

SUMMARY
Mutant isocitrate dehydrogenase (IDH) 1 and 2 play a pathogenic role in cancers including acute myeloid leukemia (AML) by producing oncometabolite 2-hydroxyglutarate (2-HG). We recently reported that tyrosine phosphorylation activates IDH1 R132H mutant in AML cells. Here we show that mutant IDH2 (mIDH2) R140Q commonly has K413-acetylation, which negatively regulates mIDH2 activity in human AML cells by attenuating dimerization and blocking binding of substrate (α-ketoglutarate) and cofactor (NADPH). Mechanistically, K413-acetylation of mitochondrial mIDH2 is achieved through a series of hierarchical phosphorylation events mediated by tyrosine kinase FLT3, which phosphorylates mIDH2 to recruit upstream mitochondrial acetyltransferase ACAT1 and simultaneously activates ACAT1 and inhibits upstream mitochondrial deacetylase SIRT3 through tyrosine-phosphorylation. Moreover, we found that the intrinsic enzyme activity of mIDH2 is much higher than mIDH1, thus the inhibitory K413-acetylation optimizes leukemogenic ability of mIDH2 in AML cells by both producing sufficient 2-HG for transformation and avoiding cytotoxic accumulation of intracellular 2-HG.
eTOC Blurb
Oncometabolite 2-hydroxyglutarate (2-HG) has been reported to suppress transformation in AML cells. In this article, Chen et al. report that different intracellular concentrations of 2-HG correlate with its different cellular functions, while inhibitory K413-acetylation optimizes leukemogenic ability of mutant IDH2 in AML cells by both producing sufficient 2-HG for transformation and avoiding cytotoxic accumulation of intracellular 2-HG.
INTRODUCTION
Isocitrate dehydrogenase isoforms 1 and 2 (IDH1 and IDH2) are highly homologous and represent two frequently mutated genes in adult acute myeloid leukemia (AML) (Cairns and Mak, 2013). IDH1 and IDH2 which are located in the cytoplasm and mitochondria, respectively, play an important role in many metabolic processes in cells including bioenergetics, biosynthesis, and redox homeostasis (Cairns and Mak, 2013; Inoue et al., 2016; Jiang et al., 2016; Metallo et al., 2011). Active IDH enzymes form homodimers and are involved in citrate metabolism with reactions that convert isocitrate to α-ketoglutarate (α-KG) and production of NADPH through reduction of cofactor NADP+ (Cairns and Mak, 2013). Besides its important metabolic function as a key intermediate in the Krebs cycle and glutaminolysis, α-KG also functions as a ligand that is required for the biological activity of diverse α-KG-dependent dioxygenases including TET2 and histone demethylases including JmjC (Inoue et al., 2016). Recently, somatic mutations of the R132 codon in the catalytic site of IDH1 were identified in glioblastoma (GBM) and AML patients. Subsequently, mutations of the corresponding codon in IDH2 (R172), as well as a novel R140Q mutation were also frequently identified in AML patients (Dang et al., 2009; Green and Beer, 2010; Gross et al., 2010; Marcucci et al., 2010; Mardis et al., 2009; Parsons et al., 2008; Ward et al., 2010; Yan et al., 2009). IDH1/2 mutations including IDH1 R132, IDH2 R172, and IDH2 R140 mutations are mutually exclusive in AML patients and usually in a heterozygous pattern with only one allele harboring an IDH1/2 mutation (Cairns and Mak, 2013). Mutant IDH has a gain-of-function activity that allows the mutant enzyme to convert α-KG to 2-hydroxyglutarate (2-HG), an oncometabolite that functions as a competitive inhibitor of α-KG-dependent enzymes such as TET2. This provides an epigenetic advantage for AML pathogenesis through DNA hypermethylation and the consequent block in differentiation that is a hallmark feature of AML (McKenney and Levine, 2013). Thus, mutant IDH1/2 represent promising therapeutic targets in the treatment of AML. Selective small-molecule inhibitors have been developed, including mutant IDH1 inhibitor ivosidenib (AG-120) and mutant IDH2 inhibitor enasidenib (AG-221/CC-90007), which bind at an allosteric site within the heterodimer interface of the mutant IDH, and evaluated in preclinical and clinical studies as monotherapy and in combination with other anti-cancer agents (Medeiros et al., 2017; Wang et al., 2013). For example, mutant IDH2 inhibitor enasidenib has been demonstrated to be effective in reversing the DNA hypermethylation and differentiation block in leukemia cells harboring mutant IDH2 R140Q, and combined treatment with tyrosine kinase FLT3 inhibitor quizartinib (AC220) and mIDH2 inhibitor enasidenib resulted in synergistic reduction of leukemic blasts in conditional IDH2+/R140QFlt3ITD mice with AML-like disease (Shih et al., 2017; Yen et al., 2017). In addition, enasidenib and ivosidenib have been FDA-approved for IDH-mutant relapsed or refractory acute myeloid leukemia.
However, it remains unclear how the transforming capability of mutant IDH1/2 and their oncometabolite product 2-HG is regulated and optimized for pathogenesis in AML cells. We recently reported that leukemogenic tyrosine kinase FLT3-JAK2 cascade promotes transforming activity of IDH1 R132H mutant in AML cells through phosphorylation of Y42 and Y391, which enhances the enzymatic activity of mutant IDH1 and consequent production of oncometabolite 2-HG (Chen et al., 2019). Interestingly, although the flanking resigns are homologous, the residues in mIDH2 that are corresponding to Y42 and Y391 in mIDH1 are phenylalanine (F82 and F432, respectively), which cannot be phosphorylated. Given that FLT3 internal tandem duplication (ITD) and tyrosine kinase domain (TKD) mutations are frequently detected in AML patients harboring IDH1/2 mutations (Boddu et al., 2017), such a difference in sequences of highly homologous IDH1 and IDH2 suggests that there likely exist exclusive isoform-specific regulatory mechanisms for mutant IDH1 and IDH2 in AML cells.
In addition, since 2-HG broadly inhibits multiple α-KG-dependent dioxygenases, accumulation of intracellular 2-HG beyond its physiologically functional levels as an oncometabolite is expected to be cytotoxic by causing genetic instability in tumor or AML cells expressing mutant IDH1/2 (Yang et al., 2012). This is supported by literature reporting that ectopic expression of mutant IDH1 in D54 glioblastoma cells resulted in attenuation of cell proliferation rate (Seltzer et al., 2010). Moreover, Su et al recently reported that 2-HG also binds to the RNA demethylase FTO, which is an α-KG-dependent dioxygenase, and competitively inhibits FTO enzymatic activity. This results in increased levels of m6A RNA modification in AML cells, leading to reduced stability of MYC and CEBPA transcripts and consequent gene expression that is important for leukemogenesis in AML cells. Consistently, treatment with cell permeable 2-HG attenuates the cell proliferation and tumor growth potential of AML cells in vitro and in vivo in xenograft mice, respectively (Su et al., 2018). Therefore, it is hypothesized that additional mechanisms may exist in mutant IDH1/2 expressing GBM and AML that offset or alleviate the toxicity of 2-HG, and/or control mutant IDH1/2 activity to avoid cytotoxic accumulation of 2-HG.
Herein we report a surprising finding that the intrinsic enzyme activity of mutant IDH2 is much higher than mutant IDH1, and that FLT3 WT or ITD mutant restricts mutant IDH2 activity in AML cells through regulation of K413-acetylation that inhibits mutant IDH2 enzyme activity. Therefore, we hypothesize that inhibitory K413-acetylation optimizes the leukemogenic ability of mutant IDH2 in AML cells by producing sufficient 2-HG for transformation and avoiding cytotoxic accumulation of intracellular 2-HG.
RESULTS
An intrinsic link between FLT3 and IDH2 mutant in AML cells: FLT3 negatively regulates IDH2 R140Q activity
We recently reported that FLT3, either wild type (WT) with basal level enzyme activity or internal tandem duplication (ITD) mutant that is a constitutively active form, activates JAK2 to enhance mutant IDH1 (mIDH1) activity in AML cells through phosphorylation of Y391 and Y42, respectively (Chen et al., 2019). Consistent with this finding, treatment with FLT3 inhibitor quizartinib resulted in decreased intracellular 2-HG levels in diverse mIDH1-expressing human primary leukemia cells from four representative AML patients (Figure 1A; left). Intriguingly, quizartinib treatment resulted in increased intracellular 2-HG levels measured by NMR in human primary leukemia cells from four mutant IDH2 (mIDH2)-positive AML patients (Figure 1A; middle and right). Consistently, we found that inhibition of FLT3 by quizartinib in primary leukemia cells from two mIDH2-harboring AML patients with expression of FLT3 WT or ITD mutant resulted in enhanced mIDH2 catalytic activity (Figure 1B), whereas, consistent with previous finding, inhibition of FLT3 WT or ITD mutant in these primary AML cells expressing mIDH2 resulted in decreased enzyme activity of endogenous IDH1 with reduced phosphorylation levels of Y42 and Y391 (Figure S1A). Moreover, in an in vitro kinase assay, purified recombinant FLT3 (rFLT3) phosphorylated recombinant IDH2 R140Q mutant but did not alter its catalytic activity (Figure 1C), whereas rFLT3 phosphorylated mutant IDH1 protein and enhanced its catalytic activity (Figure S1B). These results suggest an intrinsic link between FLT3 and mIDH2 in AML cells, where FLT3 negatively but indirectly regulates mIDH2, which is different from the previously reported FLT3-mIDH1 link (Chen et al., 2019).
Figure 1. Mutant IDH2 activity is restricted in human AML cells by FLT3 through controlling inhibitory K413-acetylation of mIDH2, which, however, is sufficient to produce comparable levels of 2-HG as cells expressing mIDH1.
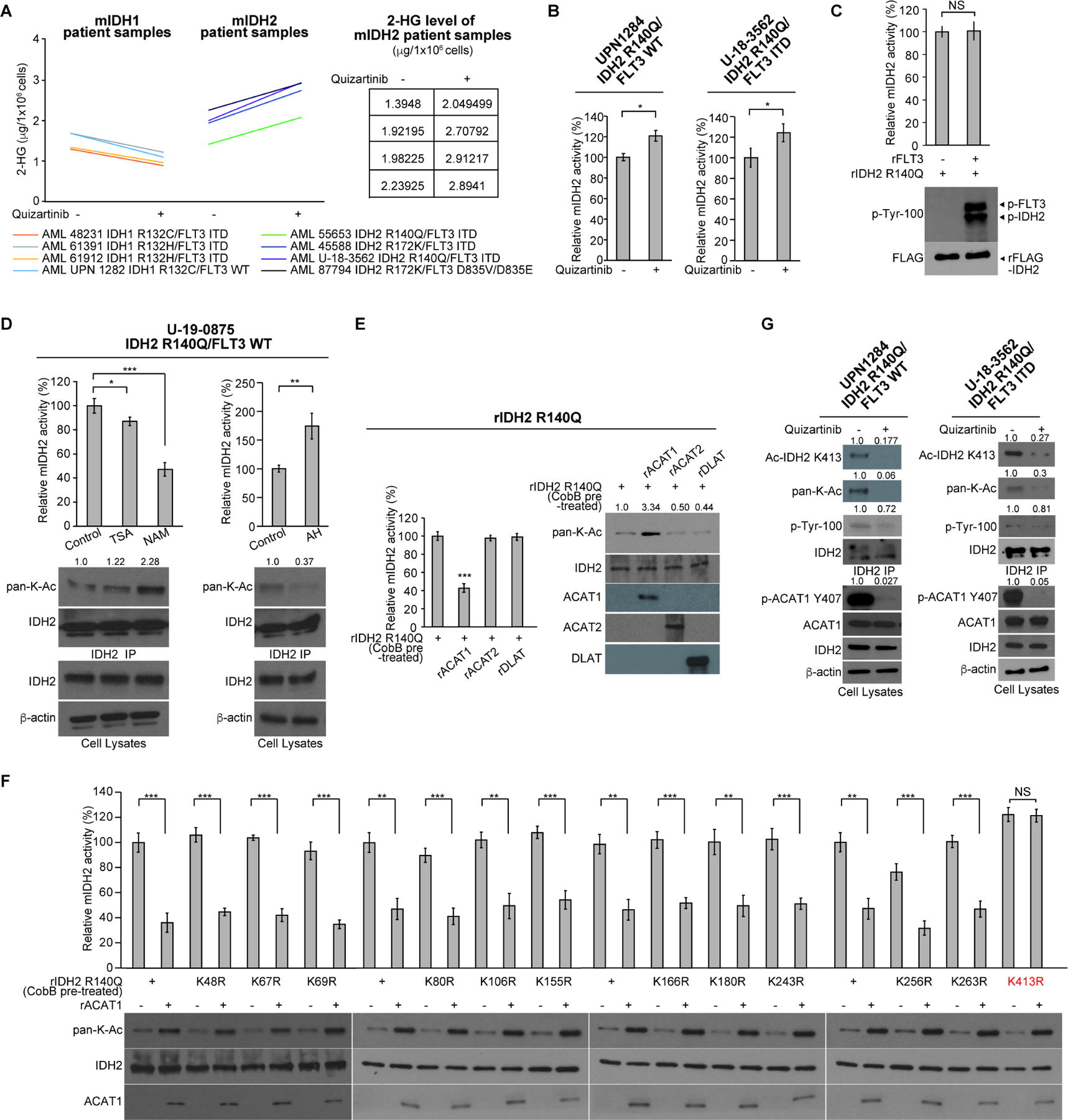
(A) Human primary AML cells expressing diverse IDH1 or IDH2 mutants were treated with or without FLT3 inhibitor quizartinib (200nM) for 72 hours, followed by NMR analysis to measure the intracellular levels of 2-HG.
(B) Human primary AML cells expressing IDH2 R140Q mutant were treated with or without FLT3 inhibitor quizartinib, followed by mIDH2 enzyme activity assay.
(C) Purified recombinant IDH2 R140Q protein was incubated with recombinant active form of rFLT3 in an in vitro kinase assay, followed by mIDH2 enzyme activity assay (upper); tyrosine phosphorylation of mIDH2 was detected by Western blotting (lower).
(D) Human primary AML cells expressing IDH2 R140Q mutant were treated with or without deacetylase inhibitors NAM or TSA (left panels) or mitochondrial acetyltransferase ACAT1 inhibitor arecoline hydrobromide (AH; right panels), followed by mIDH2 enzyme activity assay; lysine acetylation of mIDH2 was detected by Western blotting (lower panels).
(E) Purified IDH2 R140Q protein was pre-treated with cobB protein deacetylase to remove intrinsic lysine acetylation obtained in bacteria prior to treatment with recombinant acetyltransferases including mitochondrial rACAT1 or cytosolic rACAT2 or rDLAT, followed by mutant IDH2 catalytic activity assay (left) and Western blot to detect lysine acetylation (right).
(F) Purified IDH2 R140Q variants with representative individual K⭢R mutation were incubated with acetyltransferase rACAT1, followed by mIDH2 catalytic activity assay (upper) and Western blot to detect lysine acetylation (lower).
(G) Human primary AML cells expressing IDH2 R140Q mutant were treated with or without FLT3 inhibitor quizartinib, followed by Western blot to detect phosphorylation of ACAT1 (Y407) and mIDH2, and K413-acetylation of mIDH2 by a specific acetyl-IDH2 antibody (K413-Ac).
The error bars represent mean values ±SD from three replicates of each sample (*: 0.01<p<0.05; **: 0.01<p<0.001; ***: p<0.001; ns: not significant); Data are mean ±SD; p values were obtained by a two-tailed Student’s t-test.
