

Abstract
Single-cell RNA sequencing (scRNA-seq) and single-nucleus RNA-seq (snRNA-seq) allow transcriptomic profiling of thousands of cells from a renal biopsy specimen at a single-cell resolution. Both methods are promising tools to unravel the underlying pathophysiology of glomerular diseases. This review provides an overview of the technical challenges that should be addressed when designing single-cell transcriptomics experiments that focus on glomerulopathies. The isolation of glomerular cells from core needle biopsy specimens for single-cell transcriptomics remains difficult and depends upon five major factors. First, core needle biopsies generate little tissue material, and several samples are required to identify glomerular cells. Second, both fresh and frozen tissue samples may yield glomerular cells, although every experimental pipeline has different (dis)advantages. Third, enrichment for glomerular cells in human tissue before single-cell analysis is challenging because no effective standardized pipelines are available. Fourth, the current warm cell-dissociation protocols may damage glomerular cells and induce transcriptional artifacts, which can be minimized by using cold dissociation techniques at the cost of less efficient cell dissociation. Finally, snRNA-seq methods may be superior to scRNA-seq in isolating glomerular cells; however, the efficacy of snRNA-seq on core needle biopsy specimens remains to be proven. The field of single-cell omics is rapidly evolving, and the integration of these techniques in multiomics assays will undoubtedly create new insights in the complex pathophysiology of glomerular diseases.
Keywords: glomerular disease, transcriptional profiling, molecular biology, renal cell biology
Single-cell RNA sequencing (scRNA-seq) and single-nucleus RNA-seq (snRNA-seq) are powerful new methods that allow transcriptomic profiling of thousands of cells from a renal biopsy specimen at single-cell resolution.1,2 When performing scRNA-seq and snRNA-seq, four different steps can be recognized in the experimental workflow.3 First, a high-quality, single-cell or single-nucleus suspension is created from the tissue sample (Figure 1). In scRNA-seq, viable single cells are dissociated from their extracellular matrix; in snRNA-seq, single nuclei are isolated using stronger dissociation methods that lyse the cell membranes and free the nuclei.3 In the second step, the mRNA within these cells or nuclei is captured, reverse transcribed into cDNA, amplified, and sequenced (Figure 2).3 The third step consists of raw data processing, which is subsequently analyzed in the final step, by using advanced bioinformatics pipelines that translate sequencing data into expression profiles of individual cells, leading to the identification of distinct cell populations (Figure 2).3
Figure 1.
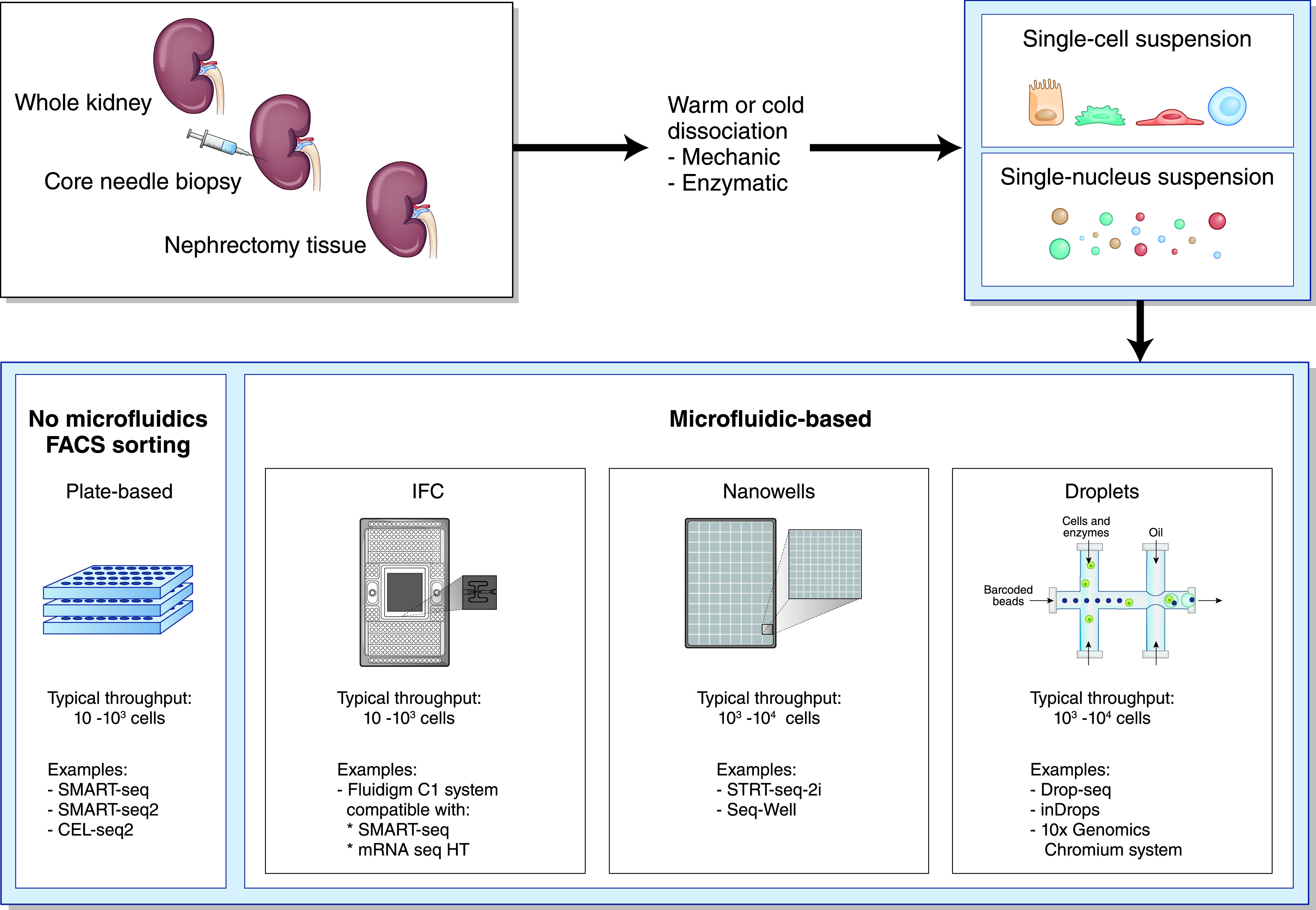
General workflow of scRNA-seq and snRNA-seq experiments. Renal tissue is mechanically and enzymatically dissociated into a single-cell or single-nucleus suspension. In scRNA-seq, viable single cells are dissociated from their extracellular matrix; in snRNA-seq, stronger dissociation techniques are used to dissociate nuclei from the cells.3 Next, cells or nuclei are sorted into a microtiter plate or loaded into a microfluidics device containing integrated fluidic circuits (IFCs), nanowells, or droplets, ready for subsequent processing steps.
Figure 2.
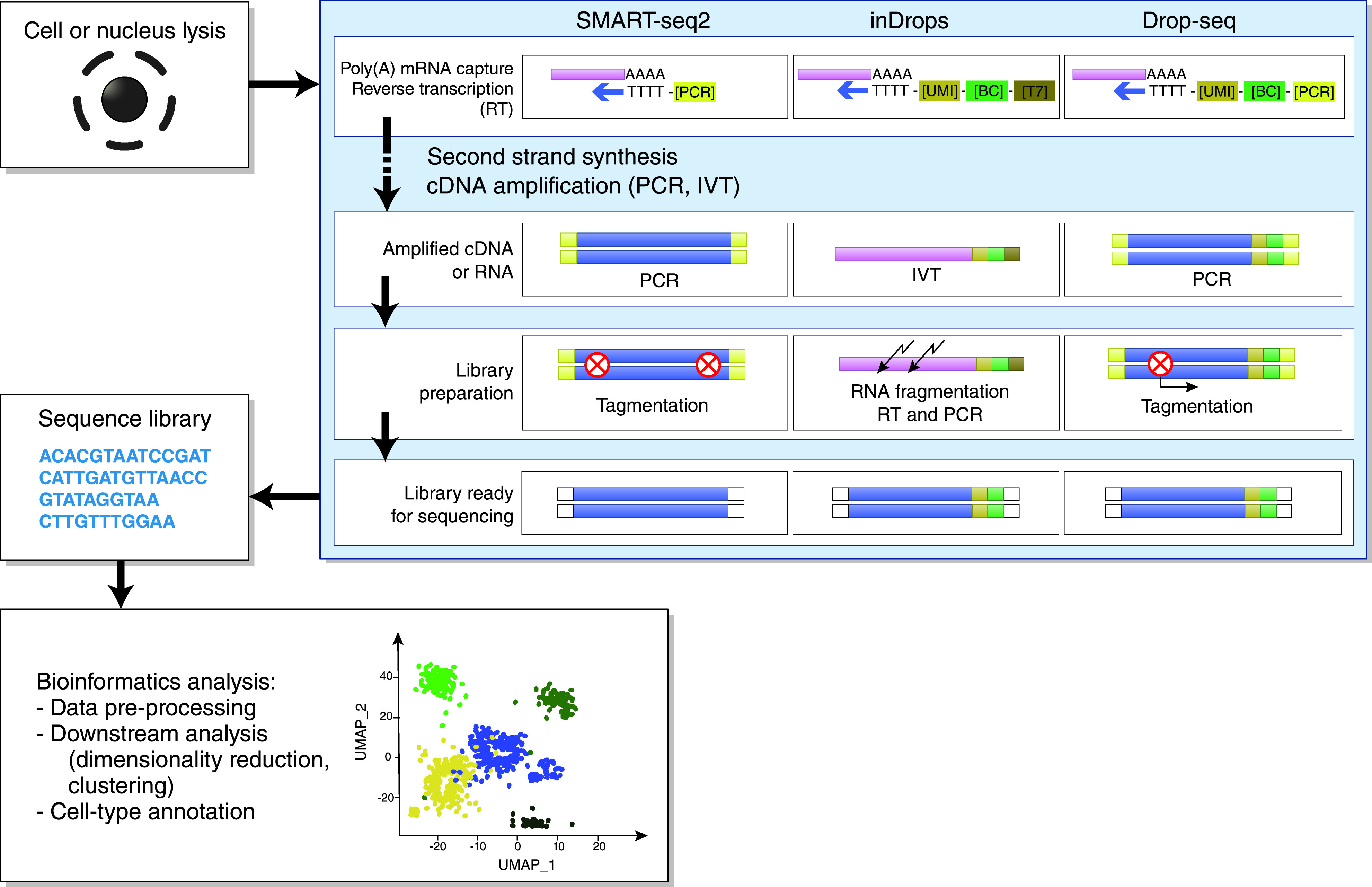
General workflow of scRNA-seq and snRNA-seq experiments, continued. Individual cells or nuclei are lysed and transcripts from single cells or single nuclei are captured using poly[T] oligonucleotide primers that hybridize with the polyadenylated (poly[A]) mRNA.3,82 These primers may also contain other oligonucleotide sequences, including a cell barcode (BC), unique molecular identifier (UMI), and adaptor sequence for PCR or RNA polymerase promotor (T7 promotor) for in vitro transcription (IVT).3 Next, a reverse transcription (RT) reaction synthesizes the first and second strand of cDNA, which is subsequently amplified by either PCR or IVT. Amplified cDNA molecules are fragmented (using enzymatic tagmentation or chemical fragmentation) and ligated with adaptors for sequencing.3 A sequence library is created and, finally, using advanced bioinformatics analysis, sequencing data are translated into cellular expression patterns, leading to the identification of distinct cell clusters.83 Adapted from refs. 3,4,81,82. UMAP, Uniform Manifold Approximation and Projection.
