

Abstract
We fine mapped the leukocyte antigen (HLA) region in 13,770 Parkinson’s disease (PD) patients, 20,214 proxy-cases, and 490,861 controls of European origin. Four HLA types were associated with PD after correction for multiple comparisons, HLA-DQA1*03:01, HLA-DQB1*03:02, HLA-DRB1*04:01, and HLA-DRB1*04:04. Haplotype analyses followed by amino acid analysis and conditional analyses suggested that the association is protective and primarily driven by three specific amino acid polymorphisms present in most HLA-DRB1*04 subtypes—11V, 13H, and 33H (OR = 0.87, 95% CI: 0.83–0.90, p < 8.23 × 10−9 for all three variants). No other effects were present after adjustment for these amino acids. Our results suggest that specific HLA-DRB1 variants are associated with reduced risk of PD, providing additional evidence for the role of the immune system in PD. Although effect size is small and has no diagnostic significance, understanding the mechanism underlying this association may lead to the identification of new targets for therapeutics development.
Subject terms: Parkinson's disease, Genomics
Introduction
Although Parkinson’s disease (PD) is primarily a neurodegenerative disorder, the role of the immune system in the pathophysiology of PD is increasingly recognized based on animal and human studies1–3. The immune system can be involved in the initiation of PD, as well as in its progression, and that this involvement can be peripheral and central3,4.
Neuropathological studies have shown evidence for microglial activation in the brains of patients. However, it was initially unclear whether this activation was a part of the disease process, a consequence, or an epiphenomenon5. Genetic evidence also links the immune system with PD, since genes such as LRRK2, the human leukocyte antigen (HLA) locus, and possibly BST1, all associated with PD6 and have a role in the immune system7–9.
The HLA region on chromosome 6 includes genes that encode components of the major histocompatibility complex (MHC)8. Several genome-wide association studies (GWASs) have shown an association between the HLA locus and the risk of PD. In the latest GWAS, an association with HLA-DRB5 has been reported, with a potential effect of the rs112485576 single nucleotide polymorphism (SNP) on the expression of HLA-DRB56. Previous studies have suggested different associations with HLA-DQA2, HLA-DQB1, HLA-DRA, HLA-DRB1, HLA-DRB5, and with haplotypes within the HLA region in Europeans10–15.
In this study, we performed HLA alleles, haplotypes, and amino acid analyses in PD on 12,137 patients, 14,422 proxy patients, and 351,953 controls. We further performed conditional analyses to fine map and identify specific drivers of the association with PD in the HLA region.
Results
Meta-analysis of HLA types in PD suggests a single association
After standard QC, a total of 12,137 patients, 14,422 proxy patients, and 351,953 controls were included in the analysis (Supplementary Data 1 details the number of individuals analyzed from each cohort for each allele, and Supplementary Data 2 provides basic demographic information for each cohort). As shown in Fig. 1A, our SNP-level meta-analysis identified the previous association for SNP, rs112485576, in the HLA locus (in the current analysis: OR = 0.87, 95% CI: 0.83–0.90, p = 5.00 × 10−13, in the previous meta-analysis: OR = 0.85, 95% CI: 0.82–0.87, p = 6.96 × 10−28)6. No other HLA SNPs were significantly associated with PD after adjusting for rs112485576 (Fig. 1B, C), indicating that the association of this locus was primarily driven by a single genetic risk factor. We also identified the previous associations for the SNPs rs17425622, rs2395163, rs3129882, and rs9275326 in the HLA locus (Supplementary Figs. 1–4). All these SNPs were reported in different previous studies, are in LD, and all represent the same main haplotype that we report in the current analysis.
Fig. 1. Validation of previously associated top HLA locus SNP (rs112485576) in our cohort.

A Forest plot describing the effect size and 95% confidence interval of rs112485576 for each cohort and fixed-effect meta-analysis. B, C Two LocusZoom plots highlighting the significant variants before (B) and after (C) the conditional analysis on rs112485576. Dashed lines correspond to the significance threshold. Linkage disequilibrium values are shown with respect to the most significant SNP in the locus.
We next performed a meta-analysis association study of all HLA types with carrier frequency above 1%. After HLA imputation, a total of 141 different HLA types across 10 HLA loci were included (setting the Bonferroni corrected threshold for statistical significance on α = 3.55 × 10−4; 0.05/141). Following these analyses, we found four HLA alleles that were associated with PD (Table 1, results for other HLA alleles are detailed in Supplementary Data 3): HLA-DQA1*03:01, HLA-DQB1*03:02, HLA-DRB1*04:01, and HLA-DRB1*04:04. These four alleles are all located within a small genomic segment and have similar odds ratios ranging between 0.84 and 0.89. Three of the four alleles have similar carrier frequencies, indicating that they could be part of the same haplotypes, with the fourth potentially representing a sub-haplotype (Table 1).
Table 1.
Meta-analyses of HLA alleles association.
| Allele | Freq cases | Freq controls | OR (95% CI) | P value1 | Direction | HetPVal |
|---|---|---|---|---|---|---|
| DQA1*03:01 | 0.168 | 0.185 | 0.86 (0.81-0.90) | 3.54e−08 | – | 0.85 |
| DQB1*03:02 | 0.182 | 0.199 | 0.87 (0.83-0.92) | 7.62e−07 | – | 0.58 |
| DRB1*04:01 | 0.187 | 0.221 | 0.89 (0.84-0.94) | 2.82e−05 | – | 0.87 |
| DRB1*04:04 | 0.068 | 0.082 | 0.84 (0.77-0.91) | 8.21e−05 | – | 0.85 |
Freq cases carrier frequency of allele in patients, Freq controls carrier frequency of an allele in controls, OR (95% CI) odds ratio and 95% confidence interval, Direction direction of beta for each cohort, HetPVal P value of heterogeneity.
