

Abstract
COVID-19, a type of viral pneumonia caused by severe acute respiratory syndrome coronavirus 2 has challenged the world as global pandemic. It has marked the identification of third generation of extremely pathogenic zoonotic coronaviruses of twenty-first century posing threat to humans and mainly targeting the lower respiratory tract. In this review, we focused on not only the structure and virology of SARS-COV-2 but have discussed in detail the molecular immunopathogenesis of this novel virus highlighting its interaction with immune system and the role of compromised or dysregulated immune response towards disease severity. We attempted to correlate the crosstalk between unregulated inflammatory outcomes with disrupted host immunity which may play a potential role towards fatal acute respiratory distress syndrome that claims to be life-threatening in COVID-19. Exploration and investigation of molecular host-virus interactions will provide a better understanding on the mechanism of fatal COVID-19 infection and also enlighten the escape routes from the same.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00284-021-02657-9.
Introduction
The inception of January 2020 marked a global outbreak of novel Coronavirus with multifarious cases of human infections being reported from December 2019 that was closely related to the South China Sea Food City Market in Hunan province of Wuhan. With respective confirmation of 2019-nCOV infections by Thailand, Japan and Korea on 13th, 16th and 20th January [1], this human infection bestrewed rapidly as World Health Organization (WHO) announced on January 31st, 2020 as COVID-19 to be listed as Public Health Emergency of International Concern (PHEIC) indicating its risk to affect multiple countries thus necessitating properly coordinated International response [2]. On February 11, 2020, WHO declared pneumonia caused by novel coronavirus as COVID-19 with simultaneous announcement from the International Virus Classification Commission regarding the emerging coronavirus to be termed as severe acute respiratory syndrome coronavirus 2 (SARS-COV-2) [3]. COVID-19 was successively proclaimed a pandemic by WHO on March 11, 2020 [4]. On the basis of recent advancement in COVID-19 research as well as the knowledge gathered from various studies on SARS-COV and MERS-COV, we attempted to review and compile the literature explaining virology, transmission, possible interactions of SARS-COV-2 with target host-cells as well as its molecular pathophysiology. We have also focused in detail on information about probable innate and adaptive host response towards SARS-COV-2 along with the molecular immunopathogenesis and inflammatory response in disease progression in case of COVID-19 pandemic.
Virology and Structure of SARS-COV-2
Being a member of the Betacoronavirus genus, SARS-COV-2 shares 50% similarity with MERS-COV sequence [5] and 79% genetic resemblance with SARS-COV [6]. However, RaTG13 (bat CoV) has been observed to be a 98% match [3] while pangolin coronavirus also possesses high sequence similarity with that of SARS-COV-2 [7].
Coronaviruses are typically enveloped and non-segmented. These single stranded (positive sense) RNA viruses are characterized by largest possible (26–32 kilobases) RNA viral genome. The nucleocapsid of virion is comprised of genomic RNA together with nucleocapsid proteins (N proteins) concealed within bilayers composed of phospholipids. It is covered by either spike glycoprotein trimmer (S) which is common in all CoVs or Hemagglutinin esterase (HE) existing in certain CoVs. S protein of viral envelop house among them the transmembrane glycoprotein (type III) known as membrane protein and also the envelope proteins (E). The distinctive crown like appearance fetches their name [8]. Ectodomain of viral S protein consists of a receptor-binding unit (S1) and a membrane fusion subunit (S2). The S1 subunit comprises of the amino-terminal domain and domain for receptor binding (RBD) which in SARS-COV specifically spans between 318 and 510 amino-acid residues [9]. The S2 subunit on the other hand is made up of one fusion peptide domain and 2 heptapads (HR 1 and 2) repeated regions [10]. The genome sequence of SARS-COV-2 have minimum of 10 Open reading frames (ORFs) with translation of ORF1a/b (the initial ORFs) that constitute for approx. two thirds viral RNA in two big polyproteins. These two polyproteins in (pp1a and pp1b) are committed in processing for non-structural proteins (Nsp1-16) which then contribute in producing the Replicase transcriptase complex (RTC) of virus [5]. ORFs, rep1a, rep1b (replication genes), 5′-UUUAAAC-3′ (the slippery sequence) and pp1a along with pp1ab (polyproteins) are the critical requirements for viral replication. The Rough endoplasmic reticulum (RER) derived membranes are further rearranged by these Nsps to form vesicles (double-membered) that assist viral replication along with transcription [11]. While Nsp15 plays a major role in coordinating viral replication and also facilitates attacking host immune system [12], the RCT complex is assembled by Nsp 1/2, 2/3 and 3/4 which helps in creating an optimum environment within body of host that augments synthesis of viral RNA along with its replication. Nsp12 encoding for RNA-dependent RNA polymerase (RdRP), RNA helicase domain together with RNA 5′-triphosphase encrypted by Nsp13 and exoribonuclease (ExoN) encoded Nsp14 are responsible for replication conformity while Nsp16 is known for encoding the activity of 2′-O-methyltransferase.The remaining SARS-COV-2 ORFs belonging on the other side of the genome (one-third) are responsible for encoding the 4 crucial structural proteins namely spike (S), nucleocapsid (N), envelop (E), and membrane (M) in addition to multiple accessory proteins having unrecognized functions with negative participation in replication process of this zoonotic virus.
