

Abstract
Cytochrome P450 2D6 (CYP2D6) is primarily expressed in the liver and in the central nervous system. It is known to be highly polymorphic in nature. It metabolizes several endogenous substrates such as anandamide (AEA). Concomitantly, it is involved in phase 1 metabolism of several antidepressants, antipsychotics, and other drugs. Research in the field of phytocannabinoids (pCBs) has recently accelerated owing to its legalization and increasing medicinal use for pain and inflammation. The primary component of cannabis is THC, which is well known for its psychotropic effects. Since CYP2D6 is an important brain and liver P450 and is known to be inhibited by CBD, we investigated the interactions of four important highly prevalent CYP2D6 polymorphisms with selected phytocannabinoids (CBD, THC, CBDV.THCV, CBN, CBG, CBC, β-carophyllene) that are rapidly gaining popularity. We show that there is differential binding of CYP2D6*17 to pCBs as compared to WT CYP2D6. We also perform a more detailed comparison of WT and *17 CYP2D6 which reveals the possible regulation of AEA metabolism by CBD. Furthermore, we use molecular dynamics to delineate the mechanism of this binding, inhibition and regulation. Taken together, we have found that the interactions of CYP2D6 with pCBs vary by polymorphism and by specific pCB class.
Keywords: Cytochrome P450, CYP2D6, cannabinoids, molecular dynamics, anandamide
INTRODUCTION
Research in the field of phytocannabinoids (pCBs) has recently accelerated owing to its legalization in several states and their potential medicinal uses. 1 The use of pCBs has been shown to alleviate pain occurring from a variety of diseases including arthritis, peripheral neuropathy, and multiple sclerosis.2, 3 In humans, pCBs have been used as anti-epileptic and anti-nausea treatments.4–9 CBD is reported to have several beneficial effects including analgesic, anti-emetic, anti-epileptic, and anxiolytic effect.10, 11 There are also numerous minor pCBs that may prove to be clinically relevant and are rapidly gaining popularity for therapeutic use including cannabigerol (CBG), cannabidiol (CBD), cannabichromene (CBC), cannabidivarin (CBDV), cannabinol (CBN), Tetrahydrocannabinol (Δ9-THC) and Δ9-Tetrahydrocannabivarin (THCV) (Figure 1A). For instance, CBDV and Δ9-THC have been shown to decrease seizures in rodents through mechanisms involving TRPV1 and CB1, respectively.12–15
Figure 1:
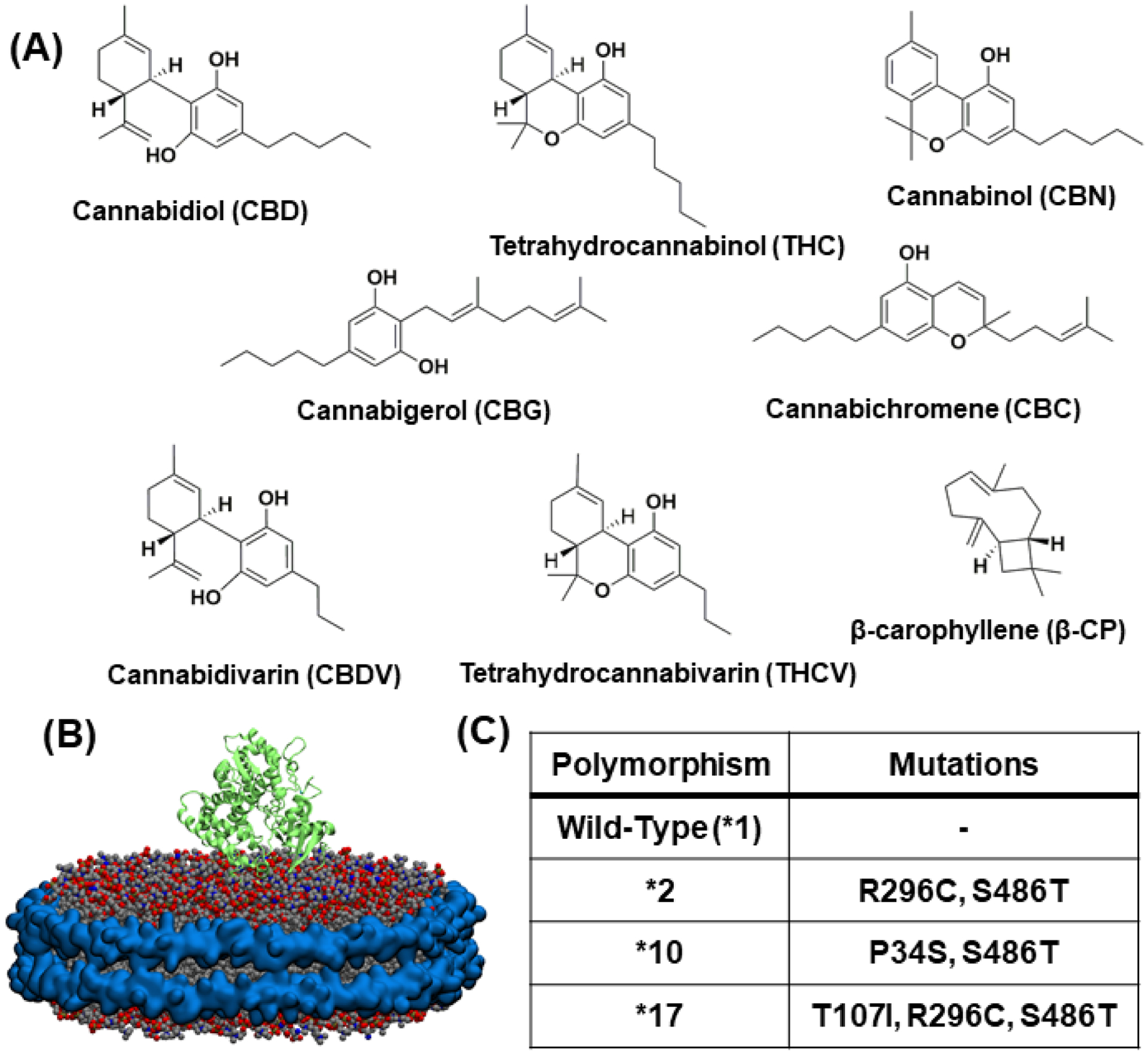
(A) Structures of the eight phytocannabinoids chosen for investigation (B) 2D6 protein (shown in green ribbon) embedded in nanodisc having 80% POPC (grey), 20% POPS (navy blue) and membrane scaffold protein around the periphery (cyan) (C) A table listing the four CYP2D6 polymorphisms and their specific mutations
Despite the emerging beneficial medical uses of the pCBs, the possibility of drug-drug interactions is a rising concern in patients with health conditions that require them to take cannabis along with other drugs. With the advent of personalized medicine, it is important to notice that the pCBs can undergo differential metabolism by different cytochrome P450s (CYP) and by different polymorphisms of the same CYP. Additionally, inhibition or activation of enzymes involved in drug metabolism by pCBs will in turn influence the metabolism of other drugs.
CYP2D6 is one of the CYPs highly expressed in the liver and brain16. It is responsible for approximately 25–30% of human drug oxidation—second only to CYP3A4.17 It is a highly polymorphic CYP and is known to have more than ~105 unique allelic polymorphisms, many of which are associated with certain ethnic populations and contribute to different metabolic profiles.17–19 For example, CYP2D6*2 is a frequent polymorphism in Caucasians (~10%), CYP2D6*10 in Asian populations (~50%) and CYP2D6*17 is most frequent in African populations (~35%) 20. These polymorphisms directly influence drug metabolism by altering the effect on drug efficacy21. Based on the different CYP2D6 functional polymorphisms, they are classified into four categories: ultra-rapid, extensive, intermediate, and poor (UM, EM, IM, and PM, respectively). Herein, we specifically focus on the following CYP2D6 polymorphisms (Figure 1C) —*1 (wild type), *2, *10, and *17—which include both extensive and intermediate metabolizers commonly occurring in a variety of ethnic groups.22 Wild-type CYP2D6 is the most common form of the gene and is an EM. CYP2D6*2 has two mutations, R296C and S486T. It is one of the more commonly multi-duplicated alleles, leading to a UM phenotype, but unduplicated is considered to be an EM.17, 23 CYP2D6*10 is an intermediate metabolizer.17 It has a P34S substitution in the N-terminus as well as an S486T substitution that ultimately result in misfolding and impaired membrane anchoring with reduced demethylation and hydroxylation activity.24 CYP2D6*17 has three substitutions, T107I, R296C, and S486T, the first of which is thought to take part in substrate recognition.17 It is also an IM, but evidence points to this being substrate dependent.17, 23, 25,26–28 Most PM phenotype polymorphisms are nonfunctional CYP2D6 variants.
CYP2D6 is abundant in brain and is involved in the metabolism of psychotropic drugs such as antidepressants, anticonvulsants, and antipsychotics. There are very few studies that have been reported on the interactions of CYP2D6 with pCBs. Hence, there is a critical need for studies that focus on the interactions of pCB with different CYP2D6 polymorphisms as these are known to affect the rate of drug metabolism and thus drug clearance.29–31 There are several studies on the metabolism of pCBs by CYPs32–34 although more mechanistic research is required to understand the involvement of pCBs in potential drug-drug interactions. Previously, it has been shown that human hepatic microsomes convert Δ9-THC, Δ8-THC, and CBN into 11-hydroxy, 8-hydroxy and 7-hydroxy products.34 The same pCBs were also supplied to microsomes from human B lymphoblastoid cells expressing specific CYPs. CYP2C9 had the highest 11-hydroxylation activity of all the CYPs tested and CYP2C19 had a low amount of activity.35 CYP3A4 catalyzed metabolism leads to several minor hydroxylation as well as epoxidation, but no other expressed CYP was an efficient pCB metabolizer.34 In a separate study CYP2J2 was tested against the pCBs CBD, Δ9-THC, Δ8-THC, CBN, CBG, and CBD, all of which were found to be substrates.32 The rate of metabolism were slower than that of pCB metabolism by CYP2C9, but CYP2J2 metabolism of CBN, CBD, and CBC was faster than CYP2C19.32, 34 Other CYPs that are implicated in pCB metabolism are CYP1A1, 1A2, 2D6, 3A5, and 3A7.36–38
For this study, we selected pCBs which would provide the representative example of CYP2D6-pCB interactions. CBD and THC were selected since they are the two major cannabinoid components in cannabis and also because both have been implicated as P450 inhibitors.39–42 CBDV, THCV, and CBN were selected to check structural relevance of the side chain length. CBG and CBC were selected for their “lipid-like” structure. β-carophyllene, an important constituent of cannabis essential oil was selected as it has been shown to activate the cannabinoid receptor 2 receptor.43, 44
The differential metabolism of pCBs by CYP is attributed to the conformational changes in the enzyme active site which affect substrate binding and the relative orientation of the substrate to the heme.28, 45 In a previous investigation, molecular docking investigations on the plasticity of CYP2D6 revealed that Phe483 is a key residue in stabilizing the binding of 7-methoxy-4-(aminomethyl)coumarin (MAMC) and the mutation of this residue to an alanine nullifies metabolism of MAMC.46, 47. Therefore, it is important to investigate the interactions of pCBs with a set of CYP2D6 polymorphisms that have mutations occurring throughout the protein.