FLT3 restricts mIDH2 activity in AML cells through regulation of inhibitory K413-acetylation of mIDH2
We previously reported that the mitochondrial fraction of FLT3 activates acetyl-coA acetyltransferase ACAT1 in mitochondria via Y407-phosphorylation to acetylate and inhibit mitochondrial pyruvate dehydrogenase A (PDHA) and PDH phosphatase 1 (PDP1) (Fan et al., 2016; Fan et al., 2014b). Since IDH2 mutant protein localizes to the mitochondria, we thus hypothesize that FLT3 might inhibit mIDH2 in AML cells through ACAT1-mediated lysine acetylation.
We found that treatment with nicotinamide (NAM), a sirtuin deacetylase (SIRT) inhibitor, effectively reduced mIDH2 activity with increased lysine acetylation of mIDH2 in human primary leukemia cells from a representative AML patient, compared to treatment with a histone deacetylase inhibitor, Trichostatin A (TSA) (Figure 1D, left), while treatment with ACAT1 inhibitor arecoline hydrobromide (AH (Fan et al., 2016)) resulted in increased mIDH2 activity with reduced lysine acetylation in primary AML cells (Figure 1D, right). Similar results were obtained using TF-1 erythroleukemia cells stably expressing FLAG-tagged IDH2 R140Q mutant (Figure S1C). Further studies confirmed that mitochondrial ACAT1, but not control acetyltransferases including cytosolic ACAT2 and DLAT, inhibited mIDH2 enzyme activity with increased lysine acetylation in an in vitro acetyltransferase assay using purified recombinant IDH2 R140Q and acetyltransferases including ACAT1, ACAT2 and DLAT (Figure 1E).
We next performed mutational analysis and generated diverse acetyl-deficient K⭢R mutants of IDH2 R140Q (Figures 1F and S2), based on the public database with 22 acetylated lysine residues of IDH2 identified in human cancer/leukemia cells (https://www.phosphosite.org/proteinAction.action?id=9667&showAllSites=true). The recombinant mIDH2 K⭢R proteins were purified and pre-treated with cobB protein deacetylase to remove intrinsic lysine acetylation obtained in bacteria prior to an in vitro acetyltransferase assay coupled with mIDH2 activity assay (Figures 1F and S2). We found that purified recombinant ACAT1 directly acetylates and inhibits diverse K⭢R mutants of IDH2 R140Q except K413R, which completely abolishes ACAT1-dependent inhibition of mIDH2 (Figure 1F). These results suggest that although ACAT1 acetylates mutant IDH2 at multiple lysine sites, acetylation at K413 is predominantly important for inhibition of mutant IDH2 by lysine acetylation. Treatment with FLT3 inhibitor quizartinib resulted in decreased K413-acetylation of mIDH2 and reduced Y407-phosphoryaltion of ACAT1 in human primary AML cells (Figure 1G), consistent with increased mIDH2 enzyme activity in the same cells treated with quizartinib (Figure 1B). These data together suggest that FLT3 might signal through ACAT1 to negatively regulate mIDH2 activity via K413-acetylation in AML cells.
Mitochondrial acetyltransferase ACAT1 and deacetylase SIRT3 are responsible for acetylation and deacetylation, respectively, at K413 of mIDH2
We found that ACAT1 inhibits IDH2 R140Q through direct K413-acetylation in an in vitro acetyltransferase assay using purified recombinant proteins of rACAT1 and rIDH2 R140Q variants, while the acetyl-deficient K413R and acetyl-mimetic K413Q mutants demonstrated increased and attenuated catalytic activity, respectively, compared to IDH2 R140Q, and both IDH2 R140Q/K413Q and IDH2 R140Q/K413R mutants were completely resistant to ACAT1 treatment (Figure 2A). Since a SIRT inhibitor NAM effectively reduced mIDH2 activity with increased lysine acetylation of mitochondrial mIDH2 in human primary AML cells (Figure 1D, left), we next examined SIRT −3, −4, and −5 that are mitochondrial deacetylases. We found that, in an in vitro deacetylase assay using purified recombinant proteins of rSIRT-3, −4 and −5 incubated with rIDH2 R140Q protein, only rSIRT3 enhanced IDH2 R140Q catalytic activity through deacetylation of K413 (Figure 2B), whereas both IDH2 R140Q/K413Q and IDH2 R140Q/K413R mutants were completely resistant to SIRT3 treatment (Figure 2C). In addition to mIDH2 R140Q mutant, ACAT1 and SIRT3 similarly acetylates and deacetylates, respectively, mIDH2 R172K at K413 (Figure S3).
Figure 2. Mitochondrial ACAT1 and SIRT3 as upstream acetyltransferase and deacetylase, respectively, regulate K413-acetylation of mIDH2, which inhibits mIDH2 by attenuating dimer formation from monomers and destabilizing dimers for conversion to monomers.
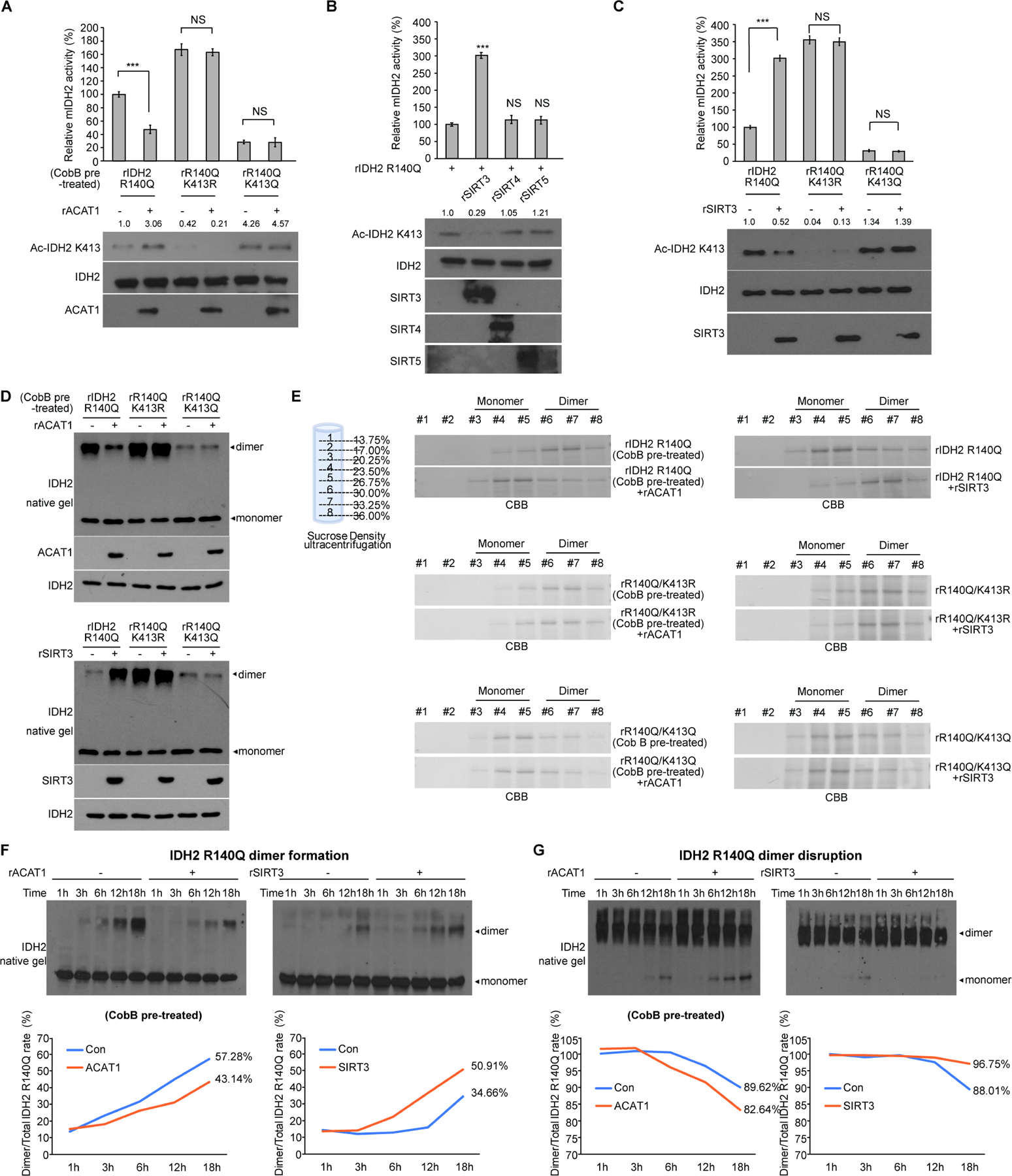
(A) Purified IDH2 R140Q variants were incubated with upstream acetyltransferase rACAT1, followed by mIDH2 catalytic activity assay (upper) and Western blot to detect K413-acetylation (lower).
(B) Purified IDH2 R140Q protein was incubated with purified recombinant deacetylases including rSIRT3, rSIRT4 or rSIRT5, followed by mIDH2 catalytic activity assay (upper) and Western blot to detect K413-acetylation (lower).
(C) Purified IDH2 R140Q variants were incubated with upstream deacetylase SIRT3, followed by mIDH2 catalytic activity assay (upper) and Western blot to detect K413-acetylation (lower).
(D) Purified IDH2 R140Q variants were either pre-treated with cobB protein deacetylase followed by incubation with acetyltransferase rACAT1 (upper) or treated with deacetylase rSIRT3 (lower), prior to being applied to native gel. Dimeric and monomeric mIDH2 proteins were determined by Western blot.
(E) Left panels: rIDH2 R140Q (top), rIDH2 R140Q/K413R (middle) and rIDH2 R140Q/K413Q (bottom) proteins were pre-treated with cobB followed by incubation with rACAT1 prior to sucrose density ultracentrifugation. Right panels: rIDH2 R140Q (top), rIDH2 R140Q/K413R (middle) and rIDH2 R140Q/K413Q (bottom) proteins were incubated with rSIRT3 prior to sucrose density ultracentrifugation. Collected fractions were applied to PAGE, followed by CBB staining.
(F) Purified monomeric IDH2 R140Q protein was either pre-treated with cobB followed by incubation with rACAT1 (left) or treated with rSIRT3 (right) in a time dependent manner, followed by native PAGE. Spontaneous dimer formation was determined by Western blotting. Lower panels show density analysis of corresponding bands to assess dimer formation in Western blotting. The ratio between homodimers and total mIDH2 proteins was quantitatively determined based on density analyses of the Western blotting.
(G) Purified dimeric IDH2 R140Q protein was either pre-treated with cobB followed by incubation with rACAT1 (left) or treated with rSIRT3 (right) in a time dependent manner, followed by native PAGE. Spontaneous monomer conversion was detected by Western blotting. Lower panels show density analysis of corresponding bands to assess dimer formation in Western blotting. The ratio between homodimers and total mIDH2 proteins was quantitatively determined based on density analyses of the Western blotting.
The error bars represent mean values ±SD from three replicates of each sample (***: p<0.001; ns: not significant); Data are mean ±SD; p values were obtained by a two-tailed Student’s t-test.
K413-acetylation inhibits mIDH2 by simultaneously attenuating dimer formation from monomers and destabilizing dimers for conversion to monomers
We next found that addition of ACAT1 to purified mIDH2 variants pre-treated with deacetylase cobB resulted in decreased amount of dimeric IDH2 R140Q protein, whereas acetyl-deficient IDH2 R140Q/K413R and acetyl-mimetic IDH2 R140Q/K413Q mutants showed increased and decreased dimer formation, respectively, compared to IDH2 R140Q, and both mutants were resistant to ACAT1 treatment (Figure 2D; upper). In contrast, addition of SIRT3 to purified mIDH2 variants led to increased amount of dimeric IDH2 R140Q protein, while both IDH2 R140Q/K413R and IDH2 R140Q/K413Q mutants were resistant to SIRT3 treatment (Figure 2D; lower). Moreover, using a sucrose density ultracentrifugation approach, we purified monomeric and dimeric proteins of mIDH2 variants that were either pre-treated with cobB followed by treatment with rACAT1 (Figure 2E, left 3 panels) or treated with SIRT3 (Figure 2E, right 3 panels). We found that the majority of rIDH2 R140Q proteins were dimers after deacetylation by pre-treatment with cobB, while acetylation by ACAT1 resulted in a shift to the monomeric form (Figure 2E, upper left). No shift was observed with ACAT1 treatment using rIDH2 R140Q/K413R (Figure 2E, middle left) and rIDH2 R140Q/K413Q (Figure 2E, lower left) proteins, which retained mostly dimeric and monomeric, respectively. In contrast, the majority of rIDH2 R140Q proteins purified from bacteria were monomers due to intrinsic lysine acetylation, while deacetylation by SIRT3 resulted in a shift to the dimeric form (Figure 2E, upper right). No shift was observed with SIRT3 treatment using rIDH2 R140Q/K413R (Figure 2E, middle right) and rIDH2 R140Q/K413Q (Figure 2E, lower right) proteins, which retained mostly dimeric and monomeric, respectively.
Finally, we sought to determine whether K413-acetylation of mIDH2 affects dimer formation from monomers and/or dimer disruption to monomers. We found that purified, recombinant monomeric IDH2 R140Q proteins with cobB pre-treatment spontaneously form dimers in a time dependent manner, while K413-acetylation of monomeric IDH2 R140Q proteins by ACAT1 resulted in reduced dimer formation (Figure 2F, left). In contrast, K413-deacetylation of monomeric IDH2 R140Q proteins without cobB pre-treatment by SIRT3 resulted in increased dimer formation (Figure 2F, right). In addition, purified, recombinant dimeric IDH2 R140Q proteins with cobB pre-treatment spontaneously disrupt to form inactive monomers in a time dependent manner, while K413-acetylation of dimeric IDH2 R140Q proteins by ACAT1 promoted dimer disruption to form monomers (Figure 2G, left). In contrast, K413-deacetylation of dimeric IDH2 R140Q proteins without cobB pre-treatment by SIRT3 resulted in reduced dimer disruption to form monomers (Figure 2G, right).
K413-acetylation attenuates substrate (α-KG) and co-factor (NADPH) binding to mIDH2
Next we found that addition of ACAT1 (Figure 3A) or SIRT3 (Figure 3B) reduced or promoted 14C-α-KG binding to rIDH2 R140Q mutant, respectively, while acetyl-deficient IDH2 R140Q/K413R and acetyl-mimetic IDH2 R140Q/K413Q mutants showed maximal and minimal binding ability to 14C-α-KG, respectively, which was not altered by ACAT1 or SIRT3 treatment. Moreover, further kinetics studies confirmed that, in the presence of increasing concentrations of α-KG as a substrate (Figure 3C; left) or NADPH as a cofactor (Figure 3C; right), K413-acetylation of cobB-pretreated IDH2 R140Q by ACAT1 resulted in decreased Vmax value with increased Km value for α-KG or NADPH, consistently suggesting that K413-acetylation inhibits IDH2 R140Q, at least in part, through attenuation of substrate and cofactor binding. In contrast, acetyl-deficient IDH2 R140Q/K413R showed comparable values of Vmax and Km for α-KG as a substrate and NADPH as a cofactor compared to IDH2 R140Q, while acetyl-mimetic IDH2 R140Q/K413Q had decreased Vmax value but increased Km value for α-KG and NADPH, and both mutants are resistant to ACAT1 treatment (Figure 3C). The exact opposite results were obtained in SIRT3-treated IDH2 R140Q variants (Figure 3D).
Figure 3. K413-acetylation attenuates substrate (α -KG) and co-factor (NADPH) binding to mIDH2.