Another discriminating trait of SARS-COV-2 is the presence of S protein consisting of furin-like cleavage zone [13]. Furin and ADAM-17/10 play a major role in promoting the cellular entry of SARS-COV-2 [14]. Transcriptional induction of furin being dependent on NOTCH [15], regulating NOTCH mediated furin activity also modulates viral entry into host-cells. Apart from furin affiliated pre-cleavage, TMPRSS2 (cell serine protease) is also necessary for proper processing of S-proteins and thus facilitates SARS-COV-2 entry inside host-cells [16].
Immune Pathogenesis During COVID-19 Infection
Any viral infection triggers a reverse host immunity response. Immune responses of individuals towards pathogens are known to vary depending on genetic diversities. Thus, an impaired immune system triggers propagation of the virus especially to the ACE2 rich organs such as lungs, kidney, intestine, heart causing massive damage to tissues. Lungs suffer from innate inflammation due to these damaged cells mediated primarily by proinflammatory factors like granulocytes and macrophages. This unrestrained immune response triggers lung inflammation with correlated detrimental pulmonary tissue, function impediment and diminished capacity of lungs insinuate life-threatening severe respiratory malfunction characterizing the last stage of COVID-19 [17]. Additionally, chemotactic factors which regulates leukocyte positions as well as dilations in host lungs are crucial for immunity in response to viral infection. Thus, spectral modifications in chemotactic factors often contributes to majorly imbalanced immune system. The host immune response thus acts as a double edged sword as its suppression or misdirection provokes replication of virus leading to tissue destruction while hyperactive immune effects impose threat of immunopathological situation [8] (Fig. 1).
Fig. 1.

Immune response (innate and adaptive) in SARS-COV-2 infection. Following entry of the virus mediated by spike (S) protein, the genomic viral RNA is released in cytoplasm and the viral replication may initiates immune response after identification of this dsRNA or ssRNA. The sensitized TLRs trigger a cascade of signaling that activates IRFs and NF-κB leading to IFN (type I) and proinflammatory cytokines production. IFN contributes majorly in increasing the secretion of antiviral proteins that helps in protecting the uninfected cells. On the other hand, CoV infection activates the macrophages and the APC primes T cells by displaying the CoV antigens, causing T cells activation. CD4 + and CD8 + T cells in humoral immunity participates in cytokine production for viral suppression. However, vigorous production of such mediators owing to persistence of virus negatively impact NK cells and CD8 + T cells activation. Moreover, T helper cells (Th17) results in secretion of an array of interferons and interleukins. On the other hand, Th2 activates B cells in humoral immunity for producing antibodies. ssRNA single stranded RNA, dsRNA double stranded RNA, TLR toll like receptors, IRF interferon receptor factor, APC antigen presenting cells, IFN interferons, NF-kB nuclear factor kappa light chain enhancer of activated B cells, TNF tumor necrosis factor, Th T helper cells, TIRAP toll/interleukin-1 receptor domain-containing adapter protein, MCP1:macrophage inflammatory protein, MAPK mitogen activated protein kinase, TRAF TNF receptor-associated factor, IL interleukins, IRKA4 interleukin-1 receptor-associated kinase 4, MyD88 myeloid differentiation primary response 8, JAK-STAT Janus kinase- signal transducers and activators of transcription
Antigen Presentation and HLA-Haplotypes in Relation to SARS-COV-2
