

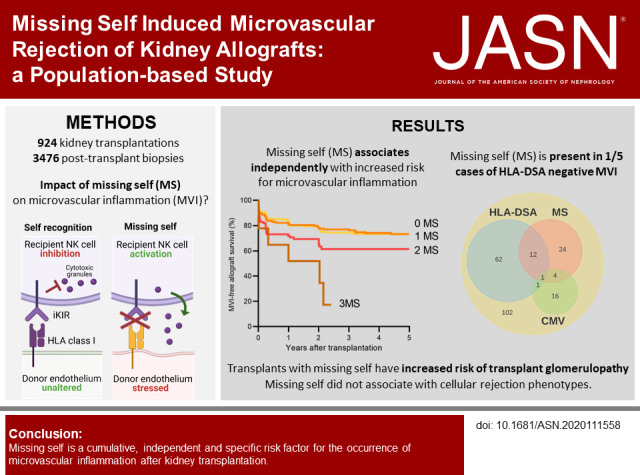
Significance Statement
Microvascular inflammation (MVI) of the kidney transplant is an important determinant of allograft outcome. Although MVI is considered a cardinal manifestation of antibody-mediated rejection, it is often encountered in the absence of circulating donor-specific antibodies, which raises uncertainty about the underlying cause. The authors used genotyping of killer cell Ig-like receptors of recipients and high-resolution HLA genotyping to assess the presence of missing self, a nonhumoral natural killer cell stimulus, in a large cohort of kidney transplantations. They found that missing self specifically increases the risk of MVI after transplantation, and could explain one fifth of patients without detectable antibodies. This study suggests systematic evaluation of missing self improves our understanding of MVI, and might be relevant for improved diagnostic classification and patient risk stratification.
Keywords: natural killer cells, kidney transplantation, missing self, microvascular inflammation, antibody-mediated rejection
Visual Abstract
Abstract
Background
Circulating anti-HLA donor-specific antibodies (HLA-DSA) are often absent in kidney transplant recipients with microvascular inflammation (MVI). Missing self, the inability of donor endothelial cells to provide HLA I–mediated signals to inhibitory killer cell Ig-like receptors (KIRs) on recipient natural killer cells, can cause endothelial damage in vitro, and has been associated with HLA-DSA–negative MVI. However, missing self’s clinical importance as a nonhumoral trigger of allograft rejection remains unclear.
Methods
In a population-based study of 924 consecutive kidney transplantations between March 2004 and February 2013, we performed high-resolution donor and recipient HLA typing and recipient KIR genotyping. Missing self was defined as the absence of A3/A11, Bw4, C1, or C2 donor genotype, with the presence of the corresponding educated recipient inhibitory KIR gene.
Results
We identified missing self in 399 of 924 transplantations. Co-occurrence of missing self types had an additive effect in increasing MVI risk, with a threshold at two concurrent types (hazard ratio [HR], 1.78; 95% confidence interval [95% CI], 1.26 to 2.53), independent of HLA-DSA (HR, 5.65; 95% CI, 4.01 to 7.96). Missing self and lesions of cellular rejection were not associated. No HLA-DSAs were detectable in 146 of 222 recipients with MVI; 28 of the 146 had at least two missing self types. Missing self associated with transplant glomerulopathy after MVI (HR, 2.51; 95% CI, 1.12 to 5.62), although allograft survival was better than with HLA-DSA–associated MVI.
Conclusion
Missing self specifically and cumulatively increases MVI risk after kidney transplantation, independent of HLA-DSA. Systematic evaluation of missing self improves understanding of HLA-DSA–negative MVI and might be relevant for improved diagnostic classification and patient risk stratification.
Kidney transplantation is the most cost-effective treatment for end-stage kidney failure, although the potential of rejection by the host immune system remains an important barrier to improve allograft outcome. Currently used immunosuppressive drugs have proven their value for the prevention and treatment of T cell–mediated rejection (TCMR).1,2 However, as the importance of acute TMCR has lessened in both incidence and functional effect in the past few decades,3–6 it has become clear that antibody-mediated rejection (ABMR) should be regarded as a distinct rejection phenotype with important implications for graft longevity.6–10
Despite increasing insights, treatment approaches targeting involved pathways or cells have so far not proven benefit in clinical studies, suggesting ABMR remains inadequately understood or represents a heterogeneous disease.11 Contributing to the latter hypothesis is that no serum anti-HLA donor-specific antibodies (HLA-DSA) are detectable in a significant number of recipients with ABMR histology, hallmarked by microvascular inflammation (MVI), associating with better outcome than its HLA-DSA–positive counterpart.12,13
Natural killer (NK) cells are pivotal effectors in the pathogenesis of ABMR, and confer an important effect on outcome after allograft infiltration.14–17 We previously demonstrated that NK cells are equally important in patients who are HLA-DSA negative with a histology of ABMR.18 Activation of NK cells is usually not determined by a single stimulus, but rather results from an imbalance between ongoing stimulatory and inhibitory signals.19 Recently, we showed that allogeneic endothelial cell damage can be induced through missing self, defined as the inability of donor endothelial cells to provide corresponding HLA I–mediated signals to specific inhibitory KIR receptors on educated recipient NK cells.20 In a follow-up study, we found that missing self synergizes with noncomplement-fixing donor-specific antibodies to increase NK cell cytotoxicity in vitro, and accelerates graft loss after MVI.21 However, the clinical relevance of missing self as a distinct nonhumoral trigger of allograft rejection remains unclear.
In this study, we investigated the hypothesis that NK cell stimulation through missing self could independently enhance or induce the onset of MVI after kidney transplantation. For this purpose, we performed high-resolution donor and recipient HLA typing and recipient KIR genotyping in an unselected cohort of 924 consecutive kidney transplantations between 2004 and 2013.
Methods
Study Population and Data Collection
All consecutive kidney transplantations in adult recipients at the University Hospitals Leuven between March 2004 and February 2013 were considered eligible. Combined transplantations, kidney transplantation after another solid organ transplant, and transplantations without recipient KIR phenotyping were excluded. Allograft protocol biopsies were performed at 3, 12, 24, and sometimes at 60 months after transplantation, in addition to clinically indicated biopsies, as reported previously.22 All clinicopathological data were prospectively collected during routine clinical follow‐up. The study was approved by the Ethics Committee of the University Hospitals Leuven (S53364, S61850, and S64006), and reported in accordance with the Strengthening the Reporting of Observational studies in Epidemiology guidelines.23
Pathologic Assessment
All post-transplant biopsies were reviewed by a single pathologist (E.L.). The severity of individual histologic lesions was scored semiquantitatively according to the Banff criteria, as described previously.12 The diagnosis of the histologic phenotypes of ABMR, TCMR, and borderline changes was established retrospectively, using the Banff 2019 criteria.24 Borderline changes were diagnosed as foci of tubulitis (t>0) with minor interstitial inflammation (i1) or moderate-severe interstitial inflammation (i2 or i3) with mild (t1) tubulitis. MVI was defined as a sum score of glomerulitis and peritubular capillaritis (i.e., MVI severity) >1. ABMR histology was termed for biopsies fulfilling the first two Banff 2019 criteria for ABMR, not taking into account acute tubular necrosis, due to insufficient clinical information, or gene expression changes.24 Biopsies considered as inadequate according to the Banff guidelines were excluded from the analysis.
HLA Genotyping
The immunologic assessment of this study cohort has been reported previously.12 Briefly, donor and recipient DNA samples were retrospectively genotyped at high resolution for HLA-A, -B, -C, -DRB1, -DRB3, -DRB4, -DRB5, -DQA1, -DQB1, -DPA1, and -DPB1 loci by next generation sequencing. Half of the donors were genotyped using the MIA I NGS FLEX 11 HLA Typing Kit (Immucor Inc., Norcross, GA) on the MiSeq sequencing instrument (Illumina Inc., San Diego, CA). The other half of the donors and all recipients were genotyped at high‐resolution level, covering the extracellular domains of all 11 HLA molecules (exon 2, 3, and 4 for HLA class I, and exon 2 and 3 for HLA class II).
Detection of Circulating HLA-DSA
Pre‐ and post-transplant anti‐HLA antibodies were systematically monitored using a sensitive Luminex-based assay in one histocompatibility laboratory (Red Cross Flanders). Anti-HLA antibodies detected in the recipient serum were analyzed for donor-specificity against HLA-A, -B, -C, -DRβ1345, -DQα1β1, and -DPα1β1 molecules. A possible presence of HLA-DSA was suspected at background-corrected mean fluorescence intensity around 500. For the final assignment of HLA-DSA, as described in detail previously,25 the sera reactivity of the patients was analyzed, taking into account both donor and recipient high-resolution HLA genotyping results. At the time of an allograft biopsy, HLA-DSA positivity was determined by presence of pretransplant HLA-DSA and/or documentation of de novo HLA-DSA during follow-up after transplantation.