Previously, we and others have shown that pCBs are metabolized by various human CYPs to form novel oxidized products.23, 48–50 Additionally, many of the pCBs tested, including CBD have been shown to inhibit CYPs.41 Herein, we explore the differences in pCB binding, metabolism, and inhibition of CYP2D6 and its mutants. Using four relevant polymorphisms of CYP2D6 we show that the spectral binding shift of CYP2D6 is dependent on the polymorphism and the pCB substrate chosen, indicating that the mutations in CYP2D6 sufficiently alter the binding pocket and that some pCBs possess structural elements essential for efficient binding. We also demonstrate that there is differential metabolism of endogenous and exogenous substrates in the presence of selected pCBs (Figure 1A). Finally, we use molecular dynamics (MD) to elucidate the molecular underpinnings of pCB interactions with CYP2D6 polymorphisms.
MATERIALS AND METHODS
Reagents
Thioridazine (14400) and all phytocannabinoids (THC - 1972-08-3, CBDV – 9001574, THCV – 18091, CBG – 15293, CBN - 521-35-7, CBC - 20675-51-8, B-CP - 21572) were purchased from Cayman Chemicals. L-histidine (H8125-25G), dextromethorphan (PHR1018-500MG), dextrorphan (PHR1974-30MG) were purchased from Millipore-Sigma. Ni-NTA resin (H-350-25), arabinose (A-300-1), and IPTG (I2481C25) were purchased from Gold Biotechnology. δ-ALA (A167) was purchased from Frontier Scientific and human liver microsome (H2D6.HA Lot No. 1710125) was purchased from Sekisui XenoTech.
CYP2D6 Growth and Purification
CYP2D6 and its mutants were grown as explained previously51 with some minor modifications. When the 500 ml cultures reached an OD600 of ~0.5, the temperature was lowered to 30°C and shaker to 190 rpm. At an OD600 of 0.7–0.8, the cultures were induced with δ-ALA, IPTG, and arabinose all at once. From this point, cultures were grown 48 hours at 30°C and 190 rpm before being spun down as done previously.
Cell pellets were resuspended at 4°C in lysozyme buffer (75 mM Tris pH 8.0, 0.25 M sucrose, 0.25 mM EDTA, 0.2 mg/ml lysozyme) at a volume of 200 ml/Lculture for 30 minutes. Spheroplasts were pelleted via centrifugation for 30 minutes at 4000 rpm and 4°C. The spheroplast pellet was resuspended in buffer 1 (0.5 M KPi, 20% glycerol, 6 mM MgCl2, 5 mM β-mercaptoethanol, 0.2 mM PMSF, 1% w/v cholate, 0.1 mM thioridazine) at a volume of 300 ml/Lculture and sonicated 6x at 40 second on/off intervals on ice. This mixture was then centrifuged for 45 minutes at 35K rpm, 4°C.
Purification proceeded as before51 with the addition of 0.05 mM thioridazine to the base column buffer. L-histidine was also added to the final wash buffer and the elution buffer at concentrations of 40 mM and 0.1 M, respectively. After elution, the protein was concentrated via Amicon spin filters and quantified on a UV-vis spectrophotometer using molar extinction coefficient value at 110 mM−1 cm−1 at 417 nm52. It was then either aliquoted and flash-frozen in liquid nitrogen or used immediately for nanodisc construction.
CYP2D6 Mutation Construction
The modified human CYP2D6*1 construct in the pCWori vector was a gift from Eric F. Johnson (The Scripps Research Institute, La Jolla, CA)53. As previously published, this construct contains an N-terminal truncation of the first 33 amino acids and was replaced with a shorter amino acid sequence, MAKKTSSKGKL, to increase solubility and reduce aggregation. To facilitate purification, a 4-histidine tag was added to the C-terminus for Ni-NTA affinity chromatography. Mutagenesis for allelic variants, CYP2D6*2, CYP2D6*10 and CYP2D6*17 was carried out using site-directed mutagenesis of the modified CYP2D6*1 plasmid, with the primers are described in Supplementary Table 121. The PCR conditions were 94°C for 2 min (1 cycle), 94°C for 30 sec, 60°C for 30 sec, 68°C for 8 min (25 cycles), 72°C for 3 min (1 cycle) and held at 4°C using high fidelity Phusion. Next, the PCR reaction mix was purified using a Qiagen PCR purification kit and subject to DpnI digest before transformation into DH5α cells. To confirm construct mutagenesis, enzyme restriction digest was performed (Nsil and HindIII) and were verified by sequencing. Specific mutations are detailed in Figure 1C.
CYP2D6 Nanodiscs
CYP2D6 nanodiscs were constructed immediately after purification for stabilization purposes. Once eluted from the Ni-NTA column, CYP2D6 was quantified and, if necessary, concentrated to a reasonable value for nanodisc construction. Lipids and MSP for nanodiscs were prepared as before. After solubilizing the lipids and incubating with MSP as previously published, CYP2D6 was added to the mixture and incubated with gentle rocking for at least 45 minutes at 4°C. BioBeads were added to the mixture and incubated for approximately 8 hours before being removed by spin filtration at 3000 rpm and 4°C for 5 minutes. The nanodiscs were left to incubate overnight with gentle rocking at 4°C before being concentrated with an Amicon concentrator and quantified via UV-vis. Glycerol was added to final concentration of 20% v/v and nanodiscs were flash frozen in small aliquots and stored at −80°C.
Soret Titration
Soret titrations were performed similar to a previous description with some modifications.32, 54 Substrates were dried under a stream of N2 gas and dissolved in DMSO as 1mg/ml stocks. The total titrated volume was kept below 2.5% of the final volume. 1 μM CYP2D6 was incubated at room temperature during the course of the experiment. Data points were taken at set concentrations of each pCB from 1–75 μM. The data was processed in OriginPro 2019 by fitting to the Michaelis-Menten or tight binding equation.
Direct Metabolism of Phytocannabinoids
Direct metabolism assays were set up in 1 ml reactions containing 0.1 M KPi, 0.2 μM 2D6 nanodiscs, 0.6 μM CPR, 40 μM pCB, and either 40 μM DXM or 40 μM AEA. Reactions were incubated for 5 minutes at room temperature before being initiated with 100 μl 10 mM NADPH (1 mM final concentration). Reactions were incubated 30 minutes at 37°C and were then quenched with an equal volume of ethyl acetate. For metabolism study using human liver microsome, 2D6 microsome (containing 0.210 nmol/mg CYP P450 protein and 143 ±6 nmol/mg protein/min NADPH-cytochrome c reductase) was incubated with THC and CBD (final concentration for both pCB were 40 μM) separately for 30 minutes at 37°C in 0.1 M KPi. The reactions were quenched and extracted using ethyl acetate.
Metabolism Assays
Dextromethorphan metabolism studies were carried out in 0.1 M KPi, pH 7.4, containing 0.2 μM CYP2D6 nanodiscs, 0.6 μM CPR, 1 mM NADPH, and substrate in 250 μl total volume. All components except NADPH were added together and incubated for 5 minutes at room temperature. Reactions were initiated with NADPH and terminated after 2 minutes by the addition of an equal volume of ACN. Phytocannabinoid metabolism was carried out in the same manner with the exceptions of the reactions being scaled up to 1 ml. Ethyl acetate was used to quench pCB metabolisms to facilitate subsequent extraction for analysis.
Inhibition of CYP2D6 Assays
For preliminary inhibition assays, 250 μl reactions were set up containing 0.1 M KPi, 0.2 μM 2D6 nanodiscs, 0.6 μM CPR, 40 μM pCB, and either 40 μM DXM or 40 μM AEA. Reactions were incubated for 5 minutes at room temperature before being initiated with 100 ul 10 mM NADPH (1 mM final concentration). Reactions were allowed to proceed for 2 minutes for DXM and 10 minutes for AEA after which they were quenched with an equal volume of ACN (DXM) or ethyl acetate (AEA). AEA samples were extracted as detailed below. DXM samples quenched in ACN were spun down for 5 minutes at 3000 rpm, 4°C and directly injected on the HPLC after filtration.
Extractions of Metabolites
Extractions were carried out as before.55 After reaction quenching, tubes were vortexed to emulsify the contents, and centrifuged for 5 minutes at 3000 rpm, 4°C. The organic layer was removed to a clean tube and the process repeated two more times from the addition of ethyl acetate, for a total of 3 extractions. The combined organic layers were then dried on a rotary evaporator and resolubilized appropriately for subsequent applications.
HPLC Analysis
Analysis of DXM metabolism was carried out via an Agilent series 1100 HPLC. A Luna 5 μm C18 column was used with an isocratic mobile phase consisting of 50% 0.01 M KPi, 50% 50:50 MeOH:ACN (pH 3.4), which ran for 30 minutes. Curve integrations were analyzed using OpenLab software and further data analysis was performed in OriginPro 2019.
LC-MS/MS Analysis
Direct metabolism reactions with pCBs qualitatively analyzed using the same method as previous and a 250×4.6 mm Luna 5 μm C18(2) 100 Å column.32 All reactions with AEA were quantified using LC-MS/MS as described previously.56
Molecular Dynamics Simulations
The crystal structure of the globular domain of CYP2D6 (PDB ID: 3TDA)57was embedded in a membrane patch consisting of 100 palmitoyloleoylphosphatidylcholine (POPC) molecules in each leaflet. The membrane patch was built using CHARMM-GUI Membrane Builder58 and the initial orientation of CYP2D6 in the membrane was modeled according to the results shown by Fischer, et. al.59 This structure was then solvated with TIP3P water60 and neutralized. The simulation box created was 100 × 100 × 140 Å3. The MUT2 (R296C/S486T), MUT10 (P34S/S486T), and MUT17 (T107I/R296C/S486T) variants were generated using the Mutator plugin of VMD61 and each initial membrane bound system was prepared using the protocol described above. Each system was first minimized for 10,000 steps and then equilibrated for 10 ns with the heavy atoms of the protein restrained (1 kcal•mol-1Å-2). Each system was then simulated for 100 ns without restraints.
The last 50 ns of the production run of each system was used for further investigation of binding poses of the phytocannabinoids. The structure of the protein from each system was saved every 100 ps to generate 500 protein structures to dock to for each system. Then, AutoDock Vina62 was used to perform ensemble docking of CBD CBDV, CBC, BCP, CBG and CBN to these protein structures. Drugs were docked to a grid box of 25 × 25 × 25 Å3 centered at the active site of each protein structure. The 10 most favorable binding poses were then stored for each protein structure, generating 5000 poses for each system.
The poses of CBD and THC docked to WT and *17 were then clustered using the root mean square deviation (RMSD) of the heavy atoms, which resulted in five clusters. The pose within each cluster with the lowest binding affinity was then chosen for further simulations. Each of these protein-pCB poses was embedded back into the POPC bilayer system (as used for apo simulation), minimized for 10,000 steps and equilibrated for 10 ns with heavy atoms of the protein and drug restrained (force constant of 1 kcal•mol-1Å-2). Then restraints were removed from each system and each system was equilibrated for an additional 50 ns.
