

Significance Statement
In autosomal dominant polycystic kidney disease (ADPKD), interstitial inflammation promotes cyst progression. TWEAK is a TNF superfamily cytokine that regulates inflammatory responses, and its receptor, Fn14, is expressed in nephron epithelium. This paper describes TWEAK’s role in ADPKD and its potential as a therapeutic target. The Fn14/TWEAK axis is upregulated in human and mouse polycystic kidneys, and TWEAK administration in mice accelerates cyst progression, whereas anti-TWEAK treatment slows cyst growth, improving kidney function and survival. Anti-TWEAK antibodies restore several ADPKD-related pathways, such as proliferation and NF-κB; slightly reduces fibrosis and apoptosis; and indirectly decreases macrophage recruitment. These findings identify the TWEAK signaling pathway as a new disease mechanism in ADPKD and a new possible therapeutic approach.
Keywords: TWEAK, autosomal dominant polycystic kidney disease, Pkd1, Fn14, inflammation
Abstract
Background
In autosomal dominant polycystic kidney disease (ADPKD), cyst development and enlargement lead to ESKD. Macrophage recruitment and interstitial inflammation promote cyst growth. TWEAK is a TNF superfamily (TNFSF) cytokine that regulates inflammatory responses, cell proliferation, and cell death, and its receptor Fn14 (TNFRSF12a) is expressed in macrophage and nephron epithelia.
Methods
To evaluate the role of the TWEAK signaling pathway in cystic disease, we evaluated Fn14 expression in human and in an orthologous murine model of ADPKD. We also explored the cystic response to TWEAK signaling pathway activation and inhibition by peritoneal injection.
Results
Meta-analysis of published animal-model data of cystic disease reveals mRNA upregulation of several components of the TWEAK signaling pathway. We also observed that TWEAK and Fn14 were overexpressed in mouse ADPKD kidney cysts, and TWEAK was significantly high in urine and cystic fluid from patients with ADPKD. TWEAK administration induced cystogenesis and increased cystic growth, worsening the phenotype in a murine ADPKD model. Anti-TWEAK antibodies significantly slowed the progression of ADPKD, preserved renal function, and improved survival. Furthermore, the anti-TWEAK cystogenesis reduction is related to decreased cell proliferation–related MAPK signaling, decreased NF-κB pathway activation, a slight reduction of fibrosis and apoptosis, and an indirect decrease in macrophage recruitment.
Conclusions
This study identifies the TWEAK signaling pathway as a new disease mechanism involved in cystogenesis and cystic growth and may lead to a new therapeutic approach in ADPKD.
Visual Abstract

Autosomal dominant polycystic kidney disease (ADPKD) is the most common renal genetic disorder, with an estimated prevalence of 1:1000 individuals.1 ADPKD is characterized by the formation and progressive growth of multiple cysts that lead to ESKD at the age of 50–60 years. The principal extrarenal manifestation is polycystic liver disease. ADPKD is caused by mutations in the polycystic kidney disease 1 (PKD1) or PKD2 genes, which encode polycystin-1 (PC1) and PC2, respectively.2 Studies involving cell and animal models have described multiple signaling pathways implicated in the molecular basis of ADPKD; however, the mechanisms underlying cystogenesis remain to be elucidated.
TNF-like weak inducer of apoptosis (TWEAK; TNF superfamily member 12 [TNFSF12]) is a type 2 transmembrane glycoprotein that can be proteolytically processed, resulting in soluble TWEAK. Both full-length TWEAK and soluble TWEAK bind to and activate the TWEAK receptor known as fibroblast growth factor–inducible 14 (Fn14; TNF receptor superfamily member 12a [TNFRSF12a]). TWEAK belongs to the TNFSF of cytokines, the members of which modulate immune responses and inflammation.3 TWEAK contributes to kidney inflammation, promoting chemokine secretion by renal cells, and regulates kidney cell proliferation, apoptosis, differentiation, and activation of NF-κB.4–7 Anti-TWEAK neutralizing antibodies are protective in experimental models of AKI and proteinuric and obstructive CKD,4,8 and have reached the clinical development stage.6 ADPKD is not primarily an inflammatory disease; however, AKI and interstitial inflammation promote cyst progression through macrophage recruitment and upregulation of proinflammatory cytokines.9,10
Our study focuses on the role of the TWEAK signaling pathway in ADPKD. First, we studied the status of the TWEAK/Fn14 axis in ADPKD using human and animal samples from an orthologous mouse ADPKD model. Second, we evaluated the role of TWEAK in different developmental windows of the animal model. On the basis of our results, we tested the therapeutic potential of anti-TWEAK strategies for ADPKD.
Methods
Data Mining
We searched PKD transcriptomics databases for TNFSF and TNFRSF members that were differentially expressed, and searched Gene Expression Omnibus and PubMed for members that showed an increased expression in PKD. For high sensitivity, we considered P<0.05 to be significant for screening purposes.
Mouse Models
We studied specific Pkd1-deletion mouse models, C57/BL6 Pkd1cond/cond;Tam-Cre− and C57/BL6 Pkd1cond/cond;Tam-Cre+, which represented the Wild Type (WT) and Pkd1 mutant models, respectively. Genotypes were confirmed by standard PCR, following the conditions and primers described by Piontek et al.11 Pkd1 was inactivated at postnatal day 10 (p10) and p11 (cystic window) or p15 and p16 (noncystic window), using the CreLoxP-tamoxifen system, resulting in the loss of Pkd1. For this, Cre recombinase was activated by intraperitoneally injecting tamoxifen (10 mg/40 g; catalog number T5648; Sigma) in corn oil (catalog number C8267; Sigma-Aldrich) into the mother of the litter to induce Pkd1 deletion in lactating pups, as described by Piontek et al.12 Experiments were conducted in strict accordance with the protocols approved by the Ethical Committee of the University of Santiago de Compostela (code, 15010/2020/002).
Kidney samples of slowly progressive C57/BL6 Pkd1cond/cond;Tam-Cre mice11,12 (Pkd1 deletion on p45, euthanized at >14 weeks) were generated as previously described.13 The C57/BL6 Pkd1cond/condTam-KSP-Cre mice were generated using the same floxable allele13 intercrossed with the Tm-KspCreERT2 (kindly provided by Dr. Peter Igarashi, University of Texas Southwestern Medical Center).14 Cre recombinase activity was induced by a single injection of tamoxifen at p45, for the first line, or at p10–11, for the second line, and the animals were harvested at p175 or p38–39, respectively . Tamoxifen, freshly prepared and dissolved in corn oil by continuous shaking at 37°C for 8 hours, was injected intraperitoneally. Similarly, kidney samples of C57/BL6 Pkd1cond;Pax8;TET-OCre mice12,15 (Pkd1 deletion on p27, euthanized at p150–160) were obtained from P.O. and T.W. (University of Maryland School of Medicine and Baltimore PKD center). Pkd1 inactivation was induced by a single injection of doxycycline at p27.
TWEAK and Anti-TWEAK Administration
Mice were injected intraperitoneally with TWEAK (30 ng/g mouse per 48 h), neutralizing anti-TWEAK mAb (IgG1 BIIB023, clone P2D10; 10 mg/kg mouse, three times weekly),8 or isotype IgG (mouse IgG2A; 10 mg/kg mouse, three times weekly) for the control groups, generously provided by Biogen, Inc. Doses were chosen on the basis of prior experience assessing the effect of TWEAK and anti-TWEAK antibodies in experimental murine kidney disease, in which in vivo–tested doses were, in turn, derived from cell culture studies.16–21 All mice were euthanized using isoflurane, and blood (by intravascular puncture) and tissues were collected. Animals were euthanized at different time points (Supplemental Figure 1). At least two litters and four animals, in a 1:1 ratio of males and females (no differences between males and females were found), were used for each treatment.
Histology
Right kidneys were fixed in 4% paraformaldehyde-buffered solution, pH=7, overnight at 4°C. Kidneys were then bisected and dehydrated, through a series of ethanol and xylene, and embedded in paraffin. Kidney slices were 4.5 µm thick.
A standard protocol was used for hematoxylin and eosin staining. For immunofluorescence, the antigen was retrieved using sodium citrate buffer (pH=6; catalog number S2369; Agilent Dako). Tissue was blocked with antibody diluent (catalog number K8006; Agilent Dako), and then incubated with lotus tetragonolobus lectin (LTL), dolichos biflorus agglutinin (DBA; 1:100; Vector Laboratories), and anti–Tamm–Horsfall protein (anti-THP; 1:250; Bio-Rad). Sections were washed three times with PBS1x, and then (if necessary) incubated with appropriate secondary antibodies conjugated to rhodamine (1:100; catalog number sc-2492; Santa Cruz Biotechnology, Inc.) and 4′,6-diamidino-2-phenylindole (1:10000; catalog number D1306; Life Technologies, Thermo Fisher Scientific). Images of longitudinal sections of both kidney and liver were captured with a loupe Leica DMD 108 (Leica) for sections stained with hematoxylin and eosin. Immunofluorescence was visualized using a Zeiss Axio Vert.A1 microscope (Carl Zeiss).
Immunohistochemistry was performed using a PT-link device with a low pH solution. Sections were then washed with PBS for 5 minutes; blocked by incubation with 4% PBS/BSA and 6% serum; and incubated with anti-Fn14 antibody (1:40; catalog number 4403; Cell Signaling), anti-F4/80 antibody (1:100; catalog number MCA497GA; Bio-Rad), anti-p65 antibody (1:800; catalog number sc-372 X; Santa Cruz Biotechnology), anti–smooth muscle actin (1:10000; catalog number GA611; DAKO, Agilent), or anti-fibronectin (1:5000; catalog number A0245; DAKO, Agilent) overnight. Sections were washed three times with PBS1x, and then incubated with biotin-conjugated, anti-rat secondary antibody (1:200; Invitrogen) and with the Vectastain Elite ABC-HRP Kit (Vector). Sections were counterstained with Carazzi hematoxylin. Images were captured using an iScan Coreo Au (Ventana Medical Systems, Inc.). F4/80-positive macrophages in medulla or cortex were quantified in 25 randomly chosen fields (×20) in a blinded manner. At least n=3 samples were used for the histologic experiments.
Terminal Deoxynucleotidyl Transferase–Mediated Digoxigenin-Deoxyuridine Nick-End Labeling
Terminal deoxynucleotidyl transferase–mediated digoxigenin-deoxyuridine nick-end labeling (TUNEL) was performed in 3-mm-thick sections of paraffin-embedded tissue using the In Situ Cell Death Detection Kit, Fluorescein (Roche), according to the manufacturer’s instructions. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole, and images were captured using fluorescence microscopy (Nikon E600). TUNEL-positive cells were quantified in 15 randomly chosen fields (×400) in a blinded manner.
Measurement of Cystic Index
Slides of longitudinal sections from the inner part of kidney (with papilla present) were taken as representative images, which were used for assessing cystic indexes. At least six microscopic images were used for quantification of the cystic index and number of cysts. Both parameters were quantified using the automatic cyst recognition software tool, CystAnalyser, as previously described.22 Cyst size was assessed by the number of pixels and cysts, classified into three bin sizes as follows: shorter cysts, <250 pixels; medium cysts, 250–750 pixels; and largest cysts, >750 pixels.
Clinical Chemistry
Blood was collected before euthanasia, and serum was obtained using BD Vacutainer SST II Advance tubes. Serum Blood Urea Nitrogen (BUN) were measured using the ADVIA 2400 Chemistry System (Siemens Healthineers).
Mouse TNFSF12/TWEAK ELISA
Serum from WT and mutant Pkd1cond/cond;Tam-Cre mice was used to assess TWEAK levels using the Mouse TNFSF12/TWEAK Sandwich High Sensitivity ELISA kit (catalog number EK1176; Boster Biological Technology).
Protein and RNA Extractions
After euthanasia, left kidneys were stored at −80°C. Whole frozen kidneys were ground in liquid nitrogen, and half of the tissue was assigned to protein extraction and the rest to RNA extraction.
Protein extracts were prepared using radioimmunoprecipitation assay lysis buffer (10 mM Tris, 5 mM EDTA, 150 mM sodium chloride, 0.1% SDS, 1% Triton X-100, and 1% sodium deoxycholate), along with 1% protease and phosphatase inhibitors (catalog numbers P8340 and P0044, respectively; Sigma). This cell or tissue lysate was centrifuged (30 minutes at 4°C and 14,000 rpm). Then, supernatants were collected and quantified using the Bradford protein assay (catalog number 5000001; Bio-Rad).