KIR Genotyping, Haplotyping, and Definition of Missing Self
Recipients were retrospectively genotyped for the presence of the 14 KIR genes (2DL1, 2DL2, 2DL3, 2DL4, 2DL5, 2DS1, 2DS2, 2DS3, 2DS4, 2DS5, 3DL1, 3DL2, 3DL3, 3DS1) and two pseudogenes (2DP1, 3DP1) at Histogenetics (Ossining, NY) on a MiSeq platform (Illumina Inc., San Diego, CA). KIR A and B haplotypes were defined respectively as the absence or presence of any of the following genes: 2DL2, 2DL5, 2DS1, 2DS2, 2DS3, 2DS5, or 3DS1. Recipient NK cells were considered as educated for an inhibitory KIR gene (2DL1, 2DL2, 2DL3, 3DL1, and 3DL2) only when the corresponding HLA class I antigen was expressed in the recipient as well (i.e., 2DL1+/C2+, 2DL2+/C1+, 2DL3+/C1+, 3DL1+/Bw4+, 3DL2+/A3+, or 3DL2+/A11+). Bw4, C1, and C2 represent cross-reactive groups across different HLA class I antigens. Missing self was defined as the absence of a corresponding donor HLA class I antigen in combination with a specific inhibitory KIR gene in an educated recipient (e.g., donor C2-/recipient 2DL1+C2+). “High” missing self was defined as the co-occurrence of two or more missing self types. An overview of the definitions used for missing self evaluation is provided in Supplemental Table 1 and Figure 1A.
Statistical Analysis
Chi-squared or Fisher’s exact test, where appropriate, were used to compare nominal variables, with Bonferroni correction in the case of multiple pairwise comparisons. Comparison of continuous variables between two or more groups was performed by t test or ANOVA for normally distributed data, and Mann–Whitney U/Kruskal–Wallis test for non-normal distributions, and post-hoc analysis was performed by Tukey test and Dunn’s multiple comparison test, respectively. Kaplan–Meier survival curves were used to plot kidney allograft survival, and survival between groups was compared using log-rank testing, with Bonferroni correction for post-hoc pairwise comparisons. Univariable and multivariable Cox proportional hazard analyses were used to quantify the risk of rejection or allograft failure. Tacrolimus exposure in the first 3 post-transplant months was estimated by calculating the average trough levels before the biopsy performed within the first 3 months after transplantation, weighted for the duration of the preceding interval between measurements. Graft failure was defined as loss of kidney transplant function, that is, return to dialysis or retransplantation, and in the case of death with a functioning graft, we censored at the time of death. When histologic endpoints were used in survival analyses, the last biopsy was considered as the end of follow-up. Logistic mixed-effect regression models were used to evaluate the association of missing self, pretransplant or de novo HLA-DSA, cold ischemia time, and cytomegalovirus (CMV) disease after transplantation with MVI or individual histologic lesions. We included random intercepts and a linear fixed effect of post-transplant time. An ordinal logistic regression model was applied to investigate the association between number of missing self types and MVI severity. A landmark survival analysis was performed at 3 months post-transplant, with a composite endpoint of graft failure or persisting eGFR reduction to 50% of the 3-month value. We used two-sided hypothesis tests with a significance level of 0.05. SAS Software was used for statistical analysis (version 9.4, SAS Insititute Inc., Cary, NC), GraphPad Prism (version 8.0.1, GraphPad Software, San Diego, CA) and Biorender were used for graphical presentation.
Results
Prevalence of Missing Self in the Study Population
Among 924 transplantations between 2004 and 2013, missing self was identified in 399 donor-recipient pairs (43.2%) (Supplemental Figure 1). A3/A11 missing self was present in 127 transplantations, Bw4 in 124 patients, C1 in 90 patients, and C2 in 149 patients. As shown in Figure 1, A and B, multiple missing self types could exist within one recipient-donor pair, with 110 transplantations having two or more mismatches. C1 and C2 missing self were mutually exclusive by definition. Notably, for C1 missing self, two mismatches could occur simultaneously when both KIR2DL2 and KIR2DL3 were expressed by the allograft recipient. No demographic parameters differed between transplants with and without missing self (Table 1). Inherent to the definition, the number of missing self types correlated with class I HLA antigen mismatches (ρ=0.16, P<0.001), although a weaker association was also found with class II mismatches (ρ=0.07, P=0.04).
Figure 1.
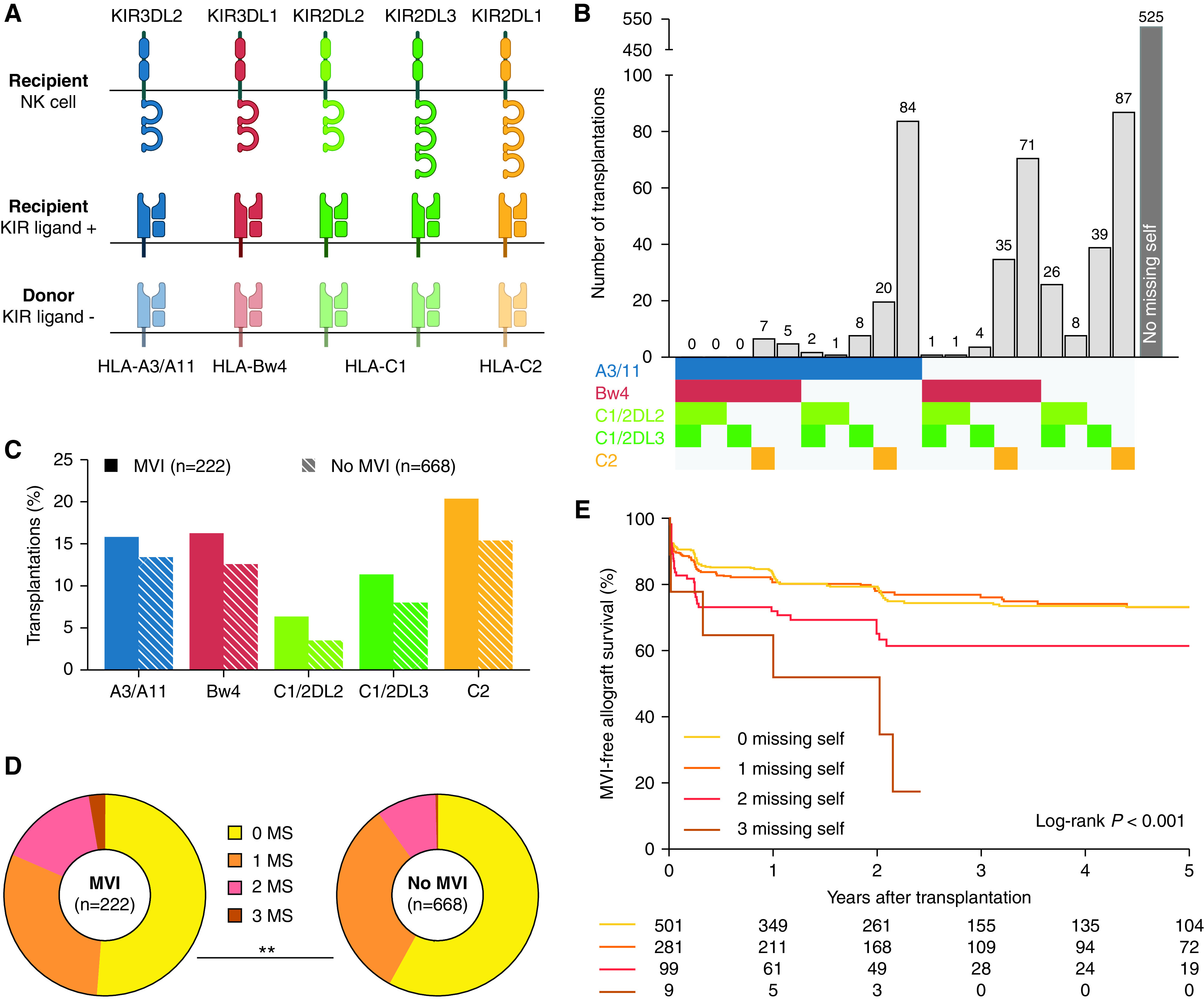
Missing self types cumulatively associate with risk of MVI. (A) Definition of missing self types. (B) Histogram of the possible combinations of missing self types within the study population (n=924). C1 and C2 missing self are mutually exclusive by definition. C1/2DL3 and C1/2DL2 indicate C1 missing self in presence of the KIR2DL2 and KIR2DL3 gene, respectively. (C) Prevalence of specific missing self types in transplantations with and without MVI during histologic follow-up after the baseline biopsy (n=890 transplantations, with 3476 posttransplant biopsies). (D) Number of missing self types in transplantations with and without MVI, chi-squared test for comparison. (E) Kaplan–Meier survival curves for incidence of MVI, censored for recipient death and allograft failure, with groups stratified according to the number of missing self types present within a donor-recipient pair. MS, missing self type. *P<0.05; **P<0.01.
Table 1.
