

Abstract
Platelets promote tumor metastasis by inducing promalignant phenotypes in cancer cells and directly contributing to cancer-related thrombotic complications. Platelet-derived extracellular vesicles (EVs) can promote epithelial-mesenchymal transition (EMT) in cancer cells, which confers high-grade malignancy. 12S-hydroxyeicosatetraenoic acid (12-HETE) generated by platelet-type 12-lipoxygenase (12-LOX) is considered a key modulator of cancer metastasis through unknown mechanisms. In platelets, 12-HETE can be esterified into plasma membrane phospholipids (PLs), which drive thrombosis. Using cocultures of human platelets and human colon adenocarcinoma cells (line HT29) and LC-MS/MS, we investigated the impact of platelets on cancer cell biosynthesis of 12S-HETE and its esterification into PLs and whether platelet ability to transfer its molecular cargo might play a role. To this aim, we performed coculture experiments with CFSE[5-(and-6)-carboxyfluorescein diacetate, succinimidyl ester]-loaded platelets. HT29 cells did not generate 12S-HETE or express 12-LOX. However, they acquired the capacity to produce 12S-HETE mainly esterified in plasmalogen phospholipid forms following the uptake of platelet-derived medium-sized EVs (mEVs) expressing 12-LOX. 12-LOX was detected in plasma mEV of patients with adenomas/adenocarcinomas, implying their potential to deliver the protein to cancer cells in vivo. In cancer cells exposed to platelets, endogenous but not exogenous 12S-HETE contributed to changes in EMT gene expression, mitigated by three structurally unrelated 12-LOX inhibitors. In conclusion, we showed that platelets induce the generation of primarily esterified 12-HETE in colon cancer cells following mEV-mediated delivery of 12-LOX. The modification of cancer cell phospholipids by 12-HETE may functionally impact cancer cell biology and represent a novel target for anticancer agent development.
Supplementary key words: blood platelets, 12-HETE, 12-Lipoxygenase, extracellular vesicles, colorectal cancer, plasma membrane phospholipids, EMT, LC-MS/MS, platelet coculture
Abbreviations: AA, arachidonic acid; ACD, acid citrate dextrose; CDC, cinnamyl-3,4-dihydroxy-α-cyanocinnamate; DMPC, di-14:0-phosphatidylcholine; DMPE, di-14:0-phosphatidylethanolamine; EMT, epithelial-mesenchymal transition; EV, extracellular vesicle; OD, optical density; mEV, medium-sized EV; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PL, phospholipid; PRP, platelet-rich plasma; 12S-HETE, 12S-hydroxyeicosatetraenoic acid; 12S-HpETE, hydroperoxyeicosatetraenoic acid; 12-LOX, 12-lipoxygenase
Arachidonic acid (AA) can be oxygenated by platelet-type 12S-lipoxygenase (12-LOX) (1) to 12S-HpETE (hydroperoxyeicosatetraenoic acid), which is quickly reduced by glutathione peroxidase in the cell to form 12S-HETE (hydroxyeicosatetranoic acid) (2, 3). 12-LOX is expressed principally in platelets (1), keratinocytes (4), and tumor cells (such as prostate cancer, breast cancer, colorectal cancer, and lung cancer) (5, 6).
The role of 12S-HETE in the control of platelet responses is not entirely elucidated. 12S-HETE has both anti- and prothrombotic functions (7). It can potentiate platelet responses via NADPH oxidase activation to generate reactive oxygen species (8). Moreover, 12-HETE has been shown to contribute to dense granule secretion (9). While thrombin and collagen activate human platelets to release high levels of free 12S-HETE, significant amounts are also found esterified to membrane phospholipids (PLs), such as phosphatidylethanolamine (PE) and phosphatidylcholine (PC) (10). Esterified 12-HETE has been shown to directly enhance tissue-factor-dependent thrombin generation through promoting coagulation factor binding to membranes and is upregulated in human disease associated with venous thrombosis (10, 11, 12).
12S-HETE facilitates the invasion and metastasis of tumors by different mechanisms, such as enhancing tumor cell motility, proteinase secretion, and angiogenesis (13, 14, 15). In prostate carcinoma cells, 12-HETE binds the orphan receptor GPR31, also known as the 12S-HETE receptor, and induces a promigratory/invasive cell response (16).
It has been proposed that 12-LOX is a marker for cancer progression within the melanoma system (17). Moreover, in a small clinical study performed in patients with esophageal squamous cell carcinoma who underwent esophagectomy, the extent of tissue 12-LOX expression was an independent prognostic factor for overall survival (18).
The esterification of 12S-HETE in membrane PLs of cancer cells and their potential role in cancer development and progression have not been studied, and it is also unknown whether HETE-PLs may play a role in cancer-associated venous thrombosis. It is noteworthy that alterations in membrane PL composition have been reported in cancer (19, 20, 21, 22, 23, 24, 25). For example, in breast tumors, an increase in membrane PC and PE is detected versus adjacent normal tissue (23, 24). The PC content was found to be enriched in saturated fatty acids and correlated with high tumor grade and lower overall survival (23) and was associated with increased chemotherapy resistance (26). Furthermore, PE and PC plasmalogens, a subclass of PLs characterized by a vinyl ether at the Sn1 position and an ester bond at the Sn2 position of the glycerol backbone, have been described to be altered in gastrointestinal cancer (27). The assessment of plasmalogens in patients has been suggested to represent potential cancer biomarkers (28).
Several lines of evidence support the central role of platelets in the metastatic dissemination of cancer cells (29, 30, 31). It has been reported that platelet-derived products induce the acquisition of a mesenchymal phenotype in cancer cells (as demonstrated by the expression of mesenchymal markers, including vimentin, fibronectin, the transcription factors Twist1, Snail, and Zeb) and the downregulation of epithelial marker expression, such as E-cadherin (32, 33, 34). Platelet-induced mesenchymal tumor cells are characterized by enhanced migratory capacities translating into increased metastatic potential (32, 34).
Platelets release extracellular vesicles (EVs), a heterogeneous population of small membrane-enclosed vesicles, both in vivo and in vitro (35, 36). EVs pelleted at intermediate centrifugation speed (lower than 20,000 g) are known as “medium size EVs” (mEVs, including microvesicles/ microparticles and ectosomes) (37). Platelet-derived mEVs are nano-sized fragments (100–1,000 nm) released from platelets under various physiological and pathological conditions (36). EV containing molecular cargo (including microRNAs, miRs) can be delivered into recipient cells, thus influencing their phenotype and functions (31, 38, 39, 40). Platelet-derived mEVs can infiltrate solid tumors in humans and mice, resulting in tumor cell apoptosis via the transfer of platelet-derived RNAs with miR-24 as a major species (40). Platelet-derived mEVs can promote epithelial-mesenchymal transition (EMT) in cancer cells (39, 41) associated with the delivery of miR-939 (39).
EMT confers on cancer cells traits associated with high-grade malignancy (42). In esophageal cancer cell lines, the knockdown of the expression of 12-LOX or the use of baicalein, an inhibitor of the 12- LOX pathway (43, 44), was associated with reduced expression of EMT markers and cell migration (18).
Here, we have tested the hypothesis that the cancer cell uptake of mEVs expressing 12-LOX released from platelets induces the biosynthesis of 12-HETE and its esterification into membrane PLs. Also, we have addressed whether the pharmacological inhibition of 12-LOX restrains platelet-dependent changes of EMT marker genes in HT29 colon cancer cells (32, 33).
Materials and Methods
Coculture experiments with human colon carcinoma cell line HT29 and isolated human platelets
The human colon adenocarcinoma cell line HT29 (HTB-38; a moderately well-differentiated cell line) was obtained from the American Type Culture Collection (ATCC, Manassas, VA). Cells were cultured, as previously described (33, 34), in McCoy's 5A medium (Invitrogen, Milan, Italy) supplemented with FBS 10%, 1% penicillin/streptomycin (P/S), and 2 mM L-glutamine. In all experiments, HT29 cells (1–2 × 106) were seeded in 6-multiwell plates containing 2 ml of McCoy's 5A supplemented with 0.5% FBS and polymixin B sulfate 10 μg/ml (Sigma-Aldrich, Milan, Italy) and 2 mM L-glutamine. Human platelets were freshly isolated from 10 healthy volunteers (from 23 and 45 years of age) who donated blood on different occasions and had not taken any medications, including nonsteroidal anti-inflammatory drugs, for at least two weeks before blood collection. This study was carried out following the Declaration of Helsinki's recommendations after approval by the local Ethics Committee of “G. d'Annunzio” University of Chieti-Pescara, and informed consent was obtained from each subject. Venous blood was collected in a syringe containing Acid-Citrate-Dextrose (ACD, Sigma-Aldrich) (ACD/blood ratio was 1:5 v/v) and then transferred to polypropylene tubes containing 0.4 U/ml of apyrase VII (Sigma-Aldrich) (45). The blood was centrifuged at 220 g for 12 min without the break, and then the platelet-rich plasma (PRP) was recovered. Phosphate-buffered saline (PBS) (Sigma-Aldrich), pH 5.9 (3:2, v/v) was then added to PRP (3:2, v/v) and centrifuged (700 g, 15 min, room temperature) (33, 34); pelleted platelets were washed in a wash Buffer (PBS pH 5.9/NaCl 0.9%, 1:1 v/v) and then resuspended in the culture medium (33, 34). Two hundred microliter of platelet suspension (containing 1 or 2 × 108 cells) was added to the 6-multiwell plate without or with HT29 cells (1 or 2 × 106 cells) (HT29 cells: platelets was 1:100) (33, 34); HT29 cells and platelets were also cultured alone. After 2 or 20 h, the conditioned media of platelets and HT29 cells cultured alone or cocultured were collected, centrifuged at 970 g for 15 min at 4°C; platelet pellets were collected from the samples of platelets cultured alone; the supernatants were respun at 16,000 g for 40 min at 4°C and supernatants, and the pellets containing mEVs were collected. HT29 cells cultured alone or with platelets were washed twice with PBS and harvested by trypsin as previously reported (33, 34). Cell pellets, mEV pellets, and supernatants were immediately frozen in liquid nitrogen and stored at –80°C until use.
Effect of 12-LOX inhibitors on 12-HETE generation
Platelets (1 × 108), isolated as reported above, were resuspended in HEPES buffer (containing 5 mM HEPES, 137 mM NaCl, 2 mM KCl, 1 mM MgCl2, 12 mM NaHCO3, 0.3 mM NaH2PO4, and 5.5 mM glucose, pH 7.4, 3.5 mg/ml BSA). They were incubated with DMSO (vehicle) or cinnamyl-3,4-dihydroxy-α-cyanocinnamate (CDC) (46) (30 μM; Enzo Life science, Milan, Italy) for 15 min at room temperature before the stimulation with thrombin (1 U/ml) for 30 min. CaCl2 2 mM and MgSO4 1 mM were added to the platelet suspension 2 min before the stimulation with thrombin. The reaction was stopped by keeping the tubes at 4°C before the centrifugation at 970 g for 15 min at 4°C. CDC (3–30 μM), baicalein (5,6,7-trihydroxy-2-phenylchromen-4-one) (43, 44) (3 μM), esculetin (6,7-dihydroxychromen-2-one) (3 μM) (44, 47), and vehicle (DMSO) were incubated with platelet-HT29 cultures for 20 h (100 platelets:1 HT29 cell); supernatants were then collected and centrifugation at 970 g for 15 min at 4°C. In some experiments, HT29 cells cultured with platelets for 20 h were extensively washed with PBS, harvested by trypsin, and resuspended in culture medium (McCoy containing 0.5% FBS, 1% P/S and 2 mM L-glutamine) at the density of 1.5 × 106 cells/ml; then 200 μl of aliquots of the cell suspension was placed in 96-multiwell and preincubated with vehicle (DMSO) or CDC, 30 μM, for 15 min; then arachidonate (AA, Sigma-Aldrich) 30 μM was added and incubated at 37°C for 30 min. Supernatants were collected centrifuged at 276 g for 5 min at 4°C (to eliminate possible HT29 cells). All supernatants were respun at 16,000 g for 40 min at 4°C; then, the supernatants were collected, frozen in liquid nitrogen, and stored at –80°C until the assessment of 12-HETE by LC-MS/MS.