1Bonferroni correction for multiple comparisons sets the threshold for statistical significance to α = 3.55 × 10−4.
HLA haplotype analysis
For haplotype analysis, we allowed for up to three genes to be included (Table 2) in each haplotype, since including more than three genes generated multiple haplotypes with low allele frequency that could not be analyzed at the current sample size. A total of 84 different HLA haplotypes (Supplementary Data 4) with allele frequency >1% were identified, setting the cut-off Bonferroni corrected p value for statistical significance at α = 5.95 × 10−4. Three different HLA haplotypes were associated with PD after correction for multiple comparisons: DQA1*03:01–DQB1*03:02, DRB1*04:01–DQA1*03:03, and DRB1*04:04–DQA1*03:01. Upon further examination, this association was found to be driven by several well-known sub-haplotypes, DRB1*04:04–DQA1*03:01–DQB1*03:02, and DRB1*04:01–DQA1*03:01/3–DQB1*03:01/2 (Supplementary Data 4). Because both DQA1*03:01 and DQA1*03:03 as well as DQB1*03:01 and DQB1*03:03 are present within the extended DRB1*04:01 haplotype, it is likely that these associations are driven by DRB1.
Table 2.
Meta-analyses of HLA haplotype association.
| Haplotype | Freq cases | Freq controls | OR (95% CI) | P value1 | Direction | HetPVal |
|---|---|---|---|---|---|---|
| DQA1*03:01–DQB1*03:02 | 0.157 | 0.173 | 0.87 (0.82–0.93) | 7.21e−05 | ++ | 0.62 |
| DRB1*04:04–DQA1*03:01 | 0.067 | 0.081 | 0.83 (0.76–0.91) | 9.82e−05 | + | 0.34 |
| DRB1*04:01–DQA1*03:03 | 0.115 | 0.143 | 0.87 (0.81–0.94) | 4.96e−04 | + | 0.71 |
Freq cases carrier frequency of a haplotype in patients, Freq controls carrier frequency of a haplotype in controls, OR (95% CI) odds ratio and 95% confidence interval, Direction direction of beta for each cohort, HetPVal P value of heterogeneity.
1Bonferroni correction for multiple comparisons sets the threshold for statistical significance to α = 5.95 × 10−4.
Meta-analysis of the association of HLA amino acid changes with PD
To further identify the specific source of the association in the HLA locus, we performed an analysis of 636 amino acid changes in the HLA genes, setting the cut-off Bonferroni corrected p value for statistical significance at α = 7.86 × 10−5. Ten amino acid changes were significantly associated with a reduced risk of PD (Supplementary Data 5). The top three associated variants are linked amino acids 11V, 13H, and 33H (Table 3) present in all DRB1*04 subtypes, complementing the HLA haplotype analysis. Four other variants, 26S, 47Q, 56R, and 76V, in the DQA1 gene, are in perfect LD with each other (r2 = 1, D′ =1, Supplementary Data 5) and in partial LD (r2 = 0.38, D′ = 0.85) with the DRB1 variants. The association of these DQA1 variants is weaker than the DRB1 variants in terms of both effect size and statistical association (Supplementary Data 5). Three other variants, 71T, 74E, and 75L, are in the DQB1 gene, are also in perfect LD with each other (r2 = 1, D′ = 1, Supplementary Data 5) and in partial LD (r2 = 0.16, D′ = 0.99) with DRB1 13H and 33H. As an additional quality control step, we repeated all the analyses reported above without the proxy cases. In these analyses, the results did not change, with all associations having the same direction of effect and with the same magnitude. No additional associations were found.
Table 3.
Meta-analyses of HLA amino acid changes association.
| Amino acid | Freq cases | Freq controls | OR (95% CI) | P value1 | Direction | HetPVal |
|---|---|---|---|---|---|---|
| DRB1 13H | 0.289 | 0.331 | 0.87 (0.83–0.91) | 4.32e−09 | – | 0.60 |
| DRB1 33H | 0.289 | 0.331 | 0.87 (0.83–0.91) | 4.32e−09 | – | 0.60 |
| DRB1 11V | 0.302 | 0.342 | 0.87 (0.83–0.91) | 8.22e−09 | – | 0.45 |
Freq cases carrier frequency of an amino acid in patients, Freq controls carrier frequency of an amino acid in controls; OR (95% CI) odds ratio and 95% confidence interval, Direction direction of beta for each cohort, HetPVal P value of heterogeneity. P value of heterogeneity.
1Bonferroni correction for multiple comparisons sets the threshold for statistical significance to α = 7.86 × 10−5.