Pre- and peritransplant characteristics of the study population (n=924 transplantations)
| Parameter | No Missing Self (n=525) | Missing Self (n=399) | P |
|---|---|---|---|
| Recipient age, yr, mean (SD) | 53.8 (12.9) | 53.5 (13.5) | 0.68 |
| Recipient female sex | 208 (39.6) | 161 (40.4) | 0.82 |
| Recipient ethnicity | 0.73 | ||
| African | 8 (1.5) | 4 (1.0) | |
| Asian | 1 (0.2) | 1 (0.3) | |
| Caucasian | 515 (98.1) | 394 (98.5) | |
| Hispanic | 1 (0.2) | 0 (0.0) | |
| Donor age, yr, mean (SD) | 47.4 (15.0) | 48.3 (14.7) | 0.36 |
| Donor female sex | 245 (46.7) | 187 (46.9) | 0.95 |
| Donor type | 0.70 | ||
| Living donation | 25 (4.8) | 17 (4.3) | |
| Donation after circulatory death | 83 (15.8) | 71 (17.8) | |
| Donation after brain death | 417 (79.4) | 311 (77.9) | |
| Donor-recipient CMV IgG | 0.35 | ||
| D-R- | 152 (29.0) | 99 (24.8) | |
| D-R+ | 149 (28.4) | 122 (30.6) | |
| D+R- | 112 (21.3) | 79 (19.8) | |
| D+R+ | 112 (21.3) | 97 (24.8) | |
| Cold ischemia time, h, mean (SD) | 14.3 (5.5) | 14.5 (5.5) | 0.68 |
| Delayed graft function | 97 (18.5) | 79 (19.8) | 0.61 |
| Repeat transplantation | 75 (14.3) | 57 (14.3) | 1.000 |
| HLA antigen mismatches, n, mean (SD) | |||
| HLA-A/B/DR | 2.5 (1.3) | 2.9 (1.3) | <0.001a |
| HLA-A/B/DR/DQ | 3.2 (1.6) | 3.7 (1.6) | <0.001a |
| HLA-A | 0.9 (0.7) | 1.1 (0.7) | <0.001a |
| HLA-B | 0.9 (0.6) | 1.1 (0.6) | <0.001a |
| HLA-C | 0.9 (0.7) | 1.1 (0.7) | 0.003a |
| HLA-DQB1 | 0.7 (0.6) | 0.7 (0.6) | 0.23 |
| HLA-DRB1 | 0.7 (0.5) | 0.7 (0.6) | 0.08 |
| Pretransplant HLA-DSA class I | 30 (5.7) | 24 (6.0) | 0.85 |
| A | 11 (2.1) | 9 (2.3) | 0.87 |
| B | 17 (3.2) | 14 (3.5) | 0.82 |
| Cw | 7 (1.3) | 4 (1.0) | 0.77 |
| Pretransplant HLA-DSA class II | 34 (6.5) | 27 (6.8) | 0.86 |
| DRβ1345 | 13 (2.5) | 10 (2.5) | 0.98 |
| DQα1β1 | 14 (2.7) | 17 (4.3) | 0.18 |
| DPα1β1 | 12 (2.3) | 9 (2.3) | 0.98 |
| Induction therapy | 220 (41.9) | 161 (40.4) | 0.64 |
| Immunosuppressive regimen | |||
| Tacrolimus | 484 (92.2) | 361 (90.5) | 0.36 |
| Cyclosporine | 36 (6.9) | 29 (7.3) | 0.81 |
| Mycophenolate acid | 494 (94.1) | 382 (95.7) | 0.27 |
| Azathioprine | 1 (0.2) | 0 (0.0) | 0.38 |
| mTOR inhibitor | 6 (1.1) | 8 (2.0) | 0.29 |
| Corticosteroids | 523 (99.6) | 399 (100.0) | 0.22 |
| Other | 28 (5.3) | 17 (4.3) | 0.45 |
| Causes of kidney disease | 0.76 | ||
| Glomerulonephritis | 132 (25.1) | 107 (26.8) | |
| Cystic nephropathy | 101 (19.2) | 73 (18.3) | |
| Tubulo-interstitial disease | 87 (16.6) | 62 (15.5) | |
| Diabetic nephropathy | 49 (9.3) | 44 (11.0) | |
| Vascular | 39 (7.4) | 24 (6.0) | |
| Congenital | 15 (2.9) | 16 (4.0) | |
| Other | 40 (7.6) | 35 (8.8) | |
| Unknown | 62 (11.8) | 38 (9.5) |
Values are n (%) unless otherwise specified. Percentages may not add up to 100% due to rounding. mTOR: mammalian target of rapamycin.
P values indicate statistical significance.
Association Between Missing Self and MVI
The median follow-up after transplantation was 7.54 years (interquartile range, 5.18–9.93). Histologic follow-up after the baseline biopsy was available for 890 transplantations, comprising 2574 protocol and 902 indication biopsies. In 222 of 890 transplantations (24.9%), MVI was detected in at least one post-transplant biopsy. A comparison of specific missing self types in transplantations with and without MVI showed a trend toward higher prevalence of in the MVI group (Figure 1C). More specifically, the prevalence of two and three co-occurring missing self types was higher in the MVI group (15.7% and 2.7% versus 9.6% and 0.5%, P=0.001, Figure 1D). In a univariable model, the risk of MVI was significantly higher in transplantations with two missing self types (hazard ratio [HR], 1.66; 95% confidence interval [95% CI], 1.13 to 2.42, P=0.009) and 3 missing self types (HR 3.95; 95% CI, 1.74 to 8.98, P<0.001) compared with no missing self, suggesting these mismatches are additive predictors (Figure 1E). One missing self was not associated with an increased risk of MVI (HR, 0.98; 95% CI, 0.72 to 1.32, P=0.87), and the recipient KIR haplotype did not associate with MVI. Other univariable associations of demographic and immunologic factors are shown in Supplemental Table 2. Next to missing self, also female sex, previous transplantation, pretransplant HLA-DSA (Figure 2A), higher number of HLA mismatches and older donor age associated with the incidence of MVI. Donor (D)–recipient (R) CMV IgG status associated with MVI, with D-/R+, D+/R- and D+/R+ transplantations having a higher risk of MVI than D-/R- transplantations (Figure 2B). In a multivariable analysis, high missing self (i.e., 2–3 types) remained independently associated with MVI, together with pretransplant HLA-DSA, D-R CMV IgG status, and the number of HLA class I and class II mismatches (Table 2). Although the prognostic effect of HLA-DSA appeared more important than missing self (HR, 5.65; 95% CI, 4.01 to 7.96, P<0.001 versus HR, 1.78; 95% CI, 1.26 to 2.53, P=0.001), missing self thus conferred an independent risk for the incidence of MVI (Figure 2, C, E, and F). Similarly, in D-/R+, D+/R-, and D+/R+ CMV IgG transplants, missing self posed an independent effect (Figure 2, D and E).
Figure 2.
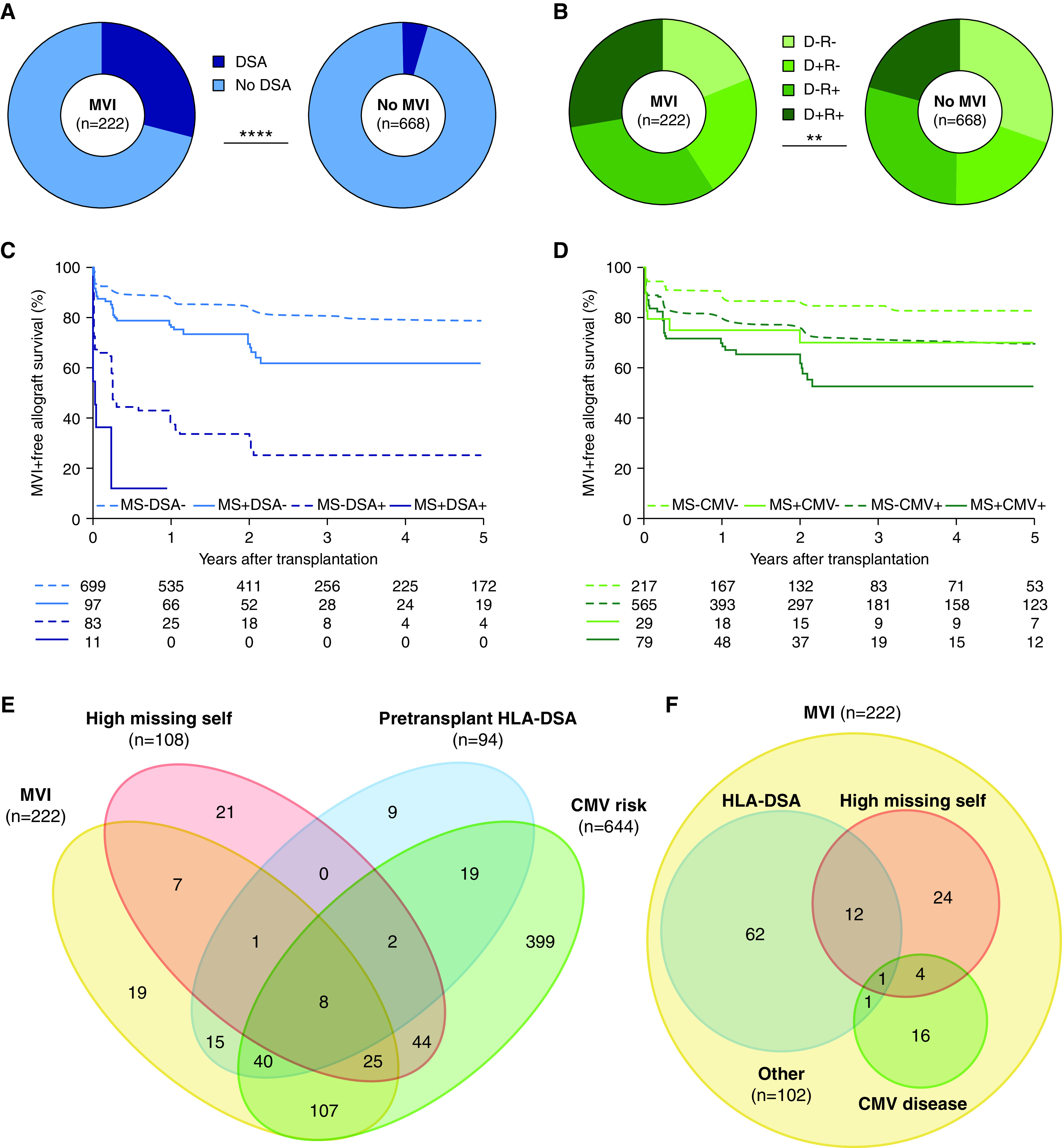
Missing self independently associates with risk of MVI (n=890 transplantations with 3476 post-transplant biopsies). (A) Prevalence of pretransplant HLA-DSA in transplantations with and without MVI during histologic follow-up, chi-squared test for comparison. (B) Pretransplant donor/recipient CMV IgG status in transplantations with and without MVI during follow-up, chi-squared test for comparison. (C) Kaplan–Meier survival curves for incidence of MVI, censored for recipient death and allograft failure, stratified according to high versus low missing self (2–3 versus 0–1 types) and pretransplant HLA-DSA. (D) Survival curves for incidence of MVI, stratified according to high missing self and post-transplant risk for CMV disease (high risk: donor and/or recipient CMV seropositive; low risk: donor and recipient CMV seronegative). (E) Venn diagram depicting the independence of high missing self, pretransplant HLA-DSA and CMV disease risk as pretransplant predictors for MVI (occurring in n=222 transplantations), and the proportion of MVI incidence associated with each predictor. (F) Venn diagram depicting the prevalence of high missing self, prior symptomatic CMV disease and HLA-DSA, either current or previous, at the first moment of diagnosis in the 222 transplantations with MVI. MS, high missing self; +/−, CMV IgG positive/negative. **P<0.01; ****P<0.0001.