The first two steps of the workflow are crucial, because they are prone to several methodologic pitfalls. First, the total number of cells that needs to be profiled increases significantly when investigating less abundant cell populations, such as podocytes.4 Second, the choice of the dissociation procedure comes with a certain degree of “dissociation bias,” especially in scRNA-seq protocols.5,6 Indeed, in mild dissociation protocols, cells strongly embedded in the extracellular matrix remain attached and are, therefore, under-represented in the scRNA-seq data.7–9 In contrast, with stronger dissociation protocols, fragile cells may become damaged, resulting in a reduction in cell viability.7–9 In snRNA-seq, difficult-to-dissociate cell types are more easily isolated, because stronger dissociation protocols are used and only nuclei are isolated, as opposed to viable cells in scRNA-seq.3 Finally, the dissociation process itself could also result in transcriptional stress responses and subsequent RNA degradation, which could mask true biologic signals within the data.7,10
Theoretically, all cells from the glomerular microenvironment—such as podocytes, glomerular endothelial cells, mesangial cells, and leukocytes—can be studied with single-cell transcriptomics. In reality, however, isolation and analysis of glomerular cells is very challenging.11 On the basis of the current literature on scRNA-seq and snRNA-seq experiments on renal tissue, this review aims to answer five key questions to optimize the design of single-cell transcriptomics experiments that focus on glomerular pathology (Figures 3 and 4; search strategy is outlined in Supplemental Figures 1 and 2).
Figure 3.
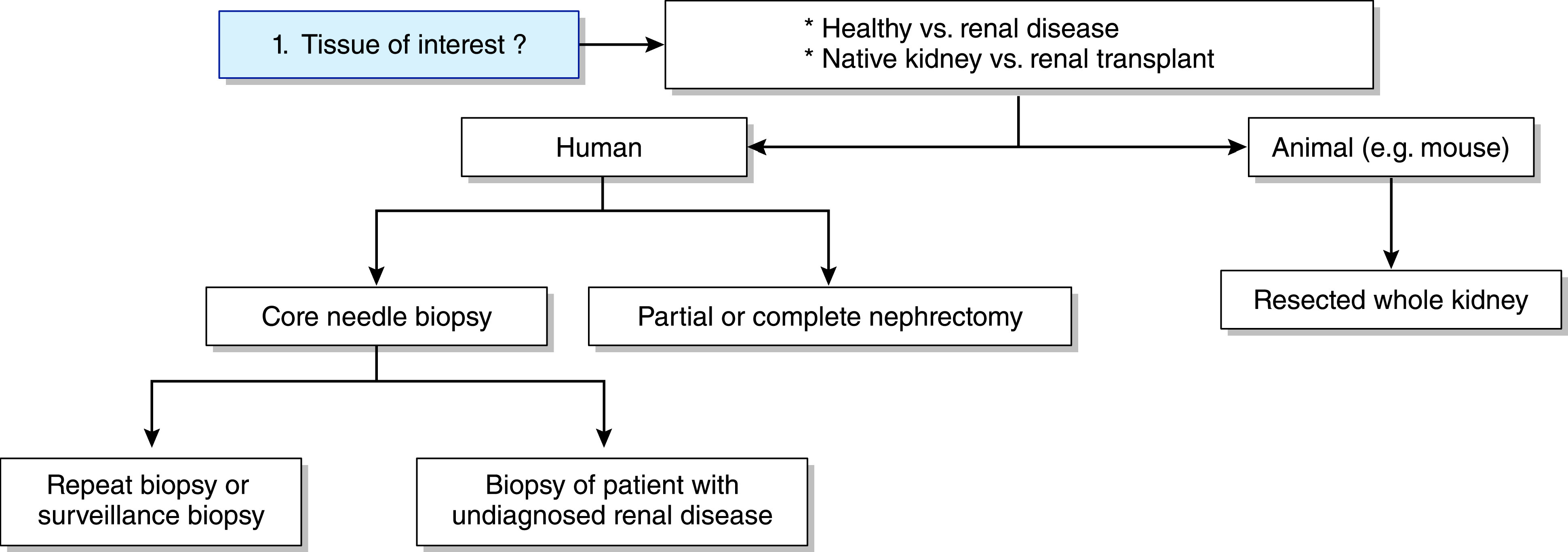
Technical flowchart of scRNA-seq and snRNA-seq experiments on renal tissue of humans or mice. The first step involves choosing the renal tissue of interest. Human renal tissue can be derived from nephrectomy tissue or core needle biopsies.
Figure 4.
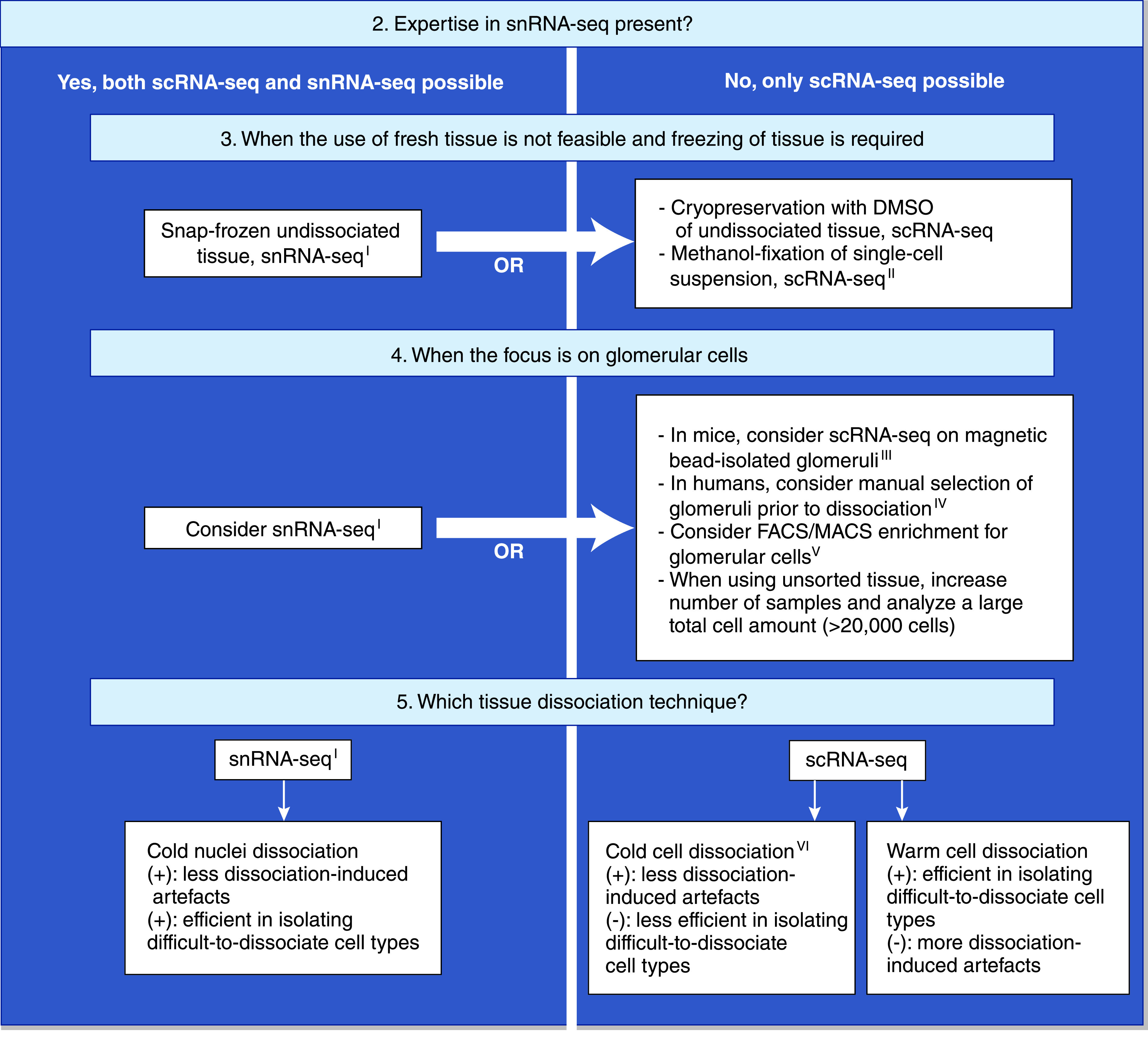
Technical flowchart of scRNA-seq and snRNA-seq experiments on renal tissue of humans or mice, continued. IsnRNA-seq has not yet been performed on human core needle biopsies. IIMethanol fixation of single cells has not yet been performed on human renal tissue. IIIMagnetic bead isolation of glomeruli is not possible in human patients/tissue. IVManual selection of glomeruli before tissue dissociation has only been performed in one scRNA-seq study and still requires validation.18 VFACS/MACS have successfully been used to enrich for glomerular endothelial cells in scRNA-seq experiments on mice,44,50 but not yet for podocytes or mesangial cells. VICold dissociation protocols have been successfully used on renal tissue of mice,33,34,45,52 but studies on human renal tissue have yet to be published. (+), advantage; (-), disadvantage.
QUESTION 1: WHICH RENAL BIOPSY SPECIMEN CAN BE USED FOR THE ISOLATION OF GLOMERULAR CELLS?