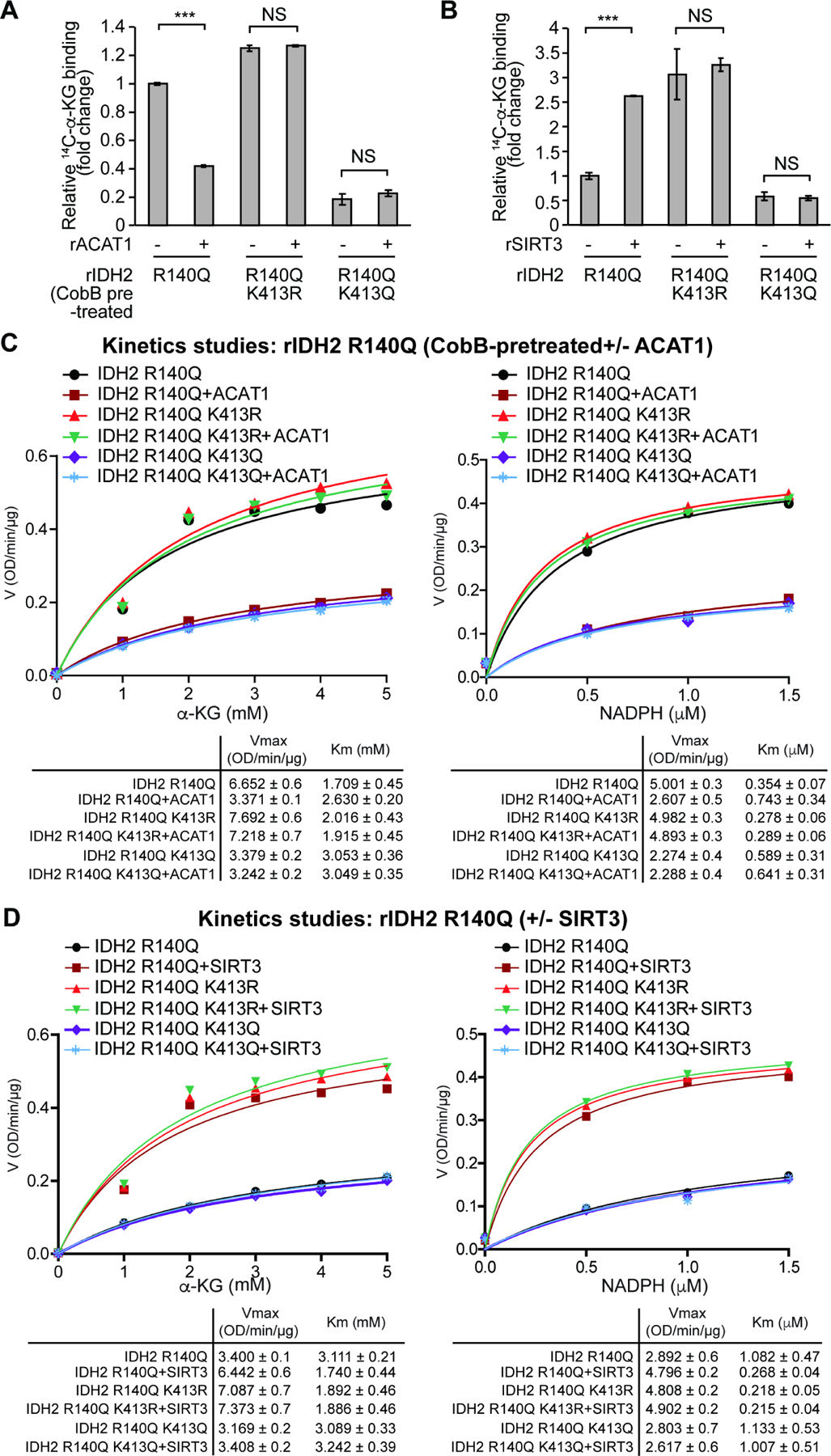
(A-B) Purified rIDH2 R140Q variants were either pre-treated with cobB followed by incubation with rACAT1 (A) or treated with rSIRT3 (B), prior to 14C-labeled α-KG binding assay.
(C-D) Vmax and Km of rIDH2 R140Q variants were measured using purified rIDH2 R140Q proteins that were either pre-treated with cobB followed by incubation with rACAT1 (C) or treated with rSIRT3 (D), followed by mIDH2 enzyme activity assay in the presence of increasing concentrations of either substrate α-KG (left panels) or cofactor NADPH (right panels). Vmax and Km values of each treated group were calculated (lower panels) and plotted (upper panels). Please note that the comparable Vmax values of IDH2 R140Q and acetyl-deficient R140Q/K413R are due to the condition that purified mIDH2 proteins were pre-treated with cobB to remove intrinsic lysine acetylation obtained in bacteria prior to rACAT1 incubation.
The error bars represent mean values ±SD from three replicates of each sample (***: p<0.001; ns: not significant); Data are mean ± SD; p values were obtained by a two-tailed Student’s t-test.
FLT3 promotes mIDH2 acetylation through Y107-phosphorylation of mIDH2 that enhances ACAT1 recruitment, while, simultaneously, FLT3 inhibits SIRT3 through Y226 phosphorylation
We sought to determine the stoichiometry of K413-acetylation of mIDH2 in human primary AML cells. At most 52–55% of total IDH2 protein was estimated to be K413-acetylated in human primary AML cells, which was cleared from cell lysates by immunoprecipitation (IP) using a specific acetyl-IDH2 (K413-ac) antibody (Figure 4A). Moreover, treatment with FLT3 inhibitor decreased K413-acetylation of endogenous IDH2 R140Q in human primary AML cells, leading to enhanced mIDH2 activity and dimerization (Figure 4B).
Figure 4. FLT3 promotes mIDH2 acetylation through Y107-phosphorylation of mIDH2 that enhances ACAT1 recruitment, while, simultaneously, FLT3 inhibits SIRT3 through Y226 phosphorylation.
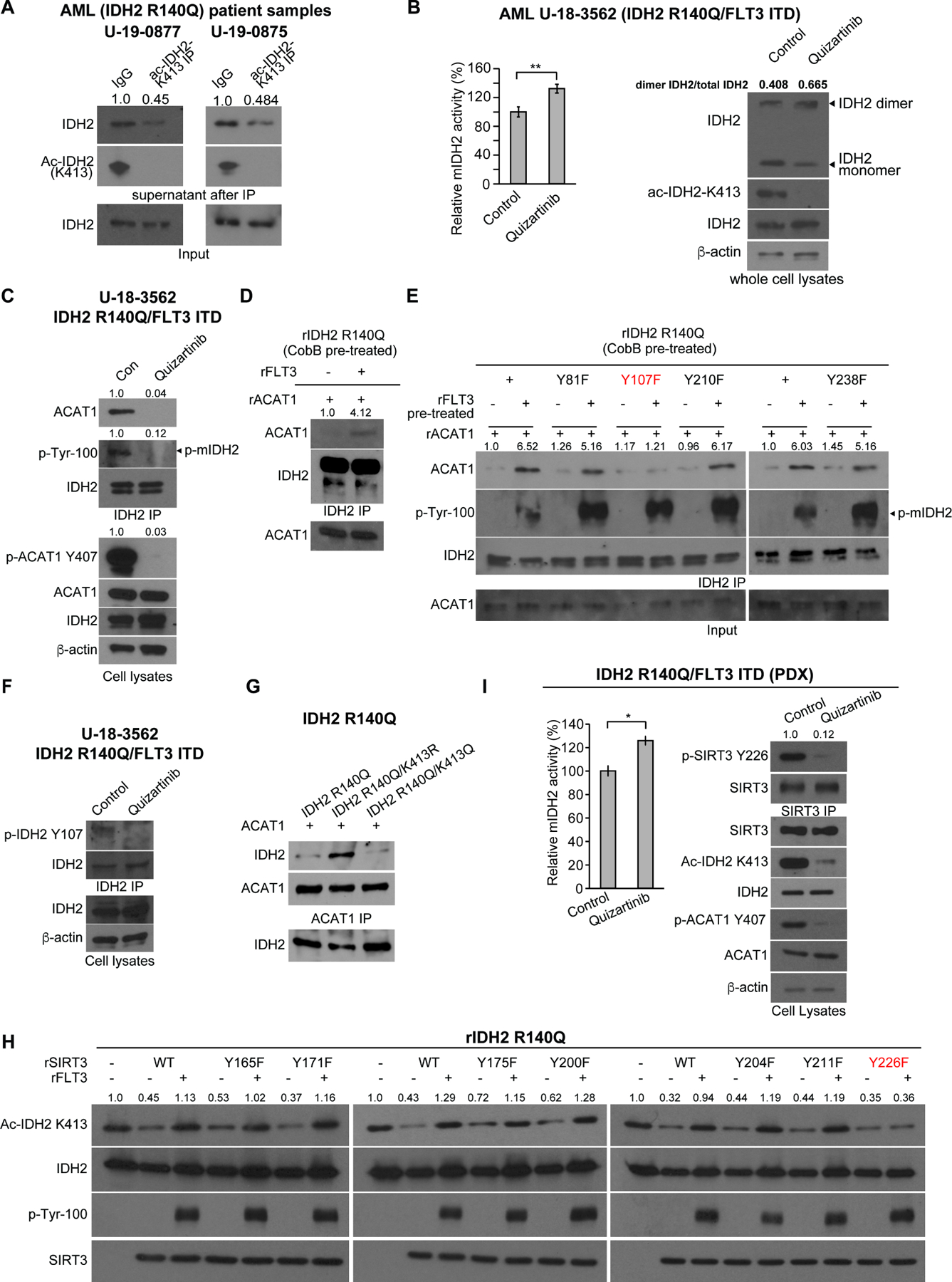
(A) Determination of stoichiometry of K413-acetylation of mutant IDH2 in human primary leukemia cells from two AML patients. K413-acetylated IDH2 protein was cleared from cell lysates by immunoprecipitation (IP) using specific acetyl-IDH2 (ac-K413) antibody, followed by Western blotting to determine protein amount of residual non-acetylated IDH2 in supernatant after IP.
(B) Human primary AML cells expressing IDH2 R140Q and FLT3 ITD mutants were treated with or without FLT3 inhibitor quizartinib, followed by mIDH2 activity assay (left) and PAGE by native gel and Western blotting to detect dimeric and monomeric IDH2, and K413-acetylation levels of IDH2 (right).
(C) Human primary AML cells expressing IDH2 R140Q and FLT3 ITD mutants were treated with or without FLT3 inhibitor quizartinib, followed by co-immunoprecipitation analysis. Binding between endogenous IDH2 and ACAT1, as well as tyrosine phosphorylation of IDH2 and ACAT1 (Y407) in human primary AML cells were detected by Western blotting.
(D) Purified rIDH2 R140Q protein was pre-treated with cobB followed by treatment with active rFLT3, prior to incubation with rACAT1. Co-immunoprecipitation (IDH2 IP) was performed followed by Western blotting to detect binding between mIDH2 and ACAT1.
(E) Purified rIDH2 R140Q variants with individual Y⭢F mutations were pre-treated with cobB followed by treatment with active rFLT3, prior to incubation with rACAT1. Coimmunoprecipitation (IDH2 IP) was performed followed by Western blotting to detect binding between mIDH2 and ACAT1.
(F) Human primary AML cells expressing IDH2 R140Q and FLT3 ITD mutants were treated with or without FLT3 inhibitor quizartinib, followed by IDH2 immunoprecipitation prior to Western blotting to detect Y107 phosphorylation level of endogenous IDH2 using a specific phosphor-IDH2 antibody (p-Y107).
(G) Purified rIDH2 R140Q variants were pre-treated with cobB followed by treatment with active rFLT3, prior to incubation with rACAT1. Co-immunoprecipitation (ACAT1 IP) was performed followed by Western blotting to detect binding between mIDH2 and ACAT1.
(H) Purified SIRT3 variants with individual Y⭢F mutations were pre-treated with rFLT3 prior to incubation with purified rIDH2 R140Q protein as a substrate in an in vitro deacetylase activity assay. K143 acetylation of IDH2 was detected by Western blotting.
(I) Human primary AML cells expressing IDH2 R140Q and FLT3 ITD mutants expanded in xenograft mice (PDX) were treated with or without FLT3 inhibitor quizartinib, followed by mIDH2 enzyme activity assay (left) and immunoprecipitation using SIRT3 antibody (right) prior to Western blotting to detect Y226 phosphorylation level of endogenous SIRT3 using a specific phosphor-SIRT3 antibody (p-Y226).
The error bars represent mean values ±SD from three replicates of each sample (*:0.01<P<0.05); Data are mean ± SD; p values were obtained by a two-tailed Student’s t-test.
We previously reported that mitochondrial PDP1 as an ACAT1 substrate needs to be tyrosine phosphorylated to recruit ACAT1 (Fan et al., 2014a). We thus hypothesize that, although tyrosine phosphorylation of mIDH2 by FLT3 does not directly affect mIDH2 enzyme activity (Figure 1C), it may be indirectly involved in mIDH2 activation by recruiting ACAT1. We found that, using primary AML cells expressing both IDH2 R140Q and FLT3 ITD mutants, treatment with FLT3 inhibitor resulted in decreased tyrosine phosphorylation levels of IDH2 with reduced endogenous binding between IDH2 and ACAT1 assessed by co-immunoprecipitation (Figure 4C). In addition, phosphorylation of rIDH2 R140Q by FLT3 resulted in increased ACAT1 binding to mIDH2 (Figure 4D), whereas although FLT3 mediated Y407 phosphorylation of ACAT1 resulted in increased ACAT1 activity assessed by increased K413 acetylation of mutant IDH2 (Figure S4A, left), rACAT1 or a phosphor-deficient rACAT1 Y407F mutants treated with rFLT3 did not affect the binding ability of ACAT1 and ACAT1 Y407F to rIDH2 R140Q (Figure S4A, middle and right, respectively). Our mutational studies on the tyrosine sites including Y81, Y107, Y210, and Y238 that are exclusive in mIDH2 but not in mIDH1 revealed that Y107-phosphorylation by FLT3 is solely responsible for enhanced ACAT1 binding to mIDH2 (Figure 4E).
We developed a specific phosphor-IDH2 antibody (p-Y107). Using this antibody, we detected Y107-phosphorylation of IDH2 R140Q mutant by rFLT3 in an in vitro kinase assay using purified recombinant FLT3 and mIDH2 proteins, but not in control mIDH2 Y107F mutant (Figure S4B). Moreover, Y107-phosphorylation of endogenous IDH2 R140Q was detected in human primary AML cells, which was reduced by treatment with FLT3 inhibitor (Figure 4F). In addition, ACAT1 binds to acetyl-deficient IDH2 R140Q/K413R mutant in a stronger manner compared to IDH2 R140Q and acetyl-mimetic IDH2 R140Q/K413Q mutant, suggesting that K413-acetylation status is also involved in ACAT1 recruitment (Figure 4G), where ACAT1 preferentially binds to mIDH2 with non-acetylated K413 while K413-acetylation of mIDH2 dissociates ACAT1.
We also hypothesize that, besides activating ACAT1 through Y407-phosphorylation (Fan et al., 2016), FLT3 might simultaneously phosphorylate and regulate SIRT3. Our mutational studies on all of the 7 tyrosine sites of SIRT3 revealed that purified rFLT3 directly phosphorylated purified rSIRT3 in an in vitro kinase assay, leading to decreased SIRT3 deacetylase activity that was assessed by ability to deacetylate K413 of mutant IDH2 (Figure 4H, first three samples in left, middle and right panels), whereas replacement of Y226 completely abolished inhibition of SIRT3 by FLT3 (Figure 4H, right). We thus developed a specific phosphor-SIRT3 antibody (p-Y226). Using this antibody, we detected Y226-phosphorylation of endogenous SIRT3 (Figure 4I, right) in human AML cells expressing IDH2 R140Q and FLT3 ITD expanded in NSG mice (Figure S4C), which was reduced by treatment with FLT3 inhibitor along with reduced K413-acetylation of mIDH2 and Y407-phosporylation of ACAT1 (Figure 4I, right) and increased mIDH2 activity (Figure 4I, left).
