

Summary
Patient-derived organoids (PDOs) recapitulate the cellular heterogeneity of the original colorectal tumor tissue. Here, we describe a protocol to generate genetically modified PDOs to investigate cancer stem cells. This protocol uses the CRISPR-Cas9 system to knock-in the IRES-EGFP-P2A-iCaspase9 cassette into the 3′ UTR of the potential cancer stem cell marker gene, which allows us to investigate their potential for self-replication and pluripotency. We describe the procedure for generating mutant PDOs and their application for stem cell research.
For complete details on the generation and use of this protocol, please refer to Okamoto et al. Okamoto et al. (2021).
Subject areas: Cancer, CRISPR, Stem cells, Organoids
Graphical abstract
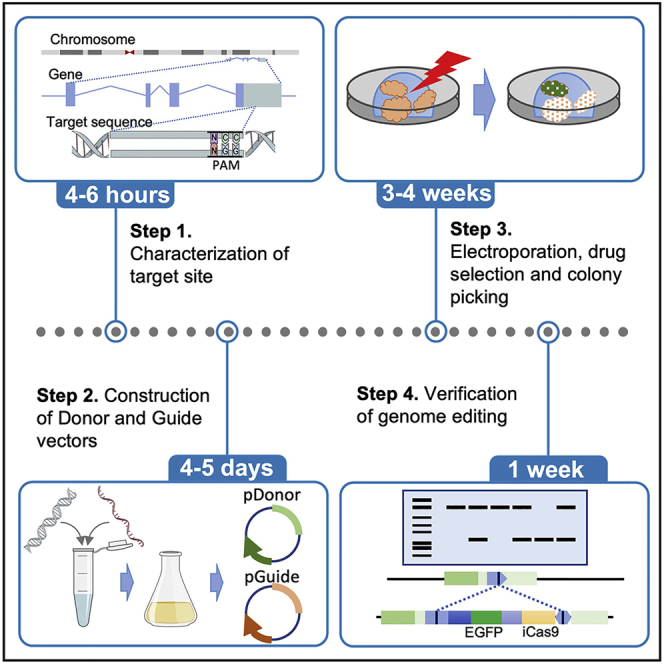
Highlights
-
•
A detailed protocol for efficient CRISPR-Cas9 mediated knock-in in PDOs
-
•
Generation of IRES-EGFP-P2A-iCaspase9 cassette for simple preparation of donor vector
-
•
Visualization of cancer stem cells in PDOs using time-lapse imaging microscopy
-
•
Specific elimination of cancer stem cells by adding AP20187
Patient-derived organoids (PDOs) recapitulate the cellular heterogeneity of the original colorectal tumor tissue. Here, we describe a protocol to generate genetically modified PDOs to investigate cancer stem cells. This protocol uses the CRISPR-Cas9 system to knock-in the IRES-EGFP-P2A-iCaspase9 cassette into the 3′ UTR of the potential cancer stem cell marker gene, which allows us to investigate their potential for self-replication and pluripotency. We describe the procedure for generating mutant PDOs and their application for stem cell research.
Before you begin
The protocol below describes the general procedure to culture PDOs. PDOs were established according to procedures, which were approved by the Research Ethics Board at the JFCR Cancer Institute (Tokyo, Japan). The procedure can be used to culture PDOs derived from colorectal cancer (CRC) patients of various ages and gender.
Preparation of Noggin conditioned medium
Note: We generated Noggin-expressing L1 cells to prepare conditioned medium. For details on the generation of Noggin-expressing L1 cells, please refer to Sakahara et al. (Sakahara et al., 2019). The Noggin conditioned medium can be replaced by commercially available recombinant protein (Preprotec, Cat# 120-10C).
-
1.
Cryopreserved Noggin-expressing L1 cells are thawed and cultured in a 100-mm dish in 10% fetal bovine serum (FBS)/DMEM. G418 is added to the medium starting from three days after thawing at a concentration of 800 μg/mL.
-
2.
The cells are trypsinized withTrypLE enzyme and are split at a ratio of 1:10 into 100-mm dishes in 10% FBS/DMEM. The medium at this and following steps does not contain G418. Typically, we use 100 plates to prepare the conditioned medium.
-
3.
Cells are cultured until reaching confluence.
-
4.
The medium is removed, and 10 mL of Advanced DMEM/F12 Supplemented with 2 mM GlutaMAX, 10 mM HEPES, 100 μg/mL each of Pen/Strep (basal medium) is added to each 100-mm dish.
-
5.
The conditioned medium is collected after incubation for 4 days, then spun at R.T. at 500 × g for 5 min. The supernatant is saved as the 1st conditioned medium at −20°C.
-
6.
Ten milliliters of basal medium are added to each 100-mm dish.
-
7.
The conditioned medium is collected as described in step 5 and stored as the 2nd conditioned medium.
Note: The 1st and 2nd conditioned media are reserved separately until their Noggin activities are determined. To avoid repeated freeze/thaw cycles, 1ml of each media is set aside and is used for the luciferase assay as described below. The remainder is dispensed into 50ml centrifuge tubes. All conditioned media are stored at −20°C.
-
8.
The activity of Noggin is checked by a dual-luciferase reporter assay using the BMP promoter.
-
9.
293T cells are plated at 1×10ˆ4 cells/well in collagen-coated 96-well plates and incubated at 37°C overnight.
Note: We use collagen-coated dish to prevent 293T cells from detaching the plate during the transfection of reporter plasmids。
-
10.
