0.25 (0.13 to 0.36) | 1.69E-05 | 0.48 | 0.17 (−0.10 to 0.44) | 0.23 | 0.32 | 0.19 (0.15 to 0.22) | 1.44E-14 | 0 |
| C4 | rs17208153-C | 0.08 | 0.28 (0.20 to 0.35) | 7.15E-11 | 0.24 | 0.16 (0.04 to 0.28) | 0.01 | 0.16 | 0.01 (−0.36 to 0.38) | 0.93 | 0.11 | 0.24 (0.18 to 0.30) | 2.01E-11 | 44 |
I2, heterogeneity index for the combined meta-analysis.
In the pathway enrichment analyses on the basis of GWAS summary statistics,37 we identified 13 significantly enriched pathways (Supplemental Table 4) with “the complement and coagulation cascades” representing the top enriched pathway (empirical enrichment P=0.0002). Specific genes underlying this enrichment were CFH, complement factor B, coagulation factor III, fibrinogen β-chain, coagulation factor II thrombin receptor like 2, α-2-macroglobulin, integrin subunit-αM, and C3.
CFH Locus on chr.1q31.3
The most highly associated “index” SNP at the CFH locus, rs3753396, is a synonymous SNP in CFH that has been previously associated with systemic complement activation42 and is in moderate LD with rs10922109 (r2=0.25) associated with age-related macular degeneration.12 Another variant in high LD with the index SNP (rs1048663; r2=0.93) has previously been associated with blood protein levels.43 We also confirmed that the top SNP at this locus is independent of the CHFR1/3 deletion previously associated with protection from age-related macular degeneration44 and IgA nephropathy.14,45
The index SNP (rs3753396) is in strong LD with rs35267550 (r2=0.93) that intersects a regulatory element specific to liver tissue and in weaker LD with rs201788925 (r2=0.86) that intersects a regulatory element in multiple cell types by FUN-LDA (Supplemental Figure 5). Importantly, the index SNP has a strong eQTL effect on CFHR3 and CFHR4 transcript levels in liver, the main site of production of these proteins, wherein the C3-decreasing allele (rs3753396-G) is associated with higher mRNA expression of CFHR3 (normalized effect size [NES] =0.66; P=5.5x10-11) and CFHR4 (NES=0.42; P=1.3x10-8) (Supplemental Figure 6). This variant is also associated with a highly significant sQTL effect on CFHR3, increasing the abundance of the main isoform of CFHR3 in GTEx project liver tissue (NES=1.4; P=8.8x10-41). Additionally, rs11806293-A (r2=0.88 with rs3753396-G) was associated with increased CFH transcript level in blood cells (Z score =6.21, false discovery rate =0). Although direction consistent, this effect was not significant for rs3753396 itself (Z score =3.66, false discovery rate =0.09). Taken together, it appears that the effect of the C3-decreasing allele may be mediated by increased transcription of CFH-related proteins in liver. Because these proteins are known to function as competitive inhibitors of factor H, the key regulator of the alternative complement pathway, their upregulation could reflect higher baseline activity of the alternative pathway, potentially explaining lower blood C3 levels in the carriers of rs3753396-G allele.
C3 Locus on chr.19p13.3
The index SNP at this locus, rs11569470-G (C3-increasing allele), is an intronic SNP in the C3 gene. Another variant in tight LD with this allele, rs11569479-T (r2=0.99), has recently been identified as a pQTL for C3, associated with higher C3 protein levels in peripheral blood proteomic study of healthy individuals (n=3200; β=0.29; P=8.4x10-16).46 The index SNP rs11569470 is also in high LD (r2=0.99) with the top SNP from the latest GWAS for age-related macular degeneration (rs10408682; P=5.0x10-19), suggesting a potential shared causal variant at this locus.12 On the basis of FUN-LDA analysis, rs11569470 intersects an active enhancer region across multiple tissues and cell types (Supplemental Figure 7), but it does not appear to have significant eQTL effects in liver. The rs11569470-G allele also has a significant sQTL effect on C3 transcript, with the G allele associated with a higher level of the dominant isoform in the pancreas (NES=0.56; P=1.3x10-18). It is possible that similar effects on C3 splicing in liver might have been missed in GTEx due to methodologic limitations of sQTL detection. Moreover, the C3-increasing allele is associated with increased mRNA levels of the nearby GPR108 gene in several GTEx tissues (Supplemental Figure 8), and a direction-consistent effect on GPR108 is also observed in blood (Z score =5.11; FDR=0.001). GPR108 encodes a transmembrane protein that activates NF-κB,47 but apart from the physical proximity to C3, it has no known links to the complement system.
Variance Explained and Genetic Correlations of C3 Levels
Using genome-wide summary statistics, we estimated the SNP-based heritability of C3 levels at approximately 11% (95% CI, −0.09 to 0.17). Notably, the two genome-wide significant loci explained approximately 2% of the overall variance in C3 levels, suggesting that additional loci could be discovered by expanding the sample size of GWAS.
We next performed genome-wide genetic correlation analysis for C3 levels and several relevant complex traits previously studied by GWAS, including infectious, inflammatory, autoimmune, cardiometabolic, and renal phenotypes (Figure 3A, Supplemental Table 5). We observed a significant genetic correlation of C3 levels with essential hypertension48 (genetic correlation rg=0.36; P=0.04) and a suggestive correlation with coronary artery disease49 (rg=0.30; P=0.06) and body mass index50 (rg=0.23; P=0.07). SLE51 had a negative correlation coefficient, but this correlation was not statistically significant (rg=−0.25; P=0.14). After exclusion of the HLA locus, we did not observe any significant genetic correlations (Figure 3B, Supplemental Table 5). We note that the relatively small sample size of our GWAS represents an important limitation in genome-wide genetic correlation analyses.
Figure 3.
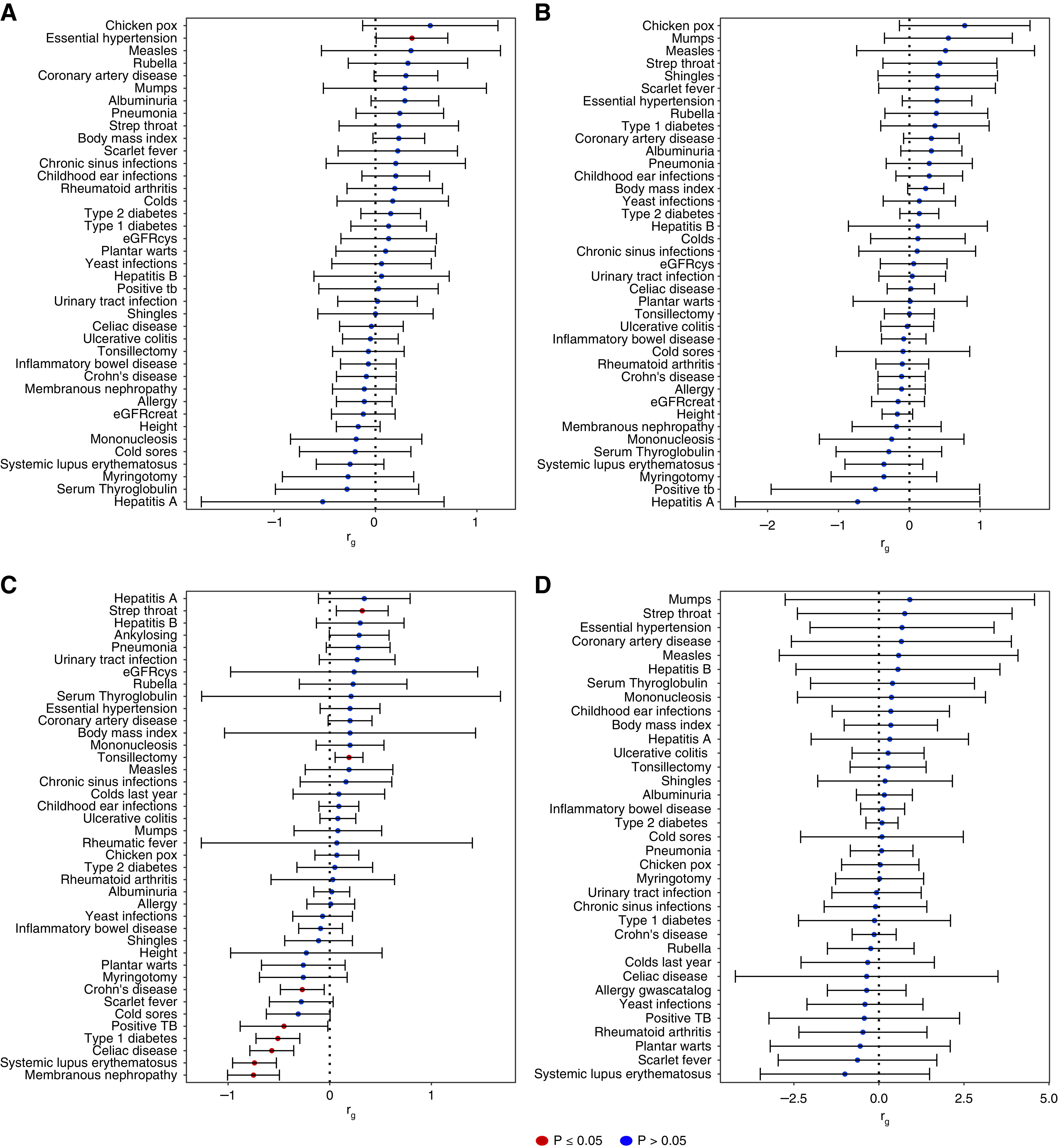
Genome-wide genetic correlations of GWAS results for C3 and C4 levels with infectious, autoimmune, and cardiometabolic traits. (A) global genetic correlations of C3 levels, (B) genetic correlations of C3 levels without the HLA region, (C) global genetic correlations of C4 levels, and (D) genetic correlations of C4 levels without the HLA region.
