

Significance Statement
Renal artery stenosis (RAS) engenders stenotic-kidney ischemia, dysfunction, and injury, but whether these are mediated by cellular senescence has not been elucidated. INK-ATTAC transgenic mice, high-resolution imaging, and unbiased single-cell RNA sequencing of murine kidneys demonstrated cellular senescence as an important mechanism of progressive injury to epithelial/stromal cells within poststenotic kidneys. Both p16-specific and broad quercetin/dasatinib interventions to blunt senescence improved renal function and structure, underscoring the central role of senescence in the pathogenesis. Furthermore, this mechanism was conserved in human subjects with RAS. These observations reveal new mechanisms that contribute to the pathogenesis of chronic ischemic renal injury, and support the development of senolytic therapy to reduce senescent cell burden and delay renal injury.
Keywords: senescence, renal artery stenosis, transcriptome, dasatinib, quercetin
Abstract
Background
Peripheral vascular diseases may induce chronic ischemia and cellular injury distal to the arterial obstruction. Cellular senescence involves proliferation arrest in response to stress, which can damage neighboring cells. Renal artery stenosis (RAS) induces stenotic-kidney dysfunction and injury, but whether these arise from cellular senescenceand their temporal pattern remain unknown.
Methods
Chronic renal ischemia was induced in transgenic INK-ATTAC and wild type C57BL/6 mice by unilateral RAS, and kidney function (in vivo micro-MRI) and tissue damage were assessed. Mouse healthy and stenotic kidneys were analyzed using unbiased single-cell RNA-sequencing. To demonstrate translational relevance, cellular senescence was studied in human stenotic kidneys.
Results
Using intraperitoneal AP20187 injections starting 1, 2, or 4 weeks after RAS, selective clearance of cells highly expressing p16Ink4a attenuated cellular senescence and improved stenotic-kidney function; however, starting treatment immediately after RAS induction was unsuccessful. Broader clearance of senescent cells, using the oral senolytic combination dasatinib and quercetin, in C57BL/6 RAS mice was more effective in clearing cells positive for p21 (Cdkn1a) and alleviating renal dysfunction and damage. Unbiased, single-cell RNA sequencing in freshly dissociated cells from healthy and stenotic mouse kidneys identified stenotic-kidney epithelial cells undergoing both mesenchymal transition and senescence. As in mice, injured human stenotic kidneys exhibited cellular senescence, suggesting this process is conserved.
Conclusions
Maladaptive tubular cell senescence, involving upregulated p16 (Cdkn2a), p19 (Cdkn2d), and p21 (Cdkn1a) expression, is associated with renal dysfunction and injury in chronic ischemia. These findings support development of senolytic strategies to delay chronic ischemic renal injury.
Cellular senescence involves irreversible proliferative arrest that develops in response to stress, and may aggravate tissue damage in kidney disease.1–3 Severe renal artery stenosis (RAS) induces stenotic-kidney hypoxia, with the activation of oxidative stress, inflammation, and fibrosis, leading to irreversible renal microvascular loss, scarring, and kidney dysfunction.4,5 Identifying the mechanisms underlying renal injury, and the kidney cell types implicated, would be useful for the development of successful treatment strategies for patients with RAS.
Senescence involves the activation of p16INK4a/Rb (Cdkn2a) or p21 (Cdkn1a) cytokine-signaling pathways.6 For example, the renal expression of p16 protein doubles in old compared with young mice, and its Cdkn2a gene is considered a biomarker for aging.7,8 In addition to p16 and p21, CellAge9 and SASP Atlas10 have also implicated Serpine1, which activates the p53-p21 pathway11 and is involved in the early stages of senescence.12,13 Therefore, senescent cells may express a combination of Cdkn2a, Serpine1, and Cdkn1a genes8,10 that enable prosurvival defenses in senescent cells. These, in turn, release senescent-associated-secretory phenotype (SASP), which are proinflammatory mediators that accelerate the pathogenesis of kidney and other diseases.
Renal tubular cells have been the primary culprits implicated in the development of cell-autonomous senescence in acute kidney ischemia.14,15 However, given that the kidney consists of many different cell types,16 additional cells might become affected as chronic damage sets in. As recent technologic advances enable the definition of global gene expression patterns of individual cells, single-cell RNA sequencing (scRNA-seq) has become a potent tool to pinpoint cells involved in pathogenesis.17–19 Although scRNA-seq is more sensitive to the tissue dissociation process than single-nucleus RNA-seq,20 the diversity and number of transcripts that it detects enable discrimination of closely related cell types and has been extensively applied in CKD (chronic kidney disease).17–21 A number of studies, including the Tabula Muris Senis Consortium7,8 and Binet et al.,22 have successfully used scRNA-seq to identify senescent cells. Therefore, we took advantage of this tool to determine the effect of chronic ischemia on specific cell types in murine stenotic kidneys.
In particular, although detected in injured kidney cells in vitro and ex vivo, the contribution of p16INK4a-dependent cellular senescence to regulation of renal function in vivo remains unclear. Therefore, we leveraged INK-ATTAC (apoptosis-through-targeted-activation) transgenic mice,23 in which a p16Ink4a (encoded by the Cdkn2a gene) promoter24 drives expression of ATTAC,25 a fusion protein comprising a mutated FK506 binding-protein element and a caspase-8 moiety. AP20187 (AP), a drug with little effect on normal cells, activates caspase-8 and selectively induces apoptosis of cells highly expressing p16Ink4a. RAS induction and AP delivery in this model enabled selective manipulation of stenotic-kidney, p16INK4a-dependent, cellular senescence. We further used this approach to determine the time point at which senolysis becomes advantageous over the course of ischemia.
However, this approach is limited to transgenic mice and, furthermore, some senescent cells may not highly express p16Ink4a. Therefore, we also tested a pharmacologic approach involving senolytic drugs that do not depend on high p16Ink4a expression in eliminating senescent cells in wild-type mice.26,27 Intermittent delivery of senolytics effectively targets antiapoptotic mechanisms that defend senescent cells from their own proapoptotic SASPs, selectively clearing them.6,28 Particularly, a senolytic combination strategy expands the range of senescent cells targeted.6 The flavonol quercetin combined with the pan-tyrosine kinase inhibitor dasatinib (DQ) have been shown to reduce senescent cell burden in mouse tissues, and to delay, prevent, or alleviate age- and senescence-related conditions.29,30 Furthermore, in the first clinical trials of this translatable approach,31,32 DQ alleviated physical dysfunction in patients with pulmonary fibrosis, and reduced senescent stem cell burden in subjects with diabetes. However, the effects of senescent cell clearance using these drugs on chronic kidney ischemic injury have not yet been assessed.
This study was designed to identify senescent cells and test the hypothesis that cellular senescence contributes to renal dysfunction in chronic ischemic kidney injury, and thereby outline a new framework for this disease. Importantly, to demonstrate its clinical relevance, we also confirmed that activation of senescence in ischemic injury is conserved in stenotic human kidneys.
Methods
In Vivo Studies
Animal experiments were approved by the Mayo Clinic Institutional Animal Care and Use Committee. The human study was approved by the Mayo Clinic Institutional Review Board and conducted in accordance with the ethical principles of the Declaration of Helsinki. Written informed consent was obtained from participants before inclusion in the study.
Mouse Models and Induction of RAS
For most experiments studying renal senescence in vivo, INK-ATTAC transgenic mice were used (developed by J.L.K., T.T., J. van Deursen, and D. Baker at the Mayo Clinic).23 Briefly, ATTAC uses a senescence-activated p16Ink4a promoter sequence to drive an FKBP-Caspase-8 gene and EGFP expression in senescent cells. These INK-ATTAC mice were bred onto a C57BL/6 genetic background as heterozygotes and genotyped (in the Kirkland laboratory). AP (B/B Homodimerizer; Clontech, Mountain View, CA) leads to dimerization and then activates FKBP-fused caspase-8 components, leading to apoptosis of cells highly expressing p16Ink4a. AP only affects cells with the ATTAC fusion protein. Male, 5- to 6-months-old, INK-ATTAC mice underwent RAS or sham surgeries. Two weeks later, mice were randomized to AP or vehicle (sham-vehicle, sham-AP, RAS-vehicle, and RAS-AP mice; n=6–8 each). AP (3.3 mg/kg) was delivered by intraperitoneal injections, three times a week, for 4 weeks (total 40 mg/kg, 12 treatments). Magnetic resonance imaging (MRI) scanning was performed to assess renal function.33
To evaluate the temporal role of senescence in renal ischemic injury progression, INK-ATTAC mice were randomly divided into RAS–day 0 (D0) vehicle, RAS–D0 AP, and RAS–D7 AP groups (n=6–8 each). AP (10 mg/kg) or vehicle was injected immediately after induction of RAS (D0) in the RAS–D0 AP and RAS–D0 vehicle groups, respectively, for 14 days (total 60 mg/kg, six treatments). In the RAS–D7 AP group, AP was delivered from D7 to D14 after RAS, at the same dose and frequency. MRI studies were conducted in all mice after 2 weeks of RAS, and mice were then euthanized. Sham-vehicle INK-ATTAC mice served as controls.
For scRNA-seq, 5- to 6-months-old, male, C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME) were euthanized 4 weeks after RAS or sham surgeries. The stenotic and right sham kidneys were collected.
Given the upregulation of Cdkn1a (p21) gene expression in INK-ATTAC stenotic kidneys detected by quantitative PCR (qPCR) and scRNA-seq, and that not every cell with high p16Ink4a expression is senescent,34 we applied an additional approach to eliminate senescent cells from wild-type mice. In C57BL/6 mice with RAS, we tested the senolytic drug combination DQ, which, together, target multiple antiapoptotic mechanisms.35 Mice were treated with DQ or saline vehicle. RAS, RAS-DQ, and sham groups included five to ten mice each. Two weeks after surgery, dasatinib (5 mg/kg; LC Laboratories, Woburn, MA) and quercetin (50 mg/kg; Sigma-Aldrich, St. Louis, MO) were delivered to RAS-DQ mice30 by oral gavage, once weekly, three times. Three days after the third DQ dosing, we studied the in vivo renal function using MRI, and mice were then euthanized.
For RAS induction, mice were anesthetized with isoflurane (2%). Through a small flank incision, the right renal artery was exposed and dissected free of the renal vein. Then, a 0.5-mm length of 0.36 mm (external diameter) × 0.20 mm (inner diameter) polytetrafluoroethylene cuff (SUBL-140; Braintree Scientific, Braintree, MA) was longitudinally cut open and positioned around it. Using 10-0 nylon, the cuff was circumferentially sutured and closed.36,37 Sham surgeries were performed in the control group, which consisted of a flank incision and isolation of the renal artery, without placement of a cuff. After recovery, mice were allowed free access to standard chow and water.