Isolation of mEVs from thrombin-stimulated washed platelets and characterization of 12-LOX activity
Washed platelets were stimulated with 1 U/ml thrombin for 30 min, and mEVs were pelleted by centrifugation for 40 min at 16,000 g at 4°C. mEV pellets were analyzed for the expression of 12-LOX by Western Blot. In some experiments, 250,000 mEVs were resuspended in 250 μl of HEPES Buffer and incubated with arachidonate (30 μM for 30 min at 37°C): the reaction was stopped immediately, keeping the samples at 4°C and by centrifuging them for 40 min at 16,000 g at 4°C. The supernatant was then collected, frozen in liquid nitrogen, and stored at –80°C until the assessment of 12-HETE by LC-MS/MS.
Isolation of platelets and mEVs from plasma of individuals with colorectal adenomas/adenocarcinomas and assessment of 12-LOX levels
Whole blood samples were collected from eight patients with colorectal adenomas or adenocarcinomas enrolled in a clinical study performed at Hospital Clinico Universitario Lozano Blesa (Zaragoza, Spain). The demographic and clinical features of the individuals studied are reported in supplemental Table S1. The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Clinical Investigation Ethics Committee of the Aragón Health Research Institute (Zaragoza, Spain) (EUDRACT number: 2013-004269-15; ClinicalTrials.gov Identifier: NCT02125409). All subjects provided written informed consent. Platelets were isolated from PRP as described above. mEVs were isolated from plasma as previously described (48). Briefly, PRP samples were centrifuged at 600 g for 5 min at room temperature (RT); then, the supernatant was centrifuged twice at 1,300 g for 5 min at RT (to remove contaminating platelets). Supernatants containing platelet mEVs were centrifuged at 18,000 g for 90 min at 18°C. Pellets containing platelet mEVs were resuspended in Tyrode's buffer (pH 7.4) with 5 mM calcium and quantified by flow cytometry with mAb against CD41 (CD41a-PerCP-Cy5.5, BD Biosciences), and the presence of whole platelets in the suspension was ruled out. Platelets and mEVs were analyzed by FacsVerse cytometer (BD Biosciences), and data were examined using FACSuite v 1.0.5 (BD Biosciences) software, as previously described (41). Pellets of platelets and mEVs were resuspended in phosphate-buffered saline (PBS, pH 7.4) and submitted to mechanical homogenization, repeated freeze–thaw cycles, and centrifugation for 10 min at 1,560 g; the supernatant was analyzed for 12-LOX by an Enzyme-Linked Immunosorbent Assay (ELISA) kit (MBS1605919 MyBioSource, San Diego, CA). Values were normalized by the corresponding protein concentration assessed using the Bradford assay (Bio-Rad, Milan, Italy).
Assessment of the expression of EMT markers, ALOX12, GRP31 in HT29 cells by real-time PCR
HT29 cells (1 × 106) were cultured alone or with platelets (1 × 108 cells) for 20 h. In some experiments, platelet-cancer cell cocultures were incubated with CDC (3–30 μM), baicalein (3 μM), or esculetin (3 μM). Moreover, 12S-HETE (10, 30, and 100 nM) (Cayman Chemical, Ann Arbor, MI) or vehicle (DMSO) was incubated with HT29 cells cultured alone for 4 and 20 h. After extensive washing, total RNA was extracted using the PureLink RNA Mini Kit (Applied Biosystems) according to the manufacturer's protocols. Two micrograms of total RNA was treated with DNAse kit (Fermentas, St. Leon-Rot, Germany) and subsequently reverse-transcribed into cDNA using Iscript cDNA Synthesis Kit (Bio-Rad) according to the manufacturer's protocols. One hundred nanograms of cDNA was used for the reaction mixture. The amplification of CDH1 (protein name: E-Cadherin), ZEB1 (protein name: Zeb1), FN1 (protein name: Fibronectin), VIM (protein name: Vimentin), ALOX12 (protein name: platelet 12-LOX), RHOA (protein name: RhoA), TWIST1 (protein name: Twist-related protein 1), GPR31 (protein name: GPR31), and GAPDH was performed using TaqMan gene expression assays (Hs01023894, Hs00232783, Hs00365052, Hs00185584, Hs00167524, Hs00357608, Hs01675818, Hs00271094, and Hs99999905, respectively) (Applied Biosystems, Foster, City, CA) according to the manufacturer's instructions using a 7900HT Real-Time PCR system (Applied Biosystems). Gene expression assays were performed by relative quantification with comparative cycle threshold (Ct) using ABI Prism, SDS 2.4 software (Applied Biosystems).
Assessment of protein levels of 12-LOX and GPR31 in HT29 cells by Western Blot
HT29 cell, platelet, and mEV pellets were lysed in lysis buffer containing 1% Triton X-100-PBS, 1 mM of phenylmethylsulfonyl fluoride (PMSF) (Sigma-Aldrich) and protease inhibitors (Thermo Scientific). Then cell lysates were put on ice for 30 min, and cell debris was removed by centrifugation (10,000 g, 5 min at 4°C). The Bradford assay (Bio-Rad, Milan, Italy) was used to assess protein concentration. Lysate samples were loaded onto 12% Sodium Dodecyl Sulfate–PolyAcrylamide Gel Electrophoresis (SDS-PAGE) and then transferred to polyvinylidene difluoride PVDF membranes (Bio-Rad). Finally, they were blocked with 5% nonfat milk in Tris-buffered saline-0.1% Tween-20 (TBS-Tween-20). In different experimental conditions, protein expression was detected using specific primary antibodies incubated overnight at 4°C: platelet 12-LOX (12-LOX, Abcam, dilution 1:1,000 in T-TBS-5% Milk), GPR31 (Sigma-Aldrich, dilution 1:1,000 in T-TBS-5% Milk), and GAPDH (Santa Cruz Biotechnology, dilution 1:1,000 in TBS-Tween 20) or β-actin (Santa Cruz Biotechnology, dilution 1:1,000 in TBS-Tween 20) were used as a loading control. Then, the membranes were washed in TBS-Tween 20 and incubated with the secondary antibodies. Quantification of optical density (OD) of different specific bands was calculated using Alliance 1 D software (UVITEC, Cambridge, UK) and normalized to the OD of GAPDH or β-actin.
Measurement of HETEs and 12-HETE esterified to membrane phospholipids
Supernatants of conditioned medium (1 ml), pellets of HT29 cells, platelets, and mEVs were resuspended in 1 ml of PBS and firstly subjected to lipid extraction as previously reported (10, 47, 49). Lipid and oxylipin standards, i.e., di-14:0-phosphatidylethanolamine (DMPE) and/or di-14:0-phosphatidylcholine (DMPC), were purchased from Avanti Polar Lipids (Alabaster, AL) and deuterated (d)HETEs were from Cayman Chemical. Five nanograms of 12-HETE-d8, 15-HETE-d8, 5-HETE-d8, DMPE, DMPC were added to samples before extraction as internal standards. In the samples of HT29 cells incubated with 12-HETE-d8, 5 ng of 5-HETE-d8 or DMPE and DMPC were added as internal standard. A volume of 2.5 ml of solvent mixture (1 M acetic acid/isopropanol/hexane [2:20:30, v/v/v]) was added to 1 ml volume of sample. Samples were vortexed for 30 s, and then 2.5 ml of hexane was added. Following vortex and centrifugation, lipids were recovered in the hexane layer. Samples were re-extracted by the addition of an equal volume of hexane, followed by vortex and centrifugation. The combined hexane layers were dried under N2 flow (10). The extracted lipids were resuspended in 200 μl of methanol and stored at –80°C until analysis. Lipid extracts were analyzed by liquid chromatography/mass spectrometry (LC-MS/MS) as previously reported (10) and the levels of 12-HETE ([M-H]- 319.2–179.1), 15-HETE ([M-H]- 319.2–219.1), 5-HETE ([M-H]- 319.2–115.1), PE 16:0p_12-HETE ([M-H]- 738.6–179.1), PE 18:1p_12-HETE ([M-H] - 764.6–179.1), PE 18:0p_12-HETE ([M-H] - 766.6–179.1), PE (18:0a) and PC (16:0a)_12-HETE ([M-H]- 782.6–179.1), PC 18:0a_12-HETE ([M-H] - 810.7–179.1) were quantified using targeted MRM runs. A typical LC-MS/MS chromatogram for analysis of these lipids is shown in supplemental Fig. S1. For PL analysis, lipid extracts were separated by reverse-phase HPLC using a Luna 3 μm C18 150 × 2-mm column (Phenomenex, Torrance, CA) with a gradient of 50%–100% B over 10 min followed by 30 min at 100% B (A, methanol:acetonitrile: water, 1 mM ammonium acetate, 60:20:20; B, methanol, 1 mM ammonium acetate) with a flow rate of 200 μl per min. Lipids were detected using multiple reaction monitoring (MRM) on a Sciex 6500 Q-Trap. Source settings were: CUR: 35, IS: -4500, TEM: 500, GS1: 40, GS2:30, CAD: -3, DP-50 V, CE-42 V, CXP: -11, EP: -10. For eicosanoid analysis, lipid extracts were separated on a C18 Spherisorb ODS2, 5 μm, 150 × 4.6-mm column (Waters, Hertfordshire, UK) using a gradient of 50%–90% B over 10 min (A, water:acetonitrile:acetic acid, 75:25:0.1; B, methanol:acetonitrile:acetic acid, 60:40:0.1) with a flow rate of 1 ml per min. Products were quantified using MRM runs using LC-MS/MS electrospray ionization on an Applied Biosystems 4000 Q-Trap. Source settings were: CUR:35, CAD: -3, IS: -4500, TEM: 650, GS1:60, GS2:30, EP: -10. Declustering potential ranged from -45 to -90, and collision energy ranged from -20 to -26 eV. Inclusion criteria for peak integration were those with an S/N ratio of at least 5/1 and at least seven points across the peak.
In some experiments of HT-29-platelet cocultures, we assessed the levels of 12R-HETE and 12S-HETE in the supernatant. To this aim, the conditioned media were extracted as reported above, and the methanol resuspended lipid extracts were analyzed for 12R- and 12S-HETE, by a modified LC-MS/MS method previously described (47, 49). Briefly, the LC-MS/MS system consists of a Waters Alliance 2795 HPLC coupled to a triple quadrupole mass spectrometer (Micromass Quattro PT, Waters), equipped with an electrospray ionization source (ESI Z-Spray), operating under negative ionization conditions. The ESI Z-Spray source's operating conditions were optimized by direct injection of the analytes into the mass spectrometer. The analysis was conducted by monitoring the precursor ion to product ion ([M-H]− 319.3–179.0 for 12-HETE and [M-H]− 327.3–184.0 for its deuterated form, used as internal standard). The chiral separation of 12S- and 12R-HETE was performed using a chiral chromatographic column (Lux® 3-m Amylose-1, 150 mm × 3.0 mm) eluting a 30-min gradient of 50%–100% solvent B (60% methanol, 40% ACN, 0.1% glacial acetic acid) and solvent A (75% water, 25% ACN, 0.1% glacial acetic acid) for 30 min (50% solvent B for 5 min; 50%–60% solvent B for 10 min; 60%–100% solvent B for 2 min; 100% solvent B from 17 to 20 min and 50% of solvent B from 21 to 30 min) with a flow rate of 0.2 ml/min. The collision energy used was 14 eV. 12R- and 12S-HETE eluted with retention times of 20.18 and 22.00 min, respectively.