All simulations were carried out with a 2 fs timestep using NAMD 2.1363 with the CHARMM36m64 and CHARMM3665 force field for proteins and lipids respectively with an NPT ensemble. Parameters for pCB molecules are obtained from CHARMM general force field.66 The temperature was maintained at 310 K using Langevin dynamics and pressure was regulated at 1.0 atm using Nosé-Hoover Langevin piston.67 The cutoff for calculating non-bonded interactions was 12 Å and a switch function was applied at 10 Å; long range electrostatics were incorporated using Particle Mesh Ewald (PME).68
RESULTS
Spectral Binding Studies of CYP2D6 Polymorphisms with Phytocannabinoids
We performed studies of pCB binding to CYP2D6 and its polymorphisms using UV–vis spectral titrations. For all these studies, CYP2D6 was incorporated into nanodiscs as it is unstable outside the membrane environment (Figure 1B).69 In order to study the perturbation of the thiol bound heme group in all the four constructs of CYP2D6, carbon monoxide (CO) binding was carried out. For this analysis, CO was added to the reduced protein (Fe II) for all the four constructs. Absorbance spectra around 450 nm suggests the thiolate group axial to the heme is retained and the P450 fold is maintained (Supplementary Figures S20). However, presence of an additional 420 nm peak for *17 might be due to the slight structural change in protein upon mutation, but prominent 450 nm signifies overall folded structure is preserved. Previous reports have indicated that change in residues in the F-G loop of CYP leads to the partial appearance of the 420 nm peak which affecting the protein structure around heme moiety.70
Increasing concentrations of pCB were titrated into CYP2D6-NDs to examine the shift in the Soret band at 417 nm and determine the binding parameters. A shift in the lower wavelength was observed upon addition of pCB in a concentration dependent manner suggesting Type I shift. The spin-state changes were substantial to see the differential binding of the pCBs to the different CYP2D6 polymorphisms. All the polymorphism-pCB combinations were fitted to either a standard Michaelis-Menten or tight-binding equation to determine their Ks and ΔAmax. Data is shown in Table 1 and described below.
Table 1.
Calculated values of Ks (in μM) and ΔA values from Soret titrations. Each of the pCBs (THC, CBD, CBG, CBC, CBDV, THCV and β-CP) were titrated against the four constraints of CYP2D6 (WT, *2, *10 and *17) The values are calculated from the means of three repeats (N=3). The error represents the standard error of the mean of three experiments.
| Cannabinoids | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| THC | CBD | CBG | CBN | CBC | CBDV | THCV | β-CP | ||
| WT | Ks (μM) | 3.41 ± 1.08 | 7.03 ± 2.24 | 13.42 ± 3.25 | 7.90 ± 2.89 | 6.09 ± 2.51 | 7.75 ± 2.76 | 3.24 ± 1.12 | 4.27 ± 1.30 |
| ΔA | 0.0486 ±.0032 | 0.0711 ±0.0060 | 0.0640 ±0.0054 | 0.0362 ±0.0038 | 0.0246 ±.0026 | 0.0217 ±0.0022 | 0.0252 ±0.0018 | 0.0281 ±0.0020 | |
| *2 | Ks (μM) | 3.46 ± 1.03 | 10.51 ± 3.67 | 10.36 ± 3.48 | 5.13 ± 0.94 | 7.94 ± 2.24 | 11.56 ± 4.36 | 11.52 ± 3.92 | 10.31 ± 4.71 |
| ΔA | 0.0142 ±0.0009 | 0.0299 ±0.0033 | 0.0377 ±0.0040 | 0.0329 ±0.0015 | 0.0377 ±0.0031 | 0.0387 ±0.0048 | 0.0392 ±0.0044 | 0.0381 ±0.0055 | |
| *10 | Ks (μM) | 8.51 ± 2.99 | 5.23 ± 2.34 | 7.28 ± 3.14 | 3.87 ± 1.26 | 9.16 ± 2.28 | 7.19 ± 2.55 | 11.50 ± 2.87 | 13.95 ± 5.27 |
| ΔA | 0.0332 ±0.0034 | 0.0320 ±0.0036 | 0.0596 ±0.0072 | 0.0203 ±0.0014 | 0.0299 ±0.0022 | 0.0385 ±0.0038 | 0.0346 ±0.0028 | 0.0297 ±0.0040 | |
| *17 | Ks (μM) | 20.10 ± 8.24 | 5.57 ± 1.22 | 11.77 ± 3.18 | 9.00 ± 2.13 | 11.64 ± 3.20 | 8.60 ± 1.86 | 7.12 ± 1.59 | 10.27 ± 3.82 |
| ΔA | 0.0737 ±0.0125 | 0.0247 ±0.0014 | 0.0745 ±0.0067 | 0.1387 ±0.0098 | 0.112 ±0.0101 | 0.162 ±0.00873 | 0.1516 ±0.0093 | 0.0809 ±0.0095 | |
Cannabidiol –
CBD was only weakly bound to WT CYP2D6, producing a Ks of 7.03 ± 2.24 μM and none of the other polymorphisms produced a substantial spin-state change. WT CYP2D6 had the greatest ΔAmax at 0.0711 ± 0.0060 while CYP2D6*17 produced the least spin-state change with a ΔAmax of 0.0247 ± 0.0014. CBD bound weakly to CYP2D6*2 with a Ks of 10.51 ± 3.67 μM (Figure 2A).
Figure 2.
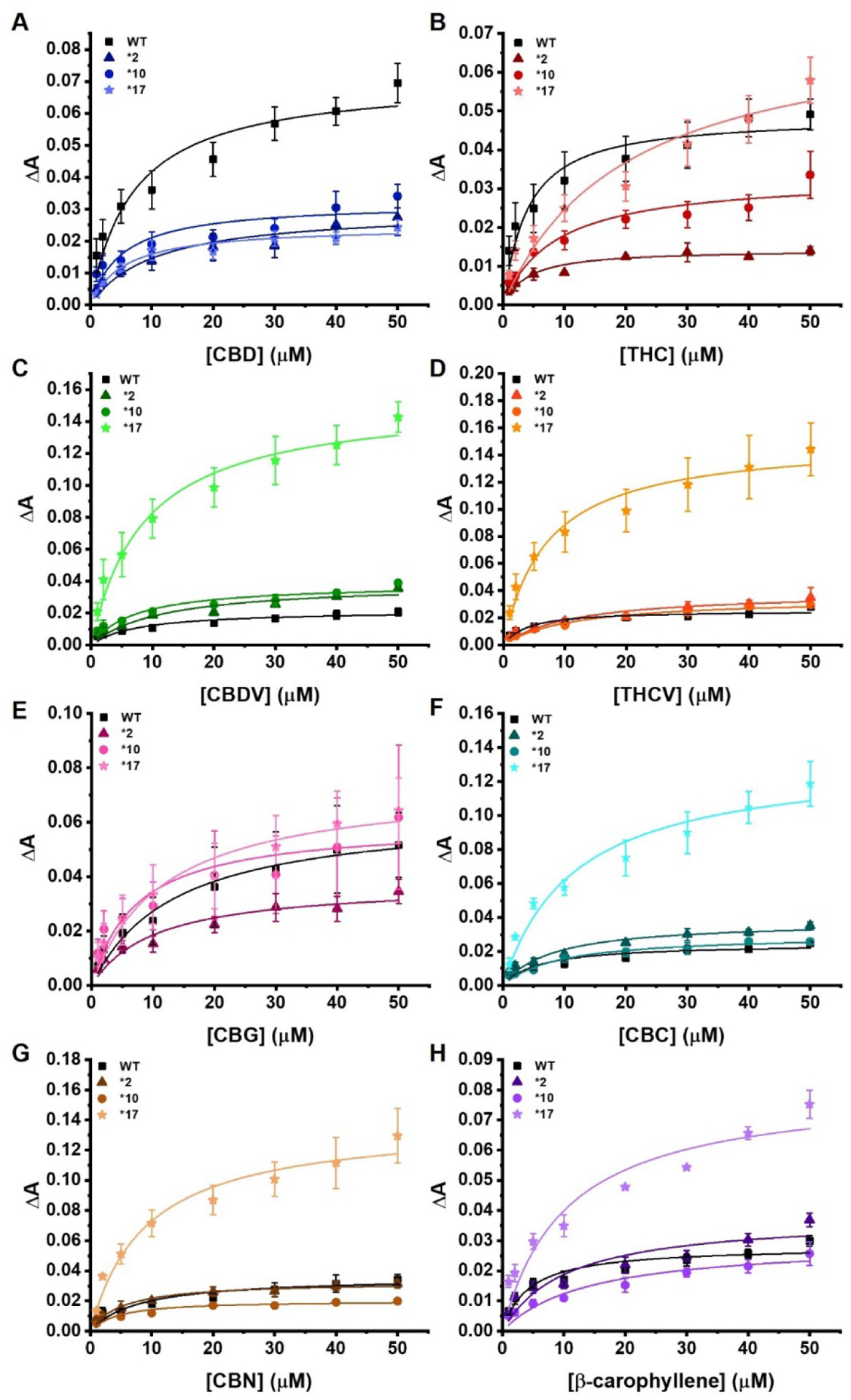
Soret titration binding curves for each of the pCBs with all four CYP2D6 polymorphisms. (A) CBD, (B) THC, (C) CBDV, (D) THCV, (E) CBG, (F) CBC, (G) CBN, and (H) β-CP with all four CYP2D6 polymorphisms. WT is shown with squares, *2 with triangles, *10 with circles, and *17 with stars. All the binding spectra were fitted to a single binding isotherm. The phytocannabinoid was added incrementally from 0 to 50 μM. The change of absorbance (ΔA) from 393 to 417 nm was calculated for each titration and plotted against the corresponding phytocannabinoids concentration (μM). Data were fitted with Origin Lab to the single binding isotherm to determine the Ks. ΔA indicates the difference in absorbance between 393 nm and 417 nm. All experiments were done in triplicates data were produced from the means of three repeats (N=3). The error represents the standard error of the mean of three experiments.
Δ9-Tetrahydrocannabinol –
With THC, the *17 mutant produced the highest spin-state change with a ΔAmax value of 0.0737 ± 0.0125. The WT and *10 exhibited slightly reduced ΔAmax values, while *2 was the lowest at 0.0142 ± 0.0009. CYP2D6*17 also has the weakest Ks value at 20.10 μM while WT CYP2D6 is the strongest at 3.41 μM (Figure 2B).
Cannabidivarin –
In the case of CBDV, WT CYP2D6 and the *10 and *17 mutants were very similar in regards to binding constants while WT CYP2D6, *2, and *10 had similar spin-state changes. CYP2D6*2 had the largest Ks of 11.56 μM. CYP2D6*17 produced a very large spin-state change approximately 6-fold higher than all other mutants. The Ks was 8.60 μM and the ΔAmax was 0.1620. The strongest binding mutant was CYP2D6*10 with a Ks of 7.19 μM (Figure 2C).
Tetrahydrocannabivarin –
CYP2D6*2 has a high Ks value of 11.52 μM, indicating weaker substrate binding. Contrary to this is CYP2D6*17, which binds THCV with a Ks of 5.88 μM. CYP2D6*17 also has the highest spin-state change with a ΔAmax of 0.147. Meanwhile, no other polymorphism has a higher ΔAmax value than 0.0367 with THCV (Figure 2D).
Cannabigerol –
CBG does not have a large difference in ΔAmax for any of the four CYP2D6 polymorphisms. CYP2D6*10 has the tightest Ks at 7.28 μM, while WT CYP2D6 is the weakest at 13.42 μM. The largest spin-shift is seen in CYP2D6*17 with an ΔAmax value of 0.0745 ± 0.0067 (Figure 2E).