Succeeding entry inside the host cells, viral antigens are subjected to the Antigen presentation cells (APC) which is a critical mechanism of antiviral host immunity. Major histocompatibility complex (MHC) and HLA (in humans) offer antigenic peptides. MHC loci (HLA) serves to be a prototypical component governing gene-related susceptibility for infections [18]. The CD4 + and CD8 + T cells consist receptors for T-cell antigens which efficiently identify conformation of antigen binding groove in association with related antigenic peptides. Thus, susceptibility to distinct diseases depends on varying HLA haplotypes [17]. This implies the importance to comprehend antigen presentation during SARS-COV-2 where previous knowledge from SARS and MERS-COV infections may be helpful. In SARS-COV, antigen presentation is majorly controlled by MHC-I [19]. While HLA-B*4601, HLA-B*0703, HLA-DR B1*1202 and HLA-Cw*0801 [20] are the polymorphisms which dictate SARS-COV vulnerability, the alleles HLA-DR0301, HLA-Cw1502 and HLA-A*0201 ensures protective functions [21]. Moreover, Mannose-binding lectin (MBL) polymorphism concurrent with Ag presentation also intensifies SARS-COV risk [22]. On the other-hand, MHCII alleles HLA-DRB1*11:01 and HLA-DQB1*02:0 confer in MERS-COV infections [23]. In cases of SARS-COV-2 patients, decrement in HLA-DR expression has been observed recently which may be related to exponentiation of hyper-inflammatory conditions [24]. Studies have pointed out the minimal potency of HLA-B*46:01 allele for presenting SARS-CoV-2 peptides resulting in enhanced susceptibility for COVID-19 infection while HLA-B*15:03 which can successfully present conserved epitopes of SARS-CoV-2 and may offer a better immune response [25].
Innate Immunity Responses During SARS-COV-2: Role of IFN1 and DC
Innate immune system comprising of conserved defense mechanism plays a pivotal role in recognizing and restricting pathogenic invasion and successively leads to adaptive immune reaction. Effective antiviral innate immunity is potentially dependent on interferon (IFN) activity and its downstream effector cascade that regulates replication of virus and effectuate successful adaptive immunity [3]. IFNs are responsible for diversified biological activities such as antiviral, immunomodulatory, antiproliferation etc. and thus potentiates the immune system in multiple ways [26]. Although all the three types of IFNs; IFNαβ (type I), IFNγ (Type II), IFNλ (Type III) account for protecting against coronavirus infections [27], type I IFN plays the predominant role of primarily in the early infection phases. Following infection, the genomic RNA (ssRNA, dsRNA) are identified by identified by Toll like receptors (TLRS) like TLR3/7 (endosomal), TLR4 or cytosolic RNA sensors [MDA5 (melanoma differentiation-associated protein 5)/Retinoic acid inducible gene1 (RIG1)] [28]. These Pattern recognition receptors (PRR) trigger interlinked signaling pathways for inducing robust IFN response. Mitochondrial antiviral signaling proteins (MAVS) are activated by MDA5/RIG1 which activates TNF receptor-associated factor 3 (TRAF3), TRAF family member-associated NF-kB activator binding kinase 1 (TBK1) and inhibitors of NF-κB kinases ɛ (IKK) that sequentially phosphorylates Interferon regulatory factor 3 (IRF3/7) and nuclear factor kappa light chain enhancer of activated B cells (NF-κB) leading to successive induction of type I IFNs [29]. The activated Type I IFN using interferon-α/β receptor (IFNAR) consecutively actuate JAK/STAT mechanistic pathway involving phosphorylation of signal transducer and activator of transcription (STAT1/2) by kinases, janus kinase (JAK) and tyrosine kinase 2 (TYK2). Phosphorylated STAT1/2 complexed IRF9 are translocated into the nucleus that results in IFN-stimulated genes (ISGs) transcription regulated by promoters bearing ISRE (IFN-stimulated response element) (Figs. 1, 2). The ISGs play essential antiviral role in innate immunity system by limiting entry as well as replication of virus within the host cells [30]. However, the heterogeneous response [31] of type I IFNs and its complex regulations emphasize the importance of critical understanding of IFN1 response dynamics in patients suffering from COVID-19.
