

Summary
Systemic lupus erythematosus (SLE) is characterized by a loss of self-tolerance, systemic inflammation, and multi-organ damage. While a variety of therapeutic interventions are available, it has become clear that an early diagnosis and treatment may be key to achieve long lasting therapeutic responses and to limit irreversible organ damage. Loss of humoral tolerance including the appearance of self-reactive antibodies can be detected years before the actual onset of the clinical autoimmune disease, representing a potential early point of intervention. Not much is known, however, about how and to what extent this pre-phase of disease impacts the onset and development of subsequent autoimmunity. By targeting the B cell compartment in the pre-disease phase of a spontaneous mouse model of SLE we now show, that resetting the humoral immune system during the clinically unapparent phase of the disease globally alters immune homeostasis delaying the downstream development of systemic autoimmunity.
Subject areas: biological sciences, immune system disorder, immune response
Graphical abstract
Highlights
-
•
The clinically unapparent pre-phase of SLE impacts clinical disease
-
•
Autoreactive IgM antibodies represent a biomarker for early therapeutic intervention
-
•
Pre-phase B cells orchestrate clinical disease
-
•
Depleting pre-phase B cells diminishes disease pathology
Biological sciences; Immune system disorder; Immune response.
Introduction
Chronic autoimmune diseases, such as systemic lupus erythematosus (SLE), multiple sclerosis (MS), and rheumatoid arthritis (RA) represent a major burden for affected individuals (Ludwig et al., 2017). While treatment options are increasing and remarkable improvements have been achieved in subgroups of patients with targeted therapies, including the blockade of pro-inflammatory cytokines, and depletion of autoreactive immune cell subsets, curative therapies are missing, and many patients show only partial responses or become refractory to therapy (Radbruch and Thiel, 2004; Thalayasingam and Isaacs, 2011). Among the different types of autoimmune diseases, SLE is characterized by its heterogeneity and breadth of organ involvement ranging from skin, kidney, blood vessels, and joints to the central nervous system, requiring an immediate and harsh therapeutic intervention to limit organ damage. As is the case for other autoimmune diseases, loss of humoral tolerance is a hallmark of SLE. Thus, immunoglobulin G (IgG) autoantibodies directed against a variety of nuclear antigens, such as DNA, RNA, and histones are diagnostic markers for SLE. While it is unclear to what extent the anti-nuclear antibody response is directly involved in disease pathology, IgG immune complex deposition in organs such as the kidney, the skin or the joints, has been shown to recruit and activate innate immune effector cells triggering organ inflammation and damage (Takai, 2002; Crampton et al., 2014).
Accordingly, targeting B cells has been an early strategy to limit disease progression and pathogenesis not only in SLE but in virtually all chronic autoimmune diseases in which an involvement of autoreactive B cells and autoantibodies has been observed (Shlomchik et al., 1994; Lipsky, 2001). For instance, depleting B cells showed great clinical benefit in the treatment of thrombocytopenia (Stasi et al., 2001; Shanafelt et al., 2003; Godeau et al., 2008; Deshayes et al., 2019), MS (Bar-Or et al., 2008; Hauser et al., 2008, 2017), and RA (Edwards et al., 2004; Cohen et al., 2006; Emery et al., 2006; Harrold et al., 2017; Gottenberg et al., 2019). However, for SLE conflicting results have been observed. While preliminary studies suggested a benefit of B lymphocyte depletion therapy (Leandro et al., 2002; Abud-Mendoza et al., 2009; Ramos-Casals et al., 2009) the final EXPLORER trial failed to confirm these previous studies and could not detect differences between placebo and rituximab treated patient groups (Merrill et al., 2010). Nonetheless, targeting B cells in earlier phases of disease development such as during the pre-clinical phase of disease, may allow achieving better clinical outcomes. For RA, for example, the PRAIRI study could demonstrate that targeting this early disease phase by a single infusion of 1000 mg of rituximab significantly delayed the development of arthritis in subjects at risk of developing RA (Gerlag et al., 2019). While the most straightforward explanation for the therapeutic activity of targeting B cells would be the depletion of autoantibody producing B cells, several studies suggest a more complex scenario (Sabahi and Anolik, 2006; Schrezenmeier et al., 2018). For example, long-lived autoantibody producing plasma cells do not express CD20 and hence may not be targeted via cytotoxic CD20-specific antibodies, suggesting that B cells may support autoimmune pathology via other pathways, such as secretion of pro- or anti-inflammatory cytokines (Hoyer et al., 2004; DiLillo et al., 2008). Indeed, it was suggested that the therapeutic benefit of Rituximab may be explained at least in part via the reduction of Interleukin (IL)-6 producing B cells (Hasegawa et al., 2006; Bosello et al., 2010; Jones et al., 2014). Of note, however, a prolonged B cell depletion during early disease may also reduce IL-10 secreting B cells that negatively regulate the development of autoimmune responses, leading to the exacerbation of disease pathology (Fillatreau et al., 2002, 2008; Matsushita et al., 2008; DiLillo et al., 2010; Haas et al., 2010; Cerqueira et al., 2019). Furthermore, the absence of secreted IgM leads to accelerated development of IgG autoantibodies and pronounced autoimmune disease in SLE-prone MRL/lpr mice, suggesting that natural IgM antibodies inhibit self-destructive antibody responses (Boes et al., 2000; Nguyen and Baumgarth, 2016). In summary, B cells and natural IgM may contribute to both, disease promoting and disease suppressing immunomodulatory pathways during different phases of an autoimmune disease, requiring a precise understanding of how the humoral immune system modulates autoimmunity.
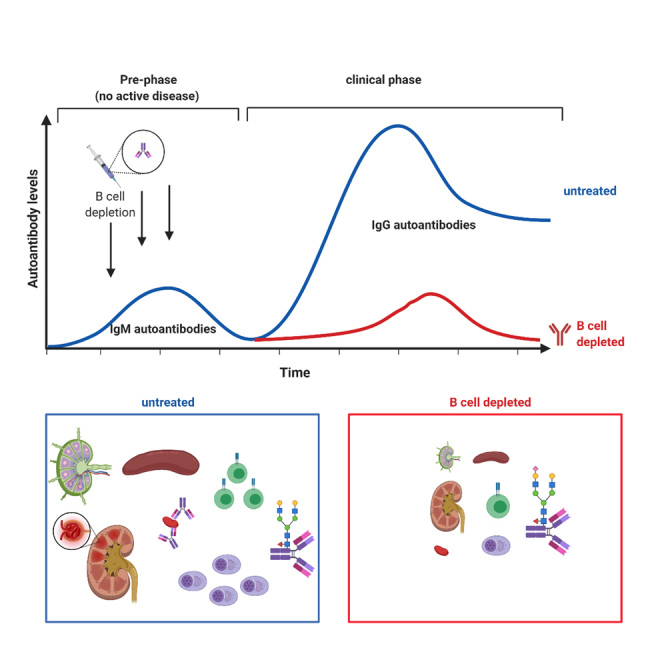
In humans it has become clear that the initial loss of humoral tolerance may occur years before the onset of clinical symptoms (Arbuckle et al., 2003). Thus, 88% of SLE patients show at least one hallmark SLE autoantibody on average three years before the diagnosis of clinical disease. Especially anti-dsDNA IgG antibodies, which can be detected in half of all SLE patients, are usually not found in the serum of healthy individuals (Wichainun et al., 2013; Jia et al., 2018). If and how this clinically unapparent phase of the disease impacts the downstream active phase and thus may represent a window of opportunity for therapeutic intervention, is largely unclear. To address this question, we used the BXSB mouse model, in which mice spontaneously develop an SLE-like chronic autoimmune disease characterized by a marked lymphadenopathy, splenomegaly, monocytosis, hemolytic anemia, hypergammaglobulinemia, proteinuria, autoantibody formation, and an immune complex dependent glomerulonephritis (Murphy and Roths, 1978, 1979; Rogers et al., 2007). Using this model, we show that a transient depletion of B cells during the pre-phase of the disease abolishes downstream disease pathology (including systemic immune system activation, lymph adenopathy and organ damage) and restores inhibitory immune checkpoints, such as serum IgG sialylation and Fc-gamma receptor IIB (FcγRIIB) expression. Our study suggests that B cell depleting strategies targeting the early pre-clinical phase of a complex systemic autoimmune disease may be able to globally restore immune homeostasis and may represent a therapeutic avenue to limit systemic autoimmune disease pathology.
Results
Defining the pre-clinical phase of systemic autoimmune disease in BXSB mice
To investigate the impact of a therapeutic intervention during the pre-phase of a spontaneously developing complex autoimmune disorder on the development of clinical disease appearing later in life, we chose BXSB mice as a model system. Compared with other spontaneous mouse models of SLE, such as MRL/lpr mice, BXSB mice develop a milder and more prolonged disease. Furthermore, BXSB mice are one of the few mouse models showing a severe monocytosis, which can also occur in human SLE. Of note however, unlike in humans, in the BXSB model predominantly male mice develop autoantibodies specific for double stranded DNA early in life. This loss of humoral tolerance is followed by an SLE-like autoimmune disease mimicking many parameters of the human disease. First clinical signs of autoimmunity can be detected around 12 weeks of age and the mean survival of affected mice is five to six months (Andrews et al., 1978; Murphy and Roths, 1979; Theofilopoulos et al., 1980). To define if a pre-clinical phase (pre-phase) of the disease, in which autoantibodies are present in the absence of autoimmune pathology, exists, we analyzed IgM, IgG, and IgA (auto) antibody production in male and in littermate female BXSB mice starting at the age of four weeks. While the total amount of IgM was higher in male mice compared with female mice at all investigated time points, IgG serum levels were not dramatically increased until eight weeks of age (Figure 1A). IgA serum antibody levels were only slightly elevated in male BXSB mice. With respect to autoantibodies, IgM anti-dsDNA autoantibodies were increasing at very early time points, showing a first peak at the age of seven weeks, while IgG anti-dsDNA antibodies were just emerging during that phase (Figure 1B). In contrast, no major increase in IgA anti-dsDNA antibodies became apparent, prompting us to focus on IgM and IgG antibody responses for the rest of our study. Furthermore, no anti-nuclear antibodies (ANAs) were detectable at this early time point (Figure 1C). Importantly, no lymphadenopathy (Figure 1D), splenomegaly (Figures 1E and 1F), thrombocytopenia (Figure 1G), kidney inflammation (Figures 1H and 1I), or elevated blood urea nitrogen (BUN) values (Figure 1J) were detectable at this age. In summary, our results suggest that a pre-clinical phase of autoimmunity exists in BXSB mice, which is characterized by a loss of humoral tolerance predominantly at the level of IgM in the absence of autoimmune pathology.
Figure 1.

Characterizing the pre-phase of autoimmunity in BXSB mice
(A and B) Depicted are the total amounts (mg/mL) of IgM, IgG and IgA serum antibodies (A) as well as IgM, IgG and IgA anti-dsDNA antibodies (in arbitrary units (AU)) (B) in the serum of female (♀) and male (♂) BXSB mice at the indicated age. Shown are the results of three independent experiments as mean +SD with a total of n = 18–22 mice (IgG and IgM) or n = 6–9 (IgA).
(C) Representative images of IgG anti-nuclear antibodies (ANA) in the serum of female (♀) and male (♂) BXSB mice at six to seven and 18 weeks of age identified by indirect immunofluorescence assay with HEp-2 slides. As a positive control serum from Scurfy mice was used. Bars indicate 50 μm.
(D–F) Depicted are representative pictures of inguinal, axial and cervical lymph nodes (D) and spleens (E) as well as the quantification of the spleen weight (F) of female (♀) and male (♂) BXSB mice at six to seven weeks of age. Differences between groups were assessed by unpaired t test, two-tailed. ns: not significant. Shown are the results of two independent experiments with a total of n = 6–10 mice per group. Symbols represent individual mice; error bars represent the mean ± SD.
(G) Shown are thrombocyte counts in the peripheral blood of female (♀) and male (♂) BXSB mice at six to seven weeks of age. Differences between groups were assessed by Mann-Whitney U test, two-tailed. ns: not significant. Shown are the results of five independent experiments with a total of n = 31–43 mice per group. Symbols represent individual mice; error bars represent the mean ± SD.
(H and I) Shown is a representative hematoxylin and eosin (H&E) staining (H) of the kidney of female (♀) and male (♂) BXSB mice at six to seven weeks of age and the quantification of the glomeruli size (I) of three independent experiments. Bars in h indicate 50 μm. Differences between groups were assessed by unpaired t test, two-tailed. ns: not significant. n = 9–12 mice per group. Symbols represent individual mice; error bars represent the mean ± SD.
(J) Depicted is the blood urea nitrogen (BUN) level (mg/dL) of six-to seven-week-old female (♀) and male (♂) BXSB mice. Differences between groups were assessed by unpaired t test, two-tailed. ns: not significant. Four independent experiments with a total of n = 31–36 mice per group. Symbols represent individual mice; error bars represent the mean ± SD.
Targeting the pre-phase of autoimmunity via B cell depletion
Depletion of B cells has become a widely used therapeutic avenue in many autoimmune diseases in humans. How a B cell depletion in the pre-phase of autoimmunity affects the severity of the active disease later in life is less well understood, however. To study this, we used a CD20-specific murine IgG2c antibody developed by Uchida et al. (Uchida et al., 2004a) to transiently deplete B cells during the pre-phase of the disease for three weeks (Figures S1 and S2A). This led to an almost complete removal of CD19+ B cells from the blood over a period of more than four weeks (Figures S2B and S2C). Moreover, the amount of B cells could be significantly decreased in the spleen four days after the first anti-CD20 antibody injection (Figure 1, Figure 2, Figure 3, Figure 4, Figure 5, Figure 6D and S2E). While immature B cells were already less abundant in isotype-treated male BXSB mice at that age and showed only a slight additional reduction, follicular and marginal zone B cells were strongly decreased after anti-CD20 antibody therapy (Figures S2F and S2G). In contrast, CD138+ TACI+ plasma blasts and plasma cells were only slightly diminished (Figures S2H and S2I). In the bone marrow, only recirculating B cells could be depleted (Figures S2J and S2K), consistent with the absence or low level of CD20 expression on pro/pre B cells, immature B cells, and plasma cells (Uchida et al., 2004b). In the peritoneal cavity, only a trend toward B cell depletion was detectable, as described before (Hamaguchi et al., 2005) (Figures S2L and S2M). In summary, our data demonstrate that treating mice with a CD20-specific antibody during the pre-phase of disease leads to a strong reduction of immature and most mature B cell populations over a period of approximately four weeks.