Phenome-Wide Associations
Because limited power of our GWAS may be contributing to negative findings in global genetic correlation analyses, we next utilized an alternative approach on the basis of phenome-wide association analysis of the polygenic predictor of C3. We first derived a genome-wide polygenic score for C3 levels using LDPred software.39 We next calculated this score for each of the 102,138 eMERGE participants and performed PheWAS for the score (Figure 4A, Supplemental Table 6). We detected strongly significant inverse associations of the score with SLE (odds ratio [OR], 0.74; 95% CI, 0.69 to 0.80; P=1.68x10-27), lupus (localized and systemic; OR, 0.75; 95% CI, 0.70 to 0.80; P=3.74x10-27), and proliferative GN (OR, 0.68; 95% CI, 0.58 to 0.78; P=1.01x10-12). This is consistent with prior studies suggesting that higher baseline complement levels may be protective from SLE and its complications.52,53 Similarly, higher C3 genetic score was associated with lower risk of coagulation defects (OR, 0.91; 95% CI, 0.88 to 0.94; P=6.43x10-09), sepsis (OR, 0.91; 95% CI, 0.88 to 0.94; P=9.79x10-08), systemic inflammatory response syndrome (OR, 0.92; 95% CI, 0.89 to 0.95; P=2.37x10-07), and shock (OR, 0.86; 95% CI, 0.81 to 0.92; P=2.85x10-07), highlighting the role of C3 in these conditions.
Figure 4.
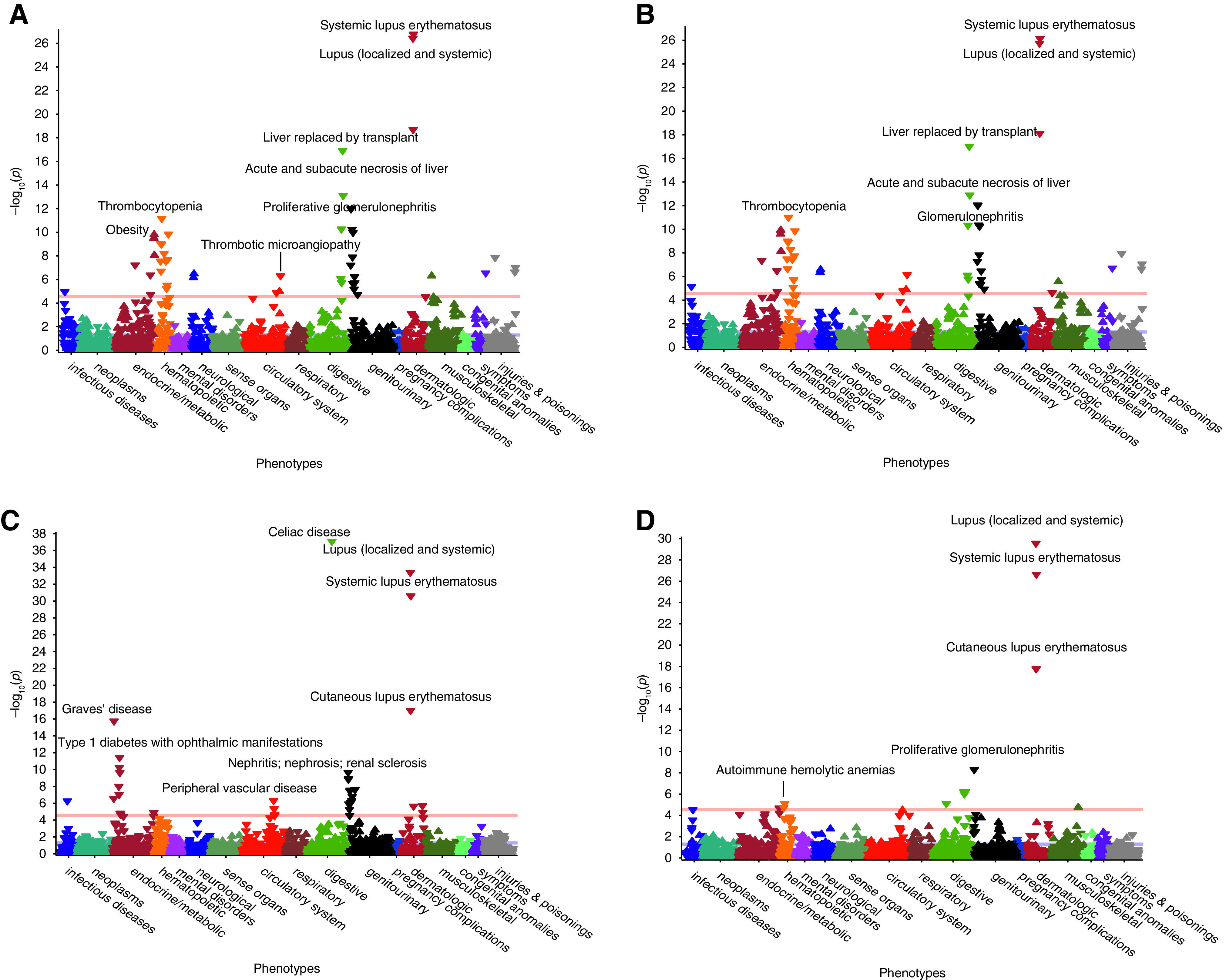
PheWAS for polygenic predictors of C3 and C4 levels in 102,138 eMERGE participants. (A and B) Phenome-wide associations of polygenic predictor of C3 levels (A) with HLA region included and (B) without HLA. (C and D) Polygenic predictor of C4 levels (C) with HLA included and (D) without HLA. Selected significant associations are labeled across various organ systems. The red horizontal lines indicate phenome-wide significance level (P=2.8x10-5) after accounting for the number of phecodes tested.
Unexpectedly but consistent with the suggestive genome-wide genetic correlation observed for body mass index, higher C3 score was significantly predictive of obesity (OR, 1.06; 95% CI, 1.04 to 1.07; P=1.57x10-10), morbid obesity (OR, 1.07; 95% CI, 1.05 to 1.10; P=8.82x10-09), and sleep apnea (OR, 1.05; 95% CI, 1.03 to 1.07; P=7.01x10-07).
