

Significance Statement
Kidney disease severity is partly determined by modifier genes. These genes can be important therapeutic targets but are difficult to identify in patient populations. Our study demonstrates a novel mouse genetic approach using Diversity Outbred mice to identify modifier genes for X-linked Alport Syndrome. We identify several candidate modifier genes and validate the candidacy of Fmn1. We show that a decrease in Fmn1 expression in Col4a5 knockout mice leads to a decrease in albuminuria and fewer podocyte protrusions in the glomerular basement membrane. Our approach can be easily adapted to identify modifier genes for other forms of kidney disease.
Keywords: albuminuria, Alport-s syndrome, gene expression, genetics and development
Abstract
Background
Mutations in COL4A5 are responsible for 80% of cases of X-linked Alport Syndrome (XLAS). Although genes that cause AS are well characterized, people with AS who have similar genetic mutations present with a wide variation in the extent of kidney impairment and age of onset, suggesting the activities of modifier genes.
Methods
We created a cohort of genetically diverse XLAS male and female mice using the Diversity Outbred mouse resource and measured albuminuria, GFR, and gene expression. Using a quantitative trait locus approach, we mapped modifier genes that can best explain the underlying phenotypic variation measured in our diverse population.
Results
Genetic analysis identified several loci associated with the variation in albuminuria and GFR, including a locus on the X chromosome associated with X inactivation and a locus on chromosome 2 containing Fmn1. Subsequent analysis of genetically reduced Fmn1 expression in Col4a5 knockout mice showed a decrease in albuminuria, podocyte effacement, and podocyte protrusions in the glomerular basement membrane, which support the candidacy of Fmn1 as a modifier gene for AS.
Conclusion
With this novel approach, we emulated the variability in the severity of kidney phenotypes found in human patients with Alport Syndrome through albuminuria and GFR measurements. This approach can identify modifier genes in kidney disease that can be used as novel therapeutic targets.
Alport Syndrome (AS) is a hereditary disease affecting one in 5000 births in the United States.1 Children and young adults with AS suffer from hearing loss, ocular abnormalities, and most notably, kidney disease.2,3 Patients with kidney disease are diagnosed with glomerular dysfunction characterized by hematuria and proteinuria. Its progression inevitably leads to ESKD, whereby patients require dialysis or transplantation.3–5 It is well known that ESKD in AS is caused by genetic mutations in the α3-, α4-, and α5-chains of type IV collagen, which are encoded by COL4A3, COL4A4, and COL4A5.2 Within the kidney, the α3-, α4-, and α5-isoforms of type IV collagen are found predominantly in the glomerular basement membrane (GBM). A dysfunction in any one of these α-chains causes inadequate assembly of the α3,4,5-trimer into the GBM. As a consequence, the GBM is susceptible to splitting, which creates the characteristic basket weave appearance with associated podocyte foot process (FP) effacement.2 COL4A5 is the only gene in this trimer that is encoded on the X chromosome and is responsible for 80% of AS diagnoses.6 Men who are hemizygous for the COL4A5 mutation have an earlier onset and increased severity of the disease, with 50% of patients requiring dialysis or kidney transplants due to ESKD by the age of 25 and almost 100% by the age of 60.7 Women who are heterozygous for the COL4A5 mutation have relatively later onset, with 12% developing ESKD by the age of 40 and 40% by the age of 80 years.6,8
It is generally acknowledged that there is a wide variability of disease severity in patients with similar AS genetic mutations and that this variability is, in part, due to the presence of underlying genetic mechanisms that modify disease progression.9,10 These modifier genes would act independently of the disease-causing mutation and have the ability to affect the expressivity of phenotypes to either extreme.11 So far, there has only been one genome-wide study on the genetic modifiers of AS. Andrews et al.9 observed variations in phenotype between incipient congenic 129X1/SvJ and C57BL/6J Col4a3 null mice, indicating the presence of modifier genes. Quantitative trait loci (QTL) analysis using “age of ESKD development” as a phenotype was performed on this cohort, and two QTL on chromosomes (Chrs) 9 and 16 were found. However, because of low mapping resolution in the F2 intercross, attributed to the limited numbers of recombination sites, candidate genes were not identified.9
In this study, we aimed to demonstrate an approach to effectively identify modifier genes of X-linked Alport Syndrome (XLAS) by introducing the Col4a5 mutation from the B6.Cg-Col4a5tm1Yseg/J, developed by Rheault et al.,12 into the diverse genetic background of the Diversity Outbred (DO) mice to model XLAS in a genetically heterogeneous environment that recapitulates the disease in human populations. This Col4a5 mutation is a transversion, 213G→T, converting codon 5 from glycine to a stop codon and is a known mutation described in a 15-year-old boy who exhibited full renal, ocular, and auditory manifestations of AS.12 In the C57BL/6J genetic background, the median life span in hemizygous males is 31 weeks and in heterozygous females, 53 weeks in our mouse facility (data not shown). Each individual DO mouse is genetically unique, derived from a randomized mating of eight founder strains consisting of five classic inbred models (A/J, C57BL/6J, 129S1/SvImJ, NOD/ShiLtJ, and NZO/HlLtJ) and three wild-derived models (CAST/EiJ, PWK/PhJ, and WSB/EiJ) capturing 45 million segregation single-nucleotide polymorphisms (SNPs) or 89% of the variation in the mouse genome. The DO mouse resource gives us the power to perform high-resolution QTL mapping of quantitative renal phenotypes to identify modifier genes in XLAS.13 The identification of modifier genes will provide new avenues for understanding the pathobiology of XLAS and for targeted therapeutic design.
Methods
Generation and Phenotyping of the F1 DO-XLAS Mouse Cohort
To generate the F1 DO-XLAS model, 100 female B6.Cg-Col4a5tm1Yseg/J (JAX #006183) mice, heterozygous for the Col4a5 mutation, were crossed with 100 male J:DO mice (JAX #009376). From each mating, one F1 female, heterozygous for the Col4a5 mutation, and one F1 male, hemizygous for the Col4a5 mutation, were used for a total cohort of 200 mice. Urine was collected at 6, 10, and 15 weeks of age, and urinary albumin and creatinine concentrations were determined using a Synchron CX5 Chemistry Analyzer (Beckman Coulter). GFR was measured at 14 weeks of age using an established protocol measuring filtration of retro-orbitally injected FITC-inulin.14 We developed a tool to automate the calculation of GFR, which can be found at https://korstanjelab.jax.org/KorstanjeLab/GFRcalc/. Animals were euthanized at 15 weeks of age after the last urine collection. The right kidney was collected, and the renal capsule, containing perinephric adipose tissue, was removed before flash freezing in liquid nitrogen. Frozen kidneys were homogenized using a ceramic mortar and pestle on dry ice and separated into three aliquots for downstream processing. The left kidney was collected in 10% neutral buffer formalin, paraffin embedded, and stained using periodic acid–Schiff. All animal experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (National Research Council) and were approved by The Jackson Laboratory’s Animal Care and Use Committee.
Col4a5 Point Mutation Genotyping
Genomic DNA from all F1 DO-XLAS mice was isolated from tail tip clippings using a nonphenol-chloroform–based DNA isolation protocol, and the Col4a5 genotype was determined using PCR with forward primer 5′-GCA-TAA-CCG-GGA-CAC-TCA-CT-3′ and reverse primer 5′-GAG-GAC-TTA-CCG-CAG-CCT-CT-3′, followed by Sanger sequencing.
Whole-Genome Diplotype Probability Construction Using Giga Mouse Universal Genotyping Array
All 200 F1 DO-XLAS mice were fully genotyped for 143,259 SNPs using the Giga Mouse Universal Genotyping Array (GigaMUGA) built on an Illumina Infinium platform (GeneSeek; Neogen Genomics).15 Founder haplotype mosaics were reconstructed using a hidden Markov model of array intensity data generated from the Illumina’s BeadStudio algorithm. To ensure the quality of genotype construction, 182 samples with call rates >90% were kept.