Effect of extracellular 12-HETE on phospholipid-esterified-12-HETE generation in HT29 cells
In some experiments, HT29 cells (1 × 106) cultured alone or with platelets (1 × 108) were incubated with 12S-HETE-d8 (Cayman Chemical; 12S-HETE-d8 contains eight deuterium atoms at the 5, 6, 8, 9, 11, 12, 14, and 15 positions) (50 ng/ml, corresponding to total 100 ng) up to 20 h. HT29 cells and the conditioned media were collected, as described above, frozen in liquid nitrogen, and stored at –80°C until the assessment of free 12-HETE-d8 and 12-HETE-d8-PLs.
mEV characterization by flow cytometry
mEV pellets isolated from the conditioned medium of HT29 cells, platelets, and platelet-HT29 cell cocultures, as reported above, were resuspended in 100 μl of Annexin buffer (BD Biosciences, Milan, Italy) and labeled with MitoStatus-APC (Thermo Fisher)/Phalloidin (Sigma-Aldrich)/ anti-CD41a-PerCP-Cy5.5(BD Biosciences) (a platelet marker: platelet GPIIb; integrin αIIb)/AnnexV-V500(BD Biosciences, Annexin V binds in a calcium-dependent manner to phosphatidylserine, PS)/anti-CD66PE (BD Biosciences) (i.e., carcinoembryonic antigen, CEA, the most commonly used tumor marker in a variety of cancers including colorectal cancer) (50–52) as reported in the manufacturer's instructions and counted by using flow cytometry. In some experiments, before the coculture, platelets were loaded with 5 μg /ml of 5-(and-6)-carboxyfluorescein diacetate, succinimidyl ester (CFSE, Sigma-Aldrich) for 15 min at 37°C, then, they were washed and added to cancer cells as previously described (53). The number of mEVs positive to CFSE was also assessed by flow cytometry. Circulating mEVs were labeled with MitoStatus-APC/Phalloidin/CD41a-PerCP-Cy5.5 and counted by flow cytometry. Phalloidin negative events (of total MPs or MitoStatus positive MPs) were analyzed for CD41 expression.
mEVs were gated based on their size (0.1–0.9 μm), and the scatter properties were analyzed by running Megamix Plus beads (Biocytex, Marseille, France, CAT 7803) at the same photomultiplier (PMT) voltages used for mEVs detection (36, 41). We also assessed mitochondria-containing mEVs (48, 54), which were positive for the platelet marker CD41. mEVs were acquired by FacsVerse cytometer (BD Biosciences), and data were analyzed using FACSuite v 1.0.5 (BD Biosciences) software.
Assessment of platelet-derived mEV internalization by HT29 cells
HT29 cells were cultured for different time points with CFSE-labeled platelets. At the end of the cocultures, HT29 cells were washed with PBS, harvested by trypsin, and centrifuged at 250 g for 5 min. After the resuspension of HT29 pellet in 100 μl of PBS (Sigma-Aldrich), cells were stained whit 7-AAD (7-amino-actinomycin D) (Thermo Fisher Scientific), as reported in the manufacturer's instructions, for the exclusion of nonviable cells in flow cytometric analysis. HT29 cells were analyzed for their positivity to CFSE signal: they represented the HT29 cells that internalized platelet-derived mEVs CFSE+. In some experiments, dm-amiloride was used for its capacity to affect micropinocytosis (55, 56). Dm-amiloride (250 μM) or DMSO was added to HT29 cells 20 min before the addition of CFSE-loaded platelets, and the cells were cocultured for 20 h. Fluorescence of HT29 cells CFSE+ was analyzed by flow cytometry after the addition of trypan blue (2 mg/ml, Sigma-Aldrich), used to quench extracellular CFSE signal (i.e., mEVs not internalized) as previously described (57).
Cell fluorescence was acquired using FacsVerse cytometer (BD Biosciences), and data were analyzed using FACSuite v 1.0.5 (BD Biosciences) software.
The uptake of platelet-derived CFSE fluorescence was assessed in HT29 cells cultured alone or cocultured with CFSE-loaded platelets for 20 h by using the ImageStream IS100 (AMNIS, Seattle, WA) imaging flow cytometer, using a 488 nm solid-state laser (70 mW), and equipped with Inspire software (v 4.1.434.0). The CFSE signal in the intracellular compartment was analyzed and quantified using FlowJoTM software v 8.8.6 (TreeStar, Ashland, OR) and IDEAS software v 5.0 (AMNIS).
Statistical analysis
All values were reported as mean ± SD (standard deviation) in the text, while shown as mean ± SEM (standard error of the mean) in the figures (the n values were reported in figure legends). 12-HETE and 12-HETE-PLs were assessed in all compartments of the cell cultures (i.e., conditioned medium [2 ml] and cellular lysates [1 ml]) and reported as the total amount (ng) detected in each compartment or the sum of all compartments. Statistical analysis was performed using GraphPad Prism Software (version 9.0 for Windows, GraphPad, San Diego, CA). The statistical tests used to calculate P-values are reported in the Figure legends. P < 0.05 values were considered statistically significant.
Results
12-HETE was generated by HT29 cells following the transfer of 12-LOX from platelets
In cancer cells cultured alone, 12-HETE in the conditioned medium was undetectable. In HT29 cells cultured with platelets for 20 h, free 12-HETE was detected in the medium (Fig. 1A). 12-HETE levels were not significantly different from those found in the supernatant of platelets cultured alone (Fig. 1A). Thus, released 12-HETE detected in platelet-cancer cell coculture was mainly of platelet origin.
Fig. 1.

Generation of free 12-HETE in the coculture between platelets and HT29 cells. HT29 cells (2 × 106) (HT) and platelets (2 × 108 cells) (PLT) were cultured alone or cocultured (HT + PLT) for 20 h. Free 12-HETE was assessed in the conditioned medium (A) or in cell pellets of HT29 cells cultured alone or with platelets (B) by LC-MS/MS technique; results are reported as the total amount (ng), all data are shown as scatter dot plots with mean + SEM, n = 6–7. C: Washed platelets (1 × 108) were stimulated with 1 U/ml Thrombin for 30 min in the presence of vehicle (DMSO) or CDC (30 μM); 12-HETE was assessed in the conditioned medium by LC-MS/MS technique; data are reported as the total amount (ng); all data are shown as scatter dot plots with mean + SEM, n = 6. D: HT29 cells (2 × 106) and platelets (2 × 108 cells) were cocultured up to 20 h in the presence of vehicle (DMSO) or CDC (3–30 μM); free 12-HETE was assessed in the conditioned medium by LC-MS/MS technique; results are reported as % of control (vehicle), all data are shown as scatter dot plots with mean + SEM, n = 3. E: HT29 cells pre-exposed to platelets for 20 h were washed and harvested by trypsin as described in Materials and Methods and resuspended in culture medium at the density of 1.5 × 106 cells/ml and then 200 μl aliquots of this cell suspension were incubated with vehicle (DMSO) or CDC (30 μM) for 15 min; then, AA 30 μM was added and incubated at 37°C for 30 min; the cells were pulled down by centrifugation and, in the supernatant 12-HETE was assessed by LC-MS/MS; results are reported as the total amount (ng), all data are shown as scatter dot plots with mean + SEM, n = 4. F: In the cell pellet of HT29 cells, not exposed or pre-exposed to platelets for 20 h, 12-LOX and the loading control β-actin were assessed by Western blot; a representative blot of 4 independent experiments is reported; densitometric analysis was also reported (all data are shown as scatter dot plots with mean + SEM, n = 4). A: ∗∗P < 0.01 versus HT alone (one-way ANOVA and Dunnett's multiple comparisons test); (B) ∗∗P < 0.01 versus HT alone, (C and E) ∗∗P < 0.01 versus vehicle (unpaired t test, two-tailed); (D) ∗∗P < 0.01 and §P < 0.01 versus all other conditions (using one-way ANOVA and Tukey's multiple comparisons test); (F) ∗∗P < 0.01 versus HT29 not exposed to platelets (one-way ANOVA and Dunnett's multiple comparisons test).
12-HETE was also assessed in the intracellular compartment of HT29 cells cultured alone or with platelets. As shown in Fig. 1B, the intracellular levels of 12-HETE in HT29 cells cultured alone were undetectable, while 12-HETE was found in cancer cells after the coincubation with platelets.
We also assessed 15-HETE levels in the conditioned medium of platelet-HT29 cell cocultures and cancer cell intracellular compartment. We found that 15-HETE was undetectable (not shown), suggesting that 12-LOX was the primary enzymatic source of 12-HETE released in the medium (mainly of platelet origin) or produced intracellularly by HT29 cells pre-exposed to platelets.
We assessed R- and S- enantiomers of 12-HETE using chiral LC-MS/MS in some experiments. In the conditioned medium of platelets cultured alone or with HT-29 cells, we detected only 12S-HETE (supplemental Fig. S2).
CDC (30 μM), an inhibitor of 12-LOX (46), significantly reduced the levels of 12-HETE detected in the medium of platelets stimulated with thrombin (Fig. 1C). In cocultures of platelets-HT-29 cells, CDC caused a concentration-dependent inhibition of 12-HETE biosynthesis (Fig. 1D). CDC (30 μM) also significantly reduced 12-HETE generated by HT29 cells exposed to platelets and, after extensive washing, incubated with AA (30 μM) for 30 min (Fig. 1E).
12-LOX protein was undetectable in HT29 cells cultured alone, while it was highly expressed in platelets (Fig. 1F). After the culture of HT29 cells with platelets for 20 h, the 12-LOX protein was detected by Western blot analysis (Fig. 1F). In contrast, 12-LOX mRNA levels were undetectable by qPCR (not shown). This excludes the possibility that platelets induce ALOX12 in HT-29 cells, instead indicating that 12-LOX was delivered to cancer cells by platelets.
12-LOX inhibitors mitigated the induction of EMT marker genes in HT29 cells induced by platelets
Incubation of platelets with HT29 cells enhanced the expression of the EMT marker genes (58, 59) ZEB1 (a transcription factor promoting EMT and loss of cell polarity), VIM (involved in changes of shape, adhesion, and motility), TWIST1 (another transcription factor promoting EMT and metastasis), FN1 (an extracellular matrix protein involved in migration and invasion) and RHOA (a GTPases implicated in migration and proliferation) (Fig. 2A). In contrast, E-cadherin mRNA levels were downregulated (E-cadherin loss is associated with EMT) (Fig. 2A). Treatment of tumor cells with CDC interfered with the platelet-dependent gene induction (Fig. 2A, B); differently, CDC did not significantly affect E-cadherin reduction (Fig. 2A, B). CDC 30 μM (causing a maximal inhibition of 12-HETE generation, as shown in Fig. 1D) significantly reduced the enhanced expression of all genes analyzed (Fig. 2A, B). At lower concentrations, the response was heterogeneous. VIM expression was very susceptible, while TWIST1 was less sensitive to CDC treatment (Fig. 2A, B). However, the CDC's 50% reduction of 12-LOX activity was sufficient to significantly mitigate gene induction except for TWIST1 (Fig. 2A, B). A significant linear relationship between % reduction of 12-HETE and gene expression of ZEB1, VIM, and RHOA by CDC was found (supplemental Table S2). We tested the effect of additional inhibitors (chemically distinct) to support the contribution of 12-LOX to EMT marker gene expression induced by platelet-cancer cell interaction. As shown in Fig. 2C, esculetin and baicalein (3 μM) significantly reduced 12-HETE generation by approximately 50%. This effect was associated with the prevention of gene induction (Fig. 2D); similarly to CDC, the two compounds did not influence the downregulation of E-cadherin expression caused by platelets (Fig. 2D).
Fig. 2.