RNA was isolated using the TRIzol protocol (TRI Reagent, catalog number T9424; Sigma), in which chloroform (catalog number C2432; Sigma) serves as the denaturation agent for cells, and isopropanol (catalog number 650447; Sigma) is the RNA-precipitating agent. The precipitate was diluted in diethylpyrocarbonate (DEPC) water (catalog number AM9920; Ambion), which also contained 1% SUPERaseIn RNase Inhibitor (catalog number AM2694; Thermo Fisher).
Western Blot
Western blotting assays were performed on whole kidney lysates, described above. Total protein content was determined using the Bradford protein assay, and the same amount of each protein was boiled at 95°C for 5 minutes and separated in 10%–12% SDS-PAGE, under reducing conditions. Proteins were then transferred onto nitrocellulose membranes (catalog number 1620115; Bio-Rad). To avoid nonspecific binding, membranes were blocked for 1 hour at room temperature, using a 2.5% BSA solution (catalog number MB046; NZYTech). Thereafter, membranes were incubated overnight at 4°C with primary antibodies and then washed in 0.3% Tween-20 in PBS. Lastly, the nitrocellulose membranes were incubated for 1 hour at room temperature with the horseradish peroxidase–conjugated secondary antibodies. Signals were developed using the SuperSignal West Pico PLUS Chemiluminescent Substrate kit (catalog number 34580; Thermo Fisher) or the Pierce ECL Western Blotting Substrate kit (catalog number 32109; Thermo Fisher) before visualization in the ChemiDoc Imaging System (Bio-Rad). Protein bands were quantified using ImageJ Lab software (version 4.0.1). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a housekeeping gene to correct for errors in protein levels.
Antibodies
The following primary antibodies were used for Western blot: anti-Fn14 (1:1000; catalog number Ab109365; Abcam), anti–phosphorylated-extracellular signal–regulated kinase (anti–p-ERK; 1:2000; catalog number 4370; Cell Signaling); anti-ERK (1:2000; catalog number 4695; Cell Signaling), anti–p-AKT (1:2000; catalog number 4060; Cell Signaling), anti-AKT (1:2000; catalog number 9272; Cell Signaling), anti-GAPDH (1:10000; catalog number mab374; Millipore), and the secondary antibody goat anti-rabbit IgG horseradish peroxidase (catalog number 31460; Thermo Fisher).
In Vitro Experiments
MCT cells are a proximal tubular epithelial cell line originally harvested from the renal cortex of SJL mice, and they have been extensively characterized.23 They were cultured in RPMI 1640 (catalog number 10379144; GIBCO), 10% decomplemented FBS, 2 mM glutamine, 100 U/ml penicillin, and 100 µg/ml streptomycin in 5% carbon dioxide at 37°C. Thereafter, subconfluent cells were rested in serum-free medium overnight and 100 ng/ml TWEAK was added for 3 hours (this concentration was determined on the basis of prior dose-response curves).16
Bone marrow–derived macrophages were obtained from 10- to 14-week-old WT mice. After euthanasia with carbon dioxide, bone marrow was extracted from the femur and tibia, and it was seeded with RPMI culture medium with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM l-glutamine, and 5 ng/ml macrophage–colony stimulating factor (macrophage-CSF; catalog number 574802; Biolegend).24 After 96 hours, macrophages were incubated with 100 ng/ml TWEAK for 3 hours.
Quantitative Real-Time PCR
RNA (1 μg) was treated with DNase I (catalog number 18068015; Invitrogen) before reverse transcription using the SuperScript III First-Strand Synthesis System kit (catalog number 18080051; Invitrogen). cDNA was used for the gene expression analysis, using FastStart Universal SYBR GREEN Master (Rox) (catalog number 04913914001; Roche) in a Mx3005P system (Agilent Technologies). The complete reactions were subjected to the following program: a denaturation step with 30 seconds at 95°C; 45 cycles of 30 seconds at 95°C, 1 minute at 60°C, and 30 seconds at 72°C; followed by a cooling step. Each sample was run in triplicate in each experiment. Primers (Supplemental Table 1) were designed using Primer 3 Plus (http://tiny.cc/pf928y), on the basis of the reference cDNA sequence published in the Ensembl genome browser (http://www.ensembl.org/index.html), with the exception of Fn14, Il-6, and regulated on activation normal T cell expressed and secreted (RANTES) primers, in which the Thermo Fisher sequence (TaqMan probe) was used.
We used the absolute quantification method to process our PCR results. Therefore, a standard curve with known concentrations was used in the assay. Additionally, each sample was normalized to a constantly expressed gene, Hprt or Gapdh (TaqMan probe; ThermoFisher), which were used as housekeeping genes.
For in vitro experiments, 1 µg RNA was reverse transcribed using the High Capacity cDNA Archive Kit (catalog number 4368814; Applied Biosystems). Quantitative PCR was performed in a 7500 Real-Time PCR System with the Prism 7000 System SDS Software, using predeveloped primers (Applied Biosystems), and the RNA expression of different genes was corrected for using GAPDH expression.
Human ADPKD Cystic Fluid/Human Cohort
The IIS–Foundation Jiménez Díaz Ethics Committee approved the protocol. Patients signed an informed consent in accordance with the European Union Directive and Spanish law. Samples from patients were obtained from the IIS–Fundación Jiménez Díaz Biobank. Urine and plasma samples were centrifuged, divided into aliquots, and stored at −80°C. Supplemental Table 2 provides the clinical characteristics of the participants. Controls included healthy individuals (two female and four males, age range of 25–63 years). Cyst fluids and cyst walls were obtained from eight superficial, clear cysts from three ADPKD kidneys (one female and two males, age range of 45–54 years) from patients who underwent nephrectomy because of volume issues. Cyst fluid was snap frozen and stored at −80°C. TWEAK levels were determined using a commercially available ELISA kit (catalog number BMS2006INST; Bender MedSystems). Cyst walls were processed as for mice kidney tissue and Fn14 was studied via immunohistochemistry. Details are provided in Supplemental Table 2.
Statistical Analyses
Data are presented as means±SEM for all cases (bar or point plots). First, normal distribution was confirmed using the Kolmogorov–Smirnov test. For comparisons between multiple groups, a one-way ANOVA test with Tukey post-test was performed, whereas the two-tailed t test was used to determine significance of differences between two groups. P<0.05 was considered statistically significant. All datasets were analyzed using GraphPad Prism software (version 5).
Results
Data Mining Identification of Fn14 as Overexpressed in PKD
Because TNFSF members and their receptors are key mediators of inflammation that also regulate cell proliferation and cell death, PKD transcriptomics databases were searched for TNFSF and TNFRSF members that were differentially expressed, with increased expression in PKD. In available microarray datasets of ADPKD in mouse, rats, and humans, we found that Fn14 (TNFRSF12a) and/or TWEAK (TNFSF12) are the only genes of the TNFSF encoding a cytokine/ligand pair that were differentially expressed and increased in the three species (Table 1).25–29 Thus, Fn14 (five datasets) and/or TWEAK (three datasets) were increased in six datasets, whereas TNFR1 or TNFR2 were increased in four datasets from the three species, but the gene encoding the cytokine TNF was not overexpressed. Interestingly, a role for TNF in PKD has already been characterized.30 No other TNFRSF/TNFSF gene was upregulated in all of the different species. These findings suggest the TWEAK/Fn14 axis could play a role in ADPKD.
Table 1.
Differentially expressed TNFSF and TNFRSF genes with increased expression in PKD
| Species | Model | Gene | FC | P Value | Reference (PMID) |
|---|---|---|---|---|---|
| Mouse | PKD1−/− embryonic kidneys at days 14.5 versus days 17.5 (GSE24352) | Fn14 | 2.85 | P<0.001 | 2151843811 |
| L3-/L3- (PKD mice) versus WT littermates; 3 weeks old | Fn14 | 3.08 | P<0.01 | 1909960312 | |
| Tnfrsf11B | 2.29 | 0.04 | |||
| Tnfrsf19 | 1.95 | 0.04 | |||
| L3−/L3− (PKD mice) versus WT littermates; 3.5 week old | Fn14 | 1.95 | 0.01 | 1909960312 | |
| Tnfrsf11B | 1.77 | 0.09 | |||
| Tnfrsf19 | 1.56 | 0.03 | |||
| Tnfr1 | 1.55 | 0.02 | |||
| Rat | Homozygous unaffected (+/+) and heterozygous affected (cy/+) Han:SPRD rats; 36 days old | Fn14 | 2.32 | P<0.001 | 2332650313 |
| Tnfr1 | 1.59 | P<0.01 | |||
| Tnfrsf9 | 1.40 | P<0.001 | |||
| Tweak | 1.22 | P<0.001 | |||
| Tnfr2 | 1.11 | P<0.01 | |||
| Homozygous unaffected (+/+) and heterozygous affected (cy/+) Han:SPRD rats; 36 days old | Fn14 | 3.07 | P<0.01 | 1910278214 | |
| Tnfr1 | 1.99 | P<0.01 | |||
| Tweak | 1.32 | P<0.001 | |||
| Human | Healthy tissue from patients with PKD versus samples with small cysts (GSE7869) | TNFR2 | 2.28 | P<0.01 | 3190766915 |
| TNFSF13B | 2.22 | P<0.01 | |||
| TNFR1 | 2.07 | P<0.01 | |||
| TNFRSF10B | 1.99 | P<0.01 | |||
| TWEAK | 1.79 | P<0.01 | |||
| TNFSF14 | 1.40 | 0.0481 | |||
| TNFSF9 | 1.36 | 0.0243 |
Transcriptomics databases were searched in PubMed. FC, fold change; PMID, PubMed identifier.
TWEAK and Its Receptor Fn14 Are Upregulated in ADPKD
Using an orthologous model of ADPKD (Pkd1cond/cond;Tam-Cre11,12), we analyzed the gene expression and protein levels of Fn14 (TWEAK receptor) at two different time points (p30 and p45). Mutant kidneys had higher Fn14 gene expression (15-fold) compared with corresponding controls, as assessed by real-time quantitative PCR (RT-qPCR) (Figure 1A). Fn14 protein levels were also increased on Western blots from ADPKD kidneys (Figure 1B, Supplemental Figure 2A), and Fn14 staining was localized to cyst-lining epithelial cells (Figure 1D). We confirmed these results in three additional ADPKD murine models. Gene expression and protein levels of Fn14 were higher in mutant Pkd1cond/cond;Tam-Cre mice with slowly progressive ADPKD (Supplemental Figure 3, A and B), in Pkd1cond/condTam-KSP-Cre14 ADPKD mice with a kidney epithelial cell promoter (Supplemental Figure 3, C and D), and in Pkd1cond;Pax8;TET-OCre+15 ADPKD mice with a tubular cell–specific promoter (Supplemental Figure 3, E and F). We assayed serum TWEAK levels by ELISA and, as in human ADPKD, there was no statistically significant increase in ADPKD mice (Figure 1C). In summary, we found that the Fn14 receptor was overexpressed in cystic kidneys of four ADPKD mouse models, and it was specifically localized in the renal cyst-lining epithelium. On the basis of published data of mouse kidney single-cell transcriptomics,31 we found that Fn14 is largely expressed in the epithelial cells of the different nephron segments, whereas TWEAK is mainly expressed in macrophages among other blood cell types and in endothelial cells, podocytes, and tubular cells (Supplemental Figure 4).
Figure 1.