The intrinsic enzyme activity of mIDH2 is much higher than mIDH1, thus the restricted mIDH2 activity by K413-acetylation is comparable to mIDH1
In order to evaluate the physiological significance of our finding that mIDH2 activity is restricted in AML cells, we thus hypothesize that the intrinsic enzyme activity of mIDH2 might be higher than that of mIDH1, and that the restricted endogenous mIDH2 activity by K413-acetylation is at least comparable to the endogenous mIDH1 enzyme activity which is enhanced by tyrosine phosphorylation. This is based on the observation that the overall intracellular levels of 2-HG in human primary AML cells expressing mutant IDH2 in the absence of FLT3 inhibitor treatment are generally comparable to or higher than the 2-HG levels in human primary AML cells expressing mutant IDH1 without FLT3 inhibition (Fig. 1A).
In an in vitro mIDH enzyme activity assay, we compared the activity of FLAG-tagged IDH1 R132H with FLAG-tagged IDH2 R140Q and IDH2 R140Q variants with either acetyl-deficient K413R or acetyl-mimetic K413Q mutation, which were overexpressed in 293T cells and purified by FLAG-pull down (Figure 5A). Consistent with our hypothesis, the results demonstrate that the intrinsic catalytic activity of mIDH2 is approximately 5-fold higher than that of mIDH1, while acetyl-deficient mIDH2 K413R mutant shows even higher catalytic activity compared to mIDH2 parental protein. In contrast, the acetyl-mimetic mIDH2 K413Q mutant exhibits residual enzyme activity compared to mIDH2 and K413R, which, however, is comparable to that of mIDH1 (Figure 5A).
Figure 5. Restricted activity of mIDH2 by K413-acetylation is comparable to mIDH1, which is sufficient to produce enough 2-HG for transformation and avoids cytotoxicity of high levels of 2-HG.
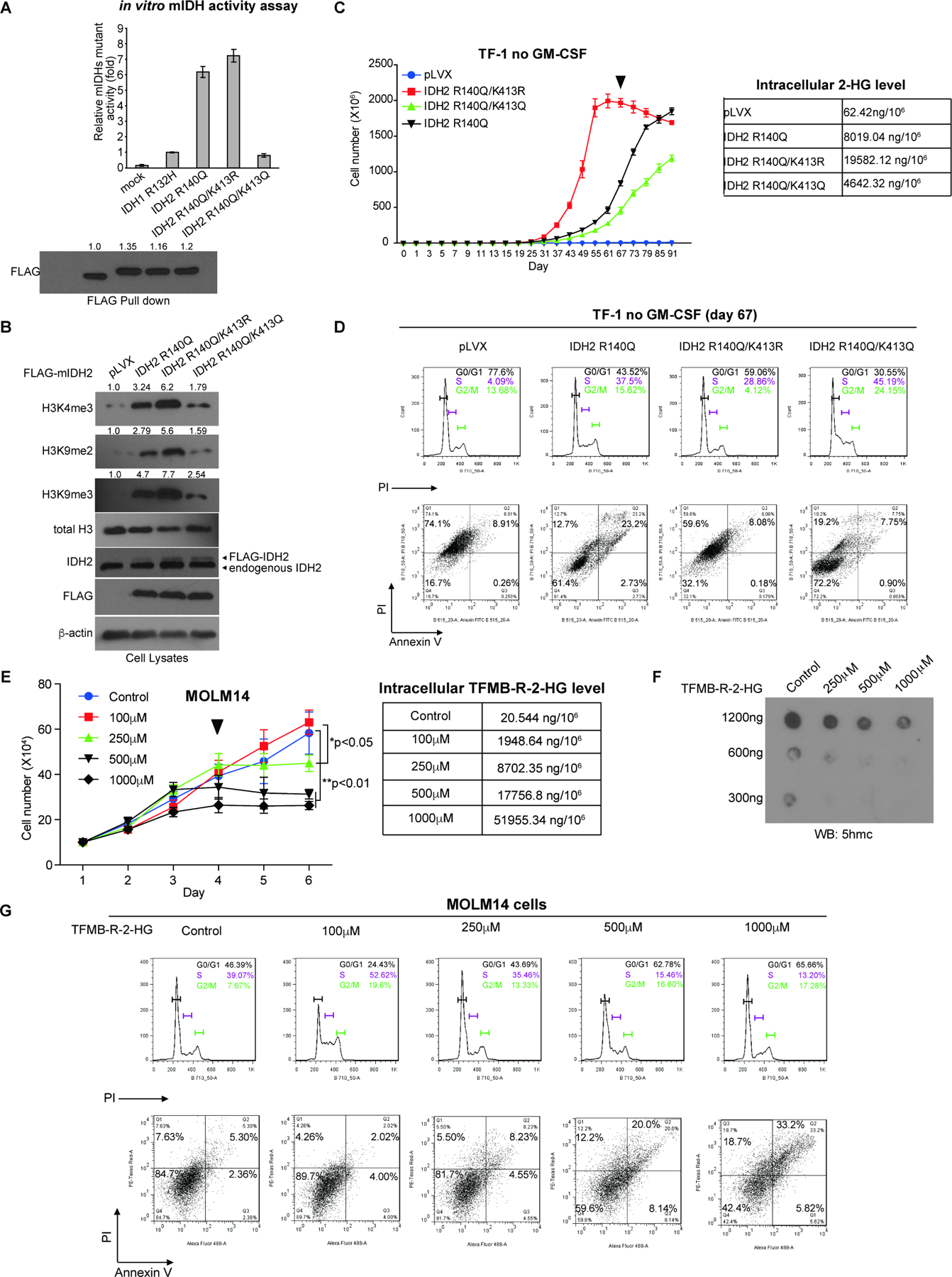
(A) FLAG-tagged IDH1 R132H mutant and IDH2 R140Q variants with acetyl-deficient K413R or acetyl-mimetic K413Q mutations were overexpressed in 293T cells. FLAG-pull down samples were applied to mutant IDH catalytic activity assay (upper) and Western blot for protein input levels (lower).
(B) TF-1 cell lines stably expressing FLAG-tagged mIDH2 variants were generated, followed by Western blot to detect histone methylation.
(C) Left: Proliferation rates of TF-1 cells stably expressing mIDH2 variants in the absence of GM-SCF. Cells were harvested at day 67 (indicated by a triangle) and cell lysates were applied to NMR for measurement of intracellular 2-HG levels (right).
(D) TF-1 cells stably expressing different mIDH2 variants were harvested at day 67 and applied to cell cycle analysis (upper panels) and cell death/apoptosis assay with PI and annexin V staining (lower panels).
(E) Left: Effects of cell-permeable TFMB-R-2-HG on MOLM14 cells. Right: MOLM14 cells treated with increasing concentrations of TFMB-R-2-HG were harvested at day 4 (indicated by a triangle) and intracellular 2-HG concentrations were measured by NMR.
(F) Effects of TFMB-R-2-HG treatments on DNA methylation in MOLM14 cells were assessed by dot blot to detect 5-hydroxymethylcytosine (5hmC). Input amounts of total genomic DNA are shown.
(G) MOLM14 cells treated with increasing concentrations of TFMB-R-2-HG were harvested at day 4 and applied to cell cycle analysis (upper panels) and cell death/apoptosis assay with PI and annexin V staining (lower panels).
The error bars represent mean values ±SD from three replicates of each sample (*: 0.01<p<0.05; **: 0.01<p<0.001); Data are mean ±SD; p values were obtained by a two-tailed Student’s t-test.
K413-acetylated mIDH2 produces sufficient 2-HG for transformation and avoids cytotoxicity of accumulation of high levels of 2-HG
In order to study whether acetylated and restricted mIDH2 remains leukemogenic, we generated TF-1 erythroleukemia cell lines with stable expression of FLAG-tagged IDH2 R140Q, acetyl-deficient R140Q/K413R or acetyl-mimetic R140Q/K413Q mutants (Figure 5B). Proliferation of TF-1 erythroleukemia cells depends on granulocyte-macrophage colony-stimulating factor (GM-CSF), but when transformed by expression of driver oncogenes such as IDH2 R140Q, TF-1 cells exhibit cytokine independent growth and are resistant to erythropoietin (EPO)-induced cell differentiation (Kernytsky et al., 2015; Losman et al., 2013). We found that expression of IDH2 R140Q resulted in increased histone hypermethylation, while expression IDH2 R140Q/K413R with enhanced mIDH2 catalytic activity led to further elevated histone hypermethylation (Figure 5B). Moreover, expression of acetyl-mimetic IDH2 R140Q/K413Q with attenuated mIDH2 activity was still sufficient to induce histone hypermethylation in TF-1 cells but to an extent that is less than the levels in cells expressing IDH2 R140Q or R140Q/K413R (Figure 5B). Consistently, we found that TF-1 cells expressing the catalytically more active IDH2 R140Q/K413R mutant demonstrated enhanced cell proliferative potential in the absence of GM-CSF, compared to control cells expressing IDH2 R140Q, while cells expressing the catalytically less active form of IDH2 R140Q/K413Q were able to proliferate in a cytokine independent manner, but at a slower rate than control cells expressing IDH2 R140Q (Figure 5C). These data together suggest that restricted mIDH2 activity by K413-acetylation remains transforming.
Importantly, IDH2 R140Q/K413R cells started showing decreased proliferative potential on day 67 with an accumulated intracellular 2-HG level as high as 19.6μg/106 cells, which is much higher than the intracellular 2-HG levels detected in control cells expressing IDH2 R140Q (8.02μg/106 cells) or the catalytically less active IDH2 R140Q/K413Q (4.64μg/106 cells), both of which were still highly proliferative at day 67 (Figure 5C). Moreover, cell cycle analysis results suggested that IDH2 R140Q/K413R cells with the highest intracellular 2-HG level showed increased G0/G1 arrest (59.06%) compared to cells with lower intracellular 2-HG levels expressing either IDH2 R140Q (43.53% in G0/G1) or IDH2 R140Q/K413Q (30.55% in G0/G1) (Figure 5D, upper).
Consistent with these results, IDH2 R140Q/K413R cells demonstrated overall increased cell death (PI+, Q1+Q2, 67.68%) compared to cells expressing either IDH2 R140Q (PI+, Q1+Q2, 35.9%) or IDH2 R140Q/K413Q (PI+, Q1+Q2, 26.95%) (Figure 5D, lower), despite that the apoptotic cell death of IDH2 R140Q/K413R cells (annexin V+, Q2+Q3, 8.96%) is comparable to that of cells expressing IDH2 R140Q/K413Q (annexin V+, Q2+Q3, 8.65%) but less than cells expressing IDH2 R140Q (annexin V+, Q2+Q3, 25.93%) (Figure 5D, lower). Results of quantitation and statistical analysis of two independent biological replicates are shown in Figure S5A. Control experiment demonstrated that treatment with increasing concentrations up to 1,000 μM of 1-(Methoxymethyl)-3-(trifluoromethyl)benzene, a TFMB derivative compound with a methoxy group replacing the hydroxy group, which improves the cell permeability and stability of TFMB, did not significantly alter the proliferation potential of MOLM14 cells (Figure S5C). This result suggest that the cytotoxicity induced by TFMB-R-2-HG is likely due to R-2-HG. These data together suggest that TF-1 cells with intracellular 2-HG levels less than 8.0μg/106 cells remain proliferative, whereas accumulated 2-HG levels as high as ~20μg/106 cells are cytotoxic to TF-1 cells and result in increased cell death.
In order to determine whether increased 2-HG level alone is responsible for the elevated cytotoxicity, we treated FLT3 ITD-expressing MOLM14 AML cells with increasing concentrations (100, 250, 500 and 1,000μM) of cell permeable TFMB-R-2-HG (Figure 5E). The results demonstrated that high concentrations of TFMB-R-2-HG (500 and 1,000μM) reduced cell proliferative potential of MOLM14 cells starting at day 3 with increased intracellular 2-HG levels (17.8 and 52.0μg/106 cells, respectively; Figure 5E) and decreased DNA demethylation assessed by reduced 5-hydroxylmethylcytosine (5hmC) levels (Figure 5F). This might be due to 2-HG-dependent inhibition of TET2, an α‒KG-dependent dioxygenase that converts 5-methylcytosine (5mC) to 5hmC, which is a prerequisite for DNA demethylation (Figure 5F). Consistent with the results obtained from TF-1 cells (Figure 5D), cell cycle and cell death analyses revealed that MOLM14 cells with accumulated 2-HG levels due to treatment with high levels of TFMB-R-2-HG (500 and 1,000μM) demonstrated increased G0/G1 arrest (62.78% and 65.66%, respectively) and elevated cell death (PI+, Q1+Q2, 32.2% and 51.9%, respectively), compared to cells treated with 100 and 250 μM of TFMB-R-2-HG and lower intracellular 2-HG levels (G0/G1, 24.43% and 43.69%, respectively; and PI+, Q1+Q2, 6.28% and 13.73%, respectively) (Fig. 5G), with increasing apoptotic cell death (annexin V+, Q2+Q3) observed among different groups of cells. Results of quantitation and statistical analysis of two independent biological replicates are shown in Figure S5B.
Cytotoxic levels of intracellular 2-HG activate apoptosis and P53 pathways and inactivate MYC signaling cascades
To delineate the mechanisms by which high levels of intracellular 2-HG induce cytotoxicity, we performed transcriptome-wide RNA-seq with MOLM14 cells similarly treated with vehicle control, lower doses (125 and 250 μM) and higher doses (500 and 1,000 μM) of TFMB-R-2-HG (Figure 6A). Principal component analysis (PCA) revealed that vehicle control, 125 and 250 μM TFMB-R-2-HG can be grouped together, separate from the two groups treated with higher doses of TFMB-R-2-HG (Figure 6B). Based on global Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005), we identified a set of increased (Figure 6C) and decreased (Figure 6D) signal pathways which are responsible for the cytotoxicity of higher intracellular 2-HG. Notably, treatment with 500 and 1,000 μM TFMB-R-2-HG, but not lower concentrations, significantly activate apoptosis pathway (Figure 6E) and P53 pathway (Figure 6F) but dramatically silence MYC pathways (Figures 6G and 6H). Collectively, these results suggest that cytotoxicity induced by high levels of intracellular 2-HG may be mediated through simultaneous activation of apoptosis and P53 pathways and inactivation of MYC signal cascades.
Figure 6. Identification of the signal pathways responsible for cytotoxicity induced by high levels of intracellular 2-HG via transcriptome-wide RNA-seq.