DNA diluent is prepared by mixing the pGL3-BRE plasmid, pRL-tk plasmid and Opti-MEM. Lipofectamine diluent is prepared by mixing Lipofectamine 2000 in Opti-MEM and incubated at R.T. for 5 min. DNA- and lipofectamine-diluent are mixed and incubated at R.T. for 15 min (Table 1).
-
11.
Next, 10 mL of 10% FBS/DMEM is added, and 150 μL is dispensed to each well of 293T-plated 96-well dishes, and incubated at 37°C overnight.
-
12.
Test conditioned medium is prepared by serial dilutions ranging from 2- to 16-fold containing 2 and 5 ng of BMP4 solution. Two different concentrations of BMP4 is used to precisely determine the Noggin activity of the conditioned media.
-
13.
The medium is changed to test the conditioned medium, and the cells are incubated at 37°C for 24 h.
-
14.
The luciferase activities are measured using the Dual-Luciferase Reporter Assay System according to the manufacturer’s instructions. Typically, x8 and x4 dilutions of conditioned medium release the inhibition of BRE promoter activity of BMP4 at 2 and 5 ng/mL, respectively (Figure 1).
-
15.
The Noggin conditioned medium can be stored at −20°C for 6 months. Repeated freezing and thawing should be avoided.
Note: The activity of 1st and 2nd conditioned media are measured separately. When both medium show sufficient activities, they are combined and the mixture is used for further study. If either medium does not have sufficient activity, only the active medium is used.
Table 1.
Preparation of transfection reagents for dual-luciferase assay
| Reagent | Final concentration | Amount (per 100 wells) | |
|---|---|---|---|
| DNA-diluent | pGL3-BRE | 10 μM | 5 μg |
| pRL-tk | 10 μM | 0.5 μg | |
| Opti-MEM | N/A | 2500 μL | |
| lipofectamine-diluent | Lipofectamine 2000 | N/A | 50 μL |
| Opti-MEM | N/A | 2500 μL |
Figure 1.

Evaluation of Noggin-conditioned medium
The value of pGL3-BRE (Firefly) was normalized to that of pRL-tk (Renilla). The activity of Noggin-conditioned medium was determined from the inhibition of BMP4-induced transcriptional activity as measured by pGL3-BRE. Cells were treated with 2 or 5 ng/mL of recombinant BMP4 and indicated dilution of Noggin conditioned medium, and data are shown as relative values compared to those obtained by twofold dilution of Noggin-conditioned medium. Data are represented as mean ± SEM.
PDOs culture
Note: This protocol is set up for a set of PDOs established from stage IV CRC, which can be cultured in ENR medium. The medium most suitable for each PDOs should be used for efficient genome editing and for the evaluation of stemness.
-
16.
The cryopreserved PDOs are placed in 10 mL of basal medium in a 15 mL conical tube.
-
17.
PDOs are spun at R.T. for 500 ×g for 5 min. The supernatant is removed and PDOs pellets are put on ice.
-
18.
PDOs are resuspended in Matrigel, and 25 μL is dispensed into each well of a prewarmed 48-well plate. Typically, 10–20 PDOs are resuspended in 25 μL of Matrigel.
-
19.
The plate is placed in a 37°C incubator for 10 min to solidify the Matrigel, and then 250 μL of ENR medium is added.
-
20.
The medium is changed every 3 days.
Note: Generally, PDOs recover their original morphology at 3–5 days after thawing. Healthy PDOs subsequently increase their sizes.
-
21.
To expand the culture, the PDOs are collected in 10 mL of basal medium in a 15 mL conical tube. Typically, PDOs are expanded every 7–10 days.
-
22.
PDOs are collected, plated and incubated as described in steps 17–19.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Stable3 | Thermo Fisher Scientific | Cat# C737303 |
| DH5alpha | Thermo Fisher Scientific | Cat# 18265017 |
| Chemicals, peptides, and recombinant proteins | ||
| Matrigel | Corning | Cat# 356231 |
| Advanced DMEM/F-12 | Thermo Fisher Scientific | Cat# 12634-028 |
| GlutaMAX | Thermo Fisher Scientific | Cat# 35050-061 |
| HEPES | Sigma-Aldrich | Cat# H3537 |
| Penicillin/Streptomycin | Thermo Fisher Scientific | Cat# 15140-122 |
| Primocin | InvivoGen | Cat# ant-pm-2 |
| B27 Supplement | Thermo Fisher Scientific | Cat# A1895601 |
| N-Acetyl-L-cysteine | Sigma-Aldrich | Cat# A9165 |
| Gastrin | Sigma-Aldrich | Cat# G9145 |
| Recombinant Mouse EGF | Thermo Fisher Scientific | Cat# PMG8043 |