Conditional analyses confirm that DRB1*04 amino acid variants likely drive the association of the HLA locus with PD
To further determine the specific genes or variants that drive these associations, we performed a set of conditional analyses and re-analyzed the allele types, haplotypes, and amino acid associations with PD. We conditioned the HLA type regression model on the following: First, we performed the conditional analysis using the top hit from the most recent PD GWAS, rs112485576, to examine the LD effect between this SNP and the different allele and amino acid associations reported in the current study. We further conditioned the HLA type regression models on DQA1*03:01, DQA1*03:03, and the DRB1 variant 13H. We have also adjusted for the PD PRS, to examine a potential polygenic effect (Supplementary Data 3 and 5). While the adjustment for the DRB1 variant 13H completely eliminated the associations in the DQA1 gene, adjustment for DQA1*03:01 and DQA1*03:03 did not completely eliminate the association of the DRB1 gene (Supplementary Data 3), again supporting this gene and these specific amino acids (11V, 13H, and 33H, Table 3) as the drivers of the association in the HLA locus. Adjustment for PRS did not change the results. It is also worth noting that the DRB1 variants 11V (r2 = 0.96, D′ = 0.99), 13H (r2 = 0.99, D′ = 0.99), and 33H (r2 = 0.99, D′ = 0.99) are in LD with rs112485576, the top GWAS hit in this locus.
Discussion
In the current study, we performed a thorough analysis of the HLA region and examined its association with PD in the European population using a total of 12,137 patients, 14,422 proxy patients, and 351,953 controls. The importance of the current study lies in its size and comprehensive approach (including validation of imputation in over 3500 individuals), which allowed us to confirm previously reported associations but also refute with a high degree of confidence other reported associations. Following a series of regression models and conditional analyses, our results indicate that the drivers of the association in the HLA region are three amino acid changes specific of HLA-DRB1*04 subtypes, 11V, 13H, and 33H (Fig. 2). Two of these amino acid changes, 13H and 33H are in perfect LD, and 11V is in very strong LD with the other two variants. This study agrees with a smaller HLA sequencing study12 in 1597 PD cases and 1606 controls which also observed a protective effect of DRB1*04 and the same amino acids, although it also reported additional associations with DRB1*01:01 and DRB1*10:01 which were not confirmed in the current study. Interestingly, the V–H–H motif at positions 11V, 13H, and 33H are central to the DRB1*04:01 heterodimer and contribute to peptide binding, notably through pocket P616 (Fig. 2).
Fig. 2. Association of the HLA-DRB1 alleles and location of associated amino acids.
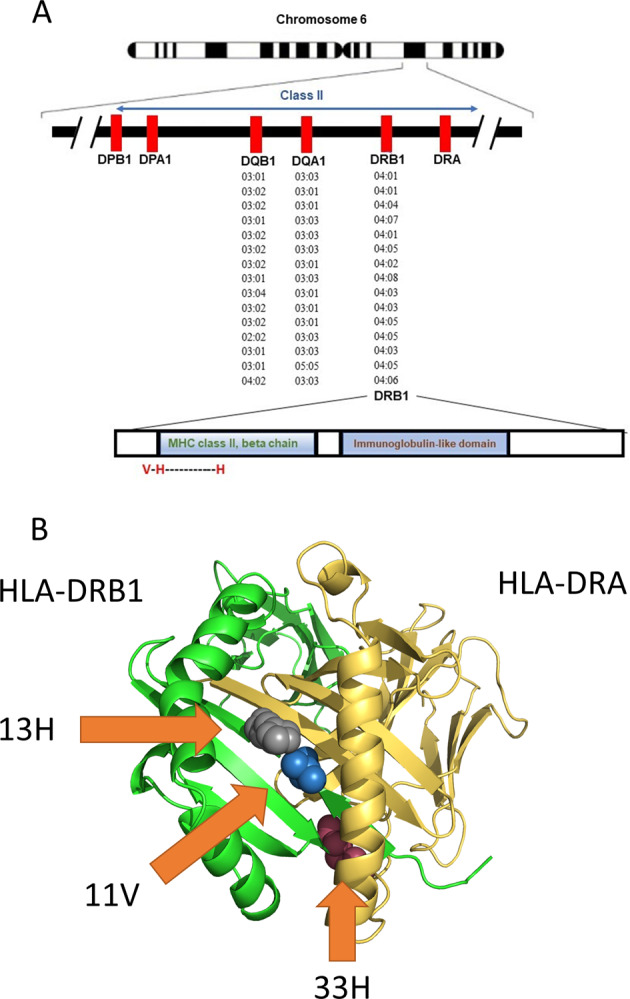
A The location of the HLA locus, alleles, and amino acids associated with Parkinson’s disease in the current study. B 3D model of HLA-DRB1–HLA-DRA and the location of the 11V, 13H, and 33H amino acids associated with PD (highlighted by arrows). The model was generated with PyMol v. 2.4.1 (pdb 4is6).