Table 2.
Pretransplant determinants of MVI: Multivariable analysis (n=890 transplantations)
| Parameter | Patients, n | Events, n | HR | 95% CI | P |
|---|---|---|---|---|---|
| Recipient sex | |||||
| Male | 538 | 118 | 1 | ||
| Female | 352 | 104 | 1.16 | 0.88 to 1.52 | 0.30 |
| Missing self types | |||||
| Low missing self (0–1) | 782 | 181 | 1 | ||
| High missing self (2–3) | 108 | 41 | 1.78 | 1.26 to 2.53 | 0.001a |
| Pretransplant HLA-DSA | |||||
| No | 796 | 158 | 1 | ||
| Yes | 94 | 64 | 5.65 | 4.01 to 7.96 | <0.001a |
| HLA class I mismatches, per 1-unit increase | 890 | 222 | 1.13 | 1.02 to 1.24 | 0.02a |
| HLA class II mismatches, per 1-unit increase | 890 | 222 | 1.18 | 1.04 to 1.34 | 0.01a |
| Retransplantation | |||||
| No | 764 | 177 | 1 | ||
| Yes | 126 | 45 | 1.13 | 0.78 to 1.66 | 0.52 |
| CMV status | |||||
| D-R- | 246 | 42 | 1 | ||
| D-R+ | 262 | 71 | 1.57 | 1.07 to 2.31 | 0.02a |
| D+R- | 182 | 48 | 1.79 | 1.17 to 2.73 | 0.007a |
| D+R+ | 200 | 61 | 1.77 | 1.18 to 2.64 | 0.005a |
| Donor age, per 1-year increment | 890 | 222 | 1.01 | 1.00 to 1.02 | 0.19 |
Multivariable Cox proportional hazards analysis of MVI incidence after transplantation.
P values indicate statistical significance.
We next investigated the association between NK cell stimuli and allograft histology in the 3476 post-transplant biopsies using logistic mixed-effect regression models (Figure 3A). Correcting for post-transplant time and the baseline covariates used in the multivariable survival analysis, we found that high missing self, defined as at least two missing self types, associated with an increased risk of MVI (odds ratio [OR], 2.16; 95% CI, 1.26 to 3.72; P=0.005) compared with low missing self (absence of, or one missing self), as was the case for prior or current HLA-DSA positivity versus no HLA-DSA (OR, 8.82; 95% CI, 5.24 to 14.87; P<0.001). Preceding CMV disease, defined as biopsy-proven tissue-invasive disease or positive CMV viremia with associated symptoms and/or laboratory alterations, associated with a significantly augmented MVI risk (OR, 2.67; 95% CI, 1.46 to 4.87; P=0.001), in contrast to the KIR BX haplotype (OR, 1.17; 95% CI, 0.77 to 1.77; P=0.48), and cold ischemia time (OR, 1.00 per hour increase; 95% CI, 0.96 to 1.03; P=0.83). High missing self also associated with ABMR histology (OR, 2.26; 95% CI, 1.28 to 4.01; P=0.005), but did not associate with the risk of TCMR (OR, 1.39; 95% CI, 0.82 to 34; P=0.22, Supplemental Table 3). Considering the individual Banff lesions, high missing self associated specifically with glomerulitis and intimal arteritis (Figure 3B). A univariable association with peritubular capillaritis and interstitial inflammation was lost in the multivariable model correcting for baseline covariates (Supplemental Table 4). We found no association with C4d deposition in HLA-DSA–negative recipients (OR, 1.14; 95% CI, 0.73 to 1.77) or HLA-DSA–positive recipients (OR, 1.09; 95% CI, 0.42 to 2.84).
Figure 3.
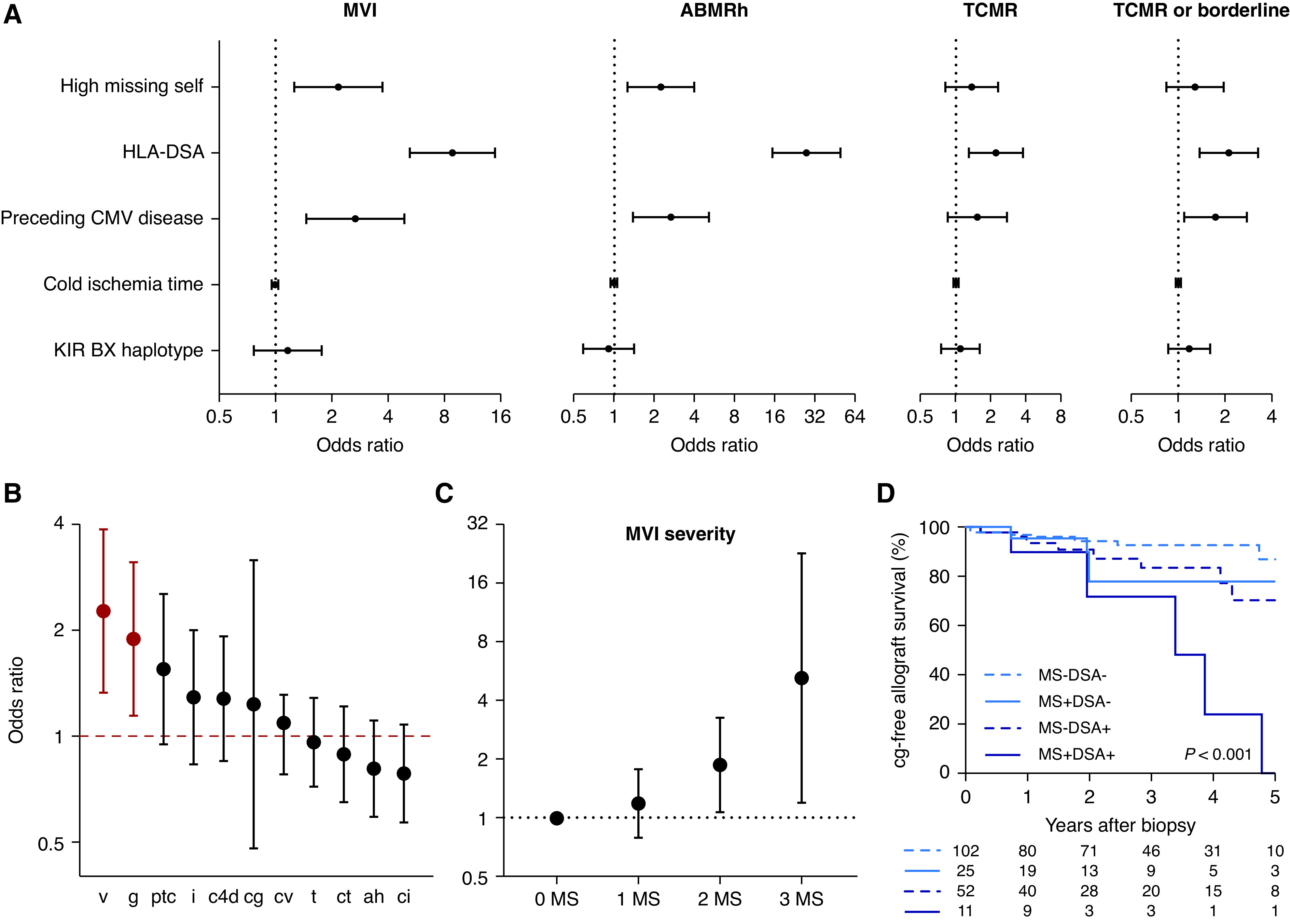
Missing self specifically associates with vascular lesions (n=890 transplantations with 3476 post-transplant biopsies). (A) Association of high missing self (2–3 types) and other NK cell stimuli with kidney allograft rejection phenotypes. The estimates and 95% CIs are on the basis of a logistic mixed-effect regression model with random intercepts and a linear fixed effect of post-transplant time, corrected for HLA class I and class II mismatch number, donor age, recipient sex, and repeat transplantation. HLA-DSA positivity was defined as presence at transplantation or de novo occurrence after transplantation. The hazard ratio for cold ischemia time is given per 1-hour increase. (B) Association of high missing self with presence of individual Banff lesions, on the basis of the logistic mixed-effect regression model described in (A), with additional correction for HLA-DSA and CMV disease. OR with 95% CI are plotted. Red color indicates statistical significance. (C) Association between number of missing self types and severity of MVI in early post-transplant biopsies (n=748 biopsies). Estimates and confidence bounds are on the basis of an ordinal logistic regression model, correcting for the covariates described in (B), in addition to tacrolimus exposure and induction therapy. (D) Kaplan–Meier survival curves for incidence of transplant glomerulopathy after the first MVI biopsy, for patients without cg at diagnosis and with available histologic follow-up (n=190). Patients were stratified according to high missing self and HLA-DSA status. ABMRh, biopsies fulfilling the first 2 Banff 2019 criteria for ABMR; v, intimal arteritis; g, glomerulitis; ptc, peritubular capillaritis; i, interstitial inflammation; c4d, c4d deposition in peritubular capillaries; cg, transplant glomerulopathy; cv, arterial fibro-intimal thickening; t, tubulitis; ct, tubular atrophy; ah, arteriolar hyalinosis; ci, interstitial fibrosis.
