

Highlights
-
•
Web server for MD-TASK and MODE-TASK, with new tools and updates.
-
•
Eight dynamic residue network centrality metrics for analyzing protein molecular dynamics, extended for static proteins.
-
•
Comparative essential dynamics for improved comparison of independent molecular dynamic simulations of related proteins.
-
•
A communication propensity tool for evaluating residue communication efficiency.
-
•
Normal mode analysis of proteins from static structures and molecular dynamic simulations.
Keywords: MD-TASK, MODE-TASK, Residue network analysis, Molecular dynamics analysis, Normal mode analysis
Abstract
The web server, MDM-TASK-web, combines the MD-TASK and MODE-TASK software suites, which are aimed at the coarse-grained analysis of static and all-atom MD-simulated proteins, using a variety of non-conventional approaches, such as dynamic residue network analysis, perturbation-response scanning, dynamic cross-correlation, essential dynamics and normal mode analysis. Altogether, these tools allow for the exploration of protein dynamics at various levels of detail, spanning single residue perturbations and weighted contact network representations, to global residue centrality measurements and the investigation of global protein motion. Typically, following molecular dynamic simulations designed to investigate intrinsic and extrinsic protein perturbations (for instance induced by allosteric and orthosteric ligands, protein binding, temperature, pH and mutations), this selection of tools can be used to further describe protein dynamics. This may lead to the discovery of key residues involved in biological processes, such as drug resistance. The server simplifies the set-up required for running these tools and visualizing their results. Several scripts from the tool suites were updated and new ones were also added and integrated with 2D/3D visualization via the web interface. An embedded work-flow, integrated documentation and visualization tools shorten the number of steps to follow, starting from calculations to result visualization. The Django-powered web server (available at https://mdmtaskweb.rubi.ru.ac.za/) is compatible with all major web browsers. All scripts implemented in the web platform are freely available at https://github.com/RUBi-ZA/MD-TASK/tree/mdm-task-web and https://github.com/RUBi-ZA/MODE-TASK/tree/mdm-task-web.
1. Introduction
Molecular dynamics (MD) simulations are a very useful method of conformational sampling to study the dynamics of proteins. Due to the large number of internal degrees of freedom and the complex set of atomic interactions found within proteins, investigating the effect of local differences within them using conventional metrics such as root mean square deviation (RMSD), root mean square fluctuation (RMSF) or the radius of gyration (Rg) may be limiting. Alternative analysis approaches may provide deeper insights.
Network analysis has the ability to abstract out such complexity while maintaining the inter-residue relationships. The term “centrality” is used as a measure of how central a residue is in the protein network, and several centrality metrics derived from the social sciences [1] may be applied to investigate protein dynamics. Many research groups have applied residue interaction network analysis on static structures, and have used multiple strategies and tools for summarizing protein interactions using various edge and node modeling approaches [2], [3], [4], [5] to minimize bias and maintain enough variance for determining topological changes in a protein. We previously proposed a post-hoc analysis approach of MD simulations using dynamic residue network (DRN) analysis to probe the impact of mutations [6], [7] and allosteric effects [8], before setting up the MD-TASK tool suite [9] in 2017. The tool introduced the concept of averaging residue network metrics over MD simulations, as an alternative to examining static networks, to consider the dynamic nature of functional proteins. This decision was made after observing that energy minimization criteria (such as the number of steps and the minimum gradient) influenced network centrality in such a way that related samples aggregated mainly according to minimization criteria. Nevertheless, these residue interaction networks are a faster way of estimating centrality as they utilize single conformations. Besides DRN, the MD-TASK tool suit also includes dynamic cross-correlation (DCC) and perturbation response scanning (PRS) techniques. Both of these techniques provide residue level analysis with different perspectives and none of them are found in commonly used MD packages. Some examples of their usage are given in the Results and Discussion section.
Our second software suite, MODE-TASK [10] was developed for the analysis of large-scale protein motions, mainly via anisotropic network model (ANM) calculations from static protein structures, and also providing various algorithms for estimating protein essential dynamics (ED) from MD simulations. These tools are helpful for the investigation of functionally-relevant changes that can vary with different orders of magnitude, and can also be more challenging to compute in the case of very large systems such as viral capsids. Normal mode analysis (NMA) and ED are two useful methods to study protein dynamics, which can assist in the analysis of structural and functional relationships [11], [12], [13].
Both MD-TASK and MODE-TASK have been highly utilized to mine protein dynamics by offering a series of novel approaches that have demonstrated their applicability in a growing number of cases [6], [13], [14], [15], [16], [17], [18], [19], [20], [21], [22], [23], [24], [25], [26], [27], [28], [29], [30]. Although both software suites are relatively easy to use, the required technical knowledge and software dependencies may act as a hurdle against more widespread usage of the tools and techniques, due to the diversity of operating systems and the relatively fast-evolving Python libraries. We aimed to bridge this gap by providing access to both tools while introducing new functionalities in MDM-TASK-web. It has been designed with a simple and intuitive interface that is supported by any recent web browser. The need for additional software, complex dependencies and command line expertise is greatly reduced. MDM-TASK-web includes new features such as additional network centrality metrics for both DRN and Residue Interaction Network (RIN) centrality calculations from single structures, a communication propensity (CP) tool [26], [31], an aggregator of weighted residue contact maps, comparative ED, an ANM workflow, NMA from MD and integrated 2D/3D visualization. All tools have been ported to Python 3.
MDM-TASK-web is not alone. There are a number of other tools and web servers aimed at facilitating structural analysis and offering features that are additional to, or variations of currently described methods. Some examples are ENCoM [32], DynaMut2 [33], LARMD [34], NAPS [2], ANCA [3], RIP-MD [4] and MDN [5]. Yet, MDM-TASK-web which uses and builds on top of algorithms previously described in MD-TASK and MODE-TASK, has many unique features compared to other web servers. These features are detailed in the rest of the article. We also provide a comparative table for some of these web servers and tools, in Section 4. Very briefly, ENCoM factors in amino acid variation in coarse-grained NMA. DynaMut2 integrates graph-based signatures and NMA to predict stabilizing and destabilizing mutations using random forest regression. LARMD is designed to run and analyze up to 4 ns (conventional and steered) MD simulations by running standard protocols mostly aimed at analyzing ligand binding and unbinding. In addition to calculating NMA from static structures, it also calculates PCA, performs community analysis from protein contact networks, MM-PBSA and dynamic cross correlation (DCC) calculations from these simulations. We note that LARMD is based on algorithms described in Bio3D [35], MDTraj [36], CAVER3.0 [37], and tools from the AMBER16 suite. We further provide a comparative table for some of these web servers and tools, in Section 4.
In this article, both the server workflow and functionalities of MDM-TASK-web are described and discussed using example data and published literature.
2. Materials and methods
2.1. Workflow and file inputs for MDM-TASK-web
MDM-TASK-web is a single-page web application powered by the Django web framework [38] and a MySQL database. It relies on the Bootstrap [39] framework and the Knockout.js [40] library for a dynamic and responsive front-end, while jobs are handled by the Job Management System (JMS) [41]. The workflow used by MDM-TASK-web is shown in Fig. 1. Compared to servers such as LARMD, CABS-flex [42] and MDWeb [43], which can be used to both run and analyze MD simulations, no molecular simulation is performed in MDM-TASK-web. Depending on the tool, user inputs mainly consist of a previously simulated protein MD trajectory file and its matching topology, or simply of the PDB file for a single structure. All of the tools perform computations on coarse-grained protein residues, whereby the protein is either entirely composed of alpha carbon atoms (Cα), or alternatively of beta carbon (Cβ) and glycine Cα atoms. The user should ensure that periodic boundary condition (PBC) corrections have been done prior to using the tools, as these will negatively impact the calculations – the embedded 3D visualizer may provide assistance in this case, and obvious cases of broken molecules will distort the cartoon representation of the protein.
Fig. 1.

Workflow for MDM-TASK-web. The flow of execution is numbered, starting with user inputs, and ends with the visualization stage, along the unidirectional arrows. Double-sided arrows denote the two-way communication handled by JMS. Internal processes are shown in gray boxes. Front-end and back-end functionalities are highlighted with a light red and yellowish background, respectively. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Inputs from the web page are handled using jQuery and Knockout.js, and job states are synchronized with a MySQL database, which is itself managed by the Django framework. The JMS API monitors job states and compute node availability in order to schedule job submission. Job status (failure or success) is reported to JMS, which then updates the job record in the MySQL database. Depending on the tool and the job state, the relevant outputs are directly displayed using the 2D figures generated by the MDM-TASK-web Python scripts, or visualized in 3D using the NGL Viewer (version 2) [44] web application.
