

Abstract
This report evaluates how herpes simplex virus (HSV) enters the brain to cause herpes simplex encephalitis (HSE) following infection at a peripheral site. We demonstrate that encephalitis regularly occurred when BALB/c mice were infected with HSV and treated daily with 2-Deoxy-D-glucose (2DG) which inhibits glucose utilization via the glycolysis pathway. The outcome of infection in the trigeminal ganglion (TG), the site to which the virus spreads, replicates and establishes latency, showed marked differences in viral and cellular events between treated and untreated animals. In control untreated mice, the replicating virus was present only during early time points, while in 2DG recipients, replicating virus remained for the 9 day observation period. This outcome correlated with significantly reduced numbers of innate inflammatory cells as well as T cells in 2DG treated animals. Moreover, T cells in the TG of treated animals were less activated and a lesser fraction expressed interferon gamma (IFN-γ) production compared to untreated controls. The breakdown of latency was accelerated when cultures of TG cells taken from mice with established HSV latency, were cultured in the presence of 2DG. Taken together the results of both in vivo and in vitro investigations demonstrate that the overall effects of 2DG therapy impaired the protective effects of one or more inflammatory cell types in the TG that normally function to control productive infection and prevent spread of virus to the brain.
Introduction
Some of the most devastating consequences of a virus infection occur when they enter and infect the brain and cause encephalitis (1). The brain is normally well protected from infection and few agents have the capacity to gain entrance. Access to the brain can occur in different ways, but the most common entry route is by crossing the blood brain barrier (BBB) (2). The BBB is a multicellular organization that is composed of vascular endothelial cells tightly connected by specialized proteins such as claudins and occludins surrounded by pericytes and supported by astrocyte cells on the central nervous system (CNS) side (3). The normally functioning BBB allows the exchanges of nutrients and metabolites between the blood circulation and the brain, but limits the passage of many soluble compounds as well as most pathogens. However, when the BBB is damaged physically or physiologically, its integrity is impaired and the brain becomes more accessible (4). Although the BBB and its function has been the topic of many studies [reviewed in (5)], it is still not clear if the host metabolic state could impact on the integrity of the BBB. Thus, changes in host metabolism could impair the barrier function of the BBB and permit viruses to cross and cause damage to CNS tissues. This might be the mechanism by which HSV infection causes encephalitis, an issue that is addressed in the present report.
Infection of the brain by HSV can result in notable damage to the brain, which if uncontrolled by antiviral drugs may have major consequences on neuronal tissues (6). This HSE syndrome can occur in a number of different circumstances. Firstly, newborns born to HSV seronegative mothers if infected with HSV-2 around the time of birth may develop encephalitis, which can have very destructive consequences unless rapidly treated with effective antiviral drugs (7). Children born with one of several genetic defects that affect their ability to produce or respond to interferons are prone to develop HSE and without modern therapies would rarely reach adulthood (6, 8). Healthy adults occasionally develop HSE and this mostly happens in those who carry the virus in a latent from, perhaps for years, after primary infection (9, 10). Such persons upon reactivation from latency, which normally causes only local lesions, can result in virus spreading to the brain resulting in HSE (11). Why this unusual event occurs and how virus passages to the brain remains unresolved, although a number of possible mechanisms have been suggested (12). These include direct passage to the brain via cranial nerves such as the olfactory or trigeminal nerves, and crossing directly through the BBB. In addition, some agents can cross the BBB into the brain within leucocytes, a circumstance resembling the Trojan horse phenomenon (12, 13).
In past studies on experimental ocular infection of adult C57BL/6 mice with HSV, we had observed that if soon after infection mice were given the drug 2DG, which interferes with glucose metabolism, they regularly succumbed to HSE, while, untreated animals did not (14). However, when therapy with 2DG was begun at a later time when ocular inflammatory lesions were developing, the mice did not develop HSE. Furthermore, 2DG treatment decreased the severity of their ocular inflammatory lesions. We advocated that the latter effect was the consequence of suppressed inflammatory T cell responses that are mainly responsible for ocular lesions (14). The cause of encephalitis in animals treated from the onset of infection with 2DG was not defined, but one possibility could be damage to the metabolism of BBB cells making the barrier more permeable to crossing by the virus. In this report, this issue is further evaluated as are other ways the virus might access the CNS. We interpret our findings to indicate that in the experimental mouse model we used, the most likely route of access to the brain was not via the BBB, made more permeable by 2DG therapy, but via the trigeminal nerve following continued and elevated viral replication in the trigeminal ganglion that occurred because 2DG treatment impaired the protective function of T cells in the TG.
Materials and Methods
Animals
Five to 6 weeks old female BALB/C mice were purchased from Envigo (US) and were used for the ocular HSV-1 infection model. Animals were kept in an animal biosafety level-2 (ABSL-2) facility where water, bedding and instruments were regularly autoclaved. All mice were housed in the American Association of Laboratory Animal Science approved facilities at the University of Tennessee, Knoxville, TN. All investigations were accomplished and followed the Institutional Animal Care and Use Committee (IACUC) guidelines and adhered to the Association for Research in Vision and Ophthalmology. Animals were visited by facility staff members and Office of Laboratory Animal Care (OLAC) veterinarians during the HSV-1 on infection by daily basis. Animals were supported with diet gel addition to their cages when they showed severe symptoms. Mice had received 24 hr watch if they presented lethargic and hunched posture and were humanely euthanized by CO2 at the end of 24 hr watch.
Virus
HSV-1 RE strain was obtained from Robert Hendricks, University of Pittsburgh PA, USA. The virus was propagated in Vero cell monolayers (ATTC, Manassas, USA) to produce viral seed stocks. Virus propagations and quantification of viral titer were described previously (15, 16). A less virulent HSV-1 strain than RE was employed for latency experiments. The virus that constructed by recombinantly to express the enhanced green fluorescein protein (EGFP) under the control of early viral promoter ICP0 and a monomeric red fluorescein protein (RFP) under the control of late viral glycoprotein C (17). HSV-1 KOS EGFP/RFP virus kindly gifted by Paul R. Kinchington, University of Pittsburgh PA, USA. The virus was propagated in Vero cell as described before (16).
Mice infection
For ocular infection, 7–8 week old BALB/c mice (n=4 with each experiment repeated 3 times) were infected under the injection of 1.25% solution of avertin anesthetic (2,2,2-Tribromoethanol; Acros Organics, Pittsburgh, PA). Ocular scratching was performed in the absence of tail and toe pinch reflexes. The epithelial surface of the anterior chamber on cornea from each mouse was scarified 15 times per side in an X-shaped pattern using the tip of 27-gauge needle without damaging the globe or iris of the eye globe. A 3 μL drop of virus inoculum containing various virus doses (HSV-1 RE; 1×103, 5×103 and 1×104 PFU/3 μL) was applied to the eye after corneal scarification. Day of infection was counted as day 0 and following days were mentioned day-post infection (dpi). A group of mice were also corneal scarified but mock-infected with PBS. For the latency experiments, 7–8 week old BALB/c mice (n=12) were subjected to ocular infection with HSV-1 KOS EGFP/RFP virus (1×106 PFU/eye).
Drug administration, sampling and clinical scoring
2DG (Sigma‐Aldrich, St. Louis, MO) was dissolved in sterile PBS and injected into mice via IP. Mice had received 250 mg/kg of 2DG in 0.2 ml volume twice a day, 10 hr interval starting from the day of infection (day 0) up to day 10 of post-infection (pi). The control group had received an equal volume of PBS. Mice were infected via ocularly 5 hr after receiving first injections for treatment and a group of mice was also either mock-infected or left untreated with a drug at day 0. HSV-1 RE ocular infected mice were regularly monitored and recorded to develop HSV-1 related lesions, including stromal keratitis (SK), angiogenesis and clinical appearances. To evaluate viral replication in the eye, swap sampling was done daily starting from day 1 up to day 8 pi. PBS soaked swabs were applied on the surface of the eye and swabs were collected individually. Swab specimens were stored in 2% FBS and antibiotic-containing PBS and kept at −80 until performing virus quantification. The SK lesion severity and angiogenesis in the eyes of mice were examined through slit-lamp bio-microscopy (Kowa Company, Nagoya, Japan) and recorded at 9 dpi along with a recording of some image examples from average score showing mice. The severity of angiogenesis and the scoring of SK in the eyes were recorded as described previously (18, 19). SK and angiogenesis were documented in a blind manner. Development of lethargy and encephalitis with the sign of neurological changes, including jumping when touch or circling itself when hanging by the tail, were also examined independently and recorded in percent changes. Mice were euthanized if they are not improved after opening a clinical case with 24 hr watch by OLAC veterinarians. Mice were recorded in survival analysis based on 24 hr watch monitoring period results. To monitor the viral replication in the TGs from ocular infected mice, mice were euthanized on day 2 and 3 pi and so on by two days intervals until the experiment’s termination on day 9 pi. TGs were excised from the brain cavity and TGs were processed individually. TGs were mechanically disrupted with tissue grinders (pellet pestle motor, Kontes). Collected and homogenized TGs were freeze-thawed three times to collect supernatants and assayed to quantify HSV-1 titer on Vero cells.
Ex vivo latency and reactivation
A latency experiment was described previously and applied to this study with some modifications (20). Latently infected TGs (5 weeks after ocular infection) were excised from HSV-1 KOS EGFP/RFP infected animals (1×106 PFU/eye). TGs were enzymatically dissociated with collagenase type I (Sigma-Aldrich, St. Louis, MO) at a 3 mg/ml concentration for 45 min at 37°C and mechanically disrupted with tissue grinder. Contaminating erythrocytes were removed using erythrocyte lysis buffer. Homogenized and pooled TGs were dispersed in wells of 24 culture plates containing the 2 TGs/well in total. To measure the reactivating virus titer in the presence of 2DG, TGs maintained for 8 days in the presence of complete DMEM media or in glucose free media (Gibco-BRL, Grand Island, NY) containing 10 U/ml recombinant murine IL-2 (PeproTech, Rocky Hill, NJ). A group of wells was supplemented with 2DG (0.5 mM) and supernatant were replaced with fresh culture media (1:20) containing the same concentration of 2DG by two days intervals and control wells were replaced with complete media. Preliminary experiments were performed with T cells to establish that this dose successfully inhibited glycolysis and was non-toxic to the cells. Collected supernatants were tested on Vero cells and plaque assay was performed followed by crystal violet staining as described before (15). Some of the wells were examined under the 4X objective lens of the EVOS FLc Imaging System (ThermoFisher Scientific, Waltham) at day 4 and images were acquired for the expression of early (EGFP-ICP0) and late (RFP-glycoprotein C) viral proteins. In a separate experiment to measure the endogenous T cell-mediated latency maintenance, T cells were depleted with the addition of 150 μg/ml anti-CD4 (BioXCell, clone GK 1.5) and anti-CD8 (BioXCell, clone 2.43) cocktail to latently infected and homogenized TG culture media as described before (20). In a comparison of the effect of 2DG on T cell activation during the reactivation, a group of wells were either supplemented with 2DG (0.5 mM) or left untreated. Reactivation was terminated at 3 day of ex-plant culture. The supernatant was tested on the Vero cell to measure reactivated virus titer in a crystal violet staining experiment.
In-vivo BBB permeability and extravasation of Evans blue
To investigate the BBB leakage, Evans blue dye (Sigma-Aldrich, St Louis, MO) was administered to mice as described previously with some modifications (21). Mice (4 dpi or 8 dpi) received 2% of Evans blue dye (4 ml/kg) that dissolved in PBS and injected to mice via IP route and left for 16 hours. A cardiac PBS perfusion was performed to mice under the effect of a lethal dose of avertin (50 mg/kg). The brain was excised out from the cranial cavity and kept on ice. To quantify leaked Evans blue from BBB, a trichloroacetic acid (TCA; Sigma-Aldrich, St Louis, MO) based dye extravasation assay was applied as described (22). To quantify the dye leakage, supernatants were diluted with 95% ethanol (1:3; vol: vol), and Evans blue fluorescein intensity was measured at 620/680 nm wavelength (BioTek, Synergy HTX) to obtain absorbance values. Absorbance values were measured with Evans blue dye standards and converted to μg/g of the brain.
H&E histological staining
For corneal histological examination, corneas were excised from mice and kept in 10% buffered formalin solution. Tissues underwent routine histologic processing and staining was performed with hematoxylin and eosin as performed previously (23).
Harvesting tissue samples for single-cell analysis and flow cytometry
On day 9 pi, individual corneas and TG were excised, suspended in complete RPMI-1640 media (10% FBS in RPMI-1640) and digested with liberase at 1.5 mg/ml (Roche Diagnostics, IN) for 45 min at 37 °C in a humidified atmosphere of 5% CO2. In a separate experiment, study was terminated at 5 dpi and tissues were processed to obtain single cells. Corneas and TG were disrupted by crushing with a 1 ml syringe plunger on a 40-micron cell strainer and a single-cell suspension was kept in a complete RPMI-1640 medium. All samples were divided or counted for the analysis of both innate and T cell responses.