| Recombinant Mouse R-Spondin1 | R&D systems | Cat# 3474-RS |
| Noggin CM | In house production | N/A |
| TrypLE Express Enzyme | Thermo Fisher Scientific | Cat# 12604-013 |
| Y-27632 | Sigma-Aldrich | Cat# Y0503 |
| Opti-MEM | Thermo Fisher Scientific | Cat# 31985-062 |
| Puromycin | Sigma-Aldrich | Cat# P9620 |
| Neomycin (G418) | Sigma-Aldrich | Cat# N1876 |
| Cell Recovery solution | Corning | Cat# 354243 |
| Recovery Cell Culture Freezing Medium | Thermo Fisher Scientific | Cat# 12648010 |
| PBS | Thermo Fisher Scientific | Cat# 14190-144 |
| AP20187 | Clontech | Cat# 635058 |
| Lipofectamine 2000 | Thermo Fisher Scientific | Cat #11668027 |
| Critical commercial assays | ||
| QIAamp DNA Mini Kit | QIAGEN | Cat# 51304 |
| PCR DIG Probe Synthesis Kit | Roche | Cat# 11636090910 |
| DIG Luminescent Detection Kit | Roche | Cat# 11363514910 |
| Dual-Luciferase Reporter Assay System | Promega | Cat# E1980 |
| DNA Ligation Kit Ver.1 | Takara | Cat# 6021 |
| Experimental models: cell lines | ||
| Noggin1-expressing L1 cells | (Sakahara et al., 2019) | N/A |
| Experimental models: organisms/strains | ||
| Patient-derived organoids | (Okamoto et al., 2021) | N/A |
| Oligonucleotides | ||
| Southern blot probe: hOLFM4-F6:CATGGTG GTGTGGTGAACATCAGC |
This paper | N/A |
| Southern blot probe: hOLFM4-R7:GCCCTCT TTCCCTGTGTTTGTGTC |
This paper | N/A |
| Southern blot probe: RFP-F1:CTCCGTGAA CGGCCACGAGTTCGAG |
This paper | N/A |
| Southern blot probe: RFP-R1:GGTGGTCTTG ACCTCGGCGTCGTAG |
This paper | N/A |
| Southern blot probe: EGFP-F1:GGCGATGC CACCTACGGCAAGCTG |
This paper | N/A |
| Southern blot probe: EGFP-R1:CTGCTGGT AGTGGTCGGCGAGCTG |
This paper | N/A |
| OLFM4-Donor-Top oligo: hOLFM4-DONOR11-5S CCGG CCTGCAGATATCTAAGTAAGTGG |
This paper | N/A |
| OLFM4-Donor-Bottom oligo: hOLFM4-DONOR11-5AS GGCC CCACTTACTTAGATATCTGCAGG |
This paper | N/A |
| OLFM4-Donor-Top oligo: hOLFM4-DONOR11-3S CCTGCAGATATCTAAGTAAGTGG |
This paper | N/A |
| OLFM4-Donor- Bottom oligo: hOLFM4-DONOR11-3AS CTAG CCACTTACTTAGATATCTGCAGG AT |
This paper | N/A |
| Recombinant DNA | ||
| pRL-tk | Promega | Cat# E2241 |
| pGL3-BRE | Addgene | Cat# 45126 |
| pCMMP-MCS-IRES-eGFP | Addgene | Cat# #36953 |
| pCMMP-MCS-IRES-mRFP | Addgene | Cat# #36972 |
| pCMMP-MCS-IRES-eGFP-P2A-iCas9 | This paper | N/A |
| pCMMP-MCS-IRES-eGFP-P2A-iCas9-OLFM4 | This paper | N/A |
| pX330 | Addgene | Cat# 42230 |
| pX330-hOLFM4 | This paper | N/A |
| pCR-hOLFM4 | This paper | N/A |
| PB-CMV-MCS-EF1a-Puro | SBI | Cat# PB510B-1 |
| PB-EF1a-MCS-IRES-Neo | SBI | Cat# PB533A-2 |
| Super piggyBac transposase expression vector | SBI | Cat# PB210PA-1 |
| Other | ||
| Super electroporator NEPA21 type II | Nepa Gene | http://www.nepagene.jp/products_nepagene_0001.htmL |
| ChemiDoc XRS+ | Bio-Rad | Cat# 1708265J1PC |
| CellVoyager CV1000 | Yokogawa | https://www.innervision.co.jp/041products/2010/p1001_14etc.htmL |
| 40 μm cell strainer | Falcon | Cat# 352340 |
| 15 mL Centrifuge tube | IWAKI | Cat# 2425-015 |
| 50 mL Centrifuge tube | IWAKI | Cat# 2445-050 |
| 48-Well plate | IWAKI | Cat# 3830-048 |
| 100 mm Dish | IWAKI | Cat# 3020-100 |
Materials and equipment (optional)
organoid culture medium (ENR medium)
| Reagent | Final concentration | Amount |
|---|---|---|
| Advanced DMEM/F12 Supplemented with 2 mM GlutaMAX, 10 mM HEPES, 100 U/mL Pen, 100 μg/mL Strep (basal medium) (store at 4°C for three months) |
N/A | 8,764 μL |
| B27 minus vitamin A (50×) (store at −20°C for 6 months) |
1× | 200 μL |
| N-acetylcysteine (500 mM) (store at −20°C for 6 months) |
1.25 mM | 25 μL |
| Gastrin (100 μM) (store at −20°C for 6 months) |
0.01 μM | 1 μL |
| EGF (100 μg/mL) (store at −20°C for 6 months) |
50 ng/mL | 5 μL |
| R-Spondin 1 (100 μg/mL) (store at −20°C for 6 months) |
50 ng/mL | 5 μL |
| Noggin CM (store at −20°C for 6 months) |
10% | 1,000 μL |
| Total | N/A | 10 mL |
Step-by-step method details
This protocol describes the knock-in of the marker cassette into the OLFM4 locus, which we identified as stem cell markers of PDOs established from CRC (Okamoto et al., 2021). However, we have also successfully used this protocol to visualize and evaluate their progeny. Thus, the main steps of the protocol can be widely adapted for analyzing various types of cells.
CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats (CRISPR)-associated protein 9) technology has been widely used to manipulate the genome of mammalian cells. To visualize stem cells, in-frame insertion of a marker gene into a gene of interest has been widely used. However, this modification inevitably disrupts the expression of genes of interest. Additionally, we have experienced a lack of expression of inserts, most likely due to epigenomic modification (Schuijers et al., 2014). Based on these observations, we inserted IRES marker gene cassettes into the 3′ UTR of the target gene.
The transfection efficiency varies among PDOs, and we adopted the method of transposon-based integration with electroporation developed by Fujii et al., which allows drug selection of transfected cells using the PiggyBac system(Fujii et al., 2015). We used the homology-independent targeted integration method (HITI) (Suzuki et al., 2016) because it allowed highly efficient knock-in of donor DNA at the target sites. Generally, 5%–10% of clones harbored the expected genome editing.
sgRNA design and preparation
Timing:1 week
-
1.
sgRNA are designed using online tools, including CHOPCHOP (http://chopchop.cbu.uib.no), CRISPOR (http://crispor.tefor.net) and CRISPick (https://portals.broadinstitute.org/gppx/crispick/public).
CRITICAL: It is important to confirm whether the crRNA recognition sequences of PDOs contain mutations or SNVs. We recommend exome sequencing because it also provides information about chromosome aneuploidy. Sanger sequencing of the PCR amplicon is an alternative choice for verification.
-
2.
To prepare the sgRNA oligonucleotides for vector insertion, the sense and antisense oligonucleotides are mixed in a 0.2 mL tube following volumes.
| Reagent | Final concentration | Amount |
|---|---|---|
| Top oligo (100 μM) | 10 μM | 1 μL |
| Bottom oligo (100 μM) | 10 μM | 1 μL |
| ddH2O | N/A | 8 μL |
| Total | 10 μL |
-
3.
The tube is placed in a thermocycler, incubated at 95°C for 5 min, and left at RT for 1 h.
-
4.
The annealed oligos are diluted into 250 μL ddH2O.
-
5.
The plasmid, pX330, is digested with Age I by mixing the reaction following reagents and volumes. Incubated at 37°C for 2 h.
| Reagent | Final concentration | Amount |
|---|---|---|
| Plasmid | 0.01 μg/μL | 10 μL |
| Buffer, 10× | 1× | 10 μL |
| ddH2O | N/A | 78 μL |
| Age I (5000 U/mL) | 0.1 U/mL | 2 μL |
| Total | 100 μL |
-
6.
Digested plasmid is purified by phenol/chloroform extraction, followed by ethanol precipitation.
-
7.
The annealed sgRNA oligonucleotide and digested plasmid are ligated and incubated at 16°C for 30 min according to the manufacturer’s instructions (DNA Ligation Kit Ver. 1).
| Reagent | Final concentration | Amount |
|---|---|---|
| Plasmid | 0.003 pmol/μL | 5 μL |
| Diluted sgRNA oligo (0.4 μM) | 6.7 nM | 1 μL |
| ddH2O | N/A | 4 μL |
| Reaction buffer (A) | N/A | 40 μL |
| Enzyme solution (B) | N/A | 10 μL |
| Total | 60 μL |
-
8.
10 μL of ligation product is transferred into 100 μL of a competent Stbl3 strain, incubated for 30 min, heat-shocked at 42°C for 60 s and incubated on ice for 3 min. Next, 500 μL of SOC medium is added and incubated at 37°C for 30 min, and 100 μL of suspension is spread onto an LB plate containing 100 μg/mL ampicillin. The plate is incubated at 37°C for 16 h.
-
9.
Colonies are picked and cultured in 3 mL LB medium, and the plasmid is purified using a QIA prep Spin Miniprep Kit.
-
10.
Plasmids are verified by using the T7sgRNA-R primer for sequencing and further prepared using a QIA prep MAXI column.
Preparing donor plasmids
The plasmids pCMMP-MCS-IRES-EGFP (Addgene #36953) and pCMMP-MCS-IRES-mRFP (Addgene #36972) are two useful resources to visualize stem cells with EGFP and RFP, respectively. We generated an EGFP-inducible Caspase9 fusion gene cassette, pCMMP-MCS-IRES-EGFP-P2A-inducible Caspase9, which allows the ablation of target cells by exposing genome-edited PDOs to AP20187 , a chemical compound that homodimerizes Caspase9 and induces apoptosis (Figure 2). All these constructs have AgeI and NotI sites at the 5′ region and PacI and NheI sites at the 3′ region, which allows the directional cloning of target sequences of sgRNA.
-
11.
Top and bottom oligonucleotides are prepared as shown in Figure 2B. Note that the top and bottom oligonucleotides are to be inserted into the 5′ cloning sites of the donor vector contain AgeI and NotI cloning sites, and those to be inserted into the 3′ cloning sites contain PacI and NheI sites.
-
12.
Anneal oligonucleotides used for insertion at the 5′ cloning site as described in steps 2–4.
-
13.
The pCMM-MCS-IRES-eGFP-iCas9 plasmid is digested with AgeI, and the fragment was purified as described in steps 5–6.
-
14.
Dissolve the digested plasmid in 90 μL of ddH2O.
-
15.
The purified AgeI-digested plasmid is digested with NotI as described in step 5.
-
16.
The AgeI/NotI double-digested plasmid is purified by nucleospin gel and PCR clean-up kits. The plasmid can be eluted twice with 20 μL of ddH2O, and the final volume of the digested plasmid should be 40 μL.
-
17.