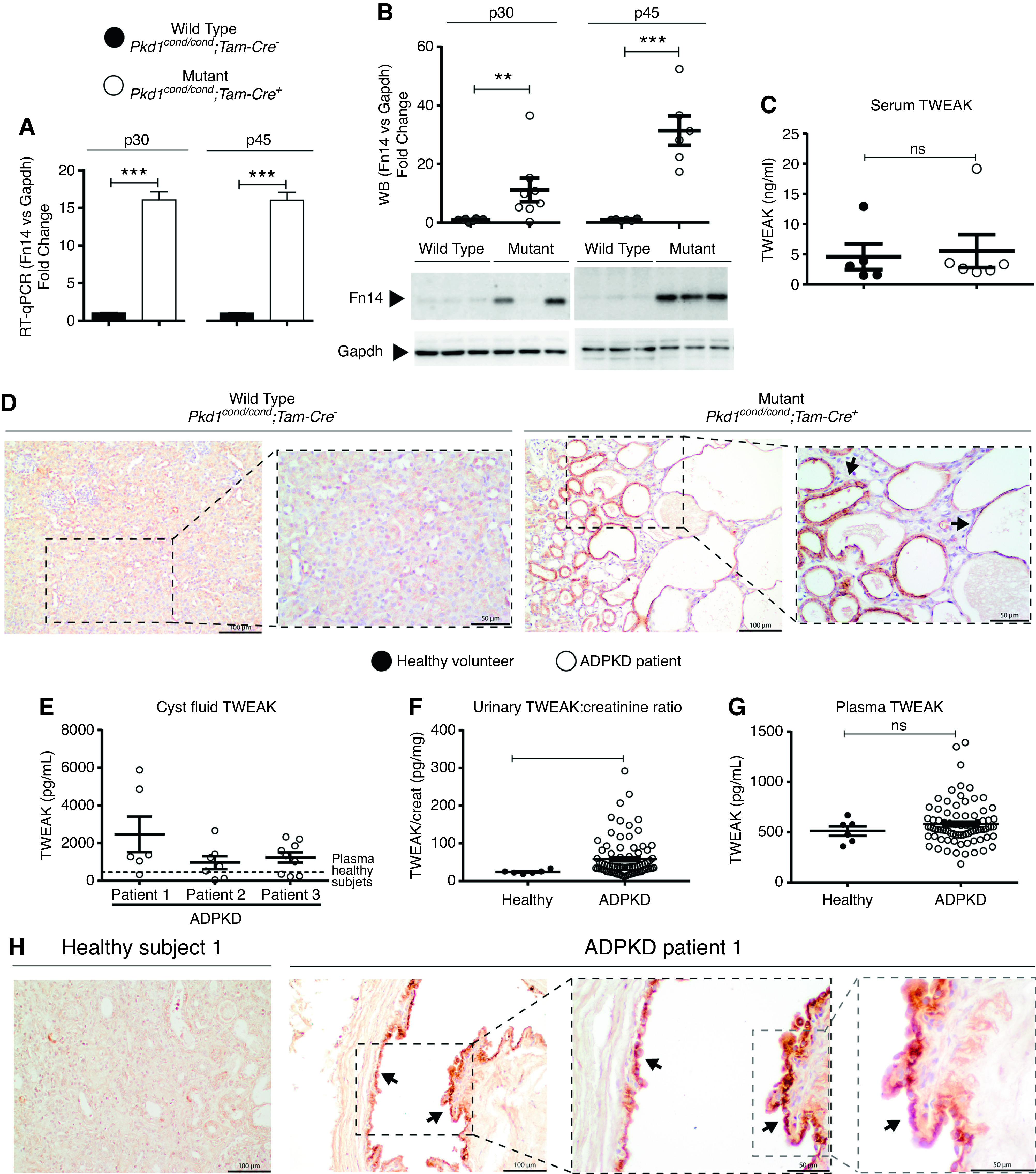
TWEAK and its receptor Fn14 are upregulated in ADPKD. Fn14 is upregulated in Pkd1cond/cond;Tam-Cre ADPKD orthologous mice (mutant), Pkd1 inactivated on p10–11. (A) Fn14 gene expression assessed by RT-qPCR in WT (n=6) and mutant kidney samples (n=6) at two different time points, p30 and p45. Gapdh was used as housekeeping control. (B) Fn14 protein level assessed by Western blot in WT (n=6) and mutant kidney samples (n=6) at p30 and p45. Upper panel, densitometry represented as fold change (WT versus mutant) and normalized to GAPDH. Lower panel, representative blots (additional information in Supplemental Figure 1). (C) No significant differences TWEAK cytokine levels were observed in PKD mouse serum by ELISA. (D) Fn14 immunohistochemistry in mouse samples at p30 (WT and mutant) shows specific staining in cyst epithelium (black arrows). Scale bars, 100 µm and 50 µm. Original magnification, ×200 and ×400. (E–G) TWEAK was assessed by ELISA in renal cyst fluid, urine, and plasma from patients with ADPKD and healthy volunteers. (E) TWEAK concentration was higher in most individual samples of ADPKD cyst fluid compared with plasma levels in healthy volunteers. (F) The urinary TWEAK/creatinine (creat) ratio is higher in patients with ADPKD compared with healthy volunteers. (G) No significant differences were observed in plasma TWEAK levels between patients with ADPKD and healthy volunteers. (H) Fn14 immunohistochemistry in human ADPKD shows staining in cyst-lining epithelial cells (arrows). Scale bars, 100 µm and 50 µm. Original magnification, ×400 and ×600. Bars represent means±SEM. *P<0.05, **P<0.01, ***P<0.001.
Next, we determined the status of the Fn14/TWEAK axis in human ADPKD. TWEAK was assayed by ELISA in cystic fluid, urine, and plasma. Average TWEAK levels were higher in cyst fluid from three patients with ADPKD compared with plasma, suggesting local production or secretion (Figure 1E). The content of the cystic fluid was heterogeneous: 16 of 22 (72.72%) cysts contained TWEAK levels above the mean TWEAK level in the plasma from healthy subjects. Similarly, urinary TWEAK levels were higher in urine from patients with ADPKD than from controls (Figure 1F). By contrast, no statistically significant differences were observed in plasma TWEAK, suggesting local TWEAK production in the kidneys (Figure 1G). Immunohistochemistry showed staining of Fn14 along the cyst epithelium (located in a perinuclear region), thus placing receptor expression at the site of TWEAK presence (Figure 1H, Supplemental Figure 2C).
TWEAK Promotes Cystogenesis and Cyst Progression in an Orthologous Model of ADPKD
Previous studies showed that early inactivation of the Pkd1 gene (before p12) triggers rapid development of polycystic disease (cystic window), whereas inactivation after p14 leads to delayed cyst formation after 4–5 months, with a mild phenotype (noncystic window).12,32 Thus, in APDKD, there is a critical developmental window for cyst formation. We studied the role of TWEAK in ADPKD in both developmental windows (cystic and noncystic) by administering tamoxifen to inactivate Pkd1 at different time points (Supplemental Figure 1).
TWEAK Induces Cystogenesis in the Noncystic Window in Pkd1-Deficient Mice
We first inactivated Pkd1 with tamoxifen in Pkd1cond/cond;Tam-Cre+ mice at p15–16, and then administered TWEAK for 2 weeks (Supplemental Figure 1A). TWEAK treatment accelerated cyst formation, but cystogenesis was focal (Figure 2A). This restricted pattern of cyst formation was not associated with increased kidney volume and kidney/body weight ratios, and renal function was similar in control and TWEAK-exposed groups (Figure 2, B and C). However, TWEAK significantly increased the cystic index and the total of number of cysts (Figure 2, D and E). Using immunofluorescence for specific nephron markers, we characterized the location of TWEAK-induced focal cystogenesis and found that most cysts were of a collecting duct origin (DBA+; Figure 2F, lower panel). Cysts also originated from the thick ascending limb (THP+; Figure 2F, center), and from dilated proximal tubules (LTL+; Figure 2F, upper panel). Our results suggest that a proinflammatory environment resulting from TWEAK administration can induce cystogenesis in the context of Pkd1 loss.
Figure 2.

TWEAK treatment during a noncystic window accelerates cyst formation. (A) Representative hematoxylin and eosin–stained kidney sections from TWEAK (n=4) and control (n=6) mice (Pkd1cond/cond;Tam-Cre+), with Pkd1 deletion induced at p15–16 and mice euthanized at p30. Schematic representation of the experimental design provided in Supplemental Figure 1A. Several cysts (Cy) are observed in a focal distribution pattern in TWEAK-treated kidneys. Arrows represent dilated tubules. Scale bars, 2 mm (upper panel) and 100 µm (lower panel). Renal function assessed by (B) BUN and the (C) kidney/body weight ratio do not change significantly. In contrast, TWEAK increased (C) the cystic index and (E) the number of cysts. (F) Immunofluorescence for specific nephron markers from proximal tubule (LTL, green, upper panel), ascending limb of the loop of Henle (THP, red, center), and collecting duct (DBA, red, lower panel). *Cyst without specific marking; yellow arrow, dilated tubule; yellow arrowhead, tubule. Scale bars, 50 µm (upper panel) and 100 µm (center and lower panels). Bars represent means±SEM. *P<0.05. DAPI, 4′,6-diamidino-2-phenylindole.
TWEAK Promotes Cystogenesis in the Cystic Window in Pkd1-Deficient Mice
Next, we tested the effect of TWEAK administration in short-, medium-, and long-term models. We administered TWEAK between p8 and p10, induced Pkd1 deletion at p10–11, and euthanized at p30 (Supplemental Figure 1B). We found that kidneys from TWEAK-exposed mice appeared larger macroscopically than control kidneys (Figure 3A), but there was no statistically significant difference in kidney/body weight ratio or renal function (Figure 3, C and E). However, the cystic index was significantly higher in TWEAK-exposed mice (Figure 3D). No differences in cyst origin were observed between control and TWEAK groups (Supplemental Figure 5).
Figure 3.
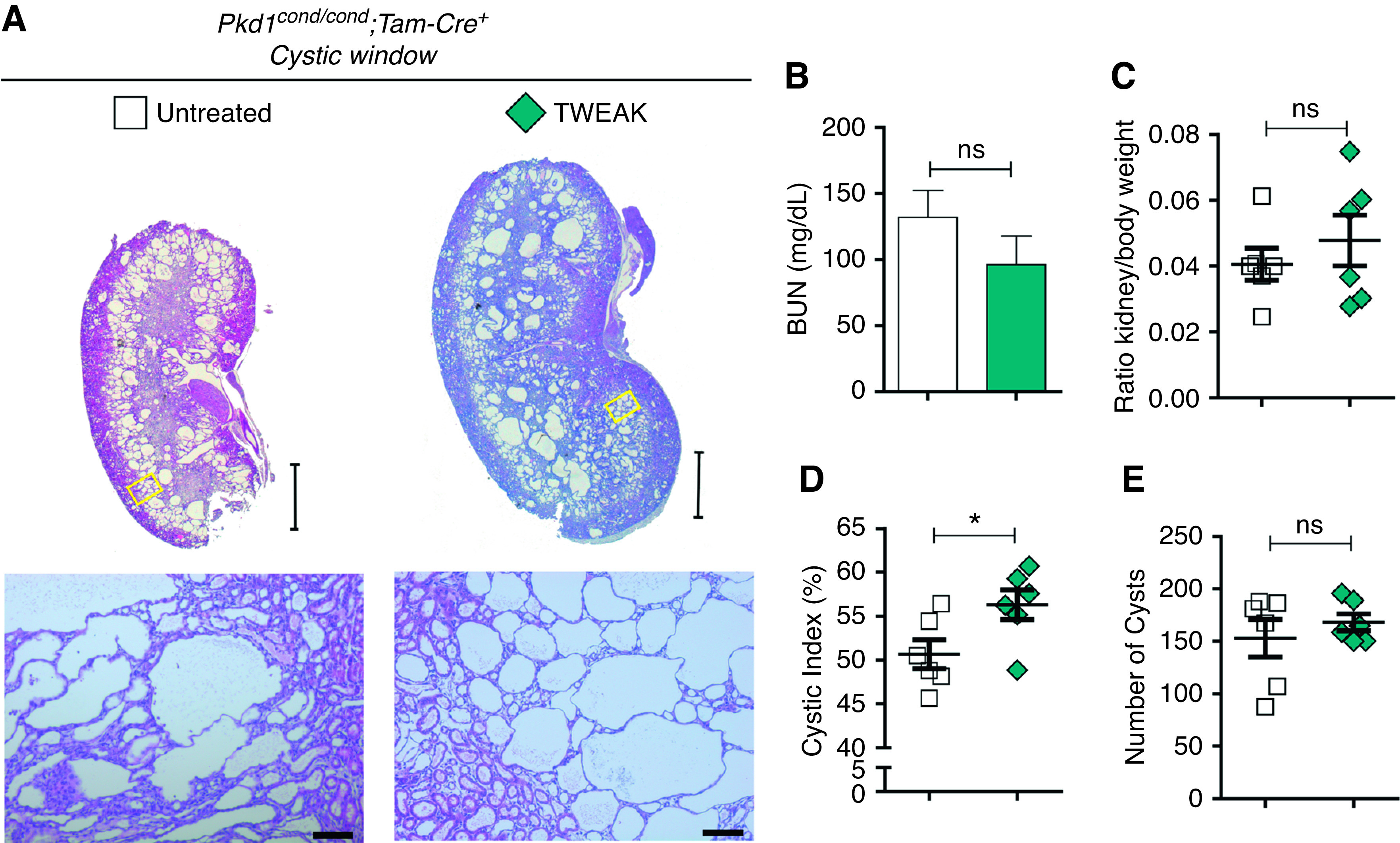
Short-term TWEAK administration before Pkd1 deletion increases the severity of cystic disease in the cystic window. Two TWEAK doses were administered before Pkd1 inactivation at p10–11. (A) Representative images of kidney histology at p30 of control (n=6) and TWEAK-exposed (n=6) mice, additional information in Supplemental Figure 1B. Scale bars, 2 mm (upper panel) and 100 µm (lower panel). (B) BUN and (C) kidney/body weight ratios of control and TWEAK-exposed mice did not change. TWEAK resulted in a significant increase in (D) the cystic index, but did not change (E) the number of cysts. Bars represent means±SEM. *P<0.05.