Single-cell transcriptomics experiments have been performed on both mouse and human kidneys (Supplemental Tables 1 and 2), and many different scRNA-seq protocols have been used (Table 1). Studies on mice allow resection of one or both kidneys, which yields ample starting tissue material. In humans, however, renal tissue is only available after core needle biopsies or nephrectomy procedures (Figure 3).
Table 1.
Overview of different scRNA-seq protocols used in studies on renal tissue
| Method | SMART-seq SMART-seq2 | Fluidigm C1 System (SMART-seq) | Fluidigm C1 System (mRNA Seq HT) | CEL-seq2 | Drop-seq | inDrops | 10x Genomics Chromium Single Cell Gene Expression Solution |
|---|---|---|---|---|---|---|---|
| Platform | Plate (FACS) | Microfluidics (IFC) | Microfluidics (IFC) | Plate (FACS) | Microfluidics (droplet) | Microfluidics (droplet) | Microfluidics (droplet) |
| Reaction volume | Microliter | Nanoliter | Nanoliter | Microliter | Nanoliter | Nanoliter | Nanoliter |
| Cell-size limitations | Independent of cell size | Three types of IFCs available: 5–10 µm, 10–17 µm, and 17–25 µm | Two types of IFCs available: 5–10 µm and 10–17 µm | Independent of cell size | Adjustable with microfluidic and droplet size | Adjustable with microfluidic and droplet size | Maximum 50–60 µm |
| Transcript coverage | Full length | Full length | 3′ end | 3′ end | 3′ end | 3′ end | 3′ end and 5′ end |
| Cell barcode incorporation | Tagmentation | Tagmentation | Oligo(T) primer 3′ | Oligo(T) primer 3′ | Oligo(T) primer 3′ | Oligo(T) primer 3′ | Oligo(T) primer 3′ |
| Use of UMI possible | No | No | No | Yes | Yes | Yes | Yes |
| cDNA amplification | PCR | PCR | PCR | IVT | PCR | IVT | PCR |
| Fragmentation method | Tagmentation | Tagmentation | Tagmentation | Random priming | Tagmentation | Chemically (KOAc, MgOAc) | Enzymatic fragmentation |
| Cellular capture rate of sample | Depending on sorting settings | Approximately 10% of cells | Approximately 10% of cells | Depending on sorting settings | <5% of cells | >75% of cells | >50% of cells |
| Typical cell output per run (microfluidics) | N/A | Maximum 96 cells per run | Maximum 800 cells per run | N/A | Thousands of cells | Thousands of cells | Thousands of cells |
| Landmark study | V184 and V285 | 86 | see Fluidigm website | 87 | 88 | 89 | 90 |
| Application on renal tissue | V148 and V229,91,92 | 13,26,40,41,46 | 11,15,43 | 14 | 9,32,34,49,52,56 | 2 | 6,14,16,17,19,21–23,25–27,33,34,39,42,44,45,47,50,51,55,58,69,91–98 |
The data included in this table was derived from previously published reviews3,4,7,82,99,100 and additional literature search. Two additional, infrequently used, plate-based protocols (Microwell-seq28 and STRT-seq18) were not included in this table. IFC, integrated fluidics circuits; UMI, unique molecular identifier; IVT, in vitro transcription; KOAc, potassium acetate; MgOAc, magnesium acetate; N/A, not available.
Human Core Needle Biopsy Specimens
Renal tissue in glomerular diseases is almost exclusively available after core needle biopsy. A major disadvantage of this is the small amount of cells contained in one core: approximately 40,000–75,000 cells in total.2 Strategies to obtain more tissue include the use of larger biopsy needles, taking one extra biopsy core per patient,12 or pooling the material of several patients.
Since 2017, eight studies have performed scRNA-seq experiments on human renal tissue obtained from core needle biopsies (Table 2).2,6,13–18 Six studies used unsorted tissue,2,6,13,15–17 one study analyzed FACS-isolated leukocytes and epithelial cells,14 whereas the most recent study used a stepwise dissociation protocol in which glomeruli were selected before tissue dissociation.18 Three of these studies were able to isolate podocytes from core needle biopsy specimens.6,17,18 The first study identified all glomerular cell types from tissue obtained from both core biopsies (preperfusion biopsy specimens from kidney donors, n=3; surveillance biopsy specimens from kidney allografts, n=5) and nephrectomy tissue (n=16).6 However, only 11 podocytes were isolated from the tissue from the eight core biopsies (11 of 7524 cells, 0.15%), whereas 159 podocytes originated from nephrectomy tissue (159 of 14,744 cells, 1.08%). A second study, by the same research group, also identified all glomerular cells from 18 core biopsy specimens from healthy kidney donors (25,163 cells) and 44 core biopsy specimens from patients with diabetic kidney disease (85,872 cells), but this study did not mention podocyte numbers or percentages.17 The last study analyzed 13 core biopsy specimens from patients with IgA nephropathy, and six nephrectomy specimens, and used a stepwise dissociation approach to enrich for glomeruli.18 The renal tissue was mildly digested, glomeruli were manually picked under a microscope with a needle and pipette, and the selected glomeruli and remaining tissue were separately dissociated further into a single-cell suspension and analyzed. Although glomerular cells were successfully isolated, the total numbers of podocytes (26 of 2875 cells, 0.9%) and mesangial cells (71 of 2875 cells, 2.5%) were still low and comparable with some scRNA-seq studies that did not use any sorting techniques.6,19 Manual picking of glomeruli is labor intensive, low throughput, and might induce transcriptional artifacts or reduce the viability of fragile cells (such as podocytes), which might explain the low cell numbers.
Table 2.
scRNA-seq studies on human renal core needle biopsy specimens
| Author | Protocol | Age of Patients | Txa | Diseaseb | Biopsyc | T°d | Cellse | Depthf | Genesg | Glomh | Remarks |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Der et al.13 | Fluidigm C1 SMART-seq | A | N | SLE (n=10) | Partial core or full core (n=16) | (+), W | 899* | 500,000 | 700 | — | *Pooling of renal and skin cells and PBMCs |
| Wu et al.2 | inDrops | A | T | Acute TCMR (n=1) | Full core (n=1) | (+), W | 4487 | 50,000 | 827 | — | Study also performed snRNA-seq on healthy tissue (Supplemental Table 2) |
| Der et al.15 | Fluidigm C1 mRNA Seq HT | A | N | SLE (n=21); healthy (Pre) (n=3) | Partial core (n=24) | (-),W, cryo | 4019* | 200,000 | ? | M | *Pooling of renal and skin cells |
| Arazi et al.14 | CEL-seq2* 10x Chromium** | A | N | SLE (n=24); healthy (Pre) (n=10) | Partial core or full core (n=34) | (-), W, cryo | 2838*; 122** | Approximately 106*; ?** | 1000–5000*; 250–3500** | — | scRNA-seq on FACS-isolated leukocytes (CD45+) versus epithelial cells (CD45-, CD10+). *CEL-seq2; **10x on two healthy donor biopsy specimens |
| Menon et al.6 | 10x Chromium | A | N; T | Healthy (Tu) (n=16); healthy (Pre) (n=3); healthy (S) (n=5) | Partial core (n=8); neph (n=16) | (-), W, cryo | 7524*; 14,744** | 3971*,***; 3089**,*** | 1339*; 1134** | P, E, M | *CNB; **nephrectomies; ***mUMIs per cell; P°, *11 cells (0.14%), **159 cells (1.08%) |
| Malone et al.16 | 10x Chromium | A | T | ABMR (n=3); nonrejection AKI (n=2) | Full core (n=5) | (+), W | 81,139 | 2497* | 1124 | — | *Mean transcripts per cell, not raw reads |
| Menon et al.17 | 10x Chromium | A | N | Healthy (Pre) (n=18); DKD (n=44) | Partial core (n=62) | (-), W, cryo | 25,163*; 85,872** | ? | 500–5000 | P, M | *Healthy; **DKD; P°, not mentioned |
| Zheng et al.18 | STRT-seq | A | N | IgAN (n=13)*; healthy (Tu)** (n=6) | Partial core (n=13); neph (n=6) | (+), W *** | 2022*; 763** | 37,875*; 38,391** | 3348*; 3105** | P, M | *IgAN;**healthy; |
| ***stepwise dissociation: | |||||||||||
| (1) glomerulus isolation (pipetting), | |||||||||||
| (2) MACS CD14+ cells, | |||||||||||
| (3) MACS CD326+ cells, | |||||||||||
| (4) unselected cells | |||||||||||
| P°, *22 cells (1%), **four cells (0.52%) |
Tx, tissue; T°, temperature; glom, glomerular cells; A, adult; N, native kidney; (+), fresh tissue; W, warm dissociation; −, no glomerular cells; T, transplant kidney; TCMR, T cell–mediated rejection; Pre, preperfusion or pretransplant core needle biopsy of living donor kidney; (-), frozen tissue; cryo, cryopreservation; ?, data could not be found in published paper; M, mesangial cells; Tu, tumor-free tissue at maximal distance of mass or tumor; S, surveillance kidney transplant biopsy; neph, biopsy from partial/total nephrectomy; P, podocytes; E, glomerular endothelial cells; CNB, core needle biopsy; mUMIs, mean unique molecular identifiers; P°, absolute and relative number of podocytes isolated in scRNA-seq studies; ABMR, antibody-mediated rejection; DKD, diabetic kidney disease; IgAN, IgA nephropathy; MACS, magnetic-activated cell sorting.
Tissue from native kidneys or transplant kidneys.
Healthy or pathologic kidney tissue. n=number of patients.
Biopsy technique. n=biopsy samples taken.
Fresh versus frozen tissue and warm versus cold dissociation.
Total number of cells isolated and analyzed after quality control.
Sequencing depth defined as “mean (raw) reads per cell,” “mean transcripts per cell,” or “mean UMIs per cell.”
Mean number of genes per cell.
Isolation of glomerular cells.
Interestingly, another scRNA-seq study analyzed a large number of cells (total of 81,139 cells) from five core needle biopsy specimens from renal allografts (antibody-mediated rejection, n=3; nonrejection AKI, n=2), but was unable to identify glomerular cells.16 A low number of glomeruli in the core biopsy samples might have played a major role (sample bias); however, differences in dissociation methods, tissue preservation, and the presence of renal inflammation might have also influenced the researchers’ ability to identify glomerular cells.