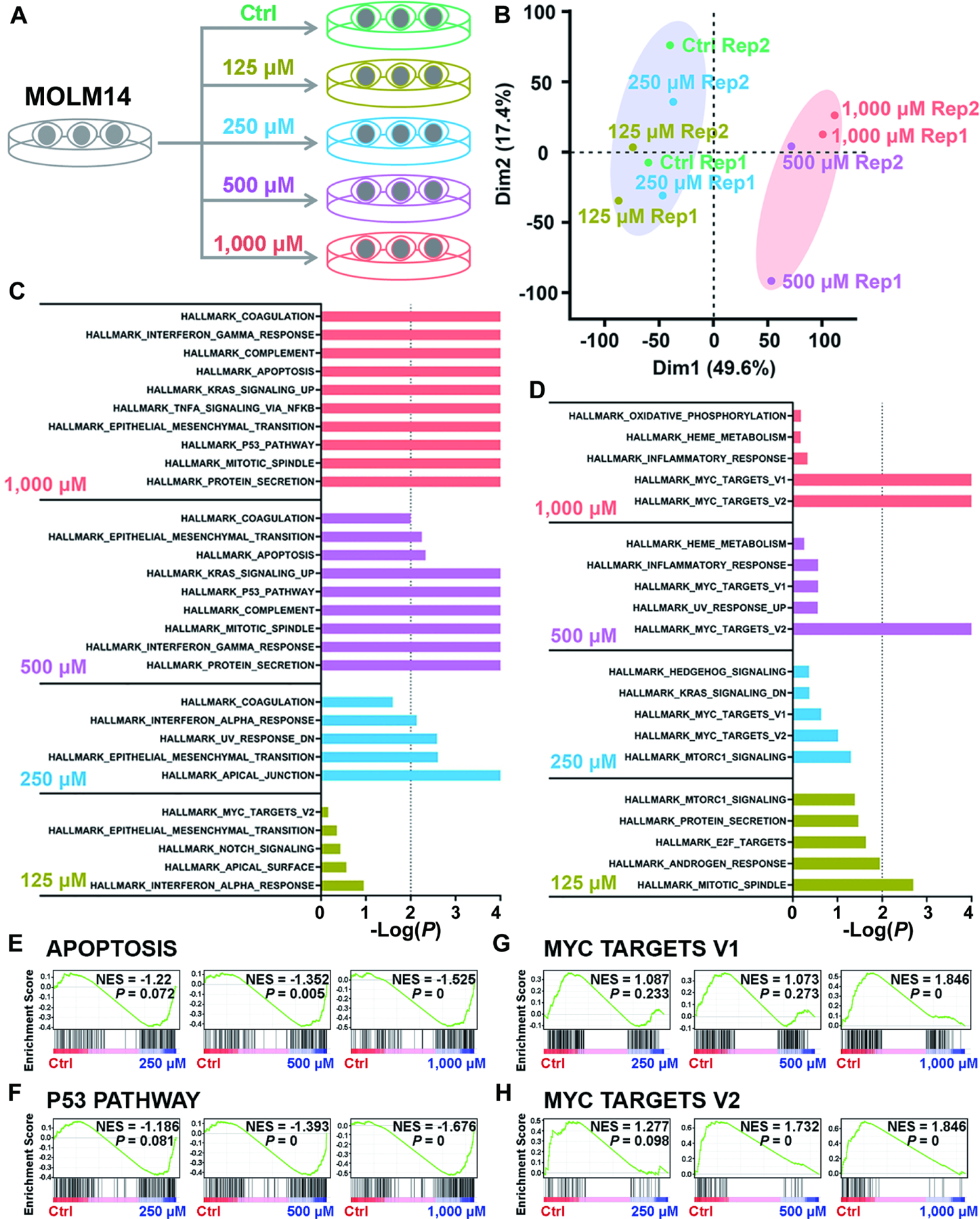
(A) Schematic diagram of RNA-seq with Molm14 cells upon treatment with 125, 250, 500, and 1,000 μM TFMB-R-2-HG.
(B) Principal component analysis (PCA) of RNA-seq data from Molm14 cells treated with TFMB-2-HG.
(C and D) The core-enriched signal pathways, including the upregulated (C) and downregulated (D) ones based on Gene Set Enrichment Analysis (GSEA), in TFMB-2HG-treated cells in contrast to vehicle control (Ctrl).
(E and F) GSEA of upregulated apoptosis (E) and P53 pathway (F) induced by TFMB-2-HG treatment in Molm14 cells. NES, Normalized Enrichment Score.
(G and H) GSEA of downregulated MYC target V1 (G) and MYC target V2 (H) induced by TFMB-2-HG treatment in Molm14 cells.
All RNA-seq experiments were conducted with 2 independent biological replicates.
DISCUSSION
Our studies for the first time demonstrate that different intracellular concentrations of 2-HG correlate with its different cellular functions. We determined a physiological range of intracellular concentrations of 2-HG that promote cell proliferation when 2-HG levels are less than 8.0μg/106 cells, which correlate with 2-HG levels of 1.4–3.0μg/106 cells assessed using human primary leukemia cells from AML patients that play a pathogenic role in AML cells (Figure 1A). In contrast, accumulation of intracellular 2-HG at and exceeding levels of 18–20μg/106 cells exhibits cytotoxicity and causes cell cycle arrest and cell death. This is conceptually similar to the different cellular functions of reactive oxygen species (ROS) at different levels, where low levels of ROS promotes cell proliferation through inhibition of protein tyrosine phosphatase, whereas high levels of ROS causes cell cycle arrest and cell death due to induction of oxidative stress (Schieber and Chandel, 2014). Moreover, transcriptome-wide RNA-seq results suggest that high levels of 2-HG in MOLM14 cells may activate apoptosis and P53 pathways to promote cell death and silence MYC signaling cascades to induce cell cycle arrest and cell growth inhibition. These findings are consistent with the recent reports that treatment with high doses of TFMB-R-2-HG suppresses glycolysis and MYC signaling cascades through inhibition of fat mass and obesity-associated protein (FTO) activity and subsequently increased global N6-methyladenosine (m6A) RNA modification (Qing et al., 2021; Su et al., 2018).
Furthermore, our studies revealed a rather surprising finding that mitochondrial mutant IDH2 activity is commonly restricted through inhibitory K413-acetylation in human AML cells. K413-acetylation was previously reported to reduce wild type (WT) IDH2 activity and deacetylase SIRT3 promotes IDH2 WT dimerization, and expression of an acetyl-mimetic mutant K413Q form of IDH2 WT in cancer cells promotes glycolysis and tumor growth (Yu et al., 2012; Zou et al., 2017). However, whether mutant IDH2 is similarly regulated in AML cells and the underlying signaling mechanisms are unclear. Most importantly, the physiological importance of K413-acetylation of oncogenic mIDH2 and subsequent production of oncometabolite 2-HG in AML cells remains unknown, which is the main focus of the current study.
Our findings provide novel insight to advance our understanding of the pathogenic role of mutant IDH2 in AML, which is optimized by K413-acetylation-mediated restriction to produce sufficient 2-HG for transformation and avoid cytotoxic accumulation of intracellular 2-HG to AML cells (Figure 7). Moreover, our findings revealed a novel mechanistic link between FLT3 and mutant IDH2. We found that FLT3 promotes mutant IDH2 lysine acetylation by phosphorylating mutant IDH2, which enhances acetyltransferase ACAT1 recruitment to mutant IDH2. In addition, FLT3 simultaneously activates ACAT1 but inhibits deacetylase SIRT3 through direct tyrosine phosphorylation, which also contributes to mutant IDH2 lysine acetylation (Figure 7). Thus, our studies showcase the beauty of complex signal transduction-based regulations of cellular processes, which provide new evidence to support a concept that hierarchical, distinct post-translational modifications act in concert to provide precise regulation of a series of sequential events, wherein mutant IDH2 is precisely controlled to achieve optimized leukemogenesis in AML.
Figure 7. Working model:
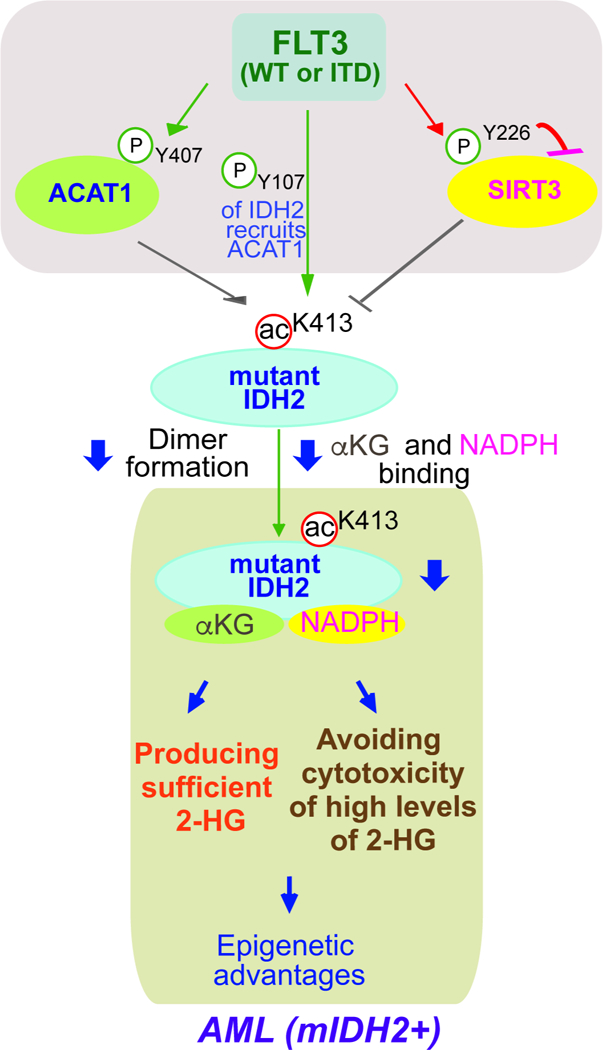
mitochondrial mutant IDH2 activity is commonly restricted through inhibitory K413-acetylation in human AML cells, which optimizes mutant IDH2-dependent transformation by producing sufficient 2-HG for transformation and avoiding cytotoxic accumulation of intracellular 2-HG to AML cells. FLT3 promotes mutant IDH2 lysine acetylation by phosphorylating mutant IDH2, which enhances acetyltransferase ACAT1 recruitment to mutant IDH2. In addition, FLT3 simultaneously activates ACAT1 but inhibits deacetylase SIRT3 through direct tyrosine phosphorylation, which also contributes to mutant IDH2 lysine acetylation.
Moreover, our findings elucidate exclusive isoform-specific regulatory mechanisms for mutant IDH1 and mutant IDH2 in AML. Mutant IDH2 cannot be similarly regulated by tyrosine phosphorylation because although the flanking regions are homologous, the corresponding residues of mIDH1 Y42 and Y391 in mIDH2 are phenylalanine (F82 and F432, respectively) that cannot be phosphorylated. In addition, K413-acetylation of mIDH2 in mitochondria is subcellular compartment-specific with mitochondrial ACAT1 and SIRT3 as the upstream acetyltransferase and deacetylase of mIDH2, respectively, which cannot modify cytosolic mIDH1.
Limitations of the Study:
Although IDH1 and IDH2 are highly homologous, our findings revealed that the intrinsic catalytic properties of mIDH1 and mIDH2 are different. The intrinsic enzyme activity of mIDH2 is much higher than mIDH1, and restricted mIDH2 activity by lysine acetylation is still comparable to that of mIDH1. Future studies are warranted to decipher the underlying molecular and structural mechanism. Moreover, understanding of such intrinsic differences between mIDH1 and mIDH2 will allow us to comprehend the differential pathogenic importance of IDH1 and IDH2 mutations during cancer evolution, including the different occurrence and frequency of IDH1 and 2 mutations in different cancer types. For example, this might help to explain why IDH2 mutations are more frequent in AML than glioblastoma (Stein, 2016). It remains possible that glioblastoma cells may be more sensitive to cytotoxicity of high 2-HG levels that is likely accumulated more efficiently in cells expressing mIDH2 due to its high intrinsic catalytic activity, which was observed in human primary AML cells expressing mIDH2 where in general the intracellular 2-HG levels are higher compared to AML cells expressing mIDH1 (Figure 1A), and/or that glioblastoma cells lack similar mechanism involving lysine acetylation in AML cells to restrict the high catalytic activity of mIDH2 activity. Future studies are warranted.
STAR+METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jing Chen (jingchen@medicine.bsd.uchicago.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Original western blot images have been deposited at Mendeley and are publicly available as of the date of publication: https://data.mendeley.com/datasets/wrt34nw5z9/draft?a=0c0cc371-dfd3–441d-a8f8-c6577c8e4caf. The DOI is listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Primary tissue samples from patients with leukemia and healthy donors
Approval of use of human specimens was given by the Institutional Review Board of Emory University School of Medicine. The clinical samples numbered with UPN were obtained with informed consent with approval by the Emory University Institutional Review Board. The clinical samples from Memorial Sloan-Kettering Cancer Center were approved from the institutional review boards of Memorial Sloan-Kettering Cancer Center, Dana-Farber Cancer Institute, and MD Anderson Cancer Center. The clinical samples from the Ohio State University were approved from institutional review board. Patients provided written informed consent in all cases at time of enrollment. Only samples from leukemia patients that were not previously treated with chemotherapy or radiation therapy were used. Mononuclear cells of peripheral blood and bone marrow samples from leukemia patients or healthy donors were isolated using lymphocyte separation medium (Cellgro). Cells were then counted and cultured in RPMI 1640 medium supplemented with 10% FBS and P/S for further indicated treatments.
Xenograft mice
Approval of the use of mice and designed experiments was given by the Institutional Animal Care and Use Committee (IACUC) of Emory University. Briefly, NSG mice (NOD scid gamma, female 6-week old, The Jackson Lab) were injected with 5X106 PDX cells (FLT3 ITD/IDH2 R140Q, Dana Farber) via tail vein. Expansion of PDX cells were monitored by staining peripheral blood samples with human anti-CD45 antibody. Once hCD45 levels in the blood reached 50%, the PDX cells were collected from the spleen.
Cells
TF-1 cells (#CRL-2003, ATCC; purchased 2018; authentication and Mycoplasma not tested) were cultured in RPMI 1640 medium with 10% FBS and 2 ng/ml recombinant human GM-CSF (R&D Systems). MOLM14 (from D. Gary Gilliand’s lab; authentication and Mycoplasma not tested) cells were cultured in RPMI 1640 medium with 10% FBS. HEK293T (from D. Gary Gilliand’s lab; authentication and Mycoplasma not tested) was cultured in Dulbecco Modified Eagle Medium (DMEM) with 10% FBS. Cells were cultured at 37 with of 5% CO2. Stable overexpression of IDH2R140Q variants in TF-1 cells was conducted by using pLVX-IRES-Hyg vector harboring FLAG-tagged IDH2 R140Q, R140Q/K413R, or R140Q/K413Q. Briefly, to produce lentivirus, each construct was co-transfected into HEK293T cells using Lenti-X™ Packaging Single shots (Clontech) according to the manufacturer’s instructions. Lentivirus-containing supernatant medium was collected 48 hours after transfection, filtered before adding to the indicated host cell lines. 24 hours after infection, target cells were subjected to hygromycin selection (Invitrogen). The overexpression of proteins was confirmed by Western blotting using antibodies against IDH2.
Antibodies
Antibodies against DYKDDDDK (FLAG) tag, p-Tyrosine (p-Tyr-100), IDH2, pan-K-Ac, SIRT3, dimethyl-histone H3 (Lys9), and histone H3 were from Cell Signaling Technology (CST). Antibody against Histone H3, trimethyl (Lys9) was from Abcam. Antibodies against FLAG, β-actin and were from Sigma-Aldrich. Antibody against IDH2 was from Santa Cruz Biotechnology. Antibody against Trimethyl-Histone H3 (Lys4) was from EMD Millipore. Antibody against IDH2 K413-Ac was from GeneTel Laboratories LLC. Antibody against ACAT1 was from Novus. Antibody against 5-Hydroxymethylcytosine (5-hmC) was from ACTIVE MOTIF. Antibody against IDH1 was from R&D SYSTEMS. FITC Annexin V Apoptosis Detection Kit was from BD Pharmingen™.Goat anti-Mouse IgG (H+L) secondary antibody and goat anti-rabbit IgG (H+L) secondary antibody were from Thermo Fisher Scientific. Antibody specially recognizing phosphorylation at tyrosine 107 of IDH2 (p-IDH2 Y107) and phosphorylation at tyrosine 226 of SIRT3 (p-SIRT3 Y226) were prepared commercially by immunizing rabbits at Shanghai HuiOu Biotechnology Co. Ltd (China).Synthesized peptides SALATQKY(p)SVAVKC as antigen for p-IDH2 Y107 and CKGLLLRLY(p)TQNIDGL as antigen for p-SIRT3 Y226 were coupled to KLH to immunize rabbits, respectively. Anti-serums were collected after five doses of immunization, and then subjected to the first step of antigen affinity purification by non-phosphorylation peptide to exclude the non-phosphorylation antibody, followed by the second step of antigen affinity purification by phosphorylation peptide for first step’s fluid penetrating fluid to get the phosphorylation antibody. ELISA assay was used to test the antibody’s titer and specificity between phosphorylation and non-phosphorylation by peptide competition.