Library Construction and RNA Sequencing
RNA from an aliquot of the homogenized whole kidney was extracted using the miRNeasy Mini Kit (Qiagen) according to the manufacturer’s protocol. Sample concentration and quality were assessed using a Nanodrop 2000 spectrophotometer (Thermo Scientific) and RNA 6000 Nano LabChip assay (Agilent Technologies), respectively. RNA quality criteria for library construction were RIN of ≥8.0 and a 260:280 ratio of ≥1.7. Poly(A) RNA sequencing (RNA-seq) libraries were constructed using the TruSeq RNA Library Prep Kit v2 (Illumina), including the addition of unique bar-code sequences to multiplex sequencing, and were quantified using quantitative PCR (Kapa Biosystems). Libraries were pooled and sequenced at 100-bp single end on the HiSeq 2500 (Illumina) using the TruSeq SBS Kit v4 at the New York Genome Center.
Allele-Specific Expression and Whole-Genome Diplotype Reconstruction Using RNA-seq
Expectation-Maximization Algorithm for Allele-Specific Expression (EMASE) and Genotyping by RNA-Seq (GBRS; http://churchill-lab.github.io/gbrs/) software were used to reconstruct the whole-genome diplotype states from the RNA-seq data to increase stringency and confidence reconstruction quality.16 EMASE was used to align multiparent allele-specific expression and gene expression simultaneously from RNA-seq data, and the diploid BAM files were used as input for GBRS. GBRS was then used to quantify multiway allele specificity considering DO generation, which quantified expression contribution from each haplotype in a diplotype. The quantified multiway gene transcript per million count was used to reconstruct genome probabilities along with an established reference transcriptome probability file that corresponds to the samples DO with generation and sex. Ten samples that did not have sufficient GigaMUGA genotype call rates were replaced with high-quality RNA-seq reconstructions. As a result, we were able to verify high-quality genotype probability for 192 samples for downstream analysis.
QTL and SNP Association Mapping
QTL mapping for an F1 model with DO background requires the use of a mixed linear regression model accounting for kinship using the DOQTL R package.13 DOQTL was used to calculate a QTL model, which accounted for sex as an additive covariate for the GFR phenotype, and a QTL model, which accounted for sex and creatinine as covariates for the albumin phenotype at the three time points. Used in conjunction with QTL mapping, DOQTL was also used to calculate SNP association with the urinary phenotypes. LOD scores (−log10[P value]) of individual SNPs under the QTL peaks of interest were calculated to further increase our resolution to the gene level. To control for inflation and to establish statistical significance from the QTL and SNP association mapping, we performed a permutation test, whereby a samples’ genotype and its corresponding phenotype were randomized and P value was calculated 1000 times. From each permutation, we identified the maximum LOD score from this randomized dataset and defined the significance threshold to be at the 95th percentile of the maximum LOD scores.
Characterization of Fmn1 Deletion
FVB.129S6-Fmn1tm2Made/J mice were obtained from The Jackson Laboratory’s repository (JAX #008665) and were previously described by Zhou et al.17 These mice were crossed with B6.Cg-Col4a5tm1Yseg/J mice (JAX #006183) to generate an experimental population of male and female mice that all had the Col4a5 mutation and either had two wild-type copies of Fmn1 or one wild-type copy of Fmn1 (heterozygous knockout for the Fmn1 mutant allele). Albuminuria was measured at 6 weeks of age, and kidneys were collected at 6 weeks of age for histology (left kidney) and RNA isolation (right kidney). Fmn1 expression was measured by quantitative PCR. The left kidney was prepared for serial block-face scanning electron microscopy as previously described,18 generating stacks of 500–1100 images per kidney. Distances and protrusion dimensions were measured in Fiji/ImageJ. Distances were measured using a grid method to obtain a minimum of 100 measurements per individual normalized to the length of the GBM. The total numbers of width measurements for each individual were (heterozygotes) 1710: n=182; 1712: n=277; and 1713: n=163 and (wild type) 2486: n=201; 2487: n=168; and 2488: n=100. Foot process (FP) effacement was calculated as the number of FPs per micrometer of GBM for each image used. Total number of FP measurements: heterozygotes: 1710: n=14; 1712: n=16; and 1713: n=12 and wild type: 2486: n=14; 2487: n=15; and 2488: n=10. Total number of protrusion measurements per individual: heterozygotes: 1710: n=86; 1712: n=62; and 1713: n=65 and wild type: 2486: n=130; 2487: n=124; and 2488: n=88. Because each measurement is not independent and we have multiple measurements per individual, we used a linear mixed effect model to determine the effect and effect size of the genotype using the lme4 package in R (version 4.0.2).
Statistical Analyses
In order to determine significant correlations between gene expression and renal phenotypes, we first excluded genes with zero counts across all samples. Pearson correlation tests were then conducted between RNA-seq expression and phenotype values to obtain both the correlation coefficients and P values. The P values were adjusted for multiple testing using Benjamini and Hochberg to control for false discovery rates. Finally, we used a threshold of adjusted P value =0.05 to identify significant correlations in each phenotype comparison. For all other statistical analyses, such as ANOVA, t test, and correlation tests, R (version 4.0.2) was used.
Data Availability
We have created an interactive DO-XLAS database (https://korstanjelab.jax.org/KorstanjeLab/Col4a5xDO/) with the ability for user-driven queries on the basis of our phenotype and RNA-seq data. RNA-seq data were deposited to the Gene Expression Omnibus under accession number GSE146660.
Results
Generation of the DO-XLAS Cohort
The Col4a5 mutant mouse (B6.Cg-Col4a5tm1Yseg/J) was originally developed in the 129X1/SvJ genetic background and subsequently backcrossed to C57BL/6J for an unknown number of generations.12 We genotyped for 143,259 SNPs using the GigaMUGA array to identify any non-B6 regions. Only the region immediately surrounding the mutation in Col4a5 contains residual 129X1/SvJ and was verified to be between 130,901,329 and 142,139,995 bp on chromosome X (Figure 1A).
Figure 1.