DNA concentration is determined using spectrophotometer.
-
18.
The annealed sgRNA oligonucleotide and digested plasmid are mixed according to the manufacturer’s instructions (DNA ligation kit Ver. 1, Takara) and incubated at 16°C for 30 min. We recommend to use 25 pmol of sgRNA oligonucleotide and 100 fmol of digested plasmid for efficient ligation.
-
19.
Competent DH5alpha is transformed with the ligated product, and the plasmid is prepared as described in steps 8–10.
-
20.
The annealed oligonucleotide used for insertion at the 3′ cloning site has appealed and is as described in step 12.
-
21.
Digest pCMM-MCS-IRES-eGFP-iCas9 in which double-stranded oligonucleotide cloned in the 5′ cloning site with PacI and purify as described in step 13–15.
-
22.
The fragment is digested from step 22 with NheI and purified as described in steps 16 and 17.
-
23.
The annealed oligonucleotide from step 21 and PacI/NheI double-digested plasmid are described in step 18.
-
24.
The competent Stbl3 strain is transformed, and the plasmid is prepared as described in step 20.
Figure 2.

Donor plasmid construction
(A) Schematic representation of a donor fragment using the pCMMP-MCS-IRES-eGFP-P2A-iCas9 plasmid. Two annealed oligonucleotides were inserted into the AgeI/NotI site at the 5′ end and the PcaI/NheI site at the 3′ end.
(B) Nucleotide sequences of 5′- and 3′-insertion sites in pCMMP-MCS-IRES-eGFP-P2A-iCas9.
PDOs electroporation
We used a NEPA21 electroporator (Nepa Gene) to transfer plasmid DNA to PDOs. The electroporation protocol was originally developed for NHEJ-mediated gene disruption and for HDR-mediated gene modification (Fujii et al., 2015). We have modified this protocol to optimize HITI-mediated knock-in of the marker cassette. For complete details of the organoid electroporation, please refer to the original protocol (Fujii et al., 2015).
-
25.
Passage PDOs 3 days before electroporation.
Note: Typically, 1 to 2×10ˆ5 cells were collected from 1-well of PDOs, and 6 wells of PDOs were sufficient for each electroporation.
-
26.
The medium is removed, and 500 μL of TrypLE solution supplemented with 10 μM Y-27632 is added to each well. PDO Matrigel is collected into a 15 mL centrifuge tube using a 1000 μL pipette.
-
27.
Cells are incubated in 37°C water bath for 15–30 min until Matrigel is properly dissolved in TrypLE solution.
-
28.
Basal medium is added up to 10 mL, and the mixture is pipetted and filtered through 40 μm cell strainer.
-
29.
The PDOs at R.T. are spun at 500 ×g for 5 min, remove the supernatant.
-
30.
One milliliter of opti-MEM is added. Pipette with 1000 μL pipette well and count the cell number.
-
31.
PDOs are washed at R.T. at 500 ×g for 5 min, and the supernatant is removed.
-
32.
Cells are resuspended at 2×10ˆ6/mL in Opti-MEM.
-
33.
Transfection components are prepared, following reagents and volumes.
| Reagent | Final concentration | Amount |
|---|---|---|
| piggyBac Vector (1 μg/mL) (PB-CMV-MCS-EF1a-Puro or PB-EF1a-MCS-IRES-Neo) |
50 μg/mL | 5 μL |
| Super piggyBac transposase expression vector (1 μg/mL) | 10 μg/mL | 1 μL |
| sgRNA expressing vector (5 μg/mL) | 100 μg/mL | 2μL |
| Donor vector (5 μg/mL) | 100 μg/mL | 2μL |
| Total | 10 μL |
-
34.
Ninety microliters of cell suspension from step 33 are added to the electroporation cuvette.
-
35.
Cells are electroporated with the following settings.
| Poring pulse | Transfer pulse | |
|---|---|---|
| Voltage | 175 V | 20 V |
| Pulse Length | 5 ms | 50 ms |
| Pulse interval | 50 ms | 50 ms |
| Number of pulses | 2 | 5 |
| Decay rate | 10% | 40% |
| Polarity | + | +/− |
-
36.
Next, 400 μL of opti-MEM supplemented with 10 μM Y-27632 is added to the electroporation cuvette, and the cells are transferred into a 15 mL centrifuge tube using the plastic pipette provided with the cuvette.
-
37.
The cells are left at R.T. for 30 min.
-
38.
A centrifuge tube at R.T. at 500 ×g for 5 min.
-
39.
Remove the supernatant and suspend the pellet in 200 μL of Matrigel.
-
40.
Plate cells in 25 μL of Matrigel onto each well of a prewarmed 48-well plate.
-
41.
The plate is incubated at 37°C for 10 min to solidify the Matrigel.
-
42.
ENR medium supplemented with 10 μM of Y-27632 is added.
Note: The stock solution of Y-27632 (10mM) is stored at −20°C for 6 months and is added to the medium just prior to use.
-
43.
The cells are incubated at 37°C for 24 h, and the medium is replaced with 250 μL of ENR medium.
-
44.
The medium is changed every 3 days.
Selection of drug resistant clones
To select the transfected cells, we used puromycin at a concentration of 2 μg/mL when using PB-CMV-MCS-EF1a-Puro. Alternatively, neomycin can be used at a concentration of 800 μg/mL when using PB-EF1a-MCS-IRES-NEO.
-
45.
The selection is started by adding a selective agent to the medium starting from day 5 after electroporation.
-
46.