Figure 2.

Effect of pre-phase B cell depletion on lymphadenopathy and splenomegaly
(A and B) Depicted are representative pictures of inguinal, axial, and cervical lymph nodes (A) and spleens (B) from isotype or anti-CD20 antibody (αCD20) treated male (♂) and female (♀) mice at 18 weeks of age of two independent experiments.
(C) Quantification of the spleen weight of 18-week-old anti-CD20 or isotype control antibody-treated BXSB mice. Significant differences between indicated groups were tested with ordinary one-way ANOVA with Tukey's multiple comparison test. Shown are the results of two independent experiments with a total of n = 5–6 mice per group. Symbols represent individual mice; error bars represent the mean ± SD. ∗∗∗∗p < 0.0001.
(D and E) Shown are representative immunofluorescence pictures of lymph node (D) and spleen (E) sections from 18-week-old female (♀) and male (♂) BXSB mice stained for the presence of B220+ B cells (depicted in green), TCRβ+ T cells (in blue) and CD138+ plasma blasts and plasma cells (in red). Size bars in D indicate 200 μm (2000 μm for isotype-treated male BXSB mice). Size bars in E indicate 500 μm.
Figure 3.

Effect of pre-phase B cell depletion on innate and adaptive immune cell subsets in autoimmune BXSB mice
(A and B) Representative FACS plots (A) and the quantification (B) of immature (Imm. B), follicular (FO), and marginal zone (MZB) B cell subsets in the spleen of 18-week-old female (♀) or male (♂) BXSB mice treated with isotype control or CD20-specific antibodies during the pre-phase of disease. Shown are two independent experiments with 5–7 mice per group. Differences between groups were determined by ordinary one-way ANOVA with Tukey's multiple comparison test. Symbols represent individual mice; error bars represent the mean ± SD. ∗∗p < 0.01; ∗∗∗p < 0.001.
(C–F) Depicted are representative FACS plots (C, E) and the quantification (D, F) of CD138+ TACI+ plasma cells in the spleen (C, D) and bone marrow (E, F) of 18-week-old female (♀) and male (♂) BXSB mice treated with isotype control or CD20-specific antibodies during the pre-phase of disease. Shown are two independent experiments with 5–7 mice per group. Differences between groups were determined by ordinary one-way ANOVA with Tukey's multiple comparison test. Symbols represent individual mice; error bars represent the mean ± SD. ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
(G–J) Shown are representative FACS plots (G, I) and the quantification (H, J) of CD4+ (G, H) and CD8+ (I, J) T cell subsets (naive, effector memory (EM), central memory (CM)) in the spleen of 18-week-old female (♀) and male (♂) BXSB mice treated with isotype control or CD20-specific antibodies during the pre-phase of disease. Shown are two independent experiments with 5–7 mice per group. Differences between groups were determined by ordinary one-way ANOVA with Tukey's multiple comparison test. Symbols represent individual mice; error bars represent the mean ± SD. ∗p < 0.05; ∗∗∗p < 0.001.
(K and L) Depicted are representative FACS plots (K) and the quantification (L) of classical and non-classical monocyte subsets in the spleen of 18-week-old female (♀) and male (♂) BXSB mice treated with isotype control or CD20-specific antibodies during the pre-phase of disease. Shown are two independent experiments with 5–7 mice per group. Differences between groups were determined by ordinary one-way ANOVA with Tukey's multiple comparison test. Symbols represent individual mice; error bars represent the mean ± SD. ∗∗p < 0.01; ∗∗∗∗p < 0.0001.
Figure 4.

Effect of pre-phase B cell depletion on humoral immunity
(A) Shown is the total amount of serum IgM, IgG1, IgG2b, IgG2c, IgG3 and IgA in 10- and 18-week-old anti-CD20 (αCD20) or isotype control antibody treated female (♀) and male (♂) BXSB mice as determined by quantitative ELISA. Significant differences between indicated groups were tested with ordinary one-way ANOVA with Tukey's multiple comparison test (normal distribution) or Kruskal-Wallis plus Dunn's multiple comparison test (no normal distribution). Two independent experiments with a total of n = 5–12 mice per group. Symbols represent individual mice; error bars represent the mean ± SD. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
(B and C) Determination of serum IgG subclass glycosylation in 10- or 18-week-old anti-CD20 (αCD20) or isotype control antibody treated female (♀) and male (♂) BXSB mice by mass spectrometry. (B) Depicted is a representative spectrum with peaks corresponding to [M+2H]2+ and [M+3H]3+ glycoforms labeled with the respective glycan structures according to Symbol Nomenclature for Graphical Representation of Glycans (Varki et al., 2015). (C) Quantification of sialylated glycan species containing one or two terminal sialic acid residues. Significant differences between indicated groups were tested with ordinary one-way ANOVA (normal distribution) or Kruskal-Wallis test (no normal distribution) and corrected for multiple testing by two-stage step-up method of Benjamini, Krieger and Yekutieli with a false discovery rate of 0.05. Two independent experiments with a total of n = 5–7 mice per group. Symbols represent individual mice; error bars represent the mean ± SD. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
(D–F) VDJ recombination in μ (D), γ1/2 (E) and α (F) heavy chains (HC) of anti-CD20 (αCD20) or isotype control antibody treated male (♂) mice at the age of 18 weeks was analyzed by high-throughput sequencing. Circos plots depicting the frequencies of VHDH, VHJH and DHJH usage and combinations of productive sequences from four individual mice are depicted. One independent experiment with a total of n = 4 mice.
Figure 5.

Effect of pre-phase B cell depletion on FcγRIIB expression
(A and B) Depicted are representative histograms of FcγRIIB expression on classical (A) and non-classical (B) monocytes in blood, spleen and bone marrow (BM) of 18-week-old anti-CD20 (αCD20) or isotype control antibody treated female (♀) and male (♂) BXSB mice.
(C–F) Quantification of the delta median fluorescence intensities (ΔMFI) of FcγRIIB on eosinophils, neutrophils and classical or non-classical monocytes in blood (C), spleen (D) and bone marrow (E) as well as peritoneal macrophages in the peritoneal cavity (PerC) (F) of 18-week-old anti-CD20 (αCD20) or isotype control antibody treated female (♀) and male (♂) BXSB mice. Significant differences between indicated groups were tested with ordinary one-way ANOVA with Tukey's multiple comparison test. Two independent experiments with a total of n = 5–7 mice per group. Symbols represent individual mice; error bars represent the mean ± SD. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
Figure 6.

Effect of pre-phase B cell depletion on systemic autoimmune pathology
(A–D) Kidney function of isotype or anti-CD20-mIgG2c (αCD20) treated female (♀) and male (♂) BXSB mice was evaluated by blood urea nitrogen (BUN) (A), immune complex and C3 deposition (B) as well as hematoxylin and eosin staining of individual kidney sections (C, D). (A) Depicted is the percentage of mice with increased BUN values (BUN >40 mg/dL) over time. p value represents the difference between isotype and αCD20-treated males. Two independent experiments with a total of n = 11–13 mice per group. (B) Representative kidney sections of IgM, IgG, IgG2b and IgG2c immune complexes and C3 depositions in the glomeruli of 18-week-old αCD20 or isotype control antibody treated female and male BXSB mice. Bars represent 50 μm. (C) Representative sections of the kidneys of 18-week-old αCD20 or isotype control antibody treated female and male BXSB mice with cell infiltrates labeled by asterisks, erythrocytes in the tubuli by non-filled arrows and protein casts by filled arrows. Bars represent 100 μm. (D) Representative sections of kidney glomeruli (left) and determined glomeruli size (right) of 18-week-old αCD20 or isotype control antibody treated female and male BXSB mice. Differences between groups were tested by ordinary one-way ANOVA with Tukey's multiple comparison test. Symbols represent individual mice; error bars represent the mean ± SD. ∗∗∗∗p < 0.0001. Two independent experiments with a total of n = 5–7 mice per group.
(E) Amount of thrombocytes per μl blood of 18-week-old anti-CD20 (αCD20) or isotype control antibody treated female (♀) and male (♂) BXSB mice. Differences between groups were tested by ordinary one-way ANOVA with Tukey's multiple comparison test. Symbols represent individual mice; error bars represent the mean ± SD. ∗p < 0.05; ∗∗∗∗p < 0.0001. Three independent experiments with a total of n = 14–19 mice per group.
(F and G) Kaplan-Meier survival curves of isotype or anti-CD20-mIgG2c (αCD20) treated female (♀) and male (♂) BXSB mice (F) and calculated mean survival (G) of respective groups. Difference between isotype and αCD20-treated males was compared by log rank test. Three independent experiments with a total of n = 9–12 mice per group.
Impact of pre-disease phase B cell depletion on lymphoid organ structure
To assess if and how a B cell depletion during the pre-phase of disease may affect clinical symptoms later in life, we studied how different immune cell subsets are altered in 18-week-old isotype or anti-CD20 antibody treated BXSB mice. At this time point, a peak in disease can be observed leading to death of around half of affected male mice (Andrews et al., 1978; Murphy and Roths, 1978; Theofilopoulos et al., 1980). As depicted in Figures 2A and 2B, the massive lymphadenopathy (Figure 2A) and splenomegaly (Figures 2B and 2C) could be completely prevented through the short-term B cell depletion in 6-week-old mice. Histological analysis of the spleen and lymph nodes revealed that the localization of B220+ B cells, TCRβ+ T cells and CD138+ plasma blasts and plasma cells in the lymph nodes of isotype-treated male BXSB mice was completely different compared with anti-CD20 antibody-treated male and female BXSB animals (Figure 2D). While female and anti-CD20-treated male BXSB mice showed a central T cell zone surrounded by B cells and almost no accumulations of CD138+ cells, the lymph nodes of isotype-treated male mice presented with structural features more closely resembling those typically found in the spleen. Thus, large accumulations of plasma blasts and plasma cells, as well as follicular structures of B and T cells were detectable. With respect to the spleen, follicular structures showed an altered appearance, characterized by diminished numbers of T cells and increased accumulations of CD138+ cells in isotype-treated male mice (Figure 2E). In anti-CD20 antibody treated male BXSB mice, however, the follicular splenic structure was more comparable to non-affected female littermate controls, suggesting that the transient B cell depletion during the pre-phase of disease interfered with the restructuring of secondary lymphoid organs during active disease.
Pre-phase B cell depletion limits innate and adaptive immune cell expansion
A more detailed cellular analysis confirmed that the increase of immature and follicular B cells (Figures 3A and 3B), as well as of CD138+ TACI+ plasma blasts and plasma cells in spleen (Figures 3C and 3D) and bone marrow (Figures 3E and 3F) could be prevented in anti-CD20 antibody treated BXSB males. Additionally, peritoneal cavity B1b cells, showed a trend toward approaching the level present in non-affected littermate controls (Figures S3I and S3J). At the level of T cells, both, CD4+ and CD8+ T cell subsets were altered in anti-CD20 antibody treated mice (Figures 3G–3J). While CD4+ T cells in isotype-treated male mice consisted of roughly 90% effector memory (EM) T cells, this T cell subset represented only about 44% of all T helper cells in anti-CD20-treated males, comparable to non-affected females (Figures 3G and 3H). Within CD8+ T cells, a similar expansion of EM T cells to 53.1% was noted in isotype-treated male BXSB mice, which could not be observed in anti-CD20 antibody treated male mice (6.3%), again comparable with the level observed in female BXSB mice (Figures 3I and 3J). Additionally, CD4+ and CD8+ T cells were expanded more than two-fold in isotype-treated males while they were only slightly increased in male BXSB mice treated with the anti-CD20 antibody (Figures 3H and 3J). Furthermore, the expansion of classical and non-classical monocytes in isotype-treated male mice was found to be reduced to levels found in female littermate controls (Figures 3K and 3L). In a similar manner, the increased level of eosinophils in the blood, spleen, and bone marrow and the expansion of peritoneal macrophages detectable in isotype-treated mice were diminished (Figures S3A–S3H). In summary, our results demonstrate that short-term B cell depletion during the pre-phase of disease prevents the systemic expansion and activation/differentiation of innate and adaptive immune cell subsets usually observed during the peak phase of clinical disease.