After the final study, all mice were euthanized by exsanguination, and blood and urine were respectively collected via inferior vena cava sampling and cystocentesis. Both kidneys were removed, weighed, and processed for flow cytometry, fresh frozen, or preserved in formalin for ex vivo studies.
scRNA-seq Analysis
We analyzed 76,000 cells from sham and RAS samples (n=4 each). A quality control was then performed on the cells to calculate the number of genes, unique molecular identifiers, and proportion of mitochondrial genes for each cell; cells with few covered genes (gene count <500) and high mitochondrial counts (mitochondrial genes >0.2) were filtered out. The matrix was then normalized on the basis of their library sizes. Of these, approximately 75% passed quality control, yielding approximately 57,000 cells for data analysis. A general statistical test was then performed to calculate gene dispersion, base mean, and cell coverage to build a gene model for performing principal component analysis (PCA) or batch alignment. Genes with high coverage (top 1413) and high dispersion (dispersion >1.5) were chosen, and combined PCA (CPCA) batch alignment analysis was performed using the iCellR R package (version 1.5.5).38–40 CPCA batch alignment automatically performs PCA analysis on the integrated or batch-aligned data. We performed t-distributed stochastic neighbor embedding and uniform manifold approximation and projection (UMAP) on the top 10 principal components, and K-nearest-neighbor-based Network graph drawing Layout (KNetL) on the top 20 principal components. KNetL map uses a force-based layout,41 and a zoom for changing the force in the system. Force-directed graph drawing algorithms assign attractive (analogous to spring force) and repulsive forces (usually described as analogous to the forces in atomic particles) to separate all pairs of nodes. Here, we extracted the nodes of the network layout and performed UMAP to create the final plot, a KNetL map.42(preprint) PhenoGraph43 clustering was then performed on the KNetL map results.
The marker genes were then found for each cluster and visualized on heat maps and bar and box plots, and marker genes were used to determine the cell types. We calculated the proportion (percentage) of the cell communities in each condition and generated Pseudotime Abstract KNetL (PAK) maps, using iCellR.38–40 In the PAK maps, each node represents a cluster, and the length and thickness of the links (edges) represent the distances between each cluster. The shorter and thicker the link, the more related (similar) are the cell communities (Supplemental Figure 1).
To assign a specific cell type to each cluster, we correlated cluster-specific differential gene expression with gene libraries obtained by filtering data from the Immunological Genome Project (ImmGen), Mouse Cell Atlas,26 Tabula Muris,44 and Tabula Muris Senis databases7,8; validating in scRNA-seq databases44 and ImmGen45; and using bulk RNA-seq data for clustering renal macrophages46 and intercalated cells47 (Supplemental Appendix 1, Supplemental Figure 2).
In Vivo Renal Function by MRI
MRI studies were performed in anesthetized (with 1%–2% isoflurane) mice on a vertical 16.4 T animal scanner equipped with a 38-mm inner diameter birdcage coil (Bruker, Billerica, MA). Electrocardiogram, respiration, and body temperature were monitored (SA Instruments, Stony Brook, NY). Renal volume was determined using a respiration-gated 3D fast imaging with steady precession sequence, and oxygenation with blood oxygen level–dependent imaging using a respiration-gated 3D multiecho gradient echo sequence.36,48 Renal blood flow (RBF) was assessed by arterial spin labeling. A total of 30 images were acquired with sampling delays after inversion between 40 and 7900 ms. Renal volumes were quantified using Analyze (version 12.0; Biomedical Imaging Resource, Mayo Clinic, MN),48 and all other MRI images using an in-house Matlab (The Mathworks, Natick, MA) module. The transverse relaxation rate R2* (1/T2*) (R2, transverse decay rate; T2, transverse relaxation parameter) served as an index of cortical and medullary hypoxia. Segmentation was performed on the perfusion map, where good contrast was observed. GFR was measured by tracing gadolinium dynamics, using our previously developed dynamic contrast–enhanced MRI technique, and calculated as normalized GFR and kidney volume.33
BP, Plasma, and Urine Studies
Blood pressure was measured in all mice, weekly or biweekly, by the tail-cuff method, using an automated oscillometric device (XBP1000 system; Kent Scientific, Torrington, CT).49 Two methods were used to assess renal function. Plasma creatinine level was assayed using the DetectX Serum Creatinine kit (Arbor Assays, Ann Arbor, MI)48 and, because Jaffe reactions may overestimate renal function in mice,50 cystatin-C levels were also measured by ELISA (catalog MSCTC0; R&D Systems, Minneapolis, MN).51 In addition, we assayed activin-A levels using a Quantikine ELISA kit (R&D Systems),52 and plasma renin content using the Angiotensin-I RIA kit (ALPCO, Salem, NH),53 according to the manufacturer’s instructions. Urinary albumin was measured using the Mouse Albumin ELISA Kit (Crystal Chem, Elk Grove Village, IL), and urinary creatinine using the Creatinine Urinary Detection Kit (Arbor Assays). The urinary albumin-creatinine ratio was calculated in individual mice.
Ex Vivo Studies
Imaging Flow Cytometry
We stained single-cell suspensions of INK-ATTAC mouse kidneys for the Live/Dead marker, followed by CD326 (EPCAM), CD324 (E-cadherin), CD45 (lymphocyte), F4/80, FCRIV (macrophages), CD11b, CD31 (endothelial cells), and lineage-positive cells (CD3, CD19, NK1.1, Ter119, Ly6G). We removed excess antibody by washing cells with MACS buffer (Miltenyi). Cells were acquired using FlowSight (Millipore), and data were analyzed using Ideas (Millipore). Live/Dead−p16+ cells were gated and evaluated for presence of the abovementioned surface markers to identify green fluorescent protein–positive (GFP+) cell types.
Real-Time PCR
Total RNA was isolated from frozen mouse kidneys using the mirVana PARIS RNA Isolation Kit, and its concentration was measured using a NanoDrop spectrophotometer (both from Thermo Fisher Scientific, Waltham, MA), and 50 μl of RNA was then treated with DNase (Thermo Fisher). The first-strand cDNA was produced from 240 ng of total RNA, using the SuperScript VILO cDNA Synthesis kit (Thermo Fisher). Relative qPCR was performed using TaqMan assays, containing 10 ng of cDNA product, using the following TaqMan probes (Thermo Fisher): Cdkn2a (mm00494449), Cdkn2d (mm00486943), Cdkn1a (mm00432448), Tp53 (mm01731290), Activin-a (mm00434339), Hif1a (mm00468869), Il1α (mm00439620), Il6 (mm00446190), Ccl2 (mm00441242), Mmp3 (mm00440295), Tnfα (mm00443258), Serpine1 (mm00435858), Hbegf (mm00439306), Spp1 (mm00436767), Tnfrsf12a (mm01302476), and Gapdh (mm99999915) or Tbp (mm01277042) (as internal control). We used the Applied Biosystems ViiA7 Real-Time PCR System for analysis. We used the 2-ΔΔCT method to calculate the fold changes of each target gene in the experimental relative to control groups.54
Renal Histology
All slides were examined in a blinded manner. To determine senescence, renal cryosections were stained for senescence-associated β-galactosidase (SA-β-gal; Cell Signaling, Boston, MA),55 with or without counterstain (Eosin-Y; Thermo Fisher). The extent of senescence (blue color) was quantified in ten random fields per section using AxioVision (Carl Zeiss, Oberkochen, Germany) and expressed as a percentage of SA-β-Gal–positive cells to total field area.56 To determine whether INK-ATTAC p16INK4a GFP+ cells are also SA-β-gal+, the same areas were successively studied by bright-field and fluorescence microscopy. An additional senescence marker, lamin-B1, was stained on renal sections using anti–Lamin-B1 antibody (catalog number ab16048; Abcam, Cambridge, MA). We manually analyzed the average fraction of lamin-B1+ cells to total cells per field.57
Cortical interstitial fibrosis was semiautomatically quantified in ten fields of Masson trichrome–stained sections, using AxioVision,58 and tubular injury was scored in sections stained with Periodic acid–Schiff, as described.59 Immunohistochemistry staining with anti-mouse F4/80 antibody (catalog number ab6640; Abcam) was used to evaluate macrophage infiltration.60 For apoptosis, we stained renal sections with terminal deoxynucleotidyl transferase–mediated digoxigenin-deoxyuridine nick-end labeling (TUNEL; catalog number G3250; Promega, Madison, WI), and ten to 15 fields per section were randomly chosen at a magnification of ×20. The degree of inflammation and apoptosis was expressed as the average fraction of F4/80+ or TUNEL+ cells per total field area.
Senescence in Human Subjects
Human kidneys were obtained for clinical indications (treatment for intractable hypertension) by surgical nephrectomy in five patients with renal artery occlusion, and compared with the seven healthy, discarded kidneys collected from four donors (LifeSource, Minneapolis, MN). Kidneys from patients with concurrent therapy with immunosuppressive or chemotherapeutic agents were excluded. Human kidneys underwent SA-β-gal staining using a fluorogenic detection kit (SPiDER-βGal; Dojindo).3 The degree of senescence (green fluorescence) was expressed as a percentage of the total field area. For mRNA expressions, relative qPCR was performed in frozen kidney tissue using TaqMan assays and the following probes (Thermo Fisher): CDKN2A (Hs00923894), CDKN1A (Hs00355782), HBEGF (Hs00181813), SPP1 (Hs00959010), TNFRSF12A (Hs00171993), SERPINE1 (Hs00167155), and TBP (Hs00427620; as internal control).
Statistical Analysis
Statistical analysis was performed using JMP version 14.1.0 (SAS Institute, Cary, NC). Results are presented as mean±SD for normally distributed variables, and as median (interquartile range) for data that did not show a normal distribution. Parametric (one-way ANOVA followed by t test) and nonparametric (Kruskal–Wallis followed by Wilcoxon or Kolmogorov–Smirnov) tests were used for comparisons among groups. Spearman correlation coefficients were used to test correlations. P≤0.05 was considered statistically significant.