Previous studies on the HLA genomic region in PD have reported associations of different genes and HLA types with PD, including HLA-DQA2, HLA-DQB1, HLA-DRA, HLA-DRB1, and HLA-DRB510–15. The largest of these studies which performed proper HLA type analysis, included 7,996 cases and 36,455 controls, compared to the current study including more than 24,000 cases and 350,000 controls, with only partial overlap of samples (more details on these and other studies are in Supplementary Data 6). Therefore, the current study is more powered to detect true positive associations and true negative results, although we cannot rule out small effects that could not be detected even in our larger sample size. Most of the previously reported associations were not replicated in the current, larger study, which allows us to focus more on the associations that were confirmed. The suggested association with HLA-DRB5 was reported in the most recent PD GWAS, in which no proper HLA imputation and analysis were performed. This reported association is based on an expression quantitative trait locus analysis, as the top associated SNP in this region, rs112485576 was also associated with differential expression of HLA-DRB6. A previous study of 2000 PD patients and 1986 controls has implicated a non-coding variant (rs3129882) within HLA-DRA as driving the association with PD and suggested that this variant affects the expression of HLA-DR and HLA-DQ genes11. Similarly, another study suggested that the same variant in HLA-DRA (rs3129882) is associated with differential expression of MHC-II on immune cells17. While our study does not rule out this possibility, since the main variants driving the association are amino acid changes in DRB1*04 that will affect epitope binding ability, it is likely that the effect on PD risk is through these variants and not due to modified expression. Additional functional studies will be required to study this hypothesis.
The current study adds further support to the hypothesis suggesting an involvement of the peripheral and central immune systems in PD. On top of the HLA locus, several other genes with potential roles in the immune system, including LRRK2 and potentially BST17,9, have been implicated in PD6. In the periphery, there are notable changes in the immune system of PD patients compared to controls, as peripheral monocytes have differential expression of immune-related proteins and markers3. Whether these changes are drivers of the disease or a result of the disease is still undetermined, but accumulating evidence suggests that they can be part of the pathogenic process of PD. In the central nervous system, pathological studies suggest that microglial cells may have a central role in PD18. Microgliosis is a prominent pathological finding in the post-mortem brains of PD patients, and evidence suggests that microglial activation occurs early in the disease process and may be involved in the pathogenesis of PD3. The specific contribution of HLA to these processes is still unclear and needs to be further studied.
One intriguing possibility that may directly involve HLA with PD is the potential interaction of HLA-DRB1*04 with α-synuclein, notably an epitope surrounding p.S129. Recent data has shown that α-synuclein fragments can bind MHC and increase T cell reactivity19. This activity is proinflammatory, involves both CD4 and CD8 cells, and may occur before the onset of motor symptoms19,20, suggesting the involvement of inflammation in early PD pathogenesis. Studying specific α-synuclein fragments has suggested that two major regions of α-synuclein may be associated with increased T cell reactivity in PD, with preferential CD4 activity: an N-terminal region involving amino acid p.Y39 and a C-terminal region surrounding amino acid p.S129, two important residues undergoing phosphorylation19. The phosphorylation of p.S129 is particularly interesting as it is well known for promoting aggregation21,22. Further analysis focused on the p.Y39 region suggested an association with α-synuclein-specific p.Y39 T cell responses and HLA DRB1*15:01 and DRB5*01:01 presentation. This association was abolished by phosphorylation, which reduced the binding of p.Y39-phosphorylated α-synuclein19,20. Other experiments by these authors have also suggested CD8+ T cells responses mediated by HLA-A11*01 presentation of epitopes in the same N-terminal region of α-synuclein. However, in the current study, we could not confirm an association of these HLA types with PD.
More interestingly in the context of our work, increased CD4+ T cell response to both p.S129 phosphorylated and unphosphorylated α-synuclein was also demonstrated, suggesting the involvement of DQB1*05:01 and DQB1*04:02, as these alleles strongly bound this α-synuclein epitopes19. These HLA alleles, however, are not associated with the risk of PD in the current study. However, the authors reported in the supplementary data that DRB1*04:01 was also a selective and strong binder of the same α-synuclein epitope with p.S129, but only when the epitope was unphosphorylated19. Notably, no other DRB1 alleles that were assayed in this study19 had increased binding affinity to the α-synuclein epitope with p.S129, except for the DRB1*04:01 allele that was a strong binder only when unphosphorylated. Binding register analysis using Immune Epitope Database (IEDB) MHC-II Binding Prediction (http://tools.iedb.org/mhcii/) suggests that this epitope binds a 9 amino acid AYEMPSEEG core, with p.S129 at the P6 position, a position postulated to be important based on our HLA-DR amino acid analysis presented above. As CD4+ T cell responses are generally stronger when epitopes are presented by HLA-DR vs. HLA-DQ23, HLA-DRB1*04 responses to the p.S129 unphosphorylated form of α-synuclein could be dominant in individuals with HLA-DRB1*04, explaining the protective effect of this HLA subtype in PD. Additional experiments will be needed to further explore this hypothesis.
Our study has several limitations. First, this study was performed on European populations, and the results may be limited to this population only. Additional studies in other populations are required. Several studies on HLA types and PD have been performed in Asian populations24–27, and the GWAS risk variant rs112485576 has a similar OR (0.85) in the largest Asian GWAS to date28, yet larger studies are required, as well as studies in other populations. An additional potential limitation of our study is its use of imputation rather than fully sequenced HLA types. Given the very high performance of the imputation tool when compared to full sequencing (Supplementary Data 7), the potential effect of imputation inaccuracies is likely small and should be diluted in our large sample size. Since different cohorts may have had different criteria for determining the presence of PD, it is important to note that for all the positive associations reported in the current study, the directionality was identical across all cohorts with similar effect magnitude, and for all meta-analyses the heterogeneity was not statistically significant (supplementary Data 3–5), indicating that the different criteria in the different cohorts did not have a major effect on the results. In addition, we cannot rule out that other, rarer HLA types that were not included in the current analysis may also have a role in PD. An additional limitation of the current study is that by adjusting for sex we eliminate potential sex-specific effects. It is possible that specific HLA types are relevant in one sex more or less than the other, and this should be studied in larger, sex-stratified cohorts.