Effects of 12-LOX inhibition on the expression of mesenchymal genes in HT29 cells cocultured with platelets. HT29 cells (HT, 1 × 106) were cultured alone or cocultured with platelets (PLT) (1 × 108) for 20 h. Twenty min before the addition of platelets, vehicle (DMSO), CDC (3–30 μM), baicalein (3 μM), and esculetin (3 μM) were added to cancer cells. The gene expression of CDH1, VIM, TWIST1, ZEB1, FIN1, and RHOA was assessed by qRT-PCR and normalized to GAPDH mRNA levels; values are reported as fold-change versus HT29 cells cultured alone, all data are shown as scatter dot plots with mean + SEM (n = 4–7). A: Concentration-dependent effects of CDC on gene expression of CDH1, VIM, TWIST1, ZEB1, FIN1, and RHOA in HT29 cells cultured with platelets: ∗∗P < 0.01 versus HT29 cells alone (multiple unpaired t test), ∗P < 0.05, #P < 0.01 versus HT + PLT (vehicle) (mixed-effects analysis followed by Tukey test for multiple comparisons). B: Heat map of % Control (vehicle) (mean) values of 12-HETE biosynthesis and gene expression versus increasing CDC concentrations. C: Reduction of 12-HETE biosynthesis in HT29 cell-platelet cocultures by baicalein and esculetin (3 μM); ∗∗P < 0.01 versus vehicle (n = 3–4) using one-way ANOVA and Dunnett's multiple comparisons test. D: Effects of baicalein or esculetin (3 μM) on gene expression of CDH1, VIM, TWIST1, ZEB1, FIN1, and RHOA in HT29 cells cultured with platelets; ∗∗P < 0.01 versus HT29 cells alone (multiple unpaired t test), #P < 0.01, ∗P < 0.05 versus vehicle (n = 4–7) (mixed-effects analysis followed by Tukey test for multiple comparisons).
Effects of exogenous 12S-HETE on EMT marker genes' expression in HT29 cells
We assessed the effects of increasing concentrations of exogenous 12S-HETE on the EMT marker genes' expression in HT29 cells at 4 and 20 h of incubation. As shown in Fig. 3A–F, 12S-HETE 10 nM (the concentration detected in the conditioned medium of platelet-cancer cell cocultures [Fig. 1A]) did not cause any significant change of EMT gene expression. In contrast, at 20 h of incubation, 100 nM of 12-HETE caused a significant reduction of CDH1 (Fig. 3A) while RHOA (Fig. 3D), FN1 (Fig. 3E), and ZEB1 (Fig. 3F) were significantly induced; the expression of VIM and TWIST1 was not affected by 12-HETE (Fig. 3B, C, respectively).
Fig. 3.

Effects of exogenous 12 S-HETE on the expression of mesenchymal genes and GPR31 in HT29 cells. HT29 cells (HT, 1 × 106) were incubated with exogenous 12S-HETE (10, 30, and 100 nM) for 4 and 20 h (A–G) The gene expression of CDH1, VIM, TWIST1, ZEB1, FIN1, RHOA, and GPR31 was assessed by qRT-PCR and normalized to GAPDH mRNA levels; values are reported as fold-change versus HT29 cells cultured alone in the presence of DMSO vehicle, all data are shown as scatter dot plots with mean + SEM (n = 6, 7); ∗P < 0.05, ∗∗P < 0.01 versus vehicle (two-way ANOVA followed by Tukey's multiple comparisons test).
In order to verify the contribution of GPR31 (considered the receptor of 12S-HETE (16)) on the modulation of cancer cell EMT gene expression by 12S-HETE, we assessed GPR31 expression at baseline (vehicle, DMSO) and after stimulation with the eicosanoid. At baseline, low gene expression levels of the receptor were found (not shown); however, a protein band with GPR31-like immunoreactivity was detected by Western blot (supplemental Fig. S3). The incubation of HT29 cells with 10 nM of 12S-HETE did not affect the expression of the receptor. However, at higher concentrations (30 and 100 nM), 12S-HETE transiently enhanced GRP31 gene expression (Fig. 3G).
Altogether these results exclude the involvement of extracellular 12S-HETE generated in the coculture of platelets and HT29 cells (approximately 10 nM) in the modulation of EMT marker genes' expression, possibly due to the low expression of GPR31. We hypothesized that cancer cell 12-LOX delivered from platelets might contribute to EMT via an endogenous pathway involving the generation of 12S-HETE and its esterification on membrane phospholipids of cancer cells.
Phospholipid-esterified-12-HETE was generated in platelet-HT29 cell cocultures
We previously showed that activated platelets generate a family of six 12-HETE containing PLs, comprising four PEs and two PCs (ie PE 16:0p_12-HETE, PE 18:1p_12-HETE, PE 18:0p_12-HETE, PE and PC (18:0a)_12-HETEs, and PC 16:0a_12-HETE) (10). The PEs include several plasmalogens, and all represent oxidized forms of the most abundant PE and PC species in platelets (10). Here, we studied the esterification of 12-HETE into membrane PLs of platelets and HT29 cells, cultured alone or with platelets. In HT29 cells cultured alone, these lipids were undetectable (not shown). However, in HT29 cells cultured with platelets, all these PL-esterified-12-HETEs were found with PE18:0p_12-HETE and PE16:0p_12-HETE being the major products (Fig. 4A). As expected, we found that platelets alone generated the six PL-esterified-12-HETEs (Fig. 4A) and that these were not influenced by culture with HT29 cells (supplemental Fig. S4A). However, the plasmalogen HETE-PEs (PE18:0p_12-HETE and PE 16:0p _12-HETE) were significantly higher in HT29 cells cultured with platelets than in platelets alone (Fig. 4A).
Fig. 4.

12-HETE-esterified in PLs of HT29 cells pre-exposed to platelets and in platelets. A: In the cell pellet of HT29 cells pre-exposed to platelets (HT29 from HT + PLT) and of platelets cultured alone (PLT), the levels of PE18:0a_12-HETE, PE18:0p_12-HETE, PE16:0p_12-HETE, PE18:1p_12-HETE, PC16:0a_12-HETE, and PC18:0a_12-HETE were assessed by LC-MS/MS; data are reported as the total amount (ng), all data are shown as scatter dot plots with mean + SEM, n = 6, 7; ∗∗P < 0.01 versus the same PL detected in HT29 cells; §P < 0.01 versus PE18:0p_12-HETE and PE16:0p_12-HETE of HT29 cells (using one-way ANOVA and Tukey's multiple comparisons test). B: The percentage of 12-HETE esterification to total 12-HETE (i.e., free and esterified forms) was calculated in HT29 cells pre-exposed to platelets (HT) and in platelets alone (PLT); all data are shown as scatter dot plots with mean + SEM, n = 6, 7; ∗∗P < 0.01 versus PLT (unpaired t test, two-tailed).
As shown in Fig. 4B, in cancer cells exposed to platelets, the percentage of 12-HETE esterified into membrane phospholipids versus the total generation of 12-HETE (extracellular and intracellular free and esterified) was significantly higher than that found in platelets.
Altogether, these results suggest that HT29 cells exposed to platelets acquire the capacity to generate 12-HETE associated with the detection of 12-LOX protein and that the eicosanoid is directly esterified into membrane phospholipids.
Extracellular 12-HETE did not contribute to phospholipid-esterified-12-HETE generation in HT29 cells
To test whether extracellular 12-HETE produced by platelets is used for 12-HETE-PL generation by HT29 cells, 12-HETE-d8 (50 ng/ml, corresponding to the total concentration of 12-HETE generated in the coculture of platelets and cancer cells as free and esterified forms) was added to cancer cells for up to 20 h; then, the levels of 12-HETE-d8 were measured by LC-MS/MS in the medium and intracellularly, both as the free acyl form or incorporated into membrane phospholipids, based on the expected mass shift due to the presence of 8 deuterium atoms. As shown in Fig. 5A, 12-HETE-d8 disappeared from the medium in a time-dependent manner. In the intracellular compartment of HT29 cells, free 12-HETE-d8 was almost undetectable throughout the time course (Fig. 5A). Furthermore, exogenously added 12-HETE-d8 was only marginally esterified into phospholipids of HT29 cells, as deuterated HETE-PLs (Fig. 5B; all HETE-PE/PC were integrated and summed together).
Fig. 5.
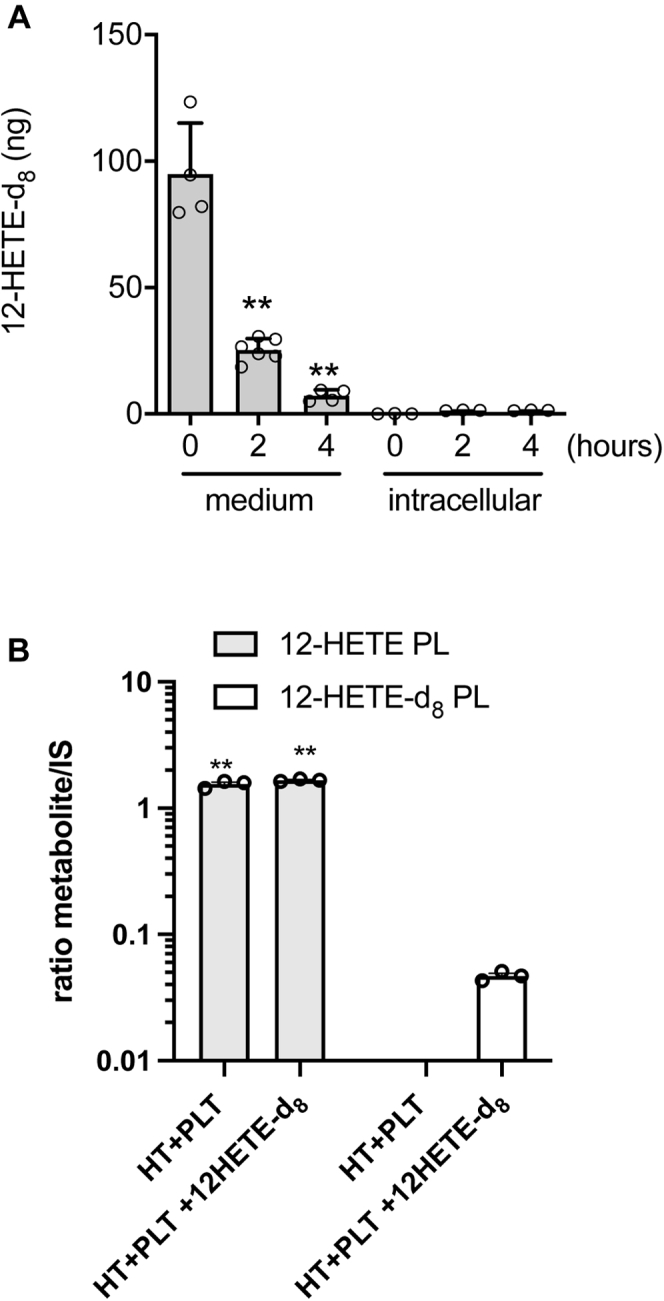
Effect of exogenous-free 12-HETE on the generation of esterified 12-HETE in PLs of HT29 cells. A: HT29 cells (1 × 106 cells) were incubated with 12-HETE-d8 (50 ng/ml, corresponding to a total 100 ng) up to 4 h, then, the levels of 12-HETE-d8 were assessed in the conditioned medium and intracellularly (in the HT29 cell pellets) by LC-MS/MS; results are reported as the total amount (ng), all data are shown as scatter dot plots with mean + SEM, n = 3–6;∗∗P < 0.01 versus time 0 (using one-way ANOVA and Tukey's multiple comparisons test). B: HT29 cells (1 × 106) were cocultured with platelets (1 × 108) for 20 h in the absence or presence of 12-HETE-d8 (50 ng/ml, corresponding to total 100 ng), and the levels of all forms of esterified 12-HETE (12-HETE PLs and 12-HETE-d8 PLs) were assessed in the cell pellet of HT29 cells by LC-MS/MS; results are reported as the sum of all species of esterified 12-HETE (as the ratio of the peak areas of the metabolite and its internal standard, IS), all data are shown as scatter dot plots with mean + SEM, n = 3, ∗∗P < 0.01 versus 12-HETE-d8 PLs (using two-way ANOVA and Tukey's multiple comparisons test).
Altogether these data suggest that the esterification of exogenous 12-HETE in phospholipids is a negligible way to generate 12-HETE-PLs in cancer cells, possibly for their capacity of colon cancer cells to rapidly metabolize HETE via β-oxidation (60). An endogenous pathway of 12S-HETE biosynthesis and its esterification in PLs involving 12-LOX delivered to cancer cells by platelet-derived mEVs is proposed.
mEVs were released from platelets in the coculture with cancer cells
As shown in Fig. 6A, during the coculture of platelets and HT29 cells, mEVs were generated, and 83.7 ± 6.5% of them expressed on their surface the platelet marker CD41 (the integrin αIIb, also known as GPIIb) (50) while only a marginal % of mEVs expressed CD66 (i.e., the carcinoembryonic antigen, CEA) (52), thus indicating their primary platelet origin.