Effect of B cell depletion on serum antibody levels, glycosylation, and B cell receptor repertoire diversity
The active phase of autoimmunity is frequently characterized by exacerbated IgG levels and an altered serum antibody glycosylation (Seeling et al., 2017). To test if the early transient B cell depletion regimen had an impact on serum antibody levels and glycosylation, we quantified serum antibody levels and determined serum IgG subclass glycosylation via mass spectrometry analysis (Figures 4 and S4). As shown in Figures 4A and 4B, B cell depletion during the pre-phase of disease limited or abrogated the increase in serum IgM, IgG1, IgG2b, IgG2c, IgG3, and IgA levels observed in the isotype-treated control cohort at ten or eighteen weeks of age. Furthermore, the decrease in sialylated IgG1 and IgG2b/IgG2c glycoforms, characteristic for the active phase of many mouse and human autoimmune diseases, could be corrected (Figures 4B and 4C), whereas no major changes were observed in galactosylated IgG glycoforms between isotype and CD20-specific antibody treated male mice (Figures S4A and S4B). To gain in depth insights into the diversity of the B cell population present in B cell depleted and isotype-treated animals at 18 weeks of age, we performed high-throughput RNA sequencing of the BCR heavy chains in splenic B cells of isotype and anti-CD20 antibody-treated male mice. By using antibody isotype-specific primers for cDNA synthesis (IgM, IgG1/2a/2b/2c and IgA; for primer sequences see Tables 1 and 2) and a separation according to individual primer sequences, BCR repertoire analysis could be performed in an antibody isotype and subclass specific manner. VHDH-, VHJH- and DHJH-pairing was analyzed for individual mice and is depicted as circos plots (Figures 4D–4F). With respect to IgM (μ HC), IgG (γ1/2 HC) and IgA (α HC) producing B cells, no obvious overrepresentations of single VHDH-, VHJH- or DHJH-combinations could be detected (Figures 4D–4F, isotype panel), suggesting that there is no restriction in the BCR repertoire in BXSB mice. For IgM- and IgA-positive B cells, this VDJ combination pattern did not show major changes in anti-CD20 antibody treated mice (Figures 4D and 4F, anti-CD20 panel, Figures S5A and S5C). With respect to IgG-expressing B cells, however, a somewhat lower level of complexity in VDJ usage was observed in B cell depleted animals (Figures 4E and S5B), which may be explained at least in part by the lower level of serum IgG and B cells at 18 weeks of age.
Table 1.
Primer for cDNA synthesis with UMI
| Name, function | Sequence 5‘-3‘ | Manufacturer |
|---|---|---|
| TAK_402, IgG1, IgG2a, IgG2b, IgG2c primer | AGATGTGTATAAGAGACAGH HHHHACAHHHHHACAHHHHN ATTCCCTTGACCAGGCA |
Integrated DNA Technologies Inc., Coralville, IA |
| TAK_IgM, IgM primer | AGATGTGTATAAGAGACAG HHHHHACAHHHHHACAHHHH NATTCCATGGCCACCAGATTCTT |
Integrated DNA Technologies Inc., Coralville, IA |
| TAK_IgA, IgA primer | AGATGTGTATAAGAGACAG HHHHHACAHHHHHACAHHH HNATTGAGCTCGTGGGAGTGTCAGTG |
Integrated DNA Technologies Inc., Coralville, IA |
Table 2.
Primer for multiplex PCR with UMI
| Name, function | Sequence 5‘-3‘ | Manufacturer |
|---|---|---|
| TAK_423, illumina adapter extension | TCGTCGGCAGCGTCAGATG TGTATAAGAGACAG |
Integrated DNA Technologies Inc., Coralville, IA |
| TAK_562 | GTCTCGTGGGCTCGGAGATG TGTATAAGAGACAGHHHHAC HHHHACHHHNGCAGGAGG TGAAGCTTCTCGAGTC |
Integrated DNA Technologies Inc., Coralville, IA |
| TAK_564 | GTCTCGTGGGCTCGGAGAT GTGTATAAGAGACAGHHHH ACHHHHACHHHNGCAGGA GGTGCAGCTTGTTGAGTC |
Integrated DNA Technologies Inc., Coralville, IA |
| TAK_568 | GTCTCGTGGGCTCGGAGAT GTGTATAAGAGACAGHHHH ACHHHHACHHHNGCAGCA GATCCAGTTGGTGCAGTC |
Integrated DNA Technologies Inc., Coralville, IA |
| TAK_575 | GTCTCGTGGGCTCGGAGATG TGTATAAGAGACAGHHHHACH HHHACHHHNGCAGGAAGT GCAGCTGTTGGAGAC |
Integrated DNA Technologies Inc., Coralville, IA |
| TAK_582 | GTCTCGTGGGCTCGGAGA TGTGTATAAGAGACAGHHHHAC HHHHACHHHNG CAGCAGGT/ideoxyI/CAGCTGCAGCAGYC |
Integrated DNA Technologies Inc., Coralville, IA |
| TAK_583 | GTCTCGTGGGCTCGGAGA TGTGTATAAGAGACAGHH \HHACHHHHACHHHNGCAG CAGGTTM/ideoxyI/GCTGCAACAGTC |
Integrated DNA Technologies Inc., Coralville, IA |
| TAK_584 | GTCTCGTGGGCTCGGAGAT GTGTATAAGAGACAGHHHH ACHHHHACHHHNGCAGCAGGTYCA/ideoxyI/CT/ideoxyI/CAGCAGTC |
Integrated DNA Technologies Inc., Coralville, IA |
| TAK_585 | GTCTCGTGGGCTCGGAGATG TGTATAAGAGACAGHHHHA CHHHHACHHHNGCAGCAGG TGCAGCTGAAGSAGTC |
Integrated DNA Technologies Inc., Coralville, IA |
| TAK_586 | GTCTCGTGGGCTCGGAGATG TGTATAAGAGACAGHHHHACH HHHACHHHNGCAGGAGGTGC AGCTTCAGGAGTC |
Integrated DNA Technologies Inc., Coralville, IA |
| TAK_587 | GTCTCGTGGGCTCGGAGATGT GTATAAGAGACAGHHHHACHH HHACHHHNGCAGGAAGTGAA/ideoxyI\CTTGAGGWGTC |
Integrated DNA Technologies Inc., Coralville, IA |
| TAK_588 | GTCTCGTGGGCTCGGAGA TGTGTATAAGAGACAGHH HHACHHHHACHHHNGCAG CAGGTTACTCTGAAAGAGT |
Integrated DNA Technologies Inc., Coralville, IA |
| TAK_589 | GTCTCGTGGGCTCGGAGATG TGTATAAGAGACAGHHHHAC HHHHACHHHNGCAGCAGAT/ideoxyI/CAGCTT/ideoxyI/AGGAGTC |
Integrated DNA Technologies Inc., Coralville, IA |
| TAK_590 | GTCTCGTGGGCTCGGAGAT GTGTATAAGAGACAGHHHH ACHHHHACHHHNGCAGGAG GTG/ideoxyI/AGCTGGTGGAGTC |
Integrated DNA Technologies Inc., Coralville, IA |
| TAK_591 | GTCTCGTGGGCTCGGAGATGT GTATAAGAGACAGHHHHACHH HHACHHHNGCAGGAGGTGCA GCTTGTAGAGAC |
Integrated DNA Technologies Inc., Coralville, IA |
| TAK_612 | GTCTCGTGGGCTCGGAGAT GTGTATAAGAGACAGHHHH ACHHHHACHHHNGCAGCAGG T/ideoxyI/CAGCTGCAGCAGcC |
Integrated DNA Technologies Inc., Coralville, IA |
Furthermore, V-, D- and J-gene usage was analyzed for all serum antibody isotypes (Figure S6). In μ HC positive B cells, D2 segments were present at a slightly higher level in anti-CD20-treated male mice (Figure S6A). In γ1/2 HC positive B cells, a higher usage of J4 segments was observed in anti-CD20-treated mice (Figure S6B). In α HC positive B cells, a higher abundance of D1 and lower level of D2 gene segments became evident (Figure S6C). Additionally, CDR3 length (Figure S7A) and individual amino acid usage in the CDR3 region (Figure S7B) was investigated. While no major differences in CDR3 length were observed in isotype and anti-CD20-treated mice at the level of IgM- and IgA-positive B cells, the presence of longer CDR3 regions in IgG positive B cells of isotype-treated mice could be reverted in anti-CD20-treated animals. In contrast, no major changes with respect to the abundance of individual amino acids could be observed (Figure S7B). In summary, depletion of B cells during the pre-phase of SLE not only significantly diminished IgG production, it also increased the frequency of anti-inflammatory sialylated IgG species and reduced the abundance of long CDR3 regions in IgG antibodies.
Restoring the threshold for immune cell activation via therapeutic targeting of indolent disease
A key factor regulating the expansion of autoreactive B cells, the production of autoantibodies, and the activation of innate immune effector cells is the inhibitory FcγRIIB (Tarasenko et al., 2007; Smith and Clatworthy, 2010; Espeli et al., 2016). Indeed, reduced levels of FcγRIIB expression on B cells were observed in patients with RA and chronic inflammatory demyelinating polyneuropathy and a functionally impaired FcγRIIB variant was suggested to be associated with the development of SLE (Pritchard and Smith, 2003; Mackay et al., 2006; Tackenberg et al., 2009; Smith and Clatworthy, 2010; Willcocks et al., 2010; Baerenwaldt et al., 2011). In line with the observations in human patient cohorts we noted a strong reduction of FcγRIIB expression in diseased male mice compared with healthy female littermate controls on innate immune effector cells, including eosinophils, neutrophils, classical monocytes in the blood, spleen and bone marrow, and on peritoneal macrophages (Figure 5). In mice that were subjected to a B cell depletion regimen during the pre-phase of disease, however, an increased FcγRIIB expression level approaching that of healthy littermate controls was observed for most innate immune effector cell subsets. Along the same lines, a reduced expression level of FcγRIIB on mature B cells in the peripheral blood, marginal zone B cells in the spleen, as well as B1 and B2 B cell subsets in the peritoneal cavity was observed in affected male mice (Figures S8A, S8B, and S8D). Interestingly, the depletion of B cells during the pre-phase of the disease normalized FcγRIIB expression on these B cell subsets, which would be in line with a reduced level of autoantibody production through increasing the threshold for B cell activation.
Effect of a pre-disease B cell depletion on autoantibody development
To analyze the impact of short-term B cell depletion therapy on autoantibody generation, we determined different types of autoantibodies in the serum of anti-CD20-treated BXSB mice. While the first rise of IgM anti-dsDNA antibodies at the age of seven weeks could be abrogated, the onset of IgG anti-dsDNA antibody production was not altered (Figures S9A and S9C). However, IgM and IgG anti-dsDNA antibodies tended to increase somewhat slower in anti-CD20-treated male BXSB mice compared with isotype-treated controls. For example, IgM but not IgG anti-dsDNA antibodies were significantly decreased at the age of 18 weeks (Figures S9B and S9D). Of note, the drop of IgM and IgG anti-dsDNA antibodies for isotype-treated males at 42 weeks of age is due to mice succumbing to terminal disease and therefore only one remaining mouse is depicted at this time point. Female BXSB littermate control mice showed no or very low amounts of IgM and IgG anti-dsDNA antibodies, which were only detectable after six months of age. To confirm the ELISA data we made use of the Crithidia luciliae indirect immunofluorescence test. Crithidia luciliae contains naked circular DNA in select subcellular regions (kinetoplasts) allowing to directly detect anti-dsDNA autoantibodies. As shown in Figure S9E, isotype-treated male BXSB mice showed weak staining of the kinetoplast starting with the age of 10 weeks, which intensified until 18 weeks of age, similar to the results obtained in the anti-dsDNA ELISA. In anti-CD20-treated male mice, a weaker staining of kinetoplasts was detectable starting from 10 weeks of age. Surprisingly, some anti-CD20-treated female BXSB mice also showed weak kinetoplast staining beginning at the age of 14 weeks, which was not detectable by anti-dsDNA IgG ELISA.
Furthermore, IgM and IgG anti-RNA autoantibodies increased over time but were absent in the pre-phase of the disease (Figures S10A and S10C). Comparable to anti-dsDNA antibodies, anti-RNA autoantibodies were increased in the isotype-treated male BXSB cohort at 18 weeks of age compared to anti-CD20-treated group. Of note, the decrease of IgG (but not IgM) anti-RNA antibodies in isotype-treated mice at 30 weeks of age (Figure S10C) is due to the death of mice with severe disease, reducing this experimental cohort to two mice with low RNA-specific autoantibody levels. In addition, ANAs were determined by an indirect immunofluorescence assay using HEp-2 slides (Figure S10E). Here, isotype-treated BXSB males showed first serum positivity already at the age of ten weeks, whereas a similar staining pattern could be detected in anti-CD20-treated mice beginning at the age of 14 weeks. In summary, our results suggest that pre-disease phase B cell depletion does not lead to an immediate but a delayed (starting at 14–16 weeks of age) reduction of autoantibodies directed against DNA or RNA.
Apart from autoantibodies, B cell activating factor (BAFF) levels have been associated with disease activity in human SLE (Stohl et al., 2003; Eilertsen et al., 2011). To analyze if there is an increase in BAFF serum levels in the pre- and early phase of SLE in BXSB mice, serum BAFF levels were determined in isotype or anti-CD20-treated BXSB mice between six (pre-phase) and 18 (active phase of disease) weeks of age (Figure S10F). While we did not observe increased serum levels of BAFF in isotype-treated male (active disease) versus isotype-treated female littermate controls in the pre- and early phase of disease, a trend toward increase BAFF levels became evident at 18 weeks of age. However, we noted a clear increase in BAFF serum levels during B cell depletion treatment with the anti-CD20 antibody in female and to an even higher level in male BXSB mice at 10 weeks of age. An increase in BAFF levels has been observed in different human autoimmune diseases where patients were treated with rituximab (Vallerskog et al., 2006; Lavie et al., 2007; Nagel et al., 2009).