2.2. 3D visualization and tool documentation
Calculated metrics (such as correlations from PRS, network centrality values and normal modes) are mapped onto a user-provided protein structure to facilitate interpretation where possible, via the NGL Viewer. A dedicated viewer is also available for generic trajectory visualization. Tool documentation is implemented via tool tips, collapsible buttons, demonstration pages and as a side panel on each of the tool input web pages.
2.3. Trajectory management
Transfer and storage of MD data can be a challenge for web servers as working with these large files can be offset by bandwidth and storage limitations. MDM-TASK-web can re-use trajectories and suggests preliminary solvent removal from the trajectory and topology files. A coarse-graining tool (https://github.com/oliserand/MD-TASK-prep) (compatible with MDTraj [36], PYTRAJ [45], MDAnalysis [46], GROMACS [47], VMD [48] and CPPTRAJ [49]) can also be used to reduce trajectory sizes by retaining only Cα and Cβ atoms. Trajectory files can be provided as URLs, and the uploaded data is re-usable. Together, these features minimize bandwidth usage and facilitate the processing of remotely simulated data without the need for specialized hardware onsite. While user data is privately stored on the server, topology and trajectory data is automatically removed after 30 days. The maximum size for a trajectory is limited to 250 Mb.
2.4. MD-TASK functionality
MD-TASK provides tools for performing DRN analysis, weighted contact network calculations, dynamic cross correlation (DCC) and perturbation response scanning (PRS) calculations. These are detailed in the sub-sections below.
2.5. DRN and RIN calculations
The previous implementation of DRN analysis has been upgraded to eight centrality metrics, and now includes betweenness centrality (BC), average shortest path lengths (L), closeness centrality (CC), eccentricity (ECC), degree centrality (DC), eigencentrality (EC), PageRank (PR) and Katz centrality (KC) that are each computed for each frame. These are then summarized for each residue as a mean, median or a standard deviation. The tool has also been adapted for using a single protein conformation (RIN centrality metrics), which is faster to compute. Details of DRN calculations are given in Table 1:
Table 1.
DRN metrics and their interpretations (adapted from [50]).
| Metric | Equation | Interpretation |
|---|---|---|
| Averaged degree centrality | Aijk is the adjacency from the 3D tensor consisting of a time series of adjacencies Aij from adjacency matrices A. It is the averaged connectivity around a residue i. The number of frames is denoted by m. A residue is more central if it has a high local connectivity. | |
| Averaged betweenness centrality | BC measures the fraction of all s-t node pairs that traverse a given node v along their geodesic distance. σi(s,t|v) denotes the number of paths bridged by residue v, while σi(s,t) is the total number of paths for the graph, both evaluated at time i. For each residue, this value is then averaged from the total m frames. | |
| Averaged farness | The farness (at time i) for a node v is the sum of its geodesic distance di to every other node u, normalised by the number of residues. The higher this number, the longer the distance to be travelled to reach other nodes of the network graph. | |
| Averaged closeness centrality | Closeness is the inverse of farness and is maximised when the latter is smallest. In other words, a node would have a high closeness when its geodesics to every other node are shortest. | |
| Averaged eigencentrality |
(Eq. i) (Eq. ii) |
Eigencentrality is an extension of degree centrality. It assigns node importance by solving for the dominant unit eigenvector EC of the adjacency matrix A. Eq. i shows the eigenvector decomposition method that can be used to determine EC. In NetworkX, EC is solved using the power iteration method. In Eq. ii, the averaged EC for residue i is determined from the time average from m frames. The converged eigenvector is a metric that recursively assigns importance, giving high centrality to nodes that have a high degree or to those connected to high importance nodes. |
| Averaged Katz centrality |
(Eq. i) (Eq. ii) |
KC is a generalization of EC, which via two constants, namely an adjacency damping coefficient α and a basal adjacency β, assigns a centrality on the basis of a node’s immediate connectivity. While β avoids adjacencies of zero, α weighs the magnitude of each centrality value. Node centrality can be dampened to various extents – larger values of α make KC tend towards EC. |
| Averaged PageRank |
(Eq. i) (Eq. ii) |
PR is an adjusted version of KC, which also assigns node centrality based on that of their neighbors. For each round of the power iteration, the centrality of each neighbor to a node is normalised by its own degree D (given the graph is undirected), and each of the resulting neighbors' centrality is summed up and assigned to the parent node. As in KC, it also includes a damping factor α and a constant β. |
| Averaged eccentricity | ECC is the longest path from a node to any other node in a graph. |
In DRN analysis, the selected network centrality metric [50] is computed for each MD frame, and the residue centrality values are aggregated as medians or time-averages. The mapped 3D structures can be directly visualized and compared in MDM-TASK-web. Mappings are also saved in the PDBx/mmCIF format, with each DRN metric stored in the B-factor field. CSV files of the DRN metrics are also generated. The default cut-off value of 6.7 Å is recommended [24], [51]. While smaller or larger values will generally work in simpler calculations such as DC, convergence problems will arise for larger values for metrics based on shortest path calculations or those that solve for eigenvectors. This will also significantly increase the computation time, due to the creation of more edges. Too small cut-off values will surely lead to disconnected nodes, and to the failure of some of metrics.
2.6. Weighted residue contact network calculations
The original R implementation from MD-TASK was ported to Python 3, with the ability to aggregate multiple residue contacts to produce a heat map. The weighted residue contact network is calculated at one selected residue locus - this would generally be a common position, or a mutation position across several related proteins [27], [52], [53].
2.7. Perturbation response scanning
The PRS back-end script has been slightly simplified from that used in MD-TASK, to require fewer parameters and structure preprocessing steps. It requires only a trajectory, an initial conformation (which is the PDB-formatted topology file) and a target conformation (also in PDB format) to generate an interactive 3D map of residue correlations. Only protein Cα atoms are used.
2.8. Dynamic cross-correlation
DCC, which shows the correlated residue motions, has been upgraded to work with protein complexes containing non-protein atoms. The speed has also been improved from the previous version.
2.9. Communication propensity
Pairwise communication propensity is computed as the mean-square fluctuation of the inter-residue distance, using Cα atoms. It relies on the fact that intra-protein signal transduction events are directly related to the distance fluctuations of communicating atoms [26], [31]. Low CP values correspond to more efficient (faster) communication compared to larger values.
2.10. MODE-TASK functionality
MODE-TASK enables the calculation of protein ED, and the estimation of normal modes both from coarse-grained static proteins under the assumptions of the elastic network model, and from MD trajectories. The coarse-graining approach used for the normal mode calculations is based on glycine Cα and Cβ atoms, and can be tuned at various levels of sampling for large proteins such that atoms are equally distributed in 3D space [13]. This approach is sequence agnostic, and differs from other web servers such as ENCoM, which accounts for amino acid differences in normal mode calculations by factoring in intramolecular residue interactions [32]; or DynaMut2, which combines conformational sampling, graph-based signatures and NMA to generate consensus predictions of the impact of mutations on protein stability and flexibility [33], [54]
2.11. Normal mode calculations from static proteins and their MD simulations
In ANM, the user is guided from the initial (optional) coarse-graining step, to solving and visualizing the normal modes. This use of a work-flow for this tool was chosen due to the need to repeat the model construction step when the cut-off value is inadequate or if the coarse-graining level is too high. A common indication of this is the lack of the leading zero-valued eigenvalues for the first six trivial modes. Arrows are colored by chain. Mean square fluctuations from all modes and from the first 20 non-trivial modes are separately displayed. NMA can now be computed from MD simulations as well, to represent the first dominant motion. For this calculation, the transpose of the reshaped Cartesian coordinate tensor is mean centered and dotted with its transpose to obtain the covariance matrix of dimension 3 N × 3 N (where N is the number of residues). This matrix is then diagonalised by eigen decomposition to retrieve the principal components [55], in descending order of eigenvalue. The percentage of explained variance is then displayed for the first 50 modes, together with the 3D mapping of the NMA using the protein topology file. A multi-PDB file is also produced to show the mode animation. Two parameters (the ignc and ignn parameters) control the number of C- and N-terminus residues to ignore from the structural alignment and the covariance matrix. These were included as a means to decrease possible technical variation from the termini. In our experience with protein MD simulations, we have often observed relatively high levels of fluctuation at the C-terminus.