Cannabichromene –
All polymorphisms except CYP2D6*17 have minimal spin-shifts with the ΔAmax values ranging from 0.0246–0.0377. WT CYP2D6 has the lowest Ks at 6.09 μM while CYP2D6*10 and *17 are the highest at 9.16 and 11.64 μM, respectively (Figure 2F).
Cannabinol –
CBN binds the most strongly to CYP2D6*10, with a Ks of 3.87 μM followed by CYP2D6*2 at 5.13 μM. In contrast, CYP2D6*17 produces the highest spin-state change with an ΔAmax of 0.1387 ± 0.0098. All three remaining mutants have similar ΔAmax values, none of them exceeding 0.04 (Figure 2G).
β-Carophyllene–
Much like several of the pCBs, β-carophyllene produces a much larger spin-state change when bound to CYP2D6 *17. Likewise, the other three polymorphisms had similar ΔAmax values, all lower than that of *17, though none of the Ks values were drastically different. Notably, the structure of β-carophyllene is vastly different from that of phytocannabinoids, as it is a sesquiterpene with no tail to mimic endogenous fatty acid substrates (Figure 2H).
β-CP bound *1 preferentially with a Ks value of 4.27 μM, while binding all other polymorphisms with a Ks higher than 10 μM. THCV followed a similar trend. CBN favorably bound *10 with a Ks of 3.87 μM, followed by *2 at 5.13 μM. THC bound *1 and *2 almost equally with Ks values of 3.41 and 3.46 μM, respectively, while binding *17 at 20.10 μM. These four pCBs also exhibit the lowest Ks values of all the pCBs tested, and all have structural similarities. This phenomenon could be linked to a combination of favorable structural interactions with the conformations of certain polymorphisms, which could shift with pCB structural changes (e.g – CBN binds *10 best rather than *1).
Molecular Modeling/Molecular Dynamics Simulations
MD simulations reveal that WT CYP2D6 and CYP2D6*10 have the strongest binding for each pCB tested (CBG, CBDV, CBC, CBN, and β-CP), which was determined by a combination of binding affinity in kcal/mol and heme distance. Examples may be seen in Supplementary Figures S1–8. CYP2D6*2 shows weaker binding (less negative binding affinity) but does possess several poses where the drug is close to the heme. Residues commonly in contact with the pCBs tested include Cys443 for *1, Lys214 for *2, Phe483 for *10, and Val308 for *17. Full graphs of the 10 most contacted residues for each mutant with each pCB can be seen in Supplementary Figures S9–S16. The only mutant whose polymorphisms came close to the most commonly contacted residue was CYP2D6 *10. Caver analysis conducted on the *17 and WT variants taken from the initial equilibrium simulation revealed a major access channel in *17 and WT proteins leading to the heme (Figure 3). The bottleneck radius was consistently smaller for the tunnel in *17 than in WT, indicating that conformational changes to *17 result in tighter access to the heme, which may explain why pCB molecules bind further from the heme in this variant. In addition, the experimentally obtained Ks values for *17 are higher compared to WT. This suggest that pCBs cannot bind effectively with the residues near the heme moiety due to smaller access channel, thereby reducing the binding affinity. The RMSD of αB, the location of the T107I mutation was largest for *17, which might indicate that the increased flexibility of this portion of the protein assists in reducing the active site and access channel size (Supplementary Figure 17). The average distance of the heavy atoms of THC and CBD to the heme iron were then computed for each simulation. For three of five clusters of THC bound to WT, the histogram showed a peak at around 5 Å, indicating strong binding of THC within the active site of the protein (Figure 4). However, in the *17 simulations, a peak existed at around 13 Å, indicating binding away from the protein active site. The average distance between the heme iron and individual atoms comprising the tail and the 3 rings of THC (Ring 1, Ring 2, and Ring 3) was also calculated for every 100 ps of each simulation. The atoms of the tail region were shown to be significantly closer to the heme than the three rings in simulations with WT, while Ring 1 and Ring 2 bound further away. This indicates that THC stably binds within the active site of WT CYP2D6 in a conformation where its hydrocarbon chain is facing towards the heme. On the other hand, in *17, while the majority of frames of THC bind far from the heme, in the few frames with close binding (around 8 Å), Ring 3 binds closest to the heme. This ring is opposite to the tail, indicating binding of the pCB opposite to in the WT system. The frames of THC bound to WT bind to the access channel; however, THC does not bind to any access channel in the *17 simulation.
Figure 3:
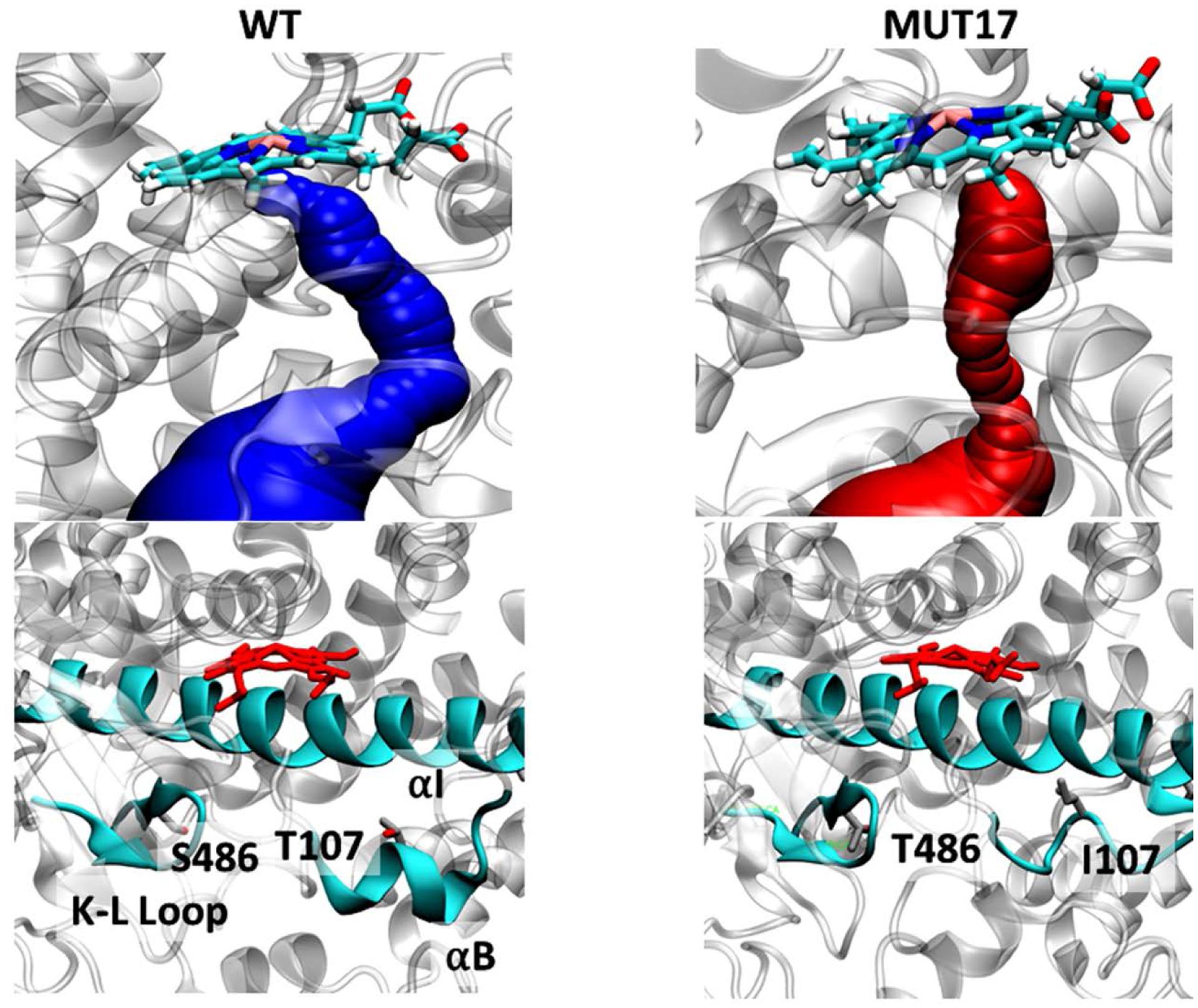
(Top) Access channel identified in WT and *17 variants of Cyp2D6. Shown is a snapshot from molecular dynamics simulations. The bottleneck radius for WT and *17 channels are 1.4 and 0.9, respectively. (Bottom) The location of mutations in *17 variant, highlighting structural changes in *17 compared to WT.
Figure 4:
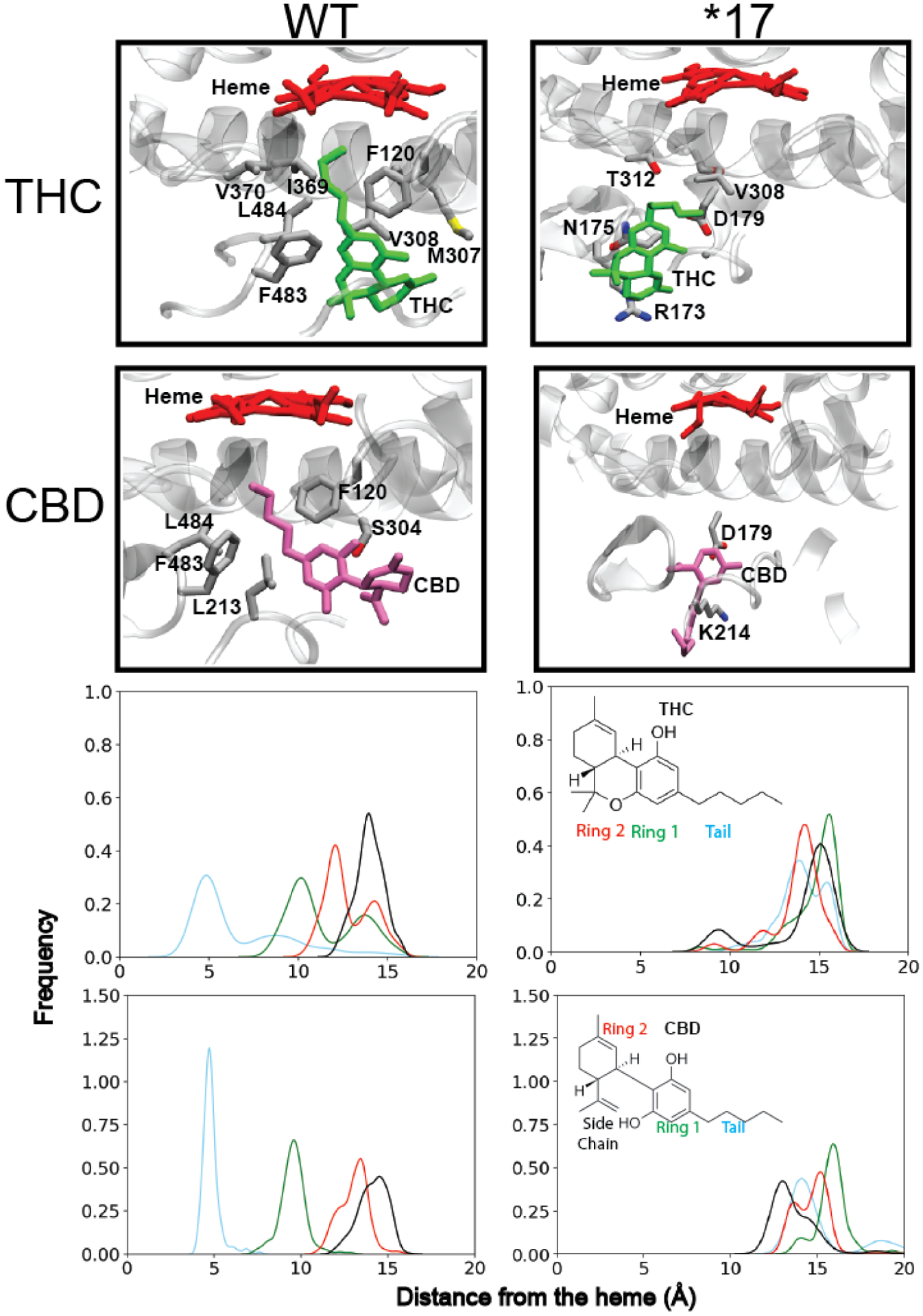
Binding conformation of THC and CBD within WT and *17 CYP2D6. (A and B) Closest binding conformation of THC and CBD to the heme. Residues commonly interacting with the pCB molecules are labeled. (C and D) Distribution of heavy atom–heme distances obtained from 50 ns MD simulations for different regions of each pCB for WT and *17 CYP2D6.