Effect of a pre-disease B cell depletion on organ pathology
One of the most severe manifestations of SLE is kidney damage and ultimately organ failure. To assess if the early B cell depletion regimen was able to limit organ destruction, we studied kidney function and structure in isotype or anti-CD20-treated BXSB mice at the age of 18 weeks. While female BXSB mice showed no increase of blood urea nitrogen (BUN) above the level of 40 mg/dL during the whole observation period, isotype-treated males displayed elevated BUN values beginning at the age of 16 weeks (Figure 6A). In anti-CD20-treated males, the rise of BUN tended to be delayed by about ten weeks. Additionally, massive immune complex and complement depositions were detectable in the kidneys of 18-week-old isotype-treated male BXSB mice, whereas anti-CD20 treatment led to considerably decreased immune complex accumulations at this time point (Figure 6B). The severe kidney destruction in isotype-treated males at this age could also be verified by hematoxylin and eosin (H&E) staining of respective kidney sections (Figures 6C and 6D). Infiltrations or the appearance of protein casts (Figure 6C), as well as glomerular expansions (Figure 6D) could be prevented by anti-CD20 treatment. Moreover, the thrombocyte count was significantly decreased in peripheral blood of isotype-treated male BXSB mice at the age of 18 weeks (Figure 6E). Of note, this thrombocytopenia could be suppressed by B cell depletion therapy during the pre-clinical phase of disease leading to almost normal platelet counts in the blood of anti-CD20-treated male mice. The decreased level of systemic autoimmunity was mirrored by a prolonged survival of anti-CD20-treated male BXSB mice. As depicted in Figures 6F and 6G short-term B cell depleted male mice lived on average 40% longer (39 weeks) as isotype-treated controls (23.5 weeks) and therefore showed a significantly prolonged survival. In contrast, no deaths were recorded for female BXSB mice throughout the whole observation period.
Discussion
B cells play a critical role in the development of many complex autoimmune diseases. Treating patients with established autoimmune diseases with monoclonal antibodies targeting the B cell survival factor BAFF (e.g. with belimumab) (Baker et al., 2003; Furie et al., 2011; Navarra et al., 2011), with proteasome inhibitors targeting long-lived plasma cells (e.g. with bortezomib) (Neubert et al., 2008; Gomez et al., 2011; Alexander et al., 2015; Jakez-Ocampo et al., 2015) or with cytotoxic antibodies specific for the pan B cell marker CD20 (e.g. with rituximab) (Grillo-Lopez et al., 2000; Abud-Mendoza et al., 2009; Ramos-Casals et al., 2009; Leandro, 2013) has shown remarkable clinical benefits. It has also become clear, however, that the treatment efficacy increases if autoimmune diseases are diagnosed early allowing a rapid therapeutic intervention (Seeling et al., 2017). In this study, we went one step further and investigated if a treatment in the earliest possible phase, the pre-phase of an autoimmune disease, in which a loss of humoral tolerance is present in the absence of active disease, affects the development and/or severity of the clinical autoimmune disease later in life.
To investigate this in an in vivo setting of a complex autoimmune disease, we made use of BXSB mice, which spontaneously develop an SLE-like chronic autoimmune disease, which replicates select aspects of the human disease (Murphy and Roths, 1978, 1979). Although it is clear that none of the spontaneous mouse models of SLE fully mimics all aspects of the human disease (which is heterogeneous even in humans), a common feature of these mouse models is the early loss of humoral tolerance, which also is a hallmark of the human disease. Our data further shows that the SLE-like disease in BXSB mice is also characterized by a pre-phase of disease during which loss of humoral tolerance occurs in the absence of autoimmune pathology. The short cycle of B cell depletion in the pre-phase of disease resulted in a predominant loss of peripheral blood and splenic (follicular and marginal zone) B cell populations, recirculating mature B cells in the bone marrow and of peritoneal B2 cells. In contrast, plasma cells were only mildly affected by CD20-specific B cell targeting as expected. With respect to the impact of B cell depletion on later disease development, this allows assessing if the already established plasma cell pool (including dsDNA and RNA-specific IgM/IgG producing plasma cells) contributes to autoimmune pathology later in life (Hamaguchi et al., 2005; Lux et al., 2014). Arguing against such a scenario, the transient depletion of immature and mature B cell subsets (but not of already established plasma cells) had a major impact on downstream disease development and severity. Thus, at eighteen weeks of age, when control mice had developed full-blown systemic disease, the mouse cohort receiving the CD20-specific antibody therapy was almost completely protected from autoimmune pathology, despite the continuing presence of serum IgG autoantibodies directed against dsDNA and RNA. However, animals with high levels of RNA-specific antibodies seemed to be more likely to die in the isotype-treated cohort of BXSB mice resulting in a marked drop of RNA reactivity in the remaining (surviving) mouse cohort after thirty weeks of age. On the other hand, RNA-specific autoantibodies steadily increased in the B cell depleted cohort, irrespective of animals succumbing to disease.
Possible explanations for these findings may be (I) that autoantibodies directed against nuclear antigens do not play a dominant role in autoimmune pathology and/or (II) that downstream effector pathways underlying autoreactive IgG activity are altered. Indeed, we noted an upregulation of the inhibitory FcγRIIB on various innate immune effector cells in blood, spleen and bone marrow. As FcγRIIB is a potent negative modulator of IgG-dependent innate immune effector cell activation, this finding would be consistent with the diminished eosinophilia in the peripheral blood and bone marrow, the reduced expansion of monocyte subsets in the spleen and blood, and the absence of peritoneal macrophage expansion (Tarasenko et al., 2007; Espeli et al., 2016). With respect to the mechanism of how an early B cell depletion may broadly restore FcγRIIB levels on innate immune effector cells and B cells, a plausible but speculative explanation may be afforded by the reduction of serum (auto)antibody levels, which may be involved in triggering inflammation resulting in down-regulation of FcγRIIB via cytokines such as IFNγ or activated complement components (Nimmerjahn and Ravetch, 2006).
Apart from innate immune effector cells, we noted an upregulation of FcγRIIB expression on B cells, which may explain the reduced level of certain autoantibody species and lower level of plasma cells in animals treated during the pre-phase of disease (Takai et al., 1996; Tarasenko et al., 2007). Furthermore, we noted an increased usage of IGHV2 and IGHD2 segments found in anti-CD20-treated animals, while B cell depletion in the pre-disease phase did not impact the use of IGHV11 and IGHV5 genes. Murine IGHV11 and the human ortholog IGHV3 are associated with so-called B1 cell responses (Nguyen and Baumgarth, 2016; Rodriguez-Zhurbenko et al., 2019) and the generation of so-called natural IgM antibodies, which were suggested to protect from autoimmune disease development (Kasaian and Casali, 1993; Choi et al., 2012). In mice, B1a cells in the peritoneal cavity (Kaveri et al., 2012) and marginal zone B cells in the spleen (Woods et al., 2011) are the main producers of natural IgM. Of note, the amount of marginal zone B cells and B1a cells was not altered, suggesting that the B cell depletion during the pre-phase of disease did not affect these protective B cell subpopulations. A potential explanation for the alterations observed in the B cell receptor repertoire in the newly developing B cells in CD20-treated mice may be afforded by age specific factors impacting B cell receptor repertoire selection such as availability of certain self-antigens.
Interestingly, we also noted an increase/normalization of sialic acid containing IgG glycoforms in the serum of CD20 antibody treated mice. We and others have demonstrated that highly sialylated IgG glycovariants can have an active anti-inflammatory activity and may help to restore immune homeostasis (Kaneko et al., 2006; Schwab and Nimmerjahn, 2013; Massoud et al., 2014; Pincetic et al., 2014; Bozza et al., 2019). Moreover, several studies suggested that sialic acid rich IgG glycovariants mediate their activity at least in part via upregulation of the inhibitory FcγRIIB on innate immune effector cells and B cells (Kaneko et al., 2006; Anthony et al., 2011; Fiebiger et al., 2015). Thus, although speculative at the moment, the increase in sialylated IgG glycoforms in the serum may be responsible at least in part for the increase in FcγRIIB expression. Apart from effects of pre-phase B cell depletion on B cells and innate immune cells, we also noted a diminished expansion of memory T cells (both CD4 and CD8) in B cell depleted animals. These findings are consistent with studies from other groups in MRL/lpr mice suggesting that B cells can play a major role in instructing autoreactive T cell responses (Chan and Shlomchik, 1998) and can further orchestrate autoimmune responses by acting as antigen-presenting cells and producers of pro-inflammatory cytokines (Shlomchik et al., 1994; Chan and Shlomchik, 1998; Chan et al., 1999; Anolik et al., 2004; Looney et al., 2004; Nashi et al., 2010; Barr et al., 2012; Getahun and Cambier, 2019).
In summary, our study suggests that pre-disease phase B cells play a critical role for the later development of systemic autoimmunity. Apart from autoantibody production, B cells may aid autoimmune pathology via cytokine and chemokine production triggering autoreactive T cells and other mononuclear cells, reduced autoantigen presentation to T cells, and/or diminished ability of dendritic cells to present autoantigens. However, the exact mechanisms still need to be elucidated. Nevertheless, therapeutic targeting of the pre-phase of autoimmunity may be a valid strategy to interfere with the development of autoimmune diseases via a global resetting of immune homeostasis leading to delayed onset of the disease.
Limitations of the study
The major limitation of the study is the focus on one mouse model of SLE, which may argue for caution when trying to translate the results to the human situation. While the use of a genetically complex mouse model characterized by spontaneous development of disease clearly is an advantage compared to other inbred mouse models of SLE, the fact that mainly male mice are affected by the disease is not recapitulated in humans. Although specific parameters of SLE development and the associated autoimmune pathology may differ between mice and humans, the fact that an early treatment of complex autoimmune diseases may lead to more durable and sustained therapeutic effects may be relevant for both species, however.
Ethics statement
The animal study was reviewed and approved by government of Lower Franconia.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse/human B220 (CD45R) PerCP (clone RA3-6B2) | BD Biosciences | Cat. #553093; RRID: AB_394622 |
| Anti-mouse/human B220 (CD45R) BV510 (clone RA3-6B2) | Biolegend | Cat. # 103248; RRID: AB_2650679 |
| Anti-mouse/human B220 (CD45R) A488 (clone RA3-6B2) | Biolegend | Cat. # 103225; RRID: AB_389308 |
| Anti-Mouse C3 FITC (clone RmC11H9) | Cederlane | Cat. # CL7503F; RRID: AB_10061294 |
| Anti-mouse CD4 PerCP (clone RM4-5) | BD Biosciences | Cat. # 553052; RRID: AB_394587 |
| Anti-mouse CD5 APC (clone 53-7.3) | Biolegend | Cat. # 100625; RRID: AB_2563928 |
| Anti-mouse CD8a A647 (clone 53-6.7) | Biolegend | Cat. # 100724; RRID: AB_389326 |
| Anti-mouse CD11b FITC (clone M1/70) | eBioscience | Cat. # 11-0112-71; RRID: AB_464933 |
| Anti-mouse/human CD11b PerCP-Cy5.5 (clone M1/70) | Biolegend | Cat. # 101228; RRID: AB_893232 |
| Anti-mouse CD19 BV510 (clone 6D5) | Biolegend | Cat. # 115546; RRID: AB_256213 |
| Anti-mouse CD21/35 FITC (clone 7G6) | BD Biosciences | Cat. # 553818; RRID: AB_395070 |
| Anti-mouse CD23 Biotin (clone B3N4) | Biolegend | Cat. # 101604; RRID: AB_312829 |
| Anti-mouse/human CD44 Biotin (clone IM7) | Biolegend | Cat. # 103003; RRID: AB_312954 |
| Anti-mouse CD45 APC/Fire750 (clone 30-F11) | Biolegend | Cat. # 103154; RRID: AB_2572116 |
| Anti-mouse CD62L PE/Cy7 (clone MEL-14) | Biolegend | Cat. # 104418: RRID: AB_313103 |
| Anti-mouse CD138 A647 (clone 281-2) | Biolegend | Cat. # 142525; RRID: AB_2566238 |
| Anti-mouse CD138 PE (clone 281-2) | Biolegend | Cat. # 553714; RRID: AB_395000 |
| Anti-mouse CD267 (TACI) PE (clone 8F10) | Biolegend | Cat. # 133403; RRID: AB_2203542 |
| Anti-mouse F4/80 Biotin | Biolegend | Cat. # 123106; RRID: AB_893501 |
| Anti-mouse FcγRIIB (clone Ly17.1) | Jeffrey Ravetch (Kimura et al., 1981) | N/A |
| Anti-mouse Gr-1 BV510 (clone RB6-8C5) | Biolegend | Cat. # 108437; RRID: AB_2562214 |
| Anti-mouse Gr-1 FITC (clone RB6-8C5) | BD Biosciences | Cat. # 553127; RRID: AB_394643 |
| Anti-mouse IgD PE/Cy7 (clone 11-26c.2a) | Biolegend | Cat. # 405719; RRID: AB_2561875 |
| Anti-mouse IgG FITC (polyclonal) | eBioscience | Cat. # 11-4011-85; RRID: AB_465218 |
| Anti-mouse F(ab')2 IgG PE (polyclonal) | Jackson ImmunoResearch | Cat. # 115-116-071; RRID: AB_2338626 |
| Anti-mouse IgG2b PE (polyclonal) | Thermo Fisher | Cat. # M32404; RRID: AB_2536646 |
| Anti-mouse IgG2c PE (polyclonal) | Jackson ImmunoResearch | Cat. # 115-005-208; RRID: AB_2338464 |
| Anti-mouse IgM PE (clone II/41) | eBioscience | Cat. # 12-5790-81; RRID: AB_465939 |
| Anti-mouse IgM PerCP/Cy5.5 (clone RMM-1) | Biolegend | Cat. # 406512; RRID: AB_2075943 |
| Anti-mouse NK1.1 FITC (clone PK136) | Biolegend | Cat. # 108706; RRID: AB_313393 |
| Anti-mouse TCRβ FITC (clone H57-597) | Biolegend | Cat. # 109206; RRID: AB_313429 |
| Anti-mouse TCRβ A647 (clone H57-597) | Biolegend | Cat. # 109218; RRID: AB_493346 |
| Streptavidin BV510 | Biolegend | Cat. # 405233 |
| Streptavidin PE/Cy7 | Biolegend | Cat. # 405206 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| AMPure XP Beads | Beckmann und Coulter | Cat. # A6388-1 |
| methylated bovine serum albumin (mBSA) | Sigma | Cat. # A1009 |
| Deoxyribonucleic acid from calf thymus | Sigma | Cat. # D4522 |
| yeast RNA | Roche | Cat. # 10109223001 |
| Critical Commercial Assays | ||
| Mouse IgA ELISA Quantitation Set | Bethyl | Cat. # E90-103 |
| Mouse IgM ELISA Quantitation Set | Bethyl | Cat. # E90-101 |
| Mouse IgG ELISA Quantitation Set | Bethyl | Cat. # E90-131 |
| Mouse IgG1 ELISA Quantitation Set | Bethyl | Cat. # E90-105 |
| Mouse IgG2b ELISA Quantitation Set | Bethyl | Cat. # E90-109 |
| Mouse IgG2c ELISA Quantitation Set | Bethyl | Cat. # E90-136 |
| Mouse IgG3 ELISA Quantitation Set | Bethyl | Cat. # E90-111 |
| Urea Nitrogen (BUN) Reagent Set | Teco Diagnostic | Cat. # B551-132 |
| Scriptum First Kit | Bio&Sell | Cat. # BS.48.100 |
| KAPA HiFi HotStart Uracil+ ReadyMix Kit | Kapa Biosystems | Cat. # KK2801 |
| Q5® High-Fidelity PCR Kit | New England BioLabs | Cat. # E0555L |
| Mouse BAFF ELISA Kit | abcam | Cat. # ab119580 |
| Deposited Data | ||
| Raw data BZR repertoire analysis | This paper | Mendeley Data: https://doi.org/10.17632/smtxktdtfc.1 |
| Data Figures and Supplementary Figures | This paper | Mendeley Data: https://doi.org/10.17632/gvxjz3twft.1 |
| Raw Data IgG Glycosylation Part 1.1 | This paper | Mendeley Data: https://doi.org/10.17632/jygxpd7g7c.1 |
| Raw Data IgG Glycosylation Part 1.2 | This paper | Mendeley Data: https://doi.org/10.17632/kjfw7cwnxf.1 |
| Raw Data IgG Glycosylation Part 2 | This paper | Mendeley Data: https://doi.org/10.17632/4s9nxvk7br.1 |
| Experimental Models: Organisms/Strains | ||
| Mouse: BXSB/MpJ | The Jackson Laboratory | RRID: IMSR_JAX:000740 |
| Software and Algorithms | ||
| AxioVision Rel. 4.9 | Carl Zeiss AG | https://www.micro-shop.zeiss.com/de/de/system/axiovision+software/software+axiovision/axiovision+basissoftware/410130-0909-000 |
| BD FACS Diva 6.1.2 | BD Biosciences | https://www.bdbiosciences.com/en-us/instruments/research-instruments/research-software/flow-cytometry-acquisition/facsdiva-software |
| Zen blue 2.5 | Carl Zeiss AG | https://www.zeiss.de/mikroskopie/produkte/mikroskopsoftware/zen-lite.html |
| GraphPad Prism 7.0 | GraphPad Software Inc. | https://www.graphpad.com/scientific-software/prism/ |
| MultiQC 1.9 | Ewels et al. (2016) | www.multiqc.info |
| IMGT® (the international ImMunoGeneTics information system®) | Alamyar et al. (2012) | www.imgt.org |
| ARGalaxy (Antigen Receptor Galaxy) | IJspeert et al., 2017 | www.bioinf-galaxian.erasmusmc.nl/argalaxy |
| Other | ||
| IIFT: Crithidia luciliae sensitive (anti-dsDNA) | Euroimmun | Cat. # FA 1572-1010-1 |
| Kallestad HEp-2 Slides | Bio-Rad | Cat. # 26103 |
Resource availability
Lead contact
The lead contact for further information and requests is Falk Nimmerjahn (falk.nimmerjahn@fau.de).