Results
Chronic Ischemia Induces Renal Senescence
In INK-ATTAC transgenic mice, cells highly expressing p16INK4a also express GFP.23 Six weeks after RAS (Figure 1A, Supplemental Table 1), the stenotic kidney had increased p16-GFP signals, which mostly colocalized with SA-β-gal staining (Figure 1B). The SA-β-gal+ area increased in stenotic kidneys (P<0.0001 versus sham), as did the number of cells positive for lamin-B1, which is a functional downstream senescence effector (P=0.003; Supplemental Figure 3A).57 Furthermore, mRNA expression of the senescence-related genes Cdkn2a (p16), Cdkn2d (p19), Cdkn1a (p21), and Activin-a was markedly elevated compared with vehicle-treated sham (all P≤0.01; Figure 1C). Expression of Hif1A, which is involved in hypoxic adaptation and antiapoptosis,27 also increased (P=0.003 versus sham), as did expression of the proinflammatory and profibrotic SASP genes Il1a, Il6, Ccl2 (MCP-1), Mmp3, Tnfa, and Serpine1 (PAI-1) (all P=0.001 versus sham; Figure 1C). Systemic plasma levels of activin-A, a biomarker of senescent cell burden,52 were also elevated in RAS mice (P<0.0001 versus sham; Figure 1D).
Figure 1.

Chronic ischemia induces senescence in the stenotic kidney (STK), and clearance of cells highly expressing p16Ink4a by AP improves renal function and oxygenation. (A) Experimental design in INK-ATTAC transgenic mice studied 6 weeks after sham or RAS surgeries. (B) SA-β-gal (cyan/blue) and p16 (green) colocalize (arrows) on mouse STK cryosections. Blue, 4′,6-diamidino-2-phenylindole. Renal gene expression of senescence and SASP factors using (C) real-time PCR (relative to Gapdh), (D) plasma levels of activin-A by ELISA, (E) creatinine levels using Jaffe reactions, and (F) SBP levels measured by the tail-cuff method using an automated oscillometric device in sham or RAS treated with vehicle (veh) or AP. *P<0.05 versus intragroup baseline, †P<0.05 versus intragroup after 2 weeks, §P<0.05 versus sham-vehicle group, #P<0.05 versus sham-AP group, ‡P<0.05 versus RAS-vehicle at the same time point. (G) Renal perfusion maps generated by arterial spin-labeling MRI (milliliters per 100 g per minute; brighter red, higher perfusion). (H) Masson trichrome staining and semiautomatic quantification on ten randomly chosen fields per section (using AxioVision) showed that increased STK fibrosis was slightly blunted by AP. Mean±SD or median±interquartile range (n=6–8). *P<0.05 versus sham-vehicle group, †P<0.05 versus RAS-vehicle group (t tests or Wilcoxon test).
AP Decreases Stenotic-Kidney Senescence
In INK-ATTAC mice, intraperitoneal AP treatment (three times a week for 4 weeks, total 40 mg/kg) significantly blunted the gene expression of Cdkn2d (P=0.05), Activin-a (P=0.02), Il6 (P=0.04), Mmp3 (P=0.02), and Tnfa (P=0.02), along with the SA-β-gal+ area (P=0.02). AP also strongly tended to decrease gene expression of Cdkn2a (P=0.07), Hif1a (P=0.08), Il1a (P=0.06), and Ccl2 (P=0.08), along with decreased lamin-B1+ cells (P=0.08), compared with vehicle-treated RAS; but this did not reach statistical significance due to high variability (Figure 1C, Supplemental Figure 3A). AP also decreased circulating activin-A levels (P=0.04 versus RAS; Figure 1D).
Senescence Is Functionally Consequential in Murine RAS
Plasma creatinine and cystatin-C levels were elevated (P=0.001 and P=0.02 versus sham, respectively; Figure 1E, Supplemental Figure 3), and systolic BP (SBP) was increased, after RAS induction in INK-ATTAC mice (all P≤0.04 versus baseline; Figure 1F). SBP in sham-AP mice was lower than that in sham-vehicle mice at 4 and 6 weeks (both P=0.01; Figure 1F). Six weeks after RAS, MRI-derived stenotic-kidney cortical perfusion was decreased (P<0.0001 versus sham; Figure 1G), and fibrosis developed (P=0.001 versus sham; Figure 1H). However, AP decreased SBP (P=0.03 versus 2 weeks) and plasma creatinine levels (P=0.03), improved stenotic-kidney cortical perfusion (P=0.006 versus RAS), and strongly tended to decrease fibrosis (P=0.06versus RAS) (Figure 1, E–H). However, a trend for a fall in plasma cystatin-C levels after AP delivery did not reached statistical significance (P=0.09; Supplemental Figure 3B).
scRNA-seq Identifies Senescent Tubular and Stromal Cells in the Stenotic Kidney
To determine which stenotic-kidney cells undergo senescence, single cells of RAS or sham kidneys (n=4 each) were purified and analyzed by scRNA-seq (Figure 2A). After quality control (Supplemental Figure 4), approximately 57,000 cells from sham and RAS mice were subjected to PCA and KNetL, followed by PhenoGraph (Figure 2B, Supplemental Appendix 3, Supplemental Figures 2 and 5). We identified 26 clusters using ImmGen61 and Mouse Cell Atlas26 (Figure 2B, Supplemental Figure 4D). All proximal tubule clusters primarily expressed typical markers (Supplemental Figure 4D, Supplemental Appendix 2). The thick ascending limb of the loop of Henle and distal convoluted tubule cells were reclustered from cluster 1 (Supplemental Figure 2A).62
Figure 2.
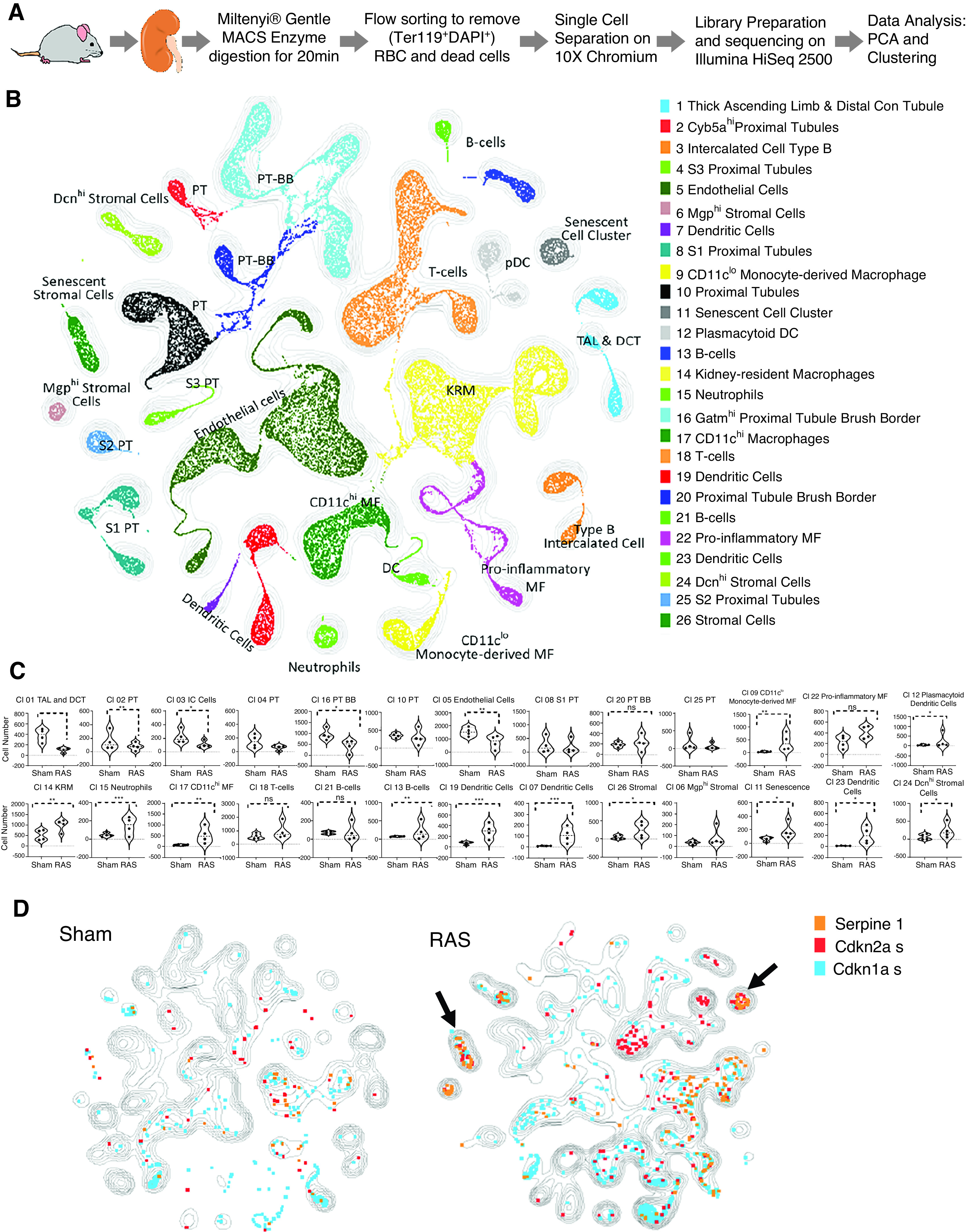
Clustering of sham and RAS kidneys by scRNA-seq illustrate cell types/clusters in sham and RAS kidneys. (A) Experimental design for isolating viable kidney cells. scRNA-seq data were analyzed together with all metadata. (B) Unsupervised KNetL followed by PhenoGraph mapping, showing 26 distinct cell clusters from 57,000 sham and RAS kidney cells. (C) Violin plots comparing the proportion of each cluster in sham and RAS samples. Individual populations were downsampled to approximately 7000 cells per sample, and proportions of individual populations per cluster were quantified (Mann–Whitney test). P<0.05 is considered significant. In RAS, cells were reduced in tubular and endothelial, but increased in myeloid and stromal clusters. (D) The contours illustrate and compare cell types/clusters between sham and RAS kidneys. Some genes are superimposed on these contours to highlight the cell types (listed in Figure 2B) that undergo senescence (Figure 2C) in RAS. Overlay of Cdkn2a+ senescent cells in sham and RAS show that most cells upregulating Cdkn2a are T and B lymphocytes, myeloid cells (macrophages [MF] and dendritic cells [DC]), stromal cells (arrow), and a senescent cell cluster (arrow). The senescent cell cluster (cluster 11) (arrow) expresses marker genes such as Serpine1, Cdkn2a, and Cdkn1a in RAS compared with sham. BB, brush border; Con tubule, convoluted tubule; DAPI, 4′,6-diamidino-2-phenylindole; DCT, distal convoluted tubule; KRM, kidney-resident macrophages; MACS, magnetic cell sorting; PT, proximal tubule; RBC, red blood cells; TAL, thick ascending limb of the loop of Henle.