Direct metabolism of pCBs by wild-type CYP2D6-ND
Next, the direct metabolism of individual pCBs was performed with wild-type CYP2D6 and the primary oxidized metabolites were identified via LC-MS. We observed that all the pCBs were metabolized mainly to monoxygenated products as determined from their masses (Supplementary Figure 18). We further investigated the fragmentation of THC and CBD metabolites in order to understand the region of the molecule undergoing oxidation. MS data suggested that hydroxylation in the alkyl chain may be a probable product for both these pCBs as observed from the fragmentation pattern. (Supplementary Figure 24 and 25). Also, CBG produced far more metabolites in a comparison of relative abundance compared to other substrates which may be due to its lipid-like long chain structure.
The probable products formed from the metabolism of THC, CBD were investigated. Fragmentation pattern from the LC/MS indicates that the alkyl chain is preferentially hydroxylated upon metabolism for the pCBs. The mass spectra and the fragmentation pattern are shown in Supplementary Figure 24 and 25. As seen from the mass fragments for THC hydroxylation in the alkyl chain seems as a probable product (Supplementary Figure 25). In addition, the MD simulation shows that the alkyl chain is oriented towards the heme moiety, which supports the experimental findings. However, for CBD, the fragments correspond to two probable products (both hydroxylation in the alkyl chain).
In order to establish a more biological relevance to the 2D6 metabolism, microsomes were used to carry out the metabolism of CBD and THC. These microsomes are isolated from cells and are rich in CYP P450. Fragmentation pattern (Supplementary Figure 29) indicate that 2D6 microsome gives similar metabolites like CYP2D6 WT protein as observed from the LC/MS data.
Reduction of CYP2D6 mediated metabolism of AEA
While pCBs are metabolized by CYPs, literature has previously shown that CYP2D6 is inhibited by pCBs. Therefore, we sought to examine the inhibition of AEA metabolism by different pCBs. CYP2D6 metabolism of AEA was carried out at constant concentration of pCB(40 μM) and constant concentration of target drug (40 μM of either AEA or DXM) and the inhibition levels compared to samples with no pCBs (Supplementary Figure S19).
AEA metabolism in presence of CBD, THC, and CBDV showed reduction in metabolism by 17.7, 13.5, and 12.3 percent, respectively as compared to WT (Supplementary Figure S19). Three other pCBs—CBC, THCV, and β-CP shows very little reduction in metabolism if at all, and even improved metabolism, though not significantly. DXM metabolism inhibition samples revealed a somewhat different trend. The most inhibitory compounds were CBD, CBC, THCV, and β-CP at 60.7, 61.6, 60.5, and 66.3 percent reduction in metabolism, respectively when compared to the complete metabolism of DXM. However, CBDV and CBG were very poor inhibitors at 13.5 and 28.0 percent.
Full inhibition experiments of AEA metabolism by WT 2D6 and 2D6*17 were performed using CBD and THC as inhibitors. The data were fitted to Hill equation (with n values around 2–3) and plotted as shown in Figure 5 A, B and the corresponding Vmax and Km values were compared (Figure 5 C, D). WT 2D6 had a much greater rate of base substrate metabolism than 2D6*17, with a Vmaxapp of ~387 compared to ~251 pmol/min/nmol (Values shown in Supplementary Information Table S7 and S8). The Kmapp of AEA to WT 2D6 was ~25 μM which is nearly similar to Km in the presence of CBD (~28 μM) or THC (~27 μM). CBD increased the rate of AEA metabolism by WT CYP2D6 to ~530 pmol/min/nmol, while the presence of THC causes small reduction. In CYP2D6*17, only AEA metabolism had the highest Vmaxapp; but CBD and THC produce no drastic rate changes. The presence of THC does reduce the binding constant of AEA from ~36 μM to ~26 μM.
Figure 5:
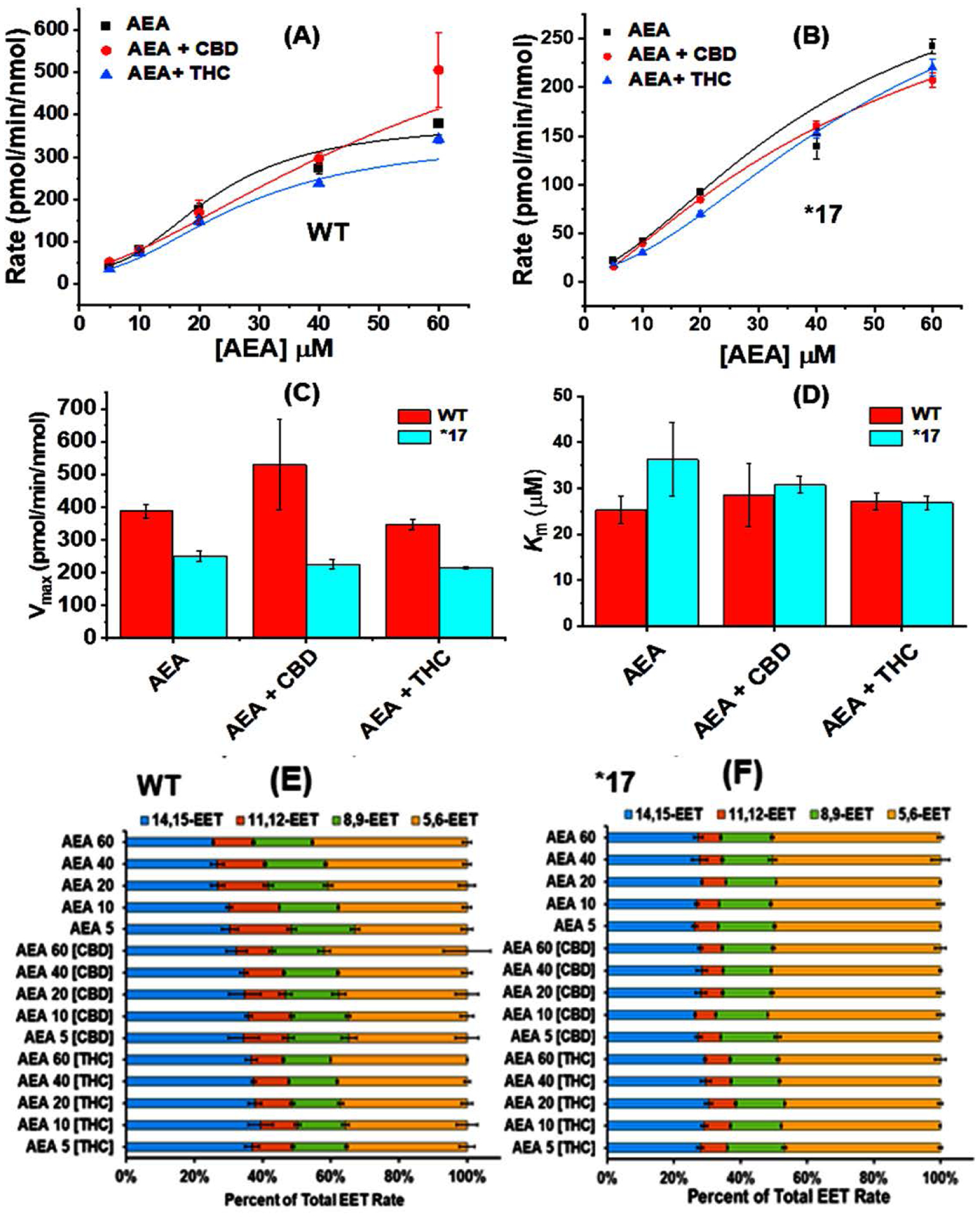
Rates of AEA metabolism in presence of (A) WT, (B) *17. The data were fitted into Hill equation where the Hill coefficient (n) for AEA, AEA +CBD and AEA + THC are 2.41 ± 0.64, 2.24 ± 1.03 and 2.35 ± 0.25 respectively for metabolism by WT and 3.43 ± 1.67, 3.08 ± 0.36 and 2.41 ± 0.31 respectively for metabolism by *17. Comparison of (C) Vmax and (D) Km for AEA metabolism in presence of WT and *17. Ratios of EET-ET production as a result of AEA metabolism by (E) WT, (F) *17. All experiments were done in triplicates data were produced from the means of three repeats (N=3). The error represents the standard error of the mean of three experiments.
We have carried out the titration of 2D6 with AEA in order to find the spin state change. AEA shows Type I shift with WT, *2, *10 and *17 with WT showing the lower affinity towards AEA as compared to the mutants. In terms of spin state change, AEA shows somewhat higher perturbation at the heme site as compared to its mutants (Figure 6 A, B). Our study shows that both AEA and pCB scan bind appreciably with 2D6. In order to understand the lower inhibition of AEA metabolism by WT 2D6 in presence of CBD we ran a molecular docking simulation of CBD-bound WT CYP2D6 and compared to the WT CYP2D6 apo structure (Figure 6 C, D). We found that in the CBD-bound structure, there was still a stable binding pose of AEA in the active site of the protein near the heme group. This indicates that AEA can bind in a region away from the CBD binding site which can facilitate the metabolism of AEA in 2D6. This observation suggest that CBD may not be a significant inhibitor for AEA metabolism by 2D6.