In total, 47 different phenotypes significantly associated with polygenic score of C3 levels, and these associations are listed in Supplemental Table 6. Similar to genetic correlation analyses, we also recalculated C3 polygenic score after excluding the HLA locus and repeated PheWAS, but the HLA exclusion did not significantly change our PheWAS results (Figure 4B, Supplemental Table 7). Next, we performed PheWAS individually for the top SNPs at the CFH and C3 loci, but these analyses did not provide additional insights beyond confirming the known association with age-related macular degeneration (Supplemental Figure 9A and B, Supplemental Tables 8 and 9).
GWAS for Complement C4 Levels
The GWAS for C4 levels was performed in 3998 individuals (Table 1). The Manhattan and regional plots are depicted in Figure 5. The genomic inflation factor (λ) was estimated at 1.01, and inspection of the QQ plot confirmed no global deviation of test statistics from the expected null distribution (Supplemental Figure 2B). We observed a strong and highly significant signal on chr.6p21.32 (rs3135353-C; β=0.40; 95% CI, 0.34 to 0.45; P=4.58x10-35) (Table 2). The second strongest signal was a suggestive locus on chr.11p13 (rs58520479-G; β=0.18; 95% CI, 0.12 to 0.23; P=1.15x10-06). Using GWAS summary statistics, we identified 11 significant pathway enrichments by VEGAS2,37 with “viral myocarditis” as the top enriched pathway (empirical P value =0.01) (Supplemental Table 4).
Figure 5.
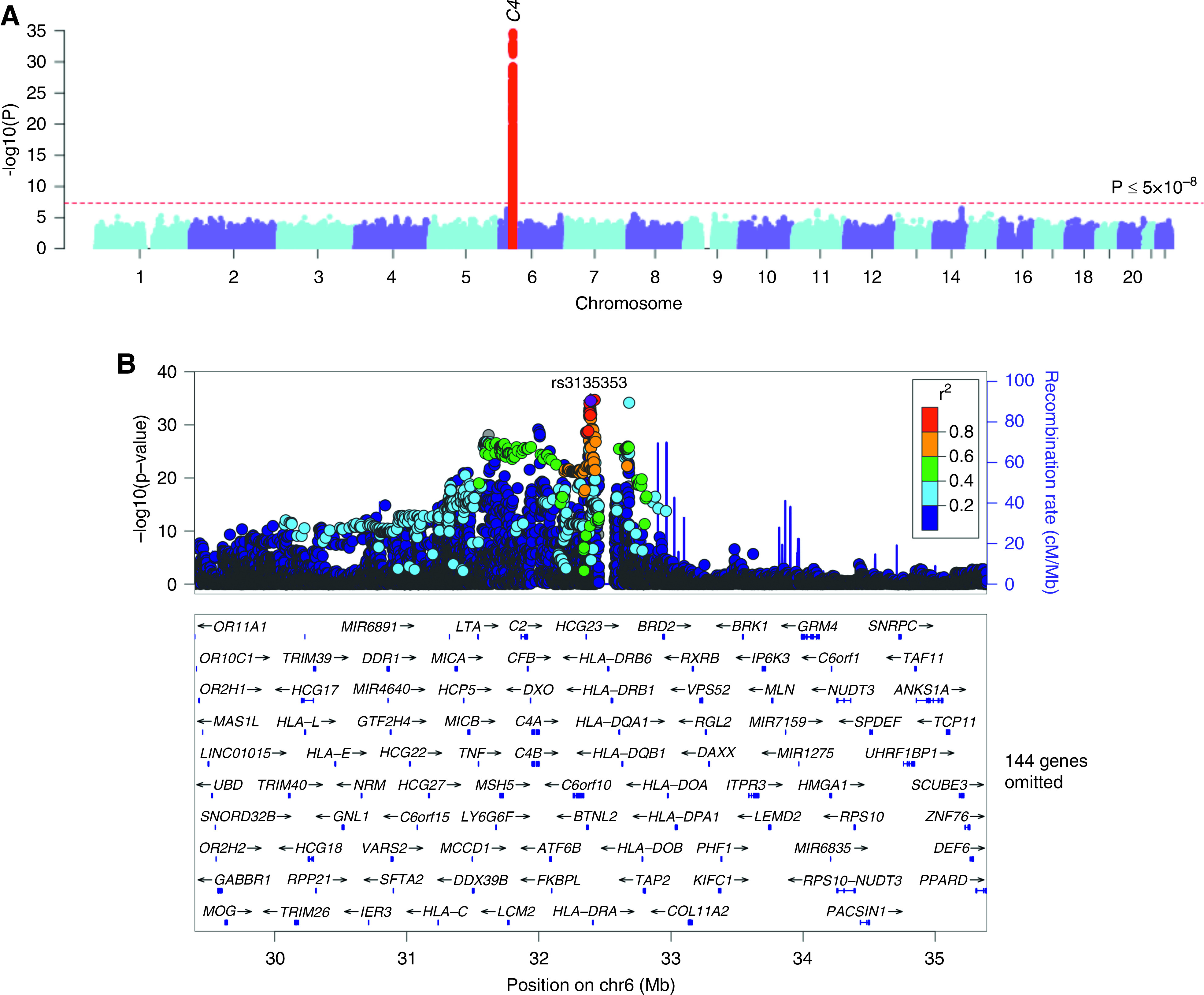
GWAS for C4 levels. (A) Manhattan plot and (B) regional plot of the C4 (MHC class 3) locus. The y axis represents −log-transformed P values, and the x axis represents genomic location. The 1000 Genome Europeans reference panel was used for LD reference.
C4 Locus on chr.6p21.32
The chr.6p21.32 signal represented by the top SNP rs3135353 was centered on the C4 gene region encoded within the HLA class 2 locus, and the signal was driven predominantly by the European (largest) ancestral group (Supplemental Figure 10). The rs3135353-C allele was associated with increased levels of C4. In stepwise conditional analyses, we identified a total of six independent genome-wide significant signals at this locus, suggesting a complex pattern of association (Table 2). The index C4-increasing allele (rs3135353-C) is in high LD with rs3129897-A (r2=0.96) associated with lower IL-21 levels,54 whereas rs1144709-T (r2=0.88 with rs805285-C, the fourth independent signal at the C4 locus) has previously been associated with decreased mathematical ability and implicated in the genetic control of brain development and neuron to neuron communication.55
The index SNP, rs3135353, is located 32 kbp away from a common multiallelic copy number variant involving the C4 gene. The C4 gene exists in two functionally distinct forms in humans, C4A and C4B, which differ by only four amino acids. This difference leads to altered binding to complement receptor type 1, such that C4A is more effective than C4B in binding to complement receptor type 1.56 C4A and C4B are arranged in a tandem array that varies in structure and copy numbers. Both long (C4AL and BL) and short (C4AS and BS) genomic forms exist and are distinguished by the presence or absence of a human endogenous retroviral insertion, which lengthens the C4 gene from 14 to 21 kb but otherwise, does not change the C4 protein sequence.57
The loci that are subject to multiallelic copy number variation are traditionally challenging to analyze, mainly because they are difficult to resolve with SNP arrays or short read sequencing. The complex multiallelic variation at the C4 locus has, therefore, been difficult to analyze in large cohorts. However, recent work by Sekar et al.32 delineated the genomic structure of the C4 gene using droplet digital PCR, allowing for a construction of reference panels that can be used for reliable imputation of C4 copy number genes from SNP data.32 Using this method, Sekar et al.32 demonstrated the association of C4 copy number variants with increased risk of schizophrenia and also showed for the first time that higher copy numbers of C4A and C4B determine brain RNA expression of C4. Therefore, we used the same approach and imputed C4 structural variants to explore which specific variant(s) may explain the observed association with blood C4 levels at this locus.