Renal ischemia markedly decreased the numbers of stenotic-kidney tubular and endothelial cells, while increasing myeloid46,63 and stromal cells (Figure 2B, Supplemental Figure 2B), which is in agreement with our previous observations.46,64–66 We identified four discrete populations of stromal cells (Figure 2, Supplemental Figure 2C). Of these, Dcn encodes decorin, which is expressed mainly by fibroblasts and is involved with matrix assembly.67 Clusters 24 and 26 were Dcn+ stromal cells that increased significantly in RAS; cluster 26 expressed Cdkn2a, Serpine1, and Cdkn1a (Figure 2, C–D, Supplemental Figure 6, A–C).
To identify senescent cells in RAS, we gated on Cdkn2a, Cdkn1a, and Serpine1 and found they increased four-fold in RAS, primarily expressed in macrophages (Supplemental Figure 7) and cluster 26 (stromal cells), but were most prominently expressed in cluster 11 (Figure 2D, Supplemental Figure 6, A–C. Therefore, we next focused on cluster 11.
Cluster 11 is a heterogeneous cluster of cells that increased significantly in RAS (Figure 2C) and differentially expresses epithelial and mesenchymal genes (Supplemental Figures 1 and 2, A–C). Interestingly, unbiased PAK analysis identified the cluster as epithelial (Supplemental Figure 2C). Reclustering cluster 11 (Figure 3A) identified five separate clusters: four of which were of an epithelial lineage, and the fifth consisted of Ankrd1+ (an important effector of fibrosis) stromal cells.68 This stromal cell cluster expressed all of the senescent genes (Cdkn2a, Serpine1, and Cdkn1a) along with the stromal markers Acta2, Map1b, and Vim, and was significantly increased in RAS versus sham mice (Figure 3A). Interestingly, Ankrd1+ cells also expressed epithelial markers, such as Lgals1, Spp1, and Cryab, and transcription factors regulating epithelial-to-mesenchymal transition (EMT), such as Zeb2, Foxc1, Tgfb2, Loxl2, and Pdpn.69
Figure 3.

Senescent cell cluster 11 is heterogenous and consists of Ankrd1+ stromal cells that express Cdkn2a, Cdkn1a, and Serpine1, suggesting senescence. (A) Reclustering of cluster 11 revealed five subclusters. Overlay of Cdkn2a+, Cdkn1a+, and Serpine1+ cells on Ankrd1+ stromal cluster (middle UMAP) shows that this cluster expresses senescent genes; overlay of RAS and sham populations shows that Ankrd1 stromal cell cluster predominantly contains RAS cells. (B) The Ankrd1+ population in RAS shows upregulation of mesenchymal genes, such as Vim, Map1b, Lgals1, Acta2, and Ankrd1, whereas epithelial cell–specific genes (e.g., Cdh16, Gpc3) are downregulated. (C) Experimental design for isolating viable epithelial cells (epi), endothelial cells, macrophages (MF), and T cells from sham and RAS kidneys. Kidneys were digested, flow sorted, and approximately 192 single cells (approximately 48 cells per population) underwent qPCR for 96 genes. (D) Data analyzed by PCA, followed by UMAP, separated cells into eight distinct clusters. Epithelial cells and macrophages from sham and RAS kidneys were compared for expression of senescence-associated genes. (E) Violin plots show that although macrophages express these genes, only epithelial cells significantly upregulate p16, p19, p21, and PAI-1 in RAS. (F) Heat map demonstrates that a portion of epithelial cells, but not macrophages, upregulate senescent genes. (G–H) Representative flow cytometry image and quantification of β-gal and p16-GFP cells expressing epithelial cell markers. IA, ischemia-associated; IFC, integrated fluidic circuits; STK, stenotic kidney.
Furthermore, we observed three separate stromal cell populations expressing higher levels of bona fide stromal markers, such as Mgp, Dcn, Col1a1, Cola2, and Has1 (Supplemental Figure 1), compared with cluster 11. Hence, the Ankrd1+ cells in cluster 11 may not be fully mature stromal cells, and it is their epithelial nature that groups them at cluster 11.
RAS upregulated mesenchymal and downregulated epithelial genes (Figure 3B, Supplemental Figure 1B). Pathway analysis using Enrichr suggested pathways such as extracellular matrix organization (Gene Ontology term 0030198) and EMT regulation (Gene Ontology term 0010717). Thus, Ankrd1+ cells are likely of epithelial lineage that are in the early stages of EMT and are undergoing senescence (Supplemental Figure 1).
Single-Cell qPCR Confirms Renal Epithelial Cell Senescence
To validate the scRNA-seq results and further probe the lineage of cluster 11, we adopted single-cell qPCR and flow sorted and lineage defined viable epithelial and endothelial cells, T cells, and macrophages (Figure 3C). Cdkn2a-expressing epithelial cells clustered independently, and we detected ischemia-associated macrophages and epithelial cells (Figure 3D). As in the scRNA-seq, only senescent epithelial cells upregulated the senescence-associated genes Cdkn2a, Cdkn1a, Spp1, Col1a1, Serpine1, and Vim. Whereas senescent epithelial cells upregulated mesenchymal markers (Vim and Yap), proinflammatory markers (Il6, Ifng, Il1b, and Irf5) remained unchanged; however, chemoattractants (Csf1, Ccl2, and Spp1) were upregulated (Figure 3, E–F).
Furthermore, we validated the epithelial lineage of senescent cells by imaging flow cytometry, where most p16-GFP+ and SA-β-gal+ cells were EPCAM+ (CD326) and E-cadherin+ (CD324) (Figure 3, G–H). Hence, most senescent cells were epithelial. Taken together, epithelial cell in early phases of EMT undergo senescence.
Few Macrophages Undergo Senescence in Ischemic Kidneys
Using scRNA-seq and single-cell PCR, we observed little upregulation of senescent gene expression in macrophages relative to epithelial cells (Figure 3E, Supplemental Figure 7C). The number of macrophages expressing senescent genes increased significantly in RAS (Supplemental Figure 7D). Unlike epithelial cells, expression of Cdkn2a and Cdkn1a was stochastic. Most Cdkn1a and Serpine1 expression was in CD11clo infiltrating macrophages that upregulated proinflammatory genes, whereas most Cdkn2a-expressing macrophages appeared to be efferocytic kidney-resident macrophages (Supplemental Figure 7, B and D). Indeed, imaging cytometry detected kidney-resident macrophages engulfing p16GFP+ particles in RAS (Supplemental Figure 7E), suggesting Cdkn2a-expressing macrophages might clear senescent cells.
DQ Reduces Stenotic-Kidney Senescence and Damage
Next, we eliminated senescent cells from wild-type mice using the broad-range senolytic drug combination DQ.35 After three doses at 1-week intervals (Figure 4A), there was a decrease in the mRNA expression of senescence and SASP markers (all P<0.02; Supplemental Figure 8A); the activity of SA-β-gal (P=0.001; Figure 4B); and the levels of plasma cystatin-C (P=0.001; Figure 4C), creatinine (P<0.0001; Supplemental Figure 8B), and activin-A (P=0.002; Supplemental Figure 8B). SBP and renin (P=0.06) remained unaffected (Figure 4C, Supplemental Figure 8C). Unlike AP, DQ also attenuated Cdkn1a (P=0.006) and Serpine1 (P=0.01) expression.
Figure 4.
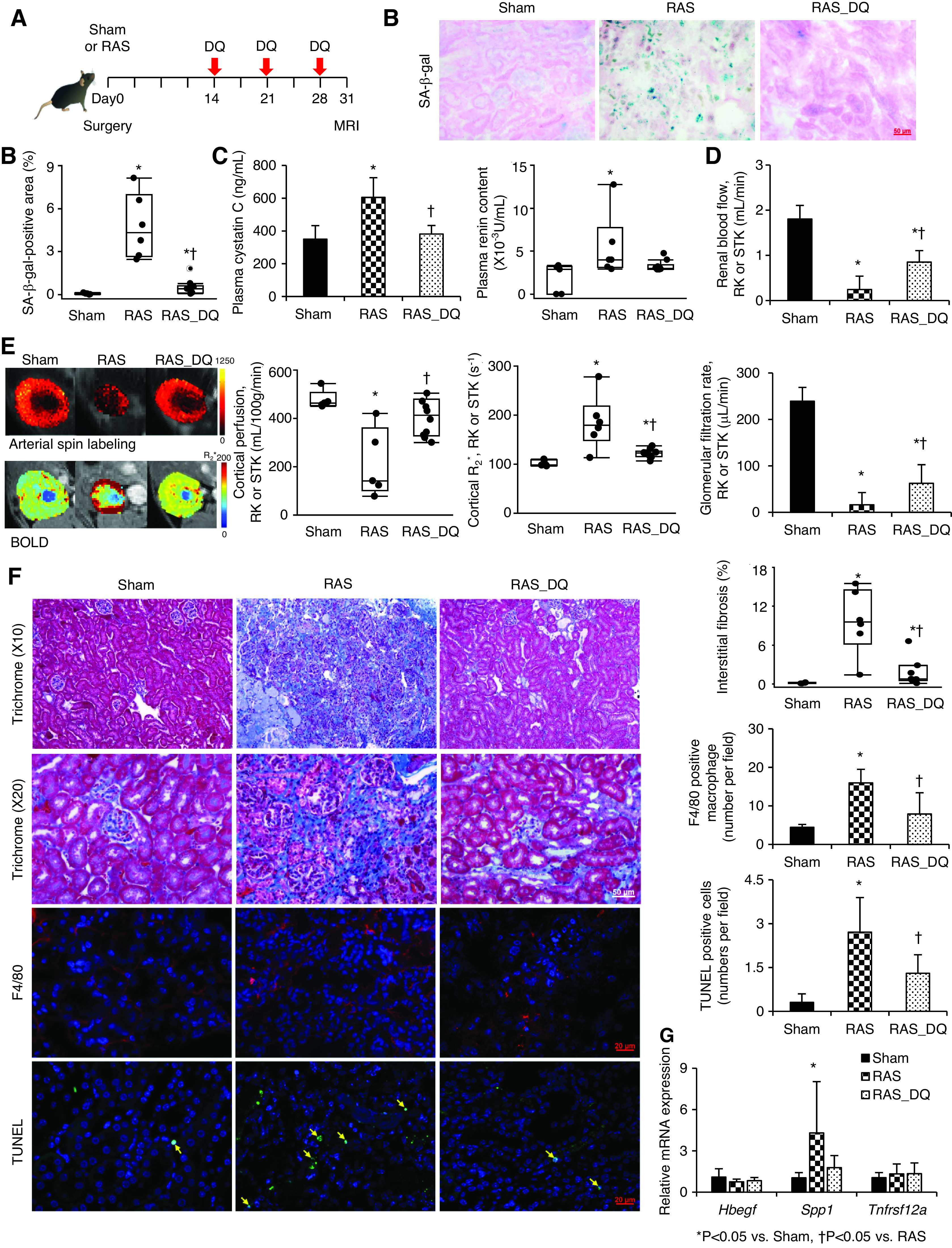
DQ alleviates stenotic-kidney (STK) cellular senescence and improves function and fibrosis in mice with RAS. (A) C57/BL6 wild-type mice treated with DQ three times weekly starting 2 weeks after surgery. (B) SA-β-gal staining (blue) and quantification on renal cryosections. (C) Plasma cystatin-C (ELISA) and renin content (RIA). (D) Single-kidney RBF (MRI). (E) Renal perfusion maps by arterial spin labeling (color bar, milliliters per 100 g per minute), hypoxia by blood oxygen level–dependent (BOLD) MRI (red, increasing hypoxia; color bar, R2* per second), and GFR (MRI). (F) Masson trichrome, F4/80 (red), and TUNEL (green, arrows) staining with semiautomatic quantifications using AxioVision. Blue, 4′,6-diamidino-2-phenylindole. (G) Renal gene expression of Hbegf, Spp1, and Tnfrsf12a quantified by real-time PCR (relative to Tbp). Mean±SD or median±interquartile range (sham, n=5; RAS, n=6; RAS-DQ, n=10). *P<0.05 versus sham, †P<0.05 versus RAS (two-tailed t tests or Wilcoxon test). RK, Right kidney.
