

Abstract
The transcription master regulator MYC plays an essential role in regulating major cellular programs and is a well-established therapeutic target in cancer. However, MYC targeting for drug discovery is challenging. New therapeutic approaches to control MYC-dependent malignancy are urgently needed. The mitogen-activated protein kinase kinase 3 (MKK3) binds and activates MYC in different cell types, and disruption of MKK3-MYC protein-protein interaction may provide a new strategy to target MYC-driven programs. However, there is no perturbagen available to interrogate and control this signaling arm. In this study, we assessed the drugability of the MKK3-MYC complex and discovered the first chemical tool to regulate MKK3-mediated MYC activation. We have designed a short 44-residue inhibitory peptide and developed a cell lysate-based time-resolved fluorescence resonance energy transfer (TR-FRET) assay to discover the first small molecule MKK3-MYC PPI inhibitor. We have optimized and miniaturized the assay into an ultra-high-throughput screening (uHTS) 1536-well plate format. The pilot screen of ~6,000 compounds of a bioactive chemical library followed by multiple secondary and orthogonal assays revealed a quinoline derivative SGI-1027 as a potent inhibitor of MKK3-MYC PPI. We have shown that SGI-1027 disrupts the MKK3-MYC complex in cells and in vitro and inhibits MYC transcriptional activity in colon and breast cancer cells. In contrast, SGI-1027 does not inhibit MKK3 kinase activity and does not interfere with well-known MKK3-p38 and MYC-MAX complexes. Together, our studies demonstrate the drugability of MKK3-MYC PPI, provide the first chemical tool to interrogate its biological functions, and establish a new uHTS assay to enable future discovery of potent and selective inhibitors to regulate this oncogenic complex.
Keywords: protein-protein interaction, chemical tool, small molecule inhibitor, oncogenic signaling, high-throughput screening, MKK3, MYC
Graphical Abstract

1. INTRODUCTION
The transcription factor MYC is a major oncogene and the master regulator of cell growth. MYC is amplified in nearly all cancers and orchestrates the expression of up to 15% of all human genes 1,2. MYC initiates and maintains tumorigenesis through the regulation of multiple cellular programs, including cell proliferation, immortalization, differentiation, metabolism, and biosynthesis as well as the expression of key immune regulatory proteins, including the programmed death-ligand 1 (PD-L1). MYC upregulation is tightly associated with poor survival and prognosis of cancer patients3–11, and regulation of MYC transcriptional activity is a highly appealing therapeutic strategy3,12–16.
However, MYC lacks known enzymatic activity and does not have a specific site for a small molecule binding. Instead, MYC is controlled by other proteins either by phosphorylation or through protein-protein interactions (PPI)17. For example, the dimerization of the MYC helix-loop-helix leucine zipper (HLH-LZ) domain with the HLH-LZ domain of MAX protein is essential for MYC binding to DNA 18,19. The Extracellular Receptor Kinase (ERK) can upregulate MYC stability through the phosphorylation of MYC at Ser62 20. Conversely, the phosphorylation of MYC at Thr58 by GSK3β promotes rapid MYC degradation 21.
The Mitogen-Activated Protein Kinase Kinase 3 (MKK3) was recently discovered as a new binding partner and activator of MYC. MKK3 is a dual-specificity serine/tyrosine kinase. It was originally discovered as the activator of its only known substrate, p38 that regulates inflammation and apoptosis 22. Accordingly, MKK3 functions have been associated solely with the regulation of the p38 signaling 23–28. We have demonstrated a novel role of MKK3 as an adaptor protein for MYC 29–31. We have shown that MKK3 directly binds to MYC, enhances MYC protein stability, and promotes MYC transcriptional activity in different cancer cell types 23–28. Interestingly, MYC can be upregulated upon its binding to truncated versions of MKK3 that lack kinase activity. It suggests that in contrast to ERK and GSK3β, MKK3 promotes MYC activity in a kinase-independent manner acting as an adaptor protein. We have also shown that MKK3 overexpression activates MYC transcriptional program and correlates with worsened clinical outcomes in African American triple-negative breast cancer (TNBC) patients. Therefore, disruption of MKK3-MYC PPI may open new clinical opportunities in TNBC and other cancers with dysregulated MYC and MKK3. However, to interrogate biological functions regulated through MKK3-MYC PPI specific chemical tools are needed.
Here, we report the design of short inhibitory peptides and the development of cell lysate-based ultra-high-throughput time-resolved fluorescence energy transfer (TR-FRET) assay to discover the first small molecule MKK3-MYC PPI inhibitors. Through a pilot screen of ~6,000 pharmacologically active compounds, we found that the MKK3-MYC complex can be selectively disrupted by small molecules avoiding the inhibition of well-defined MYC-MAX or MKK3-p38 interactions. The inhibition of the MKK3-MYC PPI correlates with the decreased MYC protein level, suppressed MYC transcriptional activity, and reduced expression of MYC-target genes, including cell growth and immune response regulators. These data support the role of MKK3 as a direct MYC activator, demonstrate the feasibility to disrupt the MKK3-MYC complex by low molecular weight compounds, and provide the first chemical tools to investigate MKK3-MYC PPI functions for therapeutic discovery.
2. RESULTS
2.1. MKK3 does not phosphorylate MYC
MKK3 is a kinase, and inhibition of its kinase activity can be a promising strategy to regulate MKK3-dependent signaling. As a dual threonine/tyrosine kinase, MKK3 specifically recognizes and phosphorylates the Thr-Gly-Tyr (TGY) motif located in the activating loop of its well-defined and only known substrate, p38 32. MYC lacks the TGY motif and is not expected to be phosphorylated by MKK3. Previously, we have shown that short MKK3 fragments that lack the kinase activity are sufficient for MYC binding and activation 31. Nevertheless, whether MKK3 can control MYC by phosphorylation remains unknown. To answer this question, we tested whether MKK3 phosphorylates MYC in the in vitro kinase assay. The radiograph obtained for MKK3 incubated with p38 (Supplementary Fig. 1, lane 5) revealed strong phosphorylation of p38 by MKK3, indicating a sufficient kinase activity of MKK3 in the assay. In contrast, no phosphorylation was observed for the MKK3-MYC reaction performed in parallel (Supplementary Fig. 1, lane 4). Moreover, incubation of MKK3 with p38 in the presence of an equal amount of MYC did not significantly change the amount of phosphorylated p38 (Supplementary Fig. 1, lane 6). This observation further supports our previous discovery that MYC and p38 bind to distinct sites on the MKK3 surface. The lack of MYC phosphorylation by MKK3 indicates that MYC is not an MKK3 substrate and supports the role of MKK3 as a MYC adaptor protein. Meanwhile, the lack of MYC phosphorylation by MKK3 suggests that MKK3 inhibitors cannot be used as potent tools to regulate MKK3-mediated MYC activation. Instead, the inhibitors of the MKK3-MYC PPI interface are needed.
2.2. MKK3-MYC PPI interface can be disrupted by short peptides
Through computational and biochemical studies, we have determined MYC Helix-Loop-Helix Leucine Zipper (HLH-LZ) domain (MYC 353–439) as the main MYC binding for MKK3 surface31. We used the computational model to characterize further the MKK3-MYC PPI interface and design short inhibitory peptides to control MKK3-MYC PPI. Based on the structural analysis, the following MYC-derived peptides have been designed and cloned into Venus-Flag (VF)-tagged expression vectors: VF-MYC 353–387, VF-MYC 387–439, VF-MYC 370–413, and VF-MYC 362–381 were generated (Fig. 1A). The GST pull-down assay revealed that besides VF-MYC HLH-LZ, GST-MKK3 strongly interacts with VF-MYC 387–439 and VF-MYC 370–413 peptides located in the middle region of the MYC HLH-LZ domain (Fig. 1B). In contrast, no interaction was observed for GST-MKK3 with VF-MYC 353–387 or VF-MYC 362–381 (Fig. 1B).