Fig. 2.
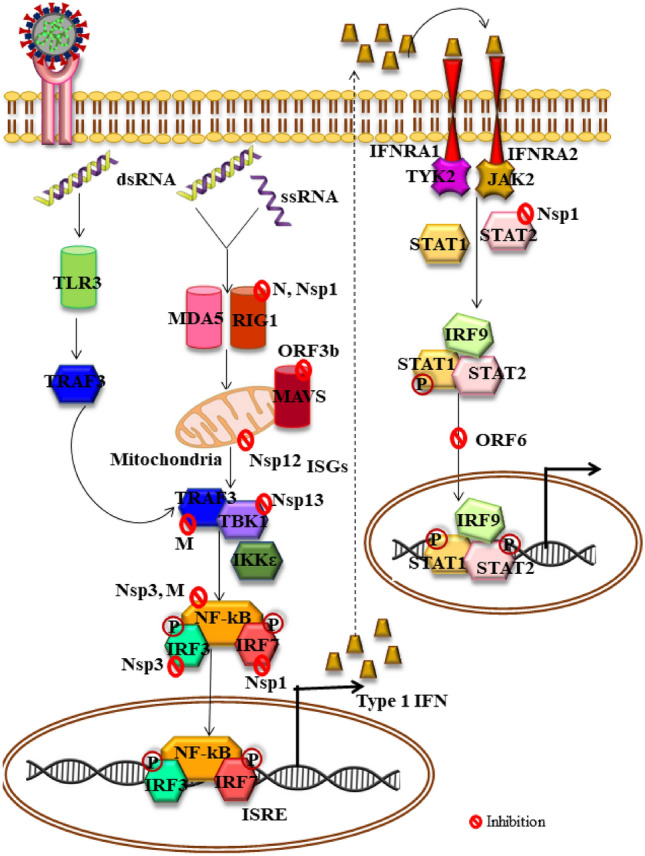
IFN signaling pathway in alarmed innate immunity and its evasion by SARS-COV-2 virus. The virus generally invades permissible cells via ACE2 receptors causing infection. The viral RNA after being recognized by TLR3 or MDA5/RIG1, triggers signaling pathways engaging TRAF3, TBK1, IKɛK which phosphorylates and activates IRFs and NF-κB leading to stimulation of the type I IFN. Sensitization of IFNAR by IFN upregulates the expression of ISGs and proteins that potentially contribute in anti-viral functions. However, various structural and non-structural SARS-COV-2 proteins along with accessory proteins have been identified that manipulate the IFN signaling and cause multi-pronged inhibition which suppresses IFN signaling increasing viral replication and severity. IFN interferon, TLR yoll like receptor, ACE2 angiotensin converting enzyme 2, IFNAR interferon-alpha/beta receptor alpha chain, TYK2 tyrosine kinase 2, TBK1 TANK-binding kinase 1, TRAF: ORF open reading frames of SARS-CoV-2, IKKε IkB kinase ε, STAT signal transducers and activators of transcription, RIG-I-like receptor dsRNA helicase enzyme, MDA5 melanoma differentiation-associated, ISG interferon-stimulated genes, ISRE: ORF open reading frames, Nsp non-structural proteins, NF-kB nuclear factor kappa light chain enhancer of activated B cells, TNF receptor-associated factor, PRR pattern recognition receptors, PAMP pathogen-associated molecular patterns
In-vitro COVID 19 infection models have revealed induction of low type I/type II IFNs only, which subsequently triggered average ISG levels and specific cytokines [interleukins (IL-1β, IL-6, TNF (Tumor necrosis factor)] as well as chemokines like CCL20, CXCL1/2/3/5/6 and 16 [32]. Early clinical studies showed either absence of type I IFNs (specially IFNβ) regardless the severity of the infection or presence of IFNα at marginal lower levels in plasma of severely ill COVID-19 patients. Successive studies further Successive studies further displayed that Peripheral blood mononuclear cells (PBMCs) did not express type I IFN encoding genes in patients with COVID-19 infection while an early transient expression of ISGs was observed in immune cells derived from blood correlating a premature IFNα (probably lung origin) burst [33]. sc-RNA-Seq also illustrated that monocytes and other cells of innate antiviral system demonstrated similar early and transitory ISG expression [34]. Moreover, patients suffering from mild or moderate COVID-19 infections revealed an early peak of IFNα/λ which subsequently declined while their levels significantly escalated in severely ill individuals during the second week [35]. Such clinical type I IFN dynamics corroborate with preclinical SARS-COV-2 murine models that unfolded the role of IFNs in significantly driving pathologic responses in-vivo but not in controlling replication of COVID-19 virus [36].