The medium is supplemented with a selective agent every 3–4 days.
-
47.
Typically, 2–3 resistant clones appear in each well. The expression of fluorescent proteins can be detected under a stereomicroscope when the expression level of the target gene is high. (Figure 2A)
-
48.
Isolate the organoids by gentle pipetting using 200 μL of pipette and transfer onto a 100-mm dish. Single organoids were picked up and transferred to each well of a 96-well U-bottom plate (Figure 3).
-
49.
Add 10 μL of Matrigel and incubate the plate in a 37°C incubator for 10 min to solidify the Matrigel.
-
50.
Add 100 μL of ENR medium supplemented with a selective agent.
-
51.
Each organoid is cultured in ENR medium supplemented with a selective agent.
-
52.
For passaging, modified PDOs are transferred into 15 mL centrifuge tubes, centrifuged at R.T. for 5 min, and carefully removed the supernatant with a 1 mL pipette.
-
53.
Then, 50 μL of Matrigel is added to each well, and a 25 μL droplet is placed in each well of a 48-well plate after pipetting well.
-
54.
Next, 250 μL of ENR medium is added, and the medium is changed every 3 days.
-
55.
To cryopreserve the PDOs, they are collected using a 1 mL pipette into 15 mL centrifuge cells.
-
56.
The centrifuge tube is spun R.T. at 500 ×g for 5 min, and the supernatant is carefully removed.
-
57.
One milliliter of Recovery Cell Culture Freezing Medium is added and transferred to 2 mL cryovials.
-
58.
The tubes are stored at −80°C for up to 1 week.
-
59.
The frozen cryovials are transferred to a liquid N2 tank.
Figure 3.

Picking up the PDOs
(A) The organoids were pipetted into 48-well plates, placed on 100-mm dishes, and observed under a stereomicroscope equipped with fluorescent light.
(B) Using a 200 μL pipette, the EGFP-positive clone was picked up and transferred to a 96-well U-bottom plate.
Isolation of genomic DNA to screen PDO clones
The clonal populations of PDOs are used for identification of genome editing. Each clone is expanded from single well in 96 well plate to multiple wells of 48 well plate. Typically, six to eight wells of a 48-well plate yield 20–40 μg of genomic DNA, which is sufficient to identify genome-edited clones.
-
60.
To isolate genomic DNA from organoids, the medium is removed from each well.
-
61.
Add 500 μL of cell recovery solution to each well, collect the organoids into a 15 mL centrifuge tube and leave on ice for 30 min with occasional pipetting.
-
62.
The PDOs are spun at R.T. at 500 ×g for 5 min to remove the supernatant.
-
63.
One milliliter of PBS is added, and the PDOs are transferred to a 1.5 mL centrifuge tube.
-
64.
The PDOs are spun at 4°C for 5 min, and the supernatant is carefully removed using a 1 mL pipette.
-
65.
The PDO pellet is frozen in liquid N2. Frozen samples can be preserved at −80°C until use.
-
66.
Genomic DNA is extracted using a QIAamp DNA mini kit following the manufacturer’s instructions.
Southern blotting
Genetic modification can be identified by PCR-based screening. However, HITI-mediated knock-in often causes unintended modification, such as tandem duplicated integration. Southern blotting validates the structure of the targeted alleles and integrations of the donor vectors.
For complete details on Southern blotting, please refer to Southern (Southern, 2006).
-
67.
Next, 30 μL of 10× restriction enzyme buffer is added to 14 μg of DNA in 270 μL of TE.
-
68.
Genomic DNA was digested by mixing the reaction following reagents and volumes. Incubated at 37°C overnight.
| Reagent | Final concentration | Amount |
|---|---|---|
| Genomic DNA | 0.01 μg/μL | 14 μg |
| Buffer, 10× | 1× | 30 μL |
| Enzyme (100,000units/mL) | 1 unit/μL | 3 μL |
| ddH2O | N/A | up to 300 μL |
| Total | 300 μL |
-
69.
Next, 1 μL of restriction enzyme is added and incubated for an additional 2 h.
-
70.
30 μL of 3 M sodium acetate (pH 7.0) and 750 μL of ethanol are added.
-
71.
DNA is spun down at 4°C at 15,000 ×g for 30 min, and the supernatant is discarded.
-
72.
Add 700 μL of 70% ethanol, spin DNA down at 4°C at 15,000 × g for 30 min, and carefully discard the supernatant.
-
73.
Air dry the pellet by leaving the tube open on the bench for 15 min.
-
74.
Dissolve the pellet in 10 μL of TE and add 2 μL of 6× loading dye.
-
75.
For a 15 cm × 15 cm gel, 150 mL of 0.7% agarose is prepared in boiling 1× TBE and cooled.
-
76.
The gel is placed in the tank and covered with 1× TBE, and the comb is removed to load the sample.
-
77.
Run overnight at 20 V.
-
78.
The gel is imaged on a UV trans illuminator after staining the gel by adding 6 μL of 10 mg/mL in a 300 mL TBE for 30 min.
-
79.
The gel is transferred to a tray for 15 min with 500 mL of 0.5 N NaOH/1.5 M NaCl, and a second 30 min wash is performed.
-
80.
The gel is rinsed three times with distilled water.
-
81.
The gel is transferred to a tray for 30 min with 500 mL of 1.5 M NaCl/0.5 M Tris-HCl (pH 7.5), and repeat a second 15 min wash.
-
82.
The transfer apparatus is assembled by placing a piece of blotting paper on a glass plate and putting the support inside a large baking dish.
