

Abstract
Cannabis use alters sperm DNA methylation, but the potential reversibility of these changes is unknown. Semen samples from cannabis users and non-user controls were collected at baseline and again following a 77-day period of cannabis abstinence (one spermatogenic cycle). Users and controls did not significantly differ by demographics or semen analyses. Whole-genome bisulfite sequencing identified 163 CpG sites with significantly different DNA methylation in sperm between groups (P < 2.94 × 10−9). Genes associated with altered CpG sites were enriched with those involved in development, including cardiogenesis and neurodevelopment. Many of the differences in sperm DNA methylation between groups were diminished after cannabis abstinence. These results indicate that sustained cannabis abstinence significantly reduces the number of sperm showing cannabis-associated alterations at genes important for early development.
Keywords: DNA methylation, cannabis, sperm, development, epigenetics
Introduction
We previously reported associations in humans between cannabis use and altered sperm DNA methylation [1]. Exposure to delta-9-tetrahydrocannabinol (THC) also alters sperm methylation in rats, with their offspring showing similar changes in somatic tissue methylation and altered gene expressions concomitant with neurobehavioral and neurochemical changes [2–5]. Prenatal exposure to cannabis has been associated with adverse birth outcomes, neurodevelopmental delays and autism-like phenotypes, congenital birth defects, and teratologies in children [6–9]. However, the father’s non-genetic contribution to these pathologies is understudied – yet highly relevant – since men are the predominant cannabis consumers [10].
Increasing studies support that the sperm epigenome is capable of intergenerational transmission of adverse health outcomes [11, 12]. It is unknown, however, if the effects of exposure on the sperm epigenome are permanent or temporary. The duration of human spermatogenesis is ∼74 days. Sperm are asynchronously produced from a pool of spermatogonial stem cells (SSCs) [13] to provide a continuous supply of mature sperm [14]. Thus, sperm at all stages of maturation are normally present throughout the seminiferous epithelium [15]. Whether environmental exposures impact the epigenome in the SSCs or in the differentiating spermatogenic cells has implications for the potential “reversibility” of the effects. It is reasonable to hypothesize that post-SSC effects are not permanent, with affected sperm removed over time by ejaculation or reabsorption. Conversely, methylation changes in immortal SSC progenitor cells would be permanent, given the high fidelity of DNA methylation maintenance during cell division. Altered methylation in SSCs is therefore expected to be propagated across all differentiated sperm cell progeny derived from those altered progenitors. The objective here was to determine if men who stop cannabis use for 11 consecutive weeks resolve cannabis-associated epigenetic changes in their sperm.
Participant Demographics and Semen Analyses
There were no significant differences in age, education level, intelligence quotient, race, ethnicity, employment status or marriage between biochemically verified cannabis users (n = 18) and controls (n = 24). There were also no significant differences between groups for semen pH, volume, concentration, motility, and morphology (Table 1).
Table 1.
participant demographics
| Users (n = 18) | Non-users (n = 24) | P-valuea | |
|---|---|---|---|
| Age, mean (SD) | 24.94 (3.70) | 28.04 (5.73) | 0.052 |
| Education, mean (SD) | 16.33 (1.53) | 16.79 (2.67) | 0.519 |
| IQ, mean (SD) | 113.78 (12.28) | 112.29 (12.07) | 0.697 |
| Race | |||
| White, n (%) | 12 (67%) | 13 (54%) | 0.406 |
| Black, n (%) | 5 (28%) | 5 (21%) | |
| Asian, n (%) | 1 (6%) | 5 (21%) | |
| Unknown, n (%) | 0 (0%) | 1 (4%) | |
| Ethnicity | |||
| Not Hispanic or Latino, n (%) | 16 (89%) | 23 (96%) | 0.387 |
| Hispanic or Latino, n (%) | 2 (11%) | 1 (4%) | |
| Employment | |||
| Full- or part-time, n (%) | 14 (78%) | 16 (67%) | 0.572 |
| Unemployed, n (%) | 0 (0%) | 1 (4%) | |
| In school, n (%) | 4 (22%) | 7 (29%) | |
| Marriage | |||
| Single/never married, n (%) | 13 (72%) | 17 (71%) | 0.466 |
| Married/partnered, n (%) | 4 (22%) | 7 (29%) | |
| Not reported, n (%) | 1 (6%) | 0 (0%) | |
| Semen analysis | |||
| Volume (ml), mean (SD) | 3.51 (1.14) | 3.87 (1.42) | 0.376 |
| pH, mean (SD) | 8.14 (0.18) | 8.23 (0.20) | 0.182 |
| Concentration (×106/ml), mean (SD) | 87.13 (64.19) | 99.29 (49.02) | 0.490 |
| Motility (%), mean (SD) | 57.69 (15.76) | 63.49 (14.91) | 0.231 |
| Morphology (% normal), mean (SD) | 4.08 (2.70) | 4.14 (1.81) | 0.932 |
ANOVA or chi-square analyses were run for continuous or categorical values, respectively.
Concordance of Whole-Genome Bisulfite Sequencing Results with an Independent Reduced Representation Bisulfite Sequencing Dataset
The top 10K differentially methylated CpG sites from whole genome bisulfite sequencing (WGBS) between cannabis users and controls prior to and after an 11-week time span during which the users abstained from cannabis use are presented in Supplemental Tables S1 and S2, respectively. Sustained cannabis abstinence was biochemically verified weekly using quantitative urine toxicology testing [16]. The threshold for Bonferroni-adjusted significance was P < 2.94 × 10−9, resulting in 163 significantly differentially methylated CpG sites before and 127 significantly differentially methylated CpG sites after cannabis abstinence (Fig. 1). Only three CpG sites were in common between these two datasets.