SARS-COV-2 has been found to deploy multipronged evasion strategies for antagonizing IFN system leading to clinical consequences of compromised type I IFN response in early studies of COVID-19 infection [28, 32, 33]. The interferon pathway can be subverted by the SARS-COV-2 virus in any one of the following stages of (a) Aversion of PRR identification of viral-RNA [37], (b) MAVS (mitochondrial antiviral-signaling protein) as well as 3/6 (TNF receptor-associated factor) ubiquitination and degeneration with inhibited nuclear transfer of IRF3 [38], (c) Blocking STAT1 mediated downstream IFN transduction cascade [39], (d) PRR signal network precluded via TANK Binding Kinase 1 (TBK1) or Inhibitor of nuclear factor-κB kinase subunit-ε (IKKε), TRAF3 or IRF3 inhibitors [37]. Other escape strategies are also currently under consideration. Till now, atleast 10 viral proteins have been recognized to interfere and manipulate the antiviral response of IFNs [40]. Some of them are depicted in Fig. 2. Thus, various evidences support the ability of SARS-COV-2 in targeting multiple phases of IFN response system which majorly deteriorate the well-orchestrated crosstalk between antiviral as well as proinflammatory mechanisms of innate and adoptive immune system.
The Dendritic cells (DC), recognized as professional Antigen presenting cells (APC) are key regulators for initiating and modulating innate as well as adaptive antiviral immune response. They can mainly be categorized as conventional DC (cDC; type 1 and 2) and plasmacytoid DC cells (pDC) [41]. In human, cDC1 and cDC2 represent CD141 + and cDC + respectively. pDCs distributed throughout the lungs are essential for type I IFN production and are thus crucial for primary response against viral infection. The DCs contribute to innate immunity via the synthesis of immunity enhancing cytokines and innate lymphocyte mobilization [natural killer (NK cells), γσT and NKT cells]. DCs exhibit efficient migration ability while constitutively expressing MHCII and other co-stimulatory molecules and are thus considered as predictors for anti-pathogenic immune response. Certain DCs are also reported to activate CD8 + T cells via MHCI through cross presentation process and thereby harbor innate immune response to adaptive response [42]. The immature DCs triggered by antigens generate mature DCs that produce a spectrum of cytokines such as IL-12,4,10, 1β along with TNF (α/β/γ). Hence, the pivotal role of DCs in inducing cytokine production, antigen presentation and specific T-cell priming signifies delayed antipathogenic immune response when they are compromised as in COVID-19 condition [43].
Previous studies have shown the potential of SARS-COV-2 to infect DCs which suppress the expression the expression of antiviral cytokines while upregulating macrophage inflammatory protein 1α (MIP1α) and other inflammatory chemokines [44]. Moreover, SARS-COV also tends to accelerate the DC mediated generation of pro-inflammatory IL-6/12 cytokines in response to secondary stimulation signals that further induced degenerative inflammatory responses [45]. In addition, pDCs are also reported for secreting higher amounts of IFNs following CoVs infections [46].
However, DCs appears to be targeted directly by SARS-COV-2 (Fig. 3). While expression of ACE2 by DCs in the interstitial lungs indicate chances of direct infection by COVID-19 virus, CD147 expression by DCs further supports this fact [47]. Recent investigations have displayed diminished levels of circulating CD1c + , CD141 + and pCDs in COVID 19 patients. Only CDc + was found to accumulate in lungs among the three DC subsets suggesting an overall decreased level [48]. While acute and convalescent COVID-19 patients revealed evident decrement of DCs in blood associated with impaired functions, significant rise in cdC: pDCs was observed in severely ill patients. Such observations can be correlated with reduced IFN production and early decline of innate immunity against the infection. Another study recently confirmed entry of virus in DCs and macrophages via DC-SIGN and furin. The increased expression of this DC-SIGN (Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin), a lectin (C-type) present on DCs and macrophages may be associated with the severity of COVID 19 pathogenesis in older patients [49].