Materials availability
The study did not generate new unique reagents.
Experimental model and subject details
Experimental models
BXSB/MpJ mice were purchased from The Jackson Laboratory (Bar Harbor, ME). All mice were held under specific pathogen-free conditions and in isolated ventilated cages in the animal facilities of the University of Erlangen, according to state and institutional guidelines and the rules and regulations of the German animal welfare laws.
Method details
In vivo experiments
The anti-CD20-mIgG2c antibody was produced by transient transfection of HEK293T cells with calcium phosphate transfection followed by purification of the secreted antibody from the culture supernatant via protein G (see Nimmerjahn et al., 2005). In brief, cells were cultured in DMEM medium supplemented with 1% Nutridoma SP (Roche), 1% glutamate (c.c. pro) and 1% penicillin-streptomycin (c.c. pro). Cell culture supernatants were harvested 7 days after transfection and proteins precipitated with ammonium sulfate precipitation, dialyzed against PBS and purified with protein G beads (Roche). Finally, the isolated antibody was dialyzed against PBS. An appropriate isotype control was purchased from BioXcell (USA).
To induce short-term B cell depletion, six- to seven-week-old BXSB mice were intravenously injected with 3 μg/g anti-CD20-mIgG2c or isotype control in PBS. Antibody treatment was repeated 3, 7, 14 and 21 days after the first injection. To determine different serum and blood parameters, mice were bled every one to two weeks. Serum was stored at -80°C until further use. Thrombocyte count was determined by analyzing whole blood (1:4 diluted with PBS) via an Advia 120 hematology system (Siemens).
Antibodies and flow cytometry
Spleen, bone marrow and peritoneal cavity flushes were passed through 70 μm cell strainer to obtain single cell suspension. Blood, spleen, and bone marrow samples underwent hydrophobic red blood cell lysis. Staining of single cell suspensions of blood, spleen, bone marrow and peritoneal cavity cells was performed in PBS, 2% FCS, 0.02% sodium azide on ice. Before staining, mouse Fc receptors were blocked with Fc block (clone 2.4G2, 0.5 μg per sample) to reduce unspecific binding to Fc receptors. When FcγRs were stained no Fc block was used. Data were acquired on a FACS Canto II (BD Biosciences) and analyzed with FACS Diva software. Analysis was restricted to viable cells, which were identified by exclusion of nucleic acid binding dye 4′,6-Diamidino-2-phenylindol (DAPI). To exclude cell aggregates from analysis, we used a gating strategy excluding events with an unproportionally large area of the forward scatter signal relating to its height. A complete list of antibodies and the gating strategy used to identify different leukocyte populations can be found in the key resources table in STAR Methods section and in Figure S1.
ELISA
Sera of BXSB mice were collected at different time points and stored at -80°C until further use. For quantification of total serum IgM, IgG and IgA the Mouse IgM, IgG and IgA ELISA Quantification Kits (Bethyl) were used according to the manufacturer's instructions: ELISA plates were coated with 100 ng/well goat anti-mouse IgM, IgG or IgA in Carbonate/Bicarbonate for 1 h at room temperature. After washing, unspecific binding was blocked with PBS, 1% BSA (200 μl/well) for 30 min at room temperature. Sera were diluted 1:2,500 to 1:15,000 in PBS, 1% BSA (50 μl/well) and incubated for 1 h at room temperature. After adequate washing, bound IgM, IgG and IgA antibodies were detected by 1:20,000 diluted (in PBS, 1% BSA) anti-IgM-HRP, anti-IgG-HRP or anti-IgA-HRP antibody (incubated for 1 h at room temperature, 50 μl/well). For detection, TMB solution was added and the reaction was stopped with 6% orthophosphoric acid (50 μl/well). OD was measured with VersaMax tunable microplate reader (Molecular Devices) at 450 and 650 nm.
For the detection of anti-dsDNA antibodies, ELISA plates were coated with 5 μg/ml (50 μl/well) methylated BSA (Sigma) in PBS for 2 h at room temperature. After washing, the plates were coated with 50 μg/ml calf thymus DNA (Sigma) in PBS (50 μl/well) at 4°C overnight. Blocking of unspecific binding was performed with PBS, 0.1% Gelatin, 3% BSA, 1 mM EDTA (200 μl/well). Sera were diluted 1:100 in the blocking solution and incubated for 1 h at room temperature (50 μl/well). Further detection was performed according to quantitative ELISA.
For detection of RNA-specific autoantibodies, ELISA plates were coated with 30 μg/ml yeast RNA (Roche) in PBS (50 μl/well) and incubated for 1 h at room temperature. After washing three times with PBS, wells were blocked with PBS, 1% BSA (200 μl/well) for 30 min. Sera were diluted 1:100 in PBS, 1% BSA (50 μl/well) and incubated for 1 h at room temperature. Further detection was performed according to quantitative ELISA.
To compare various experiments and individual ELISA plates a reference serum was included on each plate and arbitrary units (AU) were calculated according to the OD-value of the reference serum of the respective plate.
Detection of anti-dsDNA antibodies via indirect immunofluorescence test
Anti-dsDNA antibodies were determined using Crithidia luciliae sensitive slides (Euroimmun). Experiments were carried out according to the manufacturer's instructions using serum diluted 1:10 in sample buffer followed by detection with a FITC-conjugated anti-mouse IgG antibody (eBioscience). Pooled serum of old NZB/W mice served as positive, sample buffer alone as a negative control. Slides were mounted with the included mounting medium and analyzed on a Zeiss Axio Observer system with the Zen blue 2.5 software.
Detection of anti-nuclear antibodies
Autoantibodies specific for nuclear antigens were detected on HEp-2 slides (BioRad). Experiments were carried out according to the manufacturer's recommended protocol using serum diluted in PBS at 1:50 followed by detection with a FITC-conjugated anti-mouse IgG secondary antibody (eBioscience). Slides were mounted using Fluoromount imaging medium (Sigma) and analyzed on a Zeiss Axio Observer system with the Zen blue 2.5 software.
Determination of BAFF serum levels
Serum BAFF levels were quantified by ELISA using the mouse BAFF ELISA Kit (Abcam). Experiments were carried out according to the manufacturer's instruction. Serum was diluted 1:20. OD was measured with a VersaMax tunable microplate reader (Molecular Devices) at 450 and 650 nm.
Immunofluorescence microscopy
5 μm sections of frozen tissue were air-dried overnight, followed by fixation in acetone. Slides were blocked with 5% goat serum in PBS and single stained with either PE-conjugated anti-IgM, anti-IgG-Fc, anti-IgG2b, anti-IgG2c or FITC-conjugated anti-C3 antibodies, respectively (For further information see key resources table in STAR Methods section). After incubation, the excess of fluorescent dye was removed by multiple washing steps with PBS, slides were mounted using Fluoromount imaging medium (Sigma) and analyzed on a Zeiss Axio Observer microscope with the Zen blue 2.5 software.
Proteinuria assay
To evaluate kidney function, blood urea nitrogen levels were quantified in the serum of BXSB mice at the age of six weeks as well as every two weeks after isotype/anti-CD20 treatment with an enzymatic urea nitrogen kit (Teco Diagnostic) according to the manufacturer's instructions. Volume of serum and reagents was scaled down to F-bottom microplates (Greiner Bio-One) using 1 μl serum and 150 μl of enzyme reagent and color developer. OD was measured with VersaMax tunable microplate reader (Molecular Devices) at 630 nm. Percent of mice with a BUN value above 40 mg/dl were calculated and depicted as Kaplan-Meier curve.
Histological staining
Mouse kidneys were fixed in 10% neutral buffered formalin and kept in 70% ethanol until further use. Thereafter, specimens were dehydrated in an ascending ethanol series and embedded in paraffin for subsequent sectioning into 5 μm sections using a Rotary Manual Microtome (Medite). Mouse renal tissue sections were stained with hematoxylin and eosin (H&E) according to standard practice. Glomeruli size was determined by AxioVision 4.8 software (Zeiss).
B cell receptor repertoire analysis
BCR receptor repertoire analysis was performed as described by Khan et al. (Khan et al., 2016):
RNA isolation
A single cell suspension of erythrocyte lysed splenic cells was prepared and the RNA extracted using the RNeasy Plus Mini Kit (Qiagen) according to the manufacturer's protocol. RNA concentration and quality were determined on a Nanodrop 2000 Spectrophotometer.
cDNA synthesis
Next, first-strand cDNA was synthesized with SCRIPTUM FIRST cDNA synthesis Kit (Bio&Sell) using 200 – 1000 ng total RNA and 20pMol immunoglobulin specific primers (TAK_402, TAK_403, TAK_IgM, TAK_IgA) following the manufacturer’s instruction. TAK_402 binds a conserved portion of mouse IgG1, IgG2a, IgG2b, and IgG2c. TAK_403, TAK_IgM and TAK_IgA binds to mouse IgG3, IgM and IgA, respectively. Sequences for individual primers are shown in Table 1. After cDNA synthesis, samples were subjected to a bead cleanup (AMPure XP Beads, Beckman Coulter) at 1.0x, incorporating an extra ethanol wash step, followed by elution into elution buffer (Qiagen).
Multiplex PCR
Thereafter, multiplex PCR was performed: Reaction mixtures consisted of cDNA, 500 nM multiplex primer mix (TAK_562, TAK_564, TAK_568, TAK_575, TAK_582 to TAK_591, TAK_612, see Table 2), 500 nM 3′ Illumina adapter-specific primer (TAK_423) and 1x KAPA HIFI HotStart Uracil+ ReadyMix (KAPA Biosystems). The Uracil+ version enabled efficient high-fidelity amplification of TAK multiplex primer set that had deoxyinosines. Thermocycling was performed as follows: 95°C for 2 min; 9 cycles of 98°C for 20 s, 60°C for 45 s, 72°C for 70 s; 72°C for 5 min; and 4°C indefinitely. PCR reactions were then bead cleaned two times at 0.65x and eluted with elution buffer.
Adapter extension PCR
As last step, individual samples were barcoded with two individual illumina index primer i5 and i7 from the Nextera XT Index Kit v2 with Q5® High-Fidelity PCR Kit (New England Biolabs). Thermocycling was performed as follows: 94°C for 3 min; 23 cycles of 94°C for 60 s, 55°C for 60 s, 72°C for 60 s; 72°C for 10 min; and 4°C indefinitely. The PCR amplifications were separated with a 1.5% agarose gel and equimolar amounts of samples were pooled. Pooled samples were again cleaned once with beads at 0.8x and eluted with ddH2O.