To conclude, our results suggest a role for the HLA-DRB1 gene in susceptibility for PD and provide further evidence for the importance of the immune system in PD. Since the effect is small, it does not merit routine HLA typing in PD, but understanding the mechanism underlying this association may lead to a better understanding of PD in general and offer new targets for future immune-related treatment.
Methods
Study population
This study was designed as a meta-analysis of multiple cohorts, including a total of 13,770 PD patients, 20,214 proxy-patients, and 490,861 controls, as detailed in Supplementary Data 1 and 2. In brief, we included cohorts and datasets from eight independent sources: International Parkinson’s Disease Genomics Consortium (IPDGC) NeuroX dataset (dbGap phs000918.v1.p1, including datasets from multiple independent cohorts as previously described)29, McGill University (McGill)30, National Institute of Neurological Disorders and Stroke (NINDS) Genome-Wide genotyping in Parkinson’s Disease (dbGap phs000089.v4.p2)31, NeuroGenetics Research Consortium (NGRC) (dbGap phs000196.v3.p1)11, Oslo Parkinson’s Disease Study (Oslo), Parkinson’s Progression Markers Initiative (PPMI), Vance (dbGap phs000394) and PD cases and proxy-cases from the UK Biobank (UKB). Proxy-cases are first-degree relatives of PD patients, thus sharing ~50% of the patients’ genetic background and eligible to serve as proxies, as previously described32. Since they are genetically very similar, although they do not have PD, each proxy case represents a case of PD, allowing us to increase the power. However, we also analyzed the data without the proxy-cases cohort to make sure that they did not introduce any bias. All cohorts were previously included in the most recent PD GWAS6. Study protocols were approved by the Institutional Review Board at McGill University and all patients signed informed consent before participating in the studies.
Pre-imputation genotype quality control
In order to include only high-quality samples and SNPs, standard quality control (QC) was performed on all datasets individually using PLINK v1.933. Standard GWAS QC was done to filter out samples and SNPs with low call rates, heterozygote outliers along gender mismatch as previously described6. SNPs deviating from Hardy-Weinberg equilibrium were removed. Only samples of European ancestry clustering with HapMap v3 using principal component analysis were included as shown in Supplementary Fig. 5. In order to exclude related individuals, we examined relatedness in each dataset separately, followed by a relatedness test across all datasets combined, to exclude individuals who were included in more than one dataset. All individuals with pi_hat >0.125 were excluded using GCTA v1.26.034.
UK Biobank QC
For the analysis of the UKB data, unrelated participants of European ancestry (field 22006), with a low missingness rate (field 220027) were included after the exclusion of heterozygosity outliers as previously described6. PD patients from the UK Biobank were included based on self-report (field 20002) or based on their International Classification of disease diagnosis code (ICD-10, code G20, field 41270). From the remaining participants, proxy-cases were defined as first-degree relatives (parents or siblings, field 20112–20114) of patients with PD. Principal components were calculated using flashpca35 after excluding related individuals as described above. The control group was divided randomly into two groups of controls: one was included in the GWAS comparing PD patients from UKB and controls, and the second was included in the GWAS comparing proxy-cases from UKB and controls. This division was done proportionally to the size of each GWAS.
Imputation
For the SNP imputation of each dataset, we used the Michigan Imputation Server on the 1000 Genomes Project panel (Phase 3, Version 5) using Minimac3 and SHAPEIT v2.r790. Imputed UK Biobank genotyped data v3 were downloaded in July 2019. All variants with an imputation quality (r2) of >0.30 were labeled as soft calls and >0.80 were labeled as hard calls. Soft calls were only used together with the hard calls for polygenic risk score (PRS) calculation (see below); hard calls were used for all other analyses.
Association analysis of common variants on chromosome 6
Prior to determining HLA types, we performed a simple association test of all SNPs located on chromosome 6, to verify that we identified the same hit in the HLA region as previously described6. For this purpose, we generated summary statistics of chromosome 6 for each dataset and used logistic regression with an additive model adjusting for age at onset for patients and age at enrollment of controls, sex, and population stratification (first 10 principal components) with PLINK v2.00a2LM (25 Oct 2019)33. The UK Biobank data were analyzed similarly using logistic regression adjusting for age, sex, the first 10 principal components, and Townsend index to account for additional potential population stratification confounders. Finally, to harmonize effects in cases and proxy cases, summary statistics for proxy cases were rescaled based on genome-wide association study by proxy as previously described32. Each dataset was analyzed separately, followed by a meta-analysis of all datasets. To meta-analyze the different datasets, we performed a fixed-effects meta-analysis using METAL with an inverse-variance-based model and examined whether heterogeneity exists between the different cohorts36.