Reagents
β-nicotinamide adenine dinucleotide 2′-phosphate reduced tetrasodium salt hydrate(NADPH),α-ketoglutaric acid sodium salt, ATP disodium salt hydrate, arecoline hydrobromide (AH), Trichostatin A (TSA) and Nicotinamide were purchased from Sigma-Aldrich. Ketoglutaric acid sodium salt, α-[1–14C]-, 50µCi (1.85MBq) were from PerkinElmer. Inhibitors including dovitinib (TKI-258, CHIR-258), quizartinib, and AG-221 were purchased from Selleckchem. Recombinant proteins including FLT3 and FGFR1 were purchased from Thermo Fisher.
METHOD DETAILS
Cell proliferation assay
Cell proliferation assays were performed by seeding 5 × 104 cells in a 6-well plate treated with indicated concentrations of TFMB-R-2-HG or 1-(Methoxymethyl)-3-(trifluoromethyl) benzene. Cell proliferation was determined by cell numbers recorded daily after being seeded and normalized to that of each cell line at the starting time (T=0 hour) by trypan blue exclusion using TC20 Automated Cell Counter (BioRad).
Mutant IDH2 enzyme activity assay
An in vitro or in vivoIDH2 R140Q and R172K activity assay was performed using IDH2 (R140Q) Assay Kit (BPS Bioscience). To be specific, 20ng purified IDH2 R140Q and variants proteins were used for in vitro activity assay. To determine the IDH2 R140Q activity in cells, 1×106 cells were harvested and directly lysed using 100 μl1% NP-40 cell lysis buffer. 25 μl whole cell lysates were used for in vivo activity assay.
Immunoprecipitation
1–2mg of cells lysates were incubated with anti-IDH1 antibody (R&D SYSTEMS) or anti-IDH2 antibody (CST) overnight at 4 °C. After incubation, protein G-Sepharose was used for precipitation for 2 hours. The beads were then washed 3 times with 1xTBS and eluted by boiling in SDS sample buffer for Western blotting analysis.
Purification of prokaryotic recombinant proteins
6xHis-IDH2-R140Q-FLAG and variants proteins, and ACAT1 WT or Y407F mutant proteins were purified by sonicating of high expressing BL21(DE3) pLysS cells obtained from a 250 ml culture subjected to IPTG-induction for 16 hours at 30°C. Bacteria cell lysates were obtained by centrifugations and loaded onto a Ni-NTA column within 20 mM imidazole. The bound proteins were eluted with 250 mM imidazole, followed by desalting using a PD-10 column. The purified recombinant IDH2 proteins and ACAT1 proteins were examined by Coomassie Brilliant Blue staining and western blotting.
In vitro tyrosine kinase assays
In vitro kinase assays were performed as previously described(Fan et al., 2016). In brief, 1μg recombinant IDH2 R140Q, or IDH1 R132H proteins were incubated with diverse recombinant active form of tyrosine kinases for 90 min in the presence of 800 μM ATP (Sigma) at 30°C in the following assay buffer, respectively. For FGFR1 assay buffer, 10 mM HEPES (pH 7.5), 10 mM MnCl2, 150 mM NaCl, 5 mM DTT, 0.01% Triton® X-100 was used; for FLT3 assay buffer, 60 mM HEPES (pH 7.5), 3 mM MgCl2, 3 mM MnCl2, 3 μM Na3VO4, 1.2 mM DTT was used. The amount of each recombinant active kinases used for the reaction was as follows: 100 ng rFGFR1 (0.1 U), and 25 ng rFLT3 (0.06 U).
FLAG-pull down assay
200 μg total protein from whole cell lysates were incubated with 30 μl of ANTI-FLAG M2 Affinity Gel (Sigma-Aldrich) for 4 hours at 4°C, followed by washing with phosphate-buffered saline (PBS) for 3 times to remove unbound materials. The bound proteins were then eluted from beads by boiling in SDS buffer (50 mM Tris-Cl (pH 6.8), 2% (w/v) SDS, 0.1% (w/v) bromophenol blue, 100 mM DTT) for 10 minutes and visualized via immunoblotting analysis.
Native PAGE
Native gels were prepared as described (Fan et al., 2016) using 0.375M Tris-HCl, pH 8.8,10% acrylamide, without SDS. Cell lysates were mixed with 5 x native gel sample buffer (0.05% bromophenol blue, 10% glycerol, 312.5mM Tris-HCl, pH 6.8) before loading. The samples were applied to native PAGE that ran in buffer (25mM Tris-HCl, 192mM glycine), followed by Western blotting.
Sucrose density ultracentrifugation
Sucrose gradient centrifugation was performed as previously described (Fan et al., 2016). In brief, purified recombinant 6xHis-IDH2 R140Q-FLAG and variant protein was laid on a 13.75%−36% sucrose gradient and spun at 50,000 rpm for 12 hours using a Beckman MLS-50 rotor. Fractions in each section were collected and analyzed by Coomassie blue staining.
Substrate binding assay
Purified recombinant 6xHis-IDH2-FLAG R140Q and variants that are immobilized on anti-FLAG beads were treated with or without ACAT1 or SIRT3 in an in vitro assay. The beads were incubated with 0.1 mMα-ketoglutaric acid[1-14C] (Perkin Elmer) at room temperature for 1 hour and then washed twice with TBS to remove the unbound α-ketoglutaric acid[1-14C]. The IDH2-R140Qand variants proteins were eluted with 3x FLAG peptide. Bound α-ketoglutaric acid molecules to proteins were measured using a scintillation counter.
In vitro ACAT1 acetylation and SIRT3 deacetylation assay
Purified recombinant IDH2 R140Q and variants protein was mixed with 100ng ACAT1 acetyltransferase in the buffer containing 40 mM Tris-HCL pH8.0, 75 mM potassium chloride (KCl), and 10 μM acetyl CoA in a final volume of 30 μL for 45 minutes at 30 °C, and terminated by the addition of SDS-PAGE sample buffer and acetylated proteins were visualized by SDS-PAGE. Purified ACAT1 WT or Y407 mutant proteins were incubated with ACAT1antibody (CST) overnight at 4 °C and subsequently incubated with protein G-Sepharose for 2 hours at 4 °C. After three washes with TBS, purified ACAT1 WT or Y407 mutant proteins on beads were incubated with or without purified FLT3 in buffer containing 60 mM HEPES (pH 7.5), 3 mM MgCl2, 3 mM MnCl2, 3 μM Na3VO4, 1.2 mM DTT, 800 μM ATP at 30°C for 90 min, washed with TBS, and then subjected to in vitro acetyltransferase assay using purified IDH2 R140Q mutant (ACTIVE MOTIF) as an exogenous substrate in the buffer containing 40 mM Tris-HCL pH8.0, 75 mM potassium chloride (KCl), and 10 μM acetyl CoA at 30 °C. Western blot was performed to assess K413 acetylation of mutant IDH2. For deacetylation assay, 1 μg of purified recombinant R140Q and variants was mixed with 200 ng SIRT3 in the SDAC buffer (50 mM Tris-HCL PH9.0, 4 mM MgCl2, 50 mM NaCl, 0.5 mM DTT, 1mM NAD+, 0.5 μM TSA) in a final volume of 20 μL for 3 hours at 30 °C with gentle agitation. The reaction was terminated by adding SDS sample buffer. Deacetylation of substrate was detected by immunoblot analysis.
Kinetics assay
20ng purified IDH2 R140Q and variants proteins that pre-treated with or without recombinant ACAT1 or SIRT3 in an in vitro assay were incubated with various concentrations of NADPH (0–1.5 μM; Sigma-Aldrich) and 5 mMα-KG(Sigma-Aldrich) or various concentrations ofα-KG (0–5mM; Sigma-Aldrich) and 1.5 μM NADPH(Sigma-Aldrich) at 100 μl enzyme activity assay buffer (25mM Tris-HCl (pH7.5), 10 mM MgCl2, 5mM DTT). Absorbance at 340 nm was measured every 20 seconds for 5 minutes using a Spectra Max Plus spectrophotometer (Molecular Devices). Non-linear regression analysis (Michaelis-Menten) was performed in Graphpad Prism 5.03
IDH2 R140Q protein monomer conversion
1 mg IDH2 R140Q and variants protein was used for in vitro assay with ACAT1 or SIRT3, then the dimer was separated by Sucrose gradient centrifugation. Then 1μg dimer was collected as indicated time and the monomer conversion was stopped by fridge of minus 80. Then samples were injected for the Western blotting analysis with native gel.
IDH2 R140Q protein dimer formation
1 mg IDH2 R140Qand variants protein was used for in vitro assay with ACAT1 or SIRT3, then the monomer was separated by Sucrose gradient centrifugation. Then 1μg monomer was collected as indicated time and the monomer conversion was stopped by fridge of minus 80. Then samples were injected for the Western blotting analysis with native gel.
Drug treatment assay
For the Western blotting and IDH2 R140Q activity assay, treatments were performed by incubating cells with 5μM TSA, 10mM NAM, 200 nM quizartinib, or 20μM AH for 24 hours. For 2-HG measurement, treatments with tyrosine kinase inhibitors were performed by incubating cells with 200 nM quizartinib for 72 hours.
2-HG measurement in cell lines and patient samples
The sample preparation for ex vivo solid state high resolution magic angle (HRMAS)proton NMR analysis followed a previously described method (30). Briefly, 1 × 107 cells were thawed in 99.996% saline deuterium oxide (D2O, Sigma-Aldrich) in the sample holder/rotor (4 mm ZrO2). A 50 µl insert was subsequently placed in the sample holder to stabilize the sample and to provide the balance for the rotor. For NMR experiments, D2O (99.996%) containing 0.75% 3-(trimethylsilyl) propionic acid (TSP) was added to get a frequency-lock signal and to serve as an internal reference for chemical shift and metabolite quantification. Each sample was reweighed after adding TSP. All NMR samples were prepared rapidly on ice to avoid possible degradation. NMR experiments were conducted with a dedicated 4 mm HRMAS probe using a Bruker AVANCE 600 WB solid state NMR spectrometer (Bruker Instruments, Inc.). The probe-head was precooled to 4°C before loading the sample. The entire experiment was carried out at 4°C (±0.1°C) maintained via a variable temperature control unit. To ensure the spin sidebands did not distort the spectrum, the spinning rates of sample were controlled either at 2,800 KHz (±2 Hz) or at the lower spin rate of 800 Hz if the rotor-synchronized delay alternating with nutation for tailored excitation sequence was used. The pre-saturation of water was achieved with a zqpr sequence before acquisition pulses. A rotor-synchronized Carr-Purcell-Meibom-Gill pulse sequence was used to suppress broad signals from macromolecules. In all experiments, the repetition time of 5.0 s, the spectral width of 10 kHz and number of transients of 256 were used for recording the data. 95% pure 2-HG compound (Santa Cruz Biotechnology) in the solution was used as a reference for identification and assignment of the resonances from 2-HG. One-dimensional (1D) NMR spectra of a pure 2-HG compound and a mixture of 2-HG and a combination of glutamate (Glu) and glutamine (Gln) compounds (together termed Glx) prepared with 10 mM 2-HG and pH 7.0 were collected at 300 MHz at 25°C. J-coupling correlations and patterns of protons in the pure 2-HG were then analyzed and confirmed by a two-dimensional (2D) J-coupled correlated spectroscopy (COSY) method. The methyl protons of TSP at the known concentration and the methyl protons of 2-HG identified in each sample were then used for calibrating and calculating the concentration of 2-HG in the sample.
CobB treatment
For in vitro deacetylation assay, 10 μg of IDH2 R140Q and variants was incubated with 5 μg of CobB in deacetylation reaction buffer containing 50 mM Tris-HCL (pH 8.0), 135 mM NaCl, 2.5 mM KCl, and 1 mM MgCl2 in the presence of 4 mM NAD+ and 40 mM NAM.
Cell cycle and cell death/apoptosis assay
For cell cycle, 1 × 106cells were collected by centrifugation, then the cells were resuspended in 5 ml of 70% ethanol at 4°C overnight. After rinsing with PBS, the fixed cells were resuspended in PBS containing 50 μg/ml RNaseA and 50 μg/ml propidium iodide and incubated at 4°C for 4 hours. The stained cells were passed through a nylon-mesh sieve to remove cell clumps and were analyzed by a FACScan flow cytometer. For cell death/apoptosis assay, it was performed using FITC Annexin V Apoptosis Detection Kit ( BD Pharmingen™) and analyzed by a FACScan flow cytometer.
RNA-seq and relative data analysis
The RNA seq was performed as described previously (Su et al., 2018). In brief, Emory Integrated Genomics Core performs RNA extraction and QC for MOLM14 cells treated with indicated concentration of TFMB-2-HG. The RNA samples were sequenced by Novogene for 50M PE150 reads. The data have been deposited in the GEO repository with the accession number GSE173531. Gene Set Enrichment Analysis (GSEA) was used to analyze the signal pathway enrichment in different groups of samples.
Dot-blot assay
Genomic DNA was extracted by Wizard™ Genomic DNA Purification Kits (Promega Corporation) and spotted on a nitrocellulose membrane as indicated DNA amount. The membrane was placed under an ultraviolet lamp for 20 min to crosslink the DNA and then blocked with 5% milk for 1 hour, followed by incubating with the anti-5hmC (ACTIVE MOTIF) antibody at 4°C overnight. After incubation with an HRP-conjugated secondary antibody (Thermo Fisher Scientific) at room temperature for 1 hour, the membrane was washed for three times and then exposed to X-ray film.