DQ improved stenotic-kidney size (Supplemental Table 1), RBF (P=0.001), and GFR (P=0.02), and restored cortical perfusion (P=0.02) and oxygenation (P=0.03; Figure 4, D–E). DQ also alleviated interstitial fibrosis (P=0.01) and decreased the numbers of F4/80+ macrophages (P=0.006) and TUNEL+ apoptotic cells (P=0.01; Figure 4F). Interestingly, DQ-treated kidneys showed a rare population of p21+/TUNEL+ cells, suggesting apoptosis of senescent cells (Supplemental Figure 8D). Spp1 expression was upregulated in RAS (P=0.02) but restored in RAS-DQ mice (P=0.10 versus sham; Figure 4G).
Senescence Is Initially Protective, but Ultimately Drives Persistent Ischemic Injury
The expression of senescence and SASP genes increased 2 and 6 weeks after RAS (Supplemental Figure 9). The expression of Cdkn2a, Activin-a, Mmp3, and Tnfa progressively increased with duration, whereas other senescent and SASP factors remained stable.
To evaluate the temporal role of senescence during the evolution of renal ischemic injury, we initiated the same overall dose of AP delivery in INK-ATTAC mice at different time points (Figure 5A, Supplemental Table 1), starting either immediately (RAS–D0 AP) or at D7 (RAS–D7 AP) after RAS induction. Vehicle-treated RAS mice (RAS–D0 vehicle) served as controls. Compared with shams, stenotic-kidney senescence and SASP gene expression was similarly increased in both RAS–D0 vehicle and RAS–D0 AP groups, but was decreased in the RAS–D7 AP group (Supplemental Figure 10A), consistent with stenotic-kidney p16-GFP expression and plasma activin-A levels (Figure 5, B–C).
Figure 5.

Senescence in stenotic kidneys (STK) is initially protective, but ultimately drives persistent injury. (A) Experimental design. INK-ATTAC transgenic mice were studied 2 weeks after AP or vehicle started either immediately (RAS_D0AP and RAS_D0V, respectively) or 7 days (RAS_D7AP) after RAS surgery. (B) p16 (green) on renal cryosections. Blue, 4′,6-diamidino-2-phenylindole. (C) Levels of plasma activin-A by ELISA. (D) Renal perfusion by arterial spine labeling MRI (color bar, milliliters per 100 g per minute). (E) Single-kidney RBF and GFR by MRI. (F) Masson trichrome, Periodic acid–Schiff (PAS), F4/80 staining (red), and semiautomatic quantifications using AxioVision. Median±interquartile range (sham, n=5; RAS_D0V, n=6; RAS_D0AP, n=8; RAS_D7AP, n=6). *P<0.05 versus sham, †P<0.05 versus RAS_D0V; §P<0.05 versus RAS_D0AP (Wilcoxon test). RK, right kidney.
Functionally, stenotic-kidney RBF and perfusion were reduced in both RAS–D0 vehicle (P=0.02 and P=0.01, respectively) and RAS–D0 AP mice (P=0.004 and P=0.01, respectively), but improved in RAS–D7 AP mice (P≤0.05 versus both) (Figure 5, D–E). In concordance, renal fibrosis, tubular injury, macrophages, and plasma cystatin-C decreased in RAS–D7 AP mice (all P≤0.04 versus RAS–D0 vehicle and RAS–D0 AP mice; Figure 5F, Supplemental Figure 10B). Plasma creatinine and albuminuria was unaffected by timing of AP delivery (Supplemental Figure 10, C and D), possibly due to the short RAS duration and contralateral kidney compensation. SBP increased after RAS induction (P≤0.04 versus baseline for each group), but remained unchanged after AP. SBP at the first week was slightly higher in D7-AP than in D0-AP mice (P=0.04), possibly because SBPs were slightly higher at baseline in the D7-AP group, albeit not significantly (Supplemental Figure 10E). Indeed, the level (Δ) of change in SBP from baseline to 2 weeks was similar between the D0-AP and D7-AP groups (P=0.86).
Senescence Biomarkers Accumulate in Chronically Ischemic Human Kidneys
Nephrectomized stenotic kidneys from subjects with RAS showed increased positivity for SA-β-gal, and increased CDKN2A and CDKN1A gene expression (all P≤0.02 versus healthy; Figure 6, A–C). The gene expression of HBEGF was upregulated (P=0.02) in the stenotic kidney and SPP1 was downregulated (P=0.02), whereas the expression of TNFRSF12A and SERPINE1 was unchanged (P=0.33 and P=0.15, respectively).
Figure 6.
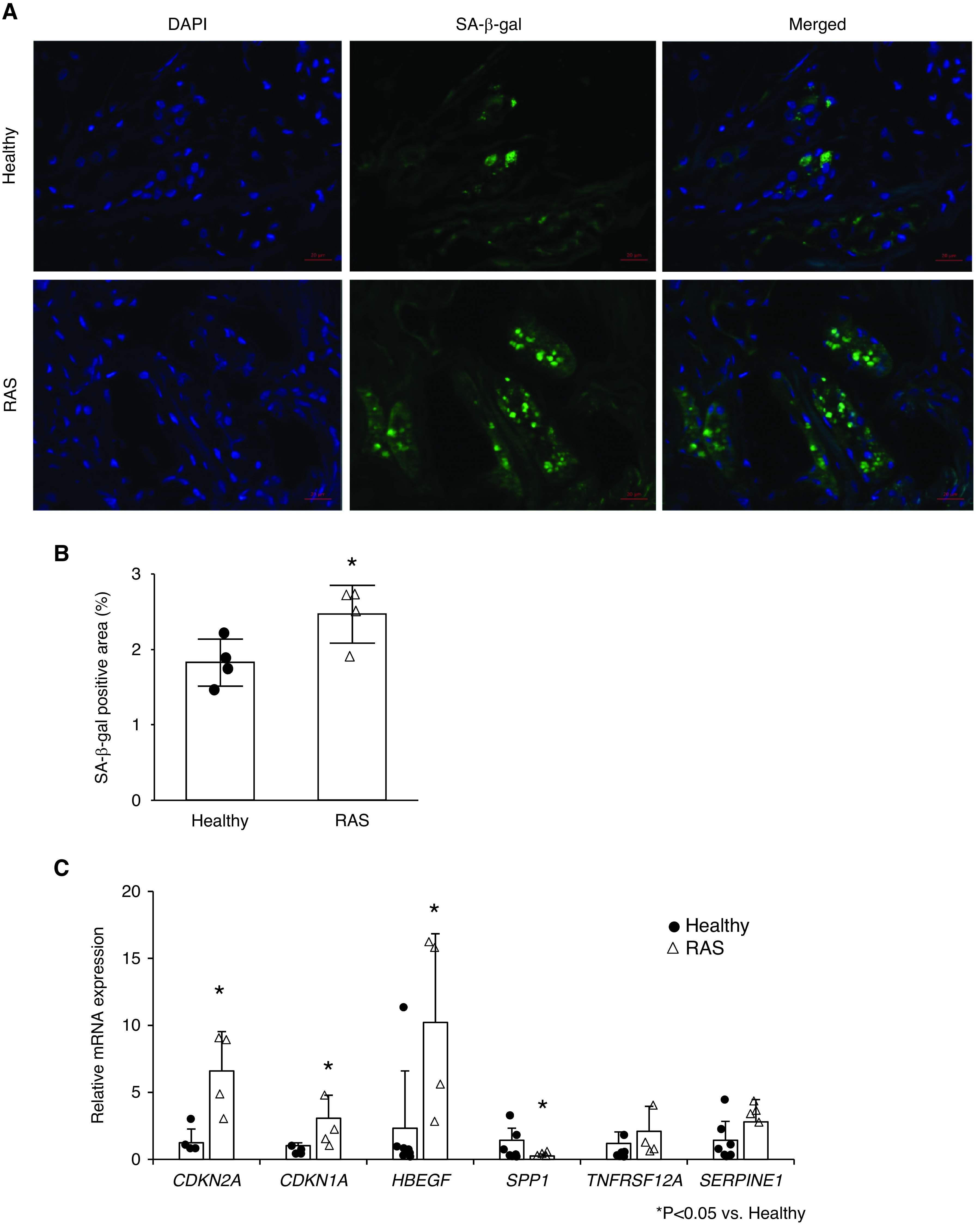
Chronic ischemia induces senescence in human kidneys distal to RAS. (A) Fluorescent staining for senescence-associated β-galactosidase (SA-β-gal; green) in healthy and RAS kidneys. Blue, 4′,6-diamidino-2-phenylindole (DAPI). (B) Semiautomatic quantification of the percentage area positive for SA-β-gal. (C) Renal gene expressions of senescence-associated genes by real-time PCR (relative to TBP). Mean±SD (healthy, n=4–7; RAS, n=4). *P<0.05 versus healthy (two-tailed t tests or Wilcoxon test).