2.12. Essential dynamics, with improvements for comparing pairs of protein simulations
Essential dynamics tools (multidimensional scaling, standard PCA, internal PCA and t-SNE) from MODE-TASK are integrated with basic default options. A new tool, which performs comparative ED aligns one trajectory to a reference trajectory before performing a single decomposition to lay out all conformations on a common set of principal axes, such that the percentage of explained variance is the one shared by both trajectories. Comparative ED features automated conformation extraction from lowest energy basins and applies k-means to sample centroid conformations from the first 2 principal components in standard PCA. N- and C-terminal residues may be deselected before the structural alignment step to reduce unwanted noise and improve performance. Residue selection is also enabled, and is applied post global fitting of the Cα atoms. This is to be used only for residue and chain selections. When more than two samples are to be compared, the stand-alone script is recommended for processing all samples at once (and not as multiple paired runs), and one should also consider the percentage of explained variance captured by the principal axes. Despite its name, the comparative ED tool can also be applied to a single trajectory, in which case it will not differ from the standard PCA algorithm, if all atoms are selected.
3. Results and discussion
For a demonstration of use cases of MDM-TASK-web, trajectory-based tools are evaluated using mutants of the dimeric HIV-1 protease (198 residues) [56] and a SARS-CoV-2 main protease (Mpro) [28], while the enterovirus 71 capsid pentamer (PDB ID: 3VBS [57]; 842 residues) is used for demonstrating the calculation of the anisotropic network model, which is based on a single protein conformation. The topology and MD trajectory of the HIV protease mutant that was used for most of the tool demonstrations was obtained from our previous work [56], which used labeled sequence data from the Stanford HIVdb [58], bearing major drug mutations (DRMs) D30N, V32I, M46I, I47V, I54L, I84V and L90M, together with accessory DRMs L10F, L33F, F53L and N88D and other mutations. In most cases, a topology and an MD trajectory are needed. In these cases, the step size parameter controls the frame sampling rate and speed of calculations, which are inversely related. Another factor that can play a potentially important role in the stability of the various trajectory-based calculations is the equilibration state of the protein. Residual effects from prior temperature and/or pressure equilibration will to some degree influence any of the aggregated metrics, and one may benefit from removing such artifacts.
3.1. The MDM-TASK-web interface
The web server tools each have a section where the job inputs are specified, as shown in Fig. 2, for the standard PCA tool. All the tools are listed on the top menu and they generally require an MDTraj-compatible topology file and its trajectory (e.g. from GROMACS or other MD simulation tools), unless specified otherwise. In addition to the documentation sources shown in Fig. 2, those of MD-TASK and those of MODE-TASK are also embedded in the “USER HELP” section, for further reference. The demonstration pages further give a use-case example of each tool.
Fig. 2.

Example of the MDM-TASK-web interface, showing the embedded sources of documentation. Documentation is mainly embedded within each input page via a drop-down button, on the side panel, as hoverable tool tips and within demonstration pages.
3.2. Dynamic residue network & residue interaction network centrality calculations
We and others, in a number of publications, showed the effectiveness of our DRN approach [6], [16], [19], [23], [24], [25], [26], [27], [28], [29], [30], [59], [60]. While MD-TASK has the functionality to calculate L and BC, MDM-TASK-web, now, provides further metric options. Additionally, the paired visualizer of MDM-TASK-web enables the comparison of related calculations by scaling the color range to span the global minimum and maximum of any two selected proteins. It may be desirable to structurally align the topology files when two homologous proteins are to be compared in order to facilitate comparison of the mapped structure. It is also worth noting that the same back-end tool (calc_network.py) can compute centrality calculations from single protein conformations when it is provided with the same topology file for both the topology and trajectory parameters (seamlessly integrated as one option under the RIN section).
Some of these metrics were previously used over single static structures. CC metric calculations, for instance, were applied to identify active site residues together with other approaches, e.g. conservation, solvent accessibility [61], [62]. MDM-TASK-web gives metric calculation options over DRN or in static form.
As a case study, we used two multi-drug resistant HIV proteases, and the results from DRN centrality calculations were mapped onto user-supplied topology files, as shown in Fig. 3. We compared the averaged BC values of a multi-drug (highly) resistant HIV protease bound to the antiretroviral (ARV) darunavir (DRV) (Fig. 3(A)) to another multi-drug resistant (but still DRV-susceptible) HIV protease bound to the drug tipranavir (TPV) (Fig. 3(B)). In the same figure, flap residue VAL54 (numbered 153; circled and highlighted in panel (B)) shows a decreased averaged BC from its homologous position in panel (A), where the residue was LEU54. Residues are labeled according to the numbering present in the provided topology file. The B-factor field of the CIF files can be used in other tools for improved visualization, as explained in the current section, for the PyMOL software.
Fig. 3.

Top views of two DRN metrics mapped in two highly drug resistant HIV protease mutants. Averaged BC is shown as a “spacefill” representation in a common color scale for panels (A) and (B); while averaged EC is shown as a “cartoon” representation in panels (C) and (D) – as obtained from MDM-TASK-web. Panel (A) shows a DRV-bound multi-drug (highly) resistant HIV protease, and panel (B) shows another, TPV-bound multi-drug resistant HIV protease, for which DRV is still effective. A color gradient ranging from pale yellow to red in the top and bottom panels, is used to represent low to high centrality values. Non-protein portions are colored blue. The flap residue 54 (numbered 153 in chain B) is circled and highlighted in the top panels, showing the decreased averaged BC in the DRV-susceptible mutant, where the residue had mutated. Panels (C) and (D) make visible inner details of averaged EC at the core of the proteases, hinting at the highly central catalytic aspartate by black arrows. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
It can further be seen from the averaged EC values in Fig. 3(C) and 3(D) that the floor of central cavity (the catalytic aspartic acid, indicated by the black arrow) has a very high average eigencentrality, indicating that it is likely connected to other well-connected residues of the active site. Additionally, this retention of centrality is very likely linked to the presence of stabilizing networks of H-bonding interactions and usage of a “fireman’s grip” segment in the protease [63].
High averaged EC is seen at the center of the protease, mainly arising from the recursively acquired centrality derived from the nearby crowd of residues close to the catalytic site. As these crowds are stably maintained, they manifest high connectivities that are typical of a compact environment. Consequently, this metric is pointing to a known critical part of the HIV protease (the catalytic residues), which relies on the stability of other nearby residues, possibly for maintained function. High connectivity (degree) nodes also tend to have a high eigencentrality, especially when surrounded by other high connectivity nodes, due to their dependence on residue neighborhood.
For more experienced users using PyMOL [64] for example, it is straightforward to use the PDBx/mmCIF-formatted files to visualize and compare related metrics for multiple related proteins using the “spectrum” command (with the B-factor values) combined with the “set grid_mode” command. Furthermore, the computed centrality metrics are all saved as a CSV file, which can be used for customized analyses by the users. For instance, one may use the data for plotting the overall centrality density distribution of a protein. It is also possible to calculate median residue centrality instead of the average, which if significantly different from the default DRN metric type may indicate the presence of skewed or multi-modal centrality distributions among the residues. In such cases, the median option could be a more robust estimate of the DRN centrality.
3.3. Weighted residue contact network and heat maps
Weighted residue contact maps are a helpful functionality for examining local residue contact frequencies over MD simulations for many different biological questions, including protein–protein interactions, and in the identification of contact changes due to mutations. MD-TASK has the functionality to calculate the contact frequencies around a single residue. A recent example to use of this functionality is the analysis of ACE2 and spike-RBD protein interaction behavior round a specific ACE2 residue, K353 [53]. This functionality has been further developed in MDM-TASK-web to be able to analyze multiple cases at the same time, and presented as heat maps. Chebon-Bore et al. [27] used this functionality to identify changes in protein-drug interactions due to malarial resistance mutations.
In our example here, Fig. 4 (A) shows the residue contact frequencies around GLN18 in an HIV protease mutant. Each map is associated with a file of weighted edges that can be aggregated and summarized using the contact heat map tool, for larger scale comparisons of a given locus across several protein samples, as shown in Fig. 4 (B). The analysis of local neighborhoods directly provides information about the conservation, and lack thereof, of residue contacts with possibly key residues associated with function. From Fig. 4 (B), one can see the noticeable gains (or maintenance) of contact between GLN18 and ILE36 in the samples labeled Demo4, Demo5 and Demo6, and its loss (or absence) in the remaining samples. One should interpret these in the light of their MD simulations, as the observed frequencies would be a direct result of the explored protein conformations.