In the multiallelic test of the six common C4 copy number variants (Figure 6A), we observed a highly significant association with C4 levels (P=9.70x10-33). In the biallelic analyses, there were two highly significant C4 copy number variants. The BS haplotype encoding a single copy of short C4B was associated with lower C4 levels (β=−0.36; 95% CI, −0.42 to −0.30; P=2.98x10-22), whereas AL-BS, a two-structure haplotype (long C4A and short C4B), was associated with higher C4 levels (β=0.25; 95% CI, 0.21 to 0.29; P=8.11x10-23) when compared with all other haplotypes. The index GWAS SNP rs3135353-T is in moderate LD (r2=0.62) with the BS haplotype (encoding a short variant of C4B and associated with lower C4). Notably, the same haplotype has recently been defined as a risk factor for SLE and a protective factor from schizophrenia.58
Figure 6.
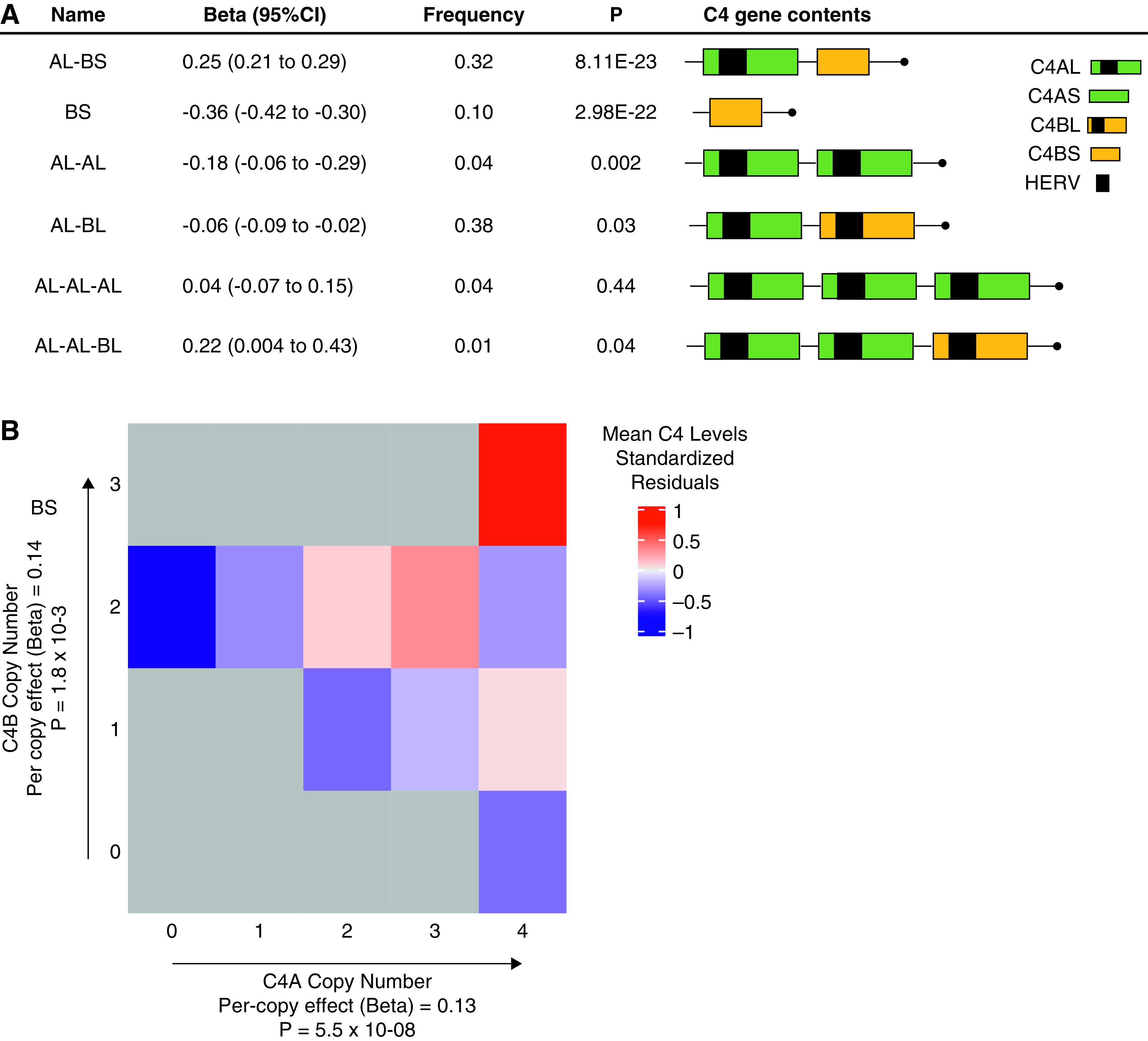
Structural forms of the C4 locus and their associations with blood C4 levels. (A) The effects of common structural C4 haplotypes on blood C4 protein levels (β; 95% CIs), haplotype frequencies, and association P values. C4A and C4B differ only by four amino acids, and the human endogenous retroviral (HERV) element is integrated in intron 9 of the C4A and C4B genes, contributing to interlocus and interallelic length heterogeneity of C4 genes. (B) Heat map of blood C4 levels as a function of ten common combinations of C4A and C4B gene copy numbers; the path from left to right on the plot corresponds to the increasing C4A gene copy, and the path from bottom to top corresponds to the increasing C4B gene copy numbers. Mutually adjusted per-copy effects on blood C4 levels and the corresponding P values are also depicted.
In GTEx, the C4-decreasing allele tagging the BS haplotype (rs3135353-T) has a cis-eQTL effect, whereby it decreases C4A gene transcript levels across multiple tissues, including whole blood (NES=−0.73; P=1.1x10-22), liver (NES=−0.66; P=4.4x10-17), and kidney (NES=−0.84; P=1.9x10-09) (Supplemental Figures 11A and 12A). The same allele is associated with increased C4B mRNA level in whole blood (NES=0.85; P=2.2x10-25) (Supplemental Figures 11B and 12B). This was further confirmed by a much larger whole-blood eQTL meta-analysis, where rs3135353-T was associated with significantly lower transcript levels of C4A (Z score =−20.45; FDR=0) while simultaneously increasing mRNA levels of C4B (Z score =25.52; FDR=0).
Lastly, we examined C4 levels as a function of both C4A and C4B gene copy numbers on the basis of the imputed haplotypes in the mutually adjusted linear regression model (Figure 6B). Each C4A gene copy was associated with an increased adjusted C4 levels of 0.13 SD units (95% CI, 0.11 to 0.15; P=5.5x10-08), whereas each C4B gene copy number was associated with independently increased adjusted C4 levels by 0.14 SD units (95% CI, 0.09 to 0.17; P=1.8x10-03).
Heritability and Genome-Wide Genetic Correlations of C4 Levels
The genome-wide significant independent SNPs at the C4 locus jointly explained approximately 13% of overall variance in C4 levels (P=2.98x10-112). In contrast, the six imputed common C4 copy number variants explained approximately 6% of variance with P=1.04x10-45. The two C4 copy number variants, BS and AL-BS, accounted for approximately 5% of variance in C4 levels, and the remaining 1% was contributed by the four other haplotypes. After excluding this locus (along with the entire HLA region), C4 levels had no appreciable residual SNP-based heritability by LD score regression (h2r=0.03; 95% CI, −0.13 to 0.19), suggesting minimal SNP contributions beyond the C4 locus.
The observation that independently significant SNPs capture more variance at this locus compared with the six C4 copy number haplotypes implies that additional genetic variants are likely contributing to the signal at this locus. For example, there could be additional nearby biallelic regulatory variants with effects on C4 production or other variation within the HLA locus related to increased C4 consumption. Because of sample size limitations, we are unable to assess the contribution of rarer copy number variations at this locus. Moreover, we could be missing Asian- and African-specific variants because the C4 reference panel is predominantly European. These factors may thus be contributing to the observed gap in variance explained.