Next generation sequencing
NGS was performed by Dr. Arif Bülent Ekici (Department of Human Genetics, Erlangen) on the Illumina MiSeq platform using a pair of overlapping paired-end reads (2x300 bp). 20% PhiX was added for improved performance. Paired-end reads were merged with the IgRepertoireConstructor (Safonova et al., 2015). Read Quality was ensured by FASTQC quality analysis by MultiQC (Ewels et al., 2016).
PCR error correction
Molecular amplification fingerprinting (MAF) error correction was achieved with unique molecular identifiers (UMI) as part of the cDNA synthesis and Multiplex Primers. PCR sequencing errors were corrected by pattern matching and filtering sequences containing both UMIS and correcting sequencing errors without introducing bias to read counts.
Analysis of B cell receptor rearrangement
Sequences were separated according to the individual immunoglobulin specific primers (TAK_402, TAK_403, TAK_IgM, TAK_IgA) and VDJ annotation was conducted with the IMGT/HighV-QUEST service (Alamyar et al., 2012). Sequences using the IgM, IgG1/2 or IgA specific primer were further analyzed with ARGalaxy immune receptor pipeline (IJspeert et al., 2017). Data of individual mice per group were summarized and presented as bar graphs or depicted as circos plots of individual mice for VDJ recombination. Raw data can be found on Mendeley Data (Mendeley Data: https://doi.org/10.17632/smtxktdtfc.1). For primer sequences see Tables 1 and 2.
Glycan analysis
IgG isolation from serum samples
IgG was isolated from 45-180 μl of mouse serum on Fast Flow Protein G Agarose beads (Merck Millipore) as follows. Protein G Agarose slurry was prewashed three times with 5x volume of 1x phosphate buffer saline (PBS, pH 7.4, prepared in house from analysis grade reagents) as follows: the beads in 1x PBS were centrifuged for 10 s at 150 g and supernatant was removed by pipetting. Beads were then diluted to a final volume with 1x PBS. Serum samples were diluted 1:7 v/v with 1x PBS in a 96-deep-well collection plate. The sample layout on the plates was determined by block randomization to distribute samples taken at two time points, from female and male animals treated with anti-CD20-specific antibody or isotype control, evenly across the columns and rows of the plate. Each plate design included one negative control, standards and duplicates to ensure quality control.
To each serum sample 100 μl of Fast Flow Protein G Agarose beads in 1x PBS (pH 7.4) was added. Samples were then incubated for 3 h at room temperature with shaking at 900 rpm. Samples were additionally resuspended by pipetting every 45 min during incubation. In the meantime, a 96-well AcroPrep Advance 0.2 μm hydrophilic polypropylene (GHP) filter plate (Pall Corporation) was preconditioned by washing with 200 μl of 70% v/v EtOH per well, followed with 200 μl of ultra-pure water per well, and finally with 300 μl of 1x PBS per well. Flow through was removed by vacuum. After incubation, the samples with beads were applied to the GHP plate and the liquid was removed with vacuum. Then the samples were washed three times with 300 μl of 1x PBS, and three times with 300 μl of ultra-pure water, liquid removed by vacuum.
After last wash, 100 μl of 0.1 M formic acid was added to each sample and the plate with samples was incubated at a shaker for 15 min. Then IgG was eluted by vacuum into a clean 96-well collection plate with 17 μl of 1 M ammonium bicarbonate per sample for neutralization.
Analysis of IgG Fc-linked N-glycosylation with nano-liquid chromatography-electrospray ionization-mass spectrometry (nano-LC-ESI-MS)
Sample preparation and nano-LC-ESI-MS analysis were performed as described previously (Pezer et al., 2016; Zaytseva et al., 2018). In brief, 15-25 μg of the isolated IgG was digested with sequencing grade trypsin (Promega Corp.), desalted with reverse-phase solid phase extraction (RP-SPE) on Chromabond C18 ec beads (Macherey-Nagel) and analyzed on a nanoACQUITY UPLC system (Waters) coupled to a Compact mass spectrometer (Bruker Daltonics). Tryptic glycopeptides corresponding to the Fc region of IgG were separated by LC in the gradient of 80% (v/v) acetonitrile (ACN, LC-MS purity, JT Baker, ThermoFischer Scientific) in 0.1% (v/v) trifluoroacetic acid (TFA, HPLC purity, Sigma-Aldrich) on a Halo C18 nano-LC column (150 mm × 100 μm i.d., 2.7 μm fused core particles, Advanced Materials Technology) and mass spectra were recorded in positive ion mode. Separation between tryptic glycopeptides corresponding to IgG2c and IgG2b subclasses was not achieved with this method and the respective N-glycoforms were quantified together. MS data was extracted using LaCyTools v1.0.1 b.7 software as described previously (Zaytseva et al., 2018). Values obtained for N-glycopeptide abundance were normalized to the total integrated area per each IgG subclass. Batch correction was performed on the log-transformed values using the ComBat method (R package “sva”, R version 4.0.3) to remove possible experimental variation due to batch effects. Following data extraction, derived traits that describe the abundances of specific N-glycan types in the Fc-linked N-glycome of mouse IgG were calculated as follows: G0 – sum of abundances of N-glycan structures without antennary galactose residues, G1 - sum of abundances of N-glycan structures with one antennary galactose residue, without sialic acid; G2 - sum of abundances of N-glycan structures with two antennary galactose residues, without sialic acid; Sialylation - sum of abundances of N-glycan structures with one or two antennary sialic acid residues. Raw data can be found on Mendeley Data (https://doi.org/10.17632/jygxpd7g7c.1, https://doi.org/10.17632/kjfw7cwnxf.1, https://doi.org/10.17632/4s9nxvk7br.1)
Quantification and statistical analysis
If not stated otherwise in the figure legend data are presented as follows: data are expressed as mean ± standard deviation. Data were analyzed and plotted with GraphPad Prism 7.0 software (GraphPad Software Inc., San Diego, CA). Outliers were removed by ROUT method. Normal distribution was tested with a Shapiro-Wilk normality test. Normally distributed data were analyzed by ordinary one-way ANOVA followed by Holm-Sidak's multiple comparison test. Non-parametric distribution was tested using Kruskal-Wallis test with Dunn's post hoc test. Differences in survival and % BUN > 40 mg/dl were analyzed with Kaplan-Meier estimates, and groups were compared with the log-rank test. p < 0.05 was considered significant. n represents individual mice.
Acknowledgments
We thank Heike Danzer for expert technical assistance and Dr. Arif Bülent Ekici, Department of Human Genetics, Erlangen, for performing the Illumina MiSeq run.
This work was funded by the German Research Foundation (Research Training Group GRK1660 to F.N. and FOR2886-B2 to A.L. and F.N.). The glycan analysis was supported by the European Structural and Investment Funds IRI (Grant #KK.01.2.1.01.0003) and Croatian Center of Research Excellence in Personalized Healthcare (Grant #KK.01.1.1.01.0010).
Author contributions
A.W. designed, performed, and analyzed the experiments. S.S. performed the evaluation of BCR repertoire analysis. O.Z. performed glycan analysis. H.A., J.K., and M.P. helped in performing experiments. A.W. and F.N. wrote the paper. O.Z., G.L., M.P., S.S., T.W., and A.L. discussed the paper. All authors read, reworked, and approved the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: September 24, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103076.
Supplemental information
Data and code availability
-
•
All raw data (Raw data BZR repertoire analysis: Mendeley Data: https://doi.org/10.17632/smtxktdtfc.1; Data Figures and Supplementary Figures: Mendeley Data: https://doi.org/10.17632/gvxjz3twft.1; Raw Data IgG Glycosylation Part 1.1: Mendeley Data: https://doi.org/10.17632/jygxpd7g7c.1; Raw Data IgG Glycosylation Part 1.2: Mendeley Data: https://doi.org/10.17632/kjfw7cwnxf.1; Raw Data IgG Glycosylation Part 2: Mendeley Data: https://doi.org/10.17632/4s9nxvk7br.1) have been deposited at Mendeley Data and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Abud-Mendoza C., Moreno-Valdes R., Cuevas-Orta E., Borjas A., Aranda F., Irazoque F., Andrade L., Vigna-Perez M., Gonzalez-Amaro R. Treating severe systemic lupus erythematosus with rituximab. An open study. Reumatol. Clin. 2009;5:147–152. doi: 10.1016/j.reuma.2008.09.008. [DOI] [PubMed] [Google Scholar]
- Alamyar E., Duroux P., Lefranc M.P., Giudicelli V. IMGT((R)) tools for the nucleotide analysis of immunoglobulin (IG) and T cell receptor (TR) V-(D)-J repertoires, polymorphisms, and IG mutations: IMGT/V-QUEST and IMGT/HighV-QUEST for NGS. Methods Mol. Biol. 2012;882:569–604. doi: 10.1007/978-1-61779-842-9_32. [DOI] [PubMed] [Google Scholar]
- Alexander T., Sarfert R., Klotsche J., Kuhl A.A., Rubbert-Roth A., Lorenz H.M., Rech J., Hoyer B.F., Cheng Q., Waka A. The proteasome inhibitior bortezomib depletes plasma cells and ameliorates clinical manifestations of refractory systemic lupus erythematosus. Ann. Rheum. Dis. 2015;74:1474–1478. doi: 10.1136/annrheumdis-2014-206016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews B.S., Eisenberg R.A., Theofilopoulos A.N., Izui S., Wilson C.B., McConahey P.J., Murphy E.D., Roths J.B., Dixon F.J. Spontaneous murine lupus-like syndromes. Clinical and immunopathological manifestations in several strains. J. Exp. Med. 1978;148:1198–1215. doi: 10.1084/jem.148.5.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anolik J.H., Barnard J., Cappione A., Pugh-Bernard A.E., Felgar R.E., Looney R.J., Sanz I. Rituximab improves peripheral B cell abnormalities in human systemic lupus erythematosus. Arthritis Rheum. 2004;50:3580–3590. doi: 10.1002/art.20592. [DOI] [PubMed] [Google Scholar]
- Anthony R.M., Kobayashi T., Wermeling F., Ravetch J.V. Intravenous gammaglobulin suppresses inflammation through a novel T(H)2 pathway. Nature. 2011;475:110–113. doi: 10.1038/nature10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbuckle M.R., McClain M.T., Rubertone M.V., Scofield R.H., Dennis G.J., James J.A., Harley J.B. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N. Engl. J. Med. 2003;349:1526–1533. doi: 10.1056/NEJMoa021933. [DOI] [PubMed] [Google Scholar]
- Baerenwaldt A., Lux A., Danzer H., Spriewald B.M., Ullrich E., Heidkamp G., Dudziak D., Nimmerjahn F. Fcgamma receptor IIB (FcgammaRIIB) maintains humoral tolerance in the human immune system in vivo. Proc. Natl. Acad. Sci. U S A. 2011;108:18772–18777. doi: 10.1073/pnas.1111810108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker K.P., Edwards B.M., Main S.H., Choi G.H., Wager R.E., Halpern W.G., Lappin P.B., Riccobene T., Abramian D., Sekut L. Generation and characterization of LymphoStat-B, a human monoclonal antibody that antagonizes the bioactivities of B lymphocyte stimulator. Arthritis Rheum. 2003;48:3253–3265. doi: 10.1002/art.11299. [DOI] [PubMed] [Google Scholar]
- Bar-Or A., Calabresi P.A., Arnold D., Markowitz C., Shafer S., Kasper L.H., Waubant E., Gazda S., Fox R.J., Panzara M. Rituximab in relapsing-remitting multiple sclerosis: a 72-week, open-label, phase I trial. Ann. Neurol. 2008;63:395–400. doi: 10.1002/ana.21363. [DOI] [PubMed] [Google Scholar]
- Barr T.A., Shen P., Brown S., Lampropoulou V., Roch T., Lawrie S., Fan B., O'Connor R.A., Anderton S.M., Bar-Or A. B cell depletion therapy ameliorates autoimmune disease through ablation of IL-6-producing B cells. J. Exp. Med. 2012;209:1001–1010. doi: 10.1084/jem.20111675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boes M., Schmidt T., Linkemann K., Beaudette B.C., Marshak-Rothstein A., Chen J. Accelerated development of IgG autoantibodies and autoimmune disease in the absence of secreted IgM. Proc. Natl. Acad. Sci. U S A. 2000;97:1184–1189. doi: 10.1073/pnas.97.3.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosello S., De Santis M., Lama G., Spano C., Angelucci C., Tolusso B., Sica G., Ferraccioli G. B cell depletion in diffuse progressive systemic sclerosis: safety, skin score modification and IL-6 modulation in an up to thirty-six months follow-up open-label trial. Arthritis Res. Ther. 2010;12:R54. doi: 10.1186/ar2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozza S., Kasermann F., Kaveri S.V., Romani L., Bayry J. Intravenous immunoglobulin protects from experimental allergic bronchopulmonary aspergillosis via a sialylation-dependent mechanism. Eur. J. Immunol. 2019;49:195–198. doi: 10.1002/eji.201847774. [DOI] [PubMed] [Google Scholar]