These results suggest a possible procystogenesis role of TWEAK that may not have become fully evident because of a short follow-up time. To extend these findings, we inactivated Pkd1 at p10–11 and administered TWEAK to the mice during the precystic window, with readout at p30 (medium term) or p45 (long term) (Supplemental Figure 1C). There was no change at p30 after TWEAK administration (data not shown), so this was considered an early time point and used to explore the cellular and molecular mechanisms involved (see below). In contrast, at p45, TWEAK aggravated cyst progression (Figure 4), resulting in decreased renal function with increased BUN levels (Figure 4B) and an increased kidney/body weight ratio (Figure 4C). Although the kidneys from control and TWEAK-exposed mice appeared to be the same size macroscopically (Figure 4A), the total weight of TWEAK-treated mice was significantly lower (Figure 4G, Supplemental Figure 6). Additionally, TWEAK also increased the renal cystic index and number of cysts (Figure 4, D and E). Finally, TWEAK-exposed mice had a decreased survival than controls (67% versus 75%, scarified at p45) (Figure 4I). In contrast to Pkd1-deficient mice, no apparent kidney damage was observed in TWEAK-exposed WT mice (Supplemental Figure 7).
Figure 4.
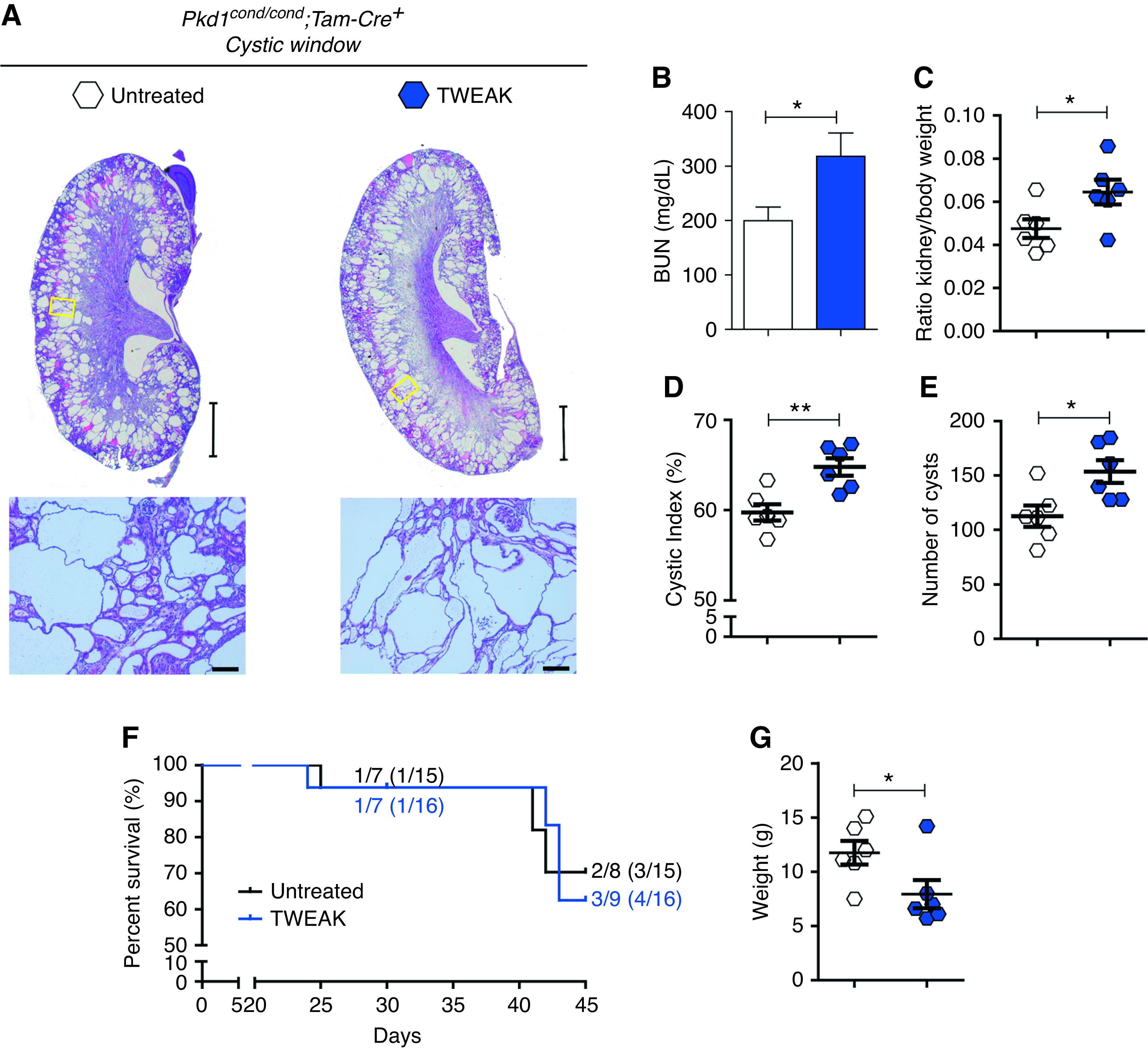
Long-term TWEAK administration increases disease progression in ADPKD. TWEAK was administered after Pkd1 inactivation, every 48 hours, until euthanasia at p45 (long term). (A) Representative hematoxylin and eosin–stained kidney sections harvested at p45 in control (n=6) and TWEAK-treated (n=6) groups. Schematic representation of the experimental design in Supplemental Figure 1C. Scale bars, 2 mm (upper panel) and 100 µm (lower panel). TWEAK increases (B) BUN, the (C) kidney/body weight ratio, (D) renal cystic index, and (E) cyst number. (F) Kaplan–Meier survival plots over time. Mice exposed to TWEAK had decreased survival compared with control mice. (G) Weight of control and TWEAK-exposed mice: TWEAK treatment slowed weight gain. Bars represent means±SEM. *P<0.05, **P<0.01.
TWEAK Promotes Inflammation in Macrophages and Epithelial Cells
To complement our in vivo findings, we investigated whether TWEAK promotes an inflammatory response in cell types relevant for ADPKD. We cultured WT bone marrow–derived macrophages and tubular epithelial cells (MCT), and then stimulated them with TWEAK for 3 hours. TWEAK promoted an inflammatory response in both cellular types, as assessed by Mcp1, RANTES, and Il-6 gene expression (Supplemental Figure 8).
Anti-TWEAK Slows Cyst Growth and Improves Renal Function in ADPKD Mice
Our results demonstrate that the Fn14/TWEAK axis is upregulated and promotes cyst progression in ADPKD. On the basis of these results, we tested whether TWEAK might be a therapeutic target. We administrated neutralizing anti-TWEAK antibodies in the cystic window of Pkd1cond/cond;Tam-Cre mice, in which Pkd1 was inactivated at p10–11 (Supplemental Figure 1C). At p18, anti-TWEAK significantly decreased the kidney/body weight ratio and number of cysts, and showed a clear trend towards significance in BUN and cystic index (Figure 5, A–E). These results demonstrated that anti-TWEAK slowed the progression of ADPKD, even in early stages of the disease. At p30, anti-TWEAK treatment resulted in smaller kidneys compared with untreated controls (Figure 5A). Kidney/body weight ratio and cystic index were remarkably lower in anti-TWEAK–treated mice, and kidney function, as assessed by BUN levels, was better preserved (Figure 5, B–D).
Figure 5.
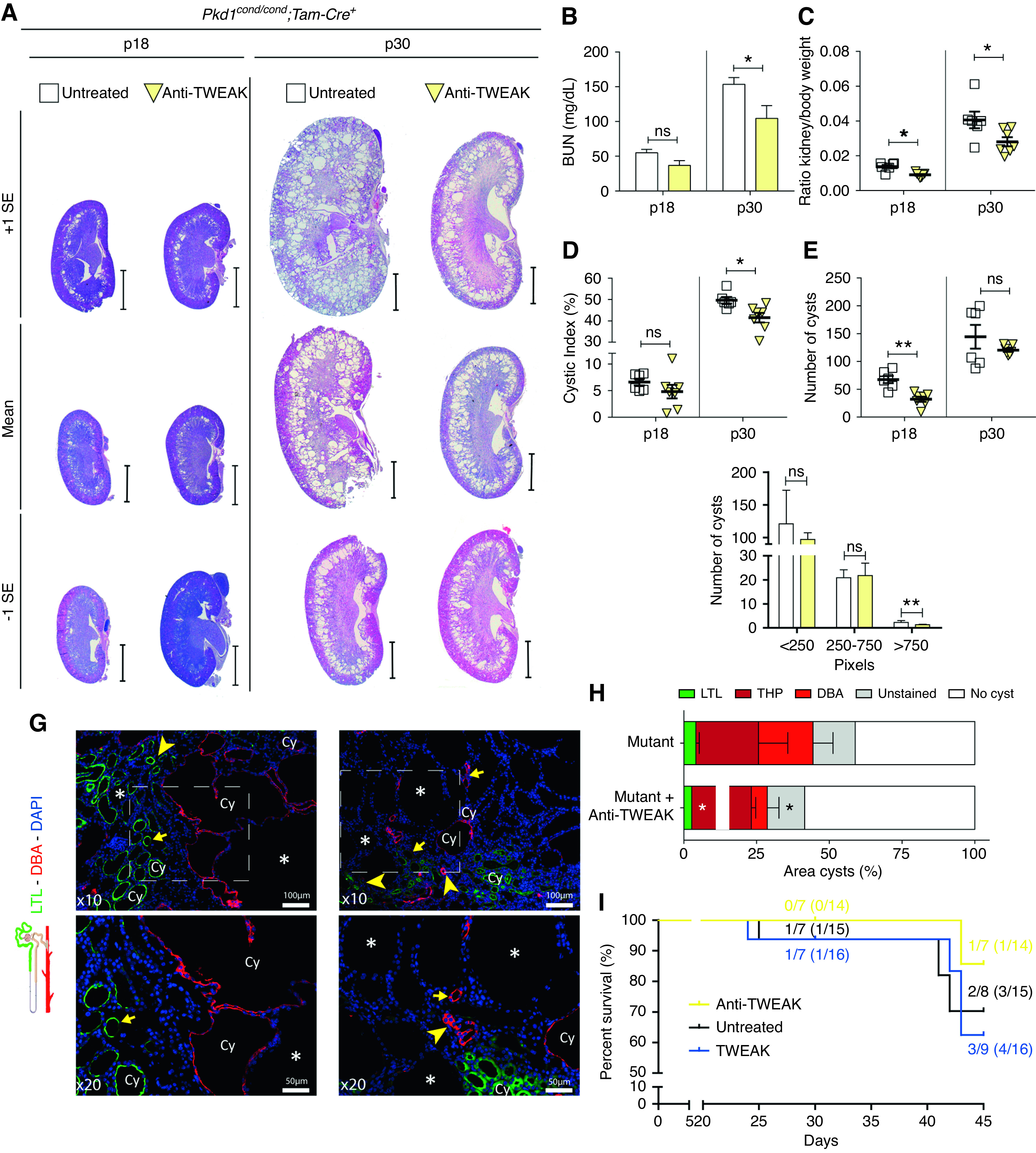
Treatment with anti-TWEAK decreases renal cyst progression. (A) hematoxylin and eosin–stained, sagittal cross-sections of kidneys treated with anti-TWEAK (n=7 at p18, n=6 at p30) and control kidneys (n=6 at p18, n=6 at p30), euthanasia occurred at p18 and p30. Representative images with values within mean±1×SEM are shown. See Supplemental Figure 1C for experimental design. Scale bars, 2 mm (upper panel) and 100 µm (lower panel). (B) BUN, (C) kidney/body weight ratio, and (D) cystic index were significantly reduced in anti-TWEAK–treated mice at p18 and p30. (E) No significant differences in cyst number were observed at p30. (F) However, classification of the number of cysts by size in three groups, according to the size estimated by number of pixels, showed that the number of largest cysts (>750 pixels) decreased with treatment. (G) Representative images of immunofluorescence staining for specific nephron markers are shown, including proximal tubule (LTL, green) and collecting duct (DBA, red). Anti-TWEAK treatment slows enlargement of LTL+ and DBA+ cysts (Cy). *Cyst without specific markers; yellow arrow, dilated tubule; yellow arrowhead, tubule. Scale bars, 100 µm (upper panel) and 50 µm (lower panel). (H) Quantification of immunofluorescence cystic area. Anti-TWEAK treatment reduces cystic area of LTL+ and DBA+ cysts. (I) Kaplan–Meier survival plots of time. Treatment with anti-TWEAK increased survival (93% survival) compared with control and TWEAK-exposed mice. Bars represent means±SEM. *P<0.05, **P<0.01.