Fig. 6.

Characterization of mEVs generated during the interaction between platelets and cancer cells. mEVs were isolated from the conditioned medium of HT29 cells cultured alone (HT) or with platelets (HT + PLT) and assessed by flow cytometry for the positivity to CD41, CD66, and Annexin V. A: The percentage of EVs positive to CD41 and CD66 is reported; all data are shown as scatter dot plots with mean + SEM, n = 3; ∗∗P < 0.01 versus CD41+ mEVs. B: The count of CD41+ mEVs in the medium of coculture at 2 and 20 h was assessed; results are reported as CD41+ mEV/μl, all data are shown as scatter dot plots with mean + SEM, n = 5–9; ∗∗P < 0.01 versus HT alone. C: The count of CD41+ AnnexinV+ MPs in the medium of coculture at 2 and 20 h was assessed; results are reported as CD41+ AnnexinV+ mEVs/μl, all data are shown as scatter dot plots with mean + SEM, n = 6–13; ∗∗P < 0.01 versus HT alone. D and E: Western Blot analysis of 12-LOX and β-actin in mEVs isolated from the conditioned medium of HT + PLT (D) or from the releasate of thrombin-stimulated platelets(E); platelets (PLT) were loaded into the gels as a positive control for the 12-LOX. F: mEVs isolated from the releasate of thrombin-stimulated platelets (250,000 mEVs in 250 μl of HEPES buffer) were incubated with and without AA (30 μM) for 30 min 37°C, and the levels of 12-HETE were assessed in the medium by LC-MS/MS; results are reported as the total amount (ng), all data are shown as scatter dot plots with mean + SEM, n = 7–9; ∗∗P < 0.01 versus mEVs. A and F: unpaired t test (two-tailed) was used; (B and C) Two-way ANOVA with Tukey's multiple comparisons test was used.
A comparable number of mEVs was detected after 2 and 20 h of coculture (Fig. 6B), suggesting a rapid mEV formation. As shown in supplemental Fig. S5, both at 2 and 20 h of cocultures, approximately 80% of mEVs showed a size range of 0.1–0.24 mm. The rest of mEVs presented a size range of 0.24–0.5 mm.
By assessing Annexin V binding (Fig. 6C), indicating the exposure of PS on the outer mEV membrane surface (51), it was found that at 2 and 20 h of coculture, mEVs AnnexinV+ were 75.9 ± 10.6 and 85.2 ± 5.2%, respectively, of total CD41+ mEVs (Fig. 6B).
Platelet mEVs expressed an active catalytic form of 12-LOX
mEVs isolated from the conditioned medium of platelet-HT29 cocultures (20 h) expressed 12-LOX (Fig. 6D). Also, mEVs released from thrombin-activated platelets expressed 12-LOX (Fig. 6E), and 12-HETE was released into the medium when mEVs were incubated with AA (30 μM) (Fig. 6F). These data show that 12-LOX expressed in platelet-derived mEVs is catalytically active.
Circulating mEVs from patients with adenomas/adenocarcinomas expressed 12-LOX
We then verified whether 12-LOX is also expressed in vivo in circulating mEVs of eight patients with colorectal adenomas/adenocarcinomas (Supplemental Table 1). Platelet-derived mEVs (CD41+) were detected in patients' plasma (1,364±442, number/μl, mean±SD, n = 8) (not shown). Circulating mEVs expressed 12-LOX that was quantified using a commercially available sensitive and specific ELISA kit (61, 62). In Fig. 7, 12-LOX levels found in circulating mEVs and platelets of patients are shown.
Fig. 7.
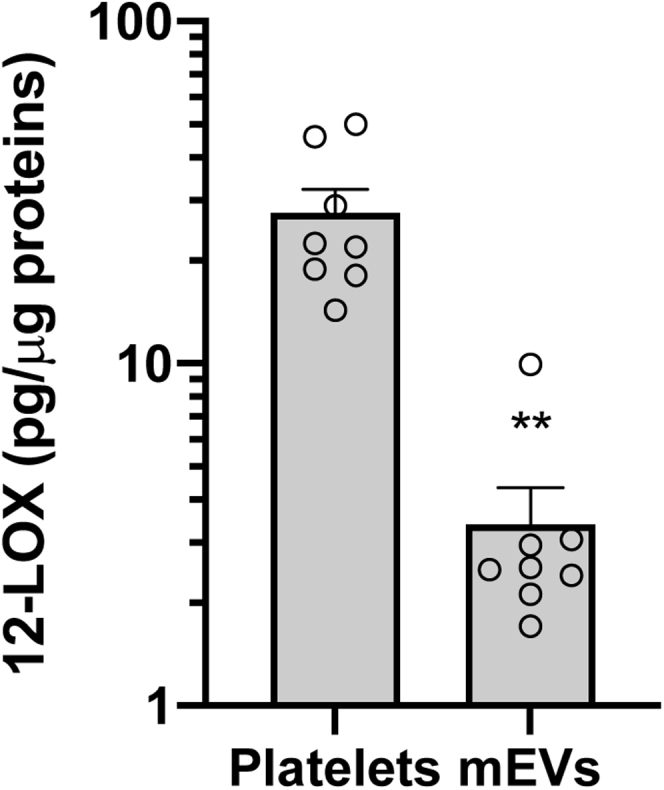
12-LOX is expressed in circulating mEVs of patients with adenomas/ adenocarcinomas. Whole blood samples were collected from eight patients with colorectal adenomas or adenocarcinomas, and mEVs were isolated from plasma as previously described (48) while washed platelets were obtained from PRP. Pellets of platelets and mEVs were homogenized and subjected to repeated freeze–thaw cycles, and after centrifugation, the supernatant was analyzed for 12-LOX by a commercially available ELISA kit. Values were normalized by the corresponding protein concentration; all data are shown as scatter dot plots with mean + SEM (n = 8), ∗∗P < 0.01 versus platelets; unpaired t test (two-tailed).
Internalization of platelet mEVs by HT29 cells
mEVs are well known to play crucial roles in cell-cell communication by transferring their contents to target cells (38, 39, 40, 41). Here, we tested whether HT29 cells internalize platelet mEVs from the medium of platelet-cancer cell cocultures. For this, HT29 cells were cultured with platelets labeled with CFSE. Upon its diffusion into the cell, intracellular esterases cleave the acetate group of CFSE, and the molecule interacts with cellular amines via its succinimidyl groups to generate a highly fluorescent green dye impermeant to the cell membrane. In the coculture of CFSE-loaded platelets and HT29 cells, CFSE positive mEVs were generated and represented 91.4 ± 5.4% of total mEVs (not shown).
As shown in Fig. 8A, HT29 cells cultured with CFSE-loaded platelets for 2 and 20 h were positive for CFSE when analyzed by flow cytometry. The internalization of platelet mEVs by cancer cells was investigated using the Amnis® imaging flow cytometry system by monitoring the CFSE fluorescent intensity in HT29 cells exposed or not to CFSE-loaded platelets for 2 and 20 h. As shown in Fig. 8B, HT29 cells cultured alone were negative to CFSE fluorescence, while cancer cells cultured with CFSE-loaded platelets showed CFSE green signal intracellularly. At 2 and 20 h, 54.5 ± 10.5 and 63 ± 7.4%, respectively, of HT29 cells were positive for CFSE signal (Fig. 8C).
Fig. 8.

Platelet-derived mEVs generated during the coculture were internalized by HT29 cells. A–E: HT29 cells (1 × 106) were incubated with CFSE-loaded platelets (1 × 108) up to 20 h. At the end of the incubation, cancer cells were washed, harvested by trypsin, and resuspended in 100 μl of PBS and analyzed for the CFSE fluorescence signal by flow-cytometry (A, D) or Amnis imaging flow cytometry (B, C). A: Flow cytometry dot plots represent CFSE negative (red) and positive (purple) HT29 cells cultured alone or with CFSE-loaded platelets for 20 h, respectively. B and C: HT29 cell suspension was analyzed by Amnis imaging flow cytometry for CFSE fluorescence; the representative images of cells, which internalized CFSE + mEVs, are reported (B), and the percentage of cells with internalized CFSE + mEVs to total CFSE+ cells are shown (C), all data are shown as scatter dot plots with mean + SEM, n = 4; ∗∗P < 0.01 versus HT alone. D: HT29 cells were cocultured with CFSE-loaded platelets in the presence of vehicle (DMSO) or dm-amiloride (250 μM) for 20 h; at the end of the incubation, cancer cells were harvested and analyzed for CFSE signal by flow-cytometry after trypan blue quenching; data are reported as % of cells with internalized CFSE + mEVs versus control (HT + PLT, vehicle), all data are shown as scatter dot plots with mean + SEM, n = 4, ∗∗P < 0.01 versus vehicle. E and F: Western blot analysis of 12-LOX in HT29 cells cultured with platelets in the presence of vehicle (DMSO) or dm-amiloride (250 μM) for 20 h; the optical density of 12-LOX bands was normalized with those of β-actin, and results are reported as % of control (vehicle), all data are shown as scatter dot plots with mean + SEM, n = 3, ∗P < 0.05 versus vehicle. C: Two-way ANOVA with Tukey's multiple comparisons test and (D and F) unpaired t test (two-tailed) were used.
Dm-amiloride affected platelet mEV internalization and reduced 12-LOX levels in HT29 cells cultured with platelets
In HT29 cells cultured with platelets for 20 h, dm-amiloride, an inhibitor of the Na+/H+ exchanger reported to affect macropinocytosis (55), significantly (P < 0.05) reduced the number of CFSE-labeled HT29 cells (Fig. 8D).
The reduction of platelet mEV internalization by dm-amiloride was associated with a significant decrease of 12-LOX levels in HT-29 cells exposed to platelets for 20 h (Fig. 8E, F). These data suggest that mEVs from platelets are internalized by cancer cells, at least in part, via the macropinocytosis pathway.
Discussion
Platelets trigger various phenotypic changes in tumor cells, recapitulating a prometastatic phenotype (29, 30, 31, 32, 33, 34). It is noteworthy the induction of EMT programs promoting cancer metastasis (32, 33, 34). 12S-HETE, a major product of AA metabolism in platelets via the activity of 12-LOX (63), has been suggested to contribute to cancer development and progression through different mechanisms (13, 14, 15, 16). This eicosanoid promotes the survival of ovarian cancer cells (64). The role of 12-LOX in EMT induction was demonstrated in human gastric cancer cells with ALOX12 overexpression (65). In esophageal cancer cell lines, the downregulation of ALOX12 expression or the pharmacological inhibition of 12-LOX activity is associated with reduced EMT markers and cell migration (18).
In platelet-cancer cell cocultures, we found that the 12-LOX inhibitor CDC (46) prevented the induction of mRNA levels of mesenchymal markers (ZEB1, TWIST1, FN1, VIM, and RHOA) in HT29 cells; this effect was associated with a significant reduction of 12-HETE generation. CDC is not a selective inhibitor of 12-LOX, also impacting 5-LOX (66). However, in HT29 cell-platelet cocultures, 5-HETE was undetectable (not shown). The finding that the extent of EMT gene changes at low CDC concentrations was heterogeneous is probably explained by the occurrence of a temporal hierarchy of complex transcriptional networks regulating EMT (67). The result that chemically distinct 12-LOX inhibitors (such as esculetin and baicalein (43, 44, 47)) also prevented the induction of EMT genes further strengthened the role of 12-LOX activity in platelet-dependent induction of EMT in cancer cells.