Figure 1. MYC 370–413 peptide binds to MKK3 and disrupt MKK3-MYC PPI.
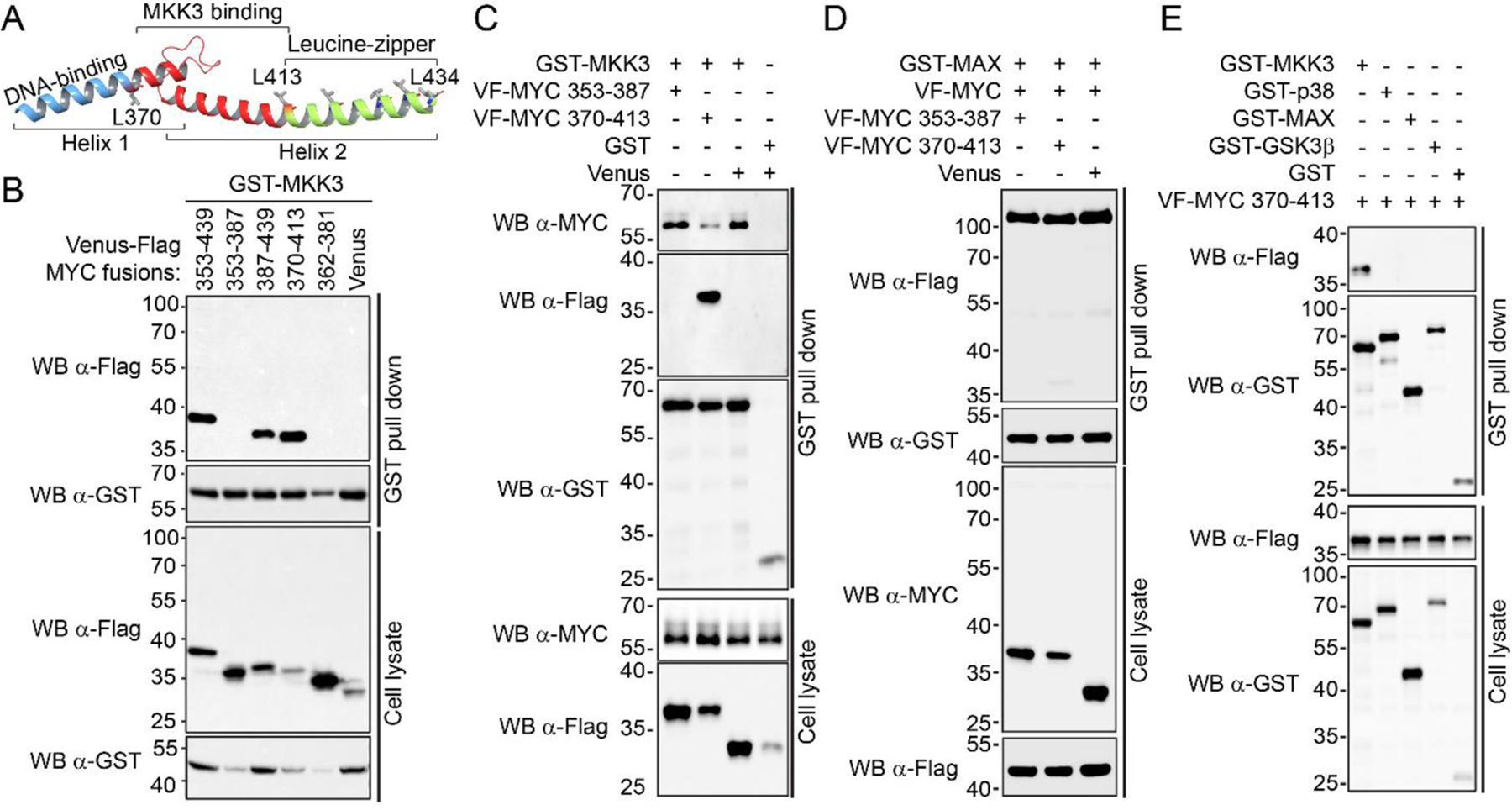
A) Crystal structure of the MYC HLH-LZ-domain; B) MYC 353–439, 387–439, and 370–413 fragments interact with GST-MKK3 in GST pull down assay. The assay was performed using the HEK293T cells co-expressing GST-MKK3 and Venus-Flag fusions of MYC fragments. Venus-Flag vector was used as a negative control. C) Venus-Flag (VF)-MYC 370–413 inhibits the interaction of endogenous MYC with GST-MKK3 in GST pull down assay. The assay was performed using the HEK293T cells co-expressing GST-MKK3 with VF-MYC 370–413. Non-binding VF-MYC 353–387 fragment and Venus-Flag vectors served as negative and vehicle controls, respectively. D) VF-MYC 370–413 peptide does not interfere with GST-MAX/VF-MYC PPI comparing to VF-MYC 353–387 and Venus-Flag vector controls in GST pull down assay performed in HEK293T cells. E) VF-MYC 370–412 demonstrates a higher affinity for GST-MKK3 comparing to GST-p38, GST-MAX, and GST-GSK3β in GST pull down assay performed in HEK293T cells. GST vector served as a negative control.
Since MYC 370–413 was identified as the shortest MKK3-binding peptide, we tested whether MYC 370–413 can compete with MYC and disrupt its interaction with MKK3 and MYC. We found that the co-expression of GST-MKK3 with VF-MYC 370–413 suppresses the binding of the endogenous MYC to GST-MKK3 as compared to the non-binding VF-MYC 353–387 or Venus vector control (Fig. 1C).
MYC HLH-LZ domain is known for its critical role in dimerization with MYC major binding partner MAX to bind the DNA. Since MYC 370–413 was derived from MYC HLH-LZ, we tested its effects on MYC-MAX PPI.
In contrast to its inhibitory effect on the MKK3-MYC PPI, MYC 370–413 did not significantly decrease MYC-MAX interaction in GST pull-down assay (Fig. 1D). This observation is in agreement with the crystallographic data available for the MYC-MAX complex 19. It has been shown that MYC binds to MAX through the leucine zipper motif, and MYC Arg424 and Arg423 are essential for MYC/MAX heterodimerization 19. MYC 370–413 does not contain these residues and thus is not expected to form a stable complex with MAX. Furthermore, we did not observe the binding of VF-MYC 370–413 to the major MKK3 substrate, p38, nor to a well-established regulator of MYC GSK3β (Fig. 1E). Together, these data demonstrate that the MKK3-MYC PPI interface can be selectively disrupted by chemical tools and provide a basis to discover small molecule inhibitors for MKK3-MYC PPI.
2.3. Development of the ultra-high-throughput screening assay for MKK3-MYC PPI inhibitors
To assess the drugability of MKK3-MYC PPI by small molecules, we have developed the cell lysate-based TR-FRET HTS assay to detect MKK3-MYC interaction directly from the cell lysates (Fig. 2). This lysate-based approach has several advantages over conventional HTS assays with recombinant purified proteins. For example, the use of lysates of cells co-expressing the binding proteins allows us to detect the PPIs under physiological conditions, including post-translational modifications. It also does not require protein purification steps that can be problematic and time-consuming, especially for disordered nuclear proteins such as MYC.
Figure 2. Development and evaluation of the MKK3-MYC PPI TR-FRET assay.