In the genetic correlation analyses that include C4 locus, we observed an inverse genetic correlation with SLE51 (rg=−0.74; P=3.58x10-12), membranous nephropathy59 (rg=−0.75; P=3.58x10-09), celiac disease (rg=−0.57; P=4.08x10-07), and type 1 diabetes (rg=−0.50; P=3.17x10-06), although it is not clear to what extent these associations are driven by the C4 locus itself versus the nearby HLA alleles (Figure 3C, Supplemental Table 10). After exclusion of the C4 and HLA locus, we observed no significant genetic correlations with other complex traits (Figure 3D, Supplemental Table 10).
Phenome-Wide Associations
We also derived a genome-wide polygenic score for C4 levels using LDPred with and without C4/HLA locus and performed a phenome-wide association of the polygenic score in all 102,138 eMERGE participants (Figure 4C). The analysis of polygenic scores that contained C4/HLA locus revealed multiple phenotypic associations (Supplemental Table 11). Some of the most robust inverse associations included SLE (OR, 0.73; 95% CI, 0.68 to 0.78; P=2.45x10-31), celiac disease (OR, 0.63; 95% CI, 0.56 to 0.70; P=8.23x10-38), type 1 diabetes with renal manifestations (OR, 0.77; 95% CI, 0.70 to 0.85; P=5.62x10-11), proliferative GN (OR, 0.72; 95% CI, 0.62 to 0.82; P=1.25x10-09), chronic hepatitis (OR, 0.81; 95% CI, 0.72 to 0.89; P=5.39x10-07), and renal failure (OR, 0.95; 95% CI, 0.93 to 0.97; P=8.29x10-07). We also observed suggestive evidence for a positive genetic correlation between C3 and C4 levels, although it was not statistically significant (rg=0.16; P=0.49). The directions of effects in the PheWAS were generally consistent with genome-wide genetic correlations of C4 levels (Figure 3C). Interestingly, after excluding the C4/HLA locus, several inverse associations persisted, such as for lupus (localized and systemic; OR, 0.74; 95% CI, 0.68 to 0.79; P=2.74x10-30), SLE (OR, 0.74; 95% CI, 0.69 to 0.80; P=2.24x10-27), proliferative GN (OR, 0.73; 95% CI, 0.62 to 0.83; P=5.09x10-09), and liver abscess and sequelae of chronic liver disease (OR, 0.73; 95% CI, 0.62 to 0.83; P=5.09x10-09) (Figure 4D, Supplemental Table 12).
Lastly, we performed phenome-wide association for the top SNP at the C4 locus. The C4-increasing allele (rs3135353-C) had similar phenotypic inverse associations to the C4 polygenic score, including with celiac disease (OR, 0.36; 95% CI, 0.22 to 0.50; P=1.80x10-43), type 1 diabetes with renal manifestations (OR, 0.54; 95% CI, 0.38 to 0.69; P=4.99x10-14), diabetic retinopathy (OR, 0.54; 95% CI, 0.40 to 0.68; P=4.28x10-17), and SLE (OR, 0.64; 95% CI, 0.52 to 0.75; P=4.57x10-14) (Supplemental Figure 9C, Supplemental Table 13).
Discussion
In this study, we take advantage of the eMERGE-III consortium biobanking resource of 102,138 genotyped participants linked to EHR to perform comprehensive GWAS and PheWAS analyses for blood complement components.
Our GWAS provides several important insights into the complement system biology. First, a common variant at the CFH locus emerges as a genome-wide significant determinant of C3 levels. The signal at this locus intersects a liver-specific regulatory element, and the C3-decreasing allele is strongly associated with increased transcript levels of CFHR3 in liver. Thus, our results support the role of CFHR3 in determining circulating levels of C3 and are consistent with the hypothesis that CFHR3 may function as a competitive inhibitor of factor H, leading to increased baseline activation of the alternative pathway. We also confirm a common functional variant at the C3 locus as a genome-wide significant determinant of C3 levels. This signal is overlapping with a known susceptibility locus for age-related macular degeneration, but it has no known association with kidney disease presently. We hypothesize that this locus could be modulating the severity of the alternative complement pathway–mediated kidney injury, but further studies are clearly needed to test this hypothesis.
Our GWAS for C4 levels points to the major role of structural variation at the C4 locus. Low copy numbers of C4A or C4B have been previously described to represent risk factors for SLE, Sjogren syndrome, Graves disease, and rheumatoid arthritis.58,60,61 At the same time, recent data suggest that although low C4 copy number variants are associated with increased risk of autoimmunity, they appear to simultaneously convey protection from schizophrenia.58 Our analysis of medical records for nearly 4000 individuals demonstrates that these copy number alleles also correlate strongly with blood C4 protein levels. Specifically, for each C4B gene copy number, greater C4A copy independently increased blood C4 levels, and for each C4A copy number, greater C4B copy also independently increased blood C4 levels. The magnitude of the independent effects on blood C4 levels was slightly greater for C4B compared with C4A copy numbers, a difference that could be potentially explained by the known differences in the binding affinity of these two gene products to the complement receptor 1. Our findings generate a new hypothesis that low dosage of C4 genes may convey increased susceptibility to kidney disorders driven by the classical complement pathway, such as lupus nephritis and other forms of immune complex–mediated GN, but this hypothesis needs to be tested in additional clinical studies.
Linkage of genetic data with EHRs allowed us to perform PheWAS for the newly discovered loci and to explore disease correlations of genome-wide polygenic predictors of C3 and C4 levels. These were associated with multiple phenotypes, highlighting the relevance of the complement system to multiple human traits. For example, the polygenic predictor of C3 levels was significantly associated with 47 phenotypes, including SLE, GN, nephropathy, coagulation defects, sepsis, shock, and many others. Similarly, the polygenic predictor of C4 levels had significant associations with 32 phenotypes phenome wide, including SLE, connective tissue diseases, and several other autoimmune conditions.
We also explored genetic correlations of C3 and C4 levels with infectious, immune, renal, and cardiometabolic traits previously studied by GWAS. As expected, genetic variants associated with increased C3 and C4 levels were inversely correlated with SLE and other autoimmune conditions. Interestingly, we also detected suggestive positive correlations with several cardiometabolic traits, such as obesity, hypertension, CAD, and hyperlipidemias. These correlations were generally consistent with our PheWAS results and are in agreement with earlier observational studies reporting increased C3 and C4 levels in association with obesity, diabetes, hypertension, and cardiovascular risk.62–66
Notably, this is the largest study for C3 and C4 levels with several strengths, including multicenter design, diverse ancestral composition, and the standardized electronic phenotyping approach on the basis of EHR data. We demonstrate that our pragmatic medical records–based phenotyping approach can be used effectively to provide new insights about the genetic regulation of the complement pathway. When expanded to even larger biobanks linked to EHR, this approach is likely to uncover additional loci that might have been missed in our study due to modest sample size.
Several additional limitations of our approach need to be recognized. The main limitation of our study is that the C3 and C4 levels analysis is on the basis of “real-life” EHR data, and thus, there is a strong ascertainment bias in the inclusion of clinically tested individuals in our GWAS. This is because complement tests are typically performed for a limited set of clinical indications, mainly as a screening test for an autoimmune or inflammatory condition, or for monitoring the activity of complement-mediated diseases, such as SLE. We have attempted to control for the indication bias in our analyses by adjusting for the known diagnoses of autoimmune and inflammatory conditions as described in the Methods section. At the same time, we recognize that this adjustment is on the basis of the coded EHR data and may thus be imperfect due to missingness or miscoded diagnoses. We feel confident that the ascertainment bias does not produce spurious GWAS peaks because we replicate the previously reported GWAS findings performed in healthy participants,22,23 and the effects of our top GWAS signals become stronger after adjustment for autoimmune diagnoses.
We also recognize that the same ascertainment bias may be skewing the range of phenotypic associations detectable by polygenic score–based PheWAS. At the same time, the polygenic scores tested in PheWAS were on the basis of GWAS adjusted for known diagnosis of autoimmune disease. In addition, our PheWAS was performed on the entire dataset of n=102,138 eMERGE participants (the sample size that is 25-fold larger compared with n=3998 patients with available complement levels used for GWAS), and we detected several associations that cannot be attributed to confounding by test indication. For example, the positive correlation of C3 polygenic score with obesity or sleep apnea is unlikely to be confounded by indication because complement levels are not routinely tested in these conditions.