QUANTIFICATION AND STATISTICAL ANALYSIS
For studies in which statistical analyses were performed, a 2-tailed Student’s t-test was used to generate p values, except a two-way ANOVA was used for cell proliferation assay. p values less than or equal to 0.05 were considered significant. Data with error bars represent mean ± SD. There is no estimate of variation in each group of data and the variance is similar between the groups. No statistical method was used to predetermine sample size. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment. All data are expected to have normal distribution. Statistical analysis and graphical presentation were performed using Prism 6.0 (GraphPad) and Microsoft Office Excel 2016.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| DYKDDDDK Tag Antibody | Cell Signaling Technology | Cat#2368S; Clone# N/A; RRID:AB_2217020 |
| Mouse monoclonal ANTI-FLAG M2 antibody | Sigma-Aldrich | Cat# F3165; Clone# M2; RRID: AB_259529 |
| Phospho-Tyrosine Mouse mAb (P-Tyr-100) antibody | Cell Signaling Technology | Cat# 9411S; Clone# N/A; RRID:AB_331228 |
| IDH2 | Santa Cruz Biotechnology | Cat# sc-374476; Clone# B-6; RRID: AB_10986415 |
| IDH2 | Cell Signaling Technology | Cat# 56439S; Clone# D8E3B; RRID: AB_2799511 |
| ac-IDH2K413 | GeneTel Laboratories | Cat# AC0004; Clone# N/A; RRID: N/A |
| ACAT1 | Novus Biologicals | Cat#NB400–141; Clone# N/A; RRID: AB_10001588 |
| p-ACAT1 Y407 | Cell Signaling Technology(Fan et al., 2016) | Cat# N/A; Clone# N/A; RRID: N/A |
| p-IDH2 Y107 | Shanghai HuiOu Biotechnology Com.LTD | Cat# N/A; Clone# N/A; RRID: N/A |
| p-SIRT3 Y226 | Shanghai HuiOu Biotechnology Com.LTD | Cat# N/A; Clone# N/A; RRID: N/A |
| ACAT1 Antibody | Cell Signaling Technology | Cat# 44276; Clone# N/A; RRID: AB_2799262 |
| p-IDH1 Y42 | SHANGHAI GENOMICS,INC.(Chen et al., 2019) | Cat# N/A; Clone# N/A; RRID: N/A |
| p-IDH1 Y391 | SHANGHAI GENOMICS,INC.(Chen et al., 2019) | Cat# N/A; Clone# N/A; RRID: N/A |
| Human Isocitrate Dehydrogenase1/IDH1 Antibody | R&D SYSTEMS | Cat# MAB7049; Clone# 843219; RRID: AB_2811299 |
| Acetylated-Lysine Antibody | Cell Signaling Technology | Cat# 9441; Clone# N/A; RRID: AB_331805 |
| ACAT2 (E1L8V) Rabbit mAb | Cell Signaling Technology | Cat# 13294; Clone# E1L8V; RRID: AB_2798172 |
| ChIPAb+ Trimethyl-Histone H3 (Lys4) antibody | Millipore | Cat# 17–614; Clone# N/A; RRID: AB_1587135 |
| Di-Methyl-Histone H3 (Lys9) Antibody | Cell Signaling Technology | Cat# 9753S; Clone# N/A; RRID: AB_659848 |
| Rabbit Anti-Histone H3, trimethyl (Lys9) ChIP Grade Polyclonal Antibody, Unconjugated | Abcam | Cat# ab8898; Clone# N/A; RRID: AB_306848 |
| Histone H3 (D1H2) XP Rabbit mAb antibody | Cell Signaling Technology | Cat# 4499S; Clone# N/A; RRID: AB_10544537 |
| PDC-E2 (H-160) antibody (DLAT) | Santa Cruz Biotechnology | Cat#sc-32925; Clone# H-160; RRID:AB_2091782 |
| SirT3 (C73E3) Rabbit mAb | Cell Signaling Technology | Cat#2627S; Clone# N/A; RRID: AB_2188622 |
| SIRT4 | Abcam | Cat#ab90485; Clone# N/A; RRID: AB_2286004 |
| APC anti-human CD45 Antibody | Biolegend | Cat# 368512 Clone# Clone 2D1; RRID: AB_2566372 |
| PE anti-mouse CD45.2 Antibody | Biolegend | Cat# 109808 Clone# Clone 104; RRID: AB_313445 |
| SIRT5 | Abcam | Cat# ab13697 Clone# N/A; RRID: AB_300574 |
| 5-Hydroxymethylcytosine (5-hmC) antibody (pAb) | ACTIVE MOTIF | Cat# 39069 Clone# N/A; RRID: N/A |
| Mouse monoclonal Anti-beta-Actin antibody | Sigma-Aldrich | Cat# A1978; Clone# AC-15; RRID: AB_476692 |
| Goat anti-Mouse IgG (H+L) Secondary Antibody, HRP | Thermo Fisher Scientific | Cat# 31430; Clone# N/A; RRID: AB_228307 |
| Goat anti-Rabbit IgG (H+L) Secondary Antibody, HRP | Thermo Fisher Scientific | Cat# 31460; Clone# N/A; RRID: AB_ 228341 |
| Bacterial and Virus Strains | ||
| BL21(DE3) Chemically Competent Cells | Sigma-Aldrich | Cat# CMC0014 |
| Biological Samples | ||
| PDX (FLT3 ITD|IDH2 R140Q) | Dana Farber | Cat#DFAM-10360-V1 |
| Patient samples | Memorial Sloan-Kettering Cancer Center, | # 55653 # 45588 # 87794 |
| Patient samples | Emory University | # UPN1284 |
| Patient samples | Ohio State University | # U-18–3562 # U-19–0875 # U-19–0877 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Myelin Basic Protein bovine | Sigma-Aldrich | Cat# M1891 |
| Coomassie Brilliant Blue R-250 Staining Solution | Bio-rad | Cat# 1610436 |
| DL-Isocitric acid trisodium salt hydrate | Sigma-Aldrich | Cat# I1252 CAS: 1637–73-6 |
| β-Nicotinamide adenine dinucleotide phosphate hydrate | Sigma-Aldrich | Cat# N5755 CAS: 53–59-8 |
| β-Nicotinamide adenine dinucleotide 2′-phosphate reduced tetrasodium salt hydrate | Sigma-Aldrich | Cat# N1630 CAS: 2646–71-1 |
| α-Ketoglutaric acid sodium salt | Sigma-Aldrich | Cat# K2010 CAS: 22202–68-2 |
| Diaphorase from Clostridium kluyveri | Sigma-Aldrich | Cat# D5540 CAS: 9001–18-7 |
| Resazurin sodium salt | Sigma-Aldrich | Cat# R7017 CAS: 62758–13-8 |
| Ketoglutaric Acid, Sodium Salt, α-[1-14C]-, 50µCi (1.85MBq) | PerkinElmer | Cat# NEC597050UC |
| Trichostatin A | Sigma-Aldrich | Cat# T1952 |
| Blue Sepharose CL-6B | Amersham Biosciences | Cat# 17–0830-01 |
| Nicotinamide | Sigma-Aldrich | Cat# N0636 |
| TFMB-R-2-HG | Cayman | Cat# 20461 CAS: 1445700–01-5 |
| 1-(Methoxymethyl)-3-(trifluoromethyl)benzene | Combi-Blocks | Cat# OT-0769–1g CAS: 380633–51-2 |
| Arecoline hydrobromide | Sigma-Aldrich | Cat# 10980 CAS: 300–08-3 |
| Quizartinib (AC220) | Selleckchem | Cat# S1526 CAS: 950769–58-1 |
| acetyl CoA | Sigma-Aldrich | Cat# S7008 CAS: 102029–73-2 |
| Recombinant IDH2 (R140Q) proteins | ACTIVE MOTIF | Cat# 31617 |
| FLT3 Recombinant Human Protein | Thermo Fisher | Cat# PV3182 |
| FLT3 [ITD] Recombinant Human Protein | Thermo Fisher | Cat# PV6190 |
| Recombinant Human GM-CSF Protein, CF | R&D SYSTEMS | 215GM050 |
| Adenosine 5′-triphosphate (ATP) disodium salt hydrate | Sigma-Aldrich | Cat# A7699 CAS: 34369–07-8 |
| Hygromycin B | Gibco | Cat# 10687010 CAS: 31282–04-9 |
| Puromycin dihydrochloride from Streptomyces alboniger | Sigma-Aldrich | Cat# P8833 CAS: 58–58-2 |
| ANTI-FLAG® M2 Affinity Gel | Sigma-Aldrich | Cat# A2220 |
| FLAG® Peptide | Sigma-Aldrich | Cat# F3290 |
| Protein G Sepharose® 4 Fast Flow | Sigma-Aldrich | Cat# GE17–0618-01 |
| Critical Commercial Assays | ||
| TransIT®-LT1 Transfection Reagent | Mirus | Cat# MIR 2305 |
| Wizard™ Genomic DNA Purification Kits | Promega | Cat# A1125 |
| Deposited Data | ||
| RNA-seq Data | This study | GEO: GSE173531 |
| Original western blot images have been deposited at Mendeley and are publicly available as of the date of publication: | This study | https://data.mendeley.com/datasets/wrt34nw5z9/draft?a=0c0cc371-dfd3–441d-a8f8-c6577c8e4caf |
| Experimental Models: Cell Lines | ||
| Human: HEK293T cells | ATCC | Cat# CRL-3216; RRID: CVCL_0063 |
| Human:TF-1 cells | ATCC | Cat# CRL-2003; RRID:CVCL_0559 |
| Human: EL-1 cells | ATCC | Cat# CRL-9854; RRID: N/A |
| Human: MOLM-14 | (Fan et al., 2016) | Cat# N/A; RRID: N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ | The Jackson Laboratory | Stock# 005557 RRID: IMSR_JAX:005557 |
| Oligonucleotides | ||
| shRNA targeting IDH2 sequence: 5’-TGCAAGAACTATGACGGAGAT −3’ |
BROAD Institute/Open Biosystems | N/A |
| Primers: IDH2 K48R mutation, Forward: 5’-aggatcaaggtggcgaggcccgtggtggagatg’ |
This paper | N/A |
| Primers: IDH2 K48R mutation, Reverse: 5’-catctccaccacgggcctcgccaccttgatcct-3’ |
This paper | N/A |
| Primers: IDH2 K67R mutation, Forward: 5’-atctggcagttcatcagggagaagctcatcctg-3’ |
This paper | N/A |
| Primers: IDH2 K67R mutation, Reverse: 5’- caggatgagcttctccctgatgaactgccagat −3’ |
This paper | N/A |
| Primers: IDH2 K69R mutation, Forward: 5’-cagttcatcaaggagaggctcatcctgccccac −3’ |
This paper | N/A |
| Primers: IDH2 K69R mutation, Reverse: 5’-gtggggcaggatgagcctctccttgatgaactg-3’ |
This paper | N/A |
| Primers: IDH2 K80R mutation, Forward: 5’-gtggacatccagctaaggtattttgacctcggg’ |
This paper | N/A |
| Primers: IDH2 K80R mutation, Reverse: 5’-cccgaggtcaaaataccttagctggatgtccac-3’ |
This paper | N/A |
| Primers: IDH2 K106R mutation, Forward: 5’-gcactggccacccagaggtacagtgtggctgtc-3’ |
This paper | N/A |
| Primers: IDH2 K106R mutation, Reverse: 5’-gacagccacactgtacctctgggtggccagtgc-3’ |
This paper | N/A |
| Primers: IDH2 K127R mutation, Forward: 5’-cgtgtggaagagttcaggctgaagaagatgtgg-3’ |
This paper | N/A |
| Primers: IDH2 K127R mutation, Reverse: 5’- ccacatcttcttcagcctgaactcttccacacg −3’ |
This paper | N/A |
| Primers: IDH2 K133R mutation, Forward: 5’-ctgaagaagatgtggagaagtcccaatggaact-3’ |
This paper | N/A |
| Primers: IDH2 K133R mutation, Reverse: 5’-agttccattgggacttctccacatcttcttcag-3’ |
This paper | N/A |
| Primers: IDH2 K155R mutation, Reverse: 5’- gagcccatcatctgcagaaacatcccacgccta −3’ |
This paper | N/A |
| Primers: IDH2 K155R mutation, Forward: 5’-taggcgtgggatgtttctgcagatgatgggctc-3’ |
This paper | N/A |
| Primers: IDH2 K166R mutation, Forward: 5’-gtccctggctggaccaggcccatcaccattggc-3’ |
This paper | N/A |
| Primers: IDH2 K166R mutation, Reverse: 5’- gccaatggtgatgggcctggtccagccagggac-3’ |
This paper | N/A |
| Primers: IDH2 K180R mutation, Forward: 5’-catggcgaccagtacagggccacagactttgtg’ |
This paper | N/A |
| Primers: IDH2 K180R mutation, Reverse: 5’-cacaaagtctgtggccctgtactggtcgccatg-3’ |
This paper | N/A |
| Primers: IDH2 K242R mutation, Forward: 5’-cagtatgccatccagaggaaatggccgctgtac-3’ |
This paper | N/A |
| Primers: IDH2 K242R mutation, Reverse: 5’-gtacagcggccatttcctctggatggcatactg −3’ |
This paper | N/A |
| Primers: IDH2 K256R mutation, Forward: 5’-aagaacaccatactgagagcctacgatgggcgt-3’ |
This paper | N/A |
| Primers: IDH2 K256R mutation, Reverse: 5’-acgcccatcgtaggctctcagtatggtgttctt-3’ |
This paper | N/A |
| Primers: IDH2 K263R mutation, Forward: 5’-tacgatgggcgtttcagggacatcttccaggag-3’ |
This paper | N/A |
| Primers: IDH2 K263R mutation, Reverse: 5’-ctcctggaagatgtccctgaaacgcccatcgta-3’ |
This paper | N/A |
| Primers: IDH2 K272R mutation, Forward: 5’-caggagatctttgacaggcactataagaccgac-3’ |
This paper | N/A |
| Primers: IDH2 K272R mutation, Reverse: 5’gtcggtcttatagtgcctgtcaaagatctcctg-3’ |
This paper | N/A |
| Primers: IDH2 K275R mutation, Forward: 5’-tttgacaagcactataggaccgacttcgacaag-3’ |
This paper | N/A |
| Primers: IDH2 K275R mutation, Reverse: 5’-cttgtcgaagtcggtcctatagtgcttgtcaaa-3’ |
This paper | N/A |
| Primers: IDH2 K280R mutation, Forward: 5’-aagaccgacttcgacaggaataagatctggtat-3’ |
This paper | N/A |
| Primers: IDH2 K280R mutation, Reverse: 5’-ataccagatcttattcctgtcgaagtcggtctt-3’ |
This paper | N/A |
| Primers: IDH2 K282R mutation, Forward: 5’-gacttcgacaagaataggatctggtatgagcac-3’ |
This paper | N/A |
| Primers: IDH2 K282R mutation, Reverse: 5’gtgctcataccagatcctattcttgtcgaagtc-3’ |
This paper | N/A |
| Primers: IDH2 K384R mutation, Forward: 5’-ctggagcaccgggggaggctggatgggaaccaa-3’ |
This paper | N/A |
| Primers: IDH2 K384R mutation, Reverse: 5’-ttggttcccatccagcctcccccggtgctccag-3’ |
This paper | N/A |
| Primers: IDH2 K442R mutation, Forward: 5’-ttcctcgacaccatcaggagcaacctggacaga-3’ |
This paper | N/A |
| Primers: IDH2 K442R mutation, Reverse: 5’ -tctgtccaggttgctcctgatggtgtcgaggaa-3’ |
This paper | N/A |
| Primers: IDH2 K413R mutation, Forward: 5’-agtggagccatgaccagggacctggcgggctgcatt-3’ |
This paper | N/A |
| Primers: IDH2 K413R mutation, Reverse: 5’-aatgcagcccgccaggtccctggtcatggctccact-3’ |
This paper | N/A |
| Primers: IDH2 K413Q mutation, Forward: 5’-agtggagccatgaccCaggacctggcgggctgcatt-3’ |
This paper | N/A |
| Primers: IDH2 K413Q mutation, Reverse: 5’ -aatgcagcccgccaggtcctGggtcatggctccact-3’ |
This paper | N/A |
| Primers: IDH2 Y81F mutation, Forward: 5’-atccagctaaagttttttgacctcgggct-3’ |
This paper | N/A |
| Primers: IDH2 Y81F mutation, Reverse: 5’-gtccacgtggggcaggatgagcttctcctt-3’ |
This paper | N/A |
| Primers: IDH2 Y107F mutation, Forward: 5’-ctggccacccagaagttcagtgtggctgtcaag-3’ |
This paper | N/A |
| Primers: IDH2 Y107F mutation, Reverse: 5’-tgcagagtcaatggtgacctggtcatcagtctg-3’ |
This paper | N/A |
| Primers: IDH2 Y210F mutation, Forward: 5’-atccagctaaagttttttgacctcgggct-3’ |
This paper | N/A |
| Primers: IDH2 Y210F mutation, Reverse: 5’-gacaccactgccatcttttggggtgaagac-3’ |
This paper | N/A |
| Primers: IDH2 Y238F mutation, Forward: 5’-cacagctgcttccagtttgccatccagaagaaa-3’ |
This paper | N/A |
| Primers: IDH2 Y238F mutation, Reverse: 5’-cgcaaaacctgagatggactcgtcggtgttgtacat-3’ |
This paper | N/A |
| Primers: SIRT3 Y165F mutation, Forward: 5’-ggagtggcctgttcagcaacctccagcag-3’ |
This paper | N/A |
| Primers: SIRT3 Y165F mutation, Reverse: 5’-ctgctggaggttgctgtacaggccactcc-3’ |
This paper | N/A |
| Primers: SIRT3 Y171F mutation, Forward: 5’-agcaacctccagcagttcgatctcccgtac-3’ |
This paper | N/A |
| Primers: SIRT3 Y171F mutation, Reverse: 5’ -agcaacctccagcagttcgatctcccgtac3’ |
This paper | N/A |
| Primers: SIRT3 Y175F mutation, Forward: 5’-cagtacgatctcccgttccccgaggccattttt-3’ |
This paper | N/A |
| Primers: SIRT3 Y175 mutation, Reverse: 5’-aaaaatggcctcggggaacgggagatcgtactg-3’ |
This paper | N/A |
| Primers: SIRT3 Y200F mutation, Forward: 5’-tggccaaggagctgttccctggaaactacaag-3’ |