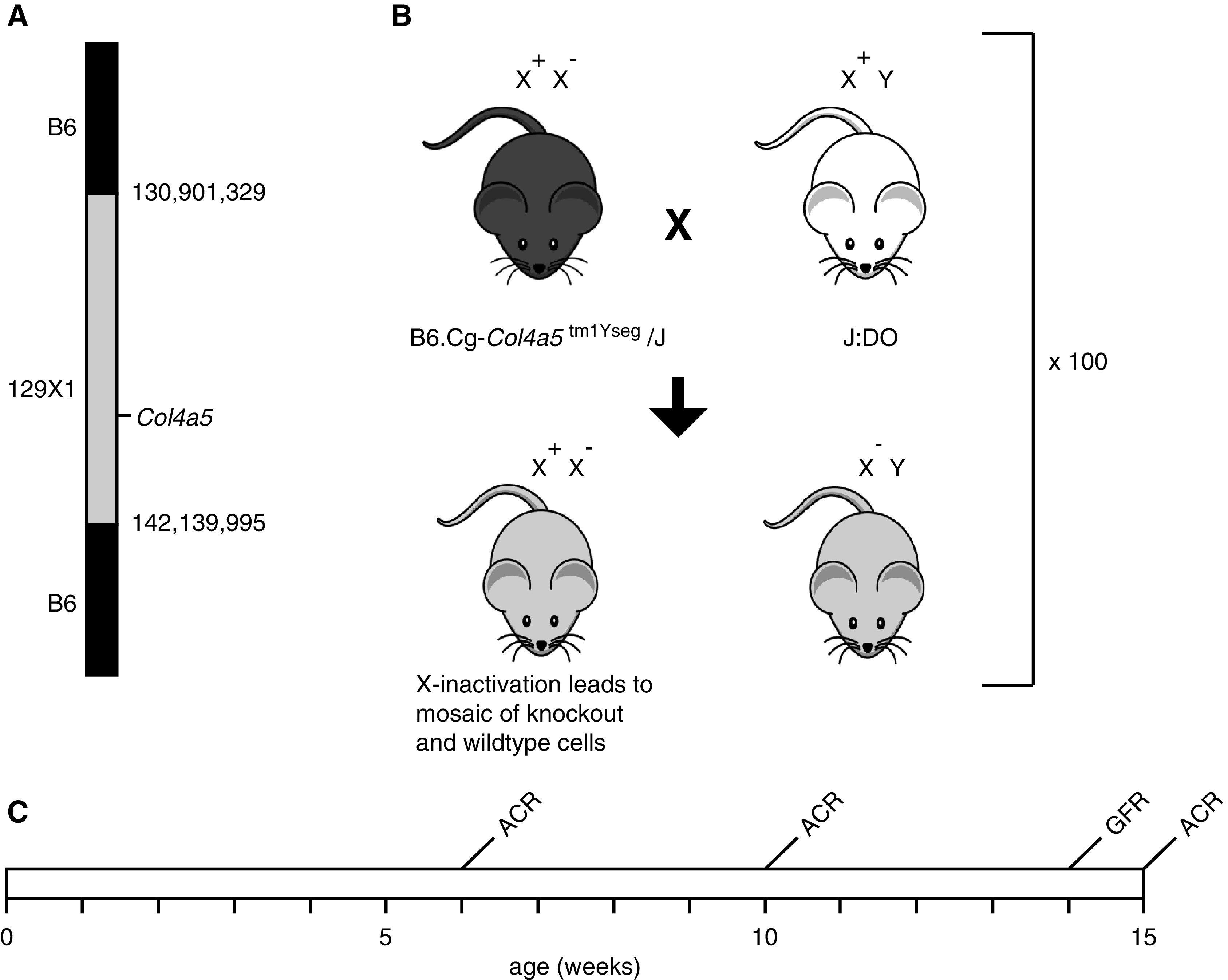
Experimental design of the study. (A) High-density SNP genotyping determined that the B6.Cg-Col4a5tm1Yseg/J JAX stock #006183 strain contains 12 Mb of 129X1/SvJ genomic sequence surrounding the Col4a5 locus. (B) Generation of DO Col4a5 mutant models (DO-XLAS); 100 heterozygous female Col4a5 mice (B6.Cg-Col4a5tm1Yseg/J) were crossed with 100 male J:DO. From each mating, we used one F1 female that was heterozygous for the Col4a5 mutation and one F1 male that was hemizygous for the Col4a5 mutation for a total of 200 mice in the experimental cohort. (C) Experimental timeline with GFR measured at 14 weeks and ACR measured at 6, 10, and 15 weeks. Tissues were collected at 15 weeks of age.
Females heterozygous for the Col4a5 mutation that leads to XLAS were crossed with male DO mice, and only the offspring with the mutation were selected to generate a cohort of 200 DO-XLAS mice (100 heterozygous females and 100 hemizygous males) for the Col4a5 mutation (Figure 1B).
The whole-genome sequence of each animal in the DO-XLAS population was reconstructed using a combination of data from GigaMUGA and RNA-seq. There are eight possible diplotype states in the population. As the DO parental line contributes eight possible haplotypes (A/J, C57BL/6J, 129S1/SvImJ, NOD/ShiLtJ, NZO/HlLtJ, CAST/EiJ, PWK/PhJ, and WSB/EiJ) and the Col4a5 mutant parental line only C57BL/6J alleles, the DO-XLAS population at any given locus has the possibility of having either a homozygous state for C57BL/6J or one of seven heterozygous states.
Recapitulating Kidney Phenotype Variation in the DO-XLAS
Spot urine was collected at 6, 10, and 15 weeks of age (Figure 1C), and renal damage was assessed by measuring albuminuria, represented by urinary albumin-creatinine ratio (ACR). ACR measured in female DO-XLAS ranged from an average of 417±549 mg/g to 1585±1204 and 2349±1406 mg/g in 6, 10, and 15 weeks of age, respectively. ACR in males ranged from an average of 1042±1343 mg/g to 3786±2381 and 5132±2609 mg/g at 6, 10, and 15 weeks of age, respectively. ANOVA tests reveal significant differences between ACR at all three time points for both females and males (P value =0.01) (Figure 2A). Renal function was assessed by measuring the GFR at 14 weeks of age. The mean GFR was 287±99 μl/min in females and 223±206 μl/min in males, significantly higher in females than males (Welch t test, P value =0.03) (Figure 2B). The males show a larger variation in GFR compared with females, as indicated by interquartile ranges on the box plot and the violin plot. A statistically significant negative correlation between ACR at 15 weeks and GFR at 14 weeks was observed in males (R2=0.41, P value =0.01) but not in females (R2=0.046, P value =0.10). Histologic analysis of kidneys at the extremes of the 15-week ACR distribution in the males shows mostly normal histology in animals with the lowest ACR and mesangial matrix expansion, protein casts, and immune cell infiltration in the animals with the highest ACR (Supplemental Figure 1). Together, these measurements are reminiscent of renal phenotypes seen in human patients with AS, as the DO-XLAS mice model shows a progressive worsening of kidney function with age and kidney function in males comparatively worse than females due to lack of a wild-type copy of Col4a5.
Figure 2.
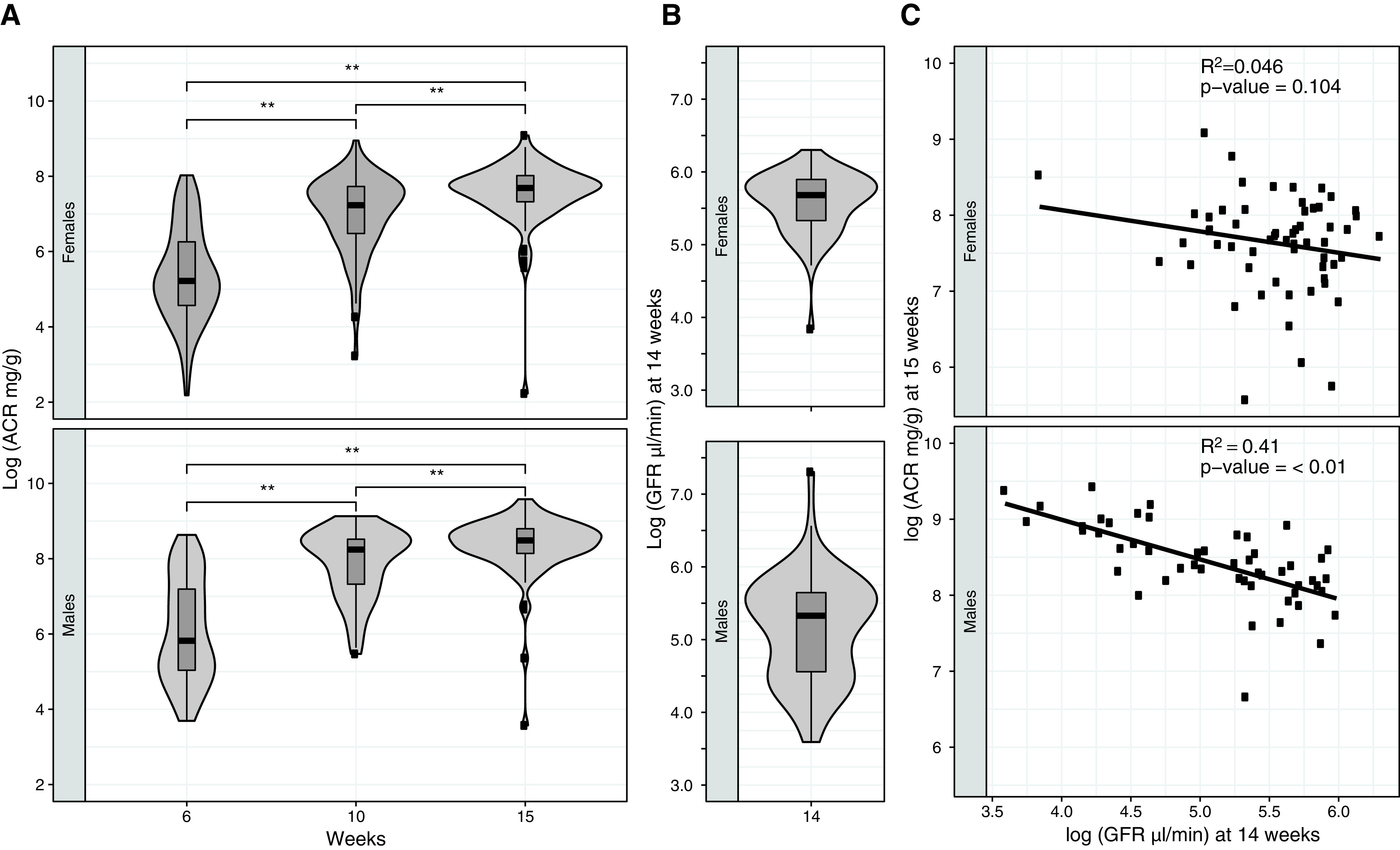
Renal phenotypes in the DO-XLAS cohort. (A) Distribution of ACR at 6, 10, and 15 weeks and (B) GFR at 14 weeks of age. Correlation between ACR at 15 weeks and GFR at 14 weeks in males shows a significant negative relationship; however, the relationship was not significant in females. **P=0.01.