We also note that our PheWAS results need to be interpreted with caution because they are correlative in nature and therefore may be prone to reverse causation. Several complex medical conditions beyond autoimmunity may have secondary effects on the complement levels, and the genetic effects on these conditions could have been partially captured by our scores. For example, complement levels are often depressed in the setting of liver disease (due to decreased production) or hypercoagulable state and sepsis (due to increased consumption). Thus, the inverse correlations of polygenic scores with sepsis, thrombosis, or chronic liver disease do not necessarily represent a causative association.
Another limitation is our inability to further dissect the phenotypic associations at the C4 locus because this locus resides within the MHC class 3 region that has extended LD with MHC class 1 and 2 regions. Thus, it is difficult to dissect the phenotypic effects of classic MHC class 1 and 2 alleles and/or common variants in other immune-related genes encoded by this locus from the effects of C4 copy number variants themselves.
In summary, CFH and C3 loci are significant genetic determinants of circulating C3 levels, whereas the C4 copy number variant locus has large effects on circulating C4 levels. The polygenic effects captured by genome-wide polygenic predictors of C3 and C4 levels are strongly associated with multiple traits from immune mediated to cardiometabolic. Further studies will be needed to experimentally dissect cause-effect relationships between genetic variants controlling C3 and C4 levels and these phenotypes. Extending our approach to much larger biobanks with genetic data linked to EHRs will empower discovery of additional variants involved in the complement system and its regulation.
Disclosures
D. Crosslin reports consultancy agreements, research funding, and scientific advisor or membership with UnitedHealth Group. M.S.V. Elkind reports research funding from BMS-Pfizer Alliance for Eliquis and Roche; scientific advisor or membership with the American Heart Association; and other interests/relationships via royalties from UpToDate. A.G. Gharavi reports consultancy agreements with AstraZeneca Center for Genomics Research and Goldfinch Bio; research funding from the Renal Research Institute; honoraria from Sanofi; and scientific advisor or membership as an editorial board member of JASN, Journal of Nephrology, and Kidney International. Because A.G. Gharavi is an editor of JASN, he was not involved in the peer review process for this manuscript. A guest editor oversaw the peer review and decision-making process for this manuscript. J. Harley reports Ownership Interest in Now Diagnostics, Inc.; Honoraria from University of Pittsburgh; and Scientific Advisor or Membership with Now Diagnostics, Inc. G. Hripcsak reports scientific advisor or membership with Journal of the American Medical Informatics Association. K. Kiryluk reports membership of a scientific advisory board for Gilead Sciences and Goldfinch Bio and other scientific partnerships with Aevi Genomics and AstraZeneca. R. Knevel reports research funding from Pfizer and honoraria from Reuma Nederland (Dutch patient organization). H. Moncrieffe is employed by Janssen: Pharmaceutical Companies of Johnson & Johnson; has ownership interest in Janssen: Pharmaceutical Companies of Johnson & Johnson; and scientific advisor or membership on the Scientific Reports Editorial Board. I.B. Stanaway reports sole ownership interest in a limited liability company called Byrell Systems registered in Washington State. Y. Shen reports Scientific Advisor or Membership with Scientific Reports. S. Raychaudhuri reports Consultancy Agreements with Mestag, Inc, Gilead, Inc, Rheos Medicines, Pfizer, Merck; Ownership Interest in Mestag, Inc; Research Funding from Biogen, Inc; and Scientific Advisor or Membership with Janssen, Immunology Advisory Committee. All remaining authors have nothing to disclose.
Funding
The eMERGE network was funded by National Human Genome Research Institute grants U01HG008657 (to Group Health Cooperative/University of Washington), U01HG008680 (to Columbia University Health Sciences), U01HG008685 (to Mass General Brigham), U01HG008672 (to Vanderbilt University Medical Center), U01HG008666 (to Cincinnati Children’s Hospital Medical Center), U01HG006379 (to Mayo Clinic), U01HG008679 (to Geisinger Clinic), U01HG008684 (to Children’s Hospital of Philadelphia), U01HG008673 (to Northwestern University), U01HG008701 (to Vanderbilt University Medical Center serving as the coordinating center), U01HG008676 (to Partners Healthcare/Broad Institute), U01HG008664 (to Baylor College of Medicine), and U54MD007593 (to Meharry Medical College).
Supplementary Material
Acknowledgments
We would like to acknowledge all study participants and members of the Electronic Medical Records and Genomics III (eMERGE-III) network. Drs. Atlas Khan and Krzysztof Kiryluk conceived the study; Dr. Krzysztof Kiryluk designed the study and provided overall supervision of the project; Drs. Atlas Khan and Krzysztof Kiryluk wrote the initial draft of the manuscript; Dr. Atlas Khan performed GWAS and PheWAS analyses, pathway analyses, functional annotations, genome-wide genetic correlation analyses, and C4 imputation; Drs. George Hripcsak, Lynn Petukhova, Ning Shang, and Chunhua Weng designed, validated, and implemented electronic phenotyping for GWAS analyses; Drs. David Crosslin and Ian B. Stanaway compiled network-wide GWAS datasets and performed genome-wide imputation analysis; Junying Zhang managed the local study database; Drs. Mitchell S.V. Elkind, Ali G. Gharavi, and Yufeng Shen consulted on the study design and critically reviewed the manuscript; Drs. Joshua C. Denny, John B. Harley, Scott J. Hebbring, Elizabeth W. Karlson, Rachel Knevel, Leah C. Kottyan, Halima Moncrieffe, Bahram Namjou-Khales, and Soumya Raychaudhuri coordinated the analysis of local medical records data and extraction of relevant phenotypes; and all authors read and approved the final version of manuscript. The National Human Genome Research Institute had no role in the design of this study, data analysis or interpretation, or writing the manuscript.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
See related editorial, “Biobanks Linked to Electronic Health Records Accelerate Genomic Discovery,” on pages 1828–1829.
Data Sharing Statement
The eMERGE-III genetic datasets with linked phenotypes are accessible through the Database of Genotype and Phenotype (dbGAP) repository (accession no. phs001584.v1.p1). The software and documentation of the eMERGE-III Autoimmune Disease Phenotype can be found on the Phenotype Knowledge Database website (https://phekb.org/phenotype/autoimmune-disease-phenotype). Genome-wide summary statistics for C3 and C4 levels are freely available for download on our laboratory website: www.columbiamedicine.org/divisions/kiryluk/resources.php.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2020091371/-/DCSupplemental.
Supplemental Table 1. Basic characteristic of study subjects in eMERGE-III by study site.
Supplemental Table 2. Distribution of eMERGE-III participants by the number of C3/C4 tests performed.
Supplemental Table 3. The overall distribution of autoimmune and inflammatory disorders among GWAS participants with C3 and C4 levels.
Supplemental Table 4. GWAS for C3- and C4-levels pathway analysis.
Supplemental Table 5. Genetic correlations of C3 levels with other complex traits.
Supplemental Table 6. Polygenic score based PheWAS analysis of C3 levels.
Supplemental Table 7. Polygenic score based PheWAS analysis of C3 levels without HLA locus.
Supplemental Table 8. PheWAS analysis of single SNP of CFH locus for C3 levels.
Supplemental Table 9. PheWAS analysis of single SNP of C3 locus for C3 levels.
Supplemental Table 10. Genetic correlation of C4 levels with other complex traits.
Supplemental Table 11. Polygenic score based PheWAS analysis of C4 levels.
Supplemental Table 12. Polygenic score based PheWAS analysis of C4 levels without C4/HLA locus.
Supplemental Table 13. PheWAS analysis of single SNP of C4 for C4 levels.
Supplemental Figure 1. Principal component analysis of the eMERGE-III participants with available C3 and C4 levels.
Supplemental Figure 2. The quantile-quantile plots for (A) GWAS results for C3 levels and (B) GWAS results for C4 levels.