Fig. 4.

Estimating contact frequencies around a single residue in a single and in multiple protein structures, using HIV protease mutants as example. The locus of interest is displayed at the center in panel (A), and is surrounded by its neighbouring residues. Additionally, the edge thicknesses and labels depict the residue contact frequencies obtained from the MD simulations. In both panels, residues are depicted by the three-letter residue code followed by the residue position, a dot and the chain label. In panel (B), multiple related contacts (gathered from the contact mapping tool) are stacked on top of each other along the y-axis, with their neighbours spanning the x-axis.
3.4. Communication propensity
By computing the CP metric, one is able to investigate residue pairs that are more or less likely to maintain their distances. This approach, for instance, was used in the analysis and identification of differences in communication efficiency between the ligand-free and the ligand bound complex of human heat shock protein 90 [26].
As an example of the interpretation of the CP metric, the topology and trajectory files of a highly (multi-drug) resistant HIV protease mutant (Fig. 5) were used, with default parameters for the tool. From the figure, it can be seen some residue pairs display relatively higher distance variations [for e.g. between residue index pairs (37, 50), (37, 135), and (40, 91)], indicating reduced stability between these loci. This can be a preliminary investigation before proceeding to more detailed analyses. It is also possible to compare CP matrices produced by the tool in two states of homologous proteins (for example a WT and a mutant) in order to deduce the increased or decreased variance in distance between residue pairs, and extract meaningful insights about the effect of mutation in a protein. At the time of writing, this feature is only available from the command line, by providing two CP matrices (each produced by the “cp.py” tool) to the “cp_analyse.py” tool found in the GitHub repository, whereby the CP matrix specified by the “--diff” parameter is subtracted from the other matrix to produce a delta CP heat map. Once more, one has to factor in the conformational sampling representativeness of the provided MD trajectories.
Fig. 5.
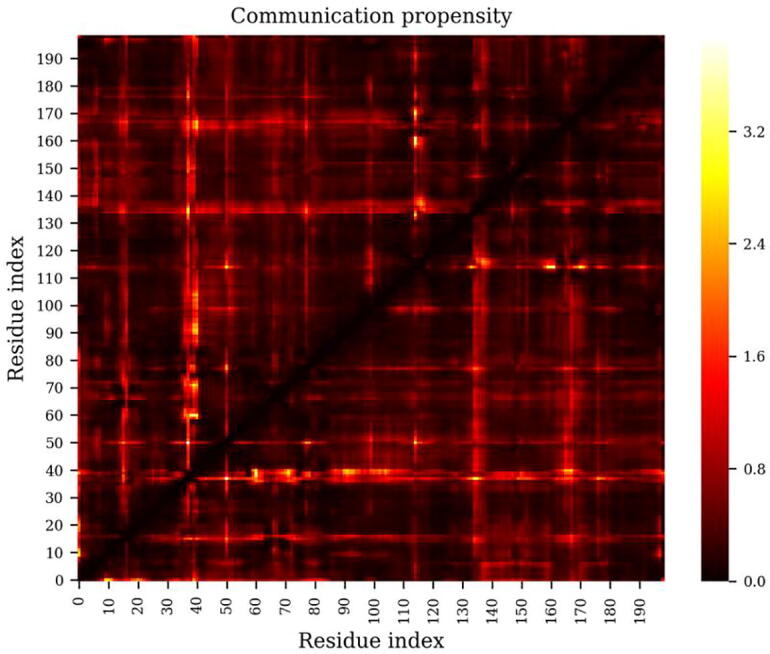
The coordination propensity calculation shows the variance in the distance between residue pairs in an HIV protease mutant.
3.5. Dynamic cross-correlation
The DCC algorithm is unchanged from that of MD-TASK, with the exception that it is now faster and supports additional atom types. Some of the recent examples from the published literature that used the MD-TASK DCC functionality include [16], [17], [18], [21].
In the current example Fig. 6, the MD simulation of a highly (multi-drug) resistant HIV protease was used. From the DCC heat map, one can inspect the trend in movement of residue pairs – these can trend together, apart or be independent. Such analyses often detect protein segments that are functionally-related. For instance, by visualizing the antiparallel regions of high cross-correlation (across the main diagonal), we find (1) residues 10–24, which form a beta hairpin known as the fulcrum; (2) residues 55–75, in which two beta hairpins (the flap and the cantilever) are connected by a common beta strand; (3) residues 45–55, which form the tip of the flap from a beta hairpin that controls substrate and inhibitor access to the catalytic site; (4) residues 95–105, which partly consist of a beta strand that composes the dimerization region. As the protein is dimeric, the patterns show a partial symmetry. It is therefore possible to set up experimental designs and monitor correlative changes associated with a given condition using DCC. While being very useful, one should also be mindful of the limitations of conformational sampling when interpreting such graphs.
Fig. 6.
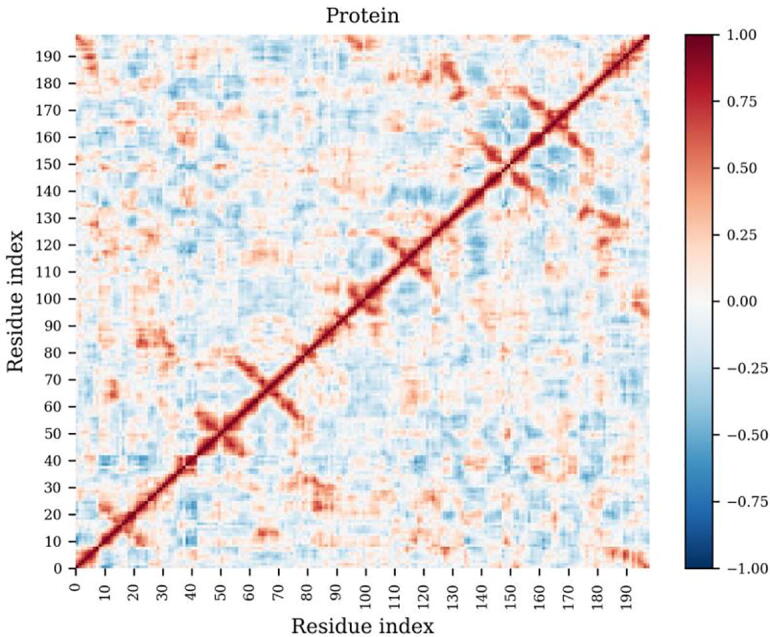
Pairwise residue correlations from an MD simulation of an HIV protease mutant. Anti-correlated and correlated movements are denoted by negative and positive DCC values, respectively, in the range [−1, 1], while uncorrelated motion has a value of zero.
Further, by a judicious choice of atom type(s) (and/or trajectory data) one can for instance investigate protein/nucleic acid complexes using a comma-separated list “CA,P”.
3.6. Normal MODE analysis (ANM and NMA from MD)
NMA can be done using either a single PDB file or multiple protein conformations from an equilibrated MD trajectory. Single conformation NMA is done according to the anisotropic network model, as implemented in MODE-TASK, while eigen decomposition of the covariance matrix is used for MD data. In the case of the ANM, a coarse-graining level of four, Cβ atoms and a cut-off distance of 24 were chosen to obtain six leading zero eigenvalues (displayed in the web server NMA workflow), corresponding to the trivial modes. By default, mode 7 (1st non-trivial) is displayed, as shown in Fig. 7 (A), but other modes can also viewed by cycling through. From the ANM, we observe rotational motions within the enterovirus 71 capsid pentamer.
Fig. 7.

Normal mode analysis using (A) the anisotropic network model obtained from a static viral capsid pentamer, and (B) the MD covariance matrix of an HIV protease mutant. In each case, each arrow is colored by its parent chain. The arrow at each residue denotes both the extent of motion and direction with respect to each of the residue.
NMA was then computed from the MD trajectory of an HIV protease mutant (Fig. 7 (B)). The decomposition differs from that used in essential dynamics in the definition of the variables. While MD frames are the variables in ED, protein residues are chosen as variables in NMA. From the first dominant motion, we observe a relatively higher amount of motion coming from the outer lateral portion of the fulcrum of the protease; internal motions are significantly smaller, with the flaps traveling only slightly more. The relative extents of motion within each chain also reveal a degree of chain asymmetry within the homodimer. While the back-end script can represent all modes, only one mode is represented in the web server at the moment – others may be shown in a future update. The dominant mode has indeed summarized an important aspect of the protease’s functional mechanics – a rotational motion of each monomer, that has been shown to be associated with the deformation and expansion of the active site [65], [66]. A similar strategy in other unknown proteins may lead to significant insights, especially for the design of inhibitors and modulators, and more generally for understanding protein motion.