A) GST-d2 and Flag-Tb fluorophore pair allow at least 5 fold over control TR-FRET assay window in 384-well plate format. The error bars demonstrate the variation between four replicates. A representative data of three independent experiments are shown; B) Temporal stability of TR-FRET signal. The TR-FRET signal was measured in the time course of 48 h; C) The TR-FRET signal is stable in the presence of the increasing amount of DMSO. D) The TR-FRET assay miniaturized into a 1,536-well uHTS format demonstrate the equally high performance as the assay performed in 384-well HTS format. E) Each compound plate used in the uHTS assay demonstrated at least a 6-fold difference between the signal and background (S/B). F) Z’ > 0.7 were determined for each compound plate used in the uHTS assay. G) The uHTS with the LOPAC, Spectrum, and EEBL libraries revealed five primary hits with more than 30% inhibition.
Previously, we successfully used the lysate-based TR-FRET assay to detect different MKK3 PPIs, including MKK3-MYC interaction in mammalian cell lysates using the Venus tag as a TR-FRET signal acceptor and GST-Tb conjugated antibody as the TR-FRET donor 31. On the other hand, we have shown that detection of MYC PPIs can be improved by using GST-d2/Flag-Tb pair of fluorophores 33. Indeed, the TR-FRET signal detected for GST-MKK3/VF-MYC PPI using the GST-d2/Flag-Tb antibody pair with concentrations at 1:500 and 1:750, respectively followed a typical TR-FRET “bell-shaped” curve34 and provided more than a five-fold signal-to-background assay window (Fig. 2A) at the lysate concentration of 0.1 mg/mL. Furthermore, GST-d2/Flag-Tb fluorophore combination provides a TR-FRET signal that is stable for at least 48 hours after the addition of antibodies (Fig. 2B) and demonstrates tolerance to up to 30% DMSO (Fig. 2C). Since GST-d2/Flag-Tb fluorophore combination demonstrated robust assay performance, we used it for the HTS assay.
To further improve the time and cost-efficiency of the assay, we have miniaturized the assay into a 1,536-well uHTS format. A direct comparison of the assay performance in 384-well (30µL lysate per well) and 1,536-well plates (5µL lysate per well) demonstrated equally high characteristics of the assay with at least 4 fold over control ratios in both plate formats (Fig. 2D).
2.4. The uHTS screening revealed SGI-1027 as the first inhibitor of the MKK3-MYC PPI
To discover the first MKK3-MYC PPI inhibitors, we have performed a pilot screen of a library of 5,296 pharmacologically active compounds, including the SPECTRUM library (MicroSource, 2000 compounds), Library of Pharmacologically Active Compounds (LOPAC, Sigma-Aldrich, 1,260 compounds), and our in-house Emory Enriched Bioactive Library (EEBL) collection of 2,036 compounds. The EEBL library is enriched in pharmacologically active compounds with known biological and pharmacological activities and contains 1,018 FDA-approved drugs. Recently, the library was successfully used to discover new IAP inhibitors as immune enhancers 35.
The Signal/Background > 6 (Fig. 2E) and the Z′ > 0.7 (Fig. 2F) and calculated across five 1,536-well plates demonstrated an excellent assay performance. A subsequent analysis of fluorescence intensity allowed us to identify ~10% of compounds as fluorescent and potentially interfering with the TR-FRET assay. Compounds that demonstrated at least 20% fold-change in fluorescence intensity comparing to DMSO control were eliminated from further analysis. After elimination of color quenching compounds, a total of 6 molecules have been prioritized with a hit cutoff of 30% inhibition at 20µM final concentration (Fig. 2G): N-[4-[(2-Amino-6-methylpyrimidin-4-yl)amino]phenyl]-4-(quinolin-4-ylamino)benzamide (SGI-1027, 37% inhibition), 5-[(3-carboxy-4-hydroxyphenyl)-(3-carboxy-4-oxocyclohexa-2,5-dien-1-ylidene)methyl]-2-hydroxybenzoic acid (Aurintricarboxylic acid or ATA, 58% inhibition), [2,6-di(propan-2-yl)phenyl] ~(N)-[2-[2,4,6-tri(propan-2-yl)phenyl]acetyl]sulfamate, (Avasimibe, 48% inhibition), and (−)-1,1’,6,6’,7,7’-Hexahydroxy-3,3’-dimethyl-5,5’-bis(1-methylethyl)-[2,2’-binaphthalene]-8,8’dicarboxaldehyde (AT101, 31% inhibition) as well as Cisplatin and Sodium nitroferricyanide that showed 55% inhibition. Since cisplatin and sodium nitroferricyanide are small inorganic molecules with a broad spectrum of pharmacological activity and limited potential for further development as chemical probes, these compounds were excluded from further investigation. SGI-1027, Avasimibe, ATA, and AT101 were selected for validation in a dose-response (DR) TR-FRET assay. AT101 did not demonstrate detectable inhibition of MKK3-MYC PPI in the DR tests performed using 0 – 100 µM compound concentrations. In contrast, the IC50 values of 8.46 µM, 14.06 µM, and 63.88 µM were determined for SGI-1027, ATA, and Avasimibe, respectively (Supplementary Fig. 2).
2.5. SGI-1027 disrupts the MKK3-MYC complex
Since SGI-1027 showed the highest potency as an MKK3-MYC PPI inhibitor in the DR TR-FRET assay, we focused on this compound as the most promising hit for further orthogonal validations and functional characterization. SGI-1027 is a widely-used micromolar inhibitor of DNA Methyltransferases (DNMT) 36. To test whether the DNMT inhibition contributes to the disruption of MKK3-MYC PPI, we used a structurally different and more potent and selective DNMT inhibitor RG108 (IC50(DNMT) = 115nM)37. (Fig. 3A). In contrast to SGI-1027, we found that RG108 does not interfere with the interaction between MKK3 and MYC at up to 100 µM concentration in the dose-response cell lysate-based TR-FRET assay (Fig. 3B).
Figure 3. Validation of SGI-1027 as the MKK3-MYC PPI inhibitor.

A) Chemical structures of DNMT inhibitors SGI-1027 and RG108. B) SGI-1027 but not RG108 inhibits the interaction between GST-MKK3 and VF-MYC in cell lysate-based TR-FRET assay. C) SGI-1027 inhibits MKK3-MYC PPI but not MKK3-p38, MYC-MAX interactions in GST pull down assays. GST- and Venus-Flag (VF)-tagged proteins were co-expressed in HEK293T cells. The cell lysates were treated with 50 µM SGI-1027. DMSO was used as vehicle control. VF vector served as a negative control for the assay. D) SGI-1027 inhibits the interaction between purified recombinant GST-MKK3 and His-MYC Helix-Loop-Helix domain in a TR-FRET assay. The percentage of inhibition was calculated for each compound compared to DMSO control. E) SGI-1027 inhibits the interaction between GST-MKK3 and endogenous MYC in a dose-dependent manner in GST-pull down assay.
Then, we have confirmed the inhibition of MKK3-MYC PPI by SGI-1027 in the orthogonal GST pull-down assay (Fig. 3C). In contrast, we did not observe a significant suppression of MKK3-p38 or MYC-MAX PPIs by SGI-1027 at up to 50 µM concentration (Fig. 3C). These data support a selectivity of SGI-1027 against the MKK3-MYC complex. Moreover, we have found that HCT116 cells treated with increased concentration of SGI-1027 demonstrate a decreased association of GST-MKK3 with endogenous MYC (Fig. 3D) in GST pull-down assay.
To further validate the on-target effect of SGI-1027 on MKK3-MYC PPI, we have monitored the disruption of MKK3-MYC PPI in TR-FRET assay performed with recombinant purified MKK3 and MYC proteins instead of cell lysates. Previously, we established a TR-FRET assay to monitor the interaction between purified recombinant MKK3 and MYC HLH-LZ domain 31. We used the assay to determine whether SGI-1027 can interfere with the MKK3-MYC HLH-LZ binding. As shown in Fig. 3E, SGI-1027 inhibits the interaction between GST-MKK3 and His-MYC-HLH-LZ with the IC50 = 9.67 ± 1.06 µM. These results are in excellent agreement with the cell lysate-based TR-FRET assay (IC50 = 8.46 ± 1.53 µM) performed for GST-MKK3 and full-length VF-MYC, and strongly support the direct effect of SGI-1027 on the MKK3-MYC complex.