Although we did not observe significant differences in the total number of cysts at p30, anti-TWEAK treatment reduced the number of larger cysts (Figure 5, E and F). Anti-TWEAK treatment reduced cysts in both proximal (LTL+) and collecting (DBA+) tubules (Figure 5, G and H, Supplemental Figure 9, upper panel), which is consistent with expression of Fn14 in both proximal and distal collecting tubules.19 There was no change in the area of cysts arising from the thick ascending limb of the loop of Henle (THP+; Supplemental Figure 9, lower panel). Anti-TWEAK also increased survival of Pkd1 mice compared with TWEAK-treated or control-untreated mice (Figure 5I). Anti-TWEAK therapy was associated with weight gain (Supplemental Figure 6), and we did not find any apparent detrimental effects of anti-TWEAK treatment (Supplemental Figure 7).
Anti-TWEAK Decreases Cell Proliferation via Inhibition of the AKT Pathway
TWEAK activates a proproliferative pathway, driven by mitogen-activated protein kinases (MAPKs)—such as ERK1/2, MAPK p38, and phosphatidylinositol 3-kinase/AKT—in different cell types, including renal cells.17,33,34 These MAPK pathways, along with mammalian target of rapamycin (mTOR), are deregulated in ADPKD.35–38 Inhibition of mTOR was beneficial in murine models of ADPKD.39–42 As has been previously reported, we found evidence of ERK1/2 activation in untreated cystic kidneys, which was unchanged by anti-TWEAK treatment at p30 (Figure 6A, Supplemental Figure 10, A and B). In contrast, anti-TWEAK reduced p38 phosphorylation, which was increased, in untreated cystic kidneys (Figure 6B, Supplemental Figure 10, C and D). Furthermore, anti-TWEAK also reduced total AKT protein levels and AKT phosphorylation, which were also increased in untreated cystic kidneys (Figure 6C, Supplemental Figure 10, E and F). Changes in Akt protein levels were not explained by changes in Akt1 gene expression (Figure 6D). In an inflammatory milieu, TWEAK induces mesangial and tubular cell apoptosis.33,43 We observed a mild reduction of proliferation (Supplemental Figure 11A) and number of apoptotic cells in cystic epithelium (Figure 6E) with anti-TWEAK treatment. In addition, we found a modest reduction of fibrosis with a reduction in fibronectin staining (Figure 6F). However, we did not find significant differences in anti–smooth muscle actin staining or Tgfβ1 and Ctgf gene expression (Supplemental Figure 11, B and C).
Figure 6.
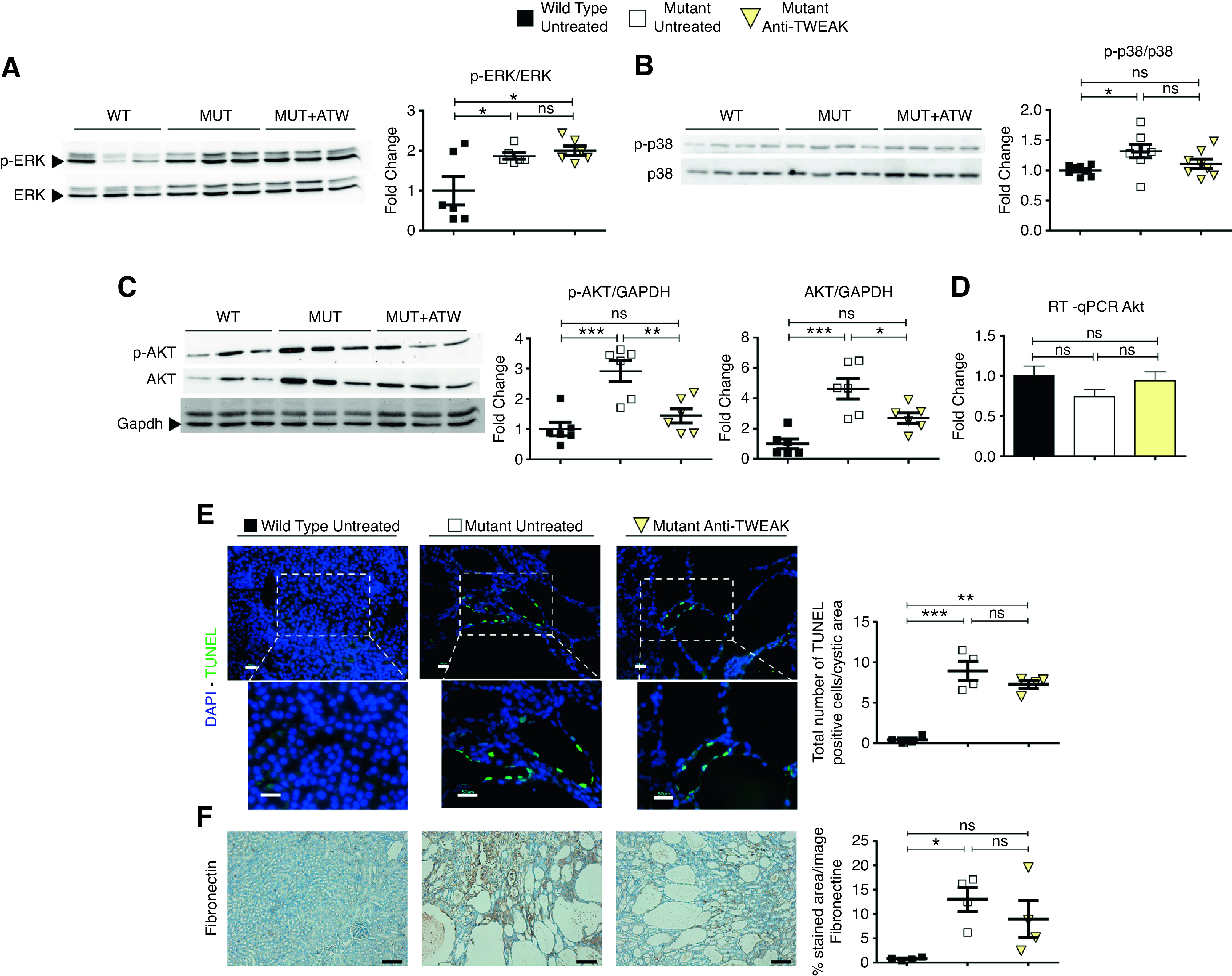
AntiTWEAK decreases cell proliferation via inhibition of AKT pathway. Anti-TWEAK treatment reduces AKT and p-AKT, but not p-ERK, at p30. (A) Western blot analysis for p-ERK and ERK of total kidney protein lysates of WT (n=6), mutant (n=6), and mutant anti-TWEAK–treated mice (n=6). (Right) p-ERK/ERK ratio. (B) Western Blot analysis for p-p38 and p38 of total kidney protein lysates of WT (n=6), mutant (n=6), and mutant anti-TWEAK–treated mice (n=6). (Right) p-p38/p38 ratio. (C) Western blot for p-AKT, AKT, and GAPDH of total kidney protein lysates of WT (n=6), mutant (n=6), and mutant anti-TWEAK–treated mice (n=6). (Right) Ratios for p-AKT/GAPDH, and AKT/GAPDH. GAPDH was used as a housekeeping gene because of changes in AKT protein levels, which precluded the use of AKT to normalize p-AKT levels. (D) RT-qPCR of Akt1 gene for WT (n=5), mutant (n=5), and mutant animals treated with anti-TWEAK (n=6). No significant differences in Akt1 mRNA were observed. (E) Anti-TWEAK decreases apoptosis in kidney anti-TWEAK–treated group, with weak significance (P<0.1), in the cystic area, as detected by TUNEL assay. Scale bars, 100 µm and 50 µm. Original magnification, ×200 and ×400. (F) Immunohistochemical analysis of fibronectin (center) in kidney slides of WT, mutant, and anti-TWEAK groups. n=4 was used per group. Fibrosis, assayed by fibronectin staining, was modestly diminished by anti-TWEAK treatment. Scale bars, 100 µm. Mice were euthanized at p30, according of the scheme shown in Supplemental Figure 4C (medium-term model). Bars represent means±SEMs. *P<0.05, **P<0.01, ***P<0.001. DAPI, 4′,6-diamidino-2-phenylindole.
Anti-TWEAK Treatment Reduces NF-κB Pathway Activation and Macrophage Recruitment
TWEAK activates the canonic and noncanonic NF-κB pathways.44 In untreated ADPKD, we observed evidence of canonic NF-κB pathway activation, because Mcp1 and Cxcl16 were upregulated, and evidence of noncanonic NF-κB pathway downregulation, because Ccl19 and Ccl21 were downregulated. Anti-TWEAK caused a moderate decrease in expression of Mcp1 (canonic pathway); however, we did not find significant differences in the noncanonic pathway NF-κB markers (Figure 7, A and B). Staining for the canonic NF-κB component p65 was significantly decreased in cystic epithelium after anti-TWEAK treatment (Figure 7C). Cystic induction by TWEAK administration in the noncystic window was related to overexpression of Fn14 (Supplemental Figure 12). Interestingly, anti-TWEAK treatment resulted in a significant reduction of Fn14, as seen by immunohistochemistry and gene expression (Figure 7, D and E).
Figure 7.

Anti-TWEAK treatment reduces NF-kB pathway activation and macrophage recruitment. Gene expression assessed by RT-qPCR of (A) Mcp1 and Cxcl16, and (B) Ccl19 and Ccl21, markers of canonic and noncanonic NF-κB pathway activation, respectively. Anti-TWEAK significantly decreases canonic NF-κB pathway activation. (C) Nuclear p65 staining is decreased in anti-TWEAK–treated mice. Representative images are shown. Mice euthanized at p30 (Supplemental Figure 4C). Anti-TWEAK decreases Fn14 expression in ADPKD. (D) Fn14 staining is decreased in cyst-lining epithelium (arrows) of mutant anti-TWEAK–treated mice. Mice euthanized at p30 (Supplemental Figure 4C). (E) Fn14 gene expression was diminished in anti-TWEAK–treated group. Mice euthanized at p30 (Supplemental Figure 4C). RT-qPCR of (F) Mcp1 (M1 markers) and (G) Arg1 (M2 markers) at p18 and p30. No differences were found at p18 (precystic stage), but anti-TWEAK treatment resulted in reduced gene expression of both proinflammatory markers at p30. Macrophage infiltration was reduced by anti-TWEAK treatment. (H) F4/80 staining in kidney tissue at p18 and p30. There were significantly reduced numbers of positive F4/80 macrophages in the medulla at p30. WT (n=6 at p18, n=5 at p30), mutant (n=6 at p18, n=5 at p30), and mutant mice treated with anti-TWEAK (n=7 at p18, n=6 at p30) were used for this RT-qPCR experiment set. Hprt was used as the housekeeping gene in all cases. For immunohistochemistry analysi s, n=4 was used per group in all instances. Bars represent means±SEM. *P<0.05, **P<0.01, ***P<0.001.