Fig. 3.

Defective dendric cells activities in SARS-COV-2 infection. The covid-19 virus can directly infect DCs which may affect both innate and adaptive immunity. SARS-COV-2 infected DCs often inhibits maturation of pDCs and consecutively antagonizes IFN type I production on one hand while compromising T cell priming due to reduced MHC molecules leading to T cell lymphopenias. Overall maladjusted DC and its subset function results in increased viral load proinflammatory condition that aggravates the severity of the infection. DC dendritic cells, TNF tumor necrosis factor, IL interleukins, IFN interferons, MHC major histocompatibility complex, DC-SIGN dendritic cell-specific intercellular adhesion molecule-3-Grabbing Non-integrin, pDC plasmacytoid dendritic cells, cDC conventional dendric cells, TCR T cell receptor
Thus, SARS-COV-2 may infect immature DCs and inhibit their maturation which successively leads to repressed type I IFN signaling and T-cell mediated adaptive responses. Also, the viral infection depicts reduced number of overall DCs (altering ratio of CDc11 + to CD123 +), suppression of maturation related factors (CD86), co-stimulatory molecules expression, attenuation of STAT1 reliant IFN1 response and switching into a proinflammatory phenotype [50] (Fig. 3). Such effects intercede with adaptive immunity causing maladjusted T cell functions which reduces viral clearance and potentiates complication of COVID 19.
Thus, the evidences reveal that innate host immunity performs a decisive role in influencing either protective or calamitous responses thereby opening a wide stretched window mediating immune intervention. Active replication of virus at later stage stimulates IFN type1 over production which favors macrophage, neutrophil infiltration that are considered as important sources of cytokines (pro-inflammatory). COVID-19 patients manifesting similar lymphocyte as well as neutrophil alterations suggest probable delayed IFN1 induction with lack of early-stage viral containment. Individuals suffering from cardiovascular disease or hypertension or diabetes are found to be more prone to COVID-19 [51]. Such co-morbidities increase the chances of susceptibility due to dysfunctional or damaged immune system that fails in successful pathogen annihilation and thus paves to disease pathology.
Adaptive Immune Responses During SARS-COV-2
Role of T-Cell Subpopulations on COVID -19 Patients
Viral clearance majorly relies on T cells. As the overall adaptive immunity is orchestrated by Th cells, direct killing of virus attacked cells largely depends on cytotoxic T cells (Fig. 1). While cytotoxic CD8 + T lymphocytes are interlinked with secretion of wide range of molecules such as IFN-γ, perforin, granzymes etc. for viral eradication from host system, CD4 + Th cells provide assistance and priming to both T and B cells improving their ability in pathogen clearance. Anti-viral adaptive immunity is dominantly expedited by Th1 type reaction. Response of T cells is also harmonized by APC generated cytokine microenvironment. Generally, 7 days following the symptoms occurrence, T-cells and B-cells response becomes prominent in the blood of COVID 19 patients. Reports have indicated that the lungs of afflicted COVID-19 patients exhibited accumulated mononuclear (T-cells/monocytes) cells with simultaneous decrement in peripheral blood T-cells (hyperactivated) levels thus causing lymphopenia and implying regulatory effect of T cells inundated from blood to infected areas [52] against infection. In case of MERS-COV, containment of disease has been effectuated by cross reactive response from T cells [53] with CD4 + T cells bearing more susceptibility. It was seen in SARS-COV that depleted CD8 + Tcells do no effect or detain replication of virus during the infection [54]. Apoptosis of T cells in MERS-COV infection has been reported. Similarly, in SARS-COV protein, cytosolic (C-terminal) domain comprises a unique BH3-alike region for its attachment to Bcl-XL that triggers the commencement of apoptosis of T cells [55]. Anti-viral T cells deficiency in the late phase of infection may lead to its protraction and assist survival of the virus [56]. Diminution of CD4 + T cells is intertwined with reduction in neutralizing antibody (NAb) and lymphocyte engagement with additional cytokine production that