2.6. A computational model of MKK3 bound to SGI-1027
To get the structural insights into the MKK3-SGI-1027 complex, we leveraged the computational modeling approach. Previously, we showed that MYC HLH-LZ binds to the MKK3 cavity located in its N-lobe around 121–135 residues31. Here, using molecular docking followed by the Molecular Mechanics/Generalized Born Surface Area (MM-GBSA) calculations, we found that SGI-1027 can also favorably bind to the same site on the MKK3 surface to interfere with MYC binding (Fig. 4A). In agreement with experimental data, the estimated binding energy obtained for SGI-1027 (MMGBSA ∆Gbind = −53.30 kcal/mol) was more favorable than for Avasimibe (MMGBSA ∆Gbind = −44.75 kcal/mol) and ATA (MMGBSA ∆Gbind = −41.68 kcal/mol). Moreover, the SGI-1027 binding energy was over two times more favorable than the binding energy obtained for RG108 docked in parallel (MMGBSA ∆Gbind = −21.08 kcal/mol).
Figure 4. A computational model of SGI-1027 bound to MKK3.

A) SGI-1027 can favorably bind to the MYC binding site. The molecular surface of SGI-1027 is shown in red. The MKK3 alpha-helices and beta-sheets are colored in blue and yellow, respectively. The predicted binding mode of the MYC HLH domain (shown in grey) is superimposed with SGI-1027 for reference. B) A The predicted binding mode of SGI-1027 at the MKK3 binding site. The carbon atoms of SGI-1027 are highlighted in green. The protein-ligand interactions are shown as dash lines: h-bonds are colored in red, Pi-cation interactions – green, and salt bridges – pink. C) A 2D representation of interactions between MKK3 and SGI-1027. The H-binds are shown as red lines, salt bridges – blue lines, and Pi-cation interactions are shown as green lines. D) GST-MKK3 1–210 demonstrates significantly (p-value < 0.01) higher affinity to VF-MYC as compared to GST MKK3 201–347 in the TR-FRET assay. The assay was performed in lysates of HEK293T cells expressing VF-MYC or Venus-Flag vector with GST-MKK3 1–210 or GST-201–347. The Fold change of the PPI TR-FRET signal over the Venus-control signal is shown. E) SGI-1027 inhibits the interaction between VF-MYC and GST-MKK3 1–210 supporting that SGI-1027 binds to the N-lobe of MKK3 as predicted by docking studies.
Based on the model obtained for SGI-1027, the quinoline moiety of the compound was located in a pocket formed by Asp63, Leu64, Ala161, Arg132, Trp137, and the quinone NH-group formed an H-bond with the carboxyl group of Asp62 (Fig. 4B, C). The backbone carbonyl oxygen atom of Phe131 formed an H-bond with the NH-group that connects the quinolone and benzene rings of SGI-1027. The benzamide aromatic ring was located near the hydrophobic side chains of Phe57 and Val59 and was involved in Pi-cation interaction with Arg113. Arg113 also formed an H-bond with the nitrogen atom of SGI-1027 pyrimidine moiety.
To validate that SGI-1027 binds to the N-terminal half of MKK3, we tested if SGI-1027 can disrupt the interaction between MYC and MKK3 1–210 4fragment. First, we confirmed that GST-MKK3 1–210 demonstrate up to 15-fold higher affinity to VF-MYC as compared to GST MKK3 201–347 (Fig. 4D). Then, we found that SGI-1027 inhibits the GST-MKK3 1–210/VF-MYC PPI in the TR-FRET assay with the IC50 = 3.8 ± 1.2µM (Fig. 4E). Moreover, the incubation of HEK293T cells with SGI-1027 did not decrease the phosphorylation of MKK3 substrate p38. This observation indicates that SGI-1027 does not compete with ATP and thus does not bind to the MKK3 ATP-binding site, further supporting the predicted binding mode This observation in (Supplementary Fig. 3).
2.7. Disruption of MKK3-MYC PPI suppresses MYC protein level and transcriptional activity
The luciferase-based MYC reporter assay was performed to determine the effect of MKK3 knockdown on MYC activation. To ensure the sufficient luminescence signal from the reporter luciferase generated by the endogenous MYC, we used the HCT116 cell line as a model system. The HCT116 cells provide both, a high level of constitutively activated MYC38 and high transfection efficiency. Previously, we have demonstrated the MKK3-MYC PPI at the endogenous level in breast cancer MCF7 and H1299 lung cancer cells.31 Here, using the co-immunoprecipitation assay we have further confirmed the endogenous interaction between MKK3 and MYC in HCT116 cells (Fig. 5A). Then, we found that the MYC level was substantially lower in the MKK3 knockdown cells than in the parent HCT116 cells (Fig. 5B). The suppression of MKK3 also correlated with the reduced MYC transcriptional activity as observed in the luciferase-based MYC reporter assay (Fig. 5C). On the other hand, the loss of MYC activity in the MKK3 knockdown cells was rescued by the MKK3 overexpression (Fig. 5C). These results further validate the critical role of MKK3 in MYC regulation.
Figure 5. Disruption of MKK3-MYC PPI correlates with inhibition of MYC activity in cancer cells.

A) Endogenous co-immunoprecipitation of MYC with MKK3 has confirmed the MKK3-MYC PPI in HCT116 colon cancer cells. B) MKK3 knockdown correlates with the suppression of MYC protein level in HCT116 colon cancer cells. Two different MAP2K3 shRNA (shRNA-1 and shRNA-2) were used. The non-target shRNA (shRNA-Ctrl) was used as a negative control. C) MKK3 knockdown correlates with the suppression of MYC transcriptional activity in Luciferase-based MYC reporter assay performed in HCT116 cells. MYC activity can be rescued by VF-MKK3 overexpression (red bars) but not Venus-Flag vector (black bars) in the knockdown cells. D) SGI-1027 but not RG108 suppresses the level of MYC and its target genes, including CDK4 and PD-L1, in HCT116 cells. E) SGI-1027 suppresses MYC, CDK4, and PD-L1 in MDA-MB-468 triple-negative breast cancer cells. F) SGI-1027 but not RG108 (G) suppresses MYC transcriptional activity in MYC reporter assay performed in HCT116 cells or MDA-MB-468 cells (H) expressing VF-MKK3 (red bars) or Venus-Flag control (black bars). Expression VF-MKK3 promotes MYC activity as compared to Venus-Flag control. This effect can be inverted by the disruption of MKK3-MYC PPI by SGI-1027. I) SGI-1027 suppresses the viability of HCT116 cells. Cell viability was determined in the Alamar Blue assay and quantified relative to DMSO control. J) SGI-1027 suppresses migration of HCT116 cells in a time- and dose-response manner. The wound healing was monitored in real-time during the time course of 18 hours in HCT116 cells treated either with DMSO control or indicated concentrations of SGI-1027. The error bars indicate the standard deviation of four replicates. K) Representative images of the wound healing obtained at the time zero and in 18 hours after the treatment of cells with DMSO or 3.0 μM SGI-1027.