A major pathologic feature of ADPKD is the development of interstitial inflammation.9 Several studies reported that macrophage activation and infiltration are associated with ADPKD cyst growth,45,46 and an increased expression of many proinflammatory markers and cytokines.30,47,48 We tested the expression of proinflammatory markers of alternative macrophage activation phenotypes, called M1 and M2, respectively. We studied four M1 macrophage markers (Nos2, Il-12, Tnfα, and Mcp1) and four M2 markers (Arg1, Mrc1, Csf1, and Csf2). At p30, there was a mixed phenotype with upregulation of two proinflammatory (Tnfα, Mcp1) and all of the alternative (Arg1, Mrc1, Csf1, Csf2) macrophage activation genes studied (Figure 7, F and G, Supplemental Figure 13, A and B). Importantly, the expression of the two most upregulated markers in untreated Pkd1 mutant mice at p30, Mcp1 and Arg1, were downregulated by anti-TWEAK treatment (Figure 7, F and G).
Finally, using F4/80, a specific marker of mouse monocytes/macrophages,49 we confirmed increased macrophage recruitment in untreated, Pkd1-deficient kidneys. F4/80-positive macrophage infiltration was significantly reduced by anti-TWEAK treatment (Figure 7H, Supplemental Figure 13C). We also studied macrophage infiltration in early disease stages to elucidate whether reduced macrophage recruitment could drive slower cyst progression in anti-TWEAK–treated mice. Anti-TWEAK decreased cyst progression in early stages of disease, but we did not find differences in F4/80 staining at this stage and there was no change in Mcp1 and Arg1 expression (Figure 7, F–H, Supplemental Figure 13C). Our data suggest that a decrease in macrophage recruitment via the TWEAK/Fn14 pathway is an indirect cause of cystic disease reduction (Figure 7H, Supplemental Figure 14).
Discussion
Renal inflammation in ADPKD has been associated with the expression and accumulation of proinflammatory cytokines, including TNFα, IL-6, IL-8, and MCP1.30,45,50–52 Our data show that the TWEAK/Fn14 axis is upregulated in Pkd1 mutant murine kidneys, both by RT-qPCR and Western Blot analysis. Similarly, we found evidence of TWEAK/Fn14 activation in human ADPKD. TWEAK was abundant in renal cyst fluid, and urine and cyst epithelium expressed Fn14; however, serum TWEAK levels in controls was similar to those in patients with ADPKD, which supports a local origin of TWEAK, likely the tubular epithelium. However, we cannot exclude a contribution by local leukocytes. In this regard, serum TWEAK is decreased in advanced CKD, but may be increased in early CKD due to inflammatory conditions.53,54 The combined human and murine data in serum TWEAK suggest that ADPKD, per se, does not modulate serum TWEAK. Finally, we provide functional in vivo evidence that the TWEAK/Fn14 axis contributes to ADPKD cyst progression, according to the “two-hit” model of cystogenesis, suggesting this pathway could serve as a potential therapeutic target (Figure 8A).
Figure 8.

Proposed model of how TWEAK accelerates cyst progression in ADPKD. (A) Mechanism of cyst formation and expansion in ADPKD. In ADPKD nephrons, all tubular epithelial cells contain one mutated and one unaffected copy of PKD1 or PKD2. According to the “two-hit” model of cystogenesis, a second mutation occurs in the unaffected allele in an individual tubular epithelial cell. When the level of functional PC1 or PC2 (products of PKD1 and PKD2, respectively) falls below a critical threshold, cyst formation is initiated. Once cysts are formed, multiple molecular pathways are deregulated, and cysts progressively increase in size. Advanced ADPKD is characterized by macrophage infiltration, proinflammatory chemoattractants, and inflammation-dependent renal injury. In this study, we report that TWEAK and Fn14 are upregulated in ADPKD and contribute to cyst progression. (B) TWEAK pathway. TWEAK is a trimeric cytokine that binds to the Fn14 receptor after trimerization. TWEAK and Fn14 are present in epithelial renal cells and regulate cell proliferation, inflammation, NF-κB activation, and other pathways. We report that targeting TWEAK with a neutralizing antibody reduced the expression of key ADPKD-related protein markers, such as AKT and p38 (proliferative way), Arg1 (M2 macrophage polarization), Mcp1 (M1 macrophage polarization and canonic NF-κB activation), and p65 (canonical NF-κB activation), and reduced macrophage infiltration. Our results suggest the TWEAK/Fn14 axis is a new potential therapeutic target for ADPKD.
It has been debated whether inflammation is a driving force for cyst formation or a consequence of disrupted renal pathology.9,55 Studies in ADPKD conditional knockout models (in which we delete the Pkd1 gene at different kidney developmental time points) have demonstrated that Pkd1 deletion in mature kidneys results in delayed cyst formation compared with deletion before p12 or with germ line knockout in utero.12,32 A series of studies have shown that “third hits,” such as cystogenesis in rodent models,56 including ischemia,57,58 renal injury,59,60 or nephrotoxicity,61 can accelerate cyst progression. Here, we find that TWEAK administration to Pkd1 mutant mice in this precystic window also accelerates cyst initiation and, likewise, can hasten more aggressive PKD when Pkd1 is inactivated before p12. These results suggest that inflammation and, specifically, the inflammatory cytokine TWEAK can play a key role in defining the rate of cyst initiation and development in ADPKD.
Therapeutic modulation of the Fn14/TWEAK axis has been successful in preclinical models of different kidney diseases, such as AKI, kidney fibrosis, lipid-induced kidney injury, or lupus nephritis.5,16,19,34,62,63 Furthermore, clinical trials targeting the Fn14/TWEAK axis were performed in rheumatoid arthritis (NCT00771329),8 lupus nephritis (ATLAS study, NCT01499355), muscle atrophy (NCT01943513), and in patients with advanced solid tumors (NCT00738764).6 We have now targeted the Fn14/TWEAK axis in an ADPKD mouse model by administering neutralizing anti-TWEAK antibodies. Our data show that anti-TWEAK slows cystic disease progression and improves renal function in a murine ADPKD mouse model. Conversely, treatment with exogenous TWEAK was clearly procystogenic in later stages (p45), and anti-TWEAK reduced cyst progression in the medium- and short-term model (p18 and p30); these time variances in the mouse can be explained by the differential effect of administrating a stimulus or a blocker, and the different inflammatory and disease states at different times. Cyst growth in ADPKD involves two main stages: cyst initiation and cyst progression (Figure 8A). Initiation is induced by a deficiency in mature PC1 and PC2 protein, caused by pathogenic mutations in the PKD1 and PKD2 genes, respectively. Disease progression incorporates other players, and interstitial inflammation was proposed as a key process in progressive cyst enlargement. Our results support that an inflammatory mediator, TWEAK, plays a role in cyst growth and progression.
We found that anti-TWEAK treatment restored normal levels of proproliferative and proinflammatory pathways related to ADPKD progression (Figure 8B). In kidney cells, TWEAK can activate MAPKs and the phosphoinositide 3-kinase/Akt pathway.4,17,64 The AKT/mTOR signaling pathway is upregulated in ADPKD kidneys, and its inhibition slows ADPKD progression.40,42,65–70 However, the ERK/MAPK signaling pathway is also upregulated, but its inhibition did not delay cyst formation.36,67,71 In our study, we confirmed upregulation of p-ERK/ERK and p-p38/p38, and of total AKT and p-AKT in ADPKD kidneys. Furthermore, we found that anti-TWEAK treatment restored p-p38/p38, total AKT, and p-AKT to control values. Our data favor the view that TWEAK has a proproliferative role via several signaling pathways and that the therapeutic modulation of TWEAK can be an effective treatment in ADPKD. These results are in line with the proproliferative activity of TWEAK in kidney tubular cells and kidney fibroblasts that was observed both in vitro and in vivo and contributed to kidney growth post–unilateral nephrectomy.17,20
TWEAK induces renal tubulointerstitial inflammation, chemokine expression,16,17,33 macrophage recruitment,16 and NF-κB pathway activation,19,34 which are signaling pathways also studied in ADPKD.55 In line with this, TWEAK or Fn14 blockade reduced inflammatory gene expression in experimental lupus nephritis,63 and reduced proinflammatory chemokines and macrophage recruitment in experimental AKI.16 We observed that anti-TWEAK effectively decreases the gene expression of Arg1 and Mcp1 and macrophage recruitment (as assessed by the total mouse macrophage marker F4/80), resulting in decreased cyst progression and improvement in BUN levels. Two different studies reported that inhibition of Arg1 or Mcp1 delays cyst growth, so it is biologically relevant that anti-TWEAK treatment reduced their expression in our study.47,48 M2 macrophages were reported to be abundant infiltrating cells in ADPKD, and pharmacologic inhibition of ARG1 slowed cyst progression and reduced proliferative indexes.48 Also, these authors enriched the characterization of the function of the macrophage population in ADPKD kidneys. Our data are in line with these findings because macrophages in advanced ADPKD present mainly an M2 phenotype rather than an M1 phenotype. Furthermore, in a further study, treatment with VEGFC reduced Arg1 expression and M2 macrophage infiltration and improved cystic disease.72 Regarding MCP1, a double knockout of both Mcp1 and Pkd1 resulted in suppressed macrophage accumulation and decreased cyst growth.47 In this regard, high urinary MCP1 was a biomarker of disease progression in patients with ADPKD who were treated with tolvaptan.73–75
Some limitations should be acknowledged. The human data we present are observational, and the murine immune system has several differences from the human system. However, our reciprocal intervention studies in an orthologous ADPKD murine model showing that TWEAK treatment worsens, whereas inactivation of the TWEAK/Fn14 axis ameliorates, cystic disease are internally consistent and aligned with the human data. Furthermore, a therapeutic tool already available and tested in humans was found to be effective in limiting cystogenesis and preserving kidney function, which may facilitate clinical translation.
In conclusion, we found that the TWEAK/Fn14 axis is upregulated in human ADPKD and both rapidly and slowly progressive murine models, where it promotes cyst initiation and progression in an orthologous Pkd1 mutant murine mice model. Furthermore, anti-TWEAK treatment effectively slows cyst growth, decreasing proproliferative, proinflammatory markers and macrophage recruitment. These results suggest that inhibition of the TWEAK/Fn14 axis and its proinflammatory component can be an effective therapeutic approach for ADPKD.
Disclosures
A. Boletta reports serving on the scientific advisory board of the Baltimore PKD Center, on the scientific advisory committee for the PKD Foundation in the United States, and on the editorial board for Scientific Reports (Nature Publishing Group); and having patents and inventions related to glycolysis inhibition for the treatment of polycystic kidney and liver disease (European patent application number 13733319.1 and US patent application number 14/413,280) and patents and inventions relating to the modulation of metabolic pathways for the treatment of polycystic kidney and liver disease (European patent application confidential). A. Carracedo reports serving on the advisory board of the not-for-profit foundations Fundación AstraZeneca (Spain) and Fundación Merck (Spain); and serving as a scientific advisor for, or member of, different not-for-profit foundations, including Fundación Ingada (as part of the board of directors) and Kaertor Foundation. M. Chiaravalli reports having patents and inventions related to glycolysis inhibition for the treatment of polycystic kidney and liver disease (European patent application number 13733319.1 and US patent application number 14/413,280). M.A. Garcia-Gonzalez reports serving on the Instituto de Investigación Sanitaria de Santiago research advisory board and receiving honoraria from Otsuka Pharmaceutical. A. Ortiz reports receiving honoraria from Amgen, Amicus, AstraZeneca, Fresenius Medical Care, Freeline, Genzyme, Mundipharma, Otsuka, and Shire; serving as scientific advisor for, or member of, the Clinical Kidney Journal, European Renal Best Practice, Journal of Nephrology, and Spanish Society of Nephrology; receiving research funding from Genzyme; and having other interests in/relationships with the European Renal Association–European Dialysis and Transplant Association and SENEFRO, and receiving speaker fees from Otsuka, and serving as a consultant for Sanofi. T. Watnick reports having patents and inventions with, and receiving royalties from, PKD DNA testing with Athena Diagnostics, and receiving royalties from UpToDate; serving on the editorial board of JASN, as a member of the medical advisory board for the National Kidney Foundation of Maryland, and on the scientific advisory committee the PKD Foundation; and receiving research funding as a study site for clinical trials sponsored by Palladio Biosciences, for the Falcon Study (Reata Pharmaceutical), and the STAGED-PKD study (Sanofi-Genzyme). All remaining authors have nothing to disclose.