Immunosuppression, CMV Disease, and Risk of MVI
We next investigated whether the level of immunosuppression could modulate the effect of missing self in the early post-transplant period. For this purpose, we restricted our analysis to the first biopsy performed within 3 months for each transplantation with available information on tacrolimus trough levels (n=748 biopsies, Supplemental Table 5). Within the first 3 months after transplantation, increasing numbers of missing self types increased the odds of a higher MVI severity compared with no missing self (OR, 1.88; 95% CI, 1.08 to 3.27; P=0.03 and OR, 5.23; 95% CI, 1.21 to 22.54; P=0.03, for two and three missing self types) whereas one missing self type did not (OR, 1.19; 95% CI, 0.79 to 1.78; P=0.41), which further corroborates our earlier results (Figure 3C). Interval-weighted average tacrolimus trough levels before the biopsy were negatively associated with the presence of MVI (OR, 0.89 per 1 ng/ml increase; 95% CI, 0.80 to 0.99; P=0.04). The effect of tacrolimus exposure was most pronounced in transplantations with high missing self (additional OR reduction 0.03 per 1 ng/ml increase; 95% CI, 0.00 to 0.07; P=0.05, Supplemental Figure 2A), whereas no interaction was found between the effect of tacrolimus exposure and presence of HLA-DSA (additional OR reduction 0.01 per 1 ng/ml; 95% CI, −0.03 to 0.04; P=0.72, Supplemental Figure 2B).
We speculated that reduction of immunosuppression might partly explain the association between CMV disease and onset of MVI. To identify risk factors for development of MVI after CMV disease, we examined all first episodes of CMV disease in recipients without MVI before infection, and with at least one follow-up biopsy (n=72 patients). All patients were treated with intravenous ganciclovir. Immunosuppressive changes were implemented in 21 of 72 patients, consisting mostly of the temporary reduction or withdrawal of mycophenolate mofetil, although this was not associated with an increased risk for MVI (HR, 1.49; 95% CI, 0.59 to 3.74; P=0.40). Other potential factors, such as presence of HLA-DSA, primo infection versus reactivation, missing self, and induction therapy were not associated with an increased risk for MVI after CMV disease either (Supplemental Table 6).
Clinicopathological Characteristics of Missing Self–associated MVI
To characterize MVI in association with missing self, we assessed the set of first biopsies per patient that showed MVI (n=222) (Table 3). No HLA-DSA were detected in 146 of 222 recipients (65.8%), of whom 28 of 146 (19.2%) had at least two missing self types (Figure 2F). Notably, 16 of 146 (11.0%) recipients had prior CMV disease in the absence of other risk factors, and 102 of 222 patients (45.9%) of MVI remained without apparent cause (no HLA-DSA, no or only one missing self, no prior CMV disease). A demographic comparison of recipients with missing self but no HLA-DSA (n=28) and HLA-DSA without missing self (n=63) showed more repeat transplantations in the latter group (49.2% versus 10.7%, Bonferroni-adjusted P<0.001). Allograft function and percentage of indication biopsies were similar between the groups. No major differences in presence or severity of individual histologic lesions were found, although MVI biopsies in transplants with high missing self without HLA-DSA tended to have less peritubular C4d deposition than biopsies in transplant with HLA-DSA with low missing self. After diagnosis, 110 of 222 patients with MVI were treated: 90 received steroids as monotherapy and 20 patients received varying combinations of plasmapheresis, ATG, rituximab and bortezomib. Between groups, no significant treatment differences were found. Despite functional and histologic similarities, the follow-up biopsy after diagnosis of MVI with high missing self but without HLA-DSA showed less persistence than HLA-DSA–positive MVI with low missing self (12.0% versus 47.4%, adjusted P=0.01). Nevertheless, high missing self associated with an increased risk of transplant glomerulopathy during histologic follow-up (HR, 2.51; 95% CI, 1.12 to 5.62; P=0.03), independently from HLA-DSA (HR, 3.55; 95% CI, 1.59 to 7.92; P=0.002; Figure 3D and Supplemental Table 7).
Table 3.
Clinicopathological characteristics of MVI in association with missing self and HLA-DSA (n=222 patients)
| Characteristics | Overall (n=222) | HLA-DSA Negative (n=146) | HLA-DSA Positive (n=76) | P | ||
|---|---|---|---|---|---|---|
| No MS (n=118) | MS (n=28) | No MS (n=63) | MS (n=13) | |||
| Transplantation | ||||||
| Repeat transplantation | 45 (20.3) | 7 (5.9)a,b | 3 (10.7)c | 31 (49.2)a,c | 4 (30.8)b | <0.001 |
| Recipient female sex | 104 (46.8) | 69 (58.5) | 14 (50.0) | 27 (42.9) | 8 (61.5) | 0.21 |
| Recipient age, yr, at transplantation, mean (SD) | 53.2 (14.2) | 53.3 (13.7) | 54.4 (15.0) | 52.6 (14.2) | 52.5 (16.4) | 0.95 |
| Living donation | 9 (4.1) | 5 (4.2) | 1 (3.6) | 3 (4.8) | 0 (0.0) | 0.88 |
| Donor female sex | 105 (47.3) | 61 (51.7) | 13 (46.4) | 37 (58.7) | 6 (46.2) | 0.65 |
| Donor age, yr, mean (SD) | 49.1 (15.3) | 49.7 (13.9) | 52.7 (15.6) | 46.5 (16.2) | 47.7 (20.9) | 0.30 |
| Cold ischemia time, h, mean (SD) | 14.9 (5.5) | 15.0 (5.8) | 14.5 (6.5) | 14.4 (4.8) | 16.4 (2.7) | 0.66 |
| Delayed graft function | 63 (28.4) | 30 (25.4) | 6 (21.4) | 21 (33.3) | 6 (46.2) | 0.27 |
| Biopsy | ||||||
| Days after transplantation, median (IQR) | 91 (7–367) | 92 (7–372) | 92 (11–374) | 90 (7–361) | 17 (7–361) | 0.76 |
| Indication biopsy | 122 (55.0) | 65 (55.1) | 13 (46.4) | 35 (55.6) | 9 (69.2) | 0.59 |
| eGFR, ml/min per 0.73 m2, mean (SD) | 33.5 (22.1) | 33.8 (23.0) | 35.6 (20.6) | 33.3 (21.9) | 27.4 (18.5) | 0.74 |
| Proteinuria, g/g creatinine, median (IQR) | 0.3 (0.1–0.6) | 0.3 (0.1–0.6) | 0.2 (0.1–0.3) | 0.3 (0.1–0.9) | 0.6 (0.3–0.7) | 0.12 |
| Preceding CMV disease | 21 (9.5) | 16 (13.6)a | 3 (10.7) | 1 (1.6)a | 1 (7.7) | 0.08 |
| Treatment after biopsy | 110 (49.5) | 58 (49.2) | 12 (42.9) | 32 (50.8) | 8 (61.5) | 0.73 |
| Steroid monotherapy | 90 (40.5) | 49 (41.5) | 11 (39.3) | 25 (39.7) | 5 (38.5) | |
| Othere | 20 (9.0) | 9(7.6) | 1 (3.6) | 7 (11.1) | 3 (23.1) | |
| Histology | ||||||
| Glomerulitis | 149 (67.2) | 73 (61.9) | 18 (64.3) | 48 (76.2) | 10 (76.9) | 0.21 |
| Peritubular capillaritis | 167 (75.3) | 88 (74.6) | 20 (71.4) | 48 (76.2) | 11 (84.6) | 0.83 |
| Intimal arteritis | 70 (31.5) | 36 (30.5) | 9 (32.1) | 21 (33.3) | 4(30.8) | 0.98 |
| C4d deposition in peritubular capillaries | 77 (34.7) | 30 (25.4)a,b | 8(28.6) | 31 (49.2)a | 8 (61.5)b | 0.002 |
| Transplant glomerulopathy | 12 (5.4) | 6 (5.1) | 0 (0.0) | 6 (9.5) | 0(0.0) | 0.22 |
| Thrombotic microangiopathy | 11 (5.1) | 5 (4.3) | 3 (10.3) | 2 (3.8) | 1 (5.9) | 0.48 |
| Tubulitis | 148 (66.6) | 86 (72.9) | 18 (64.3) | 35 (55.6) | 9 (69.2) | 0.13 |
| Interstitial inflammation | 117 (52.7) | 68 (57.6) | 14 (50.0) | 26 (41.3) | 9 (69.2) | 0.11 |
| Arteriolar hyalinosis | 75 (33.8) | 37 (31.4) | 12 (42.9) | 24 (38.1) | 2 (15.4) | 0.28 |
| Interstitial fibrosis | 66 (29.7) | 37 (31.4) | 9 (32.1) | 18 (28.6) | 2 (15.4) | 0.67 |
| Tubular atrophy | 137 (61.7) | 75 (63.6) | 20 (71.4) | 34 (54.0) | 8 (61.5) | 0.41 |
| Arterial fibrointimal thickening | 111 (50.0) | 56 (47.5) | 16 (57.1) | 34 (54.0) | 5 (38.5) | 0.58 |
| Borderline changes | 32 (14.4) | 19 (16.1) | 6 (21.4) | 5 (7.9) | 2 (15.4) | 0.32 |
| T cell-mediated rejection | 93 (41.9) | 55 (46.6) | 9 (32.1) | 22 (34.9) | 7 (53.9) | 0.24 |
| Repeat MVI on next biopsyf | 64 (31.7) | 25 (23.2)a,b | 3 (12.0)c,d | 27 (47.4)a,c | 9 (75.0)b,d | <0.001 |