Flow cytometry analysis (FACS)
To analyze T cell responses, single-cell suspensions were isolated from corneal and TG tissues and divided into half. Half of the suspensions were stimulated in a cocktail of PMA (50 ng), ionomycin (500 ng) and brefeldin A (10 μg/ml) mixture for 4 hr for T cell analysis whereas other half was left unstimulated for the investigation of innate cell responses. All staining applications were performed at 4°C. For the FACS analysis of T cell, cells were incubated with anti-CD16/CD32 (Fc Block) for 15 min at room temperature along with reagent for Live/Dead staining kit. Cell surface staining was done with respective surface fluorochrome-labeled antibodies (Abs) in FACS buffer for 30 min. For intracellular IFN-γ staining, cells were permeabilized with an intracellular staining kit (BD Bioscience, San Jose, CA) for 30 min. Afterward, cells were stained with anti-IFN-γ abs for 30 min. Finally, the cells were washed thrice with FACS buffer and re-suspended in 1% paraformaldehyde. The fluorochrome stained cells were examined to compute the cell with different phenotypes using a FACS LSR II (BD Biosciences, San Jose, CA), and the data were analyzed using FlowJo software (Tree Star, Ashland, OR). All doublet cells were gated out during the gating of cells for flow analysis, followed by gating of the live cell population. Cells were gated from live populations and defined as CD4+, CD8+, CD69+ and IFN-γ+ T cells. Proinflammatory T cells were gated as CD4+IFN-γ; activated T cells were gated either CD4+CD69+IFN-γ+ T cells or CD8+CD69+IFN-γ+ T cells. To study innate cell responses by flow cytometry, cells were blocked and stained in mixture of Live/Dead staining kit and anti-CD16/CD32. Cells were stained with a multi-staining abs cocktail containing anti-CD45, CD11b, LY6G and F4/80. T analyze the neutrophil phenotype cells, CD45+ CD11b+ LY6G+ gating was done and for the analysis of macrophages, CD45+ CD11b+ F4/80+ gating was performed.
Reagents and fluorochrome-conjugated abs for FACS analysis
The live/dead fixable near-IR staining kit was from Thermo Fisher Scientific. Rat anti-CD4 (RM4–5), anti-CD8 (53–6.7), anti-CD69 (H1.2F3), anti-IFN-γ (XMG1.2), anti-CD45 (30-F11), anti-CD11b (M1/70), anti-LY6G (1A8), protein transport inhibitor (brefeldin A) and unconjugated anti-CD16/32 (2.4G2) antibodies were obtained from BD (BD Bioscience, San Jose, CA). A fixation/permeabilization solution kit was also obtained from BD (BD Bioscience, San Jose, CA). Anti-F4/80 (BM8) was obtained from eBioscience (Invitrogen, Carlsbad, CA). Phorbol 12-myristate 13-acetate-PMA and ionomycin were purchased from Sigma-Aldrich (Sigma‐Aldrich, St. Louis, MO).
In-vitro BBB endothelial cell
To study of 2DG effect on in-vitro BBB and mRNA expressions of permeability maintaining proteins primary mouse brain microvascular endothelial (PMBVEC) (Cell Biologics, BALB-5023, Chicago, IL) was purchased and seeded in a well of 24 plate at 1×105/ml with complete mouse endothelial cell medium kit (Cell Biologics, M1168, Chicago, IL). 24 well plates were pre-coated with gelatin-based coating solution (Cell Biologics, 6950, Chicago, IL).
Real-time PCR and mRNA amplification
To quantify mRNA expressions of occludin, zonula occludens-1/tight junction protein-1 (ZO-1/Tjp-1) and claudin-5 from PMBVEC, cells were maintained in 24 well plates and total RNAs were isolated from the wells by using an RNA isolation kit (Qiagen) on daily bases up to day 4 after the seeding the cells. Cells were lysed directly in the wells after the removal of the supernatant. All procedure was followed according to kit manufacturer standard protocol. cDNA was converted from 500 ng of isolated RNA with the help of GoScript reverse transcription system kit (Promega). Taqman FAM-labeled anti-mouse specified primers were obtained and purchased from Thermo Fisher Scientific under the Applied Bio-Systems trademark and qPCR assays were performed by using 7900 Fast real-time PCR System (Applied Bio-Systems). Taqman gene expression assay probes directed to target for Ocln (Mm00500912_m1) and ZO-1/Tjp-1 (Mm00493699_m1), Cldn5 (Mm01169675_s1) gene regions were employed during the qRT-PCR amplification processes and mixed with TaqMan™ Fast Advanced Master Mix (Thermo Fisher Scientific, Waltham, MA). An actin-β (Mm00607939_s1) was included as an internal amplification control. The relative expression levels of different molecules were normalized to actin-β using a cycle threshold calculation. The relative expression between experimental groups or isolated total RNAs was calculated using the 2−ΔΔCT X 1000 formula. Results presented with relative mRNA expressions. The 2DG dose concentrations and effective dose inhibition responses evaluated in the presence of various 2DG concentration in culture medium was as described previously (14, 24).
Statistical analysis
GraphPad Prism 7 software (GraphPad Software, La Jolla, CA) was used for statistical analysis and presentations of graphs in most figures. Kaplan-Meier survival estimation analysis was used to define percent survivability and Log-rank (Mantel-Cox) tests were performed, and significant were estimated. Sidak’s multiple comparisons test was applied with two-way ANOVA to calculate significance importance between 2DG treated and untreated groups in the data analysis of viral load on eye swabs, whereas one-way of variance (ANOVA) with multiple comparison statistical analysis was performed followed by Tukey test in the Evans dye quantification analysis. A two-tailed nonparametric Student t-test analysis was performed to estimate significant differences between 2DG treated and untreated groups. The level of significance for some experiments was revealed as follows; ****p<0.0001, ***p<0.001, **p<0.01 and *p<0.05. Some results indicate p>0.05 and expressed not significant (ns). Standard deviations (SD) of each median were considered for some analysis, while it was considered as standard errors of the means (SEM) during data analysis of FACS results.
Results
Effect of 2DG therapy on outcome of ocular infection with HSV
Initial experiments were done to determine the susceptibility of young adult BALB/c mice to ocular infection with different doses of HSV-1 RE. All animals survived infection at the lowest dose tested (1×103PFU) and showed no clinical signs of encephalitis, although a few animals developed mild ocular lesions. However, with an infection dose of 1×104 PFU, 45% of mice either died or had to be terminated because of advanced signs typical of encephalitis. These included ruffled fur, hunched posture, excitability to touch and eventual lethargy. In all cases of encephalitis, virus could be cultured from brain stem samples. An intermediate dose was chosen for subsequent studies (5×103 PFU), which on average from 3 separate experiments caused encephalitis necessitating termination in around 12.5% of mice. Most of the animals infected at this dose level (including those that developed encephalitis) had ocular lesions (angiogenesis and inflammatory reactions in the corneal stroma), by the time experiments were terminated on day 9 or 10.
To evaluate the effect of inhibiting glycolysis on the outcome of HSV ocular infection, mice were infected with 5×103 HSV RE and one group received twice daily systemic therapy with 250 mg/Kg 2DG from the day of infection. A third group was treated in the same way with 2DG, but these animals were not infected with HSV. Experiments proceeded for 10 days with animals being terminated earlier if showing advanced signs of encephalitis. The combined results of three separate experiment of the same design are shown in Fig. 1A and B. They indicate that uninfected mice treated with 2DG had no detectable clinical consequences. However, the clinical outcome of infected mice was altered following treatment with 2DG. Significantly more infected mice developed encephalitis when treated with 2DG. The experiments revealed that whereas around 12.5% of control animals developed encephalitis (confirmed as virus positive after euthanasia), >85% of 2DG recipients developed encephalitis. Curiously, the time of onset of neurologic signs was usually delayed by 1 day in 2DG treated animals compared to infected control mice (Fig. 1 A), but their disease progressed more rapidly once it became evident (Fig. 1A and B).
FIGURE 1.
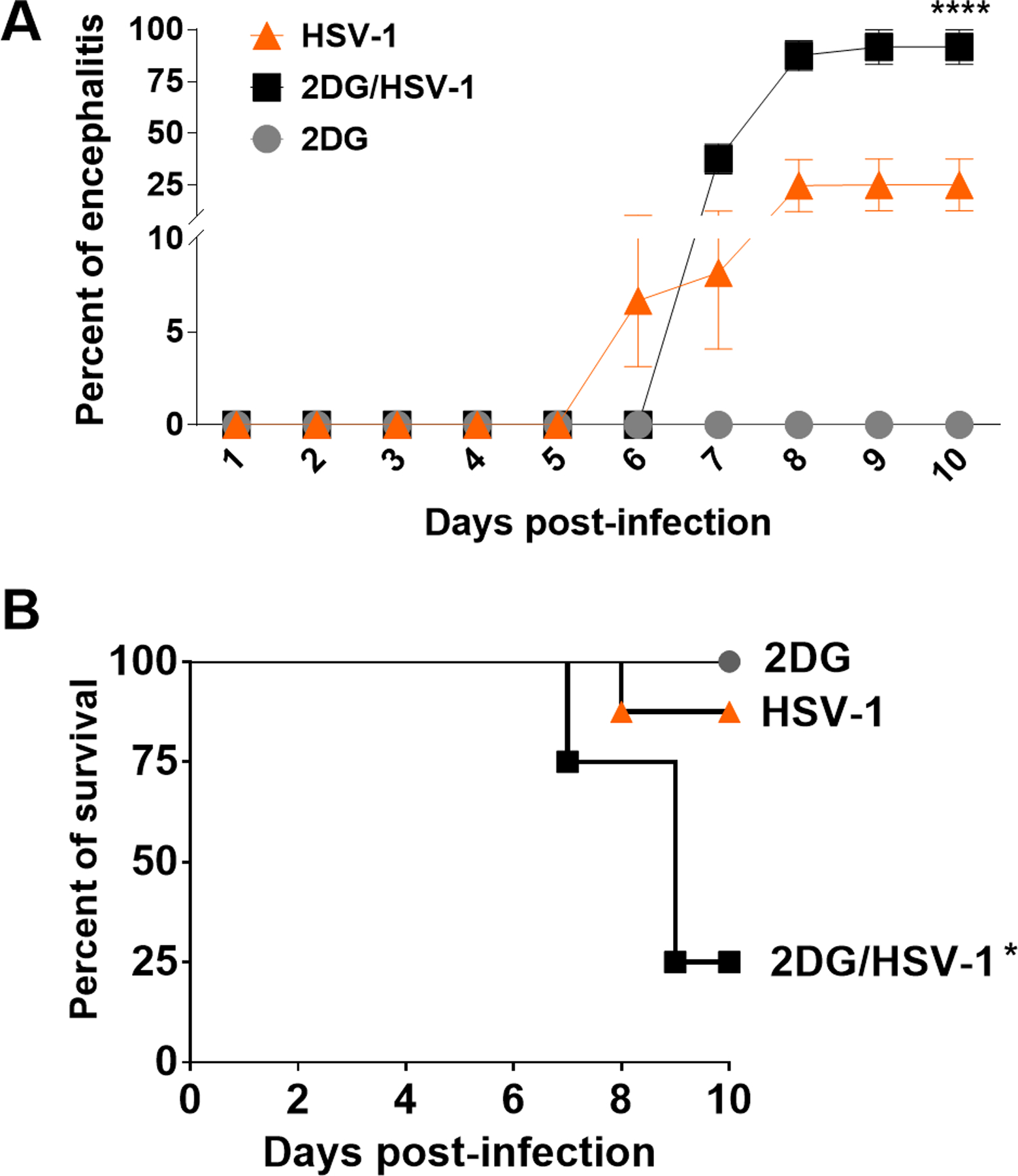
Percent of mice that developed encephalitis (A) and mortality (B). Pooled results of 3 experiments involving 12 mice per group. One group (▲) were HSV infected and were untreated. A second group (■) were HSV infected and received 2DG therapy (250mg/Kg twice a day starting from the day of ocular infection). The third group received 2DG therapy but were uninfected (●). The results show significantly increased encephalitis (A) and mortality (B) in HSV infected mice given 2DG (*p<0.05; ****p<0.0001).
Although 2DG therapy made animals more susceptible to develop encephalitis, these animals had diminished ocular inflammatory reactions. Eye swab samples were taken from animals at different times after infection to quantify levels of virus present. Peak responses were present in 48 hr samples and were on average 1 log higher in animals treated with 2DG. Animals in the drug treated group also stayed positive for a longer period than untreated controls (see Fig. 2). With respect to ocular inflammatory lesions, in one experiment whereas 16 out of 16 eyes in the untreated group showed lesions by day 9 with a mean score of 3.3, in the 2DG treated mice only 11 of 16 eyes developed detectable ocular SK and lesions were of a milder mean score of 1. The eyes of 2DG treated mice had markedly less inflammatory lesions and angiogenesis, especially at the later time periods (Fig. 3 A and B). Examples of average responses of each group, recorded by histopathology at day 9 pi, are shown in Fig. 3 C and D which shows that the inflammatory reaction in the corneal stroma was markedly reduced in 2DG treated animals.