Funding
This research was funded by Instituto de Salud Carlos III (ISCIII) under Fondo de Investigación en Salud (FIS)/Fondo Europeo de Desarrollo Regional (FEDER) grants PI16/01900 (to A.B. Sanz), PI19/00588 (to A.B. Sanz), PI19/00815 (to A. Ortiz), PI11/00069 (to M.A. Garcia-Gonzalez), PI15/01467 (to M.A. Garcia-Gonzalez), and PI18/00378 (to M.A. Garcia-Gonzalez); and by Xunta de Galicia award IN607B 2016/020 (to M.A. Garcia-Gonzalez).
Supplementary Material
Acknowledgments
The authors would like to acknowledge Dr. Linda Burkley (Biogen, Inc., Cambridge, MA) for providing the anti-TWEAK antibody. The mouse model used in this study (C57/BL6 Pkd1cond/cond,Tam-Cre) was kindly provided by the Maryland PKD Research and Translation Core Center (grant number, U54DK126114). The authors would like to thank Dr. Peter Harris for providing the Pkd1RC/RC, which could not be used for this article due to the difficulties caused by coronavirus disease 2019. The authors acknowledge Susana Carrasco for the technical support with TUNEL assay and Immunohistochemistries of Fn14.
The authors acknowledge Instituto de Salud Carlos III (SCIII) for Fondo de Investigacion SanitariaFIS/FEDER funds (CP14/00133, PI18/01386, PI18/0133, and DTS18/00032) and RETIC Red de Investigación Renal (REDinREN) funds (RD016/0009), and also European Renal Association(ERA)-PerMed-JTC2018 (KIDNEY ATTACK AC18/00064 and PERSTIGAN AC18/00071), the Sociedad Española de Nefrología (SEN), FRIAT, and the Comunidad de Madrid en Biomedicina (B2017/BMD-3686 CIFRA2-CM). Salary support was provided by a Xunta de Galicia predoctoral fellowship 2016 to Dr. A. Cordido, by ISCIII Miguel Servet to Dr. A.B. Sanz and ISCIII Sara Borrell to J.M. Martinez-Moreno .
Dr. A. Cordido performed and designed most experiments and statistical analysis, interpreted them, and wrote the manuscript; L. Nuñez-Gonzalez performed gene expression studies of inflammation markers and NF-κB markers, and interpreted the results; Dr. O. Lamas-Gonzalez performed the TWEAK administration experiment in the noncystic window; J.M. Martinez-Moreno, L. Rodriguez-Osorio, and M. Vanessa Perez-Gomez performed and interpreted Fn14/TWEAK characterization experiments in ADPKD and the p65 and F4/80 staining; D. Martin-Sanchez performed and interpreted experiments in vitro on macrophages and tubular epithelial cells; Dr. P. Outeda and Dr. T. Watnick provided the samples from the Pkd1cond,Pax8,TET-OCre mouse; M. Chiaravalli and A. Boletta provided samples of slowly progressive Pkd1cond/cond,Tam-Cre and Pkd1cond/condTam-KSP-Cre mouse models; Dr. C. Diaz and Dr. A. Carracedo reviewed the manuscript; Dr. A.B. Sanz interpreted Fn14/TWEAK characterization experiments, reviewed the manuscript, and supervised the whole project; and Dr. A. Ortiz and Dr. M.A. Garcia-Gonzalez reviewed and edited the manuscript and supervised the whole project.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2020071094/-/DCSupplemental.
Supplemental Figure 1. Schematic representation of the different experimental designs performed in the study.
Supplemental Figure 2. Western Blots and IHQs for Fn14.
Supplemental Figure 3. Fn14 is upregulated in other murine ADPKD model.
Supplemental Figure 4. Fn14 and TWEAK gene expression of renal cluster-single-cells according to Park, J et al. data.
Supplemental Figure 5. Tubule specific markers in cysts in the short treatment model of TWEAK administration.
Supplemental Figure 6. Evolution of weight over time of different mice groups.
Supplemental Figure 7. TWEAK or anti-TWEAK treatment did not alter kidney histology or related serum biochemistry values in Wild Type control mice.
Supplemental Figure 8. TWEAK promotes an inflammatory status in macrophages and tubular epithelial cells.Wild Type control mice.
Supplemental Figure 8. TWEAK promotes an inflammatory status in macrophages and tubular epithelial cells.Wild Type control mice.
Supplemental Figure 9. Cyst origin characterization in control and anti-TWEAK treated Pkd1-deficient mice, cystic window.
Supplemental Figure 10. Western blots for p-ERK and ERK, p-p38 and p38, p-AKT and AKT and GAPDH.
Supplemental Figure 11. Molecular characterization of proliferation and fibrosis.
Supplemental Figure 12. Effect of TWEAK administration in Fn14 expression.
Supplemental Figure 13. M1 and M2 inflammatory markers and F4/80 staining characterization.
Supplemental Figure 14. Macrophage infiltration of kidneys.
Supplemental Table 1. PCR primers used for real-time quantitative PCR (RT-qPCR).
Supplemental Table 2. Clinical characteristics of ADPKD patients.
References
- 1.Lanktree MB, Haghighi A, Guiard E, Iliuta IA, Song X, Harris PC, et al.: Prevalence estimates of polycystic kidney and liver disease by population sequencing. J Am Soc Nephrol 29: 2593–2600, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cornec-Le Gall E, Alam A, Perrone RD: Autosomal dominant polycystic kidney disease. Lancet 393: 919–935, 201910.1016/S0140-6736(18)32782-X [DOI] [PubMed] [Google Scholar]
- 3.Winkles JA: The TWEAK-Fn14 cytokine-receptor axis: Discovery, biology and therapeutic targeting. Nat Rev Drug Discov 7: 411–425, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sanz AB, Sanchez-Niño MD, Ortiz A: TWEAK, a multifunctional cytokine in kidney injury. Kidney Int 80: 708–718, 2011 [DOI] [PubMed] [Google Scholar]
- 5.Poveda J, Tabara LC, Fernandez-Fernandez B, Martin-Cleary C, Sanz AB, Selgas R, et al.: TWEAK/Fn14 and non-canonical NF-kappaB signaling in kidney disease. Front Immunol 4: 447, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanz AB, Izquierdo MC, Sanchez-Niño MD, Ucero AC, Egido J, Ruiz-Ortega M, et al.: TWEAK and the progression of renal disease: Clinical translation. Nephrol Dial Transplant 29[Suppl 1]: i54–i62, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cherry EM, Lee DW, Jung JU, Sitcheran R: Tumor necrosis factor-like weak inducer of apoptosis (TWEAK) promotes glioma cell invasion through induction of NF-κB-inducing kinase (NIK) and noncanonical NF-κB signaling. Mol Cancer 14: 9, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wisniacki N, Amaravadi L, Galluppi GR, Zheng TS, Zhang R, Kong J, et al.: Safety, tolerability, pharmacokinetics, and pharmacodynamics of anti-TWEAK monoclonal antibody in patients with rheumatoid arthritis. Clin Ther 35: 1137–1149, 2013 [DOI] [PubMed] [Google Scholar]
- 9.Ta MHT, Harris DCH, Rangan GK: Role of interstitial inflammation in the pathogenesis of polycystic kidney disease. Nephrology (Carlton) 18: 317–330, 2013 [DOI] [PubMed] [Google Scholar]
- 10.Kurbegovic A, Trudel M: Acute kidney injury induces hallmarks of polycystic kidney disease. Am J Physiol Renal Physiol 311: F740–F751, 2016 [DOI] [PubMed] [Google Scholar]
- 11.Piontek KB, Huso DL, Grinberg A, Liu L, Bedja D, Zhao H, et al.: A functional floxed allele of Pkd1 that can be conditionally inactivated in vivo. J Am Soc Nephrol 15: 3035–3043, 2004 [DOI] [PubMed] [Google Scholar]
- 12.Piontek K, Menezes LF, Garcia-Gonzalez MA, Huso DL, Germino GG: A critical developmental switch defines the kinetics of kidney cyst formation after loss of Pkd1. Nat Med 13: 1490–1495, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chiaravalli M, Rowe I, Mannella V, Quilici G, Canu T, Bianchi V, et al.: 2-Deoxy-d-Glucose ameliorates PKD progression. J Am Soc Nephrol 27: 1958–1969, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shao X, Somlo S, Igarashi P: Epithelial-specific Cre/lox recombination in the developing kidney and genitourinary tract. J Am Soc Nephrol 13: 1837–1846, 2002 [DOI] [PubMed] [Google Scholar]
- 15.Traykova-Brauch M, Schönig K, Greiner O, Miloud T, Jauch A, Bode M, et al.: An efficient and versatile system for acute and chronic modulation of renal tubular function in transgenic mice. Nat Med 14: 979–984, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanz AB, Justo P, Sanchez-Niño MD, Blanco-Colio LM, Winkles JA, Kreztler M, et al.: The cytokine TWEAK modulates renal tubulointerstitial inflammation. J Am Soc Nephrol 19: 695–703, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sanz AB, Sanchez-Niño MD, Izquierdo MC, Jakubowski A, Justo P, Blanco-Colio LM, et al.: Tweak induces proliferation in renal tubular epithelium: A role in uninephrectomy induced renal hyperplasia. J Cell Mol Med 13[9B]: 3329–3342, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moreno JA, Izquierdo MC, Sanchez-Niño MD, Suárez-Alvarez B, Lopez-Larrea C, Jakubowski A, et al.: The inflammatory cytokines TWEAK and TNFα reduce renal klotho expression through NFκB. J Am Soc Nephrol 22: 1315–1325, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Izquierdo MC, Sanz AB, Mezzano S, Blanco J, Carrasco S, Sanchez-Niño MD, et al.: TWEAK (tumor necrosis factor-like weak inducer of apoptosis) activates CXCL16 expression during renal tubulointerstitial inflammation. Kidney Int 81: 1098–1107, 2012 [DOI] [PubMed] [Google Scholar]
- 20.Ucero AC, Benito-Martin A, Fuentes-Calvo I, Santamaria B, Blanco J, Lopez-Novoa JM, et al.: TNF-related weak inducer of apoptosis (TWEAK) promotes kidney fibrosis and Ras-dependent proliferation of cultured renal fibroblast. Biochim Biophys Acta 1832: 1744–1755, 201310.1016/j.bbadis.2013.05.032 [DOI] [PubMed] [Google Scholar]
- 21.Sanchez-Niño MD, Poveda J, Sanz AB, Mezzano S, Carrasco S, Fernandez-Fernandez B, et al.: Fn14 in podocytes and proteinuric kidney disease. Biochim Biophys Acta 1832: 2232–2243, 2013 [DOI] [PubMed] [Google Scholar]
- 22.Cordido A, Cernadas E, Fernández-Delgado M, García-González MA: CystAnalyser: A new software tool for the automatic detection and quantification of cysts in polycystic kidney and liver disease, and other cystic disorders. PLOS Comput Biol 16: e1008337, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haverty TP, Kelly CJ, Hines WH, Amenta PS, Watanabe M, Harper RA, et al.: Characterization of a renal tubular epithelial cell line which secretes the autologous target antigen of autoimmune experimental interstitial nephritis. J Cell Biol 107: 1359–1368, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moriwaki K, Balaji S, McQuade T, Malhotra N, Kang J, Chan FKM: The necroptosis adaptor RIPK3 promotes injury-induced cytokine expression and tissue repair. Immunity 41: 567–578, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Song X, Di Giovanni V, He N, Wang K, Ingram A, Rosenblum ND, et al.: Systems biology of autosomal dominant polycystic kidney disease (ADPKD): Computational identification of gene expression pathways and integrated regulatory networks. Hum Mol Genet 18: 2328–2343, 2009 [DOI] [PubMed] [Google Scholar]
- 26.Chen WC, Tzeng YS, Li H: Gene expression in early and progression phases of autosomal dominant polycystic kidney disease. BMC Res Notes 1: 131, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dweep H, Sticht C, Kharkar A, Pandey P, Gretz N: Parallel analysis of mRNA and microRNA microarray profiles to explore functional regulatory patterns in polycystic kidney disease: Using PKD/Mhm rat model. PLoS One 8: e53780, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pandey P, Brors B, Srivastava PK, Bott A, Boehn SNE, Groene HJ, et al.: Microarray-based approach identifies microRNAs and their target functional patterns in polycystic kidney disease. BMC Genomics 9: 624, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rahimmanesh I, Fatehi R: Systems biology approaches toward autosomal dominant polycystic kidney disease (ADPKD). Clin Transl Med 9: 1–8, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li X, Magenheimer BS, Xia S, Johnson T, Wallace DP, Calvet JP, et al.: A tumor necrosis factor-alpha-mediated pathway promoting autosomal dominant polycystic kidney disease. Nat Med 14: 863–868, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park J, Shrestha R, Qiu C, Kondo A, Huang S, Werth M, et al.: Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 360: 758–763, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lantinga-van Leeuwen IS, Leonhard WN, van der Wal A, Breuning MH, de Heer E, Peters DJM: Kidney-specific inactivation of the Pkd1 gene induces rapid cyst formation in developing kidneys and a slow onset of disease in adult mice. Hum Mol Genet 16: 3188–3196, 2007 [DOI] [PubMed] [Google Scholar]