| Development of cg>0 after the first MVI biopsyg | 31 (14.9) | 10 (9.0)b | 4 (14.3) | 12 (21.1) | 5 (41.7)b | 0.01 |
Data are derived from the first biopsies with MVI during post-transplant histologic follow-up (n=222 transplants). Values are n (%) unless otherwise specified. Individual histologic lesions were considered as absent (Banff score of 0) or present (Banff score of 1, 2 or 3). P values for differences between all groups are given. MS, high missing self (1–2 types); IQR, interquartile range; cg, transplant glomerulopathy.
Significant pairwise comparisons between groups.
Varying combinations of plasmapheresis, anti-thymocyte globulin, rituximab, bortezomib and intravenous Igs.
Only patients with histologic follow-up included.
Only patients without transplant glomerulopathy and with histologic follow-up included.
Missing Self and Kidney Transplant Outcome
In a multivariable Cox proportional hazards analysis after MVI diagnosis (n=222), graft failure associated with HLA-DSA (HR, 2.31; 95% CI, 1.36 to 3.94; P=0.002) and with lower eGFR at the time of biopsy (HR, 0.96 per 1 ml/min per 1.73m2 increase; 95% CI, 0.94 to 0.99; P=0.001), whereas high missing self did not associate with the risk of graft failure (HR, 1.23; 95% CI, 0.65 to 2.33; P=0.53; Supplemental Figure 3A and Supplemental Table 8). To assess the association between MVI in the presence of missing self and allograft function in the general cohort, we next performed a landmark survival analysis starting from 3 months post-transplant, with a composite endpoint of graft failure or persisting eGFR reduction to 50% of the 3-month value. Transplantations were stratified according to the presence of MVI in the preceding trimester and, if present, substratified by high missing self and HLA-DSA status. In the absence of HLA-DSA, both in low (n=67) and high (n=19) missing self transplantations, MVI in the first 3 post-transplant months associated with an increased risk of the composite endpoint, compared with transplantations without MVI (n=709, Bonferroni-adjusted P=0.02 and P<0.001, respectively). However, transplants with MVI and high missing self, but without HLA-DSA (n=19), had better outcome than transplants with HLA-DSA–positive MVI with low missing self (n=42, adjusted P=0.005). Thus, although missing self independently associated with an increased incidence of MVI and development of transplant glomerulopathy, HLA-DSA status was the more important predictor of allograft outcome in patients with established MVI (Supplemental Figure 3B).
Discussion
In this study, we demonstrated that the risk of MVI is increased in kidney transplantations with missing self, defined as the inability of donor allograft endothelium to provide specific HLA class I binding to corresponding inhibitory KIRs on educated recipient NK cells. Importantly, the effect of multiple missing self types was additive and occurred independently from other NK cell stimuli, such as HLA-DSA and CMV disease. The biologic relevance of the strong and specific correlation with glomerulitis was supported by the increased incidence of transplant glomerulopathy after MVI diagnosis in transplantations with missing self. The clinicopathological phenotype associated with missing self but without HLA-DSA was similar to HLA-DSA–positive MVI (ABMR), although allograft survival was better.
Previous studies have also investigated the association between KIR–HLA ligand mismatches and kidney allograft rejection. Kreijveld et al. reported no difference in KIR ligand incompatibility between transplantations with or without allograft rejection after reduction of immunosuppression.26 In a single-center cohort study of 760 transplant pairs, Hanvesakul et al. did not detect a correlation between KIR ligand incompatibility and the incidence of allograft rejection after transplantation.27 However, recipient KIR genotyping was lacking or not incorporated in the analyses of these studies, thus precluding a correct definition of missing self. In line with our methodology, by combining KIR genotyping and ligand mismatch analysis, Littera et al. observed a trend toward increased prevalence of missing self among 42 kidney allograft recipients with “chronic rejection,” but not differentiating between the different histologic phenotypes of rejection.28 We found that missing self exclusively associates with the occurrence of MVI. The lack of association with cellular rejection phenotypes in our study cautions against the use of rejection as an encompassing histologic endpoint and warrants a reappraisal of previous negative association studies.
Despite similar histologic characteristics, patients with MVI and high missing self but without HLA-DSA had less recurrence and better allograft survival than MVI in patients with HLA-DSA and low missing self in our study. This could imply that missing self–induced NK cell activation is a more transient or weaker phenomenon, and help explain conflicting results reported in previous studies: whereas Bergen et al. reported a 25% decrease in 10-year allograft survival in a HLA-A, -B, and -DR matched transplantations with missing self, this was later refuted in a larger cohort study by Tran et al.29,30 Despite the ostensible lack of association with allograft function, the increased incidence of transplant glomerulopathy indicates that missing self can induce structural alterations to the allograft microvasculature. The apparent discrepancy between increased transplant glomerulopathy and lack of prognostic implications is further supported by a recent archetypical analysis of transplant glomerulopathy, where it was shown that archetypes with less frequent HLA-DSA positivity associated with better outcome than archetypes with a higher fraction of patients who were HLA-DSA positive.31
This analysis complements our recent report on NK cell–mediated endothelial cytotoxicity triggered by missing self in a series of in vitro and murine experiments, and the finding that HLA-DSA–negative recipients with MVI have a higher frequency of missing self in comparison to a matched control group.20 In the largest cohort study of missing self after kidney transplantation to date, this is the first clinical demonstration of a specific and cumulative association between missing self and post-transplant occurrence of MVI. In addition, we demonstrate this risk occurs independently from recipient HLA-DSA, which clinically translates our earlier analysis that missing self synergizes with noncomplement fixing DSA to enhance in vitro NK cell cytotoxicity.21 This interplay was further corroborated by our current finding that, after diagnosis of MVI, missing self in combination with HLA-DSA resulted in a higher degree of persistence and new transplant glomerulopathy in follow-up biopsies. However, in contrast to the Lyon cohort, missing self did not further increase the risk of graft failure in HLA-DSA–positive MVI. We speculate that this difference may be attributed to heterogeneous complement-fixing properties of the HLA-DSA in our cohort, which are known to influence graft outcome.32,33 Together with the findings from in vitro and preclinical experiments, and analogies with hematopoietic stem cell transplantation, the specific and temporal associations of missing self with MVI, which displays a biologic gradient, provide substantial arguments for a causative role for missing self in the onset of MVI. Missing self–associated MVI is thus to be considered as a true form of rejection, defined as inflammation in kidney allografts resulting from the donor-recipient genetic mismatch.