FIGURE 2.
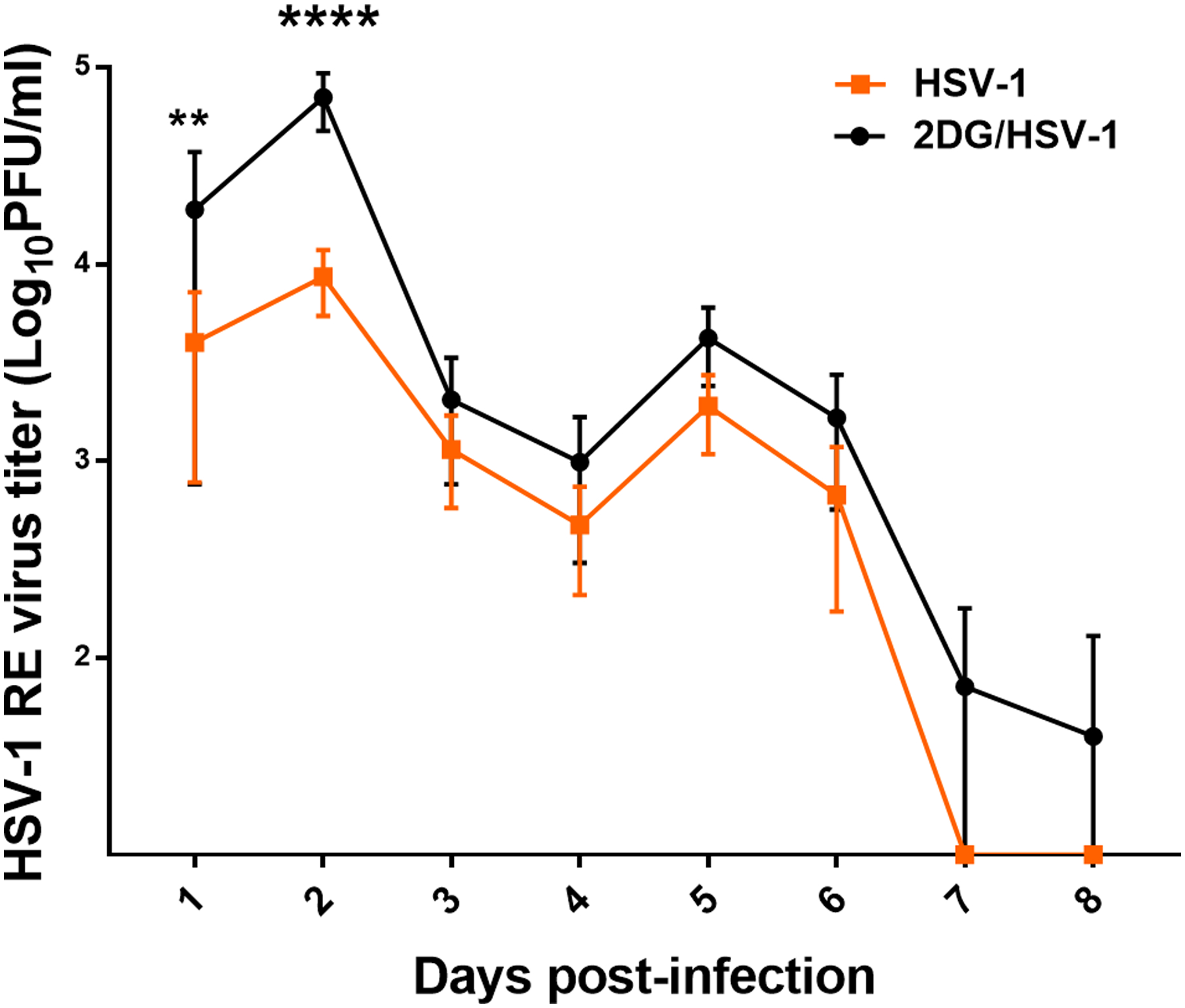
Comparison of ocular virus levels in eye swabs taken daily from HSV infected untreated and 2DG treated mice. Error bars show standard deviations of titers of swabs taken from 8 eyes. Sidak’s multiple comparisons test was applied with two-way ANOVA to calculate significance between 2DG treated and untreated groups (**p<0.01, ****p<0.0001).
FIGURE 3.
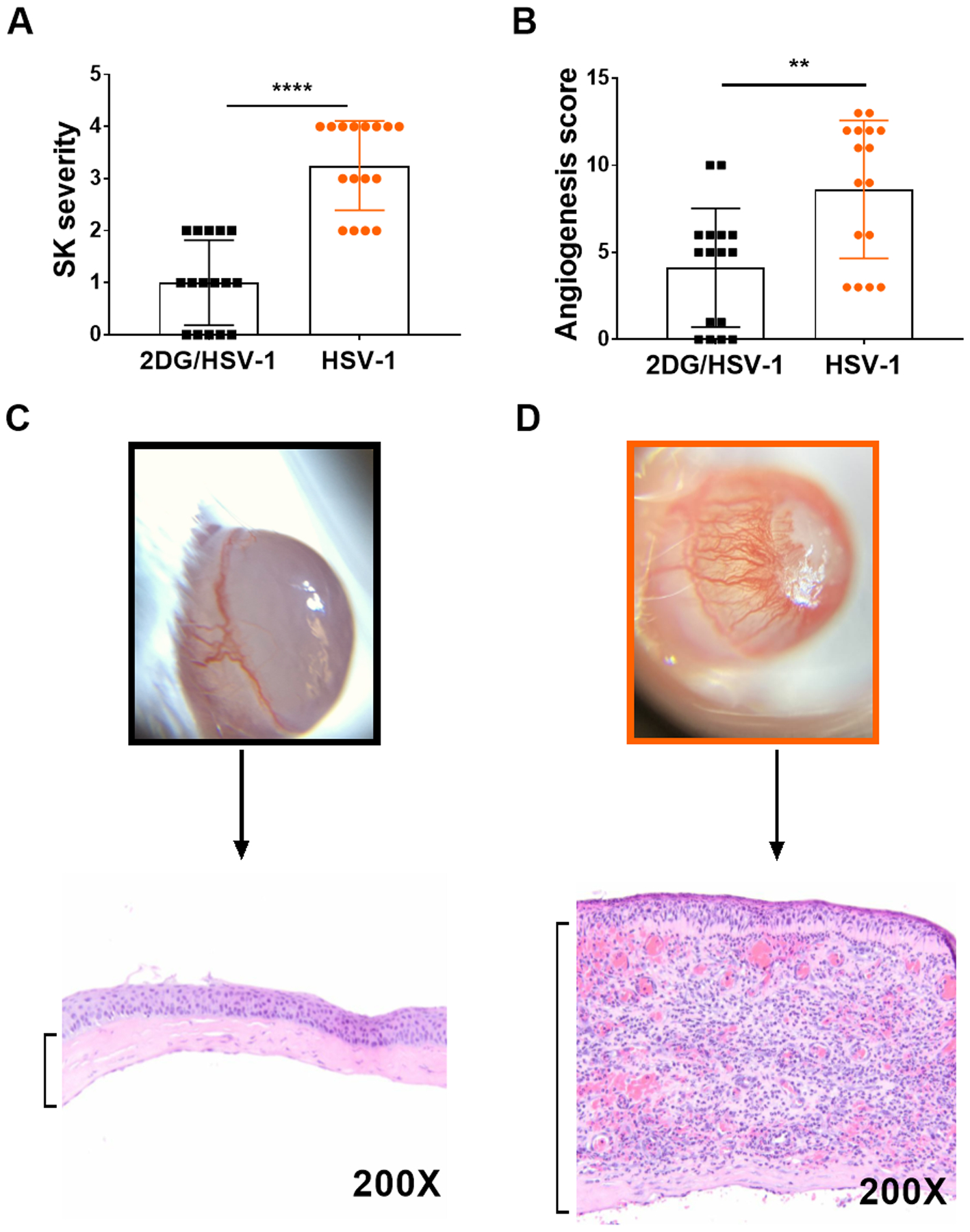
Comparison of ocular lesion severity in HSV infected untreated and 2DG treated mice. Fig 3A shows SK lesion scores of individual eyes and 3B angiogenesis scores measures as described in methods. A two-tailed nonparametric Student t-test analysis was performed to estimate significant differences between 2DG treated and untreated groups (**p<0.05; ****p<0.0001). Figs 3C and 3D show average extent of corneal inflammatory infiltrates in a 2DG (C) and control (D) untreated eye at 9 days pi.
Some corneas were also removed for liberase digestion to enumerate and characterize the cell types present in the stroma of treated and control infected mice. Samples collected on day 9 revealed the presence of CD45+ innate inflammatory cells, but there were 19 fold fewer recovered cells in the 2DG recipients (Fig. 4 A and B). Moreover, when comparing the numbers of macrophages (CD45+CD11b+F4/80+) and neutrophils (CD45+CD11b+Ly6G+) in day 9 samples, in the experiment showed that macrophage numbers were reduced 19 fold in 2DG (Fig. 4 C and D) animals and neutrophil numbers reduced by 29 fold (Fig. 4 E and F). The number of total CD4+ T cells as well as those that were IFN-γ producers (Fig. 5 A–D), the main orchestrators of SK lesions (25, 26), were also compared. Total CD4+ T cells and IFN-γ expressing CD4+ T cells present in samples from 2DG recipients were reduced 3 fold and 9.9 fold respectively compared to untreated infected controls (Fig. 5 A–D).
FIGURE 4.
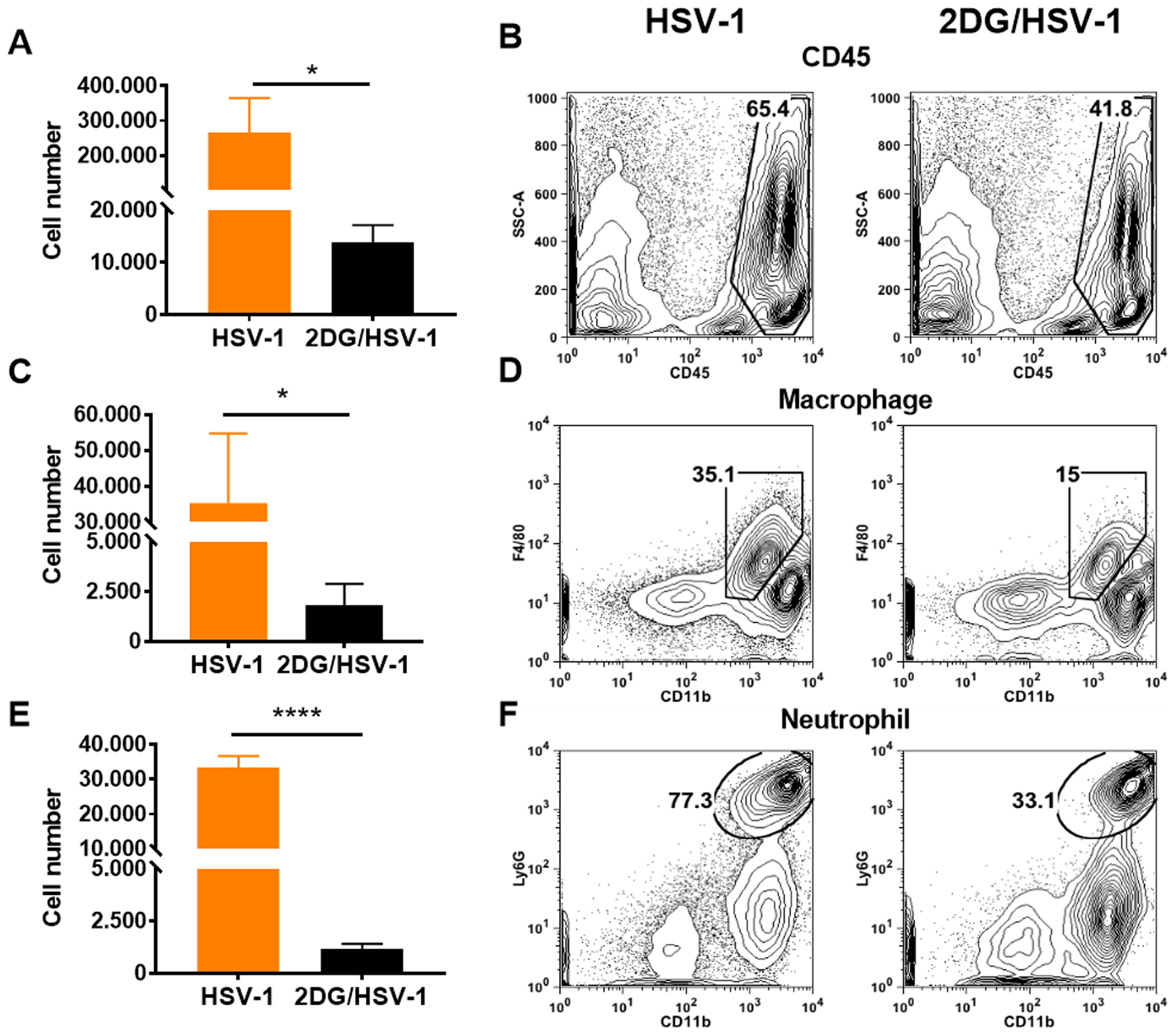
Comparison of inflammatory responses in HSV infected untreated and 2DG treated mice at 9 days pi. Liberase digested single cell samples were prepared from individual eyes and analysed by FACS to record cell numbers and frequencies of total leucocytes (CD45+) in panel A and B. Panels C and D record numbers and frequencies of macrophages ( CD45+CD11b+F4/80+) and panels E and F neutrophils ( CD45+CD11b+Ly6G+). A two-tailed nonparametric Student t-test analysis was performed to estimate significant differences between 2DG treated and untreated groups (*p<0.05; ****p<0.0001).