HLA locus analysis
To impute specific HLA types for each individual, we inferred two field resolution HLA alleles using HIBAG v1.22.0, a statistical method for HLA type imputation in R37. HIBAG was shown to be as accurate or more accurate in Europeans compared to other types of HLA imputation tools38. HIBAG provided a reference panel for Europeans (n = 2572) with high imputation accuracy for HLA-A, HLA-B, HLA-C, class I genes, and HLA-DPB1, HLA-DQA1, HLA-DQB1, and HLA-DRB1, class II genes. HLA-DRB3, HLA-DRB4, and HLA-DRB5 imputation models were trained using HIBAG37 on European origin sample training set (n = 3267) genotyped on the Illumina Infinium PsychArray-24 chip and fully sequenced at 8-digit resolution for HLA loci. These models were validated in a test set (n = 886) with high accuracy (Supplementary Data 7). Imputation accuracy for European DRB1*04 alleles was determined for DRB1*04:01 DRB1*04:02 DRB1*04:03, DRB1*04:04, DRB1*04:05, DRB1*04:07, DRB1*04:08. Alleles with an imputation probability of <0.5 were defined as undetermined and individuals with two or more undetermined alleles were excluded from the analysis (Supplementary Data 1 details the numbers included for each allele in each cohort after all quality control steps). To further examine imputation accuracy, the results of the DRB1 imputation were compared against high throughput HLA sequencing in 380 PD samples from Oslo. The combined frequency of seven different DRB1*04 alleles detected in sequence data was 0.15 with the 04:01 and 04:04 alleles being the most common (Supplementary Data 8). Imputation accuracy for DRB1*04 alleles was very high at 2-digit resolution (Supplementary Data 9).
To examine the association of HLA alleles with PD, we used R v3.6 to perform logistic regression, adjusting for age at onset, sex, and the first 10 principal components. The UK Biobank dataset was also adjusted for the Townsend index. Haplotype analyses were performed using haplo.stats in R with logistic regression as stated above. Only haplotypes with posterior probability >0.2 and a carrier frequency of >1% were included in the analysis. Amino acid association analyses were performed using HIBAG after converting P-coded alleles to amino acid sequences for exon 2, 3 of HLA class I genes, and exon 2 of class II genes. Amino acid associations were tested using logistic regression as described above. A PRS was calculated using PRSice v 2.2.11 without linkage disequilibrium (LD) clumping or P thresholding39. The beta weights from the summary statistics of the 90 genome-wide significant variants in the latest PD GWAS6 were used in the PRS. To make sure that all possible variants were included in the PRS analysis, we also performed imputation using the Haplotype Reference Consortium panel (Version r1.1 2016) with Minimac4 and Eagle v2.4. Ambiguous variants (rs6658353) and rs112485576 from the HLA region were excluded from the PRS calculation. We also used the top hit in the HLA region from this GWAS to perform adjustment in the regression models and conditional analyses to determine whether the associations reported here are in LD with the GWAS top hit. To examine whether secondary hits exist in the HLA region, we adjusted for significant HLA variants, HLA alleles, HLA amino acid changes, and PRS, by introducing significant findings from the first analyses as covariates in the regression models. Statistical analyses were only performed on alleles, haplotypes, and amino acid changes with more than 1% carrier frequency. P value significance levels were adjusted using Bonferroni correction. Meta-analysis was performed as described above. All missing data were excluded from the analyses.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Acknowledgements
We would like to thank the participants in the different cohorts. The access to part of the participants for this research has been made possible thanks to the Quebec Parkinson’s Network (http://rpq-qpn.ca/en/). This work was financially supported by grants from the Michael J. Fox Foundation, the Canadian Consortium on Neurodegeneration in Aging (CCNA), the Canada First Research Excellence Fund (CFREF), awarded to McGill University for the Healthy Brains for Healthy Lives initiative (HBHL), and Parkinson Canada. We would like to thank all members of the International Parkinson Disease Genomics Consortium (IPDGC). For a complete overview of members, acknowledgements and funding, please see http://pdgenetics.org/partners. This research was also supported in part by the Intramural Research Program of the NIH, National Institute on Aging. HLA sequencing of samples from the Oslo Parkinson’s disease study was supported by the Norwegian Parkinson Research Fund. PPMI—a public–private partnership—is funded by the Michael J. Fox Foundation for Parkinson’s Research and funding partners, including Abbvie, Allergan, Amathus Therapeutics, Avid Radiopharmaceuticals, Biogen, BioLegend, Bristol-Myers Squibb, Celgene, Denali Therapeutics, GE Healthcare, Genentech, GlaxoSmithKline, Golub Capital, Handl Therapeutics, Insitro, Janssen, Lilly, Lundbeck, Merck, Meso Scale Discovery, Neurocrine Biosciences, Pfizer, Piramal, Prevail Therapeutics, Roche, Sanofi Genzyme, Servier, Takeda, Teva, UCB, Verily and Voyager Therapeutics. This research has been conducted using the UK Biobank Resource under Application Number 35605. L.P. is supported by a grant from the National Health Association, Norway. MSA is supported by a grant from the South-Eastern Regional Health Authority, Norway. KS is supported by a postdoctoral fellowship from the Canada First Research Excellence Fund (CFREF), awarded to McGill University for the Healthy Brains for Healthy Lives initiative (HBHL). ZGO is supported by the Fonds de recherche du Québec—Santé (FRQS) Chercheurs-boursiers award.
Author contributions
E.Y., L.P., E.M., and Z.G.O. conceptualized and supervised the study. J.A.R., and F.A. performed sample preparation. E.Y., M.S.A, and L.K. performed data preparation, quality control. A.A. prepared HLA imputation panels. M.S.A, M.T., and M.K.V. performed HLA panel validation. E.Y. performed HLA imputation. E.Y. performed the statistical analysis, C.B. contributed to these analyses. M.A.E., P.S., K.S., Y.L.S., A.A.K.S., D.S., and M.S. performed additional data quality control, checks, and analysis. M.S.A, M.T., M.K.V., and L.P. provided biospecimens and clinical data. E.Y., A.A., E.M., and Z.G.O. prepared the initial paper. All authors critically reviewed, edited, and approved the article.