Then, we tested if MYC activity can also be suppressed by SGI-1027. We found that the incubation of HCT116 cells with SGI-1027 correlates with the substantial decrease in MYC level and expression of critical MYC-target genes, including cell-cycle regulating kinase CDK4 and the major immune checkpoint protein PD-L1 (Fig. 5D). In contrast, RG108, a more potent DNMT inhibitor, did not suppress the level of MYC and MYC-target genes in HCT116 cells incubated with up to 10 µM compound, which is 100 times higher concentration than the reported IC50 of RG108 against DNMT (Fig. 5D). These data support a DNMT-independent mechanism of the suppression of MKK3-induced MYC activation by SGI-1027. We also found that suppression of MYC level by SGI-1027 is not cell line-specific. For example, MYC level was decreased proportionally to the increasing concentration of SGI-1027 in MDA-MB-468 triple-negative breast cancer cells (Fig. 5E).
We have also validated the suppression of MYC activity by SGI-1027 in the luciferase-based MYC-reporter assay (Fig. 5F–H). First, we confirmed that overexpression of VF-MKK3 in HCT116 or MDA-MB-468 cells leads to a significant upregulation of MYC transcriptional activity as compared to MYC activity in cells expressing the Venus-Flag vector. This effect can be diminished by cell incubation with SGI-1027 in a dose-dependent manner Fig. 5F, H. In contrast, MYC activity was not suppressed in cells incubated with equal amounts of RG108 (Fig. 5G).
The luciferase-based MYC reporter assay relies on the luciferase enzymatic activity. To exclude non-specific effects of SGI-1027 on the luciferase activity, we tested if SGI-1027 interferes with the luciferase luminescence signal in the assay. We did not observe a decrease in the Renilla luciferase activity in cells treated with 10 µM SGI-1027 as compared to the DMSO control (Supplementary Fig. 4).
Both MYC and MKK3 play a critical role in regulating cancer cell survival and metastasis12,39–42. Therefore, we tested if the disruption of MKK3-MYC PPI and suppression of MYC transcriptional activity and by SGI-1027 correlates with decreased viability and migration of HCT116 cells. We found that SGI-1027 inhibits the viability of HCT116 cells with the IC50 = 3.2 ± 0.6 μM (Fig. 5I). These data are in agreement with the previously reported inhibitory activity of SGI-1027 against different cancer cell lines with the range of IC50 between 1.7μM and 9.1μM43. We also found that SGI-1027 suppresses the migration of HCT116 cells in a dose- and time-dependent manner (Fig. 5J). The relative wound density (RWD) of 20% determined after 18 hours of incubation of HCT116 cells with DMSO control was more than two times higher than the RWD of 8% determined for cells treated with 3 μM SGI-1027.
Discussion
Previously we have shown that MKK3 can control MYC transcriptional activity through protein-protein interaction. MKK3-MYC PPI can play a critical role in different MYC-dependent cancers and represents a promising target for therapeutic discovery in African American patients with triple-negative breast cancer44.
In this study, we have demonstrated the druggability of the MKK3-MYC PPI interface and discovered the first chemical tools to interrogate the cellular functions mediated through MKK3-MYC PPI. We found that MYC 370–413 is sufficient for the binding to MKK3 and can disrupt the interaction between MYC and MKK3 but not its major complex with MAX. Meanwhile, MYC 370–413 does not interact with other kinases, including the well-defined MKK3 substrate p38 or MYC negative regulator GSK3β. Thus this 44-residue peptide provides a valuable tool to selectively regulate MKK3-signalling transduced through its complex with MYC.
To discover small molecule MKK3-MYC PPI inhibitors, we have developed a novel cell lysate-based TR-FRET assay in ultra-HTS 1536-well plate format. Through a pilot screen of 5,889 compounds with known pharmacological activity followed by multiple validation assays, we have determined a quinoline derivative SGI-1027 as the first low molecular-weight inhibitor of MKK3-MYC PPI (IC50 = 8µM). SGI-1027 demonstrated a notable selectivity against other MKK3 and MYC PPIs, including their major MKK3-p38 and MYC-MAX PPIs. Although more detailed structural studies are needed to determine the binding mode of SGI-1027 at the MKK3-MYC PPI interface, our computational model suggests that SGI-1027 can favorably bind to the MKK3 binding site previously identified as its main binding site for MYC.
SGI-1027 is a known micromolar inhibitor of DNMTs and is widely used as a chemical probe to interrogate DNMT functions in cells45–47. Our data demonstrate that SGI-1027 also can regulate MYC transcriptional program through inhibition of MKK3-MYC PPI. We have shown that the disruption of MKK3-MYC PPI by SGI-1027 correlates with the suppression of MKK3-induced MYC activation, decreased protein level of MYC, and reduced expression of MYC-target cell growth and immune response regulators, including CDK4 and PD-L1 In contrast to SGI-1027, another and more potent nanomolar DNMT inhibitor RG108 did not inhibit the MKK3-MYC PPI and did not suppress MYC transcriptional activity in the reporter assay. These data indicate that the suppressive effect of SGI-1027 on MYC protein stability and transcriptional activity is independent of its known activity against DNMTs. In agreement with previously reported data, we showed that SGI-1027 suppresses both the viability and motility of colon cancer HCT116 cells. Our data suggest that these inhibitory effects of SGI-1027 can be mediated not only through its previously reported target DNMT but in part through the disruption of the MKK3-MYC complex. These findings further support the significance of MKK3-MYC PPI as a promising target for therapeutic discovery in cancer.
Conclusions
The master regulator transcription factor MYC is a highly attractive therapeutic target in cancer. However, MYC targeting for drug discovery is challenging, and no FDA-approved MYC inhibitors are available. Novel therapeutic approaches to control MYC dependency are urgently needed to improve clinical outcomes of patients with dysregulated MYC transcriptional programs. In this study, we demonstrated that MYC activity can be controlled by inhibiting MKK3-MYC PPI that was recently reported as a new promising target in African American patients with triple-negative breast cancer. The reported design of short inhibitory peptides and the discovery of SGI-1027 as the first molecular weight MKK3-MYC PPI inhibitor demonstrate the druggability of the MKK3-MYC PPI interface. The established cell lysate-based ultra-high-throughput screening assay can facilitate the discovery of new MKK3-MYC PPI inhibitors to inform biological studies and therapeutic strategies in breast cancer and other disorders with dysregulated MYC and MKK3.
3. METHODS
5.1. Antibodies
Tb cryptate-conjugated mouse monoclonal anti-Flag antibody (anti-Flag-Tb, catalog. no. 61FG2TLB) and d2-conjugated anti-GST antibody (anti-GST-d2, catalog. no. 61GSTDLB) were purchased from Cisbio Bioassays (Bedford, MA). Monoclonal ANTI-FLAG M2-Peroxidase (HRP) antibody (catalog. no. A8592), anti-Glutathione-S-Transferase (GST)-Peroxidase Conjugate antibody (catalog. no. A7340), monoclonal Anti-β-Actin antibody (catalog. no. A5441), and protease inhibitor (catalog. no. P8340) were purchased from Sigma-Aldrich. c-Myc rabbit monoclonal antibody (catalog. no. 5605S) was purchased from Cell Signaling. Peroxidase AffiniPure goat anti-rabbit IgG (H+L) secondary antibody (catalog. no. 111–035-003), Peroxidase AffiniPure goat anti-mouse IgG (H+L) secondary antibody (catalog. no. 115–035-003), and Peroxidase AffiniPure goat anti-rabbit IgG light chain-specific (L) secondary antibody (catalog. no. 211–032-171) were purchased from Jackson ImmunoResearch.
5.2. Cell lines and culture conditions
All cell lines were obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA). HEK293T (ATCC CRL-3216), colon cancer HCT116 (ATCC, CCL-247), and breast cancer MDA-MB-468 (ATCC HTB-132) were cultured in Dulbecco’s modified Eagle’s medium with 4.5 g/l glucose, l-glutamine, and sodium pyruvate (Corning, catalog. no. 10–013-CV) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin solution (CellGro, catalog. no. 30–002-CI). Cells were incubated at 37 °C in humidified conditions with 5% CO2.