- Cerqueira C., Manfroi B., Fillatreau S. IL-10-producing regulatory B cells and plasmocytes: molecular mechanisms and disease relevance. Semin. Immunol. 2019;44:101323. doi: 10.1016/j.smim.2019.101323. [DOI] [PubMed] [Google Scholar]
- Chan O., Shlomchik M.J. A new role for B cells in systemic autoimmunity: B cells promote spontaneous T cell activation in MRL-lpr/lpr mice. J. Immunol. 1998;160:51–59. [PubMed] [Google Scholar]
- Chan O.T., Hannum L.G., Haberman A.M., Madaio M.P., Shlomchik M.J. A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. J. Exp. Med. 1999;189:1639–1648. doi: 10.1084/jem.189.10.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y.S., Dieter J.A., Rothaeusler K., Luo Z., Baumgarth N. B-1 cells in the bone marrow are a significant source of natural IgM. Eur. J. Immunol. 2012;42:120–129. doi: 10.1002/eji.201141890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S.B., Emery P., Greenwald M.W., Dougados M., Furie R.A., Genovese M.C., Keystone E.C., Loveless J.E., Burmester G.R., Cravets M.W. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. 2006;54:2793–2806. doi: 10.1002/art.22025. [DOI] [PubMed] [Google Scholar]
- Crampton S.P., Morawski P.A., Bolland S. Linking susceptibility genes and pathogenesis mechanisms using mouse models of systemic lupus erythematosus. Dis. Model. Mech. 2014;7:1033–1046. doi: 10.1242/dmm.016451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshayes S., Khellaf M., Zarour A., Layese R., Fain O., Terriou L., Viallard J.F., Cheze S., Graveleau J., Slama B. Long-term safety and efficacy of rituximab in 248 adults with immune thrombocytopenia: results at 5 years from the French prospective registry ITP-ritux. Am. J. Hematol. 2019;94:1314–1324. doi: 10.1002/ajh.25632. [DOI] [PubMed] [Google Scholar]
- DiLillo D.J., Hamaguchi Y., Ueda Y., Yang K., Uchida J., Haas K.M., Kelsoe G., Tedder T.F. Maintenance of long-lived plasma cells and serological memory despite mature and memory B cell depletion during CD20 immunotherapy in mice. J. Immunol. 2008;180:361–371. doi: 10.4049/jimmunol.180.1.361. [DOI] [PubMed] [Google Scholar]
- DiLillo D.J., Matsushita T., Tedder T.F. B10 cells and regulatory B cells balance immune responses during inflammation, autoimmunity, and cancer. Ann. N. Y. Acad. Sci. 2010;1183:38–57. doi: 10.1111/j.1749-6632.2009.05137.x. [DOI] [PubMed] [Google Scholar]
- Edwards J.C., Szczepanski L., Szechinski J., Filipowicz-Sosnowska A., Emery P., Close D.R., Stevens R.M., Shaw T. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N. Engl. J. Med. 2004;350:2572–2581. doi: 10.1056/NEJMoa032534. [DOI] [PubMed] [Google Scholar]
- Eilertsen G.O., Van Ghelue M., Strand H., Nossent J.C. Increased levels of BAFF in patients with systemic lupus erythematosus are associated with acute-phase reactants, independent of BAFF genetics: a case-control study. Rheumatology (Oxford) 2011;50:2197–2205. doi: 10.1093/rheumatology/ker282. [DOI] [PubMed] [Google Scholar]
- Emery P., Fleischmann R., Filipowicz-Sosnowska A., Schechtman J., Szczepanski L., Kavanaugh A., Racewicz A.J., van Vollenhoven R.F., Li N.F., Agarwal S. The efficacy and safety of rituximab in patients with active rheumatoid arthritis despite methotrexate treatment: results of a phase IIB randomized, double-blind, placebo-controlled, dose-ranging trial. Arthritis Rheum. 2006;54:1390–1400. doi: 10.1002/art.21778. [DOI] [PubMed] [Google Scholar]
- Espeli M., Smith K.G., Clatworthy M.R. FcgammaRIIB and autoimmunity. Immunol. Rev. 2016;269:194–211. doi: 10.1111/imr.12368. [DOI] [PubMed] [Google Scholar]
- Ewels P., Magnusson M., Lundin S., Kaller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32:3047–3048. doi: 10.1093/bioinformatics/btw354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiebiger B.M., Maamary J., Pincetic A., Ravetch J.V. Protection in antibody- and T cell-mediated autoimmune diseases by antiinflammatory IgG Fcs requires type II FcRs. Proc. Natl. Acad. Sci. U S A. 2015;112:E2385–E2394. doi: 10.1073/pnas.1505292112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillatreau S., Gray D., Anderton S.M. Not always the bad guys: B cells as regulators of autoimmune pathology. Nat. Rev. Immunol. 2008;8:391–397. doi: 10.1038/nri2315. [DOI] [PubMed] [Google Scholar]
- Fillatreau S., Sweenie C.H., McGeachy M.J., Gray D., Anderton S.M. B cells regulate autoimmunity by provision of IL-10. Nat. Immunol. 2002;3:944–950. doi: 10.1038/ni833. [DOI] [PubMed] [Google Scholar]
- Furie R., Petri M., Zamani O., Cervera R., Wallace D.J., Tegzova D., Sanchez-Guerrero J., Schwarting A., Merrill J.T., Chatham W.W. A phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum. 2011;63:3918–3930. doi: 10.1002/art.30613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlag D.M., Safy M., Maijer K.I., Tang M.W., Tas S.W., Starmans-Kool M.J.F., van Tubergen A., Janssen M., de Hair M., Hansson M. Effects of B-cell directed therapy on the preclinical stage of rheumatoid arthritis: the PRAIRI study. Ann. Rheum. Dis. 2019;78:179–185. doi: 10.1136/annrheumdis-2017-212763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getahun A., Cambier J.C. Non-antibody-secreting functions of B cells and their contribution to autoimmune disease. Annu. Rev. Cell Dev. Biol. 2019;35:337–356. doi: 10.1146/annurev-cellbio-100617-062518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godeau B., Porcher R., Fain O., Lefrere F., Fenaux P., Cheze S., Vekhoff A., Chauveheid M.P., Stirnemann J., Galicier L. Rituximab efficacy and safety in adult splenectomy candidates with chronic immune thrombocytopenic purpura: results of a prospective multicenter phase 2 study. Blood. 2008;112:999–1004. doi: 10.1182/blood-2008-01-131029. [DOI] [PubMed] [Google Scholar]
- Gomez A.M., Vrolix K., Martinez-Martinez P., Molenaar P.C., Phernambucq M., van der Esch E., Duimel H., Verheyen F., Voll R.E., Manz R.A. Proteasome inhibition with bortezomib depletes plasma cells and autoantibodies in experimental autoimmune myasthenia gravis. J. Immunol. 2011;186:2503–2513. doi: 10.4049/jimmunol.1002539. [DOI] [PubMed] [Google Scholar]
- Gottenberg J.E., Morel J., Perrodeau E., Bardin T., Combe B., Dougados M., Flipo R.M., Saraux A., Schaeverbeke T., Sibilia J., Soubrier M., Vittecoq O., Baron G., Constantin A., Ravaud P., Mariette X., French Society of Rheumatology and the investigators participating in AIR, ORA, and REGATE registries Comparative effectiveness of rituximab, abatacept, and tocilizumab in adults with rheumatoid arthritis and inadequate response to TNF inhibitors: prospective cohort study. BMJ. 2019;364:l67. doi: 10.1136/bmj.l67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillo-Lopez A.J., White C.A., Dallaire B.K., Varns C.L., Shen C.D., Wei A., Leonard J.E., McClure A., Weaver R., Cairelli S., Rosenberg J. Rituximab: the first monoclonal antibody approved for the treatment of lymphoma. Curr. Pharm. Biotechnol. 2000;1:1–9. doi: 10.2174/1389201003379059. [DOI] [PubMed] [Google Scholar]
- Haas K.M., Watanabe R., Matsushita T., Nakashima H., Ishiura N., Okochi H., Fujimoto M., Tedder T.F. Protective and pathogenic roles for B cells during systemic autoimmunity in NZB/W F1 mice. J. Immunol. 2010;184:4789–4800. doi: 10.4049/jimmunol.0902391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamaguchi Y., Uchida J., Cain D.W., Venturi G.M., Poe J.C., Haas K.M., Tedder T.F. The peritoneal cavity provides a protective niche for B1 and conventional B lymphocytes during anti-CD20 immunotherapy in mice. J. Immunol. 2005;174:4389–4399. doi: 10.4049/jimmunol.174.7.4389. [DOI] [PubMed] [Google Scholar]
- Harrold L.R., John A., Best J., Zlotnick S., Karki C., Li Y., Greenberg J.D., Kremer J.M. Impact of rituximab on patient-reported outcomes in patients with rheumatoid arthritis from the US Corrona Registry. Clin. Rheumatol. 2017;36:2135–2140. doi: 10.1007/s10067-017-3742-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M., Hamaguchi Y., Yanaba K., Bouaziz J.D., Uchida J., Fujimoto M., Matsushita T., Matsushita Y., Horikawa M., Komura K. B-lymphocyte depletion reduces skin fibrosis and autoimmunity in the tight-skin mouse model for systemic sclerosis. Am. J. Pathol. 2006;169:954–966. doi: 10.2353/ajpath.2006.060205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser S.L., Bar-Or A., Comi G., Giovannoni G., Hartung H.P., Hemmer B., Lublin F., Montalban X., Rammohan K.W., Selmaj K. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N. Engl. J. Med. 2017;376:221–234. doi: 10.1056/NEJMoa1601277. [DOI] [PubMed] [Google Scholar]
- Hauser S.L., Waubant E., Arnold D.L., Vollmer T., Antel J., Fox R.J., Bar-Or A., Panzara M., Sarkar N., Agarwal S. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N. Engl. J. Med. 2008;358:676–688. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- Hoyer B.F., Moser K., Hauser A.E., Peddinghaus A., Voigt C., Eilat D., Radbruch A., Hiepe F., Manz R.A. Short-lived plasmablasts and long-lived plasma cells contribute to chronic humoral autoimmunity in NZB/W mice. J. Exp. Med. 2004;199:1577–1584. doi: 10.1084/jem.20040168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IJspeert H., van Schouwenburg P.A., van Zessen D., Pico-Knijnenburg I., Stubbs A.P., van der Burg M. Antigen receptor galaxy: a user-friendly, web-based tool for analysis and visualization of T and B cell receptor repertoire data. J. Immunol. 2017;198:4156–4165. doi: 10.4049/jimmunol.1601921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakez-Ocampo J., Atisha-Fregoso Y., Llorente L. Refractory primary Sjogren syndrome successfully treated with bortezomib. J. Clin. Rheumatol. 2015;21:31–32. doi: 10.1097/RHU.0000000000000210. [DOI] [PubMed] [Google Scholar]
- Jia Y., Zhao L., Wang C., Shang J., Miao Y., Dong Y., Zhao Z. Anti-double-stranded DNA isotypes and anti-C1q antibody improve the diagnostic specificity of systemic lupus erythematosus. Dis. Markers. 2018;2018:4528547. doi: 10.1155/2018/4528547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones J.D., Hamilton B.J., Skopelja S., Rigby W.F. Induction of interleukin-6 production by rituximab in human B cells. Arthritis Rheumatol. 2014;66:2938–2946. doi: 10.1002/art.38798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko Y., Nimmerjahn F., Ravetch J.V. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science. 2006;313:670–673. doi: 10.1126/science.1129594. [DOI] [PubMed] [Google Scholar]
- Kasaian M.T., Casali P. Autoimmunity-prone B-1 (CD5 B) cells, natural antibodies and self recognition. Autoimmunity. 1993;15:315–329. doi: 10.3109/08916939309115755. [DOI] [PubMed] [Google Scholar]
- Kaveri S.V., Silverman G.J., Bayry J. Natural IgM in immune equilibrium and harnessing their therapeutic potential. J. Immunol. 2012;188:939–945. doi: 10.4049/jimmunol.1102107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan T.A., Friedensohn S., Gorter de Vries A.R., Straszewski J., Ruscheweyh H.J., Reddy S.T. Accurate and predictive antibody repertoire profiling by molecular amplification fingerprinting. Sci. Adv. 2016;2:e1501371. doi: 10.1126/sciadv.1501371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S., Tada N., Nakayama E., Liu Y., Hammerling U. A new mouse cell-surface antigen (Ly-m20) controlled by a gene linked to Mls locus and defined by monoclonal antibodies. Immunogenetics. 1981;14:3–14. doi: 10.1007/BF00344295. [DOI] [PubMed] [Google Scholar]
- Lavie F., Miceli-Richard C., Ittah M., Sellam J., Gottenberg J.E., Mariette X. Increase of B cell-activating factor of the TNF family (BAFF) after rituximab treatment: insights into a new regulating system of BAFF production. Ann. Rheum. Dis. 2007;66:700–703. doi: 10.1136/ard.2006.060772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leandro M.J. B-cell subpopulations in humans and their differential susceptibility to depletion with anti-CD20 monoclonal antibodies. Arthritis Res. Ther. 2013;15 Suppl 1:S3. doi: 10.1186/ar3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leandro M.J., Edwards J.C., Cambridge G., Ehrenstein M.R., Isenberg D.A. An open study of B lymphocyte depletion in systemic lupus erythematosus. Arthritis Rheum. 2002;46:2673–2677. doi: 10.1002/art.10541. [DOI] [PubMed] [Google Scholar]
- Lipsky P.E. Systemic lupus erythematosus: an autoimmune disease of B cell hyperactivity. Nat. Immunol. 2001;2:764–766. doi: 10.1038/ni0901-764. [DOI] [PubMed] [Google Scholar]
- Looney R.J., Anolik J.H., Campbell D., Felgar R.E., Young F., Arend L.J., Sloand J.A., Rosenblatt J., Sanz I. B cell depletion as a novel treatment for systemic lupus erythematosus: a phase I/II dose-escalation trial of rituximab. Arthritis Rheum. 2004;50:2580–2589. doi: 10.1002/art.20430. [DOI] [PubMed] [Google Scholar]