We have previously found (33, 34) that platelets induce downregulation of E-cadherin (a key cell-to-cell adhesion molecule whose expression reduction potentiate tumor cell invasion and metastasis) (42), which was prevented by the selective inhibition of platelet prostaglandin (PG)E2 generation by aspirin (34) (an inhibitor of platelet cyclooxygenase-1) (31). In contrast, here, we show that the 12-LOX inhibitors did not affect E-cadherin's reduced expression in HT29 cells exposed to platelets. This suggests that PGE2 and 12-HETE differently influence the genetic program of HT29 cells involved in developing a prometastatic phenotype. Our findings may imply the possible enhanced efficacy against tumor metastasis by the coadministration of low-dose aspirin with 12-LOX inhibitors; this hypothesis warrants a further investigation in experimental animal models.
Free 12-HETE levels are not significantly different in colorectal polyps and cancer mucosa versus normal colorectal mucosa in humans (68). However, the enhanced 12-LOX expression has been demonstrated in colorectal tumors with inflammatory features (69), suggesting a possible contribution of the inflammatory tumor microenvironment on free 12S-HETE generation and action.
12-HETE is not only generated as the free acyl form but can also be found esterified in membrane PLs, particularly in platelets (10, 11, 12, 70). The formation of 12-HETE-PLs has been shown in platelets in response to different agonists and following platelet activation. These lipids can be detected on the surface of the cells, where they are proposed to enhance the ability of PS to sustain blood coagulation (10, 11, 12, 70).
Here, we assessed the platelet role in supporting the 12-HETE generation and its esterification into membrane PLs in the human colon adenocarcinoma cell line HT-29. HT29 cells cultured alone neither expressed 12-LOX nor generated 12-HETE (either as a free form or esterified in membrane PLs). However, HT29 cells cultured with platelets acquired both 12-LOX protein and the ability to generate 12-HETE. The cancer cells exposed to platelets did not express the 12-LOX mRNA, indicating that the protein was unlikely to originate from the cells' own genetic machinery. We found that platelet-released mEVs containing 12-LOX were incorporated into HT29 cells by performing experiments with fluorescence labeling platelets. This phenomenon was mitigated by dm-amiloride, which inhibits the Na+/H+ exchanger involved in macropinocytosis (55). Interestingly, in the presence of dm-amiloride, reduced levels of 12-LOX were detected in HT29 cells cocultured with platelets. The formation of macropinosomes is believed to arise from deformations of the plasma membrane known as ruffles (71), which are highly dynamic structures and are rich in actin and actin-associated proteins (72). Amiloride is not a direct inhibitor of macropinocytosis. However, it acts via the induction of submembranous acidification caused by metabolic H+ generation, unopposed by the regulatory extrusion across the membrane by Na+/H+ antiporter (55). Decreased cytosolic pH by amiloride may affect the activation of the small GTPases Rac1 known to stimulate actin filament accumulation at the plasma membrane, forming membrane ruffles (55). We have previously shown that RAC1 expression is enhanced by the interaction of HT29 cells with platelets (34). This pathway might promote platelet-derived mEV internalization and remodeling of cancer cell phospholipids with 12-HETE. Further studies are needed to address this hypothesis. The finding that mEV internalization was prevented, at least partly by amiloride, which affects macropinocytosis, seems to exclude the possibility that platelet uptake is involved in the transfer of 12-LOX into cancer cells. Studies involving the use of dynamic transmission electron microscope should be performed to clarify this issue. However, several lines of evidence show that platelets activated in vivo can deliver their cargo (also drugs) to recipient cells via the release of mEVs (38, 39, 40, 41, 73). It is noteworthy that Mir-939, transferred by platelet mEVs to ovarian epithelial cancer cells, induces their proliferation and migration (39). Platelet mEVs infiltrate solid tumors and deliver miR-24 to tumor cells in vivo and in vitro, inducing cell apoptosis (40).
In platelet-cancer cell cocultures, 12-HETE released in the medium was mainly derived from platelets. Approximately one-third of 12-HETE generated was esterified into membranes PLs of platelets. Similar to the results in thrombin-activated platelets (10), the lipids were formed from the six most abundant AA-containing PE and PC molecular species present (data not shown). Thus, the pattern of products is dictated by the substrates available in the plasma membrane. In platelets, 12-HETE was found esterified mainly in PC species with acyl-linked 16:0 or 18:0 at Sn1 and PE 18:0a. The majority of the PL-esterified 12S-HETE was retained by the cells, with only small amounts appearing in platelet-derived mEVs (supplemental Fig. 4B).
Free 12-HETE was detected intracellularly in cancer cells, but a higher % was esterified in membrane PLs. This is likely because cancer cells extensively metabolize 12-HETE via β-oxidation (60). This phenomenon also precludes the possibility of exogenous 12-HETE to be incorporated into membrane PLs of cancer cells. This result was also found in platelets (10). Thus, a very tight coupling between the endogenous generation of 12-HETE and its esterification into PLs exists.
The pattern of 12-HETE esterification into the six PE, PC species of HT29 cells was different from that found in platelets. The mechanistic clarification of this issue requires carrying out a specific study. However, it is known that cancer cells rewire their metabolism to produce more PE than healthy cells, and this is associated with enhanced expression of the genes for PE biosynthesis (74).
In HT29 cells (exposed to platelets), higher levels of plasmalogen forms were detected, specifically PE 18:0p_12-HETE and PE 16:0p_12-HETE. Our results provide the rationale to characterize the production of 12-HETE-PLs in colorectal cancer lesions and to verify whether the pattern of 12-HETE-PLs identifies patients with metastatic potential.
Overall, our findings show activated platelets' contribution to reprogram cancer cell arachidonate metabolism to 12S-HETE generation via the transfer of 12-LOX; endogenous 12S-HETE is rapidly esterified in membrane PLs of cancer cells. 12S-HETE released from platelets could influence cancer cell responses via the activation of GPR31 (16). However, the concentration detected in the coculture conditioned medium (approximately 10 nM) did not influence EMT gene expression due to the low levels of the receptor in HT29 cells. Exogenous 12-HETE can be uptaken by cancer cells but is rapidly metabolized, thus precluding its esterification to cancer cell PLs.
It has been reported that 12-LOX is regulated by the interaction with cellular proteins such as human type II keratin K5, nuclear envelope protein lamin A, integrin beta4 cytoplasmic domain, and human C8FW phosphoprotein (75). Further studies are requested to investigate the contribution of 12-LOX interacting proteins to platelet-induced EMT in cancer cells shown here.
It was previously reported that 12-LOX expressed in PRP is a promising diagnostic and prognostic biomarker of prostate cancer (61, 62). Here, we have shown that circulating mEVs collected from patients with colorectal adenomas/adenocarcinomas contain 12-LOX protein. It is noteworthy that an enhanced number of platelet-derived mEVs is detected in the bloodstream of patients with CRC and other cancer types (76, 77). Thus, the cross talk between mEV expressing 12-LOX with circulating cancer could enhance their prometastatic potential in vivo. This issue should be verified in an appropriate clinical study. If confirmed, our findings may open the way to novel therapeutic strategies to dampen tumor metastasis: (i) affecting 12-LOX activity by selective inhibitors, which are in development, such as ML-355 (78); (ii) interfering with the internalization of mEVs with macropinocytosis inhibitors (79).
In conclusion, we have shown that platelets induce 12-HETE generation in colon cancer cells and its esterification into membrane PLs via mEV-mediated delivery of 12-LOX associated with EMT marker gene expression changes. The functional role of remodeling of cancer cell PLs with 12-HETE needs verification in animal models of metastasis and in cancer patients. If confirmed, the pharmacological interference with phospholipid-esterified 12-HETE generation represents a novel target for anticancer agent development.
Data availability
All individual data are reported in the figures. There is no restriction on the availability of any data.
Supplemental data
This article contains supplemental data.
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Acknowledgments
We wish to thank the technical contribution of Ms Tania Vanessa Pierfelice (a Medical Biotechnology student of the University of Padua, performing an internship in the SpaTT Lab of “G. d'Annunzio” University for carrying out the Thesis). In addition, this study was conducted on Behalf of the Aspirin for Cancer Prevention Group (AsCaP), Wolfson Institute of Preventive Medicine, Queen Mary School of Medicine and Dentistry, and University of London, UK.
Author contributions
P. P., V. B. O., M. D., S. S., and A. C. conceptualization; P. B. data curation; M. D., S. S., C. H., R. F., S. T., R. G., A. L., and A. C. formal analysis; P. P., V. B. O., M. D., and A. L. funding acquisition; M. D., S. S., C. H., R. F., S. T., R. G., A. L., and A. C. investigation; P. L., M. M., V. J. T., and M. Z. methodology; P. P. and V. B. O. writing–original draft; P. P. and V. B. O. writing–review and editing.
Funding and additional information
The research leading to these results has received funding by Associazione Italiana per la Ricerca sul Cancro (AIRC) (IG 2017-ID. 20365 Project; Principal Investigator P. P.), by Ministero dell'Istruzione, dell'Università e della Ricerca (MIUR) (Fondi per la Ricerca Scientifica di Ateneo, [ex 60%]) to M. D. and by Instituto de Salud Carlos III, Spain (PI14/01218) to A. L. V. B. O. acknowledges funding from European Research Council (LipidArrays). V. B. O. holds a Royal Society Wolfson Research Merit Award.