3.7. Essential dynamics
ED is demonstrated via the new comparative ED tool using an early pandemic stage mutant of the SARS-CoV-2 Mpro (GISAID identifier: EPI_ISL_420610 [67]) and a reference protease, that had each been previously MD-simulated for 100 ns in our previous study [28]. In addition to uploading the trajectories and topologies to the web server, substrate binding residues were selected (as an example) using the MDTraj syntax “residue 24 25 26 41 49) or (residue 140 to 145) or (residue 163 to 168) or (residue 188 to 192)” including the quote characters. These substrate binding residues were selected from the 1.94 Å resolution crystal structure (PDB ID: 7MGR [68]) using any residue found at less than 4 Å from its bound non-structural protein substrate, using the PyMOL software. The expected number of k-means clusters was set to 3, but is not useful in this case as there is clearly one density maximum in each simulation. A very important criterion for the success and accuracy of this method lies in the correctness of structural alignment, the specification of regions of interest and the choice of equilibrated parts of trajectories. Together these decrease unwanted sources of variation, to focus on what best describes a biological event. As protomer B had displayed the non-canonical PHE140 pose, only this protomer and protomer B from the reference were prepared to be uploaded. The last 10 ns of simulation were selected in each case, using every fifth frame - due to the design implementation, the trajectories need to have the same number of frames. The ignn and ignc parameters were set to their default values of 0 and 3, respectively, to ignore three C-terminal residues, in each chain from the alignment stage onward. One can verify the number of terminal residues to ignore by independently calculating residue root mean square fluctuation (RMSF) plots – this trimming is applied to each chain. By default, the highest probability density conformations (the blue markers) are extracted from the centroids of the highest contour level using the k-nearest neighbor algorithm (where k = 1) for the points within that level, while the k-means algorithm is independently used to estimate single conformations from k possible probability density maxima, as specified from the number of clusters. In the example, conformations with the highest probability densities were observed at time t = 97560 ps and t = 98350 ps, respectively for the reference protein (Fig. 8(A)) and that from SARS-CoV-2 isolate EPI_ISL_420610 (Fig. 8(B)). The k-means prediction can be helpful as a rough estimate, when other local maxima are present and are not picked up from the maximum KDE peaks. While this algorithm is partly stochastic, this effect is mitigated by being internally parameterized with a large number of iterations (1000) and by multiple initializations (50) with different seeds.
Fig. 8.

Representations of conformational sampling from independent MD simulations of (A) a reference and (B) an early pandemic stage mutant of the dimeric SARS-CoV-2 Mpro in the same eigen subspace, using comparative essential dynamics. Dots correspond to individual protein conformations (defined by a selection) and are colored by the time of sampling. The kernel density contour plots [colored from blue (lowest density) through yellow to red (highest density)] only serve as a visual guide for the energy surface, and are independently scaled, based on the respective samples. The red labels are estimates obtained from the k-means algorithm, while the blue ones are obtained from the probability density maxima. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
By concatenating the trajectories and aligning each frame to a common reference, the tool addresses a potential problem that may arise when comparing separate MD simulations – that of the generation of distinct sets of eigenvector/eigenvalue pairs specific to each trajectory, as each set belongs to its own simulated covariance matrix. While the calculation of a single covariance matrix has higher physical memory footprint, it adequately represents the total variability shared across the trajectories, thus yielding a common set of eigenvector/eigenvalue pairs that explain the total variance. By optimizing for several of the discussed factors, a total explained variance of about 78.5% was obtained from the first 2 principal components prepared from the Mpro substrate binding residues of the two Mpro proteins, even though such performances may be influenced by the number of selected residues and protein flexibility. From the separate contours in the shared axes, as showed in Fig. 8(A) and 8(B), we clearly observe that the substrate binding residue dynamics were very different. In Fig. 8(C), which was obtained by aligning (using PyMOL) the resulting single conformations from each protease, we can see that additional conformational details associated with the non-canonical PHE140 pose were detected, with several distortions from the reference protease around the binding site. Using this tool, one can rapidly obtain equilibrium conformations from various experimental conditions, from which the user may then choose to visualize or to perform other analyses, such as those that use single conformations - for instance in centrality calculations using RIN, in ANM or as a target conformation in PRS.
3.8. Perturbation response scanning
PRS measures the tendency of an initial protein conformation to reach a known target protein conformation, by sequentially perturbing each of its residues and measuring the level of agreement via correlation calculations. The approach is very useful in determining functionally important residues (hot spots) with allosteric influence, and can therefore be used to determine candidate sites for mutagenesis studies or allosteric drug discovery approaches [59]. This method has been used for different research purposes [69], [70], [71]. We, for the first time, applied PRS to large and highly dynamic protein analysis [8], [24]. We also showed that PRS hot spot residues and the DRN metric BC are highly correlated to identify allosteric residues [24]. This correlation was, later, also identified in another study [23]. As another example, PRS findings were coupled with docking studies to investigate allosteric modulators in human heat shock proteins [24].
In this section, as an example, the interface for PRS calculation is demonstrated using the same closed conformation of HIV protease used in section 3.5, possessing high levels of (multi-drug) resistance as the starting conformation (the topology file) together with its corresponding MD trajectory and a wide-opened target conformation (PDB ID: 1TW7 [72]), which also happens to be multi-drug resistant. Applying the residue perturbation algorithm with this experimental set-up one seeks for possible trigger residues that are associated with a known conformational change of the protease flaps, proceeding from a closed to an opened state. The correlations are mapped on the starting topology, as shown in Fig. 9, and are also written to a text file. From Fig. 9, it can be seen that residues (in reddish color) from or in connection to the HIV protease flap region can be perturbed in order to make a transition from a closed to an opened conformation, seemingly with monomer asymmetry. This flap-opening movement has implications in the acceptance or release of its substrate, and similarly for inhibitors designed against it. It is known that the flaps of the protease open to accept or release its inhibitors [73], and that DRMs can reduce dimer stability and also lead to the release of a bound inhibitor [74]. Therefore, it can be insightful to study the propensity of certain mutations to lead to certain known states.
Fig. 9.

Application of PRS to scan for residues associated with the conformational shift from a closed to an opened flap conformation in HIV protease. The front (A) and (B) top views of the protease are shown, with an arrow depicting the protease flaps. The correlation mappings default to the range [0, 1], with a color gradient ranging from yellow to dark red, but can also be scaled to the range of the observed correlations by clicking on the “Min-max scaling” option when signals have a narrower range.
4. Comparing MDM-TASK-web to other existing servers for MD/protein analysis
In Table 2, we enumerate the functionalities of MDM-TASK-web and compare them against those of four existing web servers doing similar tasks.
Table 2.
Features of MDM-TASK-web and other servers used to analyse static and dynamic protein structures.