Figure 1:
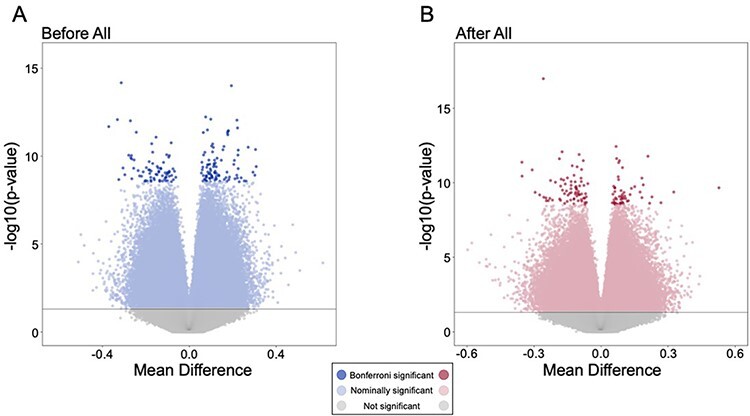
genes associated with differentially methylated CpG sites. (A) Differentially methylated CpG sites between users and controls before abstinence. Gray circles are non-significant, light blue circles are nominally significant, and dark blue circles are Bonferroni significant (n = 163, P < 2.94 × 10−9). (B) Differentially methylated CpG sites between users and controls after abstinence. Gray circles are non-significant, pink circles are nominally significant, and red circles are Bonferroni significant (n = 127, P < 2.94 × 10−9)
We compared the “before” WGBS results to those from our prior analogous reduced representation bisulfite sequencing (RRBS) study of independent cannabis users and non-user controls [1]. In that study, we identified 2151 genes associated with differentially methylated CpG sites. Among the 4123 gene names associated with the top 5000 CpG sites from the present WGBS dataset, 336 were in common between the two studies (P = 1.35 × 10−6, odds ratio = 0.75, Supplemental Fig. S1).
Functional similarities between the RRBS and WGBS findings were also confirmed. Of the 4123 unique gene names, 2789 were recognized and used in the DAVID Functional Annotation Bioinformatics platform [17], resulting in 55 significant Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways associated with the WGBS gene list (false discovery rate, FDR P < 7.83 × 10−5 to P < 0.005, Supplemental Table S3). The RRBS dataset resulted in 11 significant KEGG pathways [1]. Six pathways were identified as enriched in both independent datasets: “glutamatergic synapse,” “circadian entrainment,” “hippo signaling pathway,” “mitogen activated protein kinase signaling,” “platelet activation,” and “pathways in cancer.”
Abstinence from Cannabis Use Resolves Methylation Changes at Select Genes across the Sperm Epigenome
We determined if effect sizes of the methylation differences between users and controls for the same 163 significant CpG sites (P < 2.94 × 10−9) identified in the “before” comparison were lessened in the “after” comparison. We found the magnitude of methylation differences between users and controls was markedly diminished after cannabis abstinence (Fig. 2A). To determine if this change over time was significant, we created a contingency table using a methylation threshold previously published [1] where we binned the methylation changes to those >10% and those <10% between users and controls before and after cannabis abstinence. Distribution of the CpG sites with a >10% methylation difference between users and controls was significantly different before and after cannabis abstinence (P < 0.0001; Fisher’s exact test; Fig. 2B).
Figure 2:
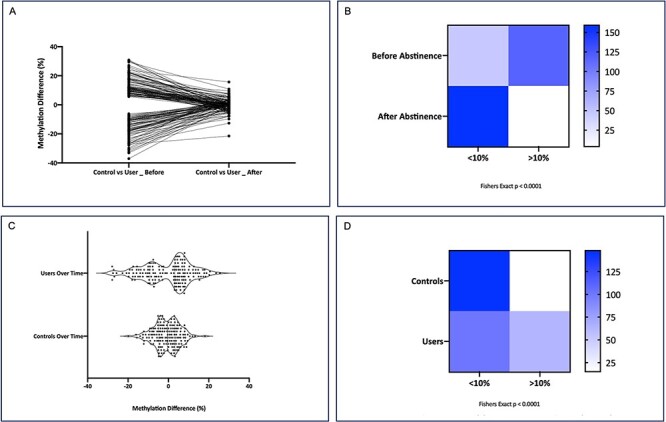
effect of abstinence on DNA methylation. (A) Methylation change over time between cannabis users (n = 18) and controls (n = 24). The methylation difference is plotted on the y-axis and the x-axis is the methylation difference at the 163 Bonferroni significant CpG sites between users and controls before and after abstinence. (B) Methylation changes were binned into <10% or >10% between users and controls both before and after abstinence. The distribution of the 163 CpG sites with a > 10% methylation difference between users and controls was significantly different before and after cannabis abstinence (Fisher’s exact P < 0.0001). (C) Violin plots show the distribution of methylation changes within groups over time. The width of the plot shows the density of the distribution of the datapoints. The median, 25th, and 75th quartiles are represented. (D) Methylation changes were binned into <10% or >10% methylation difference for users over time as well as controls over time. A Fisher’s exact test indicates that this distribution of the number of CpG sites with a > 10% methylation difference over time is significantly different between the users and controls (P < 0.0001)
We next compared mean methylation changes at the 163 significant CpG sites in both the user and non-user groups to control for an effect of time. Violin plots demonstrate that the distribution of methylation changes over time is broader in the user group (Fig. 2C). The increased centralized density of datapoints in controls indicates that more of the 163 CpG sites exhibited a smaller change in DNA methylation over time. The median methylation change in users is shifted away from zero over time at 3.22%, while the median methylation change over time in the controls was −0.43%. These results are consistent with the methylation changes being attributable to sustained cannabis abstinence in the user group rather than a simple effect of time. We statistically confirmed this using a contingency table with binning as described above within the users and controls over time. Of the 163 CpG sites in the controls, 15 showed a >10% methylation change over time. This contrasts with 62 CpG sites in the users that showed a >10% methylation change over time (P < 0.0001; Fisher’s exact test, Fig. 2D).
Alteration of Developmental Genes and Those with Neurological Relevance with Chronic Cannabis Use
We queried the functional roles of the genes annotated to the 163 significant differentially methylated CpG sites before cannabis abstinence between users and controls. Ingenuity Pathway Analysis (IPA) was used to characterize the disease and function annotations associated with this gene list and returned a list of 500 disease and function categories, each with specific disease or function annotations (Supplemental Table S4). We removed 229 cancer-associated category terms, given their overrepresentation in the IPA knowledge base and any category containing only one gene, leaving 79 categories. We focused on those with five or more disease or function annotations. This included “cell morphology,” “cellular development,” “developmental disorder,” and “nervous system development, function, and disease.” Annotations for the top 10 significant diseases or functions included “cardiogenesis,” “growth of an organism,” “morphology of B-cell follicle,” and “cerebral disorder” (Fig. 3).