5.3. Transfection
The HEK293T cells were transfected using 1 mg/mL Polyethylenimine (PEI; Polysciences, Inc., catalog no. 23966) in a ratio of 3 μl to 1 μg DNA. Cancer cells were transfected using X-tremeGENE HP (SigmaAldrich, catalog. No. 6366546001) in a ratio of 3 μl to 1 μg DNA following the manufacturer’s instructions.
DNA constructs
All GST-, VF-tagged human MKK3 and MYC plasmids for mammalian expression were generated using Gateway cloning technology (Invitrogen) as described previously31. The DNA was purified using ZymoPURE Plasmid Maxiprep Kit (Zymo Research, catalog no. D4203).
5.4. GST-pull down assay
HEK293T cells were co-transfected with GST- and Venus-Flag-tagged proteins, or empty vectors as negative controls. The proteins were expressed for 48h. Cell lysates were prepared in the 1% NP-40 lysis buffer (150 mM NaCl, 10 mM HEPES pH 7.5, 1% nonident NP-40 (Sigma-Aldrich, IGEPAL catalog. no. CA-630), 5 mM sodium pyrophosphate, 5 mM NaF, 2 mM sodium orthovanadate, 10 mg/l aprotinin, 10 mg/l leupeptin and 1 mM phenylmethylsulfonyl fluoride) for 30 min at 4 °C, followed by 15 min centrifugation at 14 000 r.p.m. at 4 °C. The cleared lysates were incubated with glutathione sepharose 4B beads (GE Healthcare, catalog no. 17–0756-05) at 4 °C for 3 hours for regular pull-down assays.
For the PPI inhibition assay, cell lysates were first pre-incubated with compounds for 30 minutes and then incubated for 75 minutes with the glutathione sepharose.
After the incubation, beads were washed three times with the 1% NP-40 lysis buffer, eluted by boiling in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS–PAGE) loading buffer, and analyzed by Western blotting.
5.5. Co-immunoprecipitation assay
The co-immunoprecipitation (co-IP) was performed as described previously31. Briefly, cells were lysed with the Lysis buffer for 30 minutes at 4 °C. Then, cell lysates were collected and cleared by centrifugation at 14,000 RPM at 4 °C for 10 min. The cleared lysates (1.5–2 mg of total proteins) were mixed with MKK3 antibody (Abcam, catalog no. ab195037) or IgG control (Abcam, catalog no.ab172730), and incubated at 4 °C overnight with end-to-end rotation. The next day, the samples were combined with 20 μL of protein A/G-beads (Emd Millipore, catalog no. LSKMAGAG02) and incubated for 2 h at 4°C. Immunoprecipitates were washed three times with 500 μl of the Lysis buffer. The immunocomplexes were eluted with SDS-PAGE sample buffer and analyzed by Western blotting with indicated MKK3 and MYC-specific antibodies. To avoid the interference of MYC bands with the IgG heavy chain, the light chain-specific secondary antibody was used for the Western blotting.
5.6. Western Blot
Proteins in sample buffer were separated by 10% SDS polyacrylamide gel electrophoresis (10% acrylamide gels) and were transferred to nitrocellulose filter membranes at 100 V for 2 h at 4 °C. After blocking the membranes in 5% nonfat dry milk in TBST buffer (20mM Tris-base, 150mM NaCl, and 0.05% Tween 20) for 30 minutes to 1 hour at room temperature, membranes were blotted with the indicated antibodies overnight at 4 °C. Membranes were washed three times for 10 minutes in the TBST buffer. The SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo, catalog no. 34580) and Dura Extended Duration Substrate (Thermo, catalog no. 34076) were used for developing membranes. The western blot imaging was performed using the ChemiDoc imaging system (Bio-Rad).
5.7. TR-FRET Measurements
Test proteins were expressed for 48 h in HEK293T cells. Cell lysates were prepared in the 1% NP-40 lysis buffer (150 mM NaCl, 10 mM HEPES pH 7.5, 1% nonident P-40 (Sigma-Aldrich, IGEPAL catalog no. CA-630), 5 mM sodium pyrophosphate, 5 mM NaF, 2 mM sodium orthovanadate, 10 mg/l aprotinin, 10 mg/l leupeptin and 1 mM phenylmethylsulfonyl fluoride) for 30 min at 4 °C, followed by 15 min centrifugation at 14 000 r.p.m. at 4 °C. The TR-FRET assay was performed in the reaction buffer (20 mM Tris-HCl pH 7.0, 50 mM NaCl, 0.01% NP-40) in 384-well or 1536-well black plates. GST-Terbium- or Flag-d2-conjugated antibodies were used to couple GST- and VF-tagged proteins as FRET donors or acceptors, respectively.
By titrating the cell lysates, we have determined 0.1 mg/mL as the optimal concentration of cell lysates that can provide the highest signal/background ratio. Cell lysates were serially diluted in FRET buffer and mixed with anti-GST-d2 and anti-Flag-Tb antibodies with the final dilution of 1:500 and 1:750, respectively.
The plates were centrifuged at 1000 RPM for 2 minutes and samples were incubated at room temperature for the indicated times. The TR-FRET signal was measured on the BMG Labtech PHERAstar FSX reader. The following settings were used for the Tb/Venus pairs: Ex 337 nm, Em1: 520 nm, Em2: 486 nm; time delay: 50 μs). For the Tb/d2 pairs, the following settings were used: Ex 337 nm, Em1: 615 nm, Em2: 665 nm; time delay: 50 μs). The TR-FRET signal is expressed as the FRET ratio 520nm/486nm × 104 or 615nm/665nm × 104 for the Tb/Venus and Tb/d2 pairs, respectively.
5.8. Miniaturization of the Assay into a 1,536-Well uHTS Format
The TR-FRET assay for uHTS was performed in a black 1,536-well plate (Corning, catalog no. 3724). The reaction mixtures contained the optimal amount of GST-MKK3/VF-MYC co-expression lysate, anti-GST-d2, and anti-Flag-Tb antibodies. The VF-MYC lysate without GST-MKK3 co-expression was used as the background control. The reaction mixtures were dispensed to 1,536-well plates (5µL/well) using a multiple-drop Combi dispenser (Thermo, catalog no. 5840320). The TR-FRET signals were measured using a BMG Labtech PHERAstar FSX plate reader.
5.9. Pilot Screening for Potential MKK3-MYC Inhibitors through uHTS in a 1536-Well Format
The uHTS experiments were performed with the LOPAC, Spectrum, and Emory Enriched EEL libraries, containing 1280, 2000, and 2609 pharmacologically active compounds, respectively. The reaction mixture containing GST-MKK3/VF-MYC lysate mixed with the anti-GST-d2 and anti-Flag-Tb antibodies was dispensed into 1536-well black solid bottom plates (5µL/well). The library compounds dissolved in 100% dimethyl sulfoxide (DMSO) were added using pintool integrated with Beckman NX (Beckman Coulter), giving a final compound concentration of 10 µM. The DMSO served as vehicle control. After centrifugation and incubation at room temperature for 2 hours, TR-FRET signals were measured using a BMG Labtech PHERAstar FSX plate reader.
The MKK3-MYC PPI inhibition was determined based on the decreased TR-FRET signal as compared to DMSO control. The 30% inhibition threshold was used to identify the primary hits.
5.10. Dose-Response TR-FRET Validation for Compound Hits
The dose-response TR-FRET assays were performed in 384-well black solid bottom plates as described above. For the lysate-based assays, the lysates containing 40 µg/mL total protein were used. For the assay with recombinant purified proteins, GST-MKK3 and His-tagged MYC Helix-Loop-Helix domain were purified as described previously 31. The final concentrations of GST-MKK3 and His-MYC-HLH-LZ were 10nM and 1µM, respectively. The reaction mixture (30 µM/well) contained 20 µL of the lysate or purified protein mixture combined with anti-GST-d2 antibody, anti-Flag-Tb antibody, and compounds at 0 to 50µM concentration range. The reaction mixtures were incubated at room temperature for 2 hours, and TR-FRET signals were measured using a BMG Labtech PHERAstar FSX plate reader.