FIGURE 5.
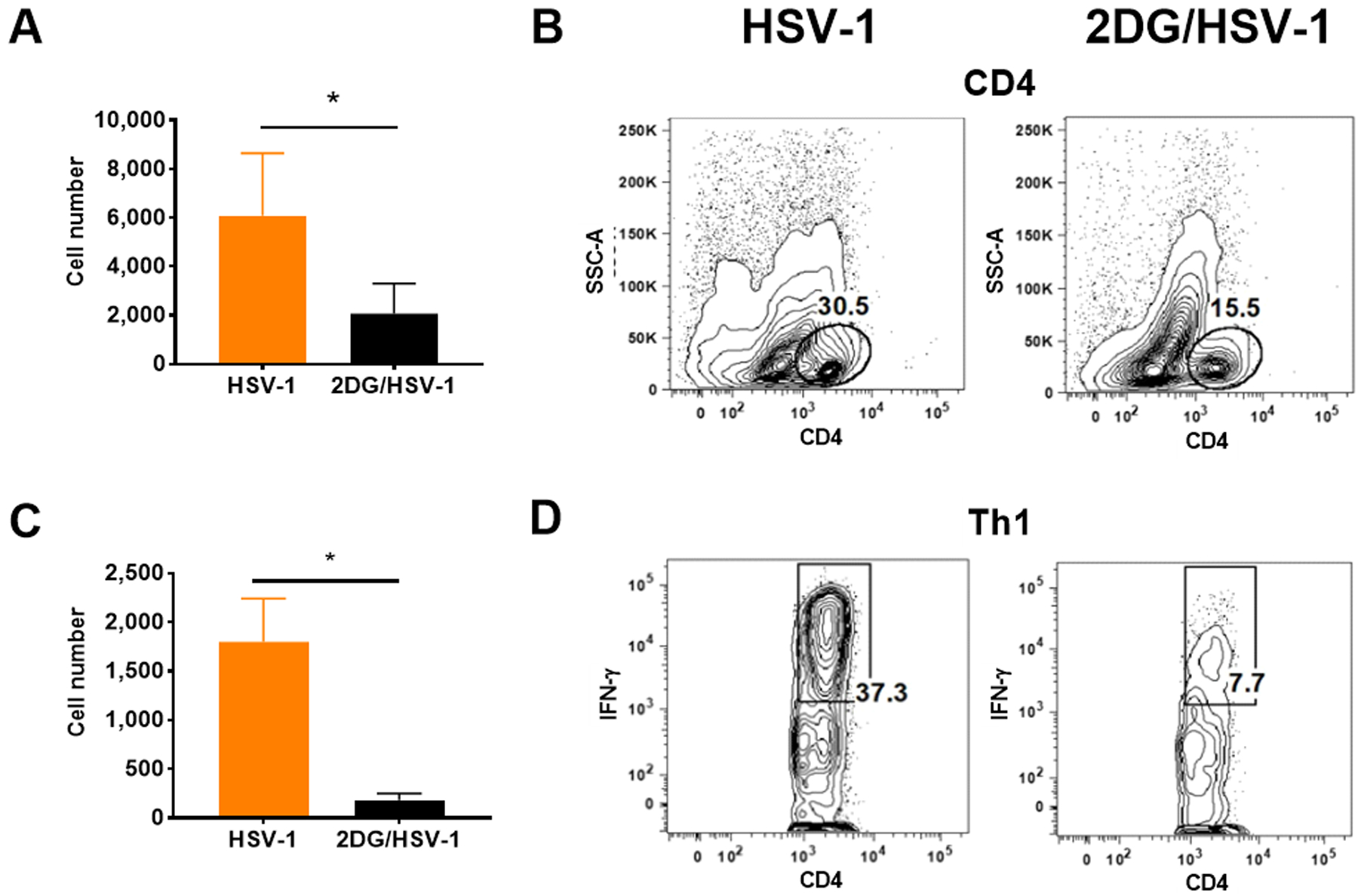
Comparison of T cell responses in HSV infected and untreated and 2DG treated mice. Liberase digested single cell samples were prepared from individual eyes and stimulated with with PMA/Ionomycin for 4 hrs to measure IFN-γ expressing CD4+ proinflammatory Th1 cell numbers and frequencies. Data analysed by FACS to record cell numbers and frequencies of total CD4+ T cells in panel A and B and IFN-γ producing Th1 cells( CD4+IFN-γ+) in panels C and D. A two-tailed nonparametric Student t-test analysis was performed to estimate significant differences between 2DG treated and untreated groups (*p<0.05).
Taken together these results demonstrate that inhibition of glucose metabolism in mice infected with HSV can result in encephalitis, but the approach reduced the inflammatory damaging reaction in the eye to HSV infection.
Responses in the TG in 2DG treated and untreated infected mice
Acute infection experiments
Experiments were also done to compare the TG response following ocular infection of 2DG treated and untreated mice. The TG is the site where HSV rapidly locates after ocular infection and where it establishes latency (27). In these experiments, BALB/c mice were infected with HSV RE (5×103 PFU/eye) and TGs were collected at different times from infected control and 2DG treated animals. The level of infectious virus was quantified after the TGs were minced and freeze thawed 3 times. At the first collection time (day 3), replicating virus was detectable in samples from both groups and titers were similar in magnitude (ns; p>0.05) (Fig. 6). However, in samples collected on day 7 and 9 replicating virus was absent in the TGs from control infected animals, but was still present in readily detectable quantities (>103 PFU) in the TG from 2DG treated animals (Fig. 6). These data indicate that after the initial period all virus in the untreated animals was in the latent form, but virus was still actively replicating throughout the observation period in the TG of 2DG recipients. This might be occurring because the inflammatory cells present in the TG, that are thought to control infection and favor latency maintenance (28–30), were too few in number and perhaps functionally impaired in mice that received the metabolic inhibitor.
FIGURE 6.
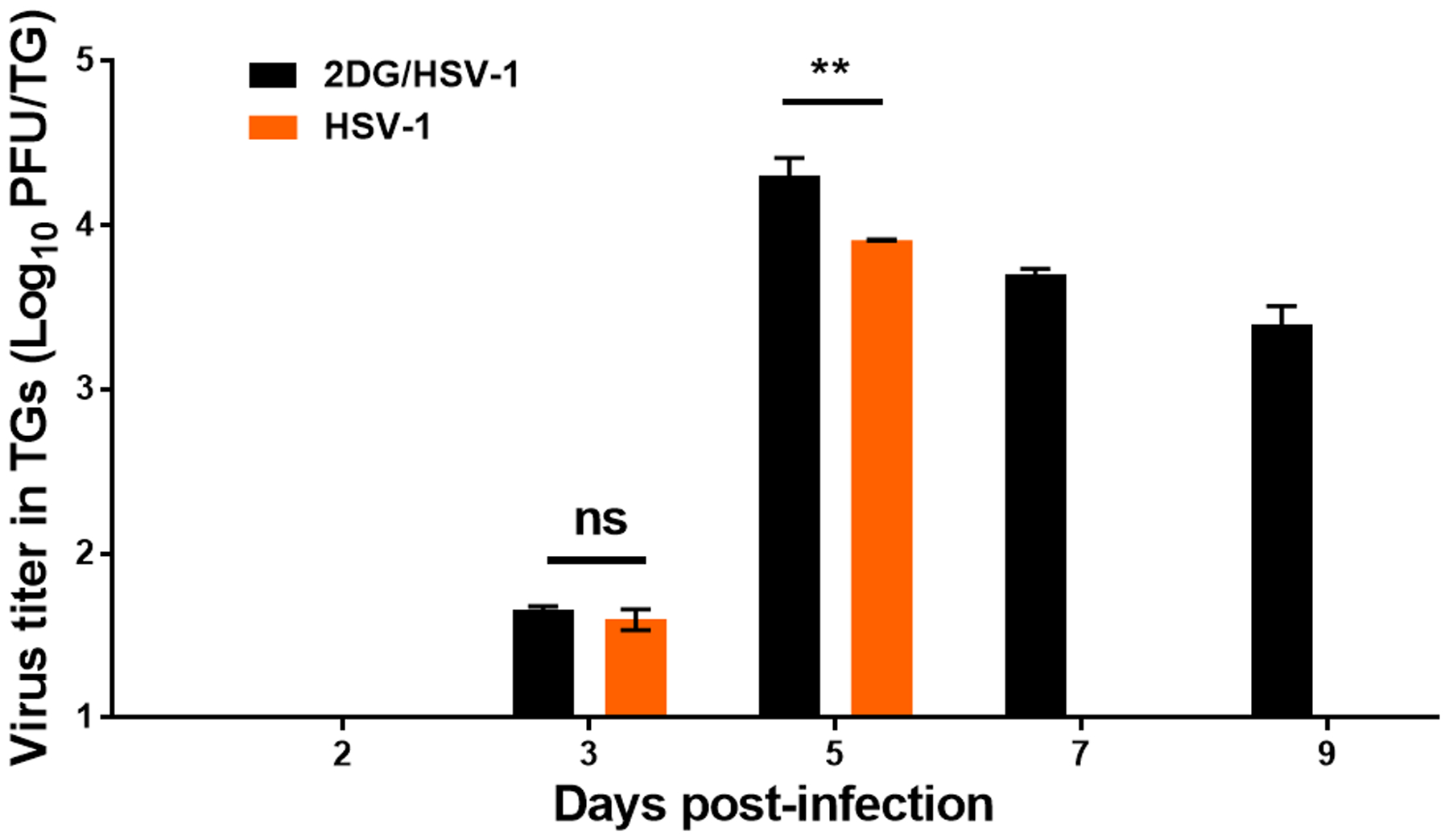
Virus levels present in individual TG from ocularly infected 2DG treated and control untreated BALB/c mice at different time post-infection. At each time point 4 TGs were harvested, freeze-thawed x3 and titrated on Vero cells. A two-way ANOVA was performed to calculate statistical differences between 2DG treated and untreated groups (**p<0.01; ns: not significant).
To gain further insight on the effects of 2DG therapy on inflammatory cells in the TG, experiments were done in which control untreated and 2DG treated BALB/c mice were ocularly infected with HSV RE and single cell suspensions were prepared from individual TGs on day 9 to record the numbers of inflammatory cells present. In samples from treated animals, the number of innate cells (CD45+) compared to untreated controls was reduced, on average, 2 fold (Fig. 7 A and B), which applied to both macrophages (CD45+,CD11b+, F4/80+) (Fig. 7 C and D) and neutrophils (CD45+,CD11b+, Ly6G+) (Fig. 7 E and F). The numbers of CD4 and CD8 T cells were also reduced in 2DG treated samples. The CD4 cells were reduced by, on average, 3 fold (Fig. 8 A and B) and CD8 by 4.2 fold (Fig. 8 B and C). Some TG cultures were stimulated with PMA/ionomycin and the number of CD4+ IFN-γ producing cells (Th1) enumerated. These were, on average, 3 fold reduced in TG from 2DG treated animals (Fig. 8 D and E).
FIGURE 7.