Figure 3:
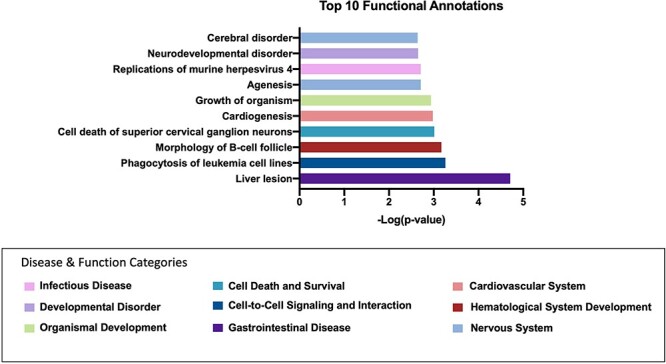
top IPA disease and function annotations for cannabis users compared to controls before abstinence. Graph of the top 10 most significant disease or function annotations associated with genes differentially methylated between cannabis users and controls before abstinence. y-Axis represents the annotation, x-axis is the−log of the P-value. The colors correspond to the category to which this disease or function was annotated (represented by the color legend at the bottom of the figure)
Alteration of Cardiovascular Genes and Those with Neurological and Reproductive Relevance Persists following Cannabis Abstinence
We performed a parallel analysis of the 127 significant CpG sites (P < 2.94 × 10−9) between cannabis users and controls after the cannabis abstinence period. Again, the top 500 diseases and functions and their associated annotations from IPA (Supplemental Table S5) were narrowed by removing cancer-associated terms and categories with only one annotated molecule, leaving 95 categories and their associated disease or function annotations. Categories with five or more disease or function annotations included “cardiovascular system development, function, and disease,” “cell death and survival,” “cell-to-cell signaling and interaction,” “nervous system development, function, and disease,” and “organ morphology.” Lastly, the top 10 significant diseases or functions from this dataset include “learning,” “formation of the hippocampus,” “cognitive impairment,” and “abnormal morphology of enlarged seminiferous tubule” (Fig. 4).
Figure 4:
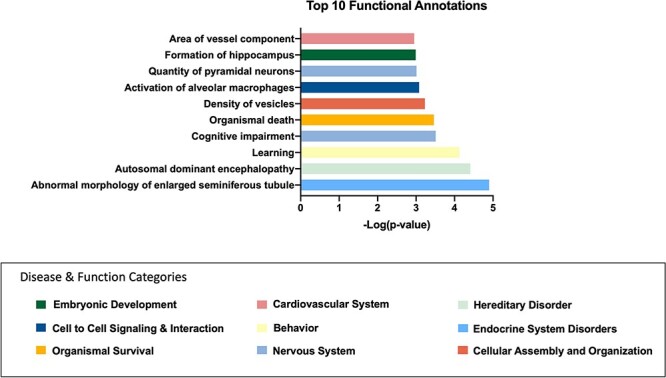
top IPA disease and function annotations for cannabis users compared to controls after abstinence. Graph of the top 10 most significant disease or function annotations associated with genes differentially methylated between cannabis users and controls after abstinence. y-Axis represents the annotation, x-axis is the−log of the P-value. The colors correspond to the category for which this disease or function was annotated to (represented by the color legend at the bottom of the figure)
Cannabis Use is Associated with Differential DNA Methylation at Genes Identified as Epimutation Biomarkers for Autism Susceptibility in Sperm
Our previous work identified hypomethylation in sperm of cannabis users relative to controls at autism candidate gene Disks-Large Associated Protein 2 (DLGAP2) and demonstrated in rats that paternal THC exposure showed heritable methylation changes at Dlgap2 [2]. We found that THC and nicotine both independently impact rat sperm DNA methylation at a select group of the same CpG sites of multiple neuroactive genes, and that these genes are enriched for bivalent chromatin as are the Simons Foundation for Autism Research curated list of autism candidate genes [18]. Recent reports have shown compelling associations between early-life exposure to cannabis and increased incidence of autism and neurodevelopmental delays in offspring [6–9]. Given that our analyses identified enrichment of epigenetically altered genes associated with neurodevelopment and developmental disorders with cannabis exposure, we assessed these genes for potential links to autism in a post hoc analysis. A sperm DNA methylation epimutation biomarker panel for transmission of autism risk was recently established [19] based on the comparison of methylation in sperm of men who had fathered children with autism to those whose children were not on the spectrum. This comparison yielded 805 differentially methylated regions (DMRs) of which 612 had gene annotations [19]. We first determined overlap between the autism epimutation biomarker panel of genes and the 163 significant CpG sites (161 genes) we identified prior to the cannabis abstinence. Reassuringly, there was no significant overlap between these two lists (Fig. 5A). However, comparison of the 127 significant CpG sites (124 genes) we identified after cannabis abstinence with the autism epimutation biomarker panel genes resulted in overlap of 10 genes (Fig. 5B, P < 0.05, odds ratio = 1.98).
Figure 5:
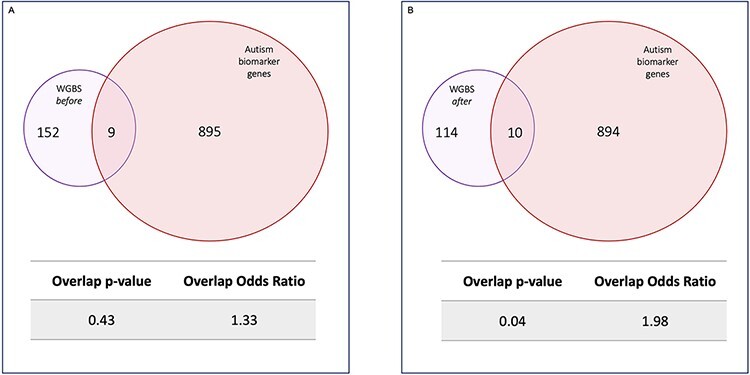
WGBS and autism epimutation biomarker gene overlap. Venn diagrams showing the number of differentially methylated genes in common between WGBS and a list of genes with methylation epimutations used as a biomarker panel for paternal risk of autism susceptibility. A nonsignificant overlap between (A) genes differentially methylated before abstinence and genes identified on the biomarker panel (P = 0.43, odds ratio = 1.33) and a significant overlap between (B) genes differentially methylated after abstinence and genes identified on the biomarker panel (P = 0.04, odds ratio = 1.98)
Discussion
We assessed the impact of cannabis use on the entirety of the sperm DNA methylome and asked whether cannabis abstinence for 11 weeks—encompassing the duration of one human spermatogenic cycle—ameliorated the cannabis-associated methylation changes in sperm. We analyzed sperm before and after the 11-week period for all participants. Users and controls did not differ demographically or by semen parameters. We were surprised at the lack of a significant difference in sperm concentration, given that our prior study had identified a significant reduction in mean sperm concentration in cannabis users as compared to controls [1]. Despite lack of significance here, mean user sperm concentrations (87.1 × 106/ml) trended lower than the mean of the controls (99.3 × 106/ml). Our data and other conflicting reports on the effects of cannabis use on sperm concentration underscore the need to assess this in a much larger population.