Human Nephrectomy Tissue
In the context of common renal pathologies (e.g., diabetic nephropathy, hypertensive nephrosclerosis), residual (tumor-free) tissue from partial or total nephrectomy specimens might be available.20,21 In contrast, nephrectomy tissue is generally unavailable in patients with rare glomerular diseases. Indeed the majority of single-cell transcriptomics studies (both scRNA-seq and snRNA-seq) on human nephrectomy tissue have involved healthy renal tissue.2,6,18,19,22–28 In all three snRNA-seq experiments, glomerular cells were present, including podocytes.2,20,24 Similarly, most of the scRNA-seq studies detected glomerular cells, including podocytes,6,18,19,21,23,25 glomerular capillary endothelial cells,6,21,23,25,28 and mesangial cells.6,18,19,28
Concluding Answer
Renal tissue in glomerular diseases is almost exclusively available after core needle biopsy. scRNA-seq on specimens from core needle biopsies is able to characterize podocytes and other glomerular cells, although cell counts remain very low.6,18 Manually enriching for glomeruli before tissue dissociation might modestly increase the chances of identifying podocytes; however, this technique is low throughput, labor intensive, and should be validated in future studies.18 The analysis of more cells per sample might increase the chance of identifying podocytes, and the inclusion of more samples might circumvent the risk of sample bias when using tissue from core needle biopsies. Whether or not snRNA-seq applied to core needle biopsy specimens is a valid alternative for studying glomerular cells remains to be investigated.
QUESTION 2: SHOULD FRESH OR FROZEN TISSUE BE USED TO ISOLATE GLOMERULAR CELLS?
When using scRNA-seq on fresh tissue, a biopsy sample should be dissociated immediately into a single-cell suspension and processed further. This does not allow for histopathologic evaluation of the biopsy sample (which takes several days) before processing and, therefore, hampers the selection of well-selected disease phenotypes.
Using a hypothermic preservation solution might be an alternative for immediate processing.29 HypoThermosol FRS (HTS-FRS) can be used to store fresh tissue at 2–8°C for 24–72 hours before starting the dissociation process.29 Mouse kidney cells that were treated with HTS-FRS and preserved for up to 72 hours showed no reduced viability, and scRNA-seq of FACS-isolated immune cells showed a similar cell type heterogeneity with an equal average number of detected genes per cell, when compared with immediately dissociated cells.29 However, to date, studies on human renal tissue have only used HTS-FRS to store samples for a few hours before downstream processing13 or cryopreservation.6,14,15
scRNA-Seq on Frozen Tissue
Snap freezing of tissue results in low-quality single-cell suspensions once the samples are thawed and subjected to standard dissociation protocols.30 Therefore, cryoprotectants are necessary to keep the cells viable and intact upon thawing. There are two preservation protocols for renal tissue: (1) methanol fixation of a single-cell suspension, and (2) cryopreservation of undissociated tissue or a single-cell suspension in DMSO.
Methanol is a coagulating fixative that does not alter the structure of nucleic acids, and dissociated, methanol-fixed cells can be frozen at −80°C and stored for several weeks.31 To date, no studies have used methanol fixation on human kidney cells, this has only been performed in mice (Supplemental Table 3).9,32–34 One study directly compared scRNA-seq on fresh tissue versus methanol-fixed frozen cells of mouse kidneys.33 The methanol-fixed cell composition was comparable with fresh tissue and included podocytes, although some cell types, especially macrophages, were slightly under-represented.33 In line with previous studies, methanol fixation appeared to damage cells, resulting in ambient RNA contamination.33,35
In the cryopreservation technique, whole tissue specimens or single-cell suspensions are immersed in a cryopreservation medium containing DMSO, and subsequently frozen and stored at −80°C.35 In one study on renal tissue from healthy mice, scRNA-seq of cryopreserved, single-cell suspensions performed poorly in isolating epithelial cells, such as proximal tubule cells, and also upregulated a cellular stress response.33 However, this result contrasts with five studies that successfully used scRNA-seq methods on cryopreserved tissue from human core needle biopsies6,14,15,17 or nephrectomy tissue,6,19 in which tissue was frozen in cryopreservation medium and only dissociated after thawing (Supplemental Table 4). These studies did not report problems with isolating tubular cells,6,15,17,19 and were also able to isolate glomerular cells, including podocytes.6,17,19
snRNA-Seq on Frozen Tissue
In the third technique, undissociated renal tissue is snap frozen without fixation or cryopreservation and stored at −80°C. This tissue appears to only be compatible with snRNA-seq pipelines. Several studies, both in mice and humans, have successfully used snRNA-seq on snap-frozen renal tissue.9,20,24,33,34,36–39 All of these studies were able to isolate podocytes and many also identified other glomerular cells, although no studies used tissue from core needle biopsies.
Concluding Answer
scRNA-seq of frozen tissue might be an attractive alternative to fresh biopsy material when studying glomerular diseases. To date, frozen, methanol-fixed, single-cell suspensions of human kidneys have not been studied. Cryopreservation with DMSO before single-cell dissociation has yielded good and consistent results in human biopsy specimens, including biopsy cores. snRNA-seq on snap-frozen tissue is efficient in capturing glomerular cells, but its efficacy in biopsy cores still needs to be proven.
QUESTION 3: CAN GLOMERULAR CELLS BE ENRICHED IN SINGLE-CELL TRANSCRIPTOMICS EXPERIMENTS?
Using selected tissue sections or cell-sorting techniques in combination with scRNA-seq or snRNA-seq to enrich for rare kidney cells is an appealing, but challenging, option. In scRNA-seq, every manipulation of tissue or cells before mRNA capture and sequencing poses the risk of inducing transcriptional artifacts or decreased cell viability of fragile cells. In snRNA-seq, cell-surface staining is not possible and alternative, complex, nuclear-staining methods are required to sort nuclei.
Selection of Glomeruli before Tissue Dissociation
In mice, glomeruli can be selectively isolated using magnetic bead perfusion. In this technique, the kidneys of a euthanized mouse are perfused with a solution containing magnetic beads that accumulate in the glomerular vessels and are used for magnetic isolation of glomeruli after partial enzymatic tissue digestion.11,32,40–43 This method effectively isolates viable glomerular cells, but is not possible in humans. Alternatively, cortical renal tissue can be dissected from the medulla before dissociation to enrich for glomeruli,34,44,45 which might be useful in nephrectomy tissue, but not in human core needle biopsy specimens. In humans, only one recent scRNA-seq study performed isolation of glomeruli before single-cell dissociation, using a labor-intensive and low-throughput method in which glomeruli were manually selected under a microscope after mild digestion of the renal tissue.18 The yield of glomerular cells remained low, and no direct comparison of this technique with scRNA-seq of unsorted tissue has been published. Taken together, to date, no validated techniques are available that efficiently enrich for glomeruli in human renal tissue before dissociation.
Glomerular Cell Sorting after Tissue Dissociation (FACS and Magnetic-Activated Cell Sorting)
FACS and magnetic-activated cell sorting (MACS) are cell-sorting techniques that have been used in dissociated renal cells to selectively enrich for a target cell population. In mouse kidneys, scRNA-seq studies enriched for collecting duct cells,46 immune cells,26,29,47 myofibroblasts,48 and endothelial cells44,49,50 (including glomerular endothelial cells44,50). One snRNA-seq study involved the creation of a reporter mouse that expressed green fluorescent protein in nephron progenitor cells, which enabled the selective enrichment for nuclei of mature nephron cells by sorting for green fluorescent protein–positive nuclei.51 In human kidneys, scRNA-seq studies enriched for immune cells,14,18,26 epithelial cells,18 proximal tubule cells,21 and myofibroblasts.21
However, to date, no studies have used cell-sorting techniques to enrich for podocytes or mesangial cells before scRNA-seq. Indeed, selective enrichment of podocytes might be very challenging. It is unclear whether cell-enrichment methods are efficient enough to isolate the few podocytes that are present in tissue from human core biopsies. FACS and MACS both require cell staining, washing, and sorting steps that might damage the podocytes, alter the transcriptional profile, and reduce viability, with low cell yield in downstream scRNA-seq analysis. However, this hypothesis should be tested in future experiments.
Concluding Answer
In mice, glomeruli can be isolated using magnetic beads, which successfully enriches for glomerular cells. In humans, current techniques to selectively enrich for glomeruli before dissociation are still low throughput and require validation and optimization. Sorting of podocytes and mesangial cells with FACS or MACS before single-cell capture and scRNA-seq has not been performed and should be the focus of future research.
QUESTION 4: WHICH DISSOCIATION PROTOCOL IS ABLE TO ISOLATE GLOMERULAR CELLS?
A single-cell suspension should meet the following ideal characteristics: the detected cell types should be representative of the sampled tissue, cell viability should be high, and the cellular transcriptomes should have as little dissociation-induced artifacts as possible.52 Therefore, an important question is how to adapt dissociation protocols when studying difficult-to-dissociate cell types, such as podocytes, in scRNA-seq experiments.
Dissociation into Single-Cell Suspension
Standard dissociation is performed at 37°C, which can trigger a cellular stress response and dissociation-induced artifacts.52 The use of cold active protease in mouse kidneys and dissociating at 6°C or 12–16°C results in less dissociation-induced artifacts.45,52 A recent study compared scRNA-seq on warm versus cold dissociated mouse renal tissue, and used this cold active protease in a dissociation protocol on ice.33 This study confirmed that warm dissociation induces a cellular stress response and that cells prone to these transcriptional changes, such as podocytes, are under-represented after warm dissociation. However, cold dissociation protocols were less efficient in isolating other difficult-to-dissociate cell types, such as cells of the loop of Henle.33 In total, four studies reported an efficient tissue dissociation of mouse kidney using cold active protease, and all studies identified podocytes.33,34,45,52 However, another study that analyzed glomeruli of mice explicitly mentioned they were unable to effectively dissociate renal cells with cold dissociation protocols, whereas an alternative warm dissociation protocol was successful.42 Furthermore, to date, no published study has successfully used cold active protease to dissociate human renal tissue.
Concluding Answer
The use of warm dissociation protocols in scRNA-seq experiments on human renal tissue is still recommended, because cold dissociation protocols using cold active protease have produced conflicting results in mice and have not yet been successfully used on human renal tissue. Therefore, future studies should test the performance of cold dissociation protocols on human renal tissue.