5.11. MYC reporter assay
The MYC transcription activity was measured using the MYC reporter assay as described previously31. The cells were grown in 6-well plates and transfected with Venus-Flag-MKK3 or Venus-Flag vector along with the Firefly luciferase reporter plasmid containing three MYC EBOX sites: GCCACGTGGCCACGTGGCCACGTGGC. The EBOX mutated that cannot be recognized by MYC (GCCTCGAGGCCTCGAGGCCTCGAGGC) was used as a negative control. Renilla luciferase expression vector served as a normalization control for protein expression. SGI-1027 or RG108 dissolved in DMSO was added to cells at 0.5 to 5 µM concentration 36 hours after transfection. DMSO was used in parallel as vehicle control. The final concentration of DMSO in cell culture media was 0.1% for all samples. Then, the cells were further incubated for 12 hours, harvested, and transferred to a 384-well plate (20 μL/well). MYC reporter assay was performed using Nano-Glo Dual-Luciferase Reporter Assay System (Promega, catalog no. N1610) following the manufacturer’s instructions. The normalized luminescence was calculated as a ratio of luminescence of Firefly luciferase to the luminescence of Renilla luciferase. The MYC activity was represented as a fold over control calculated as a ratio of the relative luminescence calculated for cells transfected with the EBOX wild type reporter to the relative luminescence calculated for cells transfected with EBOX mutant reporter plasmid.
5.12. Kinase assay
GST-MKK3 and His-MYC were purified as described previously 31. 125nM purified recombinant GST-MKK3 was mixed with 125nM His-tagged MYC or 125 nM inactive GST-p38 (positive control for MKK3 activity) purified from bacteria (Signal Chem, Cat # M39–14G-20. GST-MKK3, GST-p38, and His-MYC alone were used as negative controls for non-specific and auto-phosphorylation. The reactions were incubated with 50µM [32P]-ATP for 30 min at 37°C. The substrate phosphorylation was analyzed with the radiograph.
5.13. Scratch wound migration assay
HCT116 cells were seeded into 96-well ImageLock tissue culture plate (Essen BioScience, catalog no. 4379), 100 µL per well, and cultured until confluent. The Essen BioScience WoundMaker tool (Essen BioScience, catalog no. 4493) was used to create wounds following the manufacturer’s instructions. After wounding, the original medium was removed, and each well was gently washed with 100 µL of fresh medium. After the washing medium was removed, 100 µL of medium containing SGI-1027 at assigned concentrations or DMSO was added to each well. The assay plate was then placed into the IncuCyte S3 Live-Cell Analysis System, and repeated scanning was scheduled for every 2 hours using the IncuCyte Scratch Wound protocol.
5.14. Cell viability assay
Cells were seeded into 384-well flat clear bottom black tissue culture plate (Corning, catalog. no. 3764), 30 µL per well, and placed into the incubator. On the next day, 20 µL medium containing SGI-1027 at assigned concentrations or DMSO was added to each well, and put back into the incubator. After 72-hour incubation, 5 µL of CellTiter-Blue Cell Viability Assay reagent (Promega, catalog. no. G8081) was added to each well. After brief centrifugation, the plate was then put back for incubation at 37°C for 2 hours. Then, the plate was read by the BMG Labtech PHERAstar FSX plate reader to record fluorescence [560(20)EX/590(10)EM]. For each experiment, wells that contain medium and CellTiter-Blue without cells were used to quantify the background signal. The IC50 values were calculated using the GraphPad Prism software by fitting the four-parametric variable slope model (“non-linear regression, log (inhibitor) vs response - Variable slope (four parameters)” function).
5.15. Computational modeling
Molecular docking was performed using our previously reported model of MKK331. The structures of SGI-1027, Avasimibe, AT101, ATA and RG108 were prepared using Schrodinger LigPrep software. The molecular docking was performed using Schrodinger Glide software48. The grid box was generated around 20Å of the MKK3 121–135 residues, and the docking was performed using the Extra precision (XP) mode. The Epik state penalties, reward intramolecular hydrogen bonds and enhance planarity of conjugated pi groups as well as sample nitrogen inversions and sample ring conformations options were enabled. The binding energy was calculated using the MM-GBSA tool implemented in the Schrodinger Suite49.
5.16. Data analysis
All experiments were repeated at least three times. The data quantification was performed using the GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA, USA).
Supplementary Material
4. Acknowledgments
This research was supported in part by the National Cancer Institute of the NIH (Cancer Target Discovery and Development Network grants U01CA168449 and U01CA217875, H.F.), NCI Emory Lung Cancer SPORE (NIH P50CA217691) Career Enhancement Program awardee (A.A.I.), Emory initiative, Biological Discovery through Chemical Innovation (A.A.I), and Winship Cancer Institute (NIH 5P30CA138292).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
5 REFERENCES
- 1.Dang CV et al. The c-Myc target gene network. Semin Cancer Biol 16, 253–264, doi: 10.1016/j.semcancer.2006.07.014 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Patel JH, Loboda AP, Showe MK, Showe LC & McMahon SB Analysis of genomic targets reveals complex functions of MYC. Nature reviews. Cancer 4, 562–568, doi: 10.1038/nrc1393 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Gabay M, Li Y & Felsher DW MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harbor perspectives in medicine 4, doi: 10.1101/cshperspect.a014241 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alves Rde C, Meurer RT & Roehe AV MYC amplification is associated with poor survival in small cell lung cancer: a chromogenic in situ hybridization study. Journal of cancer research and clinical oncology 140, 2021–2025, doi: 10.1007/s00432-014-1769-1 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iwakawa R et al. MYC amplification as a prognostic marker of early-stage lung adenocarcinoma identified by whole genome copy number analysis. Clinical cancer research : an official journal of the American Association for Cancer Research 17, 1481–1489, doi: 10.1158/1078-0432.ccr-10-2484 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Wang X et al. Recurrent amplification of MYC and TNFRSF11B in 8q24 is associated with poor survival in patients with gastric cancer. Gastric cancer : official journal of the International Gastric Cancer Association and the Japanese Gastric Cancer Association 19, 116–127, doi: 10.1007/s10120-015-0467-2 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Lee HY et al. c-MYC Drives Breast Cancer Metastasis to the Brain, but Promotes Synthetic Lethality with TRAIL. Molecular cancer research : MCR 17, 544–554, doi: 10.1158/1541-7786.mcr-18-0630 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Seo AN et al. Clinicopathologic and prognostic significance of c-MYC copy number gain in lung adenocarcinomas. British journal of cancer 110, 2688–2699 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bragelmann J et al. Family matters: How MYC family oncogenes impact small cell lung cancer. Cell cycle (Georgetown, Tex.) 16, 1489–1498 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mollaoglu G et al. MYC Drives Progression of Small Cell Lung Cancer to a Variant Neuroendocrine Subtype with Vulnerability to Aurora Kinase Inhibition. Cancer Cell 31, 270–285, doi: 10.1016/j.ccell.2016.12.005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu DW et al. c-Myc suppresses microRNA-29b to promote tumor aggressiveness and poor outcomes in non-small cell lung cancer by targeting FHIT. Oncogene 34, 2072–2082, doi: 10.1038/onc.2014.152 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Chen H, Liu H & Qing G Targeting oncogenic Myc as a strategy for cancer treatment. Signal transduction and targeted therapy 3, 5, doi: 10.1038/s41392-018-0008-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Whitfield JR, Beaulieu ME & Soucek L Strategies to Inhibit Myc and Their Clinical Applicability. Front Cell Dev Biol 5, 10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Posternak V & Cole MD Strategically targeting MYC in cancer. F1000Research 5, doi: 10.12688/f1000research.7879.1 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dang CV MYC on the path to cancer. Cell 149, 22–35, doi: 10.1016/j.cell.2012.03.003 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schaub FX et al. Pan-cancer Alterations of the MYC Oncogene and Its Proximal Network across the Cancer Genome Atlas. Cell systems 6, 282–300e282 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sears RC The life cycle of C-myc: from synthesis to degradation. Cell cycle (Georgetown, Tex.) 3, 1133–1137 (2004). [PubMed] [Google Scholar]