Our study used the conservative Bonferroni correction for multiple comparisons to reduce type I errors. Given these stringent criteria and use of WGBS, 163 CpG sites significantly differed in the sperm from users and controls before cannabis abstinence. These methylation differences were reduced at these same 163 CpG sites after an 11-week period of objectively verified cannabis abstinence. The magnitude of methylation differences between users and controls was significantly greater at these CpG sites before the period of cannabis abstinence than it was after, supporting the efficacy of a cannabis abstinence period at resolving some of the methylation changes in sperm. We confirmed this was indeed attributable to the cannabis abstinence by showing there was significantly less fluctuation of these same CpG sites in the controls over time.
There are several possible reasons that only partial mitigation of DNA methylation changes was observed in the sperm of the users after sustained cannabis abstinence. First, although the spermatogenic cycle in humans is estimated to be about 74 days, the sperm in the ejaculate after the 77-day time period may represent a mixture of sperm formed after the cannabis use was stopped in addition to sperm that were formed prior to abstinence that have not yet cleared. Alternatively, the persistent methylation alterations in the user group following the period of cannabis abstinence may reflect changes that have occurred in the spermatogonia. We expect such changes would be permanent such that any asymmetrical division from these progenitors would heritably transfer methylation alterations to the spermatocytes. It is also possible that both of these scenarios occur. Follow-up studies should investigate the impact of a longer abstinence period to determine if complete resolution is possible.
The observed mitigation at a subset of genes has implications for public health interventions. Our data support the contention that refraining from cannabis use for a period of at least 11 weeks provides benefits in resolving many of the sperm methylation changes associated with cannabis use. Few studies have investigated the impacts of an intervention on dynamic changes to the sperm epigenome. Donkin et al. characterized the sperm epigenome in obese and lean men and then analyzed methylation changes in morbidly obese men who underwent bariatric surgery [20]. Sperm samples were collected 1 week before and 1 week after weight-loss surgery, and DNA methylation changes were analyzed from sperm collected at both time points. The authors observed changes to the methylation status of 1509 genes just 1 week following surgery [20]. In studies of these men 1 year after surgery, the number of CpG sites with changes in DNA methylation in sperm expanded to include an additional 2401 genes [20]. This demonstrates that the sustained weight loss experienced following bariatric surgery has cumulative effects on the sperm epigenome. It also illustrates the dynamic nature of the sperm DNA methylome and its responsiveness to the environment.
While we focused our analysis on the CpG sites that were initially identified as significantly differentially methylated between cannabis users and controls and for which methylation changes diminished with cannabis abstinence, there were still 127 significant CpG sites between users and controls after cannabis abstinence. Only three of those 127 were also identified as significantly differentially methylated between users and controls before abstinence. Thus, while abstinence was effective at resolving methylation changes at many genes, there is a distinct set that is significantly differentially methylated after abstinence. This may reflect a subset of genes whose differences became more apparent after abstinence because other stronger differences were resolved. Cannabis use and subsequent abstinence can impact a number of key physiological processes and functions, including sleep, mood, and appetite/diet. In early abstinence, withdrawal effects on these parameters could have been captured during the window of observation in this study [21]. Cannabis abstinence might also induce drug offset effects and reversal of other neurobiological adaptations [22]. Thus, it is possible that these phenomena might impact methylation at novel CpG sites that were not necessarily evident during cannabis use but became apparent following sustained cannabis abstinence. That there were fewer CpG sites that were significant after abstinence supports that abstaining from cannabis was effective at reducing the methylation signal associated with cannabis use. The precise mechanism by which some methylation changes seem to resolve with cannabis abstinence while others emerge during abstinence requires future investigation.
The sperm DNA methylation results at baseline in both groups, assessed using WGBS, support our prior results using RRBS. Both studies examined methylation in sperm from cannabis users relative to non-user controls. While WGBS identified a larger number of significant sites, there were 336 genes in common between the two methodologies, which is a significant finding. There were 11 significant pathway terms associated with the differentially methylated CpGs in the RRBS dataset and 55 for the WGBS dataset. Four terms were in common between the independent datasets, again bolstering the validity of the initial RRBS study. However, that there were an additional 49 KEGG pathway terms identified from the WGBS data not captured by the RRBS illustrates the value and power of studies encompassing the entirety of the methylome as critical for understanding the full extent of the impact of exposures, including cannabis, on the epigenome.
There were some similarities in the IPA disease and function annotations associated with the 163 CpG sites before and 127 CpG sites after cannabis abstinence that were significant between users and controls. First, one category with five or more annotations at both time points was “nervous system development, function, and disease.” Second, there were terms among the top 10 functional annotations at both time points associated with nervous system development and cardiovascular system development. Although the actual disease or function annotations were not identical, their comparable categorical associations support our prior findings showing an impact of male cannabis use on genes in sperm that are associated with early developmental processes. These findings are also concordant with our results using a rat model of paternal preconception exposure to THC and epidemiological studies associating perinatal cannabis exposure with adverse health outcomes in offspring [3, 4, 18, 23]. Specifically, rat studies demonstrated DNA methylation and gene expression changes in nucleus accumbens tissues at genes implicated in neurodevelopment [2, 24], while population-based studies have associated prenatal cannabis use with autism-like phenotypes, developmental delays, and cardiomyopathies [7, 25].