Effect of Skewed X Inactivation on ACR
Although it is commonly assumed that X inactivation is randomly determined with both X chromosomes having an equal chance to be inactivated, there is a locus (Xce) for which different alleles can shift the odds for inactivation. Among the founder strains, there are four different alleles (Xcea in 129S1/SvImJ and A/J; Xceb in C57BL/6J, NZO/HlLtJ, NOD/ShiLtJ, and WSB/EiJ; Xcec in CAST/EiJ; and Xcee in PWK/PhJ) that are segregating in our DO-XAS population. The order in strength of resistance to X inactivation is Xcea < Xcee < Xceb < Xcec.19 Because the Col4a5 mutation in our cohort is contributed by C57BL6/J (Xceb), we expect DO-XLAS females that have either Xcea or Xcee on the other X chromosome to have relatively more cells in which the mutant allele is expressed, and we expect DO-XLAS animals that have Xcec on the other X chromosome to have fewer cells in which the mutant allele is expressed. When we analyzed the albuminuria data of DO-XAS females by the Xce alleles, we observe that animals with the Col4a5 mutation carrying the Xceb allele did not differ in albuminuria levels compared with Xcea and Xcee. Interestingly, females with the Xcec allele had significantly lower albuminuria compared with those with the Col4a5 mutation carrying Xceb allele at 6 weeks (P=0.02) and 10 weeks (P=0.005) of age; however, no difference was seen at 15 weeks of age (Figure 3). This indicates that X inactivation in females may play a role in modulating albuminuria at the early stages of disease onset, but at later stages, factors other than X inactivation play a more important role in the differences in albuminuria between female mice. For GFR, we see the opposite pattern (Supplemental Figure 2), where females with the Xcec allele had significantly higher GFR compared with those with the Col4a5 mutation carrying Xceb allele (P=0.02).
Figure 3.

DO-XLAS females have one X chromosome containing an Xceb allele and one X chromosome containing an Xcea, Xceb, Xcec, or Xcee allele. Mice with an Xcec allele have lower ACR at 6 and 10 weeks of age. *P=0.05; **P=0.01.
Coexpression of Genes with ACR and GFR
In addition to ACR and GFR, we measured renal gene expression in each animal at 15 weeks of age by RNA-seq. Correlation analysis of each gene with ACR at 15 weeks and GFR at 14 weeks shows a significant correlation of 12,121 genes in the males and 307 genes in the females, with an overlap of 302 genes. As expected, the correlations with ACR are opposite from the correlations with GFR (Supplemental Figure 3A). We assume that the high number of genes in the males is caused by the severity of the disease and that, because of slower disease progression, the genes identified in the females (and that overlap with the genes in the males) are associated with earlier processes. We used this set to explore pathway enrichment using Ingenuity Pathway Analysis. The canonical pathway with the highest score [log(P value)=10.99] is leukocyte extravasation signaling (Supplemental Figure 3B), with 19 genes in this pathway being strongly positively correlated with ACR.
Identifying Modifier Genes Mediating GFR and ACR in the DO-XLAS
A QTL analysis was performed for GFR measured at 14 weeks of age to identify loci that are associated with the variation in GFR. The analysis revealed a significant locus on Chr 19 spanning a narrow ∼1-Mb Bayesian estimated interval from 26.99 to 28.04 Mb (5% significance threshold achieved from 1000 permutations) (Figure 4), which contains seven annotated genes: Vldlr, Kcnv2, Pum3, Gm24782, Rfx3, Gm8412, and Gm20407 (Supplemental Figure 4). QTL analyses were performed for all three ACR time points 6, 10, and 15 weeks of age to identify loci associated with variation in ACR (Figure 5). Six-week ACR shows suggestive QTL peaks on Chr 2 and Chr 11 with LOD scores at 6.5 and 7.13, respectively (the 5% threshold for significance is 7.31) (Figure 5A). Ten-week ACR shows suggestive QTL peaks on Chr 2 and Chr 13 with LOD scores at 6.30 and 6.62, respectively (the 5% threshold for significance is 8.52) (Figure 5B). A suggestive peak for the 15-week ACR phenotype was observed on Chr 15 with an LOD score of 10.37 (the 5% threshold for significance is 16.73) (Figure 5C). Interestingly, within the Bayesian intervals of the QTL, we identify genes such as Dgke (Chr 11), associated with the atypical hemolytic uremic syndrome,20 and Pik3r1 (Chr 13), which is shown to play an important role in podocyte ER stress during diabetic nephropathy.21 We observed a recurrent QTL (at 6 and 10 weeks) on Chr 2 between 109.12 and 113.67 Mb (Supplemental Figure 5).
Figure 4.
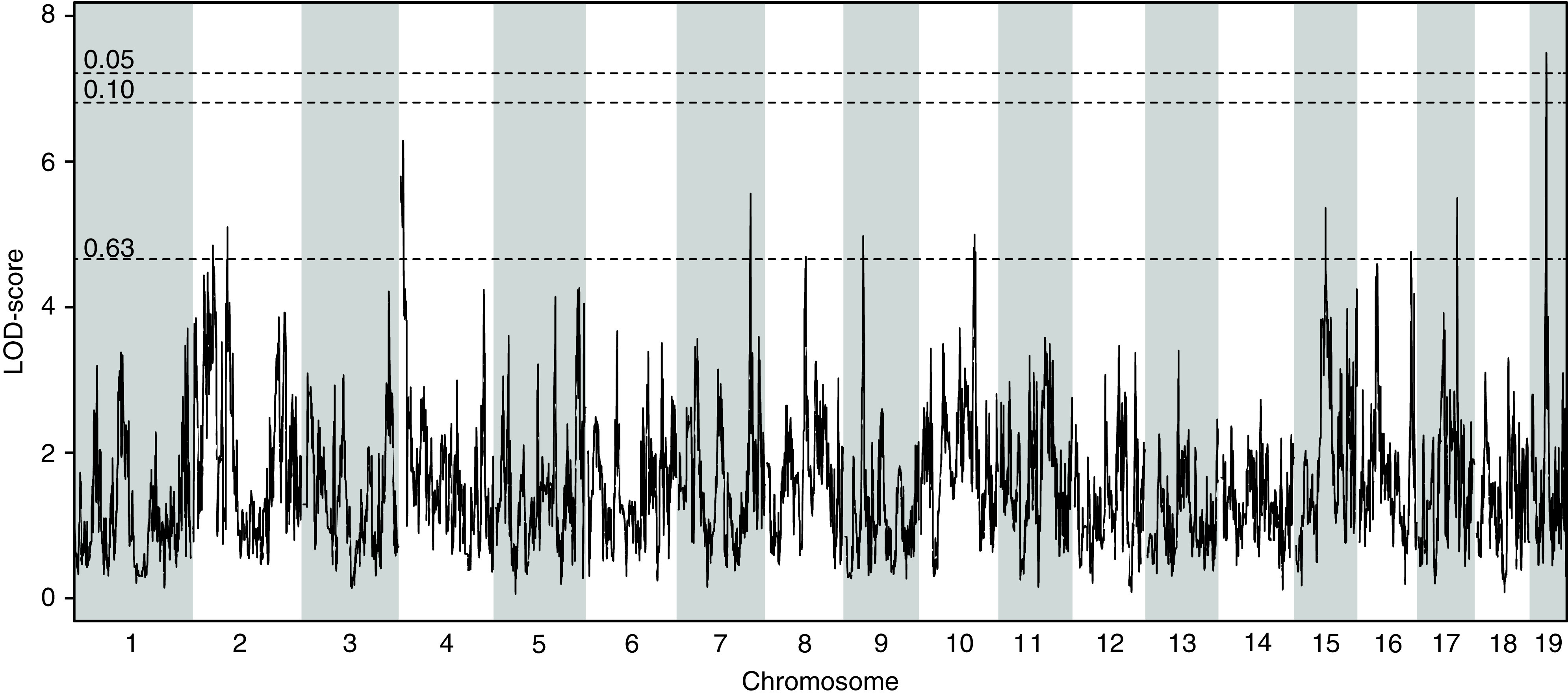
QTL analysis on the DO-XLAS cohort shows a significant association (α<0.05) for GFR with a locus on Chr 19. LOD, logarithm of the odds.