| Functionality | MDM-TASK-web | NAPS | ANCA | RIP-MD | MDN |
|---|---|---|---|---|---|
| Input file formats | Tools processing trajectories require a topology and a trajectory, both in multiple formats. PRS uses PDB files as conformations. | PDB topology and DCD trajectory file. | Single PDB files. | PDB or PSF formats. Trajectories are loaded as a multi-PDB file. | All files used by GROMACS to analyse a trajectory are needed for analyses. |
| Residue interaction network (RIN) from MD | Dynamic residue networks analysis via averaged network centralities from RINs. | Networks are aggregated to one from an MD simulation. | – | RIN for each frame | Network coupling, betweenness |
| Residue interaction network from a single structure | RIN is calculated for a single protein structure. | RIN is calculated for single protein structure. | Amino acid network (AAN) for single structure. | RIN is calculated for single protein structure. | Single network is calculated from interaction energies from MD. |
| Network metrics | Averaged network centrality metrics for betweenness, degree, eccentricity, averaged shortest paths, closeness, Katz, PageRank and eigencentrality. Same metrics are available for single conformations. |
Degree, closeness, betweenness, clustering coefficient, eccentricity, shortest paths, k-cliques, eigenspectra. | Degree, closeness, betweenness, clustering coefficient, average shortest path, edge betweenness. | – | Non-normalized and normalised node betweenness. |
| Network node types | Cβ and glycine Cα atoms. | Cβ and glycine Cα atoms, amino acids. | Cα atoms. | Cα atoms, residues. | Amino acids. |
| Network edge types | Any node < 6.7Ang to any other node. | DCC, energy, inverse distance. | Contact energy, Cα cut-off distance. | Cα contacts, H-bonds, salt-bridges, disulfide bonds, cation-π, π–π, Arg–Arg distance, Coulombic, and Lennard Jones. | Averaged inter-residue interaction energy. |
| Weighted residue contact network and heat map | Weighted contacts at a single locus. Contact heat map also aggregates residue contacts from multiple simulations for large-scale comparisons of single loci. | 2D dot matrix. | Implements a node-weighted amino acid contact energy network, and an edge-weighted amino acid contact energy network. | – | The edge weights for the network are derived from the energy involved in residue interactions. |
| Dynamic Cross Correlation | Normalised residue covariance matrix. Supports nucleic acids. | Normalised residue covariance matrix. | – | Pearson correlations of interactions | – |
| Perturbation Response Scanning | Scanning of hotspot residues leading to a target conformational change. | – | – | – | – |
| Essential dynamics (ED) | Several algorithms for visually assessing conformational distributions from MD simulations: t-SNE, internal PCA, comparative ED, multidimensional scaling. |
– | – | – | – |
| Normal Mode Analysis (NMA) | Elastic network model to extract global motions and compute MSF from a single PDB-formatted file. NMA is also computed from MD trajectories. |
– | – | – | – |
| Coordination propensity | Identifies residue pairs whose distance vary the most. | – | – | – | – |
5. Performance
Several protein structures [PDB IDs: 1HIV [75] (monomer and dimer), 2G50 [76] (monomer) and 1JYX [77] (monomer)] were used to test the performance of the averaged centrality metric calculations. It can be seen that the required time of computation of the centrality metrics (especially for averaged BC, CC, L and ECC metrics) escalates quickly (Fig. 10), ranging from minutes for 99 residues, to a few hours in the case of 1011 residues. This is mainly due to the fact that the number of potential paths to be traversed increases significantly when the number of nodes (residues) in the network graphs increases. In all cases, 101 frames were sampled from 5001 frames by specifying a step size of 50. Alternatively, the same metric can be measured from a single protein structure for a fraction of the time, but at the expense of sampling depth, using the residue interaction network option. The speed of DRN computation will be improved in a future version of the tool. The robustness of the each DRN metric was evaluated by varying the contact cut-off radius (at 6, 6.7 and 7 Å) in triplicate MD simulations, as shown in Fig. 11. We observe that the values can be influenced by its choice, however we show that centrality distributions obtained at the default value of 6.7 are very similar to those obtained at 7 Å, but experience a shift at 6 Å. Further, in our previous publication, we evaluated averaged BC values at three cut-off distances (6.7, 9 and 12 Å), and observed a change in the usage frequency for the peak BC residues by changing the cut-off, which had no observable trend, even though small values reduced the effect of flexible residues [24]. We therefore recommend a value of 6.7 Å, and reiterate the observations obtained by an earlier study [51], and which has been applied in numerous cases that used the MD-TASK software, even though values spanning 6.5–7.5 Å are also common in other studies. The reproducibility of the averaged centrality metrics is also shown in Fig. 11, using identical colors for each of the triplicate samples evaluated at each cut-off radius.
Fig. 10.

Benchmarking of the performance of MDM-TASK-web DRN metrics evaluated using triplicate MD simulations of dimeric HIV protease. Each panel shows the time taken for each of the metrics, namely the averaged (A) BC, (B) CC, (C) DC, (D) EC, (E) ECC, (F) KC, (G) L and (H) PR, for increasing protein sizes.
Fig. 11.

Reproducibility of the MDM-TASK-web DRN metrics. Probability density plots for each of the averaged network centrality metrics were evaluated from triplicate MD simulations of dimeric HIV protease, using varying residue contact cut-off radii (rc = 6, 6.7 and 7 Angstroms). Each metric is labeled along the x-axis in each of the panels (A) to (H). A cut-off value of 6 Angstroms tends to produce more divergent results.
6. Conclusion
MDM-TASK-web is a user-friendly web server for performing various types of calculations aimed at obtaining different types of insights from both static and dynamic protein data sets. By providing access to these tools in this manner also makes it available to more researchers studying proteins dynamics, without spending too much time and resources on setting up specialized hardware and software environments. The possibility of coarse-graining facilitates data transfer over the web, and tremendously reduces the data storage footprint required for the calculations, making the it more likely to be accessible for further analysis without requiring significant additional storage hardware. The novel algorithms and updates to both MD-TASK and MODE-TASK enhance the capacities of each of the tool suites.
CRediT authorship contribution statement
Olivier Sheik Amamuddy: Methodology, Software, Validation, Formal analysis, Writing – original draft, Visualization. Michael Glenister: Software, Validation. Thulani Tshabalala: Software. Özlem Tastan Bishop: Conceptualization, Methodology, Resources, Writing – original draft, Writing - review & editing, Supervision, Project administration, Funding acquisition.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Acknowledgements
We thank the Centre for High Performance Computing (CHPC); Dr D. Penkler for the CP script and RUBi members for their valuable suggestions.
Funding
This work is supported by the National Human Genome Research Institute of the National Institutes of Health under Award Number U24HG006941 to H3ABioNet; and by the Grand Challenges Africa programme [GCA/DD/rnd3/023]. Grand Challenges Africa is a programme of the African Academy of Sciences (AAS) implemented through the Alliance for Accelerating Excellence in Science in Africa (AESA) platform, an initiative of the AAS and the African Union Development Agency (AUDA-NEPAD). GC Africa is supported by the Bill & Melinda Gates Foundation (BMGF), Swedish International Development Cooperation Agency (SIDA), German Federal Ministry of Education and Research (BMBF), Medicines for Malaria Venture (MMV), and Drug Discovery and Development Centre of University of Cape Town (H3D). The content of this publication is solely the responsibility of the authors and does not necessarily represent the official views of the funders.
Conflict of interest
None declared.