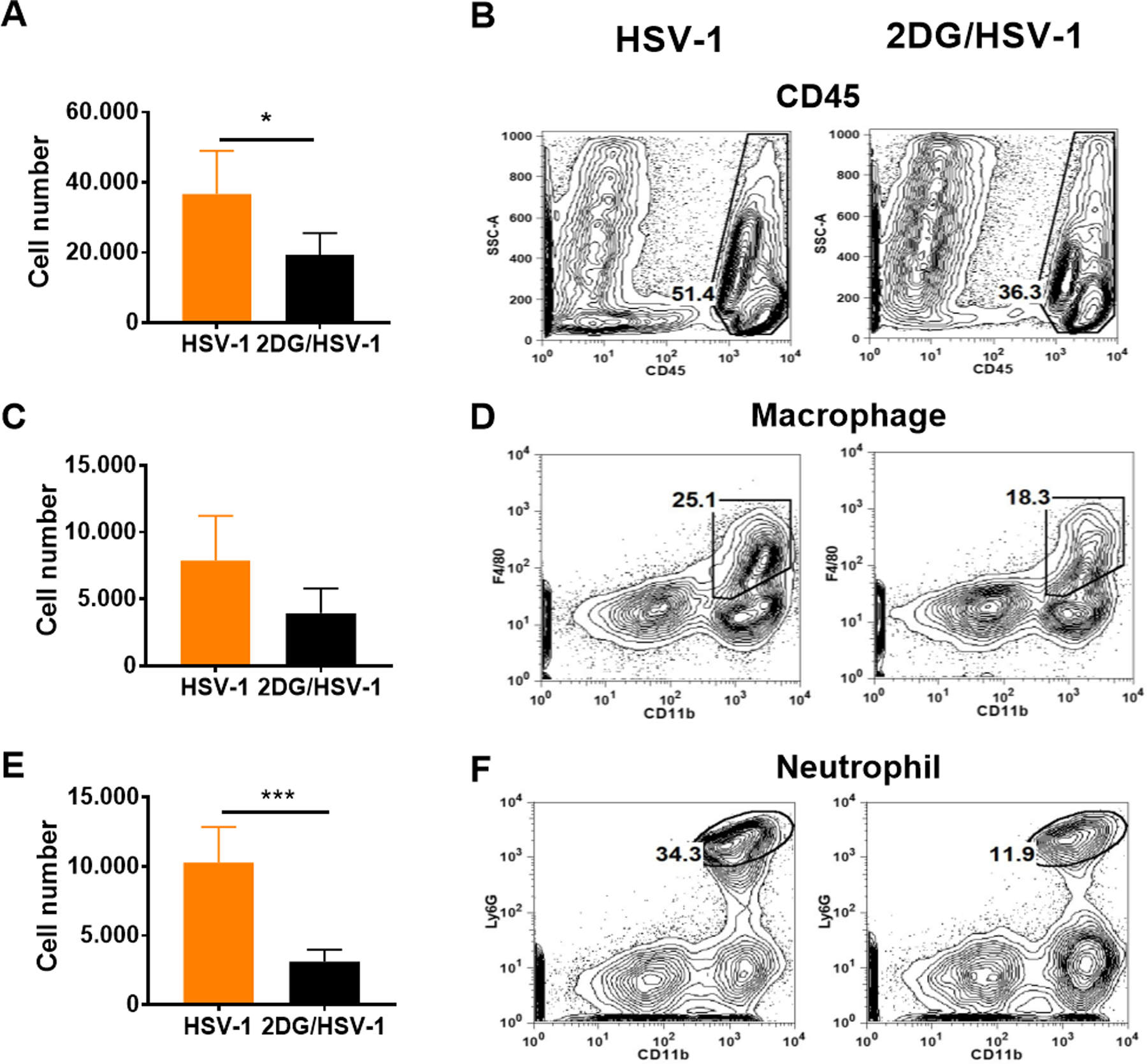
Comparison of inflammatory cell responses in the TGs from 2DG treated and untreated HSV infected mice at day 9 pi. 8 TGs were harvested from each group and enzymatically dissociated to obtain single cell suspension. Single cells were harvested and surface stained with anti-CD45 fluorochrome-labeled antibodies. Histograms represent total CD45 expressing leucocyte cell numbers and frequencies in panel A and B, respectively. Macrophages (CD45+CD11b+F4/80+; plot C and D) and neutrophils (CD45+CD11b+Ly6G+; plots E and F) were also enumerated. FACS plots represent the frequencies of total leukocytes, macrophages and neutrophils from treated and untreated TGs after gating from live cell population. A two-tailed nonparametric Student t-test analysis was performed to estimate significant differences between 2DG treated and untreated groups (*p<0.05; ***p<0.001).
FIGURE 8.
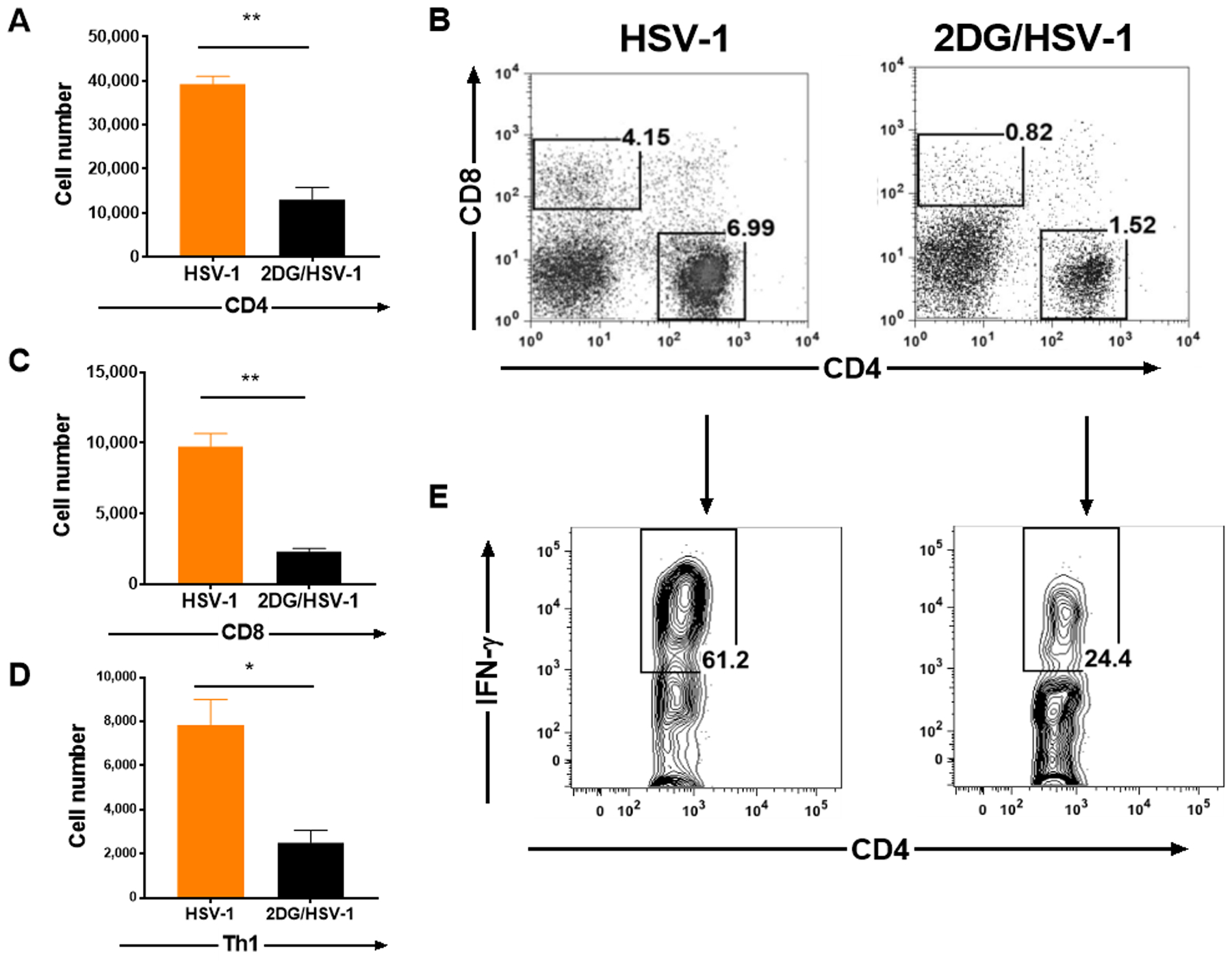
Comparison of T cell responses in the TGs from 2DG treated and untreated HSV infected mice at day 9 pi. Single cells were prepared from 8 enzymatically dissociated TGs taken from treated and untreated Balb/C mice. The number of total CD4 T cells was enumerated (A) and cell frequencies were presented in FACS plots along with CD8 T cells (B). CD8 T cells were also enumerated and presented with histograms (C). Some cultures were stimulated with PMA/Ionomycin for 4 hr to enumerate proinflammatory Th1 cell responses (CD4+IFN-γ+; panel D) and their frequencies (E). A two-tailed nonparametric Student t-test analysis was performed to estimate significant differences between 2DG treated and untreated groups (*p<0.05; **p<0.01). Error bars presenting that standard error of group medians.
We could also show using FACS analysis that the frequency of cells showing activation and the functional marker IFN-γ were diminished in 2DG recipients. Thus, in mice treated with 2DG the frequency of CD69 expressing CD8 T cells was decreased by 12% (Fig. 9 A and B) and the frequency of CD4+CD69+ by 10% (Fig. 9 C and D). CD8+ and CD4+ T cells that expressed IFN-γ were diminished by 16% (Fig. 9 E and F) and 7% respectively (Fig. 9 G and H). Taken together these data support the notion that 2DG therapy inhibits the participation of inflammatory cells in their function of maintaining latency.
FIGURE 9.
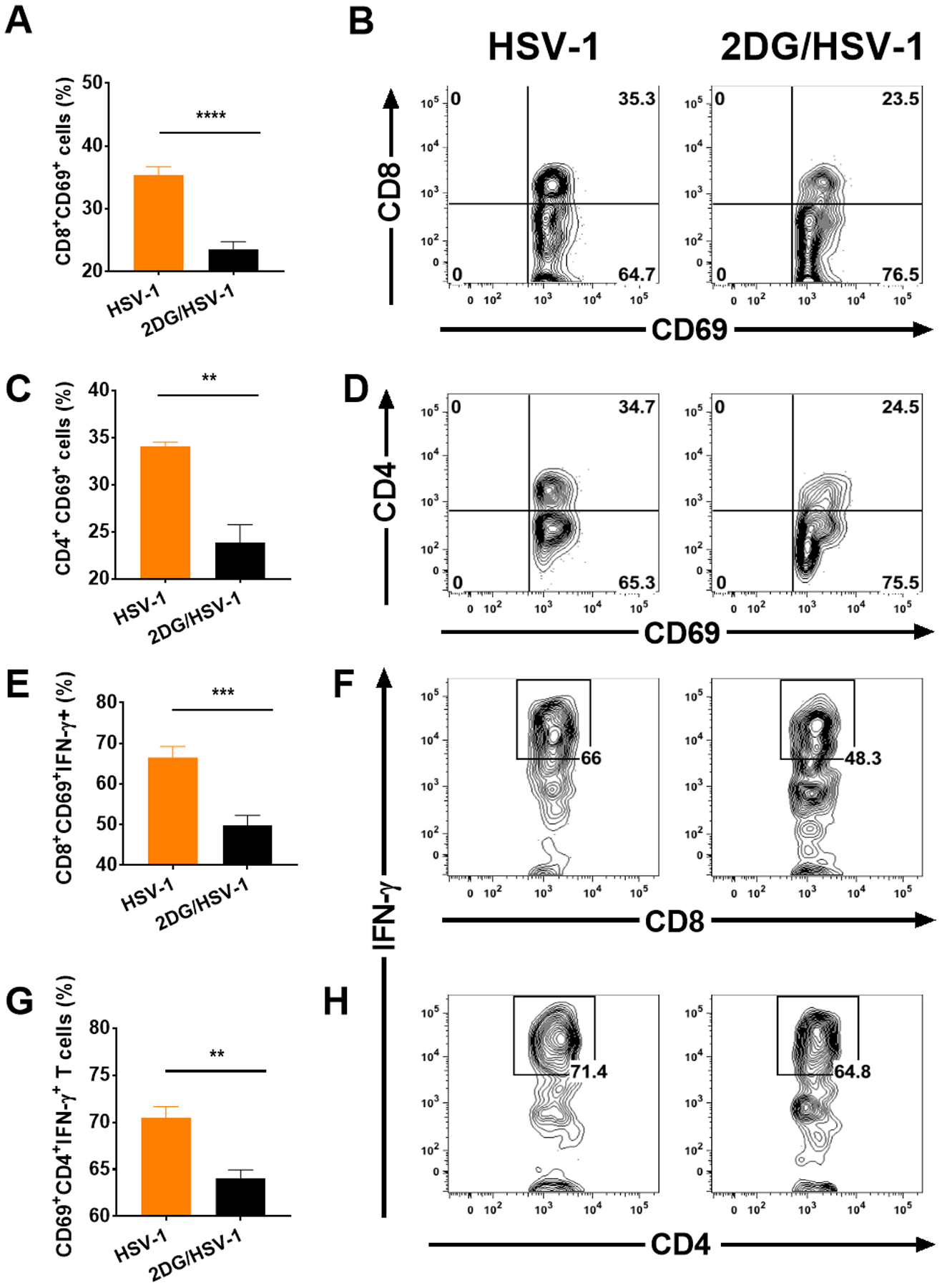
Comparison of TG cells from 2DG treated and untreated HSV infected mice at day 9 pi for T cell activation and IFN-γ producing function. Infiltrating T cells were isolated from cultures after enzymatic dissociation of 8 individual TGs. T cells were stimulated with PMA/Ionomycin and surface stained with CD4, CD8 and CD69 fluorochrome-labeled antibodies. Cells were also reacted with anti- IFN-γ to show intracellular IFN-γ as a measure of function. Panel A and B shows CD69+CD8+ differences between HSV infected untreated and 2DG treated groups. CD69 expressing total CD4 T cells were also measured (C) and the FACS plots present the percent changes within CD4 T cells (D). IFN-γ producers measured (E) and gated from activated CD8 T cells (F). IFN-γ expressing CD4 T cells were measured (G) after gating CD69+CD4+ T cells and presented with FACS plots (H). A Student t-test was used to measure statistical significant differences between treated and untreated groups (**p<0.01; ***p<0.001; ****p<0.0001).