Figure 5.
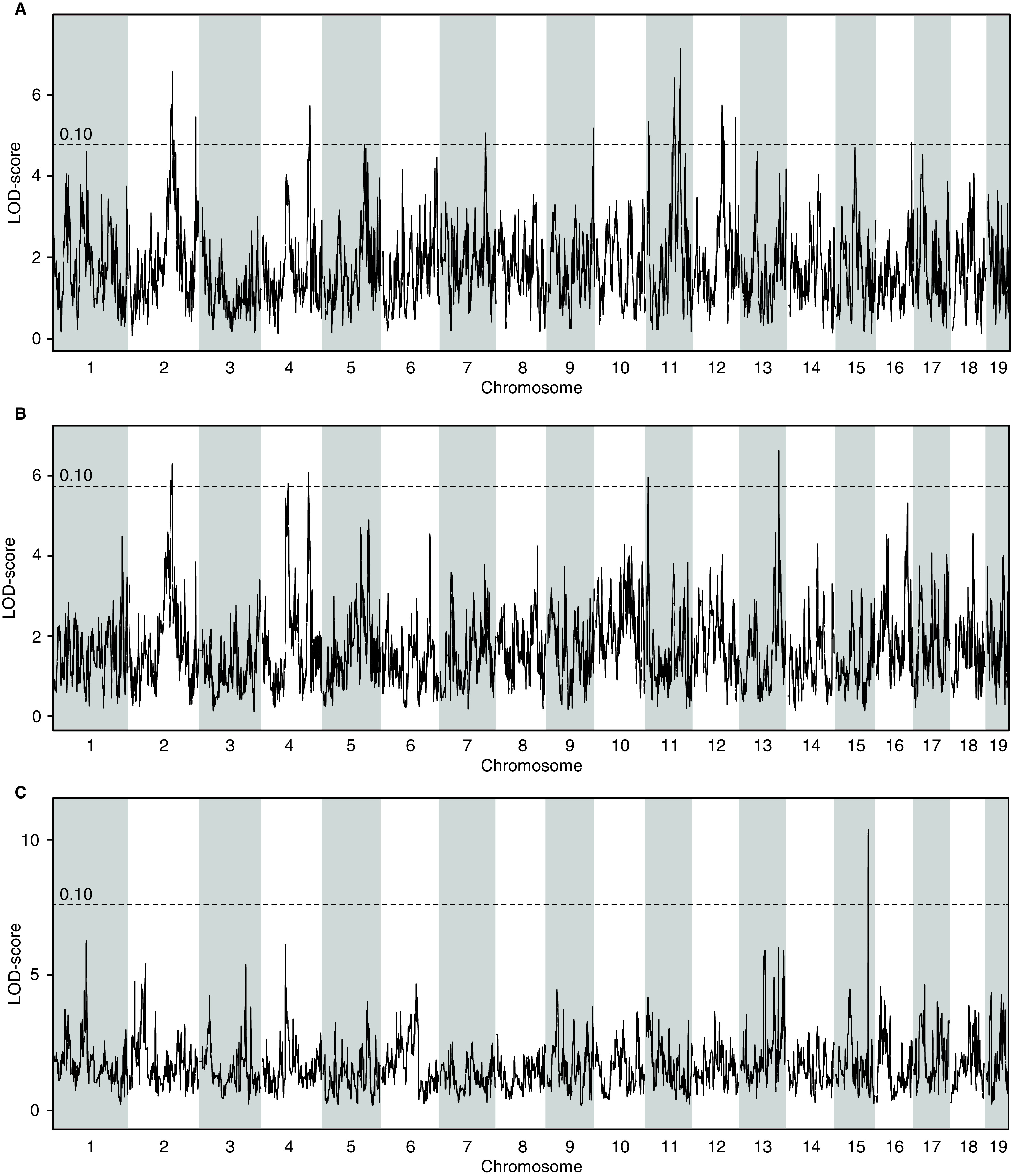
QTL analysis on the DO-XLAS cohort shows suggestive associations for ACR at (A) 6, (B) 10, and (C) 15 weeks of age. LOD, logarithm of the odds.
Fmn1 as a Candidate Modifier Gene for AS
A major advantage of using a cohort of DO mice is that we are able to determine the effects of the individual eight founder alleles at any given locus in the genome. For the Chr 2 locus, we see that animals that inherited the PWK allele have a lower ACR compared with animals that inherited an allele from any of the other founder strains, suggesting that ACR is determined by a unique variant in the PWK strain (Figure 6A). Using the complete genome sequences of all founder strains, we can identify any variant that is different in the PWK genome compared with the genomes of the other DO founder strains. Only rs252099932, a single-nucleotide variant in the Fmn1 gene, is consistent with this. FMN1 has recently been associated with albuminuria in a Japanese cohort22 and is involved in actin filament and microtubule cytoskeleton formation.23
Figure 6.