- 33.Gao H-X, Campbell SR, Burkly LC, Jakubowski A, Jarchum I, Banas B, et al.: TNF-like weak inducer of apoptosis (TWEAK) induces inflammatory and proliferative effects in human kidney cells. Cytokine 46: 24–35, 2009 [DOI] [PubMed] [Google Scholar]
- 34.Sanz AB, Sanchez-Niño MD, Izquierdo MC, Jakubowski A, Justo P, Blanco-Colio LM, et al.: TWEAK activates the non-canonical NFkappaB pathway in murine renal tubular cells: Modulation of CCL21. PLoS One 5: e8955, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu G, D’Agati V, Cai Y, Markowitz G, Park JH, Reynolds DM, et al.: Somatic inactivation of Pkd2 results in polycystic kidney disease. Cell 93: 177–188, 1998 [DOI] [PubMed] [Google Scholar]
- 36.Yamaguchi T, Pelling JC, Ramaswamy NT, Eppler JW, Wallace DP, Nagao S, et al.: cAMP stimulates the in vitro proliferation of renal cyst epithelial cells by activating the extracellular signal-regulated kinase pathway. Kidney Int 57: 1460–1471, 2000 [DOI] [PubMed] [Google Scholar]
- 37.Omori S, Hida M, Fujita H, Takahashi H, Tanimura S, Kohno M, et al.: Extracellular signal-regulated kinase inhibition slows disease progression in mice with polycystic kidney disease. J Am Soc Nephrol 17: 1604–1614, 2006 [DOI] [PubMed] [Google Scholar]
- 38.Joly D, Ishibe S, Nickel C, Yu Z, Somlo S, Cantley LG: The polycystin 1-C-terminal fragment stimulates ERK-dependent spreading of renal epithelial cells. J Biol Chem 281: 26329–26339, 2006 [DOI] [PubMed] [Google Scholar]
- 39.Wahl PR, Serra AL, Le Hir M, Molle KD, Hall MN, Wüthrich RP: Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol Dial Transplant 21: 598–604, 2006 [DOI] [PubMed] [Google Scholar]
- 40.Shillingford JM, Murcia NS, Larson CH, Low SH, Hedgepeth R, Brown N, et al.: The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci U S A 103: 5466–5471, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Novalic Z, van der Wal AM, Leonhard WN, Koehl G, Breuning MH, Geissler EK, et al.: Dose-dependent effects of sirolimus on mTOR signaling and polycystic kidney disease. J Am Soc Nephrol 23: 842–853, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ravichandran K, Zafar I, He Z, Doctor RB, Moldovan R, Mullick AE, et al.: An mTOR anti-sense oligonucleotide decreases polycystic kidney disease in mice with a targeted mutation in Pkd2. Hum Mol Genet 23: 4919–4931, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Justo P, Sanz AB, Sanchez-Niño MD, Winkles JA, Lorz C, Egido J, et al.: Cytokine cooperation in renal tubular cell injury: The role of TWEAK. Kidney Int 70: 1750–1758, 2006 [DOI] [PubMed] [Google Scholar]
- 44.Berzal S, González-Guerrero C, Rayego-Mateos S, Ucero A, Ocaña-Salceda C, Egido J, et al.: TNF-related weak inducer of apoptosis (TWEAK) regulates junctional proteins in tubular epithelial cells via canonical NF-κB pathway and ERK activation. J Cell Physiol 230: 1580–1593, 2015 [DOI] [PubMed] [Google Scholar]
- 45.Karihaloo A, Koraishy F, Huen SC, Lee Y, Merrick D, Caplan MJ, et al.: Macrophages promote cyst growth in polycystic kidney disease. J Am Soc Nephrol 22: 1809–1814, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Swenson-Fields KI, Vivian CJ, Salah SM, Peda JD, Davis BM, van Rooijen N, et al.: Macrophages promote polycystic kidney disease progression. Kidney Int 83: 855–864, 201310.1038/ki.2012.446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cassini MF, Kakade VR, Kurtz E, Sulkowski P, Glazer P, Torres R, et al.: Mcp1 promotes macrophage-dependent cyst expansion in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 29: 2471–2481, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang Y, Chen M, Zhou J, Lv J, Song S, Fu L, et al.: Interactions between macrophages and cyst-lining epithelial cells promote kidney cyst growth in Pkd1-deficient mice. J Am Soc Nephrol 29: 2310–2325, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Belliere J, Casemayou A, Ducasse L, Zakaroff-Girard A, Martins F, Iacovoni JS, et al.: Specific macrophage subtypes influence the progression of rhabdomyolysis-induced kidney injury. J Am Soc Nephrol 26: 1363–1377, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.GardnerJr.. KD, Burnside JS, Elzinga LW, Locksley RM: Cytokines in fluids from polycystic kidneys. Kidney Int 39: 718–724, 1991 [DOI] [PubMed] [Google Scholar]
- 51.Merta M, Tesar V, Zima T, Jirsa M, Rysavá R, Zabka J: Cytokine profile in autosomal dominant polycystic kidney disease. Biochem Mol Biol Int 41: 619–624, 1997 [DOI] [PubMed] [Google Scholar]
- 52.CowleyJr.. BD, Ricardo SD, Nagao S, Diamond JR: Increased renal expression of monocyte chemoattractant protein-1 and osteopontin in ADPKD in rats. Kidney Int 60: 2087–2096, 2001 [DOI] [PubMed] [Google Scholar]
- 53.Choe JY, Kim SK: Serum TWEAK as a biomarker for disease activity of systemic lupus erythematosus. Inflamm Res 65: 479–488, 2016 [DOI] [PubMed] [Google Scholar]
- 54.Yilmaz MI, Carrero JJ, Ortiz A, Martín-Ventura JL, Sonmez A, Saglam M, et al.: Soluble TWEAK plasma levels as a novel biomarker of endothelial function in patients with chronic kidney disease. Clin J Am Soc Nephrol 4: 1716–1723, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Song CJ, Zimmerman KA, Henke SJ, Yoder BK: Inflammation and fibrosis in polycystic kidney disease. Results Probl Cell Differ 60: 323–344, 2017http://link.springer.com/10.1007/978-3-319-51436-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Saigusa T, Bell PD: Molecular pathways and therapies in autosomal-dominant polycystic kidney disease. Physiology (Bethesda) 30: 195–207, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bastos AP, Piontek K, Silva AM, Martini D, Menezes LF, Fonseca JM, et al.: Pkd1 haploinsufficiency increases renal damage and induces microcyst formation following ischemia/reperfusion. J Am Soc Nephrol 20: 2389–2402, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bell PD, Fitzgibbon W, Sas K, Stenbit AE, Amria M, Houston A, et al.: Loss of primary cilia upregulates renal hypertrophic signaling and promotes cystogenesis. J Am Soc Nephrol 22: 839–848, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Patel V, Li L, Cobo-Stark P, Shao X, Somlo S, Lin F, et al.: Acute kidney injury and aberrant planar cell polarity induce cyst formation in mice lacking renal cilia. Hum Mol Genet 17: 1578–1590, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Takakura A, Contrino L, Zhou X, Bonventre JV, Sun Y, Humphreys BD, et al.: Renal injury is a third hit promoting rapid development of adult polycystic kidney disease. Hum Mol Genet 18: 2523–2531, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Happé H, Leonhard WN, van der Wal A, van de Water B, Lantinga-van Leeuwen IS, Breuning MH, et al.: Toxic tubular injury in kidneys from Pkd1-deletion mice accelerates cystogenesis accompanied by dysregulated planar cell polarity and canonical Wnt signaling pathways. Hum Mol Genet 18: 2532–2542, 2009 [DOI] [PubMed] [Google Scholar]
- 62.Hotta K, Sho M, Yamato I, Shimada K, Harada H, Akahori T, et al.: Direct targeting of fibroblast growth factor-inducible 14 protein protects against renal ischemia reperfusion injury. Kidney Int 79: 179–188, 2011 [DOI] [PubMed] [Google Scholar]
- 63.Zhao Z, Burkly LC, Campbell S, Schwartz N, Molano A, Choudhury A, et al.: TWEAK/Fn14 interactions are instrumental in the pathogenesis of nephritis in the chronic graft-versus-host model of systemic lupus erythematosus. J Immunol 179: 7949–7958, 2007 [DOI] [PubMed] [Google Scholar]
- 64.Vincent C, Findlay DM, Welldon KJ, Wijenayaka AR, Zheng TS, Haynes DR, et al.: Pro-inflammatory cytokines TNF-related weak inducer of apoptosis (TWEAK) and TNFalpha induce the mitogen-activated protein kinase (MAPK)-dependent expression of sclerostin in human osteoblasts. J Bone Miner Res 24: 1434–1449, 2009 [DOI] [PubMed] [Google Scholar]
- 65.Tao Y, Kim J, Schrier RW, Edelstein CL: Rapamycin markedly slows disease progression in a rat model of polycystic kidney disease. J Am Soc Nephrol 16: 46–51, 2005 [DOI] [PubMed] [Google Scholar]
- 66.Zafar I, Ravichandran K, Belibi FA, Doctor RB, Edelstein CL: Sirolimus attenuates disease progression in an orthologous mouse model of human autosomal dominant polycystic kidney disease. Kidney Int 78: 754–761, 2010 [DOI] [PubMed] [Google Scholar]
- 67.Boca M, Distefano G, Qian F, Bhunia AK, Germino GG, Boletta A: Polycystin-1 induces resistance to apoptosis through the phosphatidylinositol 3-kinase/Akt signaling pathway. J Am Soc Nephrol 17: 637–647, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li LX, Fan LX, Zhou JX, Grantham JJ, Calvet JP, Sage J, et al.: Lysine methyltransferase SMYD2 promotes cyst growth in autosomal dominant polycystic kidney disease. J Clin Invest 127: 2751–2764, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rowe I, Chiaravalli M, Mannella V, Ulisse V, Quilici G, Pema M, et al.: Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat Med 19: 488–493, 201310.1038/nm.3092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rogers KA, Moreno SE, Smith LA, Husson H, Bukanov NO, Ledbetter SR, et al.: Differences in the timing and magnitude of Pkd1 gene deletion determine the severity of polycystic kidney disease in an orthologous mouse model of ADPKD. Physiol Rep 4: e12846, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shibazaki S, Yu Z, Nishio S, Tian X, Thomson RB, Mitobe M, et al.: Cyst formation and activation of the extracellular regulated kinase pathway after kidney specific inactivation of Pkd1. Hum Mol Genet 17: 1505–1516, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huang JL, Woolf AS, Kolatsi-Joannou M, Baluk P, Sandford RN, Peters DJM, et al.: Vascular endothelial growth factor C for polycystic kidney diseases. J Am Soc Nephrol 27: 69–77, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zheng D, Wolfe M, Cowley BD Jr., Wallace DP, Yamaguchi T, Grantham JJ: Urinary excretion of monocyte chemoattractant protein-1 in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 14: 2588–2595, 2003 [DOI] [PubMed] [Google Scholar]
- 74.Grantham JJ, Chapman AB, Blais J, Czerwiec FS, Devuyst O, Gansevoort RT, et al.; TEMPO 3:4 Investigators: Tolvaptan suppresses monocyte chemotactic protein-1 excretion in autosomal-dominant polycystic kidney disease. Nephrol Dial Transplant 32: 969–975, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Messchendorp AL, Meijer E, Boertien WE, Engels GE, Casteleijn NF, Spithoven EM, et al.; DIPAK Consortium: Urinary biomarkers to identify autosomal dominant polycystic kidney disease patients with a high likelihood of disease progression. Kidney Int Rep 3: 291–301, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.