References
- 1.Golbeck J. Newnes; 2013. Analyzing the social web. [Google Scholar]
- 2.Chakrabarty B., Naganathan V., Garg K., Agarwal Y., Parekh N. NAPS update: network analysis of molecular dynamics data and protein–nucleic acid complexes. Nucleic Acids Res. 2019;47:W462–W470. doi: 10.1093/nar/gkz399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yan W., Yu C., Chen J., Zhou J., Shen B. ANCA: A web server for amino acid networks construction and analysis. Front Mol Biosci. 2020;7:1–8. doi: 10.3389/fmolb.2020.582702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Contreras-Riquelme S, Garate J-A, Perez-Acle T, Martin AJM, RIP-MD: a tool to study residue interaction networks in protein molecular dynamics, PeerJ. 6 (2018) e5998. https://doi.org/10.7717/peerj.5998. [DOI] [PMC free article] [PubMed]
- 5.Ribeiro A.S.T., Ortiz V. MDN: A web portal for network analysis of molecular dynamics simulations. Biophys J. 2015;109:1110–1116. doi: 10.1016/j.bpj.2015.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown D.K., Sheik Amamuddy O., Tastan Bishop Ö. Structure-based analysis of single nucleotide variants in the renin-angiotensinogen complex. Glob Heart. 2017;12:121. doi: 10.1016/j.gheart.2017.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown D.K., Tastan Bishop Ö. Role of structural bioinformatics in drug discovery by computational SNP analysis. Glob Heart. 2017;12:151–161. doi: 10.1016/j.gheart.2017.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Penkler D., Sensoy Ö., Atilgan C., Tastan Bishop Ö. Perturbation-response scanning reveals key residues for allosteric control in Hsp70. J Chem Inf Model. 2017;57:1359–1374. doi: 10.1021/acs.jcim.6b00775. [DOI] [PubMed] [Google Scholar]
- 9.Brown DK, Penkler DL, Sheik Amamuddy O, Ross C, Atilgan AR, Atilgan C, Tastan Bishop Ö. MD-TASK: a software suite for analyzing molecular dynamics trajectories, Bioinformatics. 33 (2017) 2768–2771. https://doi.org/10.1093/bioinformatics/btx349. [DOI] [PMC free article] [PubMed]
- 10.Ross C., Nizami B., Glenister M., Sheik Amamuddy O., Atilgan A.R., Atilgan C., Taştan Bishop Ö. MODE-TASK: large-scale protein motion tools. Bioinformatics. 2018;34:3759–3763. doi: 10.1093/bioinformatics/bty427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liang Z., Verkhivker G.M., Hu G. Integration of network models and evolutionary analysis into high-throughput modeling of protein dynamics and allosteric regulation: theory, tools and applications. Brief Bioinform. 2019;00 doi: 10.1093/bib/bbz029. [DOI] [PubMed] [Google Scholar]
- 12.David C.C., Jacobs D.J. Principal component analysis: a method for determining the essential dynamics of proteins. Methods Mol Biol. 2014;1084:193–226. doi: 10.1007/978-1-62703-658-0_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ross C., Atilgan A.R., Tastan Bishop Ö., Atilgan C. Unraveling the motions behind Enterovirus 71 uncoating. Biophys J. 2018;114:822–838. doi: 10.1016/j.bpj.2017.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dehury B., Tang N., Mehra R., Blundell T.L., Kepp K.P. Side-by-side comparison of Notch- and C83 binding to γ-secretase in a complete membrane model at physiological temperature. RSC Adv. 2020;10:31215–31232. doi: 10.1039/D0RA04683C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keretsu S., Ghosh S., Cho S.J. Molecular modeling study of c-KIT/PDGFRα dual inhibitors for the treatment of gastrointestinal stromal tumors. Int J Mol Sci. 2020;21:8232. doi: 10.3390/ijms21218232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischer A., Häuptli F., Lill M.A., Smieško M. Computational assessment of combination therapy of androgen receptor-targeting compounds. J Chem Inf Model. 2021;61:1001–1009. doi: 10.1021/acs.jcim.0c01194. [DOI] [PubMed] [Google Scholar]
- 17.Wang S., Xu Y., Yu X.-W. A phenylalanine dynamic switch controls the interfacial activation of Rhizopus chinensis lipase. Int J Biol Macromol. 2021;173:1–12. doi: 10.1016/j.ijbiomac.2021.01.086. [DOI] [PubMed] [Google Scholar]
- 18.Wang S., Xu Y., Yu X.-W. Propeptide in Rhizopus chinensis lipase: new insights into its mechanism of activity and substrate selectivity by computational design. J Agric Food Chem. 2021;69:4263–4275. doi: 10.1021/acs.jafc.1c00721. [DOI] [PubMed] [Google Scholar]
- 19.Sanyanga T.A., Nizami B., Tastan Bishop Ö. Mechanism of action of non-synonymous single nucleotide variations associated with α-carbonic anhydrase II deficiency. Molecules. 2019;24:3987. doi: 10.3390/molecules24213987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khairallah A., Ross C.J., Tastan Bishop Ö. Probing the structural dynamics of the Plasmodium falciparum tunneling-fold enzyme 6-pyruvoyl tetrahydropterin synthase to reveal allosteric drug targeting sites. Front Mol Biosci. 2020;7 doi: 10.3389/fmolb.2020.575196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Borges B., Gallo G., Coelho C., Negri N., Maiello F., Hardy L. Dynamic cross correlation analysis of Thermus thermophilus alkaline phosphatase and determinants of thermostability. Biochim Biophys Acta - Gen Subj. 2021;1865:129895. doi: 10.1016/j.bbagen.2021.129895. [DOI] [PubMed] [Google Scholar]
- 22.Alnami A., Norton R.S., Pena H.P., Haider S., Kozielski F. Conformational flexibility of a highly conserved helix controls cryptic pocket formation in FtsZ. J Mol Biol. 2021;433:167061. doi: 10.1016/j.jmb.2021.167061. [DOI] [PubMed] [Google Scholar]
- 23.Amusengeri A., Tastan Bishop Ö., Discorhabdin N. A South African natural compound, for Hsp72 and Hsc70 allosteric modulation: combined study of molecular modeling and dynamic residue network analysis. Molecules. 2019;24 doi: 10.3390/molecules24010188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Penkler D.L., Atilgan C., Tastan Bishop Ö. Allosteric modulation of human Hsp90α conformational dynamics. J Chem Inf Model. 2018;58:383–404. doi: 10.1021/acs.jcim.7b00630. [DOI] [PubMed] [Google Scholar]
- 25.Kimuda M.P., Laming D., Hoppe H.C., Taştan Bishop Ö. Identification of novel potential inhibitors of pteridine reductase 1 in Trypanosoma brucei via computational structure-based approaches and in vitro inhibition assays. Molecules. 2019:1–25. doi: 10.3390/molecules24010142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Penkler D.L., Tastan Bishop Ö. Modulation of human Hsp90α conformational dynamics by allosteric ligand interaction at the C-terminal domain. Sci Rep. 2019;9 doi: 10.1038/s41598-018-35835-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chebon-Bore L., Sanyanga T.A., Manyumwa C.V., Khairallah A., Tastan Bishop Ö. Decoding the molecular effects of atovaquone linked resistant mutations on Plasmodium falciparum Cytb-ISP complex in the phospholipid bilayer membrane. Int J Mol Sci. 2021;22:2138. doi: 10.3390/ijms22042138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sheik Amamuddy O., Verkhivker G.M., Tastan Bishop Ö. Impact of early pandemic stage mutations on molecular dynamics of SARS-CoV-2 M pro. J Chem Inf Model. 2020;60:5080–5102. doi: 10.1021/acs.jcim.0c00634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manyumwa C.V., Tastan Bishop Ö. In silico investigation of potential applications of gamma carbonic anhydrases as catalysts of CO2 biomineralization processes: a visit to the thermophilic bacteria Persephonella hydrogeniphila, Persephonella marina, Thermosulfidibacter takaii, and Thermu. Int J Mol Sci. 2021;22:2861. doi: 10.3390/ijms22062861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amusengeri A., Tata R.B., Tastan Bishop Ö. Understanding the pyrimethamine drug resistance mechanism via combined molecular dynamics and dynamic residue network analysis. Molecules. 2020;25:904. doi: 10.3390/molecules25040904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chennubhotla C., Bahar I., Levitt M. Signal propagation in proteins and relation to equilibrium fluctuations. PLoS Comput Biol. 2007;3:e172. doi: 10.1371/journal.pcbi.0030172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frappier V., Chartier M., Najmanovich R.J. ENCoM server: exploring protein conformational space and the effect of mutations on protein function and stability. Nucleic Acids Res. 2015;43:W395–W400. doi: 10.1093/nar/gkv343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rodrigues C.H.M., Pires D.E.V., Ascher D.B. DynaMut2: Assessing changes in stability and flexibility upon single and multiple point missense mutations. Protein Sci. 2021;30:60–69. doi: 10.1002/pro.3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang J.-F., Wang F., Chen Y.-Z., Hao G.-F., Yang G.-F. LARMD: integration of bioinformatic resources to profile ligand-driven protein dynamics with a case on the activation of estrogen receptor. Brief Bioinform. 2020;21:2206–2218. doi: 10.1093/bib/bbz141. [DOI] [PubMed] [Google Scholar]
- 35.Grant B.J., Rodrigues A.P.C., ElSawy K.M., McCammon J.A., Caves L.S.D. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics. 2006;22:2695–2696. doi: 10.1093/bioinformatics/btl461. [DOI] [PubMed] [Google Scholar]
- 36.McGibbon R.T., Beauchamp K.A., Harrigan M.P., Klein C., Swails J.M., Hernández C.X. MDTraj: A modern open library for the analysis of molecular dynamics trajectories. Biophys J. 2015;109:1528–1532. doi: 10.1016/j.bpj.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chovancova E., Pavelka A., Benes P., Strnad O., Brezovsky J., Kozlikova B. 3.0: A tool for the analysis of transport pathways in dynamic protein structures. PLoS Comput Biol. 2012;8:e1002708. doi: 10.1371/journal.pcbi.1002708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Django, [Computer Software], Django Softw. Found. (2013). https://djangoproject.com (accessed September 26, 2020).
- 39.Bootstrap, [Internet], (2020). http://getbootstrap.com (accessed September 26, 2020).
- 40.Knockout.js, [Internet], (2020). http://knockoutjs.com/ (accessed September 26, 2020).