This paper | N/A |
| Primers: SIRT3 Y200F mutation, Reverse: 5’-cttgtagtttccagggaacagctccttggcca-3’ |
This paper | N/A |
| Primers:SIRT3 Y204F mutation, Forward: 5’-ctgtaccctggaaacttcaagcccaacgtcact-3’ |
This paper | N/A |
| Primers: SIRT3 Y204F mutation, Reverse: 5’-agtgacgttgggcttgaagtttccagggtacag-3’ |
This paper | N/A |
| Primers: SIRT3 Y211F mutation, Forward: 5’-cccaacgtcactcacttctttctccggctgctt-3’ |
This paper | N/A |
| Primers:SIRT3 Y211F mutation, Reverse: 5’-aagcagccggagaaagaagtgagtgacgttggg-3’ |
This paper | N/A |
| Primers: SIRT3 Y226F mutation, Forward: 5’-ctgcttctgcggctcttcacgcagaacatcgat-3’ |
This paper | N/A |
| Primers: SIRT3 Y226F mutation, Reverse: 5’-atcgatgttctgcgtgaagagccgcagaagcag-3’ |
This paper | N/A |
| Recombinant DNA | ||
| pLVX-IDH2-FLAG | This paper | N/A |
| pLVX -IDH2-FLAG K413R | This paper | N/A |
| pLVX -IDH2-FLAG K413Q | This paper | N/A |
| pLVX-IDH2-FLAG R140Q | This paper | N/A |
| pLVX -IDH2-FLAG R140Q/K413R | This paper | N/A |
| pLVX -IDH2-FLAG R140Q/K413Q | This paper | N/A |
| pcDNA3.0-IDH2 | From Ross Levine Lab(Memorial Sloan Kettering Cancer Center) | N/A |
| pcDNA3.0-IDH2 R140Q | From Ross Levine Lab (Memorial Sloan Kettering Cancer Center) | N/A |
| pcDNA3.0-IDH2 R172K | From Ross Levine Lab (Memorial Sloan Kettering Cancer Center) | N/A |
| pET53-DEST-FLAG-SIRT3 | (Fan et al., 2016) | N/A |
| pET53-DEST-FLAG-ACAT1 | (Fan et al., 2016) | N/A |
| pET53-DEST-FLAG-ACAT1 Y407F | (Shan et al., 2014) | N/A |
| pLHCX-FLAG-ACAT1 | (Fan et al., 2016) | N/A |
| pLHCX-FLAG-ACAT1 Y407F | (Fan et al., 2016) | N/A |
| pET53-DEST-FLAG-ACAT2 | (Shan et al., 2014) | N/A |
| pET53-DEST-FLAG-DLAT | (Shan et al., 2014) | N/A |
| pET53-DEST-FLAG-CobB | (Shan et al., 2014) | N/A |
| pET53-DEST-IDH2-FLAGK48R | This paper | N/A |
| pET53-DEST -IDH2-FLAGK67R | This paper | N/A |
| pET53-DEST -IDH2-FLAGK69R | This paper | N/A |
| pET53-DEST -IDH2-FLAGK80R | This paper | N/A |
| pET53-DEST -IDH2-FLAG K106R | This paper | N/A |
| pET53-DEST -IDH2-FLAG K127R | This paper | N/A |
| pET53-DEST -IDH2-FLAG K133R | This paper | N/A |
| pET53-DEST -IDH2-FLAG K155R | This paper | N/A |
| pET53-DEST -IDH2-FLAG K166R | This paper | N/A |
| pET53-DEST -IDH2-FLAG K180R | This paper | N/A |
| pET53-DEST -IDH2-FLAG K242R | This paper | N/A |
| pET53-DEST -IDH2-FLAG K256R | This paper | N/A |
| pET53-DEST -IDH2-FLAG K263R | This paper | N/A |
| pET53-DEST -IDH2-FLAG K272R | This paper | N/A |
| pET53-DEST -IDH2-FLAG K275R | This paper | N/A |
| pET53-DEST -IDH2-FLAG K280R | This paper | N/A |
| pET53-DEST -IDH2-FLAG K282R | This paper | N/A |
| pET53-DEST -IDH2-FLAG K384R | This paper | N/A |
| pET53-DEST -IDH2-FLAG K442R | This paper | N/A |
| pET53-DEST -IDH2-FLAG K413R | This paper | N/A |
| pET53-DEST -IDH2-FLAG K413Q | This paper | N/A |
| pET53-DEST -IDH2-FLAG | This paper | N/A |
| pET53-DEST -IDH2-FLAG R140Q/K48R | This paper | N/A |
| pET53-DEST -IDH2-FLAG R140Q/K67R | This paper | N/A |
| pET53-DEST -IDH2-FLAG R140Q/K69R | This paper | N/A |
| pET53-DEST -IDH2-FLAG R140Q/K80R | This paper | N/A |
| pET53-DEST -IDH2-FLAG R140Q/K106R | This paper | N/A |
| pET53-DEST -IDH2-FLAG R140Q/K127R | This paper | N/A |
| pET53-DEST -IDH2-FLAG R140Q/K133R | This paper | N/A |
| pET53-DEST -IDH2-FLAG R140Q/K155R | This paper | N/A |
| pET53-DEST -IDH2-FLAG R140Q/K166R | This paper | N/A |
| pET53-DEST -IDH2-FLAG R140Q/K180R | This paper | N/A |
| pET53-DEST -IDH2-FLAG R140Q/K242R | This paper | N/A |
| pET53-DEST -IDH2-FLAG R140Q/K256R | This paper | N/A |
| pET53-DEST -IDH2-FLAG R140Q/K263R | This paper | N/A |
| pET53-DEST -IDH2-FLAG R140Q/K272R | This paper | N/A |
| pET53-DEST -IDH2-FLAG R140Q/K275R | This paper | N/A |
| pET53-DEST -IDH2-FLAG R140Q/K280R | This paper | N/A |
| pET53-DEST -IDH2-FLAG R140Q/K282R | This paper | N/A |
| pET53-DEST -IDH2-FLAG R140Q/K384R | This paper | N/A |
| pET53-DEST -IDH2-FLAG R140Q/K442R | This paper | N/A |
| pET53-DEST -IDH2-FLAG R140Q/K413R | This paper | N/A |
| pET53-DEST -IDH2-FLAG R140Q/K413Q | This paper | N/A |
| pET53-DEST -IDH2-FLAGR140Q | This paper | N/A |
| pET53-DEST -IDH2-FLAG Y81F | This paper | N/A |
| pET53-DEST -IDH2-FLAG Y107F | This paper | N/A |
| pET53-DEST -IDH2-FLAG Y210F | This paper | N/A |
| pET53-DEST -IDH2-FLAG Y238F | This paper | N/A |
| pET53-DEST -SIRT3 | (Shan et al., 2014) | N/A |
| pET53-DEST -SIRT3 Y165F | This paper | N/A |
| pET53-DEST -SIRT3 Y171F | This paper | N/A |
| pET53-DEST -SIRT3 Y175F | This paper | N/A |
| pET53-DEST -SIRT3 Y200F | This paper | N/A |
| pET53-DEST -SIRT3 Y204F | This paper | N/A |
| pET53-DEST -SIRT3 Y211F | This paper | N/A |
| pET53-DEST -SIRT3 Y226F | This paper | N/A |
| Software and Algorithms | ||
| GraphPad Prism 7 software | GraphPad Software | https://www.graphpad.com/ |
| Other | ||
| N/A | ||
Research Highlight.
Different intracellular concentrations of 2-HG have different cellular functions
K413 acetylation inhibits mutant IDH2 in AML cells by attenuating dimerization
Restricted mutant IDH2 produces sufficient oncometabolite 2-HG for transformation
K413-acetylation of mutant IDH2 avoids cytotoxic accumulation of intracellular 2-HG
ACKNOWLEDGEMENTS
This work was supported in part by NIH grants including CA140515, CA183594, CA174786 (J.C.), R35197594, CA173636, MSKCC Support Grant/Core Grant P30 CA008748 (R.L.L.), and CA169937 (H.M.), and Medical Scientist Training Program, The University of Chicago (A.C.Z.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental information includes five figures.
Conflict of Interest Statement: R.L.L. is on the supervisory board of Qiagen and is a scientific advisor to Loxo, Imago, C4 Therapeutics and Isoplexis, which each includes an equity interest. He receives research support from and consulted for Celgene and Roche, he has received research support from Prelude Therapeutics, and he has consulted for Incyte, Novartis, Astellas, Morphosys and Janssen. He has received honoraria from Lilly and Amgen for invited lectures and from Gilead for grant reviews.
REFERENCES
- Boddu P, Takahashi K, Pemmaraju N, Daver N, Benton CB, Pierce S, Konopleva M, Ravandi F, Cortes J, Kantarjian H, et al. (2017). Influence of IDH on FLT3-ITD status in newly diagnosed AML. Leukemia 31, 2526–2529. [DOI] [PubMed] [Google Scholar]
- Cairns RA, and Mak TW (2013). Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discov 3, 730–741. [DOI] [PubMed] [Google Scholar]
- Chen D, Xia S, Wang M, Lin R, Li Y, Mao H, Aguiar M, Famulare CA, Shih AH, Brennan CW, et al. (2019). Mutant and Wild-Type Isocitrate Dehydrogenase 1 Share Enhancing Mechanisms Involving Distinct Tyrosine Kinase Cascades in Cancer. Cancer Discov 9, 756–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. (2009). Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Lin R, Xia S, Chen D, Elf SE, Liu S, Pan Y, Xu H, Qian Z, Wang M, et al. (2016). Tetrameric Acetyl-CoA Acetyltransferase 1 Is Important for Tumor Growth. Mol Cell 64, 859–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Shan C, Kang HB, Elf S, Xie J, Tucker M, Gu TL, Aguiar M, Lonning S, Chen H, et al. (2014a). Tyr phosphorylation of PDP1 toggles recruitment between ACAT1 and SIRT3 to regulate the pyruvate dehydrogenase complex. Molecular cell 53, 534–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Shan CL, Kang HB, Elf S, Xie JX, Tucker M, Gu TL, Aguiar M, Lonning S, Chen HB, et al. (2014b). Tyr Phosphorylation of PDP1 Toggles Recruitment between ACAT1 and SIRT3 to Regulate the Pyruvate Dehydrogenase Complex. Molecular Cell 53, 534–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green A, and Beer P (2010). Somatic mutations of IDH1 and IDH2 in the leukemic transformation of myeloproliferative neoplasms. N Engl J Med 362, 369–370. [DOI] [PubMed] [Google Scholar]
- Gross S, Cairns RA, Minden MD, Driggers EM, Bittinger MA, Jang HG, Sasaki M, Jin S, Schenkein DP, Su SM, et al. (2010). Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med 207, 339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue S, Lemonnier F, and Mak TW (2016). Roles of IDH1/2 and TET2 mutations in myeloid disorders. Int J Hematol 103, 627–633. [DOI] [PubMed] [Google Scholar]
- Jiang L, Shestov AA, Swain P, Yang C, Parker SJ, Wang QA, Terada LS, Adams ND, McCabe MT, Pietrak B, et al. (2016). Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 532, 255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kernytsky A, Wang F, Hansen E, Schalm S, Straley K, Gliser C, Yang H, Travins J, Murray S, Dorsch M, et al. (2015). IDH2 mutation-induced histone and DNA hypermethylation is progressively reversed by small-molecule inhibition. Blood 125, 296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, Cowley GS, Root DE, Ebert BL, and Kaelin WG Jr. (2013). (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 339, 1621–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcucci G, Maharry K, Wu YZ, Radmacher MD, Mrozek K, Margeson D, Holland KB, Whitman SP, Becker H, Schwind S, et al. (2010). IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 28, 2348–2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, et al. (2009). Recurring mutations found by sequencing an acute myeloid leukemia genome. The New England journal of medicine 361, 1058–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenney AS, and Levine RL (2013). Isocitrate dehydrogenase mutations in leukemia. J Clin Invest 123, 3672–3677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medeiros BC, Fathi AT, DiNardo CD, Pollyea DA, Chan SM, and Swords R (2017). Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia 31, 272–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, et al. (2011). Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. (2008). An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing Y, Dong L, Gao L, Li C, Li Y, Han L, Prince E, Tan B, Deng X, Wetzel C, et al. (2021). R-2-hydroxyglutarate attenuates aerobic glycolysis in leukemia by targeting the FTO/m(6)A/PFKP/LDHB axis. Mol Cell 81, 922–939 e929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schieber M, and Chandel NS (2014). ROS function in redox signaling and oxidative stress. Curr Biol 24, R453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seltzer MJ, Bennett BD, Joshi AD, Gao P, Thomas AG, Ferraris DV, Tsukamoto T, Rojas CJ, Slusher BS, Rabinowitz JD, et al. (2010). Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res 70, 8981–8987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AH, Meydan C, Shank K, Garrett-Bakelman FE, Ward PS, Intlekofer AM, Nazir A, Stein EM, Knapp K, Glass J, et al. (2017). Combination Targeted Therapy to Disrupt Aberrant Oncogenic Signaling and Reverse Epigenetic Dysfunction in IDH2- and TET2-Mutant Acute Myeloid Leukemia. Cancer Discov 7, 494–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein EM (2016). Molecular Pathways: IDH2 Mutations-Co-opting Cellular Metabolism for Malignant Transformation. Clin Cancer Res 22, 16–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, Deng X, Wang Y, Weng X, Hu C, et al. (2018). R-2HG Exhibits Anti-tumor Activity by Targeting FTO/m(6)A/MYC/CEBPA Signaling. Cell 172, 90–105 e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, Straley K, Kernytsky A, Liu W, Gliser C, et al. (2013). Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 340, 622–626. [DOI] [PubMed] [Google Scholar]
- Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, et al. (2010). The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2hydroxyglutarate. Cancer Cell 17, 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, et al. (2009). IDH1 and IDH2 mutations in gliomas. The New England journal of medicine 360, 765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Ye D, Guan KL, and Xiong Y (2012). IDH1 and IDH2 mutations in tumorigenesis: mechanistic insights and clinical perspectives. Clin Cancer Res 18, 5562–5571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen K, Travins J, Wang F, David MD, Artin E, Straley K, Padyana A, Gross S, DeLaBarre B, Tobin E, et al. (2017). AG-221, a First-in-Class Therapy Targeting Acute Myeloid Leukemia Harboring Oncogenic IDH2 Mutations. Cancer Discov 7, 478–493. [DOI] [PubMed] [Google Scholar]
- Yu W, Dittenhafer-Reed KE, and Denu JM (2012). SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J Biol Chem 287, 14078–14086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou X, Zhu Y, Park SH, Liu G, O’Brien J, Jiang H, and Gius D (2017). SIRT3-Mediated Dimerization of IDH2 Directs Cancer Cell Metabolism and Tumor Growth. Cancer Res 77, 3990–3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Original western blot images have been deposited at Mendeley and are publicly available as of the date of publication: https://data.mendeley.com/datasets/wrt34nw5z9/draft?a=0c0cc371-dfd3–441d-a8f8-c6577c8e4caf. The DOI is listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.