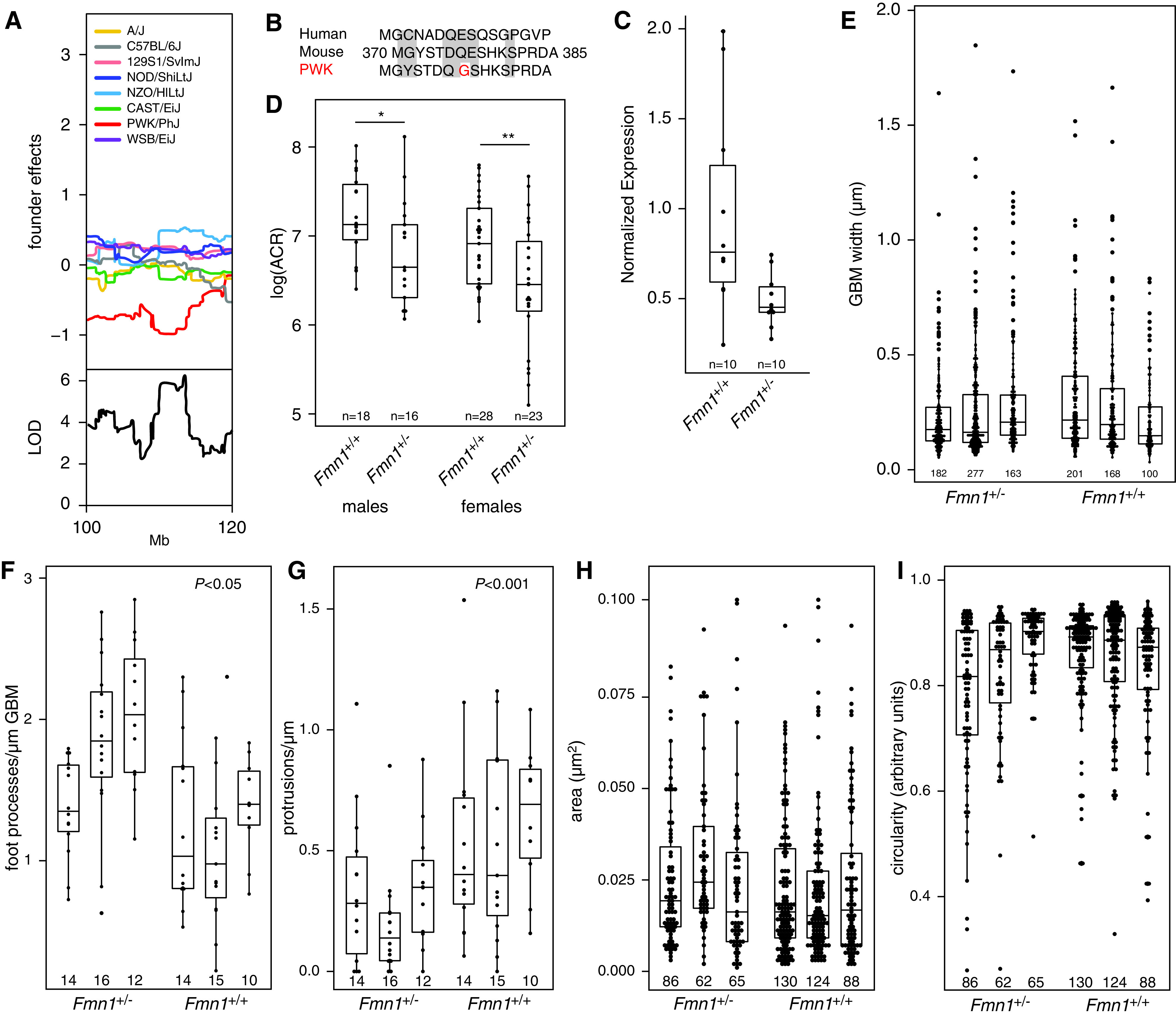
Identification and characterization of Fmn1 as a candidate gene. (A) Allele effect plot of the Chr 2 locus shows that animals that inherited the allele from the PWK founder strain have lower ACR. (B) Analysis of the founder genomes in the 95% confidence interval of the Chr 2 locus shows that the PWK genome has a genetic variant in the coding region of Fmn1 that leads to an amino acid change unique to PWK compared with the seven other founder strains of the DO-XLAS cohort and humans. (C) Quantitative PCR shows that Fmn1+/− mice have an approximately 50% lower Fmn1 expression compared with wild-type mice. (D) Col4a5 mutant mice with only one functional Fmn1 allele (Fmn1+/−) have lower ACR in both males and females compared with Col4a5 mutant mice with two functional Fmn1 alleles (Fmn1+/+). Quantification in male mice using transmission electron microscopy does not show differences in (E) GBM width, (H) protrusion area, or (I) protrusion circularity, but there are significant differences in (F) the number of FPs per micrometer of GBM and (G) the number of protrusions per micrometer of GBM. The number under each bar indicates the number of measurements in each animal. LOD, logarithm of the odds. *P=0.05; **P=0.01.
The nucleotide variant leads to an amino acid difference in the PWK FMN1 protein (a glycine at position 377) compared with FMN1 in the other strains, and the human protein (a glutamic acid at position 377) is consistent with this (Figure 6B). SIFT analysis shows that the E377G amino acid substitution is predicted to be deleterious in PWK, suggesting that the PWK allele might be dysfunctional and/or lowers Fmn1 expression, leading to lower ACR.
In order to test this hypothesis, we crossed B6.Cg-Col4a5tm1Yseg/J females with the already available FVB.129S6-Fmn1tm2Made/J, in which exon 9 of Fmn1 has been disrupted and homozygous mutant mice no longer express FMN1.17 The offspring has a hybrid B6/FVB genetic background, and with FVB being a more susceptible strain for renal damage, a more severe phenotype is expected.24 We tested Col4a5 heterozygous females with either one or two copies of Fmn1 and Col4a5 hemizygous males with either one or two copies of Fmn1. Quantitative PCR on RNA isolated from the kidneys showed that the animals with only one copy (Fmn1+/−) had a two-fold lower expression of Fmn1 (Figure 6C). As hypothesized, both males and females with one copy of Fmn1 have lower ACR at 6 weeks of age (Figure 6D).
Because Fmn1 encodes formin 1 and is thought to play a role in the polymerization of linear actin cables, we speculated that it might be involved in the GBM protrusions that have recently been identified in both patients with AS and in the Col4a3 knockout mouse model for AS.18 In order to test this, we used serial block-face scanning electron microscopy on the kidneys of Fmn1+/+ and Fmn1+/− animals. Electron microscopy showed a high degree of variability in the renal morphology regardless of the Fmn1 genotype, as expected due to deteriorating GBM caused by the Col4a5 mutation. The GBM of the Fmn1+/+ animals presented with regions of irregularly thickened, split basement membrane (also known as a basket weave appearance). These regions were consistently surrounded by extensively effaced podocyte FPs. The Fmn1+/− animals showed either regions of thin, darkly stained, uniform GBM or segments of extensive glomerulosclerosis; the two extremes were often seen within the same image, adjacent to capillary loops with normal GBM. The thin, uniform GBM of the Fmn1+/− mice was found with remarkably normal-looking podocyte FPs. The split basement membrane that was seen in the Fmn1+/+ glomeruli was occasionally seen in the Fmn1+/− kidneys but with a more regular appearance. Small, rounded, lucent regions were found in the basket weave GBM of the Fmn1+/+ mice; when traced through the image stack, these lucent regions connected back to podocyte FPs, indicating protrusion of the FPs into the damaged basement membrane. To quantify any differences between Fmn1+/+ glomeruli and Fmn1+/− glomeruli, we measured the GBM width, the number of FPs per micrometer of GBM, the number of protrusions per micrometer of GBM, the size of the protrusions (micrometers squared), and the circularity of the protrusions (score zero to one) in three males from each genotype. In order to determine whether there is a significant effect between one and two alleles of Fmn1 in Col4a5 mutant animals, we analyzed the data using a linear mixed effect model, which takes into account that not every measurement is independent (two measurements from the same animal have a different relationship than two measurements from two different animals). First, we ran a linear model with the animal identification as a random effect and assuming that genotype does not play a role (phenotype ∼(1|animal)+ error). Then, we ran the same model but adding the genotype (phenotype ∼ genotype +(1|animal)+ error). We compared the two models using an ANOVA to see if there is a significant difference between the models and to determine the effect size of the genotype. For GBM width (Figure 6E), protrusion area (Figure 6H), and protrusion circularity (Figure 6I), we did not find a significant difference. However, we observed a significant increase in the number of FPs (Figure 6F), suggesting less effacement in Fmn1+/− glomeruli (P=0.02, with an effect size of 0.5129±0.1759 FP per micrometer) and a significant decrease in protrusions in Fmn1+/− glomeruli (Figure 6G) (P<0.001, with an effect size of −0.2715±0.0698 protrusions per micrometer). Examples of these protrusions in both Fmn1+/+ glomeruli and Fmn1+/− glomeruli in mice with the Col4a5 mutation are shown in Figure 7. These data provide evidence that Fmn1 is a modifier gene for AS and that lowering Fmn1 expression ameliorates the renal phenotype in AS.
Figure 7.

Transmission electron microscopy of wild-type (Fmn1+/+) and heterozygous knockout (Fmn1+/−) male mice hemizygous for the Col4a5 mutation. Dashed lines denote the boundary of GBM. (A–I) Arrowheads indicate protrusions, which vary in size, with two good examples in B. Arrows indicate longitudinal/oblique sections through protrusions; C and D illustrate protrusions extending from FPs. Protrusions have been defined broadly here as any membrane-bound inclusion that is completely surrounded by GBM. (D, asterisk and F) This includes FPs that have become enlarged/distorted and surrounded by GBM. There is no definitive demarcation between enlarged protrusions and enlarged FPs, and G gives a good example of some of these midrange inclusions. EC, endothelial cell; P/FP, podocyte/foot process; RBC, red blood cell. Scale bars: 250 nm.