Autism spectrum disorders (ASDs) have been put forth, with epidemiologic support, as the most common form of cannabis-associated teratology [26]. Our prior work identified associations between cannabis and THC exposure in humans and rats, respectively, and altered sperm methylation at genes implicated in ASD [2, 18]. This is potentially relevant since a significant positive association between cannabis legalization and increased ASD incidence was reported [26]. Studies are increasingly examining the potential (non-genetic) role of the father in transmitting autism susceptibility. One such study identified a DNA methylation signature in sperm that distinguishes men who have fathered a child diagnosed with autism from those who have not. The researchers proposed these 805 DMRs for use as a biomarker panel to identify the potential for paternal transmission of autism susceptibility to his child [19]. We found a significant overlap between the genes within 612 of these DMRs (those with gene annotations) to the genes associated with the 127 differentially methylated CpG sites between cannabis users and controls after cannabis abstinence. Genes in common include COL14A1, FBXO25, FGF12, GALNT9, PSD3, TMCO4, AC116362.1, AC234771.2, AL732372.2, and DLGAP2; the gene we previously showed was hypomethylated in sperm of cannabis using men and that shows evidence of intergenerational epigenetic transmission from male rats exposed to THC [2]. These overlapping genes were not necessarily among those significantly differentially methylated prior to abstaining from cannabis use but rather were among those that were significantly differentially methylated after abstaining from cannabis use. The finding that DLGAP2 is among these genes—and is an imprinted gene—supports the resistance of imprinted genes to postfertilization reprogramming as well as our prior finding of heritability of altered methylation at Dlgap2 in a rodent model. Altogether, this bolsters the evidence that paternal preconception exposures can influence DNA methylation at genes that contribute to offspring neurodevelopment and further suggests cannabis use by males may increase the risk of adverse neurodevelopment in offspring through epigenetic mechanisms. The significant overlap of the autism biomarker panel genes with the genes for the present study that are differentially methylated after cannabis abstinence raises important concerns about the potential for select methylation changes to persist, or emerge, following drug cessation.
The requirement to stop cannabis use for 11 weeks was a study limitation, which may have been a barrier to participation. Indeed, several individuals were unable to complete the full 11 weeks without using cannabis, necessitating their removal from the study. This raises the possibility of potential confounding that we did not account for in that there may be inherent differences between people who can and cannot completely refrain from drug use. For example, those able to successfully abstain may have adopted other health-modifying lifestyle strategies to cope with withdrawal from use, such as a change in exercise habits, sleep schedules, or diet or perhaps experienced higher levels of stress, all of which may impact the sperm epigenome [20, 27–29]. In addition, weekly urine testing in the abstaining group could potentially miss cannabis use that occurred within several days following the prior test. The small sample size limits our ability to generalize findings, particularly due to the minimal racial and ethnic diversity of our participants. Potential confounders including the route of administration and the potency of the cannabis used were not determined. Another limitation relates to the illegal status of cannabis in North Carolina and lack of regulation regarding the purity and/or potency of cannabis and the other cannabinoids present in cannabis plant products used by the participants. In this regard, we were not able to test for contaminants (e.g. pesticides and heavy metals) in the cannabis products. The majority of cannabis users also consume nicotine [30, 31], and although nicotine users were excluded from our study, nicotine exposure also impacts methylation profiles in sperm [18]. Therefore, there is a need to examine epigenetic effects of paternal cannabis use in individuals who also use nicotine to generalize our findings to the broader population of cannabis users.
Strengths include that this is the first characterization of the effects of cannabis across the complete sperm methylome, and it supports results from our prior study assessing the impact of cannabis use on a subset of the sperm methylome [1]. We demonstrated that abstinence from cannabis use for the duration of at least one spermatogenic cycle allows partial amelioration of the observed methylation changes. Our bioinformatic analyses demonstrated differentially methylated genes are associated with early developmental processes, which supports existing literature about the effects of early-life exposure to cannabis.
There are many implications of our findings for clinical translation. Preconception counseling should advise men to stop using cannabis in advance of trying to conceive on the basis that this will promote a more normal sperm epigenome. A methylation signature of cannabis exposure could be used to screen sperm prior to conception to estimate the risk of transmission of epigenetic abnormalities. Similarly, newborns could be screened to estimate the inheritance of an altered paternal epigenome and to allow for early interventions to minimize adverse outcomes. These potential clinical benefits also necessitate a better understanding of the potential for inter- and trans-generational heritability of the epigenetic alterations and long-term effects of paternal preconception cannabis exposure in his children. Such work should involve epigenetic, developmental, and behavioral analyses of offspring.
In summary, this report is the first to characterize the impact of cannabis use on the entirety of the sperm DNA methylome. It furthermore shows that refraining from cannabis use for 11 weeks resolves a substantial proportion of the effects of cannabis on the sperm methylome. These data begin to address critical knowledge gaps about the biological effects of this exposure, which is especially important as cannabis legalization expands.
Materials and Methods
Human Subjects
Healthy males between 18 and 40 years of age (inclusive) were recruited through the Duke ADHD Clinic as part of the Cannabis-Induced Potential Heritability of Epigenetics Revisions in Sperm (CIPHERS) parent study. Of the participants, 18 were cannabis users with a self-reported frequency of cannabis use at least once weekly over the prior 6 months and 24 were non-user controls. Use at screening was measured via the gas chromatography–mass spectrometry (GC–MS) analysis of the primary urinary cannabis metabolite, 11-nor-9-carboxy-delta-9-tetrahydrocannabinol (THCCOOH) normalized to creatinine, to get a metabolite–creatinine ratio. Non-users were also verified via GC–MS. For the users, THC levels (non-creatinine-adjusted) had to be at least 50 ng/ml, THCCOOH levels had to be at least 15 ng/ml (non-creatinine-adjusted), a urine rapid screening test had to be positive, and participants had to be willing to abstain from cannabis for 11 weeks during the course of the study. For the non-users, THCCOOH levels (non-creatinine-adjusted) had to be 0 ng/ml and participants must have reported no cannabis use in the prior 6 months, fewer than 10 exposures throughout their lifetime, and have a negative urine rapid screening test. Additional inclusion criteria for both groups included willingness to provide semen samples, willingness to comply with all other study requirements, and ability to communicate verbally and in written form in English.
Participants were excluded from the study if (i) there was a positive result for any other drugs of abuse on urine rapid screening test; (ii) participants were prescribed any psychoactive medications; (iii) there was a current diagnosis of any significant psychiatric conditions (other than cannabis use disorder for the cannabis use group as long as the level of use did not require immediate clinical attention in the opinion of the study team); (iv) there was a score on the Marijuana Screening Inventory-X >10; (v) there was a score on the Alcohol Use Disorders Identification Test ≥8; (vi) expired breath carbon monoxide (CO) reading of ≥8 ppm; (vii) expired breath alcohol level of >0.000, urinary cotinine levels that are not negative or not within the non-smoker level; (viii) estimated IQ < 80; and (ix) participants were unable to comply with the study requirements or were otherwise unsuitable for participation in the opinion of the study physician and/or study psychologist.