Figure 6:
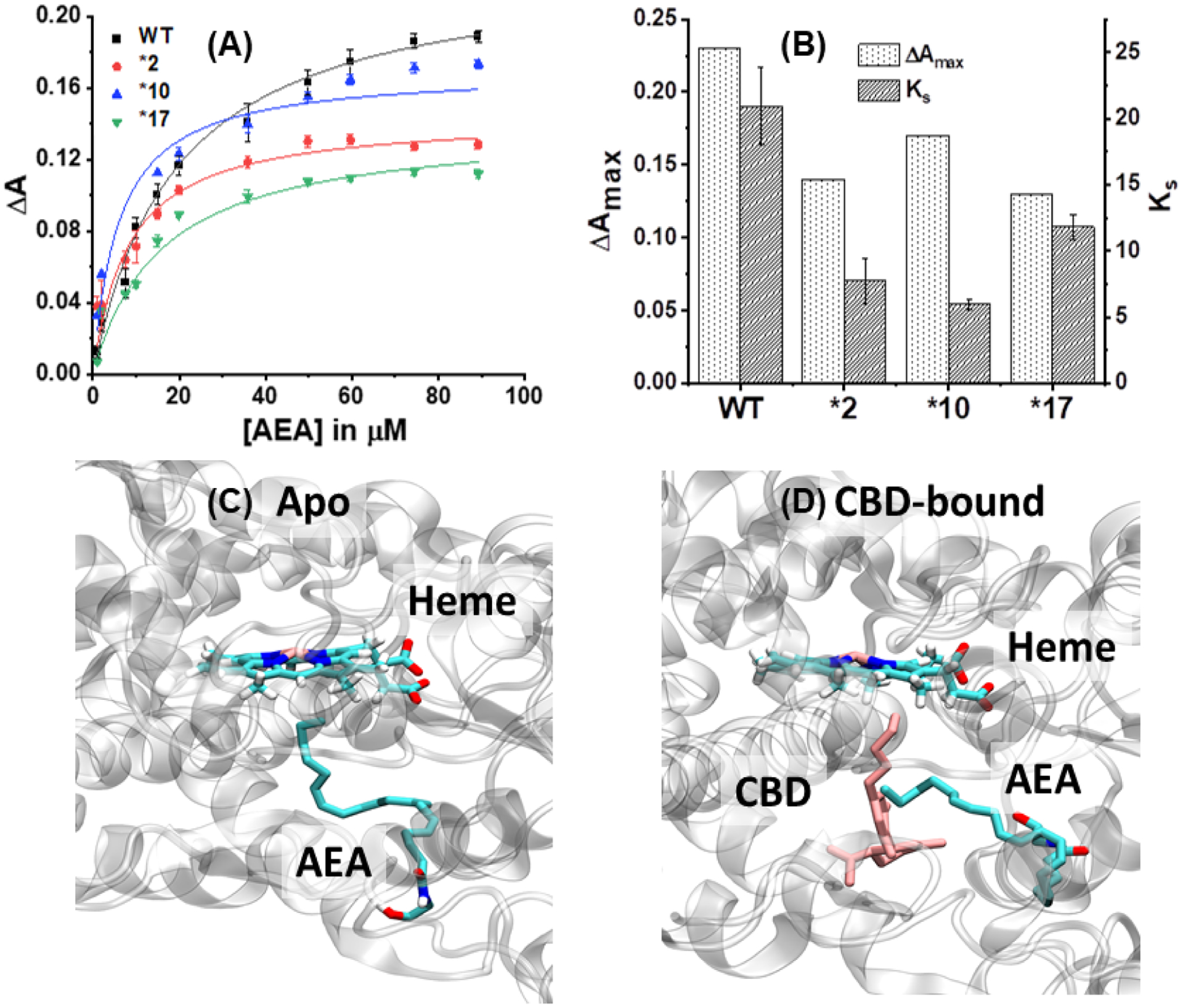
Soret titration binding curves for (A) AEA in presence of WT, *2, *10 and *17. ΔA indicates the difference in absorbance between 393 nm and 417 nm. (B) Comparison of spin state change (ΔAmax) and Ks for AEA in presence of different 2D6 constructs. Docking studies showing the binding site for anandamide (AEA) in (C) Apo: AEA bound to CYP2D6; (D) CBD-bound WT CYP2D6. The heme, AEA and CBD are highlighted.
DISCUSSION
CYP2D6 is a highly polymorphic and highly promiscuous enzyme with respect to substrate selectivity.17, 71 Herein, we chose four representative CYP2D6 polymorphisms and studied their interactions with selected phytocannabinoids in order to understand CYP2D6-pCB interactions.
We first investigated changes in substrate binding, as evidenced by the Soret shift of CYP2D6 (Figure 2). The interactions of various pCBs with CYP2D6 all exhibited a Type I shift72 in which the replacement of the active site water with pCB caused local maxima and minima at ~390 nm and ~417 nm, respectively. While some of the pCBs showed no significant differences in binding between the different polymorphisms, THC, CBN, THCV, and β-CP all demonstrated preferential binding to certain CYP2D6 polymorphisms through decreased Ks values (Table 1). Binding differences also manifested through changes in ΔAmax. Most notable were the effects on CYP2D6*17, which exhibited an increased ΔAmax when bound with CBN, CBC, CBDV, and THCV. In general, an increase in ΔA marks a greater spin-shift, in this case from low-spin to high-spin. Both CBDV and THCV have shortened alkyl chains compared to CBD and THC with no other changes. CBC has a bicyclic center with one alkyl chain on each side while CBN has a similar structure to THC, but trades two hydrogen atoms for a second aromatic ring. As the intensity of the spin-shift is indicative of relative water displacement, it can be surmised that the tightest binding substrates will also produce the greatest spin-shift73. However, in these studies this correlation was not obtained. For instance, CBD was the most tightly bound substrate to *17, but did not produce substantial spin-shift while CBDV elicited a spectral shift of 0.162—the highest of all the pCBs—indicating that it is not solely due to substrate structure.
Previous MD simulations covering the WT, *2, *4, *10, *17, and *53 variants showed that *17 has a more confined active site fold compared to the WT CYP2D6, as well as higher flexibility in the KL loop (which contains two antiparallel beta-pairs).24 Given that the proximal L-helix is one of the paired helices holding the heme, this increased flexibility could contribute to the increased spin-state changes seen in *17. (Figure 2) CYP2D6*17 has also been shown to have fewer hydrogen bonds as a result of its T107I and R296C mutations.74 CYP2D6*10 contains a P34S mutation which is known to impede membrane binding, but this would not necessarily affect substrate binding to the heme.
Modeling results indicate that both binding distance and affinity differ by mutant and pCB (Supplementary Figures 1–8). On average WT CYP2D6 has the strongest binding affinity and closest heme binding distance for all pCB tested. CYP2D6*10 is the next strongest in terms of binding affinity, followed by *17 and then *2. A possible rationale for the increased distance of pCB from the heme in CYP2D6 mutants may be alteration in the size of the access channel and active site of the protein making it difficult for pCB molecules to physically fit within the cavity. MD simulations have previously indicated that *17 has a more restricted active site, which would hinder the ability of substrates and inhibitors to bind24. Analyses conducted with the Caver software on our equilibrated WT and *17 structures additionally showed a significantly smaller bottleneck radius in *17 access channel as compared to that in WT (Supplementary Tables 2 and 3). This indicates that fewer poses dock near the heme because the pCB may not able to physically fit within the cavity. Additionally, in *17, the most common interactions between the pCB molecules and the protein lie away from the heme of the protein, indicating that the ligands are stabilized away from the active site of the protein (Supplementary Figure S9–S16).
Additional simulations of both docked THC and CBD molecules in *17 and WT structures also indicate that pCB molecules bind nearer to the heme in the WT structure. Additionally, in WT the hydrocarbon tail of both CBD THC binds in the direction of the heme, while in *17, the rings of THC tend to be oriented closer to the heme; indicating THC may orient in opposite conformations for WT and *17. Surprisingly, the residue interaction pattern of both THC and CBD in the WT simulation closely matches the residues lining the access channel as indicated by Caver analysis; this is not the case for *17 (Supplementary Tables 4 and 5). Both molecules appear to lie within the space of the access channel for a significant portion of the WT simulation (Supplementary Figure S31). This may imply that these pCBs can occupy the access channel and prevent the access of substrates to the active site of the protein. THC may experimentally perform as a weaker inhibitor of the *17 variant because it physically cannot fit in the access channel to block substrate access.
We then investigated the metabolism of AEA and DXM by CYP2D6, and the potential inhibitory effects of pCBs on these metabolisms. Previous research has shown that the pentyl side chain present on CBD played a role in the inhibition of CYP2D6. Both olivetol and CBDV were able to inhibit CYP2D6 metabolism of AMMC (3-[2-(N,N-diethyl-N-methylammonium)ethyl]-7-methoxy-4-methylcoumarin), indicating that both the side chain and hydroxyl groups of the pentylresorcinol moiety are important structural components for inhibition.41 Compounds lacking both of these features were not found to inhibit CYP2D6 metabolism. In preliminary studies with WT CYP2D6, it was shown that CBDV did not greatly inhibit DXM metabolism, though CBD, CBC, THCV, and β-CP did (Supplementary Figure S19). For AEA metabolism, CBDV along with CBD and THC showed slightly better inhibition as compared to other pCBs. This difference is likely due, at least in part, to binding at a different site, which has been seen with CYP2J2.32,75–77 In a separate study, CBD was shown to inhibit (S)-mephenytoin 4-hydroxylation by CYP2C19 as well as the O-demethylation of 3-O-methylfluorescein (OMF) and 5-hydroxylation of omeprazole.42 It is worth noting, however, that while pCBs are broadly thought of as P450 inhibitors78, all literature showing the specific inhibition of CYP2D6 by pCBs use drug substrates rather than endogenous substrates. Therefore, here we show that the inhibition of AEA metabolism by pCBs is weak.
Noting the restricted active site of *17, along with the binding differences indicated by Soret titrations, we chose to narrow our focus on the comparison of WT CYP2D6 and *17 using the endogenous substrate AEA. Standard AEA metabolism without the presence of pCBs varied as expected, with WT CYP2D6 having 1.5-fold greater metabolism compared to *17. Additionally, both forms of the enzyme had the lowest rates of metabolism in the presence of THC as well as similar Kmapp values. This agrees with the MD simulation results which suggest THC would bind well to WT CYP2D6.
Interestingly, the presence of CBD activated WT CYP2D6, increasing the Vmaxapp from 387.69 to 530.17 pmol/min/nmol (an ~1.3-fold increase). A closer look at the EET-EA regioisomer production revealed that with WT CYP2D6 14,15-EET-EA production decreases with increasing AEA concentrations while 5,6-EET-EA increases, a trend which holds true for both untreated and CBD treated CYP2D6 (Figure 5 E, F). With CBD treated WT CYP2D6, the rates of individual EET-EA production are approximately 1.7-fold and 1.2-fold higher for 14,15- and 5,6-EET-EA, respectively. There is minimal change in the presence of THC and there was no change in the regioisomeric production of EET-EAs for *17.
Conclusions
Taken together, we have found that the interactions of CYP2D6 with pCBs vary by polymorphism and by specific pCB class. We show that THC and structurally similar pCBs bind more tightly than other pCBs and that WT CYP2D6 is overall more tightly bound. We also note that CYP2D6*17 is the most prone to large spin-state changes, though the link to pCB structure is less clear. Furthermore, MD simulations show that not only do mutants have a difference in heme distance and binding affinity, but also that contacts with the I-helix have shifted to the F-helix.
Lastly, we have shown that WT CYP2D6 is remotely activated by CBD while the mutant *17 is not, which we attribute to mutations changing the shape of the substrate access channel and thus heme binding distance.