- Ludwig R.J., Vanhoorelbeke K., Leypoldt F., Kaya Z., Bieber K., McLachlan S.M., Komorowski L., Luo J., Cabral-Marques O., Hammers C.M. Mechanisms of autoantibody-induced pathology. Front. Immunol. 2017;8:603. doi: 10.3389/fimmu.2017.00603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lux A., Seeling M., Baerenwaldt A., Lehmann B., Schwab I., Repp R., Meidenbauer N., Mackensen A., Hartmann A., Heidkamp G. A humanized mouse identifies the bone marrow as a niche with low therapeutic IgG activity. Cell Rep. 2014;7:236–248. doi: 10.1016/j.celrep.2014.02.041. [DOI] [PubMed] [Google Scholar]
- Mackay M., Stanevsky A., Wang T., Aranow C., Li M., Koenig S., Ravetch J.V., Diamond B. Selective dysregulation of the FcgammaIIB receptor on memory B cells in SLE. J. Exp. Med. 2006;203:2157–2164. doi: 10.1084/jem.20051503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massoud A.H., Yona M., Xue D., Chouiali F., Alturaihi H., Ablona A., Mourad W., Piccirillo C.A., Mazer B.D. Dendritic cell immunoreceptor: a novel receptor for intravenous immunoglobulin mediates induction of regulatory T cells. J. Allergy Clin. Immunol. 2014;133:853–863 e5. doi: 10.1016/j.jaci.2013.09.029. [DOI] [PubMed] [Google Scholar]
- Matsushita T., Yanaba K., Bouaziz J.D., Fujimoto M., Tedder T.F. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J. Clin. Invest. 2008;118:3420–3430. doi: 10.1172/JCI36030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill J.T., Neuwelt C.M., Wallace D.J., Shanahan J.C., Latinis K.M., Oates J.C., Utset T.O., Gordon C., Isenberg D.A., Hsieh H.J. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010;62:222–233. doi: 10.1002/art.27233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E.D., Roths J.B. New inbred strains. Mouse News Lett. 1978;58:51. [Google Scholar]
- Murphy E.D., Roths J.B. A Y chromosome associated factor in strain BXSB producing accelerated autoimmunity and lymphoproliferation. Arthritis Rheum. 1979;22:1188–1194. doi: 10.1002/art.1780221105. [DOI] [PubMed] [Google Scholar]
- Nagel A., Podstawa E., Eickmann M., Muller H.H., Hertl M., Eming R. Rituximab mediates a strong elevation of B-cell-activating factor associated with increased pathogen-specific IgG but not autoantibodies in pemphigus vulgaris. J. Invest. Dermatol. 2009;129:2202–2210. doi: 10.1038/jid.2009.27. [DOI] [PubMed] [Google Scholar]
- Nashi E., Wang Y., Diamond B. The role of B cells in lupus pathogenesis. Int. J. Biochem. Cell Biol. 2010;42:543–550. doi: 10.1016/j.biocel.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarra S.V., Guzman R.M., Gallacher A.E., Hall S., Levy R.A., Jimenez R.E., Li E.K., Thomas M., Kim H.Y., Leon M.G. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet. 2011;377:721–731. doi: 10.1016/S0140-6736(10)61354-2. [DOI] [PubMed] [Google Scholar]
- Neubert K., Meister S., Moser K., Weisel F., Maseda D., Amann K., Wiethe C., Winkler T.H., Kalden J.R., Manz R.A., Voll R.E. The proteasome inhibitor bortezomib depletes plasma cells and protects mice with lupus-like disease from nephritis. Nat. Med. 2008;14:748–755. doi: 10.1038/nm1763. [DOI] [PubMed] [Google Scholar]
- Nguyen T.T., Baumgarth N. Natural IgM and the development of B cell-mediated autoimmune diseases. Crit. Rev. Immunol. 2016;36:163–177. doi: 10.1615/CritRevImmunol.2016018175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn F., Bruhns P., Horiuchi K., Ravetch J.V. FcgammaRIV: a novel FcR with distinct IgG subclass specificity. Immunity. 2005;23:41–51. doi: 10.1016/j.immuni.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn F., Ravetch J.V. Fcgamma receptors: old friends and new family members. Immunity. 2006;24:19–28. doi: 10.1016/j.immuni.2005.11.010. [DOI] [PubMed] [Google Scholar]
- Pezer M., Stambuk J., Perica M., Razdorov G., Banic I., Vuckovic F., Gospic A.M., Ugrina I., Vecenaj A., Bakovic M.P. Effects of allergic diseases and age on the composition of serum IgG glycome in children. Sci. Rep. 2016;6:33198. doi: 10.1038/srep33198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pincetic A., Bournazos S., DiLillo D.J., Maamary J., Wang T.T., Dahan R., Fiebiger B.M., Ravetch J.V. Type I and type II Fc receptors regulate innate and adaptive immunity. Nat. Immunol. 2014;15:707–716. doi: 10.1038/ni.2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard N.R., Smith K.G. B cell inhibitory receptors and autoimmunity. Immunology. 2003;108:263–273. doi: 10.1046/j.1365-2567.2003.01592.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radbruch A., Thiel A. Cell therapy for autoimmune diseases: does it have a future? Ann. Rheum. Dis. 2004;63 Suppl 2:ii96–ii101. doi: 10.1136/ard.2004.028340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Casals M., Soto M.J., Cuadrado M.J., Khamashta M.A. Rituximab in systemic lupus erythematosus: a systematic review of off-label use in 188 cases. Lupus. 2009;18:767–776. doi: 10.1177/0961203309106174. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Zhurbenko N., Quach T.D., Hopkins T.J., Rothstein T.L., Hernandez A.M. Human B-1 cells and B-1 cell antibodies change with advancing age. Front. Immunol. 2019;10:483. doi: 10.3389/fimmu.2019.00483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers N.J., Gabriel L., Nunes C.T., Rose S.J., Thiruudaian V., Boyle J., Morley B.J. Monocytosis in BXSB mice is due to epistasis between Yaa and the telomeric region of chromosome 1 but does not drive the disease process. Genes Immun. 2007;8:619–627. doi: 10.1038/sj.gene.6364424. [DOI] [PubMed] [Google Scholar]
- Sabahi R., Anolik J.H. B-cell-targeted therapy for systemic lupus erythematosus. Drugs. 2006;66:1933–1948. doi: 10.2165/00003495-200666150-00004. [DOI] [PubMed] [Google Scholar]
- Safonova Y., Bonissone S., Kurpilyansky E., Starostina E., Lapidus A., Stinson J., DePalatis L., Sandoval W., Lill J., Pevzner P.A. IgRepertoireConstructor: a novel algorithm for antibody repertoire construction and immunoproteogenomics analysis. Bioinformatics. 2015;31:53–61. doi: 10.1093/bioinformatics/btv238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrezenmeier E., Jayne D., Dorner T. Targeting B cells and plasma cells in glomerular diseases: translational perspectives. J. Am. Soc. Nephrol. 2018;29:741–758. doi: 10.1681/ASN.2017040367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab I., Nimmerjahn F. Intravenous immunoglobulin therapy: how does IgG modulate the immune system? Nat. Rev. Immunol. 2013;13:176–189. doi: 10.1038/nri3401. [DOI] [PubMed] [Google Scholar]
- Seeling M., Bruckner C., Nimmerjahn F. Differential antibody glycosylation in autoimmunity: sweet biomarker or modulator of disease activity? Nat. Rev. Rheumatol. 2017;13:621–630. doi: 10.1038/nrrheum.2017.146. [DOI] [PubMed] [Google Scholar]
- Shanafelt T.D., Madueme H.L., Wolf R.C., Tefferi A. Rituximab for immune cytopenia in adults: idiopathic thrombocytopenic purpura, autoimmune hemolytic anemia, and Evans syndrome. Mayo Clin. Proc. 2003;78:1340–1346. doi: 10.4065/78.11.1340. [DOI] [PubMed] [Google Scholar]
- Shlomchik M.J., Madaio M.P., Ni D., Trounstein M., Huszar D. The role of B cells in lpr/lpr-induced autoimmunity. J. Exp. Med. 1994;180:1295–1306. doi: 10.1084/jem.180.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith K.G., Clatworthy M.R. FcgammaRIIB in autoimmunity and infection: evolutionary and therapeutic implications. Nat. Rev. Immunol. 2010;10:328–343. doi: 10.1038/nri2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasi R., Pagano A., Stipa E., Amadori S. Rituximab chimeric anti-CD20 monoclonal antibody treatment for adults with chronic idiopathic thrombocytopenic purpura. Blood. 2001;98:952–957. doi: 10.1182/blood.v98.4.952. [DOI] [PubMed] [Google Scholar]
- Stohl W., Metyas S., Tan S.M., Cheema G.S., Oamar B., Xu D., Roschke V., Wu Y., Baker K.P., Hilbert D.M. B lymphocyte stimulator overexpression in patients with systemic lupus erythematosus: longitudinal observations. Arthritis Rheum. 2003;48:3475–3486. doi: 10.1002/art.11354. [DOI] [PubMed] [Google Scholar]
- Tackenberg B., Jelcic I., Baerenwaldt A., Oertel W.H., Sommer N., Nimmerjahn F., Lunemann J.D. Impaired inhibitory Fcgamma receptor IIB expression on B cells in chronic inflammatory demyelinating polyneuropathy. Proc. Natl. Acad. Sci. U S A. 2009;106:4788–4792. doi: 10.1073/pnas.0807319106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai T. Roles of Fc receptors in autoimmunity. Nat. Rev. Immunol. 2002;2:580–592. doi: 10.1038/nri856. [DOI] [PubMed] [Google Scholar]
- Takai T., Ono M., Hikida M., Ohmori H., Ravetch J.V. Augmented humoral and anaphylactic responses in Fc gamma RII-deficient mice. Nature. 1996;379:346–349. doi: 10.1038/379346a0. [DOI] [PubMed] [Google Scholar]
- Tarasenko T., Dean J.A., Bolland S. FcgammaRIIB as a modulator of autoimmune disease susceptibility. Autoimmunity. 2007;40:409–417. doi: 10.1080/08916930701464665. [DOI] [PubMed] [Google Scholar]
- Thalayasingam N., Isaacs J.D. Anti-TNF therapy. Best Pract. Res. Clin. Rheumatol. 2011;25:549–567. doi: 10.1016/j.berh.2011.10.004. [DOI] [PubMed] [Google Scholar]
- Theofilopoulos A.N., McConahey P.J., Izui S., Eisenberg R.A., Pereira A.B., Creighton W.D. A comparative immunologic analysis of several murine strains with autoimmune manifestations. Clin. Immunol. Immunopathol. 1980;15:258–278. doi: 10.1016/0090-1229(80)90039-2. [DOI] [PubMed] [Google Scholar]
- Uchida J., Hamaguchi Y., Oliver J.A., Ravetch J.V., Poe J.C., Haas K.M., Tedder T.F. The innate mononuclear phagocyte network depletes B lymphocytes through Fc receptor-dependent mechanisms during anti-CD20 antibody immunotherapy. J. Exp. Med. 2004;199:1659–1669. doi: 10.1084/jem.20040119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida J., Lee Y., Hasegawa M., Liang Y., Bradney A., Oliver J.A., Bowen K., Steeber D.A., Haas K.M., Poe J.C., Tedder T.F. Mouse CD20 expression and function. Int. Immunol. 2004;16:119–129. doi: 10.1093/intimm/dxh009. [DOI] [PubMed] [Google Scholar]
- Vallerskog T., Heimburger M., Gunnarsson I., Zhou W., Wahren-Herlenius M., Trollmo C., Malmstrom V. Differential effects on BAFF and APRIL levels in rituximab-treated patients with systemic lupus erythematosus and rheumatoid arthritis. Arthritis Res. Ther. 2006;8:R167. doi: 10.1186/ar2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A., Cummings R.D., Aebi M., Packer N.H., Seeberger P.H., Esko J.D., Stanley P., Hart G., Darvill A., Kinoshita T. Symbol nomenclature for graphical representations of glycans. Glycobiology. 2015;25:1323–1324. doi: 10.1093/glycob/cwv091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichainun R., Kasitanon N., Wangkaew S., Hongsongkiat S., Sukitawut W., Louthrenoo W. Sensitivity and specificity of ANA and anti-dsDNA in the diagnosis of systemic lupus erythematosus: a comparison using control sera obtained from healthy individuals and patients with multiple medical problems. Asian Pac. J. Allergy Immunol. 2013;31:292–298. doi: 10.12932/AP0272.31.4.2013. [DOI] [PubMed] [Google Scholar]
- Willcocks L.C., Carr E.J., Niederer H.A., Rayner T.F., Williams T.N., Yang W., Scott J.A., Urban B.C., Peshu N., Vyse T.J. A defunctioning polymorphism in FCGR2B is associated with protection against malaria but susceptibility to systemic lupus erythematosus. Proc. Natl. Acad. Sci. U S A. 2010;107:7881–7885. doi: 10.1073/pnas.0915133107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods K.M., Pope M.R., Hoffman S.M., Fleming S.D. CR2+ marginal zone B cell production of pathogenic natural antibodies is C3 independent. J. Immunol. 2011;186:1755–1762. doi: 10.4049/jimmunol.1002059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaytseva O.O., Jansen B.C., Hanic M., Mrcela M., Razdorov G., Stojkovic R., Erhardt J., Brizic I., Jonjic S., Pezer M., Lauc G. MIgGGly (mouse IgG glycosylation analysis) - a high-throughput method for studying Fc-linked IgG N-glycosylation in mice with nanoUPLC-ESI-MS. Sci. Rep. 2018;8:13688. doi: 10.1038/s41598-018-31844-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All raw data (Raw data BZR repertoire analysis: Mendeley Data: https://doi.org/10.17632/smtxktdtfc.1; Data Figures and Supplementary Figures: Mendeley Data: https://doi.org/10.17632/gvxjz3twft.1; Raw Data IgG Glycosylation Part 1.1: Mendeley Data: https://doi.org/10.17632/jygxpd7g7c.1; Raw Data IgG Glycosylation Part 1.2: Mendeley Data: https://doi.org/10.17632/kjfw7cwnxf.1; Raw Data IgG Glycosylation Part 2: Mendeley Data: https://doi.org/10.17632/4s9nxvk7br.1) have been deposited at Mendeley Data and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.