Supplemental data
References
- 1.Funk C.D., Furci L., FitzGerald G.A. Molecular cloning, primary structure, and expression of the human platelet/erythroleukemia cell 12-lipoxygenase. Proc. Natl. Acad. Sci. U. S. A. 1990;87:5638–5642. doi: 10.1073/pnas.87.15.5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Powell W.S., Rokach J. Biosynthesis, biological effects, and receptors of hydroxyeicosatetraenoic acids (HETES) and oxoeicosatetraenoic acids (oxo-ETEs) derived from arachidonic acid. Biochim. Biophys. Acta. 2015;1851:340–355. doi: 10.1016/j.bbalip.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siegel M.I., McConnell R.T., Porter N.A., Cuatrecasas P. Arachidonate metabolism via lipoxygenase and 12Lhydroperoxy-5,8,10,14-icosatetraenoic acid peroxidase sensitive to anti-inflammatory drugs. Proc. Natl. Acad. Sci. U. S. A. 1980;77:308–312. doi: 10.1073/pnas.77.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hussain H., Shornick L.P., Shannon V.R., Wilson J.D., Funk C.D., Pentland A.P., Holtzman M.J. Epidermis contains platelet-Type 12-lipoxygenase that is overexpressed in germinal layer keratinocytes in psoriasis. Am. J. Physiol. 1994;266:C243–C253. doi: 10.1152/ajpcell.1994.266.1.C243. [DOI] [PubMed] [Google Scholar]
- 5.Haeggstrom J.Z., Funk C.D. Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chem. Rev. 2011;111:5866–5898. doi: 10.1021/cr200246d. [DOI] [PubMed] [Google Scholar]
- 6.Tang K., Honn K.V. 12(S)-HETE in cancer metastasis. Adv. Exp. Med. Biol. 1999;447:181–191. doi: 10.1007/978-1-4615-4861-4_17. [DOI] [PubMed] [Google Scholar]
- 7.Yeung J., Holinstat M. 12-lipoxygenase: a potential target for novel antiplatelet therapeutics. Cardiovasc. Hematol. Agents Med. Chem. 2011;9:154–164. doi: 10.2174/187152511797037619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nardi M., Feinmark S.J., Hu L., Li Z., Karpatkin S. Complement-independent Ab-induced peroxide lysis of platelets requires 12-lipoxygenase and a platelet NADPH oxidase pathway. J. Clin. Invest. 2004;113:973–980. doi: 10.1172/JCI20726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yeung J., Apopa P.L., Vesci J., Stolla J., Rai G., Simeonov A., Jadhav A., Fernandez-Perez P., Maloney D.J., Boutaud O., Holman T.R., Holinstat M. 12-lipoxygenase activity plays an important role in PAR4 and GPVI-mediated platelet reactivity. Thromb. Haemost. 2013;110:569–581. doi: 10.1160/TH13-01-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thomas C.P., Morgan L.T., Maskrey B.H., Murphy R.C., Kühn H., Hazen S.L., Goodall A.H., Hamali H.A., Collins P.W., O'Donnell V.B. Phospholipid-esterified eicosanoids are generated in agonist-activated human platelets and enhance tissue factor-dependent thrombin generation. J. Biol. Chem. 2010;285:6891–6903. doi: 10.1074/jbc.M109.078428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Slatter D.A., Percy C.L., Allen-Redpath K., Gajsiewicz J.M., Brooks N.J., Clayton A., Tyrrell V.J., Rosas M., Lauder S.N., Watson A., Dul M., Garcia-Diaz Y., Aldrovandi M., Heurich M., Hall J. Enzymatically oxidized phospholipids restore thrombin generation in coagulation factor deficiencies. JCI Insight. 2018;3 doi: 10.1172/jci.insight.98459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lauder S.N., Allen-Redpath K., Slatter D.A., Aldrovandi M., O'Connor A., Farewell D., Percy C.L., Molhoek J.E., Rannikko S., Tyrrell V.J., Ferla S., Milne G.L., Poole A.W., Thomas C.P., Obaji S. Networks of enzymatically oxidized membrane lipids support calcium-dependent coagulation factor binding to maintain hemostasis. Sci. Signal. 2017;10 doi: 10.1126/scisignal.aan2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dilly A.K., Ekambaram P., Guo Y., Cai Y., Tucker S.C., Fridman R., Kandouz M., Honn K.V. Platelet-type 12-lipoxygenase induces MMP9 expression and cellular invasion via activation of PI3K/Akt/NF- B. Int. J. Cancer. 2013;133:1784–1791. doi: 10.1002/ijc.28165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nie D., Tang K., Diglio C., Honn K.V. Eicosanoid regulation of angiogenesis: role of endothelial arachidonate 12-lipoxygenase. Blood. 2000;95:2304–2311. [PubMed] [Google Scholar]
- 15.Schneider M., Wortmann M., Mandal P.K., Arpornchayanon W., Jannasch K., Alves F., Strieth S., Conrad M., Beck H. Absence of glutathione peroxidase 4 affects tumor angiogenesis through increased 12/15-lipoxygenase activity. Neoplasia. 2010;12:254–263. doi: 10.1593/neo.91782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo Y., Zhang W., Giroux C., Cai Y., Ekambaram P., Dilly A.K., Hsu A., Zhou S., Maddipati K.R., Liu J., Joshi S., Tucker S.C., Lee M.J., Honn K.V. Identification of the orphan G protein-coupled receptor GPR31 as a receptor for 12-(S)-hydroxyeicosatetrae-noic acid. J. Biol. Chem. 2011;286:33832–33840. doi: 10.1074/jbc.M110.216564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Winer I., Normolle D.P., Shureiqi I., Sondak V.K., Johnson T., Su L., Brenner D.E. Expression of 12-lipoxygenase as a biomarker for melanoma carcinogenesis. Melanoma Res. 2002;12:429–434. doi: 10.1097/00008390-200209000-00003. [DOI] [PubMed] [Google Scholar]
- 18.Qu Y., Wen Z., Mi S., Chen P., Wang j., Jia Y., Cheng Y. 12-lipoxygenase promotes tumor progress by TGF-β1-mediated epithelial to mesenchymal transition and predicts poor prognosis in esophageal squamous cell carcinoma. Cancer Manag. Res. 2019;11:8303–8313. doi: 10.2147/CMAR.S212478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dobrzyńska I., Szachowicz-Petelska B., Darewicz B., Figaszewski Z.A. Characterization of human bladder cell membrane during cancer transformation. J. Membr. Biol. 2015;248:301–307. doi: 10.1007/s00232-015-9770-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dobrzynska I., Szachowicz-Petelska B., Sulkowski S., Figaszewski Z. Changes in electric charge and phospholipids composition in human colorectal cancer cells. Mol. Cell Biochem. 2005;276:113–119. doi: 10.1007/s11010-005-3557-3. [DOI] [PubMed] [Google Scholar]
- 21.Szachowicz-Petelska B., Dobrzynska I., Skrodzka M., Darewicz B., Figaszewski Z.A., Kudelski J. Phospholipid composition and electric charge in healthy and cancerous parts of human kidneys. J. Membr. Biol. 2013;246:421–425. doi: 10.1007/s00232-013-9554-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sakai K., Okuyama H., Yura J., Takeyama H., Shinagawa N., Tsuruga N., Kato K., Miura K., Kawese K., Tsujimura T., Naruse T., Koike A. Composition and turnover of phospholipids and neutral lipids in human breast cancer and reference tissues. Carcinogenesis. 1992;13:579–584. doi: 10.1093/carcin/13.4.579. [DOI] [PubMed] [Google Scholar]
- 23.Hilvo M., Denkert C., Lehtinen L., Muller B., Brockmoller S., Seppanen-Laakso T., Budczies J., Bucher E., Yetukuri L., Castillo S., Berg E., Nygren H., ko Sysi-Aho M., Griffin J.L., Fiehn O. Novel theranostic opportunities offered by characterization of altered membrane lipid metabolism in breast cancer progression. Cancer Res. 2011;9:3236–3245. doi: 10.1158/0008-5472.CAN-10-3894. [DOI] [PubMed] [Google Scholar]
- 24.Guenther S., Muirhead L.J., Speller A.V.M., Golf O., Strittmatter N., Ramakrishnan R., Goldin D.R., Jones E., Veselkov K., Nicholson J., Darzi A. Spatially resolved metabolic phenotyping of breast cancer by desorption electrospray ionization mass spectrometry. Cancer Res. 2015;75:1828–1837. doi: 10.1158/0008-5472.CAN-14-2258. [DOI] [PubMed] [Google Scholar]
- 25.Beloribi-Djefaflia S., Vasseur S., Guillaumond F. Lipid metabolic reprogramming in cancer cells. Oncogenesis. 2016;5:e189. doi: 10.1038/oncsis.2015.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rysman E., Brusselmans K., Scheys K., Timmermans L., Derua R., Munck S., Veldhoven P.P.V., Waltregny D., Daniëls V.W., Machiels J., Vanderhoydonc F., Smans K., Waelkens E., Verhoeven G., Swinnen J.V. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res. 2010;70:8117–8126. doi: 10.1158/0008-5472.CAN-09-3871. [DOI] [PubMed] [Google Scholar]
- 27.Messias M.C.F., Mecatti G.C., Priolli D.G., de Oliveira Carvalho P. Plasmalogen lipids: functional mechanism and their involvement in gastrointestinal cancer. Lipids Health Dis. 2018;17:41. doi: 10.1186/s12944-018-0685-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fernandes A.M.A.P., Messias M.C.F., Duarte G.H.B., de Santis G.K.D., Mecatti G.C., Porcari A.M., Murgu M., Simionato A.V.C., Rocha T., Martinez C.A.R., Carvalho P.O. Plasma lipid profile reveals plasmalogens as potential biomarkers for colon cancer screening. Metabolites. 2020;10:262. doi: 10.3390/metabo10060262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gay L.J., Felding-Habermann B. Contribution of platelets to tumour metastasis. Nat. Rev. Cancer. 2011;11:123–134. doi: 10.1038/nrc3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Contursi A., Sacco A., Grande R., Dovizio M., Patrignani P. Platelets as crucial partners for tumor metastasis: from mechanistic aspects to pharmacological targeting. Cell Mol. Life Sci. 2017;74:3491–3507. doi: 10.1007/s00018-017-2536-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patrignani P., Patrono C. Aspirin, platelet inhibition and cancer prevention. Platelets. 2018;29:779–785. doi: 10.1080/09537104.2018.1492105. [DOI] [PubMed] [Google Scholar]
- 32.Labelle M., Begum S., Hynes R.O. Direct signaling between platelets and cancer cells induces an epithelial–mesenchymal-like transition and promotes metastasis. Cancer Cell. 2011;20:576–590. doi: 10.1016/j.ccr.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dovizio M., Maier T.J., Alberti S., Di Francesco L., Marcantoni E., Munch G., John C.M., Suess B., Sgambato A., Steinhilber D., Patrignani P. Pharmacological inhibition of platelet–tumor cell crosstalk prevents platelet-induced overexpression of cyclooxygenase-2 in HT29 human colon carcinoma cells. Mol. Pharmacol. 2013;84:25–40. doi: 10.1124/mol.113.084988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guillem-Llobat P., Dovizio M., Bruno A., Ricciotti E., Cufino V., Sacco A., Grande R., Alberti S., Arena V., Cirillo M., Patrono C., FitzGerald G.A., Steinhilber D., Sgambato A., Patrignani P. Aspirin prevents colorectal cancer metastasis in mice by splitting the crosstalk between platelets and tumor cells. Oncotarget. 2016;7:32462–32477. doi: 10.18632/oncotarget.8655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gasecka A., Nieuwland R., PSiljander P.R.M. Platelet-derived extracellular vesicles. Platelets (Fourth Edition) 2019;4:401–416. [Google Scholar]
- 36.Kailashiya J. Platelet-derived microparticles analysis: Techniques, challenges and recommendations. Anal. Biochem. 2018;546:78–85. doi: 10.1016/j.ab.2018.01.030. [DOI] [PubMed] [Google Scholar]
- 37.Mateescu B., Kowal E.J.K., Balkom B.W.M., Bartel S., Bhattacharyya S.N., Buzás E.I., Buck A.H., de Candia P., Chow F.W.N., Das S., Driedonks T.A.P., Fernández-Messina L., Haderk F., Hill A.F., Jones J.C.J. Obstacles and opportunities in the functional analysis of extracellular vesicle RNA - an ISEV position paper. J. Extracell Vesicles. 2017;6:1286095. doi: 10.1080/20013078.2017.1286095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dovizio M., Bruno A., Contursi A., Grande R., Patrignani P. Platelets and extracellular vesicles in cancer: diagnostic and therapeutic implications. Cancer Metastasis Rev. 2018;37:455–467. doi: 10.1007/s10555-018-9730-4. [DOI] [PubMed] [Google Scholar]