Data availability
Anonymized data will be shared by request from any qualified investigator. All summary statistics are found in the Supplementary Data. The accession numbers to the datasets from dbGaP (https://www.ncbi.nlm.nih.gov/gap/) used in this study are: International Parkinson’s Disease Genomics Consortium (IPDGC) NeuroX dataset (dbGap phs000918.v1.p1), National Institute of Neurological Disorders and Stroke (NINDS) Genome-Wide genotyping in Parkinson’s Disease (dbGap phs000089.v4.p2), NeuroGenetics Research Consortium (NGRC) (dbGap phs000196.v3.p1) and Vance (dbGap phs000394).
Code availability
The scripts used in this analysis is available at https://github.com/gan-orlab/HLA_HIBAG/.
Competing interests
Mathias Toft—Dr. Toft received grants from Norwegian Parkinson Research Fund and Reberg’s legacy. Lasse Pihlstrøm—Dr. Pihlstrøm received grants from National Health Association, Norway, and South-Eastern Regional Health Authority, Norway. Emmanuel Mignot—Dr. Mignot reports consultancies from Idorsia Pharmaceuticals Ltd., personal fees from Jazz Pharmaceutical, Alairion, ALPCO, INEXIA, Merck, Orexia, Rhythm, and Sunovion. Ziv Gan-Or—Dr. Gan-Or reports personal fees from Idorsia, Neuron23, Handl Therapeutics, Lysosomal Therapeutics Inc., Deerfield, Lighthouse, Prevail Therapeutics, Ono Therapeutics, Denali, and Inception Science. Eric Yu, Aditya Ambati, Maren Stolp Andersen, Lynne Krohn, Mehrdad A. Estiar, Prabhjyot Saini, Konstantin Senkevich, Yuri L. Sosero, Ashwin Ashok Kumar Sreelatha, Jennifer A. Ruskey, Farnaz Asayesh, Dan Spiegelman, Marte K. Viken, Manu Sharma, and Cornelis Blauwendraat report no relevant disclosures.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Emmanuel Mignot, Ziv Gan-Or.
Supplementary information
The online version contains supplementary material available at 10.1038/s41531-021-00231-5.
References
- 1.Sanchez-Guajardo V, Barnum CJ, Tansey MG, Romero-Ramos M. Neuroimmunological processes in Parkinson’s disease and their relation to α-synuclein: microglia as the referee between neuronal processes and peripheral immunity. ASN Neuro. 2013;5:AN20120066. doi: 10.1042/AN20120066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Q, Liu Y, Zhou J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 2015;4:19. doi: 10.1186/s40035-015-0042-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tansey MG, Romero-Ramos M. Immune system responses in Parkinson’s disease: early and dynamic. Eur. J. Neurosci. 2019;49:364–383. doi: 10.1111/ejn.14290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Devos D, et al. Colonic inflammation in Parkinson’s disease. Neurobiol. Dis. 2013;50:42–48. doi: 10.1016/j.nbd.2012.09.007. [DOI] [PubMed] [Google Scholar]
- 5.Zhang W, et al. Aggregated α‐synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J. 2005;19:533–542. doi: 10.1096/fj.04-2751com. [DOI] [PubMed] [Google Scholar]
- 6.Nalls MA, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 2019;18:1091–1102. doi: 10.1016/S1474-4422(19)30320-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malavasi F, et al. CD38 and CD157 as receptors of the immune system: a bridge between innate and adaptive immunity. Mol. Med. 2006;12:334–341. doi: 10.2119/2006-00094.Malavasi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shiina T, Hosomichi K, Inoko H, Kulski JK. The HLA genomic loci map: expression, interaction, diversity and disease. J. Hum. Genet. 2009;54:15–39. doi: 10.1038/jhg.2008.5. [DOI] [PubMed] [Google Scholar]
- 9.Wallings RL, Herrick MK, Tansey MG. LRRK2 at the interface between peripheral and central immune function in Parkinson’s. Front. Neurosci. 2020;14:443. doi: 10.3389/fnins.2020.00443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahmed I, et al. Association between Parkinson’s disease and the HLA-DRB1 locus. Mov. Disord. 2012;27:1104–1110. doi: 10.1002/mds.25035. [DOI] [PubMed] [Google Scholar]
- 11.Hamza TH, et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat. Genet. 2010;42:781–785. doi: 10.1038/ng.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hollenbach JA, et al. A specific amino acid motif of HLA-DRB1 mediates risk and interacts with smoking history in Parkinson’s disease. Proc. Natl Acad. Sci. USA. 2019;116:7419–7424. doi: 10.1073/pnas.1821778116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wissemann WT, et al. Association of Parkinson disease with structural and regulatory variants in the HLA region. Am. J. Hum. Genet. 2013;93:984–993. doi: 10.1016/j.ajhg.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bandres-Ciga S, et al. The genetic architecture of Parkinson disease in Spain: characterizing population-specific risk, differential haplotype structures, and providing etiologic insight. Mov. Disord. 2019;34:1851–1863. doi: 10.1002/mds.27864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hill-Burns EM, Factor SA, Zabetian CP, Thomson G, Payami H. Evidence for more than one Parkinson’s disease-associated variant within the HLA region. PLoS ONE. 2011;6:e27109. doi: 10.1371/journal.pone.0027109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kamoun M, et al. HLA amino acid polymorphisms and kidney allograft survival. Transplantation. 2017;101:e170–e177. doi: 10.1097/TP.0000000000001670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kannarkat, G. T. et al. Common genetic variant association with altered HLA expression, synergy with pyrethroid exposure, and risk for Parkinson’s disease: an observational and case-control study. NPJ Parkinsons Dis.10.1038/npjparkd.2015.2 (2015). [DOI] [PMC free article] [PubMed]