Supplementary Material
Acknowledgements
The authors would like to thank Dr. Lucas Li of the Roy J. Carver Biotechnology Center for performing the LC/MS analysis. We would also like to thank Dr. Ko for the use of his thermocycler when making our CYP2D6 polymorphism constructs. We want to thank Josephine Watson for making the CYP2D6 mutants and optimizing the method of protein expression. We want to thank Prof. Eric Johnson’s laboratory for the CYP2D6 construct. We want to thank Demetri Maroutsos for initial help with the project by purifying CYP2D6 and doing some initial titration experiments.
Funding Sources
Supported by National Institutes of Health Grants R01 GM1155884, R03 DA 04236502, and R21AT010761 to A.D., and R01 GM101048, U54 GM087519, and P41 GM104601 to E.T. All simulations were performed using XSEDE resources (Grant MCA06N060 to E.T.).
ABBREVIATIONS
- AEA
Anandamide
- AA
Arachidonic Acid
- β-CP
β-carophyllene
- CBC
cannabichromene
- CBD
cannabidiol
- CBDV
cannabidivarin
- CBG
cannabigerol
- CBN
cannabinol
- CYP
cytochrome P450
- CPR
cytochrome P450 reductase
- DXM
Dextromethorphane
- CB
endocannabinoid
- EET
epoxyeicosatrienoic acid
- EET-EA
epoxyeicosatrienoyl ethanolamide
- EPOX
epoxygenase
- HETE
hydroxyeicosatrienoic acid
- LC-MS/MS
liquid chromatography- tandem mass spectrometry
- MD
molecular dynamics
- ND
Nanodisc
- pCB
phytocannabinoid
- THC
tetrahydrocannabinol
- THCV
tetrahydrocannabivarin
3.9 References
- [1].Baron EP (2018) Medicinal Properties of Cannabinoids, Terpenes, and Flavonoids in Cannabis, and Benefits in Migraine, Headache, and Pain: An Update on Current Evidence and Cannabis Science, Headache 58, 1139–1186. [DOI] [PubMed] [Google Scholar]
- [2].Abrams DI, Couey P, Shade SB, Kelley ME, and Benowitz NL (2011) Cannabinoid-Opioid Interaction in Chronic Pain, Clinical Pharmacology & Therapeutics 90, 844–851. [DOI] [PubMed] [Google Scholar]
- [3].Romero-Sandoval EA, Kolano AL, and Alvarado-Vásquez PA (2017) Cannabis and Cannabinoids for Chronic Pain, Current Rheumatology Reports 19. [DOI] [PubMed] [Google Scholar]
- [4].Darmani NA (2001) Delta-9-tetrahydrocannabinol differentially supresses cisplatin-induced emesis and indices of motor function via cannabinoid CB1 receptors in the least shrew, Pharmacology Biochemistry and Behavior 69, 239–249. [DOI] [PubMed] [Google Scholar]
- [5].Darmani NA (2001) D9-Tetrahydrocannabinol and Synthetic Cannabinoids Prevent Emesis Produced by the Cannabinoid CB1 Receptor Antagonist/Inverse Agonist SR 141716A, Neuropsychopharmacology 24, 198–203. [DOI] [PubMed] [Google Scholar]
- [6].Fraguas-Sánchez AI, and Torres-Suárez AI (2018) Medical Use of Cannabinoids, Drugs 78, 1665–1703. [DOI] [PubMed] [Google Scholar]
- [7].Porter BE, and Jacobson C (2013) Report of a parent survey of cannabidiol-enriched cannabis use in pediatric treatment-resistant epilepsy, Epilepsy & Behavior 29, 574–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gofshteyn JS, Wilfong A, Devinsky O, Bluvstein J, Charuta J, Ciliberto MA, Laux L, and Marsh ED (2016) Cannabidiol as a Potential Treatment for Febrile Infection-Related Epilepsy Syndrome (FIRES) in the Acute and Chronic Phases, Journal of Child Neurology 32, 35–40. [DOI] [PubMed] [Google Scholar]
- [9].Devinsky O, Cross JH, Laux L, Marsh ED, Miller I, Nabbout R, Scheffer IE, Thiele EA, and Wright S (2017) Trial of Cannabidiol for Drug-Resistant Seizures in the Dravet Syndrome, The New England Journal of Medicine 376, 2011–2020. [DOI] [PubMed] [Google Scholar]
- [10].Williamson EM, and Evans FJ (2012) Cannabinoids in Clinical Practice, Drugs 60, 1303–1314. [DOI] [PubMed] [Google Scholar]
- [11].Abrams DI, Couey P, Shade SB, Kelly ME, and Benowitz NL (2011) Cannabinoid-Opioid Interaction in Chronic Pain, Clinical Pharmacology & Therapeutics 90, 844–851. [DOI] [PubMed] [Google Scholar]
- [12].Iannotti FA, Hill CL, Leo A, Alhusaini A, Soubrane C, Mazzarella E, Russo E, Whalley BJ, Di Marzo V, and Stephens GJ (2014) Nonpsychotropic Plant Cannabinoids, Cannabidivarin (CBDV) and Cannabidiol (CBD), Activate and Desensitize Transient Receptor Potential Vanilloid 1 (TRPV1) Channels in Vitro: Potential for the Treatment of Neuronal Hyperexcitability, ACS Chemical Neuroscience 5, 113–1141. [DOI] [PubMed] [Google Scholar]
- [13].Starowicz K, Mackuch W, Osikowicz M, Piscitelli F, Petrosino S, Di Marzo V, and Przewlocka B (2012) Spinal anandamide produces analgesia in neuropathic rats: Possible CB1- and TRPV1-mediated mechanisms, Neuropharmacology 62, 1746–1755. [DOI] [PubMed] [Google Scholar]
- [14].Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A, Azad SC, Cascio MG, Gutiérrez SO, van der Stelt M, López-Rodríguez ML, Casanova E, Schütz G, Zieglgänsberger W, Di Marzo V, Behl C, and Lutz B (2003) CB1 Cannabinoid Receptors and On-Demand Defense Against Excitotoxicity, Science 102, 84–88. [DOI] [PubMed] [Google Scholar]
- [15].Wallace MJ, Blair RE, Falenski KW, Martin BR, and DeLorenzo RJ (2003) The Endogenous Cannabinoid System Regulates Seizure Frequency and Duration in a Model of Temporal Lobe Epilepsy, The Journal of Pharmacology and Experimental Therapeutics 307, 129–137. [DOI] [PubMed] [Google Scholar]
- [16].Pan X, Ning M, and Jeong H (2017) Transcriptional Regulation of CYP2D6 Expression, Drug Metabolism and Disposition 45, 42–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhou S-F (2009) Polymorphism of Human Cytochrome P450 2D6 and Its Clinical Significance, Clinical Pharmacokinetics 48, 689–723. [DOI] [PubMed] [Google Scholar]
- [18].Gaedigk A, Sangkuhl K, Whirl-Carrillo M, Klein T, and Leeder JS (2017) Prediction of CYP2D6 phenotype from genotype across world populations, Genetics in Medicine 19, 69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Guengerich FP (2015) Human Cytochrome P450 Enzymes, In Cytochrome P450 Structure, Mechanism, and Biochemistry (Ortiz de Montellano PR, Ed.) 4 ed., pp 523–786, Springer, Cham, Switzerland. [Google Scholar]
- [20].Zhou SF (2009) Polymorphism of human cytochrome P450 2D6 and its clinical significance: Part I, Clin Pharmacokinet 48, 689–723. [DOI] [PubMed] [Google Scholar]
- [21].Yu A, Kneller BM, Rettie AE, and Haining RL (2002) Expression, purification, biochemical characterization, and comparative function of human cytochrome P450 2D6.1, 2D6.2, 2D6.10, and 2D6.17 allelic isoforms, J Pharmacol Exp Ther 303, 1291–1300. [DOI] [PubMed] [Google Scholar]
- [22].Ingelman-Sundberg M (2005) Genetic Polymorphisms of Cytochrome P450 2D6 (CYP2D6): Clinical Consequences, Evolutionary Aspects and Functional Diversity, Pharmacogenomics 5, 6–13. [DOI] [PubMed] [Google Scholar]
- [23].Yu A, Kneller BM, Rettie AE, and Haining RL (2002) Expression, Purification, Biochemical Characterization, and Comparative Function of Human Cytochrome P450 2D6.1, 2D6.2, 2D6.10 and 2D6.17 Allelic Isoforms, The Journal of Pharmacology and Experimental Therapeutics 303, 1291–1300. [DOI] [PubMed] [Google Scholar]
- [24].Don CG, and Smieško M (2018) Microsecond MD simulations of human CYP2D6 wild-type and five allelic variants reveal mechanistic insights on the function, Public Library of Science One 13, e0202534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Oscarson M, Hidestrand M, Johansson I, and Ingelman-Sundberg M (1997) A Combination of Mutations in the CYP2D6*17(CYP2D6Z) Allele Causes Alterations in Enzyme Function, Molecular Pharmacology 52, 1034–1040. [DOI] [PubMed] [Google Scholar]
- [26].Bogni A, Monshouwer M, Moscone A, Hidestrand M, Ingelman-Sundberg M, Hartung T, and Coecke S (2005) Substrate specific metabolism by polymorphic cytochrome P450 2D6 alleles, Toxicology In Vitro 19, 621–629. [DOI] [PubMed] [Google Scholar]
- [27].Cai W-M, Nikoloff DM, Pan R-M, de Leon J, Fanti P, Fairchild M, Koch WH, and Wedlund PJ (2006) CYP2D6 genetic variation in healthy adults and psychiatric African-American subjects: implications for clinical practice and genetic testing, The pharmacogenomics journal 6, 343–350. [DOI] [PubMed] [Google Scholar]
- [28].Zhou S-F, Liu J-P, and Chowbay B (2009) Polymorphism of human cytochrome P450 enzymes and its clinical impact, Drug Metabolism Reviews 41, 89–295. [DOI] [PubMed] [Google Scholar]
- [29].Bahar MA, Setiawan D, Hak E, and Wilffert B (2017) Pharmacogenetics of Drug-Drug Interaction and Drug-Drug-Gene Interaction: A Systematic Review on CYP2C9, CYP2C19, and CYP2D6, Pharmacogenomics 18, 701–739. [DOI] [PubMed] [Google Scholar]
- [30].Hu X-X, Yuan L-J, Fang P, Mao Y-H, Zhan Y-Y, Li X-Y, Dai D-P, Cai J, and Hu G (2016) Effect of CYP2D6 Genetic Polymorphism on the Metabolism of Citalopram in Vitro, Drug Metabolism and Pharmacokinetics 31, 133–138. [DOI] [PubMed] [Google Scholar]
- [31].Stingl J, and Viviani R (2014) Polymorphism in CYP2D6 and CYP2C19, members of the cytochrome P450 mixed-function oxidase system, in the metabolism of psychotropic drugs, Journal of Internal Medicine 277, 167–177. [DOI] [PubMed] [Google Scholar]
- [32].Arnold WR, Weigle AT, and Das A (2018) Cross-talk of cannabinoid and endocannabinoid metabolism is mediated via human cardiac CYP2J2, Journal of Inorganic Biochemistry 184, 88–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Su MK, Seely KA, Moran JH, and Hoffman RS (2015) Metabolism of Classical Cannabinoids and the Synthetic Cannabinoid JWH-018, Clinical Pharmacology & Therapeutics 97, 562–564. [DOI] [PubMed] [Google Scholar]
- [34].Watanabe K, Yamaori S, Funahashi T, Kimura T, and Yamamoto I (2007) Cytochrome P450 Enzymes Involved in the Metabolism of Tetrahydrocannabinols and Cannabidiol by Human Hepatic Microsomes, Life Sciences 80, 1415–1419. [DOI] [PubMed] [Google Scholar]