Ex vivo reactivation experiments
Experiments were also done to measure the effects of 2DG on the efficiency by which HSV reactivates from latency. For this purpose, BALB/c mice were ocularly infected with an HSV KOS strain of virus that expressed both the EGFP and RFP markers. This virus was less pathogenic than HSV RE with no animals succumbing to encephalitis, even when given 106 PFU/eye. Animals developed mild ocular lesions that did not cause major irritation or face scratching and the lesions resolved. Consequently, mice could be maintained for 5 weeks without needing to be culled for ill-health. The TGs were collected and pooled at 5 weeks pi and, after enzymatic dissociation, single cell suspensions were prepared and multiple cultures were established in 24 well plates. Half of the cultures received normal media (DMEM, 10% FBS) and the others the same media to which 0.5 mM 2DG was added (14). The cultures were maintained for up to 8 days with the culture fluids removed and replaced daily. The amount of virus present in the removed fluids was quantified using Vero cell monolayers. These experiments revealed that cultures that contained 2DG became virus positive one day earlier on day 3, compared those without 2DG which became positive on day 4 (Fig. 10 A). Comparisons of viral levels at day 4 were significantly higher in 2DG containing cultures, as also could be shown by confocal microscopy of some cultures (Fig. 10 B). Peak levels of virus were present on day 6 and these were similar in magnitude in both drug treated and untreated cultures. Some virus was still detectable in all cultures on day 8 (Fig. 10 A).
FIGURE 10.
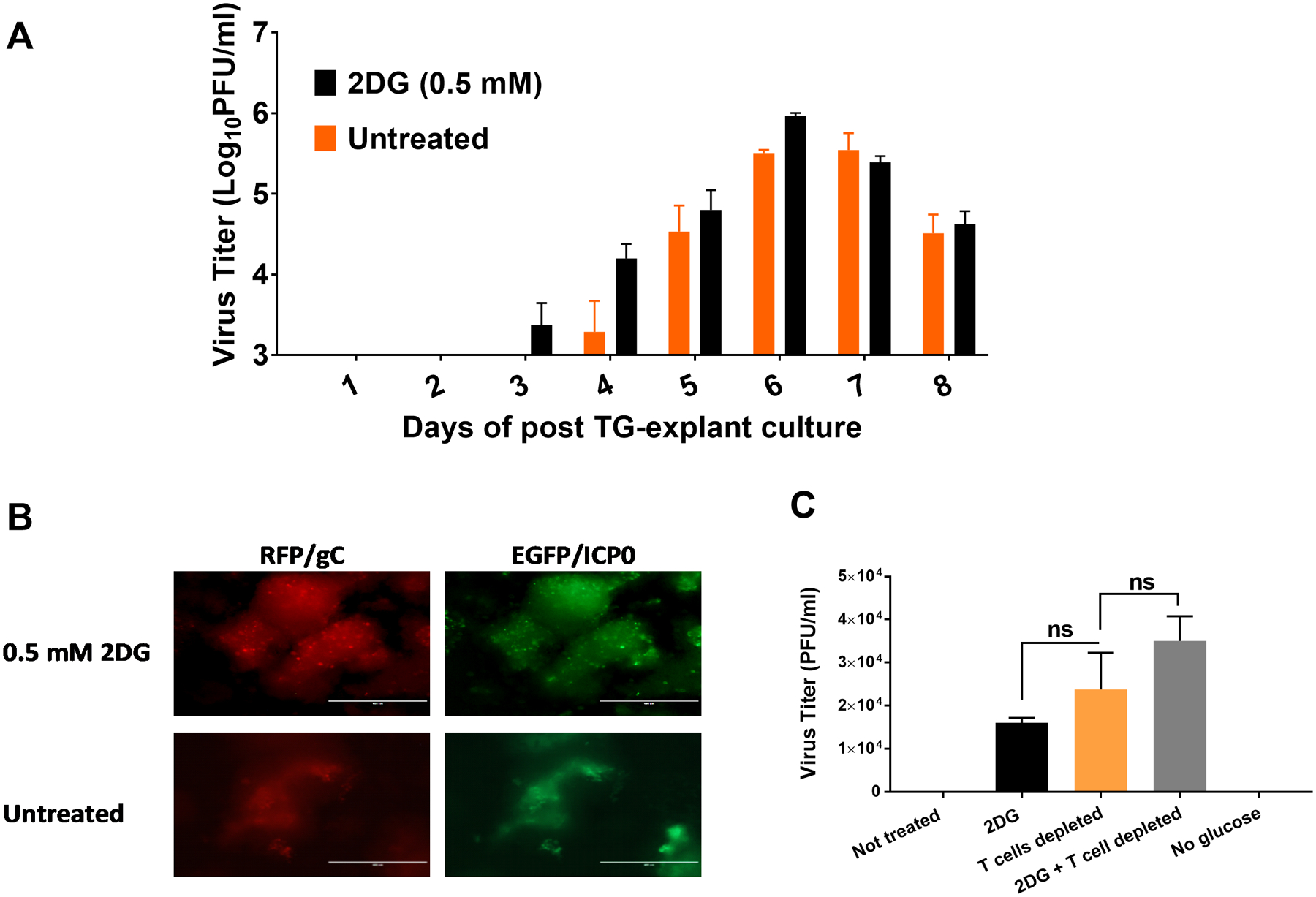
Effect of 2DG on the ability of HSV to reactivate from latently infected TG explanted cultures. BALB/c mice were infected ocularly with RFP/EGFP expressing HSV-1 KOS strain. TGs were removed from mice after the establishment of latency at 34 dpi. Explanted TGs were maintained either in complete DMEM media or media supplemented with 2DG (0.5 mM) for 8 days and the the supernatants were collected daily for viral titration and the media replaced. Panel A shows viral titer levels detected at different time points. Panel B shows viral expression differences in cultures with and without 2DG on day 4. Panel C shows viral reactivation differences on day 3 in TG cultures in normal media, media with 2DG as well as TG where both CD4 and CD8 T cells were depleted and 2DG added. A group of TGs were also cultured in glucose free media during reactivation (RFP/gC: red fluorescein protein under the control of late viral glycoprotein C of HSV-1; EGFP/ICP0: enhanced green fluorescein protein under the control of immediate early gene ICP0 of HSV-1; ns: not significant).
These experiments revealed that the presence of 2DG accelerated the breakdown of latency possible because of inhibitory effects on lymphoid cells in the TG cultures that were helping to maintain latency. Some additional support for this notion was obtained in experiments in which antibodies to both CD4 and CD8 T cells were added to some TG cultures at a concentration previously shown to inhibit T cell activity and the effect on time of viral reactivation evaluated and compared to cultures with 2DG and untreated controls (20). The results showed that by day 3 the cultures that contained the anti-T cell antibodies, as well as those treated with 2DG, showed viral reactivation by day 3 (Fig. 10 C). In contrast, in the control cultures, virus did not reactivate until day 4 (Fig. 10 A). Curiously, reactivation did not occur in the absence of glucose (Fig 10 C). These experiments indicate that 2DG likely acts by affecting the latency control activity of TG T cells. Conceivably, the effect of 2DG also could be directed at the function of latently infected neuronal cells, but experiments comparing the reactivation kinetics of TG cultures from which T cells were removed showed no differences when cells were cultured in normal media or media with added 2DG (data not shown).
Does HSV access the brain via the BBB?
Although the previously described experiments indicate that the facilitated entrance of HSV to the CNS that occurs when glucose metabolism is inhibited could be the consequence of infection control in the TG, permeability alterations in the BBB may also be involved. Experiments were performed to determine if daily 2DG therapy would damage the BBB making it more permeable for passage by a virus into the CNS. Initial experiments were done to compare leakage of the dye Evans blue from the vasculature into the brain in both uninfected controls and uninfected 2DG treated animals, as assessed at 5 and 9 days after dye injection. No significant differences were observed between treated and untreated animals (data not shown). However, in virus infected controls and infected 2DG treated animals, levels of dye in brain samples examined at 9 days were significantly elevated over that observed in uninfected mice (Fig. 11 A and B). In addition, almost 2 fold more dye was seen in the brains of daily 2DG-treated infected mice. We considered the possibility that the increased BBB leakage caused by HSV and 2DG therapy might be the consequence of hematogenous HSV affecting the BBB; however, we failed to detect virus in blood at any stage after infection (data not shown).
FIGURE 11.
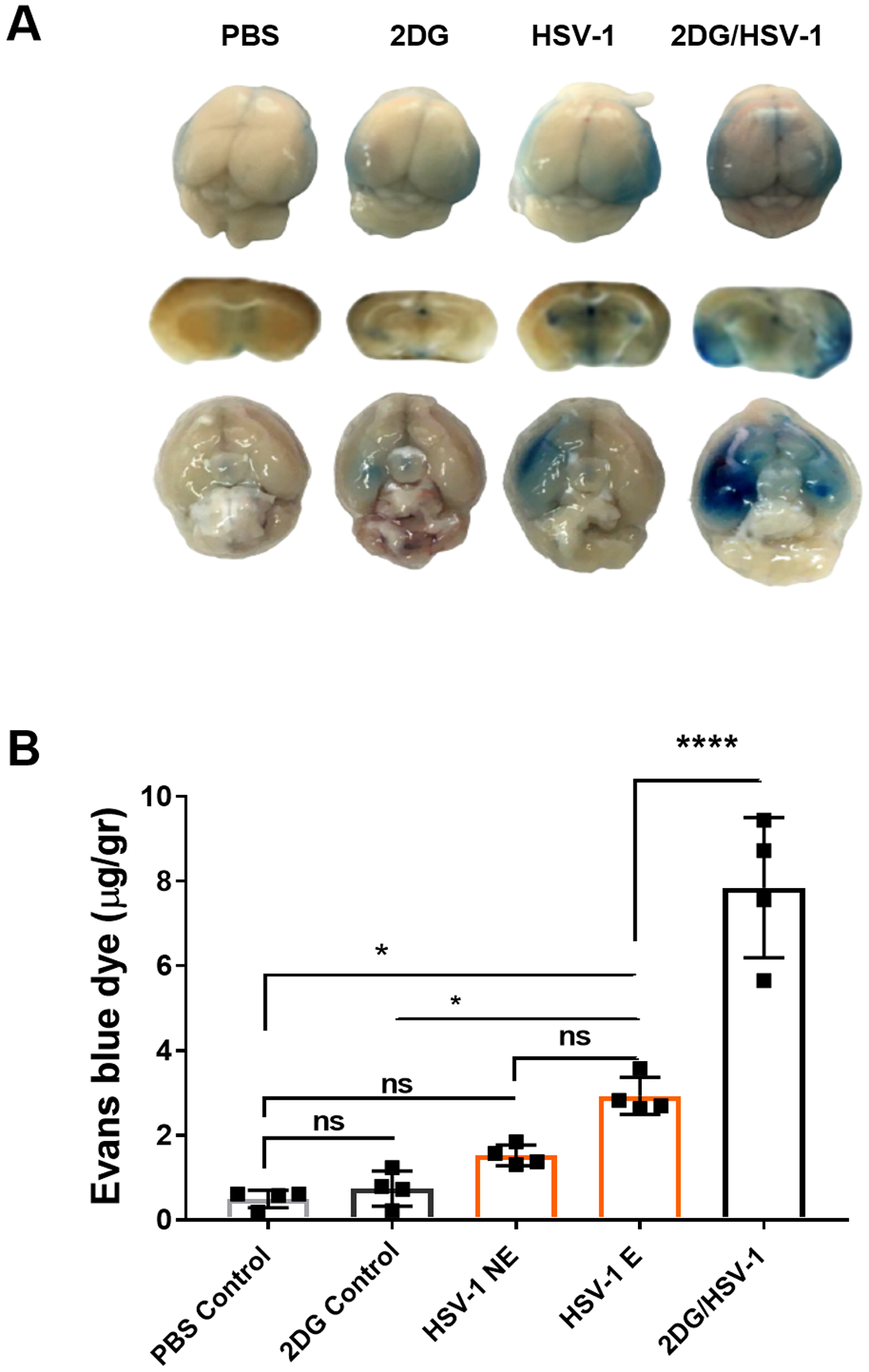
Comparison of leakage of Evans blue into the CNS in control and 2DG treated uninfected, HSV infected untreated and 2DG treated mice. Evans blue was injected IP, 16 hr prior to recording brain images and collecting brain sample to quantify dye level. BALB/c mice received Evans blue dye by IP route a day before (overnight) on the day 8 of ocular eye infection. Brains were harvested after transcardial PBS perfusion. Panel A shows Evans blue dye leakage in mice brains from dorsal, coronal and ventral positions. Dye quantification was performed from brain and homogenate tissues with fluorescence (620 nm excitation/680 nm emission) detection by a plate reader (B). Quantification of dye was calculated per gram of brains harvested. To evaluate statistical differences of dye leakage among the groups, two-way analysis of variance (ANOVA) with multiple comparison statistical analysis was performed, followed by the Tukey test (HSV-1 E: infected encephalitis developed; HSV-1 NE: HSV infected not encephalitic; ns: not significant). *p < 0.05, ****p < 0.0001.