QUESTION 5: WHICH METHOD IS PREFERRED, SCRNA-SEQ OR SNRNA-SEQ?
snRNA-seq might be an appealing alternative to scRNA-seq.2,9,20,24,33,34,36–39,51 First, some difficult-to-dissociate cell types can be detected with snRNA-seq, because dissociation methods that isolate nuclei are stronger.33 Second, nuclei can be dissociated on ice, thereby minimizing dissociation-induced artifacts.9 Third, because snap-frozen tissue can be used, snRNA-seq is compatible with historically collected biobanks of frozen samples.53 Finally, although nuclei contain lower amounts of mRNA, snRNA-seq can detect an equal average number of genes per cell when compared with scRNA-seq.9,33,54
When Podocytes Are the Focus of Research
snRNA-seq methods perform well in isolating glomerular cells, including podocytes,2,9,20,24,33,34,36–39,51 mesangial cells and/or glomerular endothelial cells.9,20,24,33,37 Three studies directly compared snRNA-seq with scRNA-seq methods.2,9,33 In the first study on human tissue, podocytes were present in the snRNA-seq experiment, but not in the scRNA-seq experiment.2 However, differences in the biopsy specimen (core needle biopsy versus nephrectomy) and renal pathophysiology (cellular rejection in renal allograft versus healthy native renal tissue) could confound this observation. A second study compared three snRNA-seq methods (sNuc-Drop-Seq, DroNC-seq, sNuc-10x) to a methanol-fixed (frozen) scRNA-seq method (Drop-seq) on healthy mouse tissue; again, all snRNA-seq methods identified podocytes, whereas the scRNA-seq method did not.9 However, Drop-seq is a suboptimal scRNA-seq method with a low cellular capture rate (<5%), and methanol fixation might have damaged cells, including podocytes. The third study systematically compared snRNA-seq with scRNA-seq on tissue from healthy adult mice using 10x Genomics.33 Both techniques identified podocytes, but, surprisingly, the percentage of podocytes was lower with the snRNA-seq technique in this study (0.7% versus 3.28%).33 So, although snRNA-seq is able to identify glomerular cells in general, scRNA-seq methods are not necessarily inferior to snRNA-seq when their experimental design is optimized. Indeed, the heterogeneity of scRNA-seq studies explains their varying performance in isolation of glomerular cells. Some studies exclusively performed scRNA-seq on magnetically isolated glomeruli from mouse kidneys and all were successful in isolating podocytes.11,32,40–43 In unsorted renal tissue from mice, all seven snRNA-seq studies9,33,34,36–39 versus half (eight of 16) of the scRNA-seq studies33,34,45,52,55–58 were able to isolate podocytes (Supplemental Table 5). Almost all successful scRNA-seq studies analyzed >20,000 cells in total. Four scRNA-seq studies used a cold dissociation protocol and all identified podocytes,33,34,45,52 whereas the unsuccessful studies all used a warm dissociation protocol. In unsorted human nephrectomy tissue, all three snRNA-seq studies2,20,24 versus half (four of eight) of the scRNA-seq studies6,19,23,25 were able to identify podocytes (Table 3). The successful human scRNA-seq studies analyzed more nephrectomy samples (n=3–20 samples) when compared with the unsuccessful studies (n=1–3 samples). Overall, in both the snRNA-seq studies and the successful scRNA-seq studies on unsorted renal tissue from humans and mice, the percentage of podocytes to total isolated cells was extremely low (0.03%–2.78% for scRNA-seq; 0.25%–4.86% for snRNA-seq).
Table 3.
scRNA-seq or snRNA-seq studies on unsorted human nephrectomy tissue
| Author | Protocol | Age of Patients | Txa | Diseaseb | Biopsyc | T°d | Cellse | Depthf | Genesg | Glomh | Remarks |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gillies et al.19,i | 10× Chromium | A | N | Healthy (Tu) (n=3) | Neph (n=3) | (-), W, cryo | 4734 | ? | ? | P, M | Study on eQTL and integration with scRNA-seq; P°, 49 cells (1%) |
| Wu et al.2,j | inDrops | A | N | Healthy (D) (n=1) | Neph (n=1) | (+), C | 4259 | 50,000 | >400 | P | P°, not mentioned |
| Young et al.23,i | 10x Chromium | F, Ch, A | N | Fetal (n=2); healthy (D) (n=1); healthy (Tu)* (n=8); tumor* (n=8) | Neph (n=20); whole (n=2); other** (n=16) | (+), W | 72,501 | ? | ? | P, E | *Same patients; **tissue from renal pelvis, ureter, and tumor; P°, 259 cells (0.3%) |
| Wilson et al.20,j | 10x Chromium | A | N | Healthy (Tu) (n=3); DKD (Tu) (n=3) | Neph (n=6) | (-), C, snap | 23,980 | 6894* | 2541 | P, E, M | *mUMIs per cell; P°, 663 cells (2.76%) |
| Lake et al.24,j | snDrop-seq | A | N | Healthy (Tu) (n=14); healthy (D) (n=2) | Neph (n=19) | (+), C; (-), C, snap | 17,659 | 1082* | 589 | P, E, M | *Mean transcripts per cell, not raw reads; P°, 859 cells (4.86%) |
| Stewart et al.25,i | 10x Chromium | F, Ch, A | N | Fetal (n=6); healthy (Tu) (n=10); healthy (D) (n=3) | Neph (n=14); whole (n=6) | (+), W | 40,268*; 27,203** | ? | ? | P, E | *Mature kidneys; **fetal kidneys; P°, 126 cells (0.3%) for F and Ch |
| Menon et al.6,i | 10x Chromium | A | N, T | Healthy (Tu) (n=16); healthy (Pre) (n=3); healthy (S) (n=5) | CNB (n=8); neph (n=16) | (-), W, cryo | 7524*; 14,744** | 3971*,***; 3089**,*** | 1339*; 1134** | P, E, M | *CNB; ** nephrectomies; *** mUMIs per cell; P°, *11 cells (0.14%), **159 cells (1.08%) |
| Liao et al.27,i | 10x Chromium | A | N | Healthy (Tu) (n=3) | Neph (n=3) | (+), W | 23,366 | Approximately 30,000 | Approximately 800 | — | |
| Liu et al.95,i | 10x Chromium | A | T | Chronic rejection (n=2) | Neph* (n=2) | (+), W | 27,197 | Approximately 2500** | 200–2500 | — | *Reason for nephrectomy unclear; **mUMIs per cell |
| Han et al.28,i | Microwell-seq | F*, A** | N | Fetal (n=4)*; healthy (D) (n=1)**; healthy (Tu) (n=2)** | Whole* (n=4); Neph** (n=3) | (+), W | 22,439*; 22,692** | Approximately 858*,***; approximately 1251**,*** | ? | P*, M* E** | Study constructing the “human cell landscape.” *Fetal kidney; **adult kidney; ***mUMIs per cell; P°, not mentioned |
| Deng et al.22,i | 10x Chromium | A | N | Healthy (Tu) (n=1) | Neph (n=1) | (+), W | 6138 | ? | ? | ? | Study also performed Sanger sequencing on FACS-isolated PTs of two additional kidneys |
Tx, tissue; T°, temperature; glom, glomerular cells; A, adult; N, native kidney; Tu, tumor-free tissue at maximal distance of mass or tumor; neph, biopsy from partial/total nephrectomy; (-), frozen tissue; W, warm dissociation; cryo, cryopreservation; ?, data could not be found in published paper; P, podocytes; M, mesangial cells; eQTL, expression quantitative trait loci; P°, absolute and relative number of podocytes isolated in scRNA-seq and snRNA-seq studies; D, healthy tissue from declined/discarded kidneys that were donated for potential transplantation; (+), fresh tissue; C, cold dissociation; F, fetal; Ch, child; tumor, tissue invaded by tumor; whole, dissection of whole fetal kidney; E, glomerular endothelial cells; DKD, diabetic kidney disease; snap, snap-frozen tissue; mUMIs, mean unique molecular identifiers; T, transplant kidney; Pre, preperfusion or pretransplant core needle biopsy of living donor kidney; S, surveillance kidney transplant biopsy; CNB, core needle biopsy; PTs, proximal tubular cells; −, no glomerular cells.
Tissue from native kidneys or transplant kidneys.
Healthy or pathologic kidney tissue. n=number of patients.
Biopsy technique. n=biopsy samples taken.
Fresh versus frozen tissue and warm versus cold dissociation.
Total number of cells isolated and analyzed after quality control.
Sequencing depth defined as “mean (raw) reads per cell,” “mean transcripts per cell,” or “mean UMIs per cell.”
Mean number of genes per cell.
Isolation of glomerular cells.
scRNA-seq experiment.
snRNA-seq experiment.
In summary, snRNA-seq methods are efficient in identifying glomerular cells, including podocytes, but their efficacy on human core needle biopsy specimens remain to be proven. scRNA-seq pipelines are less robust and are more reliant on the number of included samples, the total number of isolated cells, the dissociation protocol, and—possibly—renal pathophysiology. Because absolute podocyte counts in unsorted renal tissue with both scRNA-seq and snRNA-seq remain very low, future research should focus more on developing sorting techniques for glomeruli or individual glomerular cells to enrich for this rare cell population.
When Leukocytes Are the Focus of Research
Although efficient in identifying glomerular cells, snRNA-seq does not perform as well in isolating leukocytes, especially lymphocytes, when directly compared with scRNA-seq.2,33 Indeed, some snRNA-seq studies could not identify leukocytes,2,36,39 whereas others only identified macrophages9,24,33,34; however, nearly all of these studies used healthy renal tissue or tissue from noninflammatory kidney disease. In contrast, two large snRNA-seq studies that analyzed inflammatory renal pathologies (ischemia-reperfusion injury in mice,37 diabetic kidney disease20) successfully isolated clusters of T lymphocytes, B lymphocytes, dendritic cells, and plasma cells.20,37 It is possible that snRNA-seq underestimates the presence of leukocytes in the sample, but scRNA-seq might also artificially inflate leukocyte rates due to under-representation of other cell types due to incomplete cell dissociation.33
Concluding Answer
When human glomerular cells are the focus of research and nephrectomy tissue is available, an snRNA-seq method is the first choice. Whether or not snRNA-seq can be used on renal core needle biopsy samples remains to be investigated. When using scRNA-seq to study glomerular cells, it is advised to include many samples; analyze a large number of cells (>20,000); and use successful, previously published dissociation techniques. When leukocytes are the focus of research, scRNA-seq might be preferred over snRNA-seq.