Discussion
This study demonstrates that chronic renal ischemia induces loss of proximal, distal tubular, and endothelial cells, and triggers cellular senescence, which partly accounts for injury and dysfunction in murine stenotic kidneys. Moreover, this senescence pathway is conserved in human RAS. Unbiased scRNA-sequencing, single-cell PCR, imaging cytometry, and immunofluorescence implicated epithelial cells (possibly in early mesenchymal transition). The INK-ATTAC transgenic model revealed a role for p16, and a broad DQ intervention efficaciously reduced renal atrophy, fibrosis, and inflammation, and improved RBF. Importantly, the improvement in renal outcomes achieved by both strategies positions cellular senescence as a central mechanism driving the pathogenesis of chronic ischemic renal injury.
Our INK-ATTAC RAS mouse model developed typical senescence-associated indices, including upregulated expression of CDK-inhibitors and SASP genes, SA-β-gal staining, activin-A levels, and lamin-B1 immunoreactivity, which correlated with renal atrophy and disease duration. Despite their relatively low frequency, clearance of cells highly expressing p16Ink4a decreased several senescence indicators and improved kidney hemodynamics and function; this implicates cellular senescence in progressive stenotic-kidney injury, which is possibly attributable to the noxious factors released by these cells. Given that merely few senescent cells in healthy mouse kidneys are sufficient in inducing injury,1 senescent cells may contribute to CKD.
Analyzing similar numbers of cells per sample in scRNA-seq revealed relative cellular changes in chronic renal ischemia, including the loss of epithelial and endothelial cells, accumulation of inflammatory cells, and a surge in stromal cells. In the stenotic kidney, we observed an increase in Cdkn2a-expressing cells, with high expression of Cdkn1a and Serpine1, implicating them in kidney injury and fibrosis. Whereas Cdkn1a-, Cdkn2a-, and Serpine1-expressing stromal, myeloid, and lymphoid cells also increased, the highest expression of these genes was detected in a novel cluster of epithelial cells.
Our observations are congruent with recent studies that used the novel p16-tdtomato reporter to identify age-associated senescent renal proximal and distal tubular epithelia.70 Flow cytometry also revealed that Epcam+Sca-1+ epithelial cells increase Cdkn2a expression with age, and that CD73-expressing mesenchymal stromal cells undergo senescence.71 Taken together, renal epithelial cells are particularly susceptible to undergo senescence compared with other kidney cell types.
We observed that macrophages in severe ischemia did not undergo active senescence. Previous studies have implicated macrophages in senescence,72,73 although p16 expression in macrophages does not necessarily indicate senescence.74 Our observations suggest senescent cells release chemoattractant molecules that recruit macrophages, which may, in turn, clear senescent cells.75 This pathway may create a niche facilitating regeneration.
Indeed, the heterogeneous cell cluster that upregulated a full senescence-associated gene profile of Cdkn2a, Cdkn1a, and Serpine1 expression largely included epithelial cells expressing mesenchymal markers (confirmed by reclustering and imaging cytometry), suggesting that cells undergoing early EMT might be particularly prone to senescence and injury. Renal epithelial cell senescence has been observed in aging76; in acute,14 glomerular,77 and diabetic renal disease; and in transient ischemic injury.78,79 In concordance, we detected senescent tubular cell–derived extracellular vesicles in the urine of patients with RAS.64,80
Canonic SASP markers (Serpine1, Tnfa, and Ccl2) were upregulated in murine stenotic kidneys. We have previously detected the release of proinflammatory cytokines from human and swine stenotic kidneys, including IL-1α, IL-6, and TNF-α, possibly linked to SASPs.81,82 Indeed, our study showed that both SASPs and senescent cell accumulation directly correlated with renal fibrosis, similarly to aged mice.79 Cdkn2a+ stenotic-kidney cells upregulated transcription factors (Tsc22d1, Ankrd1, and Tgfb2) that support cell survival and inhibit proliferation.83,84 Upregulation of Tsc22d1/Ankrd1 suggests upstream activation of TGFβ signaling in addition to Yap/Taz (Hippo signaling),85 which is implicated in renal fibrosis. Therefore, development of cellular senescence in chronically ischemic kidneys may drive kidney dysfunction and scarring.
Early AP delivery failed to achieve the improvement in stenotic-kidney function and structure conferred by later (D7 to D14) delivery, suggesting cellular senescence may be initially protective. Indeed, it has several physiologic roles to mediate wound healing, repair, and antitumorigenic properties.15 Possibly, upregulation of chemoattractants, such as osteopontin (Spp1)86,87 and Ccl2, may recruit macrophages to remove cellular debris, limiting early renal damage. Furthermore, senescence increases cellular survival by conferring resistance to apoptosis,88 which is rampant in early kidney injury.89
Both AP in transgenic and DQ in wild-type RAS mice reduced renal senescence, dysfunction, inflammation, and fibrosis; however, AP reversed fibrosis only partially, possibly because some profibrotic stromal cell clusters were sustained. But senescent cells are unlikely to invariably express p16 and, therefore, AP did not fully blunt expression of Cdkn1a or SASP factors. Indeed, a broader senolytic intervention using DQ showed higher efficacy by dramatically reducing expression of senescent genes and improving functional indices. Moreover, it elicited robust improvement of renal function, reflected in both plasma creatinine and cystatin-C levels, whereas the effects of AP were less consistent. Therefore, eliminating a range of senescent cells might be more beneficial in achieving resolution of ischemic kidney damage than purging only p16+ cells. Pertinently, pharmacologic approaches that permit senolysis in non–genetically manipulated models are also clinically applicable. Indeed, targeting senescent cells using DQ is also effective in patients with pulmonary fibrosis30,31 and diabetic kidney disease.32
Importantly, the detection of cellular senescence in stenotic human kidneys identifies it as a conserved mechanism in ischemic kidney injury. Chronic ischemia upregulated CDKN2A and CDKN1A gene expression, and SERPINE1 showed a trend to increased expression. This observation establishes a unified framework for chronic ischemic kidney injury. Intriguingly, human stenotic kidneys removed for advanced RAS showed decreased SPP1 expression, possibly due to its dependence on the disease type and phase.90
Our study had several limitations. The murine model is characterized by relatively short RAS duration, yet develops prominent features that resemble chronic human renal ischemia. Given that the mean SBP was lower in sham and AP INK-ATTAC mice with 6-week RAS, we cannot rule out that systemic hemodynamic effects of AP might have affected our results. We focused our scRNA-seq analysis on identifying kidney cells undergoing senescence19,26,44,91,92; future studies will need expand our findings to elucidate the cell-specific mechanisms operating in ischemic kidneys. Podocyte markers were expressed, but podocytes or mesangial cells did not cluster uniquely in our scRNA-seq analysis.91 Severe ischemia and loss of vascular endothelial growth factor can cause podocyte injury, detachment, and, in turn, mesangiolysis93,94; however, we cannot rule out technical issues related to cell isolation. Whereas single-cell qPCR validated most of our findings, we might have missed some senescent cells during flow sorting. Our 6-month-old mice showed a relatively high proportion of immune cell clusters,19 but lack of bulk transcriptomic data on kidney monocytes limited their identification.95 With elimination t1/2s of <11 hours, intermittent (rather than continuous) DQ treatment targets nonproliferating cells and is effectively senolytic rather than anti-inflammatory, and reduces potential off-target effects.26 Finally, including a sham and DQ-treated group would strengthen our study, although we have previously shown that DQ had little effect on the healthy kidneys of control mice.96
In conclusion, RAS triggers prominent senescence in renal epithelial/stromal cells, which is characterized by a novel gene signature, and at least partly mediates kidney dysfunction and damage. Our observations reveal new mechanisms that contribute to the pathogenesis of chronic ischemic renal injury, and support the development of senolytic therapy to reduce senescent cell burden and delay renal injury.
Disclosures
B. Childs reports having ownership interest in, and patents and invention with, Unity Biotechnology. J.L. Kirkland reports being a scientific advisor for, or member of, the American Federation for Aging Research, the Elysium Scientific Advisory Board, European Institute for the Biology of Aging, and National Institutes of Health (NIH); receiving honoraria from the NIH; having other interests in/relationships with the Mayo Clinic via licensed patents to Unity Biotechnologies, which may result in future royalties or transfer to the author; and having ownership interest in Unity Biotechnologies. J.L. Kirkland and T. Tchkonia report having a financial interest related to this research. T. Tchkonia reports Ownership Interest in Unity Biotechnology Inc; Honoraria from HRI Roswell Park Division, Buffalo, NY, Pfizer; and Patents and Inventions with UNITY Biotechnology. L.O. Lerman reports being an advisor to AstraZeneca; receiving honoraria from AstraZeneca, Janssen Pharmaceuticals, and Wei Jian; receiving research funding from AstraZeneca and Novo Nordisk; having consultancy agreements with AstraZeneca, Janssen Pharmaceuticals, and Wei Jian Inc.; having patents and inventions with Cohbar and Stealth Biopharmaceuticals; and having other interests in/relationships with the American Heart Association and the NIH. T. Niewold reports having consultancy agreements with AstraZeneca, Innova, Janssen, Progentec, Roivant, and Thermo Fisher; receiving research funding from EMD Serono; serving as the editor in chief for Journal of Immunological Methods, associate editor for Translational Research, and associate editor for Lupus Science and Medicine; having ownership interest in Progentec; and being a scientific advisor for, or member of, the Progentec Scientific Advisory Board and Sjögren’s Syndrome Foundation. S. Textor reports serving as the data and safety monitoring board chair for Sentien Biopharmaceuticals and as a member of the data and safety monitoring board for a phase 1b trial with Sentien Biopharmaceuticals; having consultancy agreements with UpToDate (as section editor); and serving as a scientific advisor for, or member of, UpToDate. All remaining authors have nothing to disclose.
Funding
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases grants DK100081, DK109134, DK120292, and DK122734, and National Institute on Aging grants AG013925 and AG062104. This work was also partly supported by the Connor Group, Noaber Foundation, and the Mayo Clinic Center for Regenerative Medicine. A.S. Puranik is funded by the New York University
Supplementary Material
Acknowledgments
We thank Ziyan Lin, Applied Bioinformatics Laboratories, New York University School of Medicine for technical assistance.
Data Sharing Statement
Source data are available for Figures 1–6. On reasonable request, data, analytic methods, and study materials will be made available to other researchers for the purposes of reproducing the results. Datasets are uploaded to Gene Expression Omnibus, under accession number GSE159204 (see https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE159204).
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2020091373/-/DCSupplemental.
Supplemental Appendix 1. Cluster-specific marker genes.
Supplemental Appendix 2. Immgen-based cell call.
Supplemental Appendix 3. Detailed methods.
Supplemental Figure 1. UMAP plots representing genes preferentially expressed in Ankrd11 stromal cell cluster.
Supplemental Figure 2. Sub-clustering and psuedotime abstract KNetL (PAK) analysis in sham and RAS.