Participants who met all inclusion and no exclusion criteria were scheduled to return to the Duke Fertility Clinic to provide a baseline sperm sample as well as an additional urine sample for THCCOOH analyses. These baseline samples were collected for both the users and non-users in the study. A second semen sample was obtained from non-users at the end of 11 weeks.
For the user group, following the baseline sample collection, participants were asked to abstain from cannabis use for 11 weeks and to provide weekly urine samples for drug testing to confirm abstinence via GC–MS and rapid urinalysis. Contingency management procedures were followed to encourage cannabis abstinence for the cannabis group following an escalating schedule of reinforcement in the form of money to promote abstinence. Participants in the cannabis use attended weekly lab visits to provide urine samples to verify cannabis abstinence ranging from 4 days per week (Week 1 of cannabis abstinence) and once per week (Week 11 of cannabis abstinence only). Cannabis abstinence was assessed via self-report and was verified using two methods. First, semi-quantitative rapid urine tests were used to test illicit drugs, including THC. Results for THC were expected to be positive during the first few visits due to residual THC in their systems. If participants tested positive for any substance other than THC, they were withdrawn from the study. Second, a urine sample was analyzed in an analytical laboratory via enzyme immunoassay (EIA) and liquid chromatography with tandem mass spectrometry (LC-MS MS) to determine levels of THC and THCCOOH (the primary metabolite) present. The sample was normalized to urine creatinine concentration to obtain a metabolite–creatinine ratio. Continuous abstinence was evaluated using the results of sequential tests based on the algorithm developed by Schwilke and colleagues [16]. This helped determine if the value was due to a recent use or due to residual use. The THCCOOH creatinine-adjusted value obtained from LC-MS MS was used to calculate abstinence. LC-MS MS analysis was stopped once the rapid urine tests were negative for THC.
At the end of the 11 weeks of the study, a second semen sample was collected. All study procedures were reviewed and approved by the Duke Institutional Review Board and were conducted in accordance with the 2013 Declaration of Helsinki. Written informed consent was provided by all participants.
Semen Analyses
Semen analysis was performed at the Duke Fertility Clinic. Semen was analyzed for parameters including volume, appearance, viscosity, pH, white blood cell (WBC) concentration, sperm concentration, total motility, forward progression, and calculated total motile sperm count. Semen was allowed to liquify at room temperature prior to analysis. Volume was determined via aspirating sample into a 5- or 10-ml Falcon pipette (VWR, Randor, PA). Viscosity was determined by dispersing drops from a 5-ml pipette. pH and WBC analyses were performed with QwikCheckTM Test Strips (Medical Electronic Systems, Los Angeles, CA). Concentration and motility were quantified with 5-μl samples using a Makler Counting Chamber (Sefi-Medical Instruments, Haifa, Israel), examined at 200× with an inverted phase-contrast microscope (NIKON DIAPHOT inverted phase, Brighton, MI). Duplicate analyses with a minimum of 100 sperm were performed. Forward progression was characterized for the largest proportion of motile sperm and was defined as 2 = definite forward progression, 1 = weak/sluggish progression, and 0 = absence for forward progression. After the completion of semen analysis, samples were subjected to two-step isolate-gradient centrifugation (Irvine Scientific) to select a motile population enriched in normal morphology. Pelleted sperm were frozen at −80°C for subsequent studies.
DNA Extraction from Sperm
DNA was extracted from human sperm samples according to Qiagen’s Puregene DNA Purification Protocol (Qiagen, Germantown, MD). Briefly, sperm samples underwent cell lysis and Proteinase K digestion, followed by treatment with RNase A solution. Following protein precipitation and removal, DNA was isolated and eluted in 30 μl of nuclease-free water. Genomic DNA (gDNA) was quantified on the NanoDrop 2000 and quality was determined through measurement of the A260/280 and A260/230 ratios. gDNA was stored at −80°C for subsequent studies.
Whole-Genome Bisulfite Sequencing
Library Construction and Sequencing
For each sample, DNA concentration was measured using a Qubit assay (Thermo Fisher, Waltham, MA), and 250 ng was provided to the Duke Genomic and Computational Biology’s Sequencing and Genomic Technologies Core for library construction and sequencing. The DNA was fragmented using a Covaris E210 instrument (Covaris, Woburn, MA) to generate ∼300-bp DNA fragments. Fragmented DNA samples were then converted into Illumina libraries using the Kapa HyperPrep kit (Roche, Basel, Switzerland). In brief, DNA fragments were end repaired, A-tailed, and ligated to barcoded Illumina adapters. Each library was then bisulfite converted using the EZ DNA Methylation-Lightning Kit (Zymo Research, Irvine, CA) and amplified by polymerase chain reaction. Libraries were pooled in equimolar ratios and sequenced on an Illumina NovaSeq 6000 sequencer. Sequencing was done at 150-bp paired-end reads on S4 flow cells.
Data Processing and Quality Control
Raw data were processed using the fastp algorithm to trim low-quality bases and adapter sequences [32]. Read-pairs that were at least 200 nt in length were mapped to the hg38 version of the human genome using the bisulfite sequencing MAPping (BSMAP) algorithm [33]. Amplification artifacts were removed using the “MarkDuplicates” application from the Picard toolkit [34]. The “methRatio” application from BSMAP was then used to call the depth and percent methylation at each CpG locus in the human genome. Sites with a read depth lower than 6× or greater than 60× within any sample were considered missing in that sample. Sites that are not present in at least 50% of the cohort were excluded from the subsequent analysis. CpG sites that overlapped a common variant from the single nucleotide polymorphism database (dbSNP) were excluded from the analysis, as were invariant loci as defined by not having at least a 5% difference between the 10th and 90th percentile of percent methylation values. Median normalization was then applied across all samples before a final percent methylation value was calculated.
WGBS Sequencing Metrics
Genomic DNA from the sperm samples of cannabis users and non-user controls from baseline and Week 11 underwent WGBS to characterize DNA methylation changes across the entire methylome and to determine whether abstinence from cannabis use ameliorated these effects. The average insert size sequenced across all participants was 178.5 base pairs, and the mean coverage achieved by sequencing was 10.06×. The cytosine-guanine (CG) content of the regions sequenced was 24.18%. The distribution of methylation across the human genome for each individual is represented in Supplemental Fig. S2.