- [35].Ohyama K, Nakajima M, Suzuki M, Shimada N, Yamazaki H, and Yokoi T (2000) Inhibitory effects of amiodarone and its N-deethylated metabolite on human cytochrome P450 activities: Prediction of in vivo drug interactions, British Journal of Clinical Pharmacology 49, 244–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Patilea-Vrana GI, Anoshchenko O, and Unadkat JD (2019) Hepatic Enzymes Relevant to the Disposition of (−)-Δ9-Tetrahydrocannabinol (THC) and Its Psychoactive Metabolite, 11-OH-THC, Drug Metabolism and Disposition 47, 249–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Su MK, Seely KA, Moran JH, and Hoffman RS (2015) Metabolism of Classical Cannabinoids and the Synthetic Cannabinoid JWH-018, Clinical Pharmacology & Therapeutics 97. [DOI] [PubMed] [Google Scholar]
- [38].Stout SM, and Cimino NM (2014) Exogenous cannabinoids as substrates, inhibitors, and inducers of human drug metabolizing enzymes: a systematic review, Drug Metabolism Reviews 46, 86–95. [DOI] [PubMed] [Google Scholar]
- [39].Yamaori S, Ebisawa J, Okushima Y, Yamamoto I, and Watanabe K (2011) Potent inhibition of human cytochrome P450 3A isoforms by cannabidiol: Role of phenolic hydroxyl groups in the resorcinol moiety, Life Sciences 88, 730–736. [DOI] [PubMed] [Google Scholar]
- [40].Yamaori S, Koeda K, Kushihara M, Hada Y, Yamamoto I, and Watanabe K (2012) Comparison in the in Vitro Inhibitory Effects of Major Phytocannabinoids and Polycyclic Aromatic Hydrocarbons Contained in Marijuana Smoke on Cytochrome P450 2C9 Activity, Drug Metabolism and Pharmacokinetics 27, 294–300. [DOI] [PubMed] [Google Scholar]
- [41].Yamaori S, Okamoto Y, Yamamoto I, and Watanabe K (2011) Cannabidiol, a Major Phytocannabinoid, As a Potent Atypical Inhibitor for CYP2D6, Drug Metabolism and Disposition 39, 2049–2056. [DOI] [PubMed] [Google Scholar]
- [42].Jiang R, Yamaori S, Okamoto Y, Yamamoto I, and Watanabe K (2013) Cannabidiol is a Potent Inhibitor of the Catalytic Activity of Cytochrome P450 2C19, Drug Metabolism and Pharmacokinetics 28, 332–338. [DOI] [PubMed] [Google Scholar]
- [43].Alberti TB, Barbosa WLR, Vieira JLF, Raposo NRB, and Dutra FC (2017) (−)-b-Carophyllene, a CB2 Receptor-Selective Phytocannabinoid Suppresses Motor Paralysis and Neuroinflammation in a Murine Model of Multiple Sclerosis, International Journal fo Molecular Sciences 18, 691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Gertsch J, Leonti M, Raduner S, Racz I, Chen J-Z, Xie X-Q, Altmann K-H, Karsak M, and Zimmer A (2008) Beta-carophyllene is a dietary cannabinoid, Proceedings of the National Academy of Science of the United States of America 105, 9099–9104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hudeček J, Anzenbacherova E, Anzenbacher P, Munro AW, and Hildebrandt P (2000) Structural Similarities and Differences of the Heme Pockets of Various P450 Isoforms as Revealed by Resonance Raman Spectroscopy, Archives of Biochemistry and Biophysics 383, 70–78. [DOI] [PubMed] [Google Scholar]
- [46].Hritz J, de Ruiter A, and Oostenbrink C (2008) Impact of Plasticity and Flexibility on Docking Results for Cytochrome P450 2D6: A Combined Approach of Molecular Dynamics and Ligand Docking, Journal of Medicinal Chemistry 51, 7469–7477. [DOI] [PubMed] [Google Scholar]
- [47].Lussenberg BMA, Keizers PHJ, de Graaf C, Hidestrand M, Ingelman-Sundberg M, Vermeulen NPE, and Commandeur JNM (2005) The Role of Phenylalanine 483 in Cytochrome P450 2D6 Is Strongly Substrate Dependent, Biochemical Pharmacology 70, 1253–1261. [DOI] [PubMed] [Google Scholar]
- [48].Arafa MH, and Atteia HH (2018) Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6) are associated with long term tramadol treatment-induced oxidative damage and hepatotoxicity, Toxicology and Applied Pharmacology 346, 37–44. [DOI] [PubMed] [Google Scholar]
- [49].Fang P, Zheng X, He J, Ge H, Tang P, Cai J, and Hu G (2017) Functional characterization of wild-type and 24 CYP2D6 allelic variants on gefitinib metabolism in vitro, Drug Design, Development and Therapy 11, 1283–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Niwa T, Shizuku M, and Yamano K (2017) Effect of genetic polymorphism on the inhibition of dopamine formation from p-tyramine catalyzed by brain cytochrome P450 2D6, Archives of Biochemistry and Biophysics 620, 23–27. [DOI] [PubMed] [Google Scholar]
- [51].McDougle DR, Palaria A, Magnetta E, Meling DD, and Das A (2013) Functional studies of N-terminally modified CYP2J2 epoxygenase in model lipid bilayers, Protein Science 22, 964–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Luthra A, Denisov IG, and Sligar SG (2011) Spectroscopic features of cytochrome P450 reaction intermediates, Arch Biochem Biophys 507, 26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Wang A, Savas U, Hsu MH, Stout CD, and Johnson EF (2012) Crystal structure of human cytochrome P450 2D6 with prinomastat bound, J Biol Chem 287, 10834–10843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Schenkman JB, and Jansson I (2006) Spectral Analyses of Cytochromes P450, In Cytochrome P450 Protocols (Phillips IR, and Shephard EA, Eds.), pp 11–18, Humana Press, Totowa, NJ. [Google Scholar]
- [55].Huff HC, Maroutsos D, and Das A (2019) Lipid Composition and Macromolecular Crowding Effects on CYP2J2-Mediated Drug Metabolism in Nanodiscs, Protein Science 28, 928–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].McDougle DR, Kambalyal A, Meling DD, and Das A (2014) Endocannabinoids Anandamide and 2-Arachidonoylglycerol Are Substrates for Human CYP2J2 Epoxygenase, Journal of Pharmacology and Experimental Therapeutics 351, 616–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Wang A, Stout CD, Zhang Q, and Johnson EF (2015) Contributions of ionic interactions and protein dynamics to chytochrome P450 2D6 (CYP2D6) substrate and inhibitor binding, Journal of Biological Chemistry 290, 5092–5104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Jo S, Kim T, Iyer VG, and Im W (2008) CHARMM-GUI: a web-based graphical user interface for CHARMM, Journal of Computational Chemistry 29, 1859–1865. [DOI] [PubMed] [Google Scholar]
- [59].Fisher A, Don CG, and Smieško M (2018) Molecular Dynamics Simulations Reveal Structural Differences among Allelic Variants of Membrane-Anchored Cytochrome P450 2D6, Journal of Chemical Information and Modeling 58, 1962–1975. [DOI] [PubMed] [Google Scholar]
- [60].Jorgenson WL, Chandrasekhar J, Madura JD, Impey RW, and Klein ML (1983) Comparison of simple potential functions for simulating liquid water, The Journal of Chemical Physics 79, 926–935. [Google Scholar]
- [61].Humphrey W, Dalke A, and Schulten K (1996) VMD: visual molecular dynamics, Journal of Molecular Graphics 14, 33–38. [DOI] [PubMed] [Google Scholar]
- [62].Trott O, and Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, Journal of Computational Chemistry 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, and Schulten K (2005) Scalable molecular dynamics with NAMD, Journal of Computational Chemistry 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, De Groot BL, Grubmüller H, and MacKerell AD Jr. (2017) CHARMM36m: an improved force field for folded and intrinsically disordered proteins, Nature Methods 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD Jr., and Pastor RW (2010) Updated of the CHARMM all-atom additive force field for lipids: validation on six lipid types, The Journal of Physical Chemistry B 114, 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, and MacKerell AD Jr. (2010) CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields, Journal of Computational Chemistry 31, 671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Feller SE, Zhang Y, Pastor RW, and Brooks BR (1995) Constant pressure molecular dynamics simulation: the Langevin piston method, The Journal of Chemical Physics 103, 4613–4621. [Google Scholar]
- [68].Darden T, York D, and Pedersen L (1993) Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems, The Journal of Chemical Physics 98, 10089–10092. [Google Scholar]
- [69].Cojocaru V, Balali-Mood K, Sansom MS, and Wade RC (2011) Structure and dynamics of the membrane-bound cytochrome P450 2C9, PLoS Comput Biol 7, e1002152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].McDougle DR, Baylon JL, Meling DD, Kambalyal A, Grinkova YV, Hammernik J, Tajkhorshid E, and Das A (2015) Incorporation of charged residues in the CYP2J2 F-G loop disrupts CYP2J2-lipid bilayer interactions, Biochim Biophys Acta 1848, 2460–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Puangpetch A, Vanwong N, Nuntamool N, Hongkaew Y, Chamnanphon M, and Sukasem C (2016) CYP2D6 polymorphisms and their influence on risperidone treatment, Pharmacogenomics and Personalized Medicine 9, 131–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Luthra A, Denisov IG, and Sligar SG (2011) Spectroscopic features of cytochrome P450 reaction intermediates, Archives of Biochemistry and Biophysics 507, 26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Conner KP, Woods CM, and Atkins WM (2011) Interactions of cytochrome P450s with their ligands, Archives of Biochemistry and Biophysics 507, 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Handa K, Nakagome I, Yamaotsu N, Gouda H, and Hirono S (2014) In Silico Study on the Inhibitory Interaction of Drugs With Wild-Type CYP2D6.1 and the Natural Variant CYP2D6.17, Drug Metabolism and Pharmacokinetics 29, 52–60. [DOI] [PubMed] [Google Scholar]
- [75].Arnold WR, Baylon JL, Tajkhorshid E, and Das A (2017) Arachidonic Acid Metabolism by Human Cardiovascular CYP2J2 Is Modulated by Doxorubicin, Biochemistry 56, 6700–6712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Arnold WR, Baylon JL, Tajkhorshid E, and Das A (2016) Asymmetric Binding and Metabolism of Polyunsaturated Fatty Acids (PUFAs) by CYP2J2 Epoxygenase, Biochemistry 55, 6969–6980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Das A, Weigle AT, Arnold WR, Kim JS, Carnevale LN, and Huff HC (2020) CYP2J2 Molecular Recognition: A New Axis for Therapeutic Design, Pharmacol Ther 215, 107601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Bornheim LM, Kim KY, Chen B, and Correia MA (1993) The Effect of Cannabidiol on Mouse Hepatic Microsomal Cytochrome P450-Dependent Anandamide Metabolism, Biochemical and Biophysical Research Communications 197, 740–746. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.