Discussion
The identification of modifier genes in AS has been a focus in the AS research community due to the variation in renal phenotype severity despite the knowledge of the causal genes. Previous attempts at mapping modifier genes in mice have been limited due to low genomic resolution and lack of genetic variation in the cohort studied. In this proof of concept study, we generated a genetically diverse cohort of mice with a Col4a5 mutation, which presented renal phenotypes (measured as ACR and GFR) with wide variation. The variations in both genetics and phenotypes of the cohort closely mimic the diversity found in the human population.
We were able to emulate the increased severity of hemizygous XLAS males compared with heterozygous XLAS female mice through both an overall increase in ACR and lower GFR. For both sexes, we saw a gradual increase in ACR between 6, 10, and 15 weeks of age. We also observed a significant negative correlation between 15-week ACR and 14-week GFR measurements in males (Figure 2). We did not see this in females, but we believe that this is due to the slower progression of disease development in the females and expect there would be a correlation at a later time point.
The genetic effect on X inactivation and the presence of a locus on the X chromosome that can affect disease severity in females have been observed in Alport mice previously.25 In heterozygous female mice, on average, 50% of podocytes produce the wild-type COL4A5 protein. Genetic factors that can increase the proportion of cells that express the wild-type COL4A could lead to better disease outcomes. In our study, female mice that inherited an Xce allele from the CAST founder strain, which predicts a higher percentage of cells with inactivation of the mutant Col4a5 allele, had lower albuminuria at a young age (6 and 10 weeks). This brings up the interesting possibility of reactivating the X chromosome that harbors the wild-type COL4A5 in heterozygous women as a therapeutic option, something this is being explored in other X-linked diseases, such as Rett Syndrome.26
We obtained renal gene expression profiles of all animals in the study with the goal of identifying genes that correlate with our renal phenotypes and in this way, identify pathways that are involved in the variation of the disease severity. The difference between the number of genes in males (12,121) and females (307) is most likely due to the different timelines in disease progression where, at the time of tissue collection, the males are in a more advanced stage than the females, affecting many more pathways. We, therefore, focused on the 302 overlapping genes in our pathway analysis. The canonical pathway with the highest score [log(P value)=10.99] is leukocyte extravasation signaling, which is strongly positively correlated with ACR and negatively correlated with GFR. Recruitment of leukocytes to sites of injury is a process controlled by leukocyte interactions with the endothelial layer and the ability of the leukocyte to breach the vascular wall. In human AS and mouse models of AS, it has been shown that disease progression is associated with interstitial inflammatory leukocyte infiltrates. Blocking this process improves the survival of AS mice and is associated with fewer interstitial macrophages, apoptotic tubular epithelial cells, tubular atrophy, interstitial fibrosis, and less globally sclerotic glomeruli.27
Our study was designed as a proof of concept to investigate the effect of modifier genes, and with the relatively small cohort, the chance of identifying loci with a significant association is low. Nevertheless, using QTL analysis on the GFR data, we were able to identify a significant association with a modifier locus on Chr 19 (Figure 4) and narrow it to an approximately 1-Mb Bayesian interval between 26.99 and 28.04 Mb, which contains seven annotated genes: Vldlr, Kcnv2, Pum3, Gm24782, Rfx3, Gm8412, and Gm20407. Vldlr encodes the very low–density lipoprotein receptor, a member of the highly conserved low-density lipoprotein receptor family, which also includes Lrp2. Kcnv2 encodes a potassium voltage-gated channel modifier subfamily V member 2 that is primarily expressed in the retina and associated with cone dystrophy with supernormal rod response. Pum3 encodes a pumilio RNA binding family member 3, which recognizes double-stranded RNA or DNA without sequence specificity. Rfx3 encodes a highly conserved transcription factor, regulatory factor X3, which has been shown to regulate primary cilia formation in Mus musculus, Caenorhabditis elegans, and Drosophila melanogaster. Further studies will be needed to identify and test the genes within this interval.
Our QTL analyses on the ACR data did not result in any statistically significant associations (Figure 5). However, we identified a number of loci that were close to the statistical significance and warrant further investigation. Associations were found within a locus on Chr 11 containing Dgke, a gene associated with the atypical hemolytic uremic syndrome,20 and Chr 13 containing Pik3r1, which is shown to play an important role in podocyte ER stress during diabetic nephropathy.21 Interestingly, both genes encode enzymes that are in the same pathway and regulate the levels of diacylglycerol-containing arachidonic acid, which has been shown to modify slit diaphragm function that can contribute to proteinuria and kidney failure.20 We observed a recurrent QTL (at 6 and 10 weeks) on Chr 2 between 109.12 and 113.67 Mb and using the powerful DO mouse resource, were able to identify Fmn1 as the most likely candidate gene for this association. This gene has recently been associated with albuminuria in a Japanese cohort.22
FMN1 is involved in actin filament and microtubule cytoskeleton formation.23 This is an important process in podocyte FP effacement and possibly, in the formation of the protrusions that have recently been identified in both patients with AS and AS mouse models.18 Because animals in our cohort that inherited a predicted dysfunctional Fmn1 allele had lower albuminuria, we hypothesized that heterozygous Fmn1 knockout mice (with only one functional allele) would have lower albuminuria and fewer protrusions. Comparing Col4a5 mutant mice that had only one functional Fmn1 allele (Fmn1+/−) with Col4a5 mutant mice that had two functional Fmn1 alleles (Fmn1+/+) indeed showed significantly lower albuminuria (Figure 6D), more FPs per micrometer of GBM (Figure 6F) (suggesting less podocyte effacement), and fewer protrusions of the podocyte FPs into the GBM (Figure 6G). Together, our results indicate that Fmn1 is a modifier gene that is able to mediate the severity of XLAS in mice through reduction of ACR and lessening of podocyte FP evasion into the GBM.
A previous study by Andrews et al.9 identified AS modifier genes at Chr 9 and Chr 16, which we did not detect. There are several possible explanations. First is the difference in phenotypes used for the analysis. We use GFR and albuminuria at a specific age, whereas Andrews et al.9 use “age to renal failure.” These different phenotypes can be determined by different loci. Second, the Andrews et al.9 cohort was very small. Initial analysis was performed on only the extremes of the distribution, and then, additional genotyping and permutation testing were specifically done on Chr 9 and Chr 16, so there might not have been genome-wide statistical significance. Third, their analysis was in a cross between B6J and 129X1. Because our DO cohort does not contain the 129X1 (DO contains the 129S1/SvImJ substrain, which is genetically different), the alleles leading to the Chr 9 and Chr 16 QTL might not be present in the DO. Lastly, even if the alleles would be present, they were likely recessive in the Andrews cohort, and because each locus in our DO cohort is either heterozygous or B6 homozygous, we would miss recessive alleles.
In conclusion, this proof of concept study in which we generated a small cohort of genetically diverse mice, all carrying the Col4a5 mutation, showed that this is an effective strategy to identify modifier genes for AS. We found several loci associated with the variation in disease severity, identified several candidate genes that have previously been associated with different forms of kidney disease, and provided evidence for Fmn1 as a modifier gene for AS. The association between our candidate modifier genes with other forms of kidney disease brings up the interesting possibility that they may not be disease specific, and therapeutics developed against these targets could be applicable to a spectrum of diseases. Our approach can easily be customized to identify and study modifier genes for other forms of kidney disease, such as polycystic kidney disease or diabetic nephropathy.
Disclosures
K. Letson is employed by Regeneron Pharmaceuticals. R. Lennon reports consultancy agreements with Travere Therapeutics; scientific advisor or membership with the Kidney Research UK grants panel and the Scientific Advisory Research Network for the Alport Syndrome Foundation; and other interests/relationships via funding from Kidney Research UK and as a trustee for Alport UK and a trustee for Kidneys for Life. R. Korstanje reports membership of the Scientific Advisory Research Network for the Alport Syndrome Foundation. All remaining authors have nothing to disclose.