Validation Experiments in Single-Cell Transcriptomics
Single-cell transcriptomics experiments are prone to different forms of transcriptional bias. Tissue processing, (cryo)preservation, tissue dissociation, and cell-sorting techniques may all introduce transcriptional artifacts that can obscure true, biologically relevant, gene expression signals. Therefore, it is necessary to perform validation experiments on proposed marker genes and transcriptional signatures from single-cell transcriptomics experiments to confirm that they do indeed result from a true biologic signal and originate from the proposed cell type. Single-cell gene expression profiles can be validated using other well-established techniques that detect gene expression (e.g., bulk RNA-seq, RT-PCR, and RNA in situ hybridization [ISH]), as long as they are not subject to the same transcriptional bias. Alternatively, validation could focus on protein expression levels (e.g., immunohistochemistry [IHC], immunofluorescence, or emerging single-cell proteome technologies). A first in silico validation can be performed by comparing gene expression with previously published bulk RNA-seq datasets14 or protein atlases (e.g., The Human Protein Atlas,59 http://www.proteinatlas.org).6,24 Validation experiments using bulk RNA-seq on unsorted or undissociated tissue will help detect cell sorting– and dissociation-induced transcriptional artifacts and will also indicate whether the dissociation process introduced bias toward cell types that are dissociated more easily.32,33,46,49,58 RNA staining of renal tissue using ISH techniques is an alternative, lower-throughput method to validate the expression of a small set of marker genes.6,17,21 Additionally, this method visually identifies gene expression in cells, and can, therefore, validate whether the proposed marker gene of a cell cluster is indeed visualized in the same cell type on the tissue slide. Finally, routine protein-staining techniques (IHC, immunofluorescence) can be used to visually validate whether gene expression indeed correlates with protein synthesis in the proposed cell type.2,18,20,21,23,24 An emerging field aims to obtain high-multiplex IHC or immunofluorescence readouts by using more innovative approaches. For instance, the CODEX platform (Akoya Biosciences) is already able to detect >40 cell markers in one staining step.60 This method uses validated antibodies that are conjugated to a unique oligonucleotide, which hybridizes to “reporting fluorophores” in a cyclical process to finally image all of the cell markers in the tissue.60
Future Perspectives
The combination of several multiomics measurements (e.g., genomics, epigenomics, proteomics, spatial information) on the same single cells creates an integrated view on cellular identity, cell state, and behavior.61 The different single-cell modalities and their integration in multimodal assays have recently been reviewed.61,62 We briefly highlight three promising, multimodal, single-cell assays that combine gene expression data with (1) spatial information, (2) detection of cell surface proteins, or (3) analysis of chromatin accessibility.
Spatially Resolved Transcriptomics
Many glomerular diseases have a focal distribution and the integration of spatial information with single-cell transcriptomics might, therefore, help to identify cell (sub)populations and cell states in diseased microenvironments. Currently, there are four major spatially resolved transcriptomics techniques63: (1) computational reconstruction of the spatial information of dissociated tissue64–66; (2) single-cell analysis of cells or tissue of interest isolated through laser-capture microdissection; (3) high-throughput in situ transcriptomics (e.g., fluorescence-ISH techniques and in situ sequencing); and (4) spatial barcoding techniques (e.g., Slide-seq67 and spatial transcriptomics,68 which is now commercialized as the Visium Spatial Gene Expression Solution by 10x Genomics). To date, two studies have used a computational method (de novo Spatial Reconstruction of Single-cell Gene Expression, NovoSpaRc)66 on single-cell or single-nucleus datasets of dissociated mouse renal tissue to reconstruct the spatial position of single cells on the basis of transcriptome similarity.34,66 The landmark study that introduced the Slide-seq technique demonstrated the spatial distribution of tubular cells and podocytes in a mouse kidney,67 and another recent study used the Visium platform (10x Genomics) in a murine endotoxemia model to map the location of a proximal tubular cell subtype (S3 type 2) in the mouse kidney.69 More studies using in situ sequencing or spatial barcoding techniques on renal tissue are anticipated in the near future.
Integrated Detection of Cell Surface Proteins
The simultaneous measurement of single-cell gene expression and cell surface proteins is possible with the Cellular Indexing of Transcriptomes and Epitopes by sequencing (CITE-seq) and RNA Expression And Protein sequencing (REAP-seq) assays.70,71 Using DNA-barcoded antibodies against cell surface proteins, both the intracellular mRNA and antibody barcodes are converted into cDNA and sequenced.61 To date, no studies using these techniques on non-tumor renal tissue have been published.
Integrated Analysis of Chromatin Accessibility
The study of the epigenome and its correlation with the transcriptome on a single-cell level creates a deeper understanding of gene expression regulation.61,62 A popular technique to study chromatin accessibility in bulk cells is the assay for transposase-accessible chromatin using sequencing (ATAC-seq), in which DNA from open chromatin is tagged and excised by barcoded transposase Tn5, after which the excised DNA fragments are sequenced.72 Analogous to transcriptomics, single-cell methods for chromatin accessibility emerged with single-cell ATAC-seq, which is compatible with existing single-cell microfluidic devices, including plate- and droplet-based methods.73,74 Alternatively, a combination of barcodes can be used to identify single cells, as used in the sci-ATAC-seq protocol, which uses a single-cell combinatorial indexing (“sci”) strategy.75 One study provided a single-cell atlas of chromatin accessibility from 13 different tissues in adult mice, including renal tissue, using this sci-ATAC-seq technique.76 Integrated multiomics assays that combine single-cell transcriptomics with chromatin accessibility exist in different protocols, including sci-CAR,36 scCAT,77 Paired-seq,78 SHARE-seq,79 and SNARE-seq,80 and have been commercialized in the Chromium Single Cell Multiome ATAC + Gene Expression assay. To date, only one study has used a combined chromatin accessibility and gene expression assay (sci-CAR) on adult mouse renal tissue.36
Conclusions
Single-cell research has gained traction in the field of nephrology and has already proven to be a powerful and useful tool to dissect the (patho)physiology of healthy kidneys and glomerular diseases. This review addressed five key methodologic questions that arise when designing single-cell transcriptomics experiments that focus on glomerular cells. Currently, several challenges remain when core needle biopsy samples are used for scRNA-seq experiments, and no data are yet available on the performance of snRNA-seq protocols. Single-cell transcriptomics experiments on frozen renal tissue are feasible using three possible approaches. To date, no efficient techniques are available that can enrich for glomeruli or podocytes in human renal tissue before dissociation, and the creation of a high-quality single-cell suspension remains the Achilles heel of scRNA-seq, with cold dissociation protocols resulting in less artifacts but also less efficient cell dissociation. Finally, the choice between scRNA-seq or snRNA-seq also depends upon the cells of interest: when glomerular cells are the focus of research, an snRNA-seq method might be preferable, whereas scRNA-seq methods may be superior when profiling immune cells. The delay between biopsy and histopathologic diagnosis currently hampers the widespread use of fresh core needle biopsy specimens and future studies should, therefore, further optimize snRNA-seq and frozen-tissue pipelines and glomerular cell enrichment techniques applicable to human renal core needle biopsy specimens. The field of single-cell transcriptomics is rapidly evolving, and combining these techniques with several other single-cell measurements and multiomics assays will undoubtedly create new insights in the complex pathophysiology of glomerular diseases.
Disclosures
M. Naesens reports serving as an advisor for the European Medicines Agency (EMA), and on the editorial boards of several journals. B. Sprangers reports serving as an ad hoc expert for the EMA. All remaining authors have nothing to disclose.
Funding
D. Deleersnijder holds a KU Leuven Special Research Fund PhD scholarship (DB/19/010/BM. J. Callemeyn is supported by a Fonds Wetenschappelijk Onderzoek (Research Foundation Flanders) fellowship grant, number 1196119N. B. Sprangers is a Fonds Wetenschappelijk Onderzoek senior clinical investigator, via grant 1842919N. M. Naesens is a Fonds Wetenschappelijk Onderzoek senior clinical investigator, via grant 1844019N.
Supplementary Material
Acknowledgments
The authors would like to thank Albert Herelixka for designing the figures included in this publication.
D. Deleersnijder, B. Sprangers, A.H. Van Craenenbroeck, I. Arijs, and J. Callemeyn were responsible for the conceptualization and design of the review; D. Deleersnijder was responsible for data acquisition; D. Deleersnijder, B. Sprangers, A.H. Van Craenenbroeck, J. Callemeyn, M. Naesens, I. Arijs, and D. Lambrechts were responsible for analysis and interpretation of data; D. Deleersnijder, B. Sprangers, A.H. Van Craenenbroeck, J. Callemeyn, M. Naesens, I. Arijs, and D. Lambrechts drafted the work and revised it critically for important intellectual content; D. Deleersnijder, B. Sprangers, A.H. Van Craenenbroeck, J. Callemeyn, M. Naesens, I. Arijs, and D. Lambrechts approved this version of the manuscript to be published; and D. Deleersnijder, B. Sprangers, A.H. Van Craenenbroeck, J. Callemeyn, M. Naesens, I. Arijs, and D. Lambrechts agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2021020157/-/DCSupplemental.