Although our experiments provided no indication that inhibiting glucose metabolism with 2DG caused the BBB of uninfected mice to become more permeable to leakage of Evans blue dye, we also performed experiments in vitro using primary mouse brain microvascular endothelial (PMBVEC) cells to measure the effect of 2DG on the production of molecules known to control the permeability of the BBB. These were the proteins occludin, ZO-1/Tjp-1 and claudin-5. Cultures were terminated at different time points and the levels of mRNA for the 3 proteins as well as β-actin as a control quantified by qPCR. Initial experiments were also performed with the PMBVEC cells to measure any toxic effects of different concentrations of 2DG during a 72 hr period. The test experiments were done using a range of 2DG concentrations (up to 50% toxic level) and the concentrations of mRNA for the 3 permeability proteins and β-actin quantified at different time periods (data not shown). The result in Fig. 12 (A–C) show that the presence of 2DG at non-toxic levels resulted in significant inhibition of permeability protein mRNA levels, with the greatest effect on claudin-5. Curiously, some reports have indicated that claudin-5 can play the most relevant part in controlling the permeability of the BBB (31–33).
FIGURE 12.
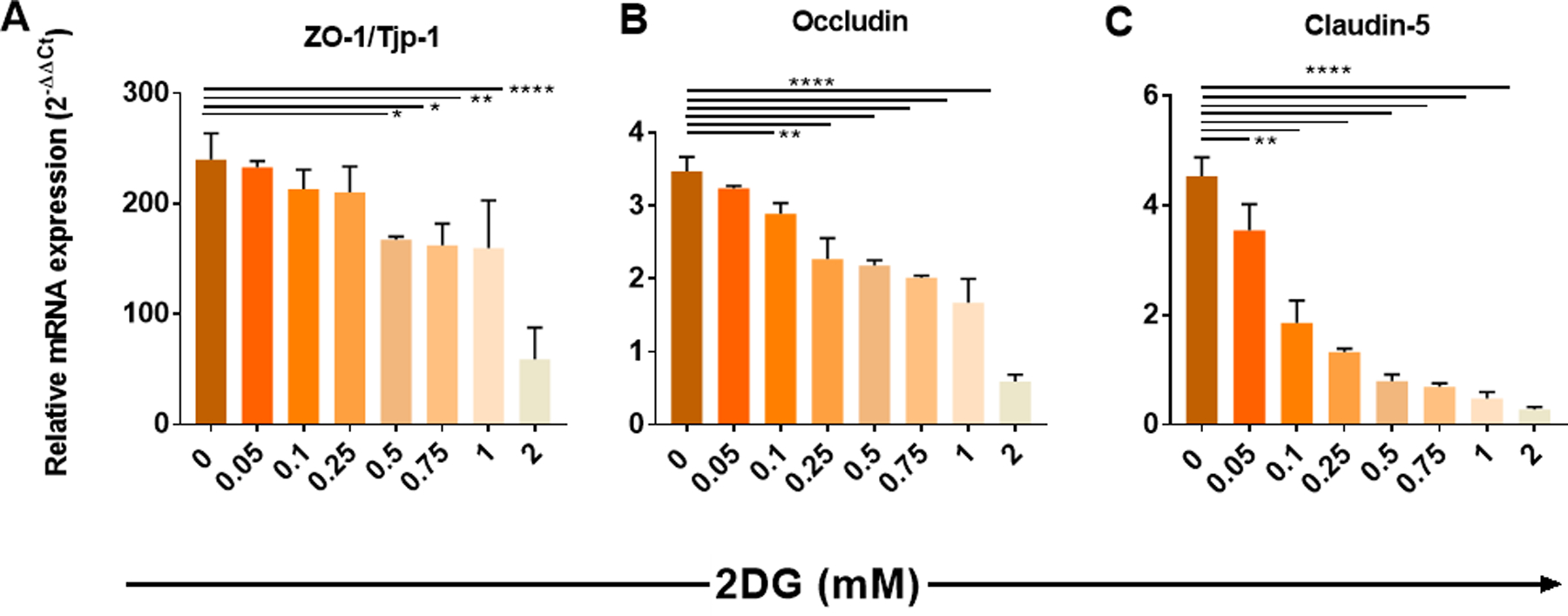
Effect of 2DG on the production levels of mRNA for ZO-1/Tjp-1, Occludin and Claudin-5 from in vitro cultures of PBMVEC cells. Cell cultures were maintained for 4 days in a range of concentrations of 2DG from zero to 2 mM. Cells were harvested at 4 days, and the levels of mRNA present measured with using qPCR and normalized to β-actin mRNA level. ZO-1/Tjp-1 (A), occludin (B) and claudin-5 (C) was measured and results were expressed in relative mRNA expression with the power of 2−ΔΔCt. One-way ANOVA analysis was performed followed by Dunnet’s multiple comparisons test (*p<0.05; **p<0.01;****p<0.0001).
Discussion
Few viruses succeed in accessing and replicating in the brain but when they do the outcome can be devastating and is often lethal. Herpes simplex virus is an occasional cause of encephalitis in adult humans and without rapid antiviral therapy this disease has debilitating consequences (10). Heterologous hosts infected with HSV, such as some primates and laboratory rodent models are more prone to develop herpes encephalitis, as is the homologous human host if immune functions are compromised (8, 34). Thus, as our own previous studies revealed, mice given 2DG, which inhibits their ability to utilize glucose, from the onset of infection were highly prone to develop encephalitis (14). In this report, we further explore this outcome and provide reasons to explain their heightened susceptibility to encephalitis. We showed using a model of BALB/c mice that were infected in the cornea with the RE strain of HSV-1 that inhibiting glucose utilization using 2DG administration resulted in most mice developing encephalitis. In contrast, the infected 2DG treated mice developed far less severe ocular lesions, a response known to be mediated by the host immune response to the infection (25, 26). Indeed, our studies showed markedly diminished inflammatory reactions in the eyes of 2DG treated mice. In comparing the outcome of infection in the TG, the site to which virus spreads, replicates and establishes latency (35, 36), marked differences in viral and cellular events were observed between treated and untreated animals. In control untreated mice replicating virus was present only during early time points, while in 2DG treated mice, replicating virus was present for the entire 9 day observation period. This outcome was likely the result of the 2DG impairing the virus controlling function of inflammatory cells, particularly T cells, in the TG. In fact, comparisons of the inflammatory cell contents in the TG from untreated and 2DG treated animals revealed that 2DG treated animals had significantly decreased numbers of innate cells as well as T cells. Moreover, we showed that T cells in the TG of treated animals were less activated and a lesser fraction expressed the protective IFN-γ producing function compared to untreated controls. Other studies revealed that inhibiting glucose metabolism, in vitro TG cultures from previously infected mice, with established latency, resulted in accelerated breakdown of latency and increased the amount of virus produced by TG especially in the early stages. In addition, the effects of inhibiting T cell activity in TG cultures were similar to 2DG treatment, further indicating that 2DG acted by compromising T cell activity in the TG. Taken together, the results of both in vivo and in vitro investigations demonstrate that 2DG therapy impairs the protective effects of one or more inflammatory cell types in the TG that normally function to control productive infection. This local inhibitory effect could allow more prolonged virus production and facilitate anterograde passage of the virus along the nerve axons to the brain, but this outcome was not formally demonstrated (37). What still needs further study is to determine the mechanisms by which the 2DG therapy mediated its effect and if indeed it was directed only at T cell function or had additional off target activities noted by others such as antiviral effects (38, 39). In our system, the latter mechanism was unlikely since the use of 2DG resulted in higher levels of virus production. We did not ascertain which metabolic pathways were primarily affected by 2DG in the system we studied, but investigations are currently underway to provide this information.
We also evaluated the alternative explanation that by inhibiting glucose metabolism, 2DG made mice more susceptibility to herpes encephalitis as a result of altered function of cells of the BBB making the barrier more permeable to viral passage into the brain. However, we failed to find evidence that the regimen used for 2DG treatment in normal untreated mice caused any significant changes to BBB permeability. Nevertheless, the permeability of the BBB was increased in HSV infected mice and this increase was even greater in HSV infected 2DG recipients. We suspect that an explanation for these findings could be that HSV once present in the brain, from access via the neural route, could be damaging the BBB from the brain side and this possibility needs to be further explored. Accordingly, once HSV has entered the CNS, it is known to infect and change the function of astrocytes which form part of the BBB (40–42). It was also curious to observe that in vitro studies using brain derived epithelial cells revealed that the presence of 2DG in cultures resulted in the diminished production of occludin, ZO-1/Tjp-1 and claudin-5, a group of molecules involved in maintaining the restrictive permeability of the BBB (31). Thus, changes in BBB permeability during HSV infection especially if treated with inhibitors of glucose metabolism was observed, but whether or not viral passage can occur via the BBB route requires further study.
Alphaherpes virus infections in their homologous hosts rarely gain access to the CNS. This is fortunate since severe neuropathology usually results (43). In humans infected with HSV, lesions are usually confined to peripheral sites and the virus travels retrograde in peripheral nerves to the local ganglion where it is maintained in a non-replicating latent form, likely for life (44). When the virus reactivates from latency, a common occurrence, almost invariably it disseminates from the ganglion to the peripheral site, but almost never ascends in axons that enter the brain (45). However, HSV infection of the brain does become a reality under some circumstances that include inadequate control by the immune system. This is most commonly seen when newborns from seronegative mothers are infected with HSV (7). However, occasionally HSE does occur in immunocompetent adults with the syndrome usually happening in persons already latently infected with HSV and whom may have experienced previous HSV lesions confined to the periphery (46). The reason HSV is usually excluded from entry to the CNS is unclear, but this report raises the possibility that concurrent metabolic imbalances at the time of infection or virus reactivation may play a role. Accordingly, metabolic imbalances could compromise the protective function of one or more components of the immune system that normally control infection and preclude the passage of virus in the axons that access the CNS. In support of this possibility we showed in a model system that mice infected with HSV that additionally received 2DG, which inhibits glucose metabolism in the cells mainly responsible for antiviral control, were highly likely to develop lethal HSE (14). Others have also advocated that the nature of the metabolic environment at the peripheral site at the time and place where HSV reactivates from latency in humans could explain why such reactivations are sometimes subclinical, but at other times symptomatic and tissue damaging (47).
Our results, in a mouse model system, provide evidence that viral dissemination to the brain was most likely the consequence of the 2DG therapy causing a reduction in the number and function of inflammatory cells in the TG that under normal circumstances act to suppress viral replication and favor the maintenance of latency. The idea that HSV replication is controlled and that latency is established and maintained in the TG by the action of inflammatory cells, particularly CD8 T cells, was initially advocated by the Hendricks group (20, 48). However, the hypothesis remains controversial and the topic of how HSV latency is established, maintained and terminated excites passionate debate amongst herpesvirologists. Our studies add support for the inflammatory cell control notion since we could show that in animals with inhibited glucose metabolism, the numbers and function of inflammatory cells present in the TGs were significantly diminished in 2DG recipients and viral replication was increased. We assume that the consequence of the lack of control by the 2DG inhibited T cells also could result in virus spilling into the trigeminal root axons with virus gaining access to the brain to cause encephalitis. However, confirmation of this pathogenesis will require further studies to detect and quantify the presence within and follow the passage of virus in nerve axons and entry sites into the brain, a challenging study for which we would need to involve talented collaborators.
A second series of experiments designed to reveal the effects of metabolic manipulation on the pathogenesis of herpes encephalitis was performed using explanted TG from animals with well-established latency. In these experiments, individual TG were collected 5 weeks after ocular infection, a time when replicating virus was no longer present and animals were viable, healthy and any mild eye lesions that initially resulted from the ocular infection had fully resolved. Such studies revealed that the inclusion of 2DG in the media during ex vivo culture accelerated the time when virus reactivated from latency by, on average, 1 day. The effect on 2DG was assumed to be the consequence of inhibiting the function of inflammatory cells, particularly T cells, still present in ganglia at that stage of infection since the effect caused by inhibiting glycolysis could be duplicated if the T cells were inhibited with specific antibodies. The observations that 2DG impairs the expansion and function of proinflammatory cells is well known from in vitro studies that include those done previously in our own laboratory (14).