In our study, two thirds of recipients with MVI had no detectable HLA-DSA at the time of diagnosis, of which one fifth could be explained by the presence of missing self. In the current Banff diagnostic categorization of kidney transplant rejection, HLA-DSA–negative MVI is considered as either ABMR (when C4d is positive) or no ABMR (C4d negative).24 The therapeutic implications of both categories can be harmful for patients with HLA-DSA–negative MVI: either they are exposed to the use of unproven and potentially harmful treatment when an undetected humoral etiology is assumed, or the possibility of ongoing nonhumoral alloimmune mechanisms is largely ignored. Because novel approaches to improve long-term kidney allograft outcome are urgently needed, our work supports the exploration of NK cell modulation as a therapeutic strategy for vascular rejection phenotypes. Additional studies are required to identify which patients with missing self–induced MVI may benefit from treatment, and which therapy should be preferentially administered. Interestingly, tacrolimus exposure associated with a decreased risk of early MVI, specifically in transplantations with high missing self, potentially suggesting targeting higher trough levels could be relevant for prevention in those at risk. Although several in vitro studies have shown that calcineurin inhibitors affect NK cell activity,34–36 prospective clinical studies will be necessary to confirm this finding. Taken together, our work highlights an unrecognized group for whom no appropriate therapy is available, and where all efforts should go into identifying the likely cause of these “unexplained” patients.37 This could potentially be addressed in future updates of the Banff classification.
Our findings are not only relevant for clinical classification of kidney transplant biopsies, but also translate basic scientific concepts of NK cell biology to the human setting. NK cell activation results from the interplay between activating and inhibitory signals.19 This is further exemplified by the strong association between post-transplant CMV disease and the occurrence of MVI. After CMV disease, NK cells can display adaptive-like properties, by the expansion and differentiation of inhibitory KIR-expressing NK cells capable of increased responsiveness, which theoretically enhances the risk for missing self.38,39 However, the effect of CMV disease was also seen in recipients without missing self and involved cellular rejection as well, in line with previous reports.40,41 In a multivariable analysis, reduction of immunosuppression after CMV disease did not account for the increased risk of MVI. Further dedicated statistical studies will be necessary to disentangle the time-dependent relationships between CMV disease, NK cell activation, and allograft rejection.
This study has several limitations. First, we did not test for non–HLA-DSA, as their evaluation is not standardized, cutoff values are unclear, and there is no consensus on which non-HLA-DSA are clinically relevant. Therefore, it is possible certain patients with unexplained MVI in this cohort were caused by non–HLA-DSA.42–44 Second, we adopted a reductionist view on NK cell activation by focusing solely on inhibitory KIR signaling. As mentioned earlier, NK cell triggering depends on the interplay of a plethora of membrane-bound receptors, many of which have unknown ligands or functions. We speculate that the threshold of necessary missing self types for NK cell activation fluctuates in function of the proinflammatory character of the cellular environment, which in turn varies in response to infection, ischemia, or other causes of allograft stress, although we did not observe associations with donor characteristics or organ storage conditions. Third, NK cells are characterized by stochastic gene expression, and cellular subsets exhibit great variability in absolute numbers between individuals with a similar genotype.45 Therefore, although missing self can be predicted within a recipient-donor pair, the range of inhibitory KIR expressing NK cells that are present is subject to interindividual variation. These dynamic and probabilistic aspects of NK cell activation could not be addressed in this observational study. Targeted experiments with deep spatiotemporal phenotyping of circulating NK cells are needed to further refine our results.
In conclusion, we have identified a type of kidney transplant rejection that is related to missing self. The associated clinicopathological phenotype is similar to HLA-DSA–positive MVI, although transplant survival is better. Systematic evaluation of missing self improves our understanding of HLA-DSA–negative MVI, may refine personalized risk stratification and lead to the development of new treatment strategies for patients who undergo kidney transplantation.
Disclosures
B. Sprangers reports being a scientific advisor or member as an expert ad hoc for the European Medicines Agency. D. Kuypers reports having consultancy agreements with Astellas, CSL Behring, and UCB; reports receiving research funding from Astellas; reports receiving honoraria from Astellas, CSL Behring, and UCB; reports being a scientific advisor or member as Associate Editor of Transplantation, editorial board member of Current Clinical Pharmacology, Therapeutic Drug Monitoring, and Transplantation Reviews; and reports receiving speakers bureau from Astellas. M. Emonds reports being a scientific advisor or member with the Eurotransplant Tissue Typing Advisory Committee. M. Naesens reports being a scientific advisor or member via the editorial boards for several journals and is an advisor for the European Medicines Agency. O. Thaunat reports having consultancy agreements from Novartis; reports receiving research funding from Biomerieux, BMS, Immucor, and Novartis; and reports being a scientific advisor or member of the European Society for Organ Transplantation. All remaining authors have nothing to disclose.
Funding
This project was supported by a project grant from Research Foundation Flanders grants G087620N, 1196119N (to J. Callemeyn), and 1844019N and 1842919N (to M. Naesens and B. Sprangers).
Supplementary Material
Acknowledgments
J. Callemeyn, M.-P. Emonds, and M. Naesens designed the study; J. Callemeyn, A. Koenig, O. Thaunat, and M. Naesens performed data analysis and interpretation; J. Callemeyn, E. Lerut, A. Senev, M. Coemans, D. Kuypers, B. Sprangers, M.-P. Emonds, and M. Naesens were responsible for data and sample collection; J. Callemeyn and M. Naesens wrote the original draft; and all authors reviewed and edited the manuscript.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2020111558/-/DCSupplemental.
Supplemental Table 1. Definitions of epitopes, KIR haplotypes and missing self.
Supplemental Table 2. Pretransplant determinants of MVI: univariable analysis (n=890 transplantations).
Supplemental Table 3. The association between NK cell stimuli and kidney transplant rejection phenotypes (n=3476 biopsies).
Supplemental Table 4. The association between missing self and individual histological lesions (n=3476 biopsies).
Supplemental Table 5. Association of missing self and peritransplantation factors with early occurrence of MVI (n=748 biopsies).
Supplemental Table 6. Risk factors for development of MVI after CMV disease (n=72 transplantations).
Supplemental Table 7. Determinants of transplant glomerulopathy after diagnosis of MVI (n=190 patients).
Supplemental Table 8. Predictors of allograft failure after diagnosis of MVI (n=222 patients).
Supplemental Figure 1. Study cohort overview.
Supplemental Figure 2. Tacrolimus exposure associates with decreased risk of early MVI in transplantations with missing self (n=748 biopsies).
Supplemental Figure 3. Missing self does not influence allograft function after MVI diagnosis.
References
- 1.Samaniego M, Becker BN, Djamali A: Drug insight: maintenance immunosuppression in kidney transplant recipients. Nat Clin Pract Nephrol 2: 688–699, 2006 [DOI] [PubMed] [Google Scholar]
- 2.Halloran PF: Immunosuppressive drugs for kidney transplantation. N Engl J Med 351: 2715–2729, 2004 [DOI] [PubMed] [Google Scholar]
- 3.Coemans M, Süsal C, Döhler B, Anglicheau D, Giral M, Bestard O, et al.: Analyses of the short- and long-term graft survival after kidney transplantation in Europe between 1986 and 2015. Kidney Int 94: 964–973, 2018 [DOI] [PubMed] [Google Scholar]
- 4.Lamb KE, Lodhi S, Meier-Kriesche HU: Long-term renal allograft survival in the United States: A critical reappraisal. Am J Transplant 11: 450–462, 2011 [DOI] [PubMed] [Google Scholar]
- 5.Meier-Kriesche HU, Schold JD, Srinivas TR, Kaplan B: Lack of improvement in renal allograft survival despite a marked decrease in acute rejection rates over the most recent era. Am J Transplant 4: 378–383, 2004 [DOI] [PubMed] [Google Scholar]
- 6.Halloran PF, Chang J, Famulski K, Hidalgo LG, Salazar ID, Merino Lopez M, et al.: Disappearance of T cell-mediated rejection despite continued antibody-mediated rejection in late kidney transplant recipients. J Am Soc Nephrol 26: 1711–1720, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sellarés J, de Freitas DG, Mengel M, Reeve J, Einecke G, Sis B, et al.: Understanding the causes of kidney transplant failure: The dominant role of antibody-mediated rejection and nonadherence. Am J Transplant 12: 388–399, 2012 [DOI] [PubMed] [Google Scholar]
- 8.Naesens M, Kuypers DRJ, De Vusser K, Evenepoel P, Claes K, Bammens B, et al.: The histology of kidney transplant failure: A long-term follow-up study. Transplantation 98: 427–435, 2014 [DOI] [PubMed] [Google Scholar]