Differentially Methylated CpG Sites
To identify potentially differentially methylated CpG sites, a beta-binomial linear regression model with an arc-sine link function was performed on each CpG site individually utilizing the DSS Bioconductor package in R [35–37]. Controls and users were compared at the before and after time points in separate model runs.
Ingenuity Pathway Analysis
The top 10K differentially methylated CpG sites were initially identified for each exposure group—before abstinence and after abstinence—relative to controls, and each CpG site was mapped to the nearest gene. The IPA software was used to perform a bioinformatic analysis on annotated genes from the lists of the 10K CpG sites that were significant following Bonferroni-adjusted significance (P < 2.94 × 10−9). Gene lists were uploaded to the IPA platform, and top diseases and functions associated with these genes were analyzed.
KEGG Pathway Analysis
KEGG pathway analysis was performed for comparison with previously generated RRBS data [1]. The 4123 unique gene names associated with the top 5000 differentially methylated CpG sites identified via WGBS were entered into the Database for Annotation, Visualization, and Integrated Discovery (DAVID) V6.8. Of those, 2789 genes were recognized by DAVID. Their associated KEGG pathways were identified and used in downstream analyses.
Statistical Analysis
Statistics were run in GraphPad Prism Version 9 (GraphPad Software, San Diego, CA). Chi-square or analysis of variance (ANOVA) was run for the analysis of participant demographics for categorical or continuous values, respectively. For semen analyses, ANOVA was performed to detect significant differences. A Fisher’s exact test was run to determine methylation changes over time. Additionally, a Fisher’s exact test was run to determine the overlap and odds ratios between the WGBS and RRBS datasets as well as between the WGBS and the autism biomarker dataset.
Supplementary Material
Acknowledgements
We thank Carole Grenier for her assistance with the laboratory-based portion of the project. We are grateful to the CIPHERS participants for their willingness to take part in the study.
Contributor Information
Rose Schrott, Duke University Program in Environmental Health, Nicholas School of the Environment, Duke University, 9 Circuit Drive, Durham, NC, USA.
Susan K Murphy, Duke University Program in Environmental Health, Nicholas School of the Environment, Duke University, 9 Circuit Drive, Durham, NC, USA; Department of Obstetrics and Gynecology, Division of Reproductive Sciences, Duke University Medical Center, 701 W. Main Street, Durham, NC, USA.
Jennifer L Modliszewski, Duke Center for Genomic and Computational Biology, Duke University Medical Center, 101 Science Drive, Durham, NC, USA.
Dillon E King, Duke University Program in Environmental Health, Nicholas School of the Environment, Duke University, 9 Circuit Drive, Durham, NC, USA.
Bendu Hill, Department of Psychiatry and Behavioral Sciences, Duke University Medical Center, 2608 Erwin Road, Durham, NC, USA.
Nilda Itchon-Ramos, Department of Psychiatry and Behavioral Sciences, Duke University Medical Center, 2608 Erwin Road, Durham, NC, USA.
Douglas Raburn, Department of Obstetrics and Gynecology, Division of Reproductive Endocrinology and Infertility, Duke University Medical Center, 5704 Fayetteville Road, Durham, NC, USA.
Thomas Price, Department of Obstetrics and Gynecology, Division of Reproductive Endocrinology and Infertility, Duke University Medical Center, 5704 Fayetteville Road, Durham, NC, USA.
Edward D Levin, Department of Psychiatry and Behavioral Sciences, Duke University Medical Center, 2608 Erwin Road, Durham, NC, USA.
Ryan Vandrey, Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, 5510 Nathan Shock Drive, Baltimore, MD, USA.
David L Corcoran, Duke Center for Genomic and Computational Biology, Duke University Medical Center, 101 Science Drive, Durham, NC, USA.
Scott H Kollins, Department of Psychiatry and Behavioral Sciences, Duke University Medical Center, 2608 Erwin Road, Durham, NC, USA.
John T Mitchell, Department of Psychiatry and Behavioral Sciences, Duke University Medical Center, 2608 Erwin Road, Durham, NC, USA.
Data Availability
Data are available upon request. WGBS data are available through the GEO Sequence Read Archives (accession number PRJNA754049).
Supplementary data
Supplementary data is available at EnvEpig online.
Funding
The John Templeton Foundation (grants 60564 and 60957 to S.K.M.); the National Institute of Environmental Health Sciences (grant T32ES021432 to R.S. and D.E.K.).
Author contributions
Conceptualization: S.K.M. and S.H.K.; methodology: R.S., S.K.M., J.L.M., D.R., T.P., E.D.L., R.V., D.L.C., S.H.K., and J.T.M.; investigation: R.S., S.K.M., J.L.M., D.E.K., B.H., N.I.R., D.R., T.P., D.C., S.H.K., and J.T.M.; visualization: R.S., S.K.M., J.L.M., and D.E.K.; funding acquisition: S.K.M., E.D.L., and S.H.K.; project administration: S.K.M.; supervision: S.K.M., D.C., T.P., D.C., S.H.K., and J.T.M.; writing—original draft: R.S. and S.K.M.; writing—review and editing: R.S., S.K.M., J.L.M., D.E.K., B.H., N.I.R., D.R., T.P., E.D.L., R.V., D.C., S.H.K., and J.T.M.
Conflict of interest statement. The authors declare that they have no competing interests.