- 41.Brown D.K., Penkler D.L., Musyoka T.M., Bishop Ö.T., Lisacek F. An open source workflow management system and web-based cluster front-end for high performance computing. PLoS ONE. 2015;10:e0134273. doi: 10.1371/journal.pone.0134273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jamroz M., Kolinski A., Kmiecik S. CABS-flex: server for fast simulation of protein structure fluctuations. Nucleic Acids Res. 2013;41:W427–W431. doi: 10.1093/nar/gkt332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hospital A., Andrio P., Fenollosa C., Cicin-Sain D., Orozco M., Gelpí J.L. MDWeb and MDMoby: an integrated web-based platform for molecular dynamics simulations. Bioinformatics. 2012;28:1278–1279. doi: 10.1093/bioinformatics/bts139. [DOI] [PubMed] [Google Scholar]
- 44.Rose A.S., Bradley A.R., Valasatava Y., Duarte J.M., Prlić A., Rose P.W. NGL viewer: web-based molecular graphics for large complexes. Bioinformatics. 2018;34:3755–3758. doi: 10.1093/bioinformatics/bty419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nguyen H, Roe DR, Swails J, Case DA, PYTRAJ: Interactive data analysis for molecular dynamics simulations, (2016). https://doi.org/10.5281/zenodo.44612.
- 46.Michaud-Agrawal N., Denning E.J., Woolf T.B., Beckstein O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J Comput Chem. 2011;32:2319–2327. doi: 10.1002/jcc.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abraham M.J., Murtola T., Schulz R., Páll S., Smith J.C., Hess B. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1-2:19–25. doi: 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- 48.Humphrey W., Dalke A., Schulten K. VMD: Visual molecular dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 49.Roe D.R., Cheatham T.E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- 50.Hagberg A.A., Schult D.A., Swart P.J. In: Proc. 7th Python Sci. Conf., Pasadena, CA USA. Varoquaux G., Vaught T., Millman J., editors. 2008. Exploring network structure, dynamics, and function using NetworkX; pp. 11–15. http://www.osti.gov/energycitations/product.biblio.jsp?osti_id=960616. [Google Scholar]
- 51.Atilgan A.R., Akan P., Baysal C. Small-world communication of residues and significance for protein dynamics. Biophys J. 2004;86:85–91. doi: 10.1016/S0006-3495(04)74086-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nyamai D.W., Tastan Bishop Ö. Identification of selective novel hits against Plasmodium falciparum Prolyl tRNA synthetase active site and a predicted allosteric site using in silico approaches. Int J Mol Sci. 2020;21:3803. doi: 10.3390/ijms21113803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ma S., Li H., Yang J., Yu K. Molecular simulation studies of the interactions between the human/pangolin/cat/bat ACE2 and the receptor binding domain of the SARS-CoV-2 spike protein. Biochimie. 2021;187:1–13. doi: 10.1016/j.biochi.2021.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rodrigues C.H.M., Pires D.E.V., Ascher D.B. DynaMut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 2018;46:W350–W355. doi: 10.1093/nar/gky300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harris C.R., Millman K.J., van der Walt S.J., Gommers R., Virtanen P., Cournapeau D. Array programming with NumPy. Nature. 2020;585:357–362. doi: 10.1038/s41586-020-2649-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sheik Amamuddy O., Bishop N.T., Tastan Bishop Ö. Characterizing early drug resistance-related events using geometric ensembles from HIV protease dynamics. Sci Rep. 2018;8:1–11. doi: 10.1038/s41598-018-36041-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang X., Peng W., Ren J., Hu Z., Xu J., Lou Z. A sensor-adaptor mechanism for enterovirus uncoating from structures of EV71. Nat Struct Mol Biol. 2012;19:424–429. doi: 10.1038/nsmb.2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rhee S.-Y. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res. 2003;31:298–303. doi: 10.1093/nar/gkg100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sheik Amamuddy O., Veldman W., Manyumwa C., Khairallah A., Agajanian S., Oluyemi O. Integrated computational approaches and tools for allosteric drug discovery. Int J Mol Sci. 2020;21:847. doi: 10.3390/ijms21030847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xiao F., Song X., Tian P., Gan M., Verkhivker G.M., Hu G. Comparative dynamics and functional mechanisms of the CYP17A1 tunnels regulated by ligand binding. J Chem Inf Model. 2020;60:3632–3647. doi: 10.1021/acs.jcim.0c00447. [DOI] [PubMed] [Google Scholar]
- 61.Amitai G., Shemesh A., Sitbon E., Shklar M., Netanely D., Venger I. Network analysis of protein structures identifies functional residues. J Mol Biol. 2004;344:1135–1146. doi: 10.1016/j.jmb.2004.10.055. [DOI] [PubMed] [Google Scholar]
- 62.Thibert B., Bredesen D.E., del Rio G. Improved prediction of critical residues for protein function based on network and phylogenetic analyses. BMC Bioinf. 2005;6:1–15. doi: 10.1186/1471-2105-6-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wlodawer A., Miller M., Jaskolski M., Sathyanarayana B., Baldwin E., Weber I. Conserved folding in retroviral proteases: crystal structure of a synthetic HIV-1 protease. Science. 1989;245:616–621. doi: 10.1126/science:2548279. [DOI] [PubMed] [Google Scholar]
- 64.Schrödinger L, The PyMOL Molecular Graphics System, Version 2.4.0a0, (2015).
- 65.Batista P.R., Robert C.H., Maréchal J.-D., Hamida-Rebaï M.B., Pascutti P.G., Bisch P.M. Consensus modes, a robust description of protein collective motions from multiple-minima normal mode analysis—application to the HIV-1 protease. Phys Chem Chem Phys. 2010;12:2850. doi: 10.1039/b919148h. [DOI] [PubMed] [Google Scholar]
- 66.Hornak V., Okur A., Rizzo R.C., Simmerling C. HIV-1 protease flaps spontaneously open and reclose in molecular dynamics simulations. Proc Natl Acad Sci. 2006;103:915–920. doi: 10.1073/pnas.0508452103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Elbe S., Buckland-Merrett G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob Challenges. 2017;1:33–46. doi: 10.1002/gch2.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Macdonald E.A., Frey G., Namchuk M.N., Harrison S.C., Hinshaw S.M., Windsor I.W. Recognition of divergent viral substrates by the SARS-CoV-2 main protease. BioRxiv. 2021 doi: 10.1101/2021.04.20.440716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Atilgan C., Atilgan A.R., Nussinov R. Perturbation-response scanning reveals ligand entry-exit mechanisms of ferric binding protein. PLoS Comput Biol. 2009;5:e1000544. doi: 10.1371/journal.pcbi.1000544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Atilgan C., Gerek Z.N., Ozkan S.B., Atilgan A.R. Manipulation of conformational change in proteins by single-residue perturbations. Biophys J. 2010;99:933–943. doi: 10.1016/j.bpj.2010.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gerek Z.N., Ozkan S.B., Nussinov R. Change in allosteric network affects binding affinities of PDZ domains: analysis through perturbation response scanning. PLoS Comput Biol. 2011;7:e1002154. doi: 10.1371/journal.pcbi.1002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Martin P., Vickrey J.F., Proteasa G., Jimenez Y.L., Wawrzak Z., Winters M.A. “Wide-Open” 1.3 Å structure of a multidrug-resistant HIV-1 protease as a drug target. Structure. 2005;13:1887–1895. doi: 10.1016/j.str.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 73.Weber I.T., Kneller D.W., Wong-Sam A. Highly resistant HIV-1 proteases and strategies for their inhibition. Future Med Chem. 2015;7:1023–1038. doi: 10.4155/fmc.15.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Weber I., Agniswamy J. HIV-1 protease: structural perspectives on drug resistance. Viruses. 2009;1:1110–1136. doi: 10.3390/v1031110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Thanki N., Rao J.K.M., Foundling S.I., Wlodawer A., Howe W.J., Moon J.B. Crystal structure of a complex of HIV-1 protease with a dihydroxyethylene-containing inhibitor: comparisons with molecular modeling. Protein Sci. 1992;1:1061–1072. doi: 10.1002/pro.v1:810.1002/pro.5560010811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Williams R., Holyoak T., McDonald G., Gui C., Fenton A.W. Differentiating a ligand’s chemical requirements for allosteric interactions from those for protein binding. Phenylalanine inhibition of pyruvate kinase. Biochemistry. 2006;45:5421–5429. doi: 10.1021/bi0524262. [DOI] [PubMed] [Google Scholar]
- 77.Juers D.H., Heightman T.D., Vasella A., McCarter J.D., Mackenzie L., Withers S.G. A structural view of the action of Escherichia coli (lac Z) β-galactosidase. Biochemistry. 2001;40:14781–14794. doi: 10.1021/bi011727i. [DOI] [PubMed] [Google Scholar]