- 9.Loupy A, Vernerey D, Tinel C, Aubert O, Duong van Huyen JP, Rabant M, et al.: Subclinical rejection phenotypes at 1 year post-transplant and outcome of kidney allografts. J Am Soc Nephrol 26: 1721–1731, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Loon E, Senev A, Lerut E, Coemans M, Callemeyn J, Van Keer JM, et al.: Assessing the complex causes of kidney allograft loss. Transplantation 104: 2557–2566, 2020 [DOI] [PubMed] [Google Scholar]
- 11.Schinstock CA, Mannon RB, Budde K, Chong AS, Haas M, Knechtle S, et al.: Recommended treatment for antibody-mediated rejection after kidney transplantation: The 2019 Expert Consensus From the Transplantion Society Working Group. Transplantation 104: 911–922, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Senev A, Coemans M, Lerut E, Van Sandt V, Daniëls L, Kuypers D, et al.: Histological picture of antibody-mediated rejection without donor-specific anti-HLA antibodies: Clinical presentation and implications for outcome. Am J Transplant 19: 763–780, 2019 [DOI] [PubMed] [Google Scholar]
- 13.Luque S, Lúcia M, Melilli E, Lefaucheur C, Crespo M, Loupy A, et al.: Value of monitoring circulating donor-reactive memory B cells to characterize antibody-mediated rejection after kidney transplantation. Am J Transplant 19: 368–380, 2019 [DOI] [PubMed] [Google Scholar]
- 14.Yazdani S, Callemeyn J, Gazut S, Lerut E, de Loor H, Wevers M, et al.: Natural killer cell infiltration is discriminative for antibody-mediated rejection and predicts outcome after kidney transplantation. Kidney Int 95: 188–198, 2019 [DOI] [PubMed] [Google Scholar]
- 15.Kohei N, Tanaka T, Tanabe K, Masumori N, Dvorina N, Valujskikh A, et al.: Natural killer cells play a critical role in mediating inflammation and graft failure during antibody-mediated rejection of kidney allografts. Kidney Int 89: 1293–1306, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parkes MD, Halloran PF, Hidalgo LG: Evidence for CD16a-mediated NK cell stimulation in antibody-mediated kidney transplant rejection. Transplantation 101: e102–e111, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hidalgo LG, Sis B, Sellares J, Campbell PM, Mengel M, Einecke G, et al.: NK cell transcripts and NK cells in kidney biopsies from patients with donor-specific antibodies: Evidence for NK cell involvement in antibody-mediated rejection. Am J Transplant 10: 1812–1822, 2010 [DOI] [PubMed] [Google Scholar]
- 18.Callemeyn J, Lerut E, de Loor H, Arijs I, Thaunat O, Koenig A, et al.: Transcriptional changes in kidney allografts with histology of antibody-mediated rejection without anti-HLA donor-specific antibodies. J Am Soc Nephrol 31: 2168–2183, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lanier LL: Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol 9: 495–502, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koenig A, Chen CC, Marçais A, Barba T, Mathias V, Sicard A, et al.: Missing self triggers NK cell-mediated chronic vascular rejection of solid organ transplants. Nat Commun 10: 5350, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koenig A, Mezaache S, Callemeyn J, Barba T, Mathias V, Sicard A, et al.: Missing self-induced activation of NK cells combines with non-complement-fixing donor-specific antibodies to accelerate kidney transplant loss in chronic antibody-mediated rejection. J Am Soc Nephrol 32: 479–494, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coemans M, Van Loon E, Lerut E, Gillard P, Sprangers B, Senev A, et al.: Occurrence of diabetic nephropathy after renal transplantation despite intensive glycemic control: An observational cohort study. Diabetes Care 42: 625–634, 2019 [DOI] [PubMed] [Google Scholar]
- 23.Vandenbroucke JP, von Elm E, Altman DG, Gøtzsche PC, Mulrow CD, Pocock SJ, et al.; STROBE Initiative: Strengthening the Reporting of Observational Studies in Epidemiology (STROBE): Explanation and elaboration. PLoS Med 4: e297, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loupy A, Haas M, Roufosse C, Naesens M, Adam B, Afrouzian M, et al.: The Banff 2019 Kidney Meeting Report (I): Updates on and clarification of criteria for T cell- and antibody-mediated rejection. Am J Transplant 20: 2318–2331, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Senev A, Emonds MP, Van Sandt V, Lerut E, Coemans M, Sprangers B, et al.: Clinical importance of extended second field high-resolution HLA genotyping for kidney transplantation. Am J Transplant 20: 3367–3378, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kreijveld E, van der Meer A, Tijssen HJ, Hilbrands LB, Joosten I: KIR gene and KIR ligand analysis to predict graft rejection after renal transplantation. Transplantation 84: 1045–1051, 2007 [DOI] [PubMed] [Google Scholar]
- 27.Hanvesakul R, Kubal C, Moore J, Neil D, Cook M, Ball S, et al.: KIR and HLA-C interactions promote differential dendritic cell maturation and is a major determinant of graft failure following kidney transplantation. PLoS One 6: e23631, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Littera R, Piredda G, Argiolas D, Lai S, Congeddu E, Ragatzu P, et al.: KIR and their HLA Class I ligands: Two more pieces towards completing the puzzle of chronic rejection and graft loss in kidney transplantation. PLoS One 12: e0180831, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Bergen J, Thompson A, Haasnoot GW, Roodnat JI, de Fijter JW, Claas FH, et al.: KIR-ligand mismatches are associated with reduced long-term graft survival in HLA-compatible kidney transplantation. Am J Transplant 11: 1959–1964, 2011 [DOI] [PubMed] [Google Scholar]
- 30.Tran TH, Unterrainer C, Fiedler G, Döhler B, Scherer S, Ruhenstroth A, et al.: No impact of KIR-ligand mismatch on allograft outcome in HLA-compatible kidney transplantation. Am J Transplant 13: 1063–1068, 2013 [DOI] [PubMed] [Google Scholar]
- 31.Aubert O, Higgins S, Bouatou Y, Yoo D, Raynaud M, Viglietti D, et al.: Archetype analysis identifies distinct profiles in renal transplant recipients with transplant glomerulopathy associated with allograft survival. J Am Soc Nephrol 30: 625–639, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sicard A, Ducreux S, Rabeyrin M, Couzi L, McGregor B, Badet L, et al.: Detection of C3d-binding donor-specific anti-HLA antibodies at diagnosis of humoral rejection predicts renal graft loss. J Am Soc Nephrol 26: 457–467, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Loupy A, Lefaucheur C, Vernerey D, Prugger C, Duong van Huyen JP, Mooney N, et al.: Complement-binding anti-HLA antibodies and kidney-allograft survival. N Engl J Med 369: 1215–1226, 2013 [DOI] [PubMed] [Google Scholar]
- 34.Morteau O, Blundell S, Chakera A, Bennett S, Christou CM, Mason PD, et al.: Renal transplant immunosuppression impairs natural killer cell function in vitro and in vivo. PLoS One 5: e13294, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meehan AC, Mifsud NA, Nguyen THO, Levvey BJ, Snell GI, Kotsimbos TC, et al.: Impact of commonly used transplant immunosuppressive drugs on human NK cell function is dependent upon stimulation condition. PLoS One 8: e60144, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pradier A, Papaserafeim M, Li N, Rietveld A, Kaestel C, Gruaz L, et al.: Small-molecule immunosuppressive drugs and therapeutic immunoglobulins differentially inhibit NK cell effector functions in vitro. Front Immunol 10: 556, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Callemeyn J, Ameye H, Lerut E, Senev A, Coemans M, Van Loon E, et al.: Revisiting the changes in the Banff classification for antibody-mediated rejection after kidney transplantation [published online ahead of print, December 31, 2020]. Am J Transplant 2020 [DOI] [PubMed] [Google Scholar]
- 38.Foley B, Cooley S, Verneris MR, Pitt M, Curtsinger J, Luo X, et al.: Cytomegalovirus reactivation after allogeneic transplantation promotes a lasting increase in educated NKG2C+ natural killer cells with potent function. Blood 119: 2665–2674, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Béziat V, Liu LL, Malmberg JA, Ivarsson MA, Sohlberg E, Björklund AT, et al.: NK cell responses to cytomegalovirus infection lead to stable imprints in the human KIR repertoire and involve activating KIRs. Blood 121: 2678–2688, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stern M, Hirsch H, Cusini A, van Delden C, Manuel O, Meylan P, et al.; Members of Swiss Transplant Cohort Study: Cytomegalovirus serology and replication remain associated with solid organ graft rejection and graft loss in the era of prophylactic treatment. Transplantation 98: 1013–1018, 2014 [DOI] [PubMed] [Google Scholar]
- 41.Hasanzamani B, Hami M, Zolfaghari V, Torkamani M, Ghorban Sabagh M, Ahmadi Simab S: The effect of cytomegalovirus infection on acute rejection in kidney transplanted patients. J Renal Inj Prev 5: 85–88, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reindl-Schwaighofer R, Heinzel A, Kainz A, van Setten J, Jelencsics K, Hu K, et al.; iGeneTRAiN consortium: Contribution of non-HLA incompatibility between donor and recipient to kidney allograft survival: genome-wide analysis in a prospective cohort. Lancet 393: 910–917, 2019 [DOI] [PubMed] [Google Scholar]
- 43.Steers NJ, Li Y, Drace Z, D’Addario JA, Fischman C, Liu L, et al.: Genomic mismatch at LIMS1 locus and kidney allograft rejection. N Engl J Med 380: 1918–1928, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Delville M, Lamarthée B, Pagie S, See SB, Rabant M, Burger C, et al.: Early acute microvascular kidney transplant rejection in the absence of anti-HLA antibodies is associated with preformed IgG antibodies against diverse glomerular endothelial cell antigens. J Am Soc Nephrol 30: 692–709, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Husain Z, Alper CA, Yunis EJ, Dubey DP: Complex expression of natural killer receptor genes in single natural killer cells. Immunology 106: 373–380, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.