- 18.Levesque S, et al. Reactive microgliosis: extracellular micro-calpain and microglia-mediated dopaminergic neurotoxicity. Brain. 2010;133:808–821. doi: 10.1093/brain/awp333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sulzer D, et al. T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature. 2017;546:656–661. doi: 10.1038/nature22815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lindestam Arlehamn CS, et al. α-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson’s disease. Nat. Commun. 2020;11:1875. doi: 10.1038/s41467-020-15626-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fayyad M, et al. Investigating the presence of doubly phosphorylated α-synuclein at tyrosine 125 and serine 129 in idiopathic Lewy body diseases. Brain Pathol. 2020;30:831–843. doi: 10.1111/bpa.12845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li XY, et al. Phosphorylated alpha-synuclein in red blood cells as a potential diagnostic biomarker for multiple system atrophy: a pilot study. Parkinsons Dis. 2020;2020:8740419. doi: 10.1155/2020/8740419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grifoni A, et al. Characterization of magnitude and antigen specificity of HLA-DP, DQ, and DRB3/4/5 Restricted DENV-Specific CD4+ T Cell Responses. Front. Immunol. 2019;10:1568. doi: 10.3389/fimmu.2019.01568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang KH, Wu YR, Chen YC, Fung HC, Chen CM. Association of genetic variants within HLA-DR region with Parkinson’s disease in Taiwan. Neurobiol. Aging. 2020;87:140 e113–140 e118. doi: 10.1016/j.neurobiolaging.2019.11.002. [DOI] [PubMed] [Google Scholar]
- 25.Ma ZG, Liu TW, Bo YL. HLA-DRA rs3129882 A/G polymorphism was not a risk factor for Parkinson’s disease in Chinese-based populations: a meta-analysis. Int. J. Neurosci. 2015;125:241–246. doi: 10.3109/00207454.2014.926349. [DOI] [PubMed] [Google Scholar]
- 26.Sun C, et al. HLA-DRB1 alleles are associated with the susceptibility to sporadic Parkinson’s disease in Chinese Han population. PLoS ONE. 2012;7:e48594. doi: 10.1371/journal.pone.0048594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao Y, et al. Association of HLA locus variant in Parkinson’s disease. Clin. Genet. 2013;84:501–504. doi: 10.1111/cge.12024. [DOI] [PubMed] [Google Scholar]
- 28.Foo JN, et al. Identification of risk loci for Parkinson disease in Asians and comparison of risk between Asians and Europeans: a genome-wide association study. JAMA Neurol. 2020;77:746–754. doi: 10.1001/jamaneurol.2020.0428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nalls MA, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 2014;46:989–993. doi: 10.1038/ng.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gan-Or Z, et al. The Quebec Parkinson network: a researcher-patient matching platform and multimodal biorepository. J. Parkinson’s Dis. 2020;10:301–313. doi: 10.3233/JPD-191775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simón-Sánchez J, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 2009;41:1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu JZ, Erlich Y, Pickrell JK. Case-control association mapping by proxy using family history of disease. Nat. Genet. 2017;49:325–331. doi: 10.1038/ng.3766. [DOI] [PubMed] [Google Scholar]
- 33.Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience10.1186/s13742-015-0047-8 (2015). [DOI] [PMC free article] [PubMed]
- 34.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abraham G, Inouye M. Fast principal component analysis of large-scale genome-wide data. PLoS ONE. 2014;9:e93766. doi: 10.1371/journal.pone.0093766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng X, et al. HIBAG—HLA genotype imputation with attribute bagging. Pharmacogenomics J. 2014;14:192–200. doi: 10.1038/tpj.2013.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karnes JH, et al. Comparison of HLA allelic imputation programs. PLoS ONE. 2017;12:e0172444. doi: 10.1371/journal.pone.0172444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choi SW, O’Reilly PF. PRSice-2: polygenic risk score software for biobank-scale data. Gigascience. 2019;8:giz082. doi: 10.1093/gigascience/giz082. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Anonymized data will be shared by request from any qualified investigator. All summary statistics are found in the Supplementary Data. The accession numbers to the datasets from dbGaP (https://www.ncbi.nlm.nih.gov/gap/) used in this study are: International Parkinson’s Disease Genomics Consortium (IPDGC) NeuroX dataset (dbGap phs000918.v1.p1), National Institute of Neurological Disorders and Stroke (NINDS) Genome-Wide genotyping in Parkinson’s Disease (dbGap phs000089.v4.p2), NeuroGenetics Research Consortium (NGRC) (dbGap phs000196.v3.p1) and Vance (dbGap phs000394).
The scripts used in this analysis is available at https://github.com/gan-orlab/HLA_HIBAG/.