Supplemental Figure 3. Senescence staining and plasma cystatin C levelin 6-week RAS.
Supplemental Figure 4. Quality control of the Single-cell RNA sequencingdata.
Supplemental Figure 5. Comparison of UMAP, tSNE, KNetL plot of single-cell RNA sequencing data.
Supplemental Figure 6. Mapping senescent cell clusters in Sham andRAS.
Supplemental Figure 7. Macrophages expressing senescent genes significantly increase in RAS.
Supplemental Figure 8. DQ alleviates stenotic kidney cellular senescence in RAS.
Supplemental Figure 9. Renal gene expression of senescence and SASP factors in sham, 2-week RAS, and 6-week RAS, quantified by RT-PCR relative to GAPDH.
Supplemental Figure 10. Senescence in STK is initially protective but ultimately drives persistent injury.
Supplemental Table 1. Characteristics in mice.
References
- 1.Kim SR, Jiang K, Ferguson CM, Tang H, Chen X, Zhu X, et al.: Transplanted senescent renal scattered tubular-like cells induce injury in the mouse kidney. Am J Physiol Renal Physiol 318: F1167–F1176, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim SR, Jiang K, Ogrodnik M, Chen X, Zhu XY, Lohmeier H, et al.: Increased renal cellular senescence in murine high-fat diet: effect of the senolytic drug quercetin. Transl Res 213: 112–123, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim SR, Eirin A, Zhang X, Lerman A, Lerman LO: Mitochondrial protection partly mitigates kidney cellular senescence in swine atherosclerotic renal artery stenosis. Cell Physiol Biochem 52: 617–632, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lerman LO, Textor SC, Grande JP: Mechanisms of tissue injury in renal artery stenosis: ischemia and beyond. Prog Cardiovasc Dis 52: 196–203, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kwon SH, Lerman LO: Atherosclerotic renal artery stenosis: current status. Adv Chronic Kidney Dis 22: 224–231, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kirkland JL, Tchkonia T: Cellular senescence: A translational perspective. EBioMedicine 21: 21–28, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schaum N, Lehallier B, Hahn O, Pálovics R, Hosseinzadeh S, Lee SE, et al.; Tabula Muris Consortium: Ageing hallmarks exhibit organ-specific temporal signatures. Nature 583: 596–602, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tabula Muris C; Tabula Muris Consortium: A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 583: 590–595, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Avelar RA, Ortega JG, Tacutu R, Tyler EJ, Bennett D, Binetti P, et al.: A multidimensional systems biology analysis of cellular senescence in aging and disease. Genome Biol 21: 91, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Basisty N, Kale A, Jeon OH, Kuehnemann C, Payne T, Rao C, et al.: A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol 18: e3000599, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang C, Liu G, Luckhardt T, Antony V, Zhou Y, Carter AB, et al.: Serpine 1 induces alveolar type II cell senescence through activating p53-p21-Rb pathway in fibrotic lung disease. Aging Cell 16: 1114–1124, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alcorta DA, Xiong Y, Phelps D, Hannon G, Beach D, Barrett JC: Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc Natl Acad Sci U S A 93: 13742–13747, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stein GH, Drullinger LF, Soulard A, Dulić V: Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol Cell Biol 19: 2109–2117, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jin H, Zhang Y, Ding Q, Wang SS, Rastogi P, Dai DF, et al.: Epithelial innate immunity mediates tubular cell senescence after kidney injury. JCI Insight 4: e125490, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Docherty MH, O’Sullivan ED, Bonventre JV, Ferenbach DA: Cellular senescence in the kidney. J Am Soc Nephrol 30: 726–736, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Al-Awqati Q, Oliver JA: Stem cells in the kidney. Kidney Int 61: 387–395, 2002 [DOI] [PubMed] [Google Scholar]
- 17.Stewart BJ, Ferdinand JR, Clatworthy MR: Using single-cell technologies to map the human immune system - implications for nephrology. Nat Rev Nephrol 16: 112–128, 2020 [DOI] [PubMed] [Google Scholar]
- 18.Stewart BJ, Ferdinand JR, Young MD, Mitchell TJ, Loudon KW, Riding AM, et al.: Spatiotemporal immune zonation of the human kidney. Science 365: 1461–1466, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park J, Shrestha R, Qiu C, Kondo A, Huang S, Werth M, et al.: Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 360: 758–763, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bakken TE, Hodge RD, Miller JA, Yao Z, Nguyen TN, Aevermann B, et al.: Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS One 13: e0209648, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Young MD, Mitchell TJ, Vieira Braga FA, Tran MGB, Stewart BJ, Ferdinand JR, et al.: Single-cell transcriptomes from human kidneys reveal the cellular identity of renal tumors. Science 361: 594–599, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Binet F, Cagnone G, Crespo-Garcia S, Hata M, Neault M, Dejda A, et al.: Neutrophil extracellular traps target senescent vasculature for tissue remodeling in retinopathy. Science 369: eaay5356, 2020 [DOI] [PubMed] [Google Scholar]
- 23.Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, et al.: Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479: 232–236, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang W, Wu J, Zhang Z, Tong T: Characterization of regulatory elements on the promoter region of p16(INK4a) that contribute to overexpression of p16 in senescent fibroblasts. J Biol Chem 276: 48655–48661, 2001 [DOI] [PubMed] [Google Scholar]
- 25.Pajvani UB, Trujillo ME, Combs TP, Iyengar P, Jelicks L, Roth KA, et al.: Fat apoptosis through targeted activation of caspase 8: a new mouse model of inducible and reversible lipoatrophy. Nat Med 11: 797–803, 2005 [DOI] [PubMed] [Google Scholar]
- 26.Han X, Wang R, Zhou Y, Fei L, Sun H, Lai S, et al.: Mapping the mouse cell atlas by microwell-seq. Cell 173: 1307, 2018 [DOI] [PubMed] [Google Scholar]
- 27.Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, et al.: The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 14: 644–658, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tchkonia T, Kirkland JL: Aging, cell senescence, and chronic disease: Emerging therapeutic strategies. JAMA 320: 1319–1320, 2018 [DOI] [PubMed] [Google Scholar]
- 29.Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, Lahat A, et al.: Cellular senescence drives age-dependent hepatic steatosis. Nat Commun 8: 15691, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, et al.: Cellular senescence mediates fibrotic pulmonary disease. Nat Commun 8: 14532, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, Hashmi SK, et al.: Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 40: 554–563, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, et al.: Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of dasatinib plus quercetin in individuals with diabetic kidney disease. EBioMedicine 47: 446–456, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang K, Ponzo TA, Tang H, Mishra PK, Macura SI, Lerman LO: Multiparametric MRI detects longitudinal evolution of folic acid-induced nephropathy in mice. Am J Physiol Renal Physiol 315: F1252–F1260, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hall BM, Balan V, Gleiberman AS, Strom E, Krasnov P, Virtuoso LP, et al.: p16(Ink4a) and senescence-associated β-galactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging (Albany NY) 9: 1867–1884, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, et al.: Senolytics improve physical function and increase lifespan in old age. Nat Med 24: 1246–1256, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ebrahimi B, Crane JA, Knudsen BE, Macura SI, Grande JP, Lerman LO: Evolution of cardiac and renal impairment detected by high-field cardiovascular magnetic resonance in mice with renal artery stenosis. J Cardiovasc Magn Reson 15: 98, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Warner GM, Cheng J, Knudsen BE, Gray CE, Deibel A, Juskewitch JE, et al.: Genetic deficiency of Smad3 protects the kidneys from atrophy and interstitial fibrosis in 2K1C hypertension. Am J Physiol Renal Physiol 302: F1455–F1464, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li F, Huang Q, Luster TA, Hu H, Zhang H, Ng WL, et al.: In vivo epigenetic CRISPR screen identifies Asf1a as an immunotherapeutic target in Kras-mutant lung adenocarcinoma. Cancer Discov 10: 270–287, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Giro-Perafita A, Luo L, Khodadadi-Jamayran A, Thompson M, Akgol Oksuz B, Tsirigos A, et al.: LncRNA RP11-19E11 is an E2F1 target required for proliferation and survival of basal breast cancer. NPJ Breast Cancer 6: 1, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frazzette N, Khodadadi-Jamayran A, Doudican N, Santana A, Felsen D, Pavlick AC, et al.: Decreased cytotoxic T cells and TCR clonality in organ transplant recipients with squamous cell carcinoma. NPJ Precis Oncol 4: 13, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fruchterman TMJ, Reingold EM: Graph drawing by force-directed placement. Softw Pract Exp 21: 1129–1164, 1991 [Google Scholar]
- 42.Khodadadi-Jamayran A, Tsirigos A: Graph drawing-based dimensionality reduction to identify hidden communities in single-cell sequencing spatial representation. bioRxiv. 10.1101/2020.05.05.078550 (Preprint posted May 5, 2020) [DOI]
- 43.Levine JH, Simonds EF, Bendall SC, Davis KL, Amir AD, Tadmor MD, et al.: Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell 162: 184–197, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.The Tabula Muris Consortium: Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562: 367–372, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heng TS, Painter MW; Immunological Genome Project Consortium: The Immunological Genome Project: Networks of gene expression in immune cells. Nat Immunol 9: 1091–1094, 2008 [DOI] [PubMed] [Google Scholar]
- 46.Puranik AS, Leaf IA, Jensen MA, Hedayat AF, Saad A, Kim KW, et al.: Kidney-resident macrophages promote a proangiogenic environment in the normal and chronically ischemic mouse kidney. Sci Rep 8: 13948, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen L, Lee JW, Chou CL, Nair AV, Battistone MA, Păunescu TG, et al.: Transcriptomes of major renal collecting duct cell types in mouse identified by single-cell RNA-seq. Proc Natl Acad Sci U S A 114: E9989–E9998, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiang K, Ferguson CM, Ebrahimi B, Tang H, Kline TL, Burningham TA, et al.: Noninvasive assessment of renal fibrosis with magnetization transfer MR imaging: Validation and evaluation in murine renal artery stenosis. Radiology 283: 77–86, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheng J, Zhou W, Warner GM, Knudsen BE, Garovic VD, Gray CE, et al.: Temporal analysis of signaling pathways activated in a murine model of two-kidney, one-clip hypertension. Am J Physiol Renal Physiol 297: F1055–F1068, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Keppler A, Gretz N, Schmidt R, Kloetzer HM, Groene HJ, Lelongt B, et al.: Plasma creatinine determination in mice and rats: an enzymatic method compares favorably with a high-performance liquid chromatography assay. Kidney Int 71: 74–78, 2007 [DOI] [PubMed] [Google Scholar]