Funding
This work was supported by an Alport Syndrome Foundation grant (to R. Korstanje) and Wellcome Trust Senior Fellowship award 202860/Z/16/Z (to R. Lennon).
Supplementary Material
Acknowledgments
The authors thank the staff in the EM Core Facility in the Faculty of Biology, Medicine and Health at the University of Manchester for their assistance, in particular David Smith for collecting the serial block-face scanning electron microscopy images, and the Wellcome Trust for equipment grant support to the Electron Microscopy Core Facility. We acknowledge the contribution of Heidi Munger and the Genome Technologies Service at The Jackson Laboratory for expert assistance with RNAseq.
R. Korstanje and Y. Takemon designed the study; H.S. Savage, K. Letson, Y. Takemon, and V. Wright generated the mice; B. Davenport, D.M. Gatti, K. Letson, H.S. Savage, S.M. Sheehan, Y. Takemon, and V. Wright generated the data; D.M. Gatti, R. Korstanje, Y. Takemon, and V. Wright analyzed the data; and R. Korstanje, R. Lennon, and Y. Takemon wrote the manuscript.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2020060777/-/DCSupplemental.
Supplemental Figure 1. Histologic analysis of kidneys at the extremes of the 15-week ACR distribution in the males.
Supplemental Figure 2. Allele effects of the Xce locus in females.
Supplemental Figure 3. Correlation of gene expression with (A) ACR and GFR and (B) pathway enrichment using Ingenuity Pathway Analysis.
Supplemental Figure 4. Bayesian estimated interval on Chr 19 associated with GFR.
Supplemental Figure 5. Bayesian estimated interval on Chr 2 associated with ACR.
References
- 1.Hertz JM, Thomassen M, Storey H, Flinter F: Clinical utility gene card for: Alport syndrome. Eur J Hum Genet 20: 713, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kruegel J, Rubel D, Gross O: Alport syndrome--insights from basic and clinical research. Nat Rev Nephrol 9: 170–178, 2013 [DOI] [PubMed] [Google Scholar]
- 3.Savige J, Gregory M, Gross O, Kashtan C, Ding J, Flinter F: Expert guidelines for the management of Alport syndrome and thin basement membrane nephropathy. J Am Soc Nephrol 24: 364–375, 2013 [DOI] [PubMed] [Google Scholar]
- 4.Kelly YP, Patil A, Wallis L, Murray S, Kant S, Kaballo MA, et al.: Outcomes of kidney transplantation in Alport syndrome compared with other forms of renal disease. Ren Fail 39: 290–293, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cosgrove D, Liu S: Collagen IV diseases: A focus on the glomerular basement membrane in Alport syndrome. Matrix Biol 57-58: 45–54, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rheault MN: Women and Alport syndrome. Pediatr Nephrol 27: 41–46, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kashtan CE, Ding J, Gregory M, Gross O, Heidet L, Knebelmann B, et al.; Alport Syndrome Research Collaborative: Clinical practice recommendations for the treatment of Alport syndrome: A statement of the Alport Syndrome Research Collaborative. Pediatr Nephrol 28: 5–11, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A, et al.: X-linked Alport syndrome: Natural history and genotype-phenotype correlations in girls and women belonging to 195 families: A “European Community Alport Syndrome Concerted Action” study. J Am Soc Nephrol 14: 2603–2610, 2003 [DOI] [PubMed] [Google Scholar]
- 9.Andrews KL, Mudd JL, Li C, Miner JH: Quantitative trait loci influence renal disease progression in a mouse model of Alport syndrome. Am J Pathol 160: 721–730, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Korstanje R, Caputo CR, Doty RA, Cook SA, Bronson RT, Davisson MT, et al.: A mouse Col4a4 mutation causing Alport glomerulosclerosis with abnormal collagen α3α4α5(IV) trimers. Kidney Int 85: 1461–1468, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nadeau JH: Modifier genes in mice and humans. Nat Rev Genet 2: 165–174, 2001 [DOI] [PubMed] [Google Scholar]
- 12.Rheault MN, Kren SM, Thielen BK, Mesa HA, Crosson JT, Thomas W, et al.: Mouse model of X-linked Alport syndrome. J Am Soc Nephrol 15: 1466–1474, 2004 [DOI] [PubMed] [Google Scholar]
- 13.Gatti DM, Svenson KL, Shabalin A, Wu LY, Valdar W, Simecek P, et al.: Quantitative trait locus mapping methods for diversity outbred mice. G3 (Bethesda) 4: 1623–1633, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Backer G, Eddy S, Sheehan SM, Takemon Y, Reznichenko A, Savage HS, et al.: FAR2 is associated with kidney disease in mice and humans. Physiol Genomics 50: 543–552, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morgan AP, Fu CP, Kao CY, Welsh CE, Didion JP, Yadgary L, et al.: The Mouse universal genotyping array: From substrains to subspecies. G3 (Bethesda) 6: 263–279, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raghupathy N, Choi K, Vincent MJ, Beane GL, Sheppard KS, Munger SC, et al.: Hierarchical analysis of RNA-seq reads improves the accuracy of allele-specific expression. Bioinformatics 34: 2177–2184, 201810.1093/bioinformatics/bty078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou F, Leder P, Zuniga A, Dettenhofer M: Formin1 disruption confers oligodactylism and alters Bmp signaling. Hum Mol Genet 18: 2472–2482, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Randles MJ, Collinson S, Starborg T, Mironov A, Krendel M, Königshausen E, et al.: Three-dimensional electron microscopy reveals the evolution of glomerular barrier injury. Sci Rep 6: 35068, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Calaway JD, Lenarcic AB, Didion JP, Wang JR, Searle JB, McMillan L, et al.: Genetic architecture of skewed X inactivation in the laboratory mouse. PLoS Genet 9: e1003853, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lemaire M, Frémeaux-Bacchi V, Schaefer F, Choi M, Tang WH, Le Quintrec M, et al.: Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat Genet 45: 531–536, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Madhusudhan T, Wang H, Dong W, Ghosh S, Bock F, Thangapandi VR, et al.: Defective podocyte insulin signalling through p85-XBP1 promotes ATF6-dependent maladaptive ER-stress response in diabetic nephropathy. Nat Commun 6: 6496, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okuda H, Okamoto K, Abe M, Ishizawa K, Makino S, Tanabe O, et al.: Genome-wide association study identifies new loci for albuminuria in the Japanese population. Clin Exp Nephrol 24: 1–9, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dettenhofer M, Zhou F, Leder P: Formin 1-isoform IV deficient cells exhibit defects in cell spreading and focal adhesion formation. PLoS One 3: e2497, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Randles MJ, Woolf AS, Huang JL, Byron A, Humphries JD, Price KL, et al.: Genetic background is a key determinant of glomerular extracellular matrix composition and organization. J Am Soc Nephrol 26: 3021–3034, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rheault MN, Kren SM, Hartich LA, Wall M, Thomas W, Mesa HA, et al.: X-inactivation modifies disease severity in female carriers of murine X-linked Alport syndrome. Nephrol Dial Transplant 25: 764–769, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carrette LLG, Wang CY, Wei C, Press W, Ma W, Kelleher RJ 3rd, et al.: A mixed modality approach towards Xi reactivation for Rett syndrome and other X-linked disorders. Proc Natl Acad Sci U S A 115: E668–E675, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ninichuk V, Gross O, Reichel C, Khandoga A, Pawar RD, Ciubar R, et al.: Delayed chemokine receptor 1 blockade prolongs survival in collagen 4A3-deficient mice with Alport disease. J Am Soc Nephrol 16: 977–985, 2005 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We have created an interactive DO-XLAS database (https://korstanjelab.jax.org/KorstanjeLab/Col4a5xDO/) with the ability for user-driven queries on the basis of our phenotype and RNA-seq data. RNA-seq data were deposited to the Gene Expression Omnibus under accession number GSE146660.