- 18.Walhout AJ, Gubbels JM, Bernards R, van der Vliet PC & Timmers HT c-Myc/Max heterodimers bind cooperatively to the E-box sequences located in the first intron of the rat ornithine decarboxylase (ODC) gene. Nucleic acids research 25, 1493–1501 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nair SK & Burley SK X-ray structures of Myc-Max and Mad-Max recognizing DNA. Molecular bases of regulation by proto-oncogenic transcription factors. Cell 112, 193–205 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Sears R et al. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes & development 14, 2501–2514 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gregory MA, Qi Y & Hann SR Phosphorylation by glycogen synthase kinase-3 controls c-myc proteolysis and subnuclear localization. The Journal of biological chemistry 278, 51606–51612, doi: 10.1074/jbc.M310722200 (2003). [DOI] [PubMed] [Google Scholar]
- 22.Schieven GL The biology of p38 kinase: a central role in inflammation. Current topics in medicinal chemistry 5, 921–928 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Holand T et al. A role for mitogen kinase kinase 3 in pulmonary inflammation validated from a proteomic approach. Pulmonary pharmacology & therapeutics 27, 156–163, doi: 10.1016/j.pupt.2014.01.006 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Inoue T et al. Mitogen-activated protein kinase kinase 3 is a pivotal pathway regulating p38 activation in inflammatory arthritis. Proceedings of the National Academy of Sciences of the United States of America 103, 5484–5489, doi: 10.1073/pnas.0509188103 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kang Y et al. MAPK kinase 3 potentiates Chlamydia HSP60-induced inflammatory response through distinct activation of NF-kappaB. Journal of immunology (Baltimore, Md. : 1950) 191, 386–394, doi: 10.4049/jimmunol.1300481 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Kim EK & Choi EJ Pathological roles of MAPK signaling pathways in human diseases. Biochimica et biophysica acta 1802, 396–405, doi: 10.1016/j.bbadis.2009.12.009 (2010). [DOI] [PubMed] [Google Scholar]
- 27.Dong C, Davis RJ & Flavell RA MAP kinases in the immune response. Annual review of immunology 20, 55–72, doi: 10.1146/annurev.immunol.20.091301.131133 (2002). [DOI] [PubMed] [Google Scholar]
- 28.Zhang W & Liu HT MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell research 12, 9–18, doi: 10.1038/sj.cr.7290105 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Mo XL et al. AKT1, LKB1, and YAP1 revealed as MYC interactors with NanoLuc-based protein-fragment complementation assay. Molecular pharmacology 91, 339–347, doi: 10.1124/mol.116.107623 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Z et al. The OncoPPi network of cancer-focused protein-protein interactions to inform biological insights and therapeutic strategies. Nat Commun 8, 14356, 14310.11038/ncomms14356 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ivanov AA et al. OncoPPi-informed discovery of Mitogen-Activated Protein Kinase Kinase 3 as a novel binding partner of c-Myc. Oncogene 36, 5852–5860, doi:doi: 10.1038/onc.2017.180 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cargnello M & Roux PP Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiology and molecular biology reviews : MMBR 75, 50–83, doi: 10.1128/mmbr.00031-10 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiong J et al. Development of a Time-Resolved Fluorescence Resonance Energy Transfer Ultrahigh-Throughput Screening Assay for Targeting the NSD3 and MYC Interaction. Assay Drug Dev Technol 16, 96–106, doi: 10.1089/adt.2017.835 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Acker MG & Auld DS Considerations for the design and reporting of enzyme assays in high-throughput screening applications. Perspectives in Science 1, 56–73, doi: 10.1016/j.pisc.2013.12.001 (2014). [DOI] [Google Scholar]
- 35.Mo X et al. HTiP: High-Throughput Immunomodulator Phenotypic Screening Platform to Reveal IAP Antagonists as Anti-cancer Immune Enhancers. Cell chemical biology, doi: 10.1016/j.chembiol.2018.11.011 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gros C et al. New insights on the mechanism of quinoline-based DNA Methyltransferase inhibitors. The Journal of biological chemistry 290, 6293–6302, doi: 10.1074/jbc.M114.594671 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brueckner B et al. Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer research 65, 6305–6311, doi: 10.1158/0008-5472.CAN-04-2957 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Yochum GS et al. Serial analysis of chromatin occupancy identifies beta-catenin target genes in colorectal carcinoma cells. Proceedings of the National Academy of Sciences of the United States of America 104, 3324–3329, doi: 10.1073/pnas.0611576104 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou M, Yu X, Jing Z, Wu W & Lu C Overexpression of microRNA21 inhibits the growth and metastasis of melanoma cells by targeting MKK3. Molecular medicine reports 20, 1797–1807, doi: 10.3892/mmr.2019.10408 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun Y et al. MKK3 modulates JNK-dependent cell migration and invasion. Cell death & disease 10, 149, doi: 10.1038/s41419-019-1350-6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baldari S, Ubertini V, Garufi A, D’Orazi G & Bossi G Targeting MKK3 as a novel anticancer strategy: molecular mechanisms and therapeutical implications. Cell death & disease 6, e1621, doi: 10.1038/cddis.2014.591 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meskyte EM, Keskas S & Ciribilli Y MYC as a Multifaceted Regulator of Tumor Microenvironment Leading to Metastasis. Int J Mol Sci 21, doi: 10.3390/ijms21207710 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valente S et al. Selective non-nucleoside inhibitors of human DNA methyltransferases active in cancer including in cancer stem cells. Journal of medicinal chemistry 57, 701–713, doi: 10.1021/jm4012627 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang X et al. High expression of MKK3 is associated with worse clinical outcomes in African American breast cancer patients. Journal of translational medicine 18, 334, doi: 10.1186/s12967-020-02502-w (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Datta J et al. A new class of quinoline-based DNA hypomethylating agents reactivates tumor suppressor genes by blocking DNA methyltransferase 1 activity and inducing its degradation. Cancer research 69, 4277–4285, doi: 10.1158/0008-5472.CAN-08-3669 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 46.Garcia-Dominguez P, Dell’aversana C, Alvarez R, Altucci L & de Lera AR Synthetic approaches to DNMT inhibitor SGI-1027 and effects on the U937 leukemia cell line. Bioorganic & medicinal chemistry letters 23, 1631–1635, doi: 10.1016/j.bmcl.2013.01.085 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Sun N, Zhang J, Zhang C, Zhao B & Jiao A DNMTs inhibitor SGI-1027 induces apoptosis in Huh7 human hepatocellular carcinoma cells. Oncology letters 16, 5799–5806, doi: 10.3892/ol.2018.9390 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Friesner RA et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. Journal of medicinal chemistry 49, 6177–6196, doi: 10.1021/jm051256o (2006). [DOI] [PubMed] [Google Scholar]
- 49.Greenidge PA, Kramer C, Mozziconacci JC & Sherman W Improving docking results via reranking of ensembles of ligand poses in multiple X-ray protein conformations with MM-GBSA. Journal of chemical information and modeling 54, 2697–2717, doi: 10.1021/ci5003735 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.