- 51.Ougaard MKE, Kvist PH, Jensen HE, Hess C, Rune I, Søndergaard H: Murine nephrotoxic nephritis as a model of chronic kidney disease. Int J Nephrol 2018: 8424502, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu M, Palmer AK, Ding H, Weivoda MM, Pirtskhalava T, White TA, et al.: Targeting senescent cells enhances adipogenesis and metabolic function in old age. eLife 4: e12997, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Menard J, Catt KJ: Measurement of renin activity, concentration and substrate in rat plasma by radioimmunoassay of angiotensin I. Endocrinology 90: 422–430, 1972 [DOI] [PubMed] [Google Scholar]
- 54.Livak KJ, Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 25: 402–408, 2001 [DOI] [PubMed] [Google Scholar]
- 55.Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O: Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc 4: 1798–1806, 2009 [DOI] [PubMed] [Google Scholar]
- 56.Melk A, Kittikowit W, Sandhu I, Halloran KM, Grimm P, Schmidt BM, et al.: Cell senescence in rat kidneys in vivo increases with growth and age despite lack of telomere shortening. Kidney Int 63: 2134–2143, 2003 [DOI] [PubMed] [Google Scholar]
- 57.Radspieler MM, Schindeldecker M, Stenzel P, Försch S, Tagscherer KE, Herpel E, et al.: Lamin-B1 is a senescence-associated biomarker in clear-cell renal cell carcinoma. Oncol Lett 18: 2654–2660, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zou X, Kwon SH, Jiang K, Ferguson CM, Puranik AS, Zhu X, et al.: Renal scattered tubular-like cells confer protective effects in the stenotic murine kidney mediated by release of extracellular vesicles. Sci Rep 8: 1263, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang X, Krier JD, Amador Carrascal C, Greenleaf JF, Ebrahimi B, Hedayat AF, et al.: Low-energy shockwave therapy improves ischemic kidney microcirculation. J Am Soc Nephrol 27: 3715–3724, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hou J, Cai S, Kitajima Y, Fujino M, Ito H, Takahashi K, et al.: 5-Aminolevulinic acid combined with ferrous iron induces carbon monoxide generation in mouse kidneys and protects from renal ischemia-reperfusion injury. Am J Physiol Renal Physiol 305: F1149–F1157, 2013 [DOI] [PubMed] [Google Scholar]
- 61.ImmGen Consortium: Open-source ImmGen: Mononuclear phagocytes. Nat Immunol 17: 741, 2016 [DOI] [PubMed] [Google Scholar]
- 62.Chen L, Clark JZ, Nelson JW, Kaissling B, Ellison DH, Knepper MA: Renal-tubule epithelial cell nomenclature for single-cell rna-sequencing studies. J Am Soc Nephrol 30: 1358–1364, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zimmerman KA, Bentley MR, Lever JM, Li Z, Crossman DK, Song CJ, et al.: Single-cell RNA sequencing identifies candidate renal resident macrophage gene expression signatures across species. J Am Soc Nephrol 30: 767–781, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sun IO, Santelli A, Abumoawad A, Eirin A, Ferguson CM, Woollard JR, et al.: Loss of renal peritubular capillaries in hypertensive patients is detectable by urinary endothelial microparticle levels. Hypertension 72: 1180–1188, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhu XY, Chade AR, Rodriguez-Porcel M, Bentley MD, Ritman EL, Lerman A, et al.: Cortical microvascular remodeling in the stenotic kidney: role of increased oxidative stress. Arterioscler Thromb Vasc Biol 24: 1854–1859, 2004 [DOI] [PubMed] [Google Scholar]
- 66.Santelli A, Sun IO, Eirin A, Abumoawad AM, Woollard JR, Lerman A, et al.: Senescent kidney cells in hypertensive patients release urinary extracellular vesicles. J Am Heart Assoc 8: e012584, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schaefer L: Small leucine-rich proteoglycans in kidney disease. J Am Soc Nephrol 22: 1200–1207, 2011 [DOI] [PubMed] [Google Scholar]
- 68.Samaras SE, Almodóvar-García K, Wu N, Yu F, Davidson JM: Global deletion of Ankrd1 results in a wound-healing phenotype associated with dermal fibroblast dysfunction. Am J Pathol 185: 96–109, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vasaikar SV, Deshmukh AP, den Hollander P, Addanki S, Kuburich NA, Kudaravalli S, et al.: EMTome: A resource for pan-cancer analysis of epithelial-mesenchymal transition genes and signatures. Br J Cancer 124: 259–269, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Omori S, Wang TW, Johmura Y, Kanai T, Nakano Y, Kido T, et al.: Generation of a p16 reporter mouse and its use to characterize and target p16high cells in vivo. Cell Metab 32: 814–828.e6, 2020 [DOI] [PubMed] [Google Scholar]
- 71.Lefevre L, Iacovoni JS, Martini H, Belliere J, Maggiorani D, Dutaur M, et al.: Kidney inflammaging is promoted by CCR2(+) macrophages and tissue-derived micro-environmental factors. Cell Mol Life Sci 78: 3485–3501, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, Baker DJ: Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 562: 578–582, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM: Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 354: 472–477, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Di Mitri D, Azevedo RI, Henson SM, Libri V, Riddell NE, Macaulay R, et al.: Reversible senescence in human CD4+CD45RA+CD27- memory T cells. J Immunol 187: 2093–2100, 2011 [DOI] [PubMed] [Google Scholar]
- 75.Elder SS, Emmerson E: Senescent cells and macrophages: key players for regeneration? Open Biol 10: 200309, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Berkenkamp B, Susnik N, Baisantry A, Kuznetsova I, Jacobi C, Sörensen-Zender I, et al.: In vivo and in vitro analysis of age-associated changes and somatic cellular senescence in renal epithelial cells. PLoS One 9: e88071, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sis B, Tasanarong A, Khoshjou F, Dadras F, Solez K, Halloran PF: Accelerated expression of senescence associated cell cycle inhibitor p16INK4A in kidneys with glomerular disease. Kidney Int 71: 218–226, 2007 [DOI] [PubMed] [Google Scholar]
- 78.Verzola D, Gandolfo MT, Gaetani G, Ferraris A, Mangerini R, Ferrario F, et al.: Accelerated senescence in the kidneys of patients with type 2 diabetic nephropathy. Am J Physiol Renal Physiol 295: F1563–F1573, 2008 [DOI] [PubMed] [Google Scholar]
- 79.Clements ME, Chaber CJ, Ledbetter SR, Zuk A: Increased cellular senescence and vascular rarefaction exacerbate the progression of kidney fibrosis in aged mice following transient ischemic injury. PLoS One 8: e70464, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang LH, Zhu XY, Eirin A, Nargesi AA, Woollard JR, Santelli A, et al.: Early podocyte injury and elevated levels of urinary podocyte-derived extracellular vesicles in swine with metabolic syndrome: role of podocyte mitochondria. Am J Physiol Renal Physiol 317: F12–F22, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Eirin A, Gloviczki ML, Tang H, Gössl M, Jordan KL, Woollard JR, et al.: Inflammatory and injury signals released from the post-stenotic human kidney. Eur Heart J 34: 540–548a, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Eirin A, Zhang X, Zhu XY, Tang H, Jordan KL, Grande JP, et al.: Renal vein cytokine release as an index of renal parenchymal inflammation in chronic experimental renal artery stenosis. Nephrol Dial Transplant 29: 274–282, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wu X, Yamada-Mabuchi M, Morris EJ, Tanwar PS, Dobens L, Gluderer S, et al.: The Drosophila homolog of human tumor suppressor TSC-22 promotes cellular growth, proliferation, and survival. Proc Natl Acad Sci U S A 105: 5414–5419, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nguyen HT, Bride SH, Badawy AB, Adam RM, Lin J, Orsola A, et al.: Heparin-binding EGF-like growth factor is up-regulated in the obstructed kidney in a cell- and region-specific manner and acts to inhibit apoptosis. Am J Pathol 156: 889–898, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jiménez AP, Traum A, Boettger T, Hackstein H, Richter AM, Dammann RH: The tumor suppressor RASSF1A induces the YAP1 target gene ANKRD1 that is epigenetically inactivated in human cancers and inhibits tumor growth. Oncotarget 8: 88437–88452, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sawaki D, Czibik G, Pini M, Ternacle J, Suffee N, Mercedes R, et al.: Visceral adipose tissue drives cardiac aging through modulation of fibroblast senescence by osteopontin production. Circulation 138: 809–822, 2018 [DOI] [PubMed] [Google Scholar]
- 87.Pedraza CE, Nikolcheva LG, Kaartinen MT, Barralet JE, McKee MD: Osteopontin functions as an opsonin and facilitates phagocytosis by macrophages of hydroxyapatite-coated microspheres: Implications for bone wound healing. Bone 43: 708–716, 2008 [DOI] [PubMed] [Google Scholar]
- 88.Li Y, Lerman LO: Cellular senescence: A new player in kidney injury. Hypertension 76: 1069–1075, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Havasi A, Borkan SC: Apoptosis and acute kidney injury. Kidney Int 80: 29–40, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Toyonaga T, Nakase H, Ueno S, Matsuura M, Yoshino T, Honzawa Y, et al.: Osteopontin deficiency accelerates spontaneous colitis in mice with disrupted gut microbiota and macrophage phagocytic activity. PLoS One 10: e0135552, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wu H, Malone AF, Donnelly EL, Kirita Y, Uchimura K, Ramakrishnan SM, et al.: Single-cell transcriptomics of a human kidney allograft biopsy specimen defines a diverse inflammatory response. J Am Soc Nephrol 29: 2069–2080, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Karaiskos N, Rahmatollahi M, Boltengagen A, Liu H, Hoehne M, Rinschen M, et al.: A single-cell transcriptome atlas of the mouse glomerulus. J Am Soc Nephrol 29: 2060–2068, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Eremina V, Cui S, Gerber H, Ferrara N, Haigh J, Nagy A, et al.: Vascular endothelial growth factor a signaling in the podocyte-endothelial compartment is required for mesangial cell migration and survival. J Am Soc Nephrol 17: 724–735, 2006 [DOI] [PubMed] [Google Scholar]
- 94.Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M, Weisstuch J, et al.: VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med 358: 1129–1136, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Caldemeyer L, Dugan M, Edwards J, Akard L: Long-term side effects of tyrosine kinase inhibitors in chronic myeloid leukemia. Curr Hematol Malig Rep 11: 71–79, 2016 [DOI] [PubMed] [Google Scholar]
- 96.Palmer AK, Xu M, Zhu Y, Pirtskhalava T, Weivoda MM, Hachfeld CM, et al.: Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 18: e12950, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.