References
- 1.Murphy SK, Itchon-Ramos N, Visco Z. et al. Cannabinoid exposure and altered DNA methylation in rat and human sperm. Epigenetics 2018;13:1208–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schrott R, Acharya K, Itchon-Ramos N. et al. Cannabis use is associated with potentially heritable widespread changes in autism candidate gene DLGAP2 DNA methylation in sperm. Epigenetics 2020;15:161–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holloway ZR, Hawkey AB, Pippin E. et al. Paternal factors in neurodevelopmental toxicology: THC exposure of male rats causes long-lasting neurobehavioral effects in their offspring. Neurotoxicology 2020;78:57–63. [DOI] [PubMed] [Google Scholar]
- 4.Levin ED, Hawkey AB, Hall BJ. et al. Paternal THC exposure in rats causes long-lasting neurobehavioral effects in the offspring. Neurotoxicol Teratol 2019;74:106806. [DOI] [PubMed] [Google Scholar]
- 5.Slotkin TA, Skavicus S, Levin ED. et al. Paternal Δ9-tetrahydrocannabinol exposure prior to mating elicits deficits in cholinergic synaptic function in the offspring. Toxicol Sci 2020;174:210–7. [DOI] [PubMed] [Google Scholar]
- 6.Dong C, Chen J, Harrington A. et al. Cannabinoid exposure during pregnancy and its impact on immune function. Cell Mol Life Sci 2018;76:729–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reece AS, Hulse GK. Cannabis teratology explains current patterns of Coloradan congenital defects: the contribution of increased cannabinoid exposure to rising teratological trends. Clin Pediatr 2019;58:1085–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corsi DJ, Donelle J, Sucha E. et al. Maternal cannabis use in pregnancy and child neurodevelopmental outcomes. Nat Med 2020;26:1536–40. [DOI] [PubMed] [Google Scholar]
- 9.Gunn JK, Rosales CB, Center KE. et al. Prenatal exposure to cannabis and maternal and child health outcomes: a systematic review and meta-analysis. BMJ Open 2016;6:e009986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cuttler C, Mischley LK, Sexton M. Sex differences in cannabis use and effects: a cross-sectional survey of cannabis users. Cannabis Cannabinoid Res 2016;1:166–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morkve Knudsen GT, Rezwan FI, Johannessen A. et al. Epigenome-wide association of father’s smoking with offspring DNA methylation: a hypothesis-generating study. Environ Epigenet 2019;5:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Soubry A, Schildkraut JM, Murtha A. et al. Paternal obesity is associated with IGF2 hypomethylation in newborns: results from a Newborn Epigenetics Study (NEST) cohort. BMC Med 2013;11:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Rooij DG. The spermatogonial stem cell niche. Microsc Res Tech 2009;72:580–5. [DOI] [PubMed] [Google Scholar]
- 14.Neto FT, Bach PV, Najari BB. et al. Spermatogenesis in humans and its affecting factors. Semin Cell Dev Biol 2016;59:10–26. [DOI] [PubMed] [Google Scholar]
- 15.Amann RP. The cycle of the seminiferous epithelium in humans: a need to revisit? J Androl 2008;29:469–87. [DOI] [PubMed] [Google Scholar]
- 16.Schwilke EW, Gullberg RG, Darwin WD. et al. Differentiating new cannabis use from residual urinary cannabinoid excretion in chronic, daily cannabis users. Addiction 2011;106:499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sherman BT, Huang Da W, Tan Q. et al. DAVID Knowledgebase: a gene-centered database integrating heterogeneous gene annotation resources to facilitate high-throughput gene functional analysis. BMC Bioinform 2007;8:426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schrott R, Rajavel M, Acharya K. et al. Sperm DNA methylation altered by THC and nicotine: vulnerability of neurodevelopmental genes with bivalent chromatin. Sci Rep 2020;10:16022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garrido N, Cruz F, Egea RR. et al. Sperm DNA methylation epimutation biomarker for paternal offspring autism susceptibility. Clin Epigenetics 2021;13:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Donkin I, Versteyhe S, Ingerslev LR. et al. Obesity and bariatric surgery drive epigenetic variation of spermatozoa in humans. Cell Metab 2016;23:369–78. [DOI] [PubMed] [Google Scholar]
- 21.Bonnet U, Preuss UW. The cannabis withdrawal syndrome: current insights. Subst Abuse Rehabil 2017;8:9–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vandrey R, Umbricht A, Strain EC. Increased blood pressure after abrupt cessation of daily cannabis use. J Addict Med 2011;5:16–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holloway ZR, Hawkey AB, Torres AK. et al. Paternal cannabis extract exposure in rats: preconception timing effects on neurodevelopmental behavior in offspring. Neurotoxicology 2020;81:180–8. [DOI] [PubMed] [Google Scholar]
- 24.Watson CT, Szutorisz H, Garg P. et al. Genome-wide DNA methylation profiling reveals epigenetic changes in the rat nucleus accumbens associated with cross-generational effects of adolescent THC exposure. Neuropsychopharmacology 2015;40:2993–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reece AS, Hulse GK. Canadian cannabis consumption and patterns of congenital anomalies: an ecological geospatial analysis. J Addict Med 2020;14:e195–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reece AS, Hulse GK. Effect of cannabis legalization on US autism incidence and medium term projections. Clin PediatrOA 2019;4:1–17. [Google Scholar]
- 27.Denham J, O’Brien BJ, Harvey JT. et al. Genome-wide sperm DNA methylation changes after 3 months of exercise training in humans. Epigenomics 2015;7:717–31. [DOI] [PubMed] [Google Scholar]
- 28.Franzago M, Sabovic I, Franchi S. et al. Sperm DNA methylation at metabolism-related genes in vegan subjects. Front Endocrinol 2021;12:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salas-Huetos A, James ER, Salas-Salvado J. et al. Sperm DNA methylation changes after short-term nut supplementation in healthy men consuming a Western-style diet. Andrology 2021;9:260–8. [DOI] [PubMed] [Google Scholar]
- 30.Schauer GL, Berg CJ, Kegler MC. et al. Assessing the overlap between tobacco and marijuana: trends in patterns of co-use of tobacco and marijuana in adults from 2003–2012. Addict Behav 2015;49:26–32. [DOI] [PubMed] [Google Scholar]
- 31.Cohn AM, Abudayyeh H, Perreras L. et al. Patterns and correlates of the co-use of marijuana with any tobacco and individual tobacco products in young adults from Wave 2 of the PATH Study. Addict Behav 2019;92:122–7. [DOI] [PubMed] [Google Scholar]
- 32.Chen S, Zhou Y, Chen Y. et al. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018;34:i884–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xi Y, Li W. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinform 2009;10:232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Broad Institute . Picard Tools. http://broadinstitute.github.io/picard (16 August 2021, date last accessed).
- 35.Computing TRFfS . R: A Language and Environment for Statistical Computing. 2020. http://www.R-project.org/ (16 August 2021, date last accessed).
- 36.Wu H, Xu T, Feng H. et al. Detection of differentially methylated regions from whole-genome bisulfite sequencing data without replicates. Nucleic Acids Res 2015;43:e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park Y, Wu H. Differential methylation analysis for BS-seq data under general experimental design. Bioinformatics 2016;32:1446–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available upon request. WGBS data are available through the GEO Sequence Read Archives (accession number PRJNA754049).