Supplemental Table 1. Overview of all studies reporting on scRNA-seq and/or snRNA-seq on mouse tissue
Supplemental Table 2. Overview of all studies reporting on scRNA-seq and/or snRNA-seq on human tissue
Supplemental Table 3. scRNA-seq studies on methanol-fixed or cryopreserved tissue of mouse kidneys
Supplemental Table 4. scRNA-seq studies on cryopreserved human renal tissue
Supplemental Table 5. scRNA-seq or snRNA-seq studies on unsorted mouse renal tissue
Supplemental Figure 1. Literature search strategy
Supplemental Figure 2. PRISMA 2009 flow diagram of the literature search strategy
References
- 1.Wilson PC, Humphreys BD: Single-cell genomics and gene editing: Implications for nephrology. Nat Rev Nephrol 15: 63–64, 2019 [DOI] [PubMed] [Google Scholar]
- 2.Wu H, Malone AF, Donnelly EL, Kirita Y, Uchimura K, Ramakrishnan SM, et al.: Single-cell transcriptomics of a human kidney allograft biopsy specimen defines a diverse inflammatory response. J Am Soc Nephrol 29: 2069–2080, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lafzi A, Moutinho C, Picelli S, Heyn H: Tutorial: Guidelines for the experimental design of single-cell RNA sequencing studies. Nat Protoc 13: 2742–2757, 2018 [DOI] [PubMed] [Google Scholar]
- 4.Haque A, Engel J, Teichmann SA, Lönnberg T: A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med 9: 75, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilson PC, Humphreys BD: Kidney and organoid single-cell transcriptomics: The end of the beginning. Pediatr Nephrol 35: 191–197, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Menon R, Otto EA, Hoover P, Eddy S, Mariani L, Godfrey B, et al.; Nephrotic Syndrome Study Network (NEPTUNE): Single cell transcriptomics identifies focal segmental glomerulosclerosis remission endothelial biomarker. JCI Insight 5: e133267, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu H, Humphreys BD: The promise of single-cell RNA sequencing for kidney disease investigation. Kidney Int 92: 1334–1342, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kirita Y, Chang-Panesso M, Humphreys BD: Recent insights into kidney injury and repair from transcriptomic analyses. Nephron 143: 162–165, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu H, Kirita Y, Donnelly EL, Humphreys BD: Advantages of single-nucleus over single-cell RNA sequencing of adult kidney: Rare cell types and novel cell states revealed in fibrosis. J Am Soc Nephrol 30: 23–32, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stewart BJ, Ferdinand JR, Clatworthy MR: Using single-cell technologies to map the human immune system - implications for nephrology. Nat Rev Nephrol 16: 112–128, 2020 [DOI] [PubMed] [Google Scholar]
- 11.Fu J, Akat KM, Sun Z, Zhang W, Schlondorff D, Liu Z, et al.: Single-cell RNA profiling of glomerular cells shows dynamic changes in experimental diabetic kidney disease. J Am Soc Nephrol 30: 533–545, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rao DA, Arazi A, Wofsy D, Diamond B: Design and application of single-cell RNA sequencing to study kidney immune cells in lupus nephritis. Nat Rev Nephrol 16: 238–250, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Der E, Ranabothu S, Suryawanshi H, Akat KM, Clancy R, Morozov P, et al.: Single cell RNA sequencing to dissect the molecular heterogeneity in lupus nephritis. JCI Insight 2: 118–138, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arazi A, Rao DA, Berthier CC, Davidson A, Liu Y, Hoover PJ, et al.; Accelerating Medicines Partnership in SLE network: The immune cell landscape in kidneys of patients with lupus nephritis [published correction appears in Nat Immunol 20: 1404, 2019 10.1038/s41590-019-0473-3]. Nat Immunol 20: 902–914, 2019. 31209404 [Google Scholar]
- 15.Der E, Suryawanshi H, Morozov P, Kustagi M, Goilav B, Ranabothu S, et al.; Accelerating Medicines Partnership Rheumatoid Arthritis and Systemic Lupus Erythematosus (AMP RA/SLE) Consortium: Tubular cell and keratinocyte single-cell transcriptomics applied to lupus nephritis reveal type I IFN and fibrosis relevant pathways [published correction appears in Nat Immunol 20: 1556, 2019 10.1038/s41590-019-0529-4]. Nat Immunol 20: 915–927, 2019. 31110316 [Google Scholar]
- 16.Malone AF, Wu H, Fronick C, Fulton R, Gaut JP, Humphreys BD: Harnessing expressed single nucleotide variation and single cell RNA sequencing to define immune cell chimerism in the rejecting kidney transplant. J Am Soc Nephrol 31: 1977–1986, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Menon R, Otto EA, Sealfon R, Nair V, Wong AK, Theesfeld CL, et al.: SARS-CoV-2 receptor networks in diabetic and COVID-19-associated kidney disease. Kidney Int 98: 1502–1518, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zheng Y, Lu P, Deng Y, Wen L, Wang Y, Ma X, et al.: Single-cell transcriptomics reveal immune mechanisms of the onset and progression of IgA nephropathy. Cell Rep 33: 108525, 2020 [DOI] [PubMed] [Google Scholar]
- 19.Gillies CE, Putler R, Menon R, Otto E, Yasutake K, Nair V, et al.; Nephrotic Syndrome Study Network (NEPTUNE): An eQTL landscape of kidney tissue in human nephrotic syndrome. Am J Hum Genet 103: 232–244, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilson PC, Wu H, Kirita Y, Uchimura K, Ledru N, Rennke HG, et al.: The single-cell transcriptomic landscape of early human diabetic nephropathy. Proc Natl Acad Sci U S A 116: 19619–19625, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuppe C, Ibrahim MM, Kranz J, Zhang X, Ziegler S, Perales-Patón J, et al.: Decoding myofibroblast origins in human kidney fibrosis. Nature 589: 281–286, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deng Z, Wang X, Liu Y, Tian X, Deng S, Sun Y, et al.: Single-cell RNA sequencing confirms IgG transcription and limited diversity of VHDJH rearrangements in proximal tubular epithelial cells. Sci Rep 10: 19657, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Young MD, Mitchell TJ, Vieira Braga FA, Tran MGB, Stewart BJ, Ferdinand JR, et al.: Single-cell transcriptomes from human kidneys reveal the cellular identity of renal tumors. Science 361: 594–599, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lake BB, Chen S, Hoshi M, Plongthongkum N, Salamon D, Knoten A, et al.: A single-nucleus RNA-sequencing pipeline to decipher the molecular anatomy and pathophysiology of human kidneys. Nat Commun 10: 2832, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stewart BJ, Ferdinand JR, Young MD, Mitchell TJ, Loudon KW, Riding AM, et al.: Spatiotemporal immune zonation of the human kidney. Science 365: 1461–1466, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zimmerman KA, Bentley MR, Lever JM, Li Z, Crossman DK, Song CJ, et al.: Single-cell RNA sequencing identifies candidate renal resident macrophage gene expression signatures across species. J Am Soc Nephrol 30: 767–781, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liao J, Yu Z, Chen Y, Bao M, Zou C, Zhang H, et al.: Single-cell RNA sequencing of human kidney. Sci Data 7: 4, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han X, Zhou Z, Fei L, Sun H, Wang R, Chen Y, et al.: Construction of a human cell landscape at single-cell level. Nature 581: 303–309, 2020 [DOI] [PubMed] [Google Scholar]
- 29.Wang W, Penland L, Gokce O, Croote D, Quake SR: High fidelity hypothermic preservation of primary tissues in organ transplant preservative for single cell transcriptome analysis. BMC Genomics 19: 140, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guillaumet-Adkins A, Rodríguez-Esteban G, Mereu E, Mendez-Lago M, Jaitin DA, Villanueva A, et al.: Single-cell transcriptome conservation in cryopreserved cells and tissues. Genome Biol 18: 45, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alles J, Karaiskos N, Praktiknjo SD, Grosswendt S, Wahle P, Ruffault PL, et al.: Cell fixation and preservation for droplet-based single-cell transcriptomics. BMC Biol 15: 44, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karaiskos N, Rahmatollahi M, Boltengagen A, Liu H, Hoehne M, Rinschen M, et al.: A single-cell transcriptome atlas of the mouse glomerulus. J Am Soc Nephrol 29: 2060–2068, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Denisenko E, Guo BB, Jones M, Hou R, de Kock L, Lassmann T, et al.: Systematic assessment of tissue dissociation and storage biases in single-cell and single-nucleus RNA-seq workflows. Genome Biol 21: 130, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hinze C, Karaiskos N, Boltengagen A, Walentin K, Redo K, Himmerkus N, et al.: Kidney single-cell transcriptomes predict spatial corticomedullary gene expression and tissue osmolality gradients. J Am Soc Nephrol 32: 291–306, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wohnhaas CT, Leparc GG, Fernandez-Albert F, Kind D, Gantner F, Viollet C, et al.: DMSO cryopreservation is the method of choice to preserve cells for droplet-based single-cell RNA sequencing. Sci Rep 9: 10699, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cao J, Cusanovich DA, Ramani V, Aghamirzaie D, Pliner HA, Hill AJ, et al.: Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 361: 1380–1385, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kirita Y, Wu H, Uchimura K, Wilson PC, Humphreys BD: Cell profiling of mouse acute kidney injury reveals conserved cellular responses to injury. Proc Natl Acad Sci U S A 117: 15874–15883, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hyndman KA, Speed JS, Mendoza LD, Allan JM, Colson J, Sedaka R, et al.: Fluid-electrolyte homeostasis requires histone deacetylase function. JCI Insight 5: e137792, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sidhom EH, Kim C, Kost-Alimova M, Ting MT, Keller K, Avila-Pacheco J, et al.: Targeting a Braf/Mapk pathway rescues podocyte lipid peroxidation in CoQ-deficiency kidney disease. J Clin Invest 131: e141380, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu Y, Ye Y, Bao W, Yang Q, Wang J, Liu Z, et al.: Genome-wide identification of genes essential for podocyte cytoskeletons based on single-cell RNA sequencing. Kidney Int 92: 1119–1129, 2017 [DOI] [PubMed] [Google Scholar]
- 41.Lu Y, Ye Y, Yang Q, Shi S: Single-cell RNA-sequence analysis of mouse glomerular mesangial cells uncovers mesangial cell essential genes. Kidney Int 92: 504–513, 2017 [DOI] [PubMed] [Google Scholar]