Many virus infections that do access the brain accomplish the effect by crossing the BBB, which in many instances needs to be made more permeable for the viral passage to occur (2). Consequently, we expected that the HSE observed in the HSV infected mice that received 2DG might be explained by the metabolic inhibitor causing damage to the permeability function of the BBB. However, we failed to find evidence in vivo that treating uninfected mice with 2DG caused their BBB to become more permeable, at least as measured by the intravascular leakage into the brain of Evans blue dye. However, there could have been subtle effects on the function of the BBB that could not be detected, since in an in vitro model system of brain endothelial cells we could show that the presence of 2DG in the culture media under conditions that inhibited glycolysis did significantly reduce the expression of three proteins involved in controlling the permeability function of the BBB. Moreover, the inhibitory effects of 2DG were more apparent against the tight junction protein claudin-5, the protein which some studies have advocated is mostly responsible for maintaining impermeability of the BBB (32, 49). Currently, we are exploring the use of improved in vitro approaches to evaluate the influence of metabolic inhibitors on BBB function, but at the present stage of our investigations we favor the hypothesis that encephalitis in the mouse model of encephalitis is more likely to be the consequence of virus passing to the brain via innervating nerve tracts rather than passage via the BBB.
KEY POINTS.
Therapy with 2DG reduce the expression of ocular eye lesions by HSV.
Inhibiting glucose metabolism results in encephalitis in mice.
HSE occurred in response to breakdown of immune control in the TG.
Acknowledgments
We thank Manikannan Mathayan, Sujata Agarwal and Siddheshvar Bhela for contribution to the study. We thank Paul R. Kinchington for providing a stock of HSV-1 KOS-ICP0-EGFP/gC-RFP. We also would like to thank University of Tennessee, Knoxville, TN ABSL-2 facility staff members and OLAC technician and veterinarians for visits and clinical inspections.
This work was supported by National Institutes of Health Grant R21AI142862 and R01EY005093.
Abbreviations used in this article:
- BBB
blood brain barrier
- dpi
day-post infection
- EGFP
enhanced green fluorescein protein
- gC
glycoprotein C
- HSE
herpes simplex encephalitis
- ICP0
immediate early gene
- mM
millimolar
- pi
post-infection
- PMBVEC
primary mouse brain microvascular endothelial cells
- RFP
red fluorescein protein
- SK
stromal keratitis
- TG
trigeminal ganglion
- ZO-1/Tjp-1
zonula occludens-1/tight junction protein 1
Footnotes
Disclosures
The authors disclose no financial conflicts of interest exist.
References
- 1.Tyler KL. 2018. Acute viral encephalitis. N. Engl. J. Med 379: 557–566. [DOI] [PubMed] [Google Scholar]
- 2.Spindler KR, and Hsu T-H. 2012. Viral disruption of the blood–brain barrier. Trends Microbiol. 20: 282–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sweeney MD, Zhao Z, Montagne A, Nelson AR, and Zlokovic BV. 2019. Blood-brain barrier: From physiology to disease and back. Physiol. Rev 99: 21–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, and Begley DJ. 2010. Structure and function of the blood-brain barrier. Neurobiol. Dis 37: 13–25. [DOI] [PubMed] [Google Scholar]
- 5.Hawkins BT, and Davis TP. 2005. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev 57: 173–185. [DOI] [PubMed] [Google Scholar]
- 6.Bradshaw MJ, and Venkatesan A. 2016. Herpes simplex virus-1 encephalitis in adults: pathophysiology, diagnosis, and management. Neurotherapeutics 13: 493–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.James SH, Sheffield JS, and Kimberlin DW. 2014. Mother-to-child transmission of herpes simplex virus. JPIDS 3Suppl1: S19–S23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Casanova J-L 2015. Human genetic basis of interindividual variability in the course of infection. PNAS 112: E7118–E7127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Whitley RJ, and Gnann JW. 2002. Viral encephalitis: familiar infections and emerging pathogens. The Lancet 359: 507–513. [DOI] [PubMed] [Google Scholar]
- 10.Gnann JW, and Whitley RJ. 2017. Herpes simplex encephalitis: An update. Infect. Dis. Rep 19: 13. [DOI] [PubMed] [Google Scholar]
- 11.Steiner I 2011. Herpes simplex virus encephalitis: new infection or reactivation? Curr. Opin. Neurol 24: 268–274. [DOI] [PubMed] [Google Scholar]
- 12.Dando SJ, Mackay-Sim A, Norton R, Currie BJ, St. John JA, Ekberg JAK, Batzloff M, Ulett GC, and Beacham IR. 2014. Pathogens penetrating the central nervous system: Infection pathways and the cellular and molecular mechanisms of invasion. Clin. Microbiol. Rev 27: 691–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peluso R, Haase A, Stowring L, Edwards M, and Ventura P. 1985. A Trojan Horse mechanism for the spread of visna virus in monocytes. Virology 147: 231–236. [DOI] [PubMed] [Google Scholar]
- 14.Varanasi SK, Donohoe D, Jaggi U, and Rouse BT. 2017. Manipulating glucose metabolism during different stages of viral pathogenesis can have either detrimental or beneficial effects. J. Immunol 199: 1748–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bhela S, Mulik S, Reddy PB, Richardson RL, Gimenez F, Rajasagi NK, Veiga-Parga T, Osmand AP, and Rouse BT. 2014. Critical role of microRNA-155 in herpes simplex encephalitis. J. Immunol 192: 2734–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sumbria D, Berber E, and Rouse BT. 2021. Supplementing the diet with sodium propionate suppresses the severity of viral immuno-inflammatory lesions. J. Virol 95: e02056–02020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramachandran S, Davoli KA, Yee MB, Hendricks RL, and Kinchington PR. 2010. Delaying the expression of herpes simplex virus type 1 glycoprotein B (gB) to a true late gene alters neurovirulence and inhibits the gB-CD8+ T-cell response in the trigeminal ganglion. J. Virol 84: 8811–8820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jaggi U, Varanasi SK, Bhela S, and Rouse BT. 2018. On the role of retinoic acid in virus induced inflammatory response in cornea. Microb. Infec 20: 337–345. [DOI] [PubMed] [Google Scholar]
- 19.Bhela S, Mulik S, Gimenez F, Reddy PBJ, Richardson RL, Varanasi SK, Jaggi U, Xu J, Lu PY, and Rouse BT. 2015. Role of miR-155 in the pathogenesis of herpetic stromal keratitis. Am. J. Pathol 185: 1073–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu T, Khanna KM, Chen X, Fink DJ, and Hendricks RL. 2000. CD8(+) T cells can block herpes simplex virus type 1 (HSV-1) reactivation from latency in sensory neurons. J. Exp. Med 191: 1459–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manaenko A, Chen H, Kammer J, Zhang JH, and Tang J. 2011. Comparison Evans Blue injection routes: Intravenous versus intraperitoneal, for measurement of blood-brain barrier in a mice hemorrhage model. J. Neurosci. Methods 195: 206–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wick MJ, Harral JW, Loomis ZL, and Dempsey EC. 2018. An optimized Evans Blue protocol to assess vascular leak in the mouse. J. Vis. Exp: 57037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Biswas PS, Banerjee K, Zheng M, and Rouse BT. 2004. Counteracting corneal immunoinflammatory lesion with interleukin-1 receptor antagonist protein. J. Leukoc. Biol 76: 868–875. [DOI] [PubMed] [Google Scholar]
- 24.Vichai V, and Kirtikara K. 2006. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc 1: 1112–1116. [DOI] [PubMed] [Google Scholar]
- 25.Doymaz MZ, and Rouse BT. 1992. Herpetic stromal keratitis: an immunopathologic disease mediated by CD4+ T lymphocytes. Investig. Ophthalmol. Vis. Sci 33: 2165–2173. [PubMed] [Google Scholar]
- 26.Thomas J, Gangappa S, Kanangat S, and Rouse BT. 1997. On the essential involvement of neutrophils in the immunopathologic disease: herpetic stromal keratitis. J. Immunol 158: 1383–1391. [PubMed] [Google Scholar]
- 27.Roizman B, and Whitley RJ. 2013. An inquiry into the molecular basis of hsv latency and reactivation. Annu. Rev. Microbiol 67: 355–374. [DOI] [PubMed] [Google Scholar]
- 28.Frank GM, Lepisto AJ, Freeman ML, Sheridan BS, Cherpes TL, and Hendricks RL. 2010. Early CD4+ T cell help prevents partial CD8+ T cell exhaustion and promotes maintenance of herpes simplex virus 1 latency. J. Immunol 184: 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Decman V, Freeman ML, Kinchington PR, and Hendricks RL. 2005. Immune control of HSV-1 latency. Viral Immunol. 18: 466–473. [DOI] [PubMed] [Google Scholar]
- 30.Rajasagi NK, and Rouse BT. 2019. The role of t cells in herpes stromal keratitis. Front. Immunol 10: 512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiao H, Wang Z, Liu Y, Wang P, and Xue Y. 2011. Specific role of tight junction proteins claudin-5, occludin, and zo-1 of the blood–brain barrier in a focal cerebral ischemic insult. J. Mol. Neurosci 44: 130–139. [DOI] [PubMed] [Google Scholar]
- 32.Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, Furuse M, and Tsukita S. 2003. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J. Cell Biol 161: 653–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greene C, Hanley N, and Campbell M. 2019. Claudin-5: Gatekeeper of neurological function. FBCNS 16: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feldman LT, Ellison AR, Voytek CC, Yang L, Krause P, and Margolis TP. 2002. Spontaneous molecular reactivation of herpes simplex virus type 1 latency in mice. PNAS 99: 978–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stevens JG, Nesburn AB, and Cook ML. 1972. Latent herpes simplex virus from trigeminal ganglia of rabbits with recurrent eye infection. Nat. New Biol 235: 216–217. [DOI] [PubMed] [Google Scholar]
- 36.Knotts FB, Cook ML, and Stevens JG 1973. Latent herpes simplex virus in the central nervous system of rabbits and mice. J. Exp. Med 138: 740–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wojaczynski GJ, Engel EA, Steren KE, Enquist LW, and Patrick Card J. 2015. The neuroinvasive profiles of H129 (herpes simplex virus type 1) recombinants with putative anterograde-only transneuronal spread properties. Brain Struct. Funct 220: 1395–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang F, Zhang S, Vuckovic I, Jeon R, Lerman A, Folmes CD, Dzeja PP, and Herrmann J. 2018. Glycolytic stimulation is not a requirement for M2 macrophage differentiation. Cell Metab. 28: 463–475.e464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Courtney RJ, Steiner SM, and Benyesh-Melnick M. 1973. Effects of 2-deoxy-D-glucose on herpes simplex virus replication. Virology 52: 447–455. [DOI] [PubMed] [Google Scholar]
- 40.Michael BD, Bricio-Moreno L, Sorensen EW, Miyabe Y, Lian J, Solomon T, Kurt-Jones EA, and Luster AD. 2020. Astrocyte- and neuron-derived CXCL1 drives neutrophil transmigration and blood-brain barrier permeability in viral encephalitis. Cell Rep. 32: 108150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCarthy M, Norenberg MD, Norenberg LO, and Dix RD. 1990. Herpes simplex virus type 1 infection of rat astrocytes in primary culture: effects of dibutyryl cyclic AMP. J. Neuropathol. Exp. Neurol 49: 3–20. [DOI] [PubMed] [Google Scholar]
- 42.Hensel N, Raker V, Förthmann B, Detering NT, Kubinski S, Buch A, Katzilieris-Petras G, Spanier J, Gudi V, Wagenknecht S, Kopfnagel V, Werfel TA, Stangel M, Beineke A, Kalinke U, Paludan SR, Sodeik B, and Claus P. 2019. HSV-1 triggers paracrine fibroblast growth factor response from cortical brain cells via immediate-early protein ICP0. J. Neuroinflammation 16: 248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Banerjee K, and Rouse BT. 2007. Immunopathological aspects of HSV infection. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, and Yamanishi K, eds. Cambridge University Press, Cambridge. [PubMed] [Google Scholar]
- 44.Marshall KR, Lachmann RH, Efstathiou S, Rinaldi A, and Preston CM. 2000. Long-term transgene expression in mice infected with a herpes simplex virus type 1 mutant severely impaired for immediate-early gene expression. J. Virol 74: 956–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koyuncu OO, Hogue IB, and Enquist LW. 2013. Virus infections in the nervous system. Cell Host Microbe 13: 379–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tyler KL 2004. Herpes simplex virus infections of the central nervous system: Encephalitis and meningitis, including Mollaret’s. Herpes-Cambridge 11: 57A–64A. [PubMed] [Google Scholar]
- 47.Stevens JG, Haarr L, Porter DD, Cook ML, and Wagner EK. 1988. Prominence of the herpes simplex virus latency-associated transcript in trigeminal ganglia from seropositive humans. J. Infect. Dis 158: 117–123. [DOI] [PubMed] [Google Scholar]
- 48.Liu T, Tang Q, and Hendricks RL. 1996. Inflammatory infiltration of the trigeminal ganglion after herpes simplex virus type 1 corneal infection. J. Virol 70: 264–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Itallie CMV, and Anderson JM. 2006. Claudins and epithelial paracellular transport. Annu. Rev. Physiol 68: 403–429. [DOI] [PubMed] [Google Scholar]