Supplemental Figure 3. Regional plots of the CFH locus: (A) European, (B) African, and (C) East Asian ancestry depicted with LocusZoom using 1000 Genomes European, African, and East Asian LD reference panels, respectively.
Supplemental Figure 4. Regional plots of the C3 locus by (A) European, (B) African, and (C) East Asian ancestry depicted with LocusZoom using 1000 Genomes European, African, and East Asian LD reference panels, respectively.
Supplemental Figure 5. Tissue-specific functional predictions for variants at the CFH locus.
Supplemental Figure 6. Expression QTL effects for rs3753396-G, the top signal on chr.1q31.3 (GTEx version 8), demonstrating a strong and highly specific liver eQTL effect on (A) CFHR3 and (B) CFHR4 transcripts.
Supplemental Figure 7. Tissue-specific functional predictions for the C3 locus variants.
Supplemental Figure 8. The top rs11569470-A on chr.19p13.3 demonstrating significant eQTLs for GPR108 transcript in several tissues based on data from GTEx version 8.
Supplemental Figure 9. PheWAS plots for the top SNPs at the following loci: (A) CFH, (B) C3, and (C) C4.
Supplemental Figure 10. Regional plots of the C4 locus: (A) European, (B) African, and (C) East Asian ancestry depicted with LocusZoom using 1000 Genomes European, African, and East Asian LD reference panels, respectively.
Supplemental Figure 11. The top rs3135353-T on chr.6p21.32 demonstrating significant eQTLs for (A) C4A and (B) C4B transcripts in multiple tissues on the basis of GTEx version 8.
Supplemental Figure 12. The opposed cis-eQTL effects of rs3135353 on (A) C4A and (B) C4B transcript levels in whole blood (GTEx version 8).
References
- 1.Walport MJ: Complement. First of two parts. N Engl J Med 344: 1058–1066, 2001 [DOI] [PubMed] [Google Scholar]
- 2.Walport MJ: Complement. Second of two parts. N Engl J Med 344: 1140–1144, 2001 [DOI] [PubMed] [Google Scholar]
- 3.Noris M, Benigni A, Remuzzi G: The case of complement activation in COVID-19 multiorgan impact. Kidney Int 98: 314–322, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Song W-C, FitzGerald GA: COVID-19, microangiopathy, hemostatic activation, and complement. J Clin Invest 130: 3950–3953, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Risitano AM, Mastellos DC, Huber-Lang M, Yancopoulou D, Garlanda C, Ciceri F, et al.: Complement as a target in COVID-19? [published correction appears in Nat Rev Immunol 20: 448, 2020 0.1038/s41577-020-0366-6]. Nat Rev Immunol 20: 343–344, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bomback AS, Markowitz GS, Appel GB: Complement-mediated glomerular diseases: A tale of 3 pathways. Kidney Int Rep 1: 148–155, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park DH, Connor KM, Lambris JD: The challenges and promise of complement therapeutics for ocular diseases. Front Immunol 10: 1007, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zipfel PF, Wiech T, Stea ED, Skerka C: CFHR gene variations provide insights in the pathogenesis of the kidney diseases atypical hemolytic uremic syndrome and C3 glomerulopathy. J Am Soc Nephrol 31: 241–256, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith RJH, Appel GB, Blom AM, Cook HT, D’Agati VD, Fakhouri F, et al.: C3 glomerulopathy - understanding a rare complement-driven renal disease. Nat Rev Nephrol 15: 129–143, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raychaudhuri S, Iartchouk O, Chin K, Tan PL, Tai AK, Ripke S, et al.: A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nat Genet 43: 1232–1236, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seddon JM, Yu Y, Miller EC, Reynolds R, Tan PL, Gowrisankar S, et al.: Rare variants in CFI, C3 and C9 are associated with high risk of advanced age-related macular degeneration. Nat Genet 45: 1366–1370, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fritsche LG, Igl W, Bailey JN, Grassmann F, Sengupta S, Bragg-Gresham JL, et al.: A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat Genet 48: 134–143, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Julià A, López-Longo FJ, Pérez Venegas JJ, Bonàs-Guarch S, Olivé À, Andreu JL, et al.: Genome-wide association study meta-analysis identifies five new loci for systemic lupus erythematosus. Arthritis Res Ther 20: 100, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kiryluk K, Li Y, Scolari F, Sanna-Cherchi S, Choi M, Verbitsky M, et al.: Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat Genet 46: 1187–1196, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tian C, Hromatka BS, Kiefer AK, Eriksson N, Noble SM, Tung JY, et al.: Genome-wide association and HLA region fine-mapping studies identify susceptibility loci for multiple common infections. Nat Commun 8: 599, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Denny JC, Ritchie MD, Basford MA, Pulley JM, Bastarache L, Brown-Gentry K, et al.: PheWAS: Demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics 26: 1205–1210, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Unsworth DJ: Complement deficiency and disease. J Clin Pathol 61: 1013–1017, 2008 [DOI] [PubMed] [Google Scholar]
- 18.Seppänen M, Lokki ML, Timonen T, Lappalainen M, Jarva H, Järvinen A, et al.: Complement C4 deficiency and HLA homozygosity in patients with frequent intraoral herpes simplex virus type 1 infections. Clin Infect Dis 33: 1604–1607, 2001 [DOI] [PubMed] [Google Scholar]
- 19.Soto K, Wu YL, Ortiz A, Aparício SR, Yu CY: Familial C4B deficiency and immune complex glomerulonephritis. Clin Immunol 137: 166–175, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hunnangkul S, Nitsch D, Rhodes B, Chadha S, Roberton CA, Pessôa-Lopes P, et al.: Familial clustering of non-nuclear autoantibodies and C3 and C4 complement components in systemic lupus erythematosus. Arthritis Rheum 58: 1116–1124, 2008 [DOI] [PubMed] [Google Scholar]
- 21.Rhodes B, Hunnangkul S, Morris DL, Hsaio LC, Graham DS, Nitsch D, et al.: The heritability and genetics of complement C3 expression in UK SLE families. Genes Immun 10: 525–530, 2009 [DOI] [PubMed] [Google Scholar]
- 22.Yang X, Sun J, Gao Y, Tan A, Zhang H, Hu Y, et al.: Genome-wide association study for serum complement C3 and C4 levels in healthy Chinese subjects. PLoS Genet 8: e1002916, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borné Y, Muhammad IF, Lorés-Motta L, Hedblad B, Nilsson PM, Melander O, et al.: Complement C3 associates with incidence of diabetes, but no evidence of a causal relationship. The Journal of Clinical Endocrinology & Metabolism 102: 4477–4485 [DOI] [PubMed] [Google Scholar]
- 24.eMERGE-Consortium: Lessons learned from the eMERGE Network: Balancing genomics in discovery and practice. HGG Advances 2: 100018, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kirby JC, Speltz P, Rasmussen LV, Basford M, Gottesman O, Peissig PL, et al.: PheKB: A catalog and workflow for creating electronic phenotype algorithms for transportability. J Am Med Inform Assoc 23: 1046–1052, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stanaway IB, Hall TO, Rosenthal EA, Palmer M, Naranbhai V, Knevel R, et al.; eMERGE Network: The eMERGE genotype set of 83,717 subjects imputed to ∼40 million variants genome wide and association with the herpes zoster medical record phenotype. Genet Epidemiol 43: 63–81, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abraham G, Inouye M: Fast principal component analysis of large-scale genome-wide data. PLoS One 9: e93766, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen WM: Robust relationship inference in genome-wide association studies. Bioinformatics 26: 2867–2873, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Willer CJ, Li Y, Abecasis GR: METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26: 2190–2191, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al.: PLINK: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81: 559–575, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al.; 1000 Genomes Project Analysis Group: The variant call format and VCFtools. Bioinformatics 27: 2156–2158, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, et al.; Schizophrenia Working Group of the Psychiatric Genomics Consortium: Schizophrenia risk from complex variation of complement component 4. Nature 530: 177–183, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Browning BL, Zhou Y, Browning SR: A one-penny imputed genome from next-generation reference panels. Am J Hum Genet 103: 338–348, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Das S, Forer L, Schönherr S, Sidore C, Locke AE, Kwong A, et al.: Next-generation genotype imputation service and methods. Nat Genet 48: 1284–1287, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loh PR, Danecek P, Palamara PF, Fuchsberger C, A Reshef Y, K Finucane H, et al.: Reference-based phasing using the Haplotype Reference Consortium panel. Nat Genet 48: 1443–1448, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bulik-Sullivan BK, Loh PR, Finucane HK, Ripke S, Yang J, Patterson N, et al.; Schizophrenia Working Group of the Psychiatric Genomics Consortium: LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet 47: 291–295, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mishra A, Macgregor S: VEGAS2: Software for more flexible gene-based testing. Twin Res Hum Genet 18: 86–91, 2015 [DOI] [PubMed] [Google Scholar]