- 42.Chung J-J, Goldstein L, Chen YJ, Lee J, Webster JD, Roose-Girma M, et al.: Single-cell transcriptome profiling of the kidney glomerulus identifies key cell types and reactions to injury. J Am Soc Nephrol 31: 2341–2354, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ge X, Zhang T, Yu X, Muwonge AN, Anandakrishnan N, Wong NJ, et al.: LIM-nebulette reinforces podocyte structural integrity by linking actin and vimentin filaments. J Am Soc Nephrol 31: 2372–2391, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dumas SJ, Meta E, Borri M, Goveia J, Rohlenova K, Conchinha NV, et al.: Single-cell RNA sequencing reveals renal endothelium heterogeneity and metabolic adaptation to water deprivation. J Am Soc Nephrol 31: 118–138, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ransick A, Lindström NO, Liu J, Zhu Q, Guo JJ, Alvarado GF, et al.: Single-cell profiling reveals sex, lineage, and regional diversity in the mouse kidney. Dev Cell 51: 399–413.e7, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen L, Lee JW, Chou CL, Nair AV, Battistone MA, Păunescu TG, et al.: Transcriptomes of major renal collecting duct cell types in mouse identified by single-cell RNA-seq. Proc Natl Acad Sci U S A 114: E9989–E9998, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.do Valle Duraes F, Lafont A, Beibel M, Martin K, Darribat K, Cuttat R, et al.: Immune cell landscaping reveals a protective role for regulatory T cells during kidney injury and fibrosis. JCI Insight 5: e130651, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kramann R, Machado F, Wu H, Kusaba T, Hoeft K, Schneider RK, et al.: Parabiosis and single-cell RNA sequencing reveal a limited contribution of monocytes to myofibroblasts in kidney fibrosis. JCI Insight 3: e99561, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barry DM, McMillan EA, Kunar B, Lis R, Zhang T, Lu T, et al.: Molecular determinants of nephron vascular specialization in the kidney. Nat Commun 10: 5705, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kalucka J, de Rooij LPMH, Goveia J, Rohlenova K, Dumas SJ, Meta E, et al.: Single-cell transcriptome atlas of murine endothelial cells. Cell 180: 764–779.e20, 2020 [DOI] [PubMed] [Google Scholar]
- 51.Legouis D, Ricksten SE, Faivre A, Verissimo T, Gariani K, Verney C, et al.: Altered proximal tubular cell glucose metabolism during acute kidney injury is associated with mortality. Nat Metab 2: 732–743, 2020 [DOI] [PubMed] [Google Scholar]
- 52.Adam M, Potter AS, Potter SS: Psychrophilic proteases dramatically reduce single-cell RNA-seq artifacts: A molecular atlas of kidney development. Development 144: 3625–3632, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Habib N, Avraham-Davidi I, Basu A, Burks T, Shekhar K, Hofree M, et al.: Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat Methods 14: 955–958, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grindberg RV, Yee-Greenbaum JL, McConnell MJ, Novotny M, O’Shaughnessy AL, Lambert GM, et al.: RNA-sequencing from single nuclei. Proc Natl Acad Sci U S A 110: 19802–19807, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Park J, Shrestha R, Qiu C, Kondo A, Huang S, Werth M, et al.: Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 360: 758–763, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rudman-Melnick V, Adam M, Potter A, Chokshi SM, Ma Q, Drake KA, et al.: Single-cell profiling of AKI in a murine model reveals novel transcriptional signatures, profibrotic phenotype, and epithelial-to-stromal crosstalk. J Am Soc Nephrol 31: 2793–2814, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marshall JL, Doughty BR, Subramanian V, Guckelberger P, Wang Q, Chen LM, et al.: HyPR-seq: Single-cell quantification of chosen RNAs via hybridization and sequencing of DNA probes. Proc Natl Acad Sci U S A 117: 33404–33413, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dhillon P, Park J, Hurtado Del Pozo C, Li L, Doke T, Huang S, et al.: The nuclear receptor ESRRA protects from kidney disease by coupling metabolism and differentiation. Cell Metab 33: 379–394.e8, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al.: Proteomics. Tissue-based map of the human proteome. Science 347: 1260419, 2015 [DOI] [PubMed] [Google Scholar]
- 60.Goltsev Y, Samusik N, Kennedy-Darling J, Bhate S, Hale M, Vazquez G, et al.: Deep profiling of mouse splenic architecture with CODEX multiplexed imaging. Cell 174: 968–981.e15, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stuart T, Satija R: Integrative single-cell analysis. Nat Rev Genet 20: 257–272, 2019 [DOI] [PubMed] [Google Scholar]
- 62.Carter B, Zhao K: The epigenetic basis of cellular heterogeneity. Nat Rev Genet 22: 235–250, 202110.1038/s41576-020-00300-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liao J, Lu X, Shao X, Zhu L, Fan X: Uncovering an organ’s molecular architecture at single-cell resolution by spatially resolved transcriptomics. Trends Biotechnol 39: 43–58, 2021 [DOI] [PubMed] [Google Scholar]
- 64.Satija R, Farrell JA, Gennert D, Schier AF, Regev A: Spatial reconstruction of single-cell gene expression data. Nat Biotechnol 33: 495–502, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Achim K, Pettit JB, Saraiva LR, Gavriouchkina D, Larsson T, Arendt D, et al.: High-throughput spatial mapping of single-cell RNA-seq data to tissue of origin. Nat Biotechnol 33: 503–509, 2015 [DOI] [PubMed] [Google Scholar]
- 66.Nitzan M, Karaiskos N, Friedman N, Rajewsky N: Gene expression cartography. Nature 576: 132–137, 2019 [DOI] [PubMed] [Google Scholar]
- 67.Rodriques SG, Stickels RR, Goeva A, Martin CA, Murray E, Vanderburg CR, et al.: Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 363: 1463–1467, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ståhl PL, Salmén F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, et al.: Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353: 78–82, 2016 [DOI] [PubMed] [Google Scholar]
- 69.Janosevic D, Myslinski J, McCarthy TW, Zollman A, Syed F, Xuei X, et al.: The orchestrated cellular and molecular responses of the kidney to endotoxin define a precise sepsis timeline. eLife 10: e62270, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, et al.: Simultaneous epitope and transcriptome measurement in single cells. Nat Methods 14: 865–868, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Peterson VM, Zhang KX, Kumar N, Wong J, Li L, Wilson DC, et al.: Multiplexed quantification of proteins and transcripts in single cells. Nat Biotechnol 35: 936–939, 2017 [DOI] [PubMed] [Google Scholar]
- 72.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ: Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10: 1213–1218, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, et al.: Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523: 486–490, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lareau CA, Duarte FM, Chew JG, Kartha VK, Burkett ZD, Kohlway AS, et al.: Droplet-based combinatorial indexing for massive-scale single-cell chromatin accessibility. Nat Biotechnol 37: 916–924, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cusanovich DA, Daza R, Adey A, Pliner HA, Christiansen L, Gunderson KL, et al. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348: 910–914, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cusanovich DA, Hill AJ, Aghamirzaie D, Daza RM, Pliner HA, Berletch JB, et al.: A single-cell atlas of in vivo mammalian chromatin accessibility. Cell 174: 1309–1324.e18, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu L, Liu C, Quintero A, Wu L, Yuan Y, Wang M, et al.: Deconvolution of single-cell multi-omics layers reveals regulatory heterogeneity. Nat Commun 10: 470, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhu C, Yu M, Huang H, Juric I, Abnousi A, Hu R, et al.: An ultra high-throughput method for single-cell joint analysis of open chromatin and transcriptome. Nat Struct Mol Biol 26: 1063–1070, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ma S, Zhang B, LaFave LM, Earl AS, Chiang Z, Hu Y, et al.: Chromatin potential identified by shared single-cell profiling of RNA and chromatin. Cell 183: 1103–1116.e20, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen S, Lake BB, Zhang K: High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nat Biotechnol 37: 1452–1457, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ding J, Adiconis X, Simmons SK, Kowalczyk MS, Hession CC, Marjanovic ND, et al.: Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nat Biotechnol 38: 737–746, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Picelli S: Single-cell RNA-sequencing: The future of genome biology is now. RNA Biol 14: 637–650, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wu Y, Zhang K: Tools for the analysis of high-dimensional single-cell RNA sequencing data. Nat Rev Nephrol 16: 408–421, 2020 [DOI] [PubMed] [Google Scholar]
- 84.Ramsköld D, Luo S, Wang YC, Li R, Deng Q, Faridani OR, et al.: Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat Biotechnol 30: 777–782, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Picelli S, Björklund ÅK, Faridani OR, Sagasser S, Winberg G, Sandberg R: Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods 10: 1096–1098, 2013 [DOI] [PubMed] [Google Scholar]
- 86.Tan SJ, Phan H, Gerry BM, Kuhn A, Hong LZ, Min Ong Y, et al.: A microfluidic device for preparing next generation DNA sequencing libraries and for automating other laboratory protocols that require one or more column chromatography steps. PLoS One 8: e64084, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hashimshony T, Senderovich N, Avital G, Klochendler A, de Leeuw Y, Anavy L, et al.: CEL-Seq2: Sensitive highly-multiplexed single-cell RNA-seq. Genome Biol 17: 77, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, et al.: Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161: 1202–1214, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, et al.: Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161: 1187–1201, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, et al.: Massively parallel digital transcriptional profiling of single cells. Nat Commun 8: 14049, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Conway BR, O’Sullivan ED, Cairns C, O’Sullivan J, Simpson DJ, Salzano A, et al.: Kidney single-cell atlas reveals myeloid heterogeneity in progression and regression of kidney disease. J Am Soc Nephrol 31: 2833–2854, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.The Tabula Muris Consortium: Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562: 367–372, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kreimann K, Jang MS, Rong S, Greite R, von Vietinghoff S, Schmitt R, et al.: Ischemia reperfusion injury triggers CXCL13 release and B-cell recruitment after allogenic kidney transplantation. Front Immunol 11: 1204, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhao Z, Wu J, Xu H, Zhou C, Han B, Zhu H, et al.: XJB-5-131 inhibited ferroptosis in tubular epithelial cells after ischemia-reperfusion injury. Cell Death Dis 11: 629, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liu Y, Hu J, Liu D, Zhou S, Liao J, Liao G, et al.: Single-cell analysis reveals immune landscape in kidneys of patients with chronic transplant rejection. Theranostics 10: 8851–8862, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dangi A, Natesh NR, Husain I, Ji Z, Barisoni L, Kwun J, et al.: Single cell transcriptomics of mouse kidney transplants reveals a myeloid cell pathway for transplant rejection. JCI Insight 5: e141321, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Omori S, Wang TW, Johmura Y, Kanai T, Nakano Y, Kido T, et al.: Generation of a p16 reporter mouse and its use to characterize and target p16high cells in vivo. Cell Metab 32: 814–828.e6, 2020 [DOI] [PubMed] [Google Scholar]
- 98.Ni P, Clinkenbeard EL, Noonan ML, Richardville JM, McClintick J, Hato T, et al.: Targeting fibroblast growth factor 23-responsive pathways uncovers controlling genes in kidney mineral metabolism. Kidney Int 99: 598–608, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.See P, Lum J, Chen J, Ginhoux F: A single-cell sequencing guide for immunologists. Front Immunol 9: 2425, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hedlund E, Deng Q: Single-cell RNA sequencing: Technical advancements and biological applications. Mol Aspects Med 59: 36–46, 2018 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.