-
83.
The dish is filled with 10× SSC.
-
84.
The Hybond-N+ membrane is soaked in 10× SSC for 5 min.
-
85.
The gel from step 82 is removed and inverted so that it underside is uppermost.
-
86.
The membrane is placed on top of the gel and any bubbles removed.
-
87.
Two thick blotting papers are placed on top of the membrane.
-
88.
Paper towels are placed on the blotting papers.
-
89.
A glass plate is placed on the top of the stack and weighed to 500 g.
-
90.
Allow the transfer of DNA overnight.
-
91.
The paper towels and the blotting paper are removed, the position of the gel slots on the membrane is marked, and the gel is peeled from the membrane.
-
92.
the membrane is soaked in 6× SSC for 5 min and air dried, and DNA is fixed to membrane by UV irradiation (0.12 J/cm2).
Detection of target allele and inserted fragment
Digoxigenin (DIG)-labeled probes are sensitive enough to detect wild-type and mutant alleles of PDOs. A segment corresponding to the genomic sequence of OLFM4 (hOLFM4 probe) is employed to validate the insertion into the expected site, and that corresponding to the EGFP sequence (EGFP probe) is used to validate the copy number of the donor vector (Figure 4). We generally validate the insertion of the donor vector into the OLFM4 locus first and then evaluate the copy number of EGFP after reprobing the first probe.
-
93.
The DIG-labeled probe is prepared using a PCR DIG Probe Synthesis kit (Roche) following conditions. pCR2.1-hOLFM4 and pCMMP-MCS-IRES-EGFP are used as templates for OLFM4 and EGFP probes, respectively. Probes can be stored at −20°C for at least two months.
| Steps | Temperature | Time | Cycles |
|---|---|---|---|
| Initial Denaturation | 95°C | 2 min | 1 |
| Denaturation | 95°C | 10 s | 10 cycles |
| Annealing | 58°C (hOLFM4) 67°C (EGFP) |
30 s | |
| Extension | 72°C | 2 min | |
| Final extension | 72°C | 7 min | 1 |
| Hold | 4°C | Forever | |
-
94.
The hybridization buffer (6× SSC, 5× Denhardt’s solution, 100 μg/mL salmon testis DNA, 0.5% SDS, 50% formamide) is prepared and preheated at 42°C.
-
95.
Membrane is placed in a plastic bag, into which 250 mL of preheated-hybridization buffer is poured. The bag is sealed and incubated for 1 h.
-
96.
Twenty microliters of DIG-labeled probe is placed in a 1.5 mL tube, and 30 μL of ddH2O is added.
-
97.
The probe is denatured by placing it in a heat block heated at 100°C for 5 min and immediately placing on ice for 3 min.
-
98.
Probe solution is added into 10 mL of hybridization buffer preheated at 42°C.
-
99.
The prehybridization buffer is replaced with hybridization buffer containing a DIG-labeled probe, the bag is sealed, and hybridization is conducted at 42°C overnight.
-
100.
The membrane is removed and put into a clean plastic tray with a lid.
-
101.
The membrane is washed for 5 min with 0.1% SDS, 2× SSC at R.T. twice.
-
102.
The membrane is washed for 30 min with 0.1% SDS and 2× SSC at 65°C once.
-
103.
The membrane is washed for 5 min with 0.1% SDS, 0.2 × SSC at R.T. once.
-
104.
The probe is detected using a DIG Luminescent Detection Kit (Roche) according to the manufacturer’s instructions.
-
105.
To visualize the signal, the membrane is placed with the DNA facing up on Saran wrap. We use a ChemiDoc XRC plus system (Bio-Rad) for visualization, setting the first image to 30, the last image to 120 and the number of images to 4.
-
106.
To strip the membrane, it is washed with alkaline buffer (0.2 M NaOH, 0.1% SDS) twice for 5 min at R.T., followed by washing with 2× SSC twice.
-
107.
The process from step 94 is repeated to hybridize with the EGFP probe and detect the signal.
Figure 4.

Southern blot analysis of HITI-mediated genome editing
(A) Schematic representation of knock-in of the IRES-EGFP-P2A-iCas9 cassette into the 3′ UTR of the OLFM4 gene. The hOLFM4 probe detected 11.8 kb and 5.1 kb fragments of wild-type and KI alleles, respectively. The EGFP probe identified a 9.4 kb fragment of KI-allele.
(B) Analysis of 16 puromycin-resistant clones. Five clones contained monoallelic mutation (shown by yellow numbers), and five clones harbored biallelic mutations (shown by red numbers).
Expected outcomes
An example of screening of the HITI-mediated IRES-EGFP-P2A-iCas9 cassette into the 3′UTR of the OLFM4 gene is shown in Figure 4. In this experiment, 62.5% of clones harbored the expected integration, including five biallelic mutations.
The signal intensity of EGFP in PDOs mirrors the expression level of endogenous OLFM4 in the original clone (Okamoto et al., 2021). OLFM4-positive cells were detected in a 3-dimensional image under confocal laser scanning microscopy (Figure 5A). They are also suited for time-lapse imaging analysis (Figure 5B). Note that inducible Caspase 9 effectively induced apoptosis in response to AP20178 exposure.
Figure 5.

PDOs harboring the IRES-EGFP-P2A-iCas9 fragment in the 3′ UTR of the OLFM4 gene
(A) Organoids released from Matrigel were incubated with Hoechst 33342 and examined under confocal laser microscopy.