- 38.Backenroth D, He Z, Kiryluk K, Boeva V, Pethukova L, Khurana E, et al.: FUN-LDA: A latent dirichlet allocation model for predicting tissue-specific functional effects of noncoding variation: Methods and applications. Am J Hum Genet 102: 920–942, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vilhjálmsson BJ, Yang J, Finucane HK, Gusev A, Lindström S, Ripke S, et al.; Schizophrenia Working Group of the Psychiatric Genomics Consortium, Discovery, Biology, and Risk of Inherited Variants in Breast Cancer (DRIVE) study: Modeling linkage disequilibrium increases accuracy of polygenic risk scores. Am J Hum Genet 97: 576–592, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vosa U, Claringbouid A, Westra H-J, Jan Bonder M, Deelen P, Zeng B, et al.: Unraveling the polygenic architecture of complex traits using blood eQTL metaanalysis. bioRxiv. 10.1101/447367 (Preprint posted October 19, 2018)
- 41.Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, et al.; 1000 Genomes Project Consortium: An integrated map of structural variation in 2,504 human genomes. Nature 526: 75–81, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lorés-Motta L, Paun CC, Corominas J, Pauper M, Geerlings MJ, Altay L, et al.: Genome-wide association study reveals variants in CFH and CFHR4 associated with systemic complement activation: Implications in age-related macular degeneration. Ophthalmology 125: 1064–1074, 2018 [DOI] [PubMed] [Google Scholar]
- 43.Suhre K, Arnold M, Bhagwat AM, Cotton RJ, Engelke R, Raffler J, et al.: Connecting genetic risk to disease end points through the human blood plasma proteome [published correction appears in Nat Commun 8: 15345, 2017 10.1038/ncomms15345]. Nat Commun 8: 14357, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hughes AE, Orr N, Esfandiary H, Diaz-Torres M, Goodship T, Chakravarthy U: A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration [published correction appears in Nat Genet 39: 567, 2007]. Nat Genet 38: 1173–1177, 2006 [DOI] [PubMed] [Google Scholar]
- 45.Gharavi AG, Kiryluk K, Choi M, Li Y, Hou P, Xie J, et al.: Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat Genet 43: 321–327, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Emilsson V, Ilkov M, Lamb JR, Finkel N, Gudmundsson EF, Pitts R, et al.: Co-regulatory networks of human serum proteins link genetics to disease. Science 361: 769–773, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dong D, Zhou H, Na SY, Niedra R, Peng Y, Wang H, et al.: GPR108, an NF-κB activator suppressed by TIRAP, negatively regulates TLR-triggered immune responses. PLoS One 13: e0205303, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou W, Nielsen JB, Fritsche LG, Dey R, Gabrielsen ME, Wolford BN, et al.: Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat Genet 50: 1335–1341, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, et al.: A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet 47: 1121–1130, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, et al.; LifeLines Cohort Study; ADIPOGen Consortium; AGEN-BMI Working Group; CARDIOGRAMplusC4D Consortium; CKDGen Consortium; GLGC; ICBP; MAGIC Investigators; MuTHER Consortium; MIGen Consortium; PAGE Consortium; ReproGen Consortium; GENIE Consortium; International Endogene Consortium: Genetic studies of body mass index yield new insights for obesity biology. Nature 518: 197–206, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bentham J, Morris DL, Graham DSC, Pinder CL, Tombleson P, Behrens TW, et al.: Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat Genet 47: 1457–1464, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Walport MJ: Complement and systemic lupus erythematosus. Arthritis Res 4[Suppl 3]: S279–S293, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Einav S, Pozdnyakova OO, Ma M, Carroll MC: Complement C4 is protective for lupus disease independent of C3. J Immunol 168: 1036–1041, 2002 [DOI] [PubMed] [Google Scholar]
- 54.Sun BB, Maranville JC, Peters JE, Stacey D, Staley JR, Blackshaw J, et al.: Genomic atlas of the human plasma proteome. Nature 558: 73–79, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee JJ, Wedow R, Okbay A, Kong E, Maghzian O, Zacher M, et al.; 23andMe Research Team; COGENT (Cognitive Genomics Consortium); Social Science Genetic Association Consortium: Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat Genet 50: 1112–1121, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gatenby PA, Barbosa JE, Lachmann PJ: Differences between C4A and C4B in the handling of immune complexes: The enhancement of CR1 binding is more important than the inhibition of immunoprecipitation. Clin Exp Immunol 79: 158–163, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dangel AW, Mendoza AR, Baker BJ, Daniel CM, Carroll MC, Wu LC, et al.: The dichotomous size variation of human complement C4 genes is mediated by a novel family of endogenous retroviruses, which also establishes species-specific genomic patterns among Old World primates. Immunogenetics 40: 425–436, 1994 [DOI] [PubMed] [Google Scholar]
- 58.Kamitaki N, Sekar A, Handsaker RE, de Rivera H, Tooley K, Morris DL, et al.; Schizophrenia Working Group of the Psychiatric Genomics Consortium: Complement genes contribute sex-biased vulnerability in diverse disorders. Nature 582: 577–581, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xie J, Liu L, Mladkova N, Li Y, Ren H, Wang W, et al.: The genetic architecture of membranous nephropathy and its potential to improve non-invasive diagnosis. Nat Commun 11: 1600, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lv Y, He S, Zhang Z, Li Y, Hu D, Zhu K, et al.: Confirmation of C4 gene copy number variation and the association with systemic lupus erythematosus in Chinese Han population. Rheumatol Int 32: 3047–3053, 2012 [DOI] [PubMed] [Google Scholar]
- 61.Yang Y, Chung EK, Wu YL, Savelli SL, Nagaraja HN, Zhou B, et al.: Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): Low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. Am J Hum Genet 80: 1037–1054, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gabrielsson BG, Johansson JM, Lönn M, Jernås M, Olbers T, Peltonen M, et al.: High expression of complement components in omental adipose tissue in obese men. Obes Res 11: 699–708, 2003 [DOI] [PubMed] [Google Scholar]
- 63.Muscari A, Massarelli G, Bastagli L, Poggiopollini G, Tomassetti V, Drago G, et al.: Relationship of serum C3 to fasting insulin, risk factors and previous ischaemic events in middle-aged men. Eur Heart J 21: 1081–1090, 2000 [DOI] [PubMed] [Google Scholar]
- 64.Onat A, Uzunlar B, Hergenç G, Yazici M, Sari I, Uyarel H, et al.: Cross-sectional study of complement C3 as a coronary risk factor among men and women. Clin Sci (Lond) 108: 129–135, 2005 [DOI] [PubMed] [Google Scholar]
- 65.Engström G, Hedblad B, Eriksson KF, Janzon L, Lindgärde F: Complement C3 is a risk factor for the development of diabetes: A population-based cohort study. Diabetes 54: 570–575, 2005 [DOI] [PubMed] [Google Scholar]
- 66.Weyer C, Tataranni PA, Pratley RE: Insulin action and insulinemia are closely related to the fasting complement C3, but not acylation stimulating protein concentration. Diabetes Care 23: 779–785, 2000 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.