- 39.Tang M., Jiang L., Lin Y., Wu X., Wang K., He Q., Wang X., Li W. Platelet microparticle-mediated transfer of miR-939 to epithelial ovarian cancer cells promotes epithelial to mesenchymal transition. Oncotarget. 2017;8:97464–97475. doi: 10.18632/oncotarget.22136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Michael J.V., Wurtzel J.G.T., Mao G.F.M., Rao A.K., Kolpakov M.A., Sabri A., Hoffman N.E., Rajan S., Tomar D., Madesh M., Nieman M.T., Yu J., Edelstein L.C., Rowley J.W., Weyrich A.S. Platelet microparticles infiltrating solid tumors transfer miRNAs that suppress tumor growth. Blood. 2017;130:567–580. doi: 10.1182/blood-2016-11-751099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grande R., Dovizio M., Marcone S., Szklanna P.B., Bruno A., Ebhardt H.A., Cassidy H., Ní Áinle F., Caprodossi A., Lanuti P., Marchisio M., Mingrone G., Maguire P.B., Patrignani P. Platelet-derived microparticles from obese individuals: characterization of number, size, proteomics, and crosstalk with cancer and endothelial cells. Front. Pharmacol. 2019;10:7. doi: 10.3389/fphar.2019.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 43.Deschamps J.D., Kenyon V.A., Holman T.R. Baicalein is a potent in vitro inhibitor against both reticulocyte 15-human and platelet 12-human lipoxygenases. Bioorg. Med. Chem. 2006;14:4295–4443. doi: 10.1016/j.bmc.2006.01.057. [DOI] [PubMed] [Google Scholar]
- 44.Sekiya K., Okuda H. Selective inhibition of platelet lipoxygenase by baicalein. Biochem. Biophys. Res. Comm. 1982;105:1090–1095. doi: 10.1016/0006-291x(82)91081-6. [DOI] [PubMed] [Google Scholar]
- 45.Minuz P., Meneguzzi A., Fumagalli L., Degan M., Calabria S., Ferraro R., Ricci M., Veneri D., Berton G. Calcium-Dependent Src Phosphorylation and reactive oxygen species generation are implicated in the activation of human platelet induced by thromboxane A2 analogs. Front. Pharmacol. 2018;9:1081. doi: 10.3389/fphar.2018.01081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cho H., Ueda M., Tamaoka M., Hamaguchi M., Aisaka K., Kiso Y., Inoue T., Ogino R., Tatsuoka T., Ishihara T., Noguchi T., Morita I., Murota S. Novel caffeic acid derivatives: extremely potent inhibitors of 12-lipoxygenase. J. Med. Chem. 1991;34:1503–1505. doi: 10.1021/jm00108a039. [DOI] [PubMed] [Google Scholar]
- 47.Tacconelli S., Fullone R., Dovizio M., Pizzicoli G., Marschler S., Bruno A., Zucchelli M., Contursi A., Ballerini P., Patrignani P. Pharmacological characterization of the biosynthesis of prostanoids and hydroxyeicosatetraenoic acids in human whole blood and platelets by targeted chiral lipidomics analysis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids. 2020;1865:158804. doi: 10.1016/j.bbalip.2020.158804. [DOI] [PubMed] [Google Scholar]
- 48.Duchez A.C., Boudreau L.H., Naika G.S., Bollinger J., Belleannée C., Cloutier N., Laffont B., Mendoza-Villarroel R.E., Lévesque T., Rollet-Labelle E., Rousseau M., Allaeys I., Tremblay J.J., Poubelle P.E., Lambeau G. Platelet microparticles are internalized in neutrophils via the concerted activity of 12-lipoxygenase and secreted phospholipase A2-IIA. Proc. Natl. Acad. Sci. U. S. A. 2015;112:E3564–E3573. doi: 10.1073/pnas.1507905112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mazaleuskaya L.L., Salamatipour A., Sarantopoulou D., Weng L., FitzGerald G.A., Blair I.A., Mesaros C. Analysis of HETEs in human whole blood by chiral UHPLC-ECAPCI/HRMS. J. Lipid Res. 2018;59:564–575. doi: 10.1194/jlr.D081414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bagamery K., Kvell K., Landau R., Graham J. Flow cytometric analysis of CD41-labeled platelets isolated by the rapid, one-step OptiPrep method from human blood. Cytometry A. 2005;65:84–87. doi: 10.1002/cyto.a.20133. [DOI] [PubMed] [Google Scholar]
- 51.van Genderen H.O., Kenis H., Hofstra L., Narula J., Reutelingsperger C.P. Extracellular annexin A5: functions of phosphatidylserine-binding and two-dimensional crystallization. Biochim. Biophys. Acta. 2008;1783:953–963. doi: 10.1016/j.bbamcr.2008.01.030. [DOI] [PubMed] [Google Scholar]
- 52.Locker G.Y., Hamilton S., Harris J., Jessup J.M., Kemeny N., Macdonald J.S., Somerfield M.R., Hayes D.F., Bast R.C., Jr. ASCO 2006 update of recommendations for the use of tumor markers in gastrointestinal cancer. J. Clin. Oncol. 2006;24:5313–5327. doi: 10.1200/JCO.2006.08.2644. [DOI] [PubMed] [Google Scholar]
- 53.Yu L.X., Yan L., Yang W., Wu F.Q., LinG Y., Chen S.Z., Tang L., Tan Y.X., Cao D., Wu M.C., Yan H.X., Wang H.Y. Platelets promote tumour metastasis via interaction between TLR4 and tumour cell-released high-mobility group box1 protein. Nat. Commun. 2014;5:5256. doi: 10.1038/ncomms6256. [DOI] [PubMed] [Google Scholar]
- 54.Boudreau L.H., Duchez A.C., Cloutier N., Soulet D., Martin N., Bollinger J., Paré A., Rousseau M., Naika G.S., Lévesque T., Laflamme C., Marcoux G., Lambeau G., Farndale R.W., Pouliot M. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood. 2014;124:2173–2183. doi: 10.1182/blood-2014-05-573543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koivusalo M., Welch C., Hayashi H., Scott C.C., Kim M., Alexander T., Touret N., Hahn K.M., Grinstein S. Amiloride inhibits macropinocytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. J. Cell Biol. 2010;188:547–563. doi: 10.1083/jcb.200908086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Faille D., El-Assaad F., Mitchell A.J., Alessi M.C., Chimini G., Fusai T., Grau G.E., Combes V. Endocytosis and intracellular processing of platelet microparticles by brain endothelial cells. J. Cell Mol. Med. 2012;16:1731–1738. doi: 10.1111/j.1582-4934.2011.01434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Patiño T., Soriano J., Barrios L., Ibáñez E., Nogués C. Surface modification of microparticles causes differential uptake responses in normal and tumoral human breast epithelial cells. Sci. Rep. 2015;5:11371. doi: 10.1038/srep11371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kalluri R., Weinberg R.A. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lamouille S., Xu J., Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Riehl T.E., Turk J., Stenson W.F. Metabolism of oxygenated derivatives of arachidonic acid by Caco-2 cells. J. Lipid Res. 1992;33:323–331. [PubMed] [Google Scholar]
- 61.Gondek T., Szajewski M., Szefel J., Aleksandrowicz-Wrona E., Skrzypczak-Jankun E., Jankun J., Lysiak-Szydlowska W. Evaluation of 12-lipoxygenase (12-LOX) and plasminogen activator inhibitor 1 (PAI-1) as prognostic markers in prostate cancer. Biomed. Res. Int. 2014;2014:102478. doi: 10.1155/2014/102478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Piotrowska M., Szefel J., Skrzypczak-Jankun E., Lysiak-Szydłowska W., Szajewski M., Aleksandrowicz-Wrona E., Jankun J. The concentration of 12-lipoxygenase in platelet rich plasma as an indication of cancer of the prostate. Contemp. Oncol. (Pozn) 2013;17:389–393. doi: 10.5114/wo.2013.37221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tourdot B.E., Holinstat M. Targeting 12-lipoxygenase as a potential novel antiplatelet therapy. Trends Pharmacol. Sci. 2017;38:1006–1015. doi: 10.1016/j.tips.2017.08.001. [DOI] [PubMed] [Google Scholar]
- 64.Liu Q., Tan W., Che J., Yuan D., Zhang L., Sun Y., Yue X., Xiao L., Jin Y. 12-HETE facilitates cell survival by activating the integrin-linked kinase/NF-κB pathway in ovarian cancer. Cancer Manag. Res. 2018;10:5825–5838. doi: 10.2147/CMAR.S180334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhong C., Zhuang M., Wang X., Li J., Chen Z., Huang Y., Chen F. 12-Lipoxygenase promotes invasion and metastasis of human gastric cancer cells via epithelial-mesenchymal transition. Oncol. Lett. 2018;16:1455–1462. doi: 10.3892/ol.2018.8808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pergola C., Jazzar B., Rossi A., Buehring U., Luderer S., Dehm F., Northoff H., Sautebin L., Werz O. Cinnamyl-3,4-dihydroxy-{alpha}-cyanocinnamate (CDC) is a potent inhibitor of 5-lipoxygenase. J. Pharmacol. Exp. Ther. 2011;338:205–213. doi: 10.1124/jpet.111.180794. [DOI] [PubMed] [Google Scholar]
- 67.Meyer-Schaller N., Cardner M., Diepenbruck M., Saxena M., Tiede S., Lüönd F., Ivanek R., Beerenwinkel N., Christofori G. A hierarchical regulatory landscape during the multiple stages of EMT. Dev. Cell. 2019;48:539–553.e6. doi: 10.1016/j.devcel.2018.12.023. [DOI] [PubMed] [Google Scholar]
- 68.Shureiqi I., Chen D., Day R.S., Zuo X., Hochman F.L., Ross W.A., Cole R.A., Moy O., Morris J.S., Xiao L., Newman R.A., Yang P., Lippman S.M. Profiling lipoxygenase metabolism in specific steps of colorectal tumorigenesis. Cancer Prev. Res. (Phila) 2010;3:829–838. doi: 10.1158/1940-6207.CAPR-09-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Klampfl T., Bogner E., Bednar W., Mager L., Massudom D., Kalny I., Heinzle C., Berger W., Stättner S., Karner J., Klimpfinger M., Fürstenberger G., Krieg P., Marian B. Up-regulation of 12(S)-lipoxygenase induces a migratory phenotype in colorectal cancer cells. Exp. Cell Res. 2012;318:768–778. doi: 10.1016/j.yexcr.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hammond V.J., O'Donnell V.B. Esterified eicosanoids: generation, characterization and function. Biochim. Biophys. Acta. 2012;1818:2403–2412. doi: 10.1016/j.bbamem.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.West M.A., Antoniou A.N., Prescott A.R., Azuma T., Kwiatkowski D.J., Watts C. Membrane ruffling, macropinocytosis and antigen presentation in the absence of gelsolin in murine dendritic cells. Eur. J. Immunol. 1999;29:3450–3455. doi: 10.1002/(SICI)1521-4141(199911)29:11<3450::AID-IMMU3450>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 72.Sallusto F., Cella M., Danieli C., Lanzavecchia A. Dendritic cells use macropinocytosis and the mannose receptor to concentrate macromolecules in the major histocompatibility complex class II compartment: downregulation by cytokines and bacterial products. J. Exp. Med. 1995;182:389–400. doi: 10.1084/jem.182.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lazar S., Goldfinger L.E. Platelet microparticles and miRNA transfer in cancer progression: many targets, modes of action, and effects across cancer stages. Front. Cardiovasc. Med. 2018;5:13. doi: 10.3389/fcvm.2018.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lesko J., Triebl A., Stacher-Priehse E., Fink-Neuböck N., Lindenmann J., Smolle- Jüttner F.M., Köfeler H.C., Hrzenjak A., Olschewski H., Leithner K. Phospholipid dynamics in ex vivo lung cancer and normal lung explants. Exp. Mol. Med. 2021;53:81–90. doi: 10.1038/s12276-020-00547-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tang K., Finley R.L., Jr., Nie D., Honn K.V. Identification of 12-lipoxygenase interaction with cellular proteins by yeast two-hybrid screening. Biochemistry. 2000;39:3185–3191. doi: 10.1021/bi992664v. [DOI] [PubMed] [Google Scholar]
- 76.Kim H.K., Song K.S., Park Y.S., Kang Y.H., Lee Y.J., Lee K.R., Kim H.K., Ryu K.W., Bae J.M., Kim S. Elevated levels of circulating platelet microparticles, VEGF, IL-6 and RANTES in patients with gastric cancer: possible role of a metastasis predictor. Eur. J. Cancer. 2003;39:184–191. doi: 10.1016/s0959-8049(02)00596-8. [DOI] [PubMed] [Google Scholar]
- 77.Wang C.C., Tseng C.C., Chang H.C., Huang K.T., Fang W.F., Chen Y.M., Yang C.T., Hsiao C.C., Lin M.C., Ho C.K., Yip H.K. Circulating microparticles are prognostic biomarkers in advanced non-small cell lung cancer patients. Oncotarget. 2017;8:75952–75967. doi: 10.18632/oncotarget.18372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Luci D., Jameson J.B., II, Yasgar A., Diaz G., Joshi N., Kantz A., Markham K., Perry S., Kuhn N., Yeung J., Schultz L., Holinstat M., Nadler J., Taylor-Fishwick D.A., Jadhav A. Probe Reports from the NIH Molecular Libraries Program. National Center for Biotechnology Information (US); Bethesda, MD: 2013. Discovery of ML355, a potent and selective inhibitor of human 12-lipoxygenase. [PubMed] [Google Scholar]
- 79.Lin H.P., Singla B., Ghoshal P., Faulkner J.L., Cherian-Shaw M., O'Connor P.M., She J.X., de Chantemele E.J.B., Csányi G. Identification of novel micropinocytosis inhibitors using a rational screen of Food and Drug Administration-approved drugs. Br. J. Pharmacol. 2018;175:3640–3655. doi: 10.1111/bph.14429. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All individual data are reported in the figures. There is no restriction on the availability of any data.