(B and C) Time-lapse imaging of PDO. PDOs were embedded into Matrigel on glass-bottom plates and cultured for three days with (B) or without AP20147 (C). Note that EGFP expression of PDOs was decreased (B), because OLFM4-postive stem cells reduced their self-replication potential late after passage (Okamoto et al., 2021).
EGFP-positive clones can be isolated using fluorescence-activated cell sorting (FACS), and the ability of self-replication and multiple differentiation to evaluate stemness can be tested (Okamoto et al., 2021).
Limitations
This protocol suffers from limitations that are inherent to the expression of knock-in genes. The major limitation is that the expression of marker genes is affected by the integration site. It depends on several factors, such as the stability, splicing variants and ternary structure of mRNA transcribed from the target gene. Despite the large number of knock-in studies, it is still challenging to predict an integration site that faithfully mirrors the expression of target genes. Thus, we strongly recommend testing whether the expression of fluorescent proteins recapitulates that of endogenous before proceeding to in-depth analyses.
Troubleshooting
Problem 1
Low genome editing activity of sgRNA (step 1)
Potential solution
Low efficiency of genome editing occurs due to several reasons. Most likely, the poor potential of sgRNA to induce DSB is the cause of low efficiency. The efficiency of genome editing also depends on PDOs. We recommend designing and synthesizing additional sgRNAs. The efficiency of sgRNA can be tested by the induction of in-dels after introducing sgRNA plasmids into the PDOs of interest.
Problem 2
Considerable cell death after electroporation (step 35).
Potential solution
Up to 2 × 10ˆ6 cells can be used per electroporation. If electroporation kills all cells, reduce the concentration of plasmids, while the ratio of each vectors remains constant. If still all cells are killed, the poring voltage and poring pulse length can be reduced, but this often reduces the efficiency of electroporation.
Problem 3
PDOs do not grow after electroporation (step 47).
Potential solution
Organoid cells require high activities of niche factors for clonal outgrowth. Check the activities of Noggin conditioned medium and use those having high activity. It should be noted that the R-Spondin activity of recombinant protein might vary among the lots. Several recombinant proteins as well as R-Spondin-expressing cells are commercially available (Merk, Cat#SCC111), and these are the alternatives resources of R-Spondin.
Problem 4
Too many colonies are appeared after drug selection (step 47).
Potential solution
The optimal concentration of puromycin sometimes varies among the PDOs. In our hands, 2 μg/mL is the optimal concentration, but it can be increased up to 10 μg/mL. Optimal concentration on PDOs of interest can be determined by titrating different concentrations of drug and selecting the lowest concentration that kills all cells after 5–7 days.
Problem 5
Loss of organoids while transferring to 96 well plate (step 48).
Potential solution
Try to remove the Matrigel as much as possible by repeated gentle pipetting. The use of low retention tip (Cat# 2069-05, Thermo Fisher Scientific) prevents organoids from sticking them to the tip.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ryoji Yao (ryao@jfcr.or.jp).
Materials availability
Reagents generated within this study are available upon request.
Acknowledgments
We thank all members of the Yao lab for valuable discussions and practical input, in particular Mayuko Yamamoto, for her help in culturing PDOs. This work was supported in part by MEXT/JSPS KAKENHI grant numbers 16H06276 (R.Y.), 16K14620 (R.Y.), 17H063333 (R.Y.), 18H02684 (R.Y), and 16H06279 (PAGS).
Author contributions
T.O. and R.Y. conceived and designed experiments; Y.N., H.Y., and M.F. performed experiments; H. Y. and R.Y. analyzed the data; R.Y. wrote the manuscript; all authors reviewed and edited the manuscript.
Declaration of interests
The authors declare no competing interests.
Data and code availability
This study did not generate any unique datasets or codes.
References
- Fujii M., Matano M., Nanki K., Sato T. Efficient genetic engineering of human intestinal organoids using electroporation. Nat. Protoc. 2015;10:1474–1485. doi: 10.1038/nprot.2015.088. [DOI] [PubMed] [Google Scholar]
- Okamoto T., duVerle D., Yaginuma K., Natsume Y., Yamanaka H., Kusama D., Fukuda M., Yamamoto M., Perraudeau F., Srivastava U. Comparative analysis of patient-matched PDOs revealed a reduction in OLFM4-associated clusters in metastatic lesions in colorectal Cancer. Stem Cell Rep. 2021;16:1–14. doi: 10.1016/j.stemcr.2021.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakahara M., Okamoto T., Oyanagi J., Takano H., Natsume Y., Yamanaka H., Kusama D., Fusejima M., Tanaka N., Mori S. IFN/STAT signaling controls tumorigenesis and the drug response in colorectal cancer. Cancer Sci. 2019;110:1293–1305. doi: 10.1111/cas.13964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuijers J., van der Flier L.G., van Es J., Clevers H. Robust cre-mediated recombination in small intestinal stem cells utilizing the olfm4 locus. Stem Cell Rep. 2014;3:234–241. doi: 10.1016/j.stemcr.2014.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southern E. Southern blotting. Nat. Protoc. 2006;1:518–525. doi: 10.1038/nprot.2006.73. [DOI] [PubMed] [Google Scholar]
- Suzuki K., Tsunekawa Y., Hernandez-Benitez R., Wu J., Zhu J., Kim E.J., Hatanaka F., Yamamoto M., Araoka T., Li Z. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature. 2016;540:144–149. doi: 10.1038/nature20565. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate any unique datasets or codes.