is concomitant with immune directed pneumonitis (interstitial) and retarded pulmonary SARS-COV clearance [57]. Inflamed infection site recruits by neutrophils and monocytes via IL-17 that leads to incitement of other chemokines and cytokines like TNFβ, IL-6, IL-21, IL-1, IL-8 and macrophage inflammatory protein 1 (MCP-1) [58]. When compared to the mild and moderately affected group, the patients facing severe viral attack demonstrated T-cells mainly memory phenotype with prominent upsurged frequency of TNFα, IL-2, IFNγ (belonging to polyfunctional, CD4 + T-cells) together with TNFα, IFNγ and degranulated state (CD8 + Tcells types). NAb titres were escalated remarkably in response to dominant T cells expression in the severe groups of patients who illustrated enhanced serum levels of IL-10, IL-4, IL-5 (Th2) cytokines [59]. MERS-COV infection severity is consistent with early increment in CD8 + Tcells while dominancy of Th1 cells is evident in the convalescent phase [60]. As neutrophils are known for its degenerative activity in any type of infection, the contribution of Th17 in coronavirus pathogenesis, whether preventive or detrimental should be keenly investigated. The standard activation of T cells is hindered by IL-6 that may explain the apparent lymphopenia [61]. In severe cases of COVID-19 patients, there occurs an apparent decrease in number of CD4 + Tcells, CD8 + T cells, NK cells and B-cells in peripheral blood cells. This declined CD4 + T-cells and CD8 + T-cells count is associated with concurrent drop in TNF-α and IL-6 thus indicating a negative correlation between these factors [62]. However, those patients responding to treatment demonstrates an increase in in CD8 + T cells and B cells. As the prodromal condition in SARS-COV-2 infection deteriorates into symptomatic phase, patients manifests gradually increasing subpopulation of exhausted or terminally differentiated CD8 + T cells with higher expression of receptors Programmed cell death protein 1 (PD1), T Cell immunoglobulin mucin 3 (TIM3), Lymphocyte-activation gene 3 (LAG3), NKG2A, Cytotoxic T-lymphocyte-associated protein 4 (CTLA4) and CD39. Increased expression of these inhibitory receptors might be an outcome of either T cell exhaustion or its extensive activation. SARS-COV-2 spike protein specific CD4 + Tcells associated with a Th1 cytokine profile is observed in cases of acute infection. Although the response of Th2 cells are greatly altered in severe COVID 19 conditions, they retain their normal response in mild symptomatic patients [63]. Also, patients suffering from severe conditions of SARS-COV-2 infection showed higher percentage of CD45RA + naïve T cells while CD3 + naïve T cells and CD45RO + memory T cells count was lower [51].
B Cell Responses in SARS-COV-2 Infection
As already mentioned, B-cells activity becomes apparent in concurrence of follicular helper T cells (Tfh) approximately a week following onset of symptoms. Recent reports indicate majority of COVID-19 patients exhibits development of IgG and/or IgM antibodies shortly after the viral infection. Various clinical studies have pointed out the duration of these antibodies production (approximately 4–22 days) may vary from patient to patient [64], and elevated IgG/IgM titres are generally associated with the severe cases. It has also been accounted that 2 weeks following disease onset, majority of patients showed development of anti-RBD IgG/IgM antibodies while anti NP-IgG/IgM antibodies were detected in comparatively smaller population of infected patients. The co-existence of virus specific IgG Abs in patients along with SARS-COV-2 virus for a long period of time may indicate that viroid clearance may not depend on such Abs [65].
Although the functional prospects of antibody mediated protection are not clarified till now, however, viral neutralization by Abs is considered to be one primary mechanistic approach. Previously in SARS and MERS-COV infections, Neutralizing antibodies (NAbs) are detected that block spike protein RBD interaction with their receptor. NAbs target primarily S-protein RBD122 of SARS-COV that constitute amino acid spanning region 318–510 which undergoes independent association with ACE2 host-receptor target [9]. It has been noticed that except a few, most monoclonal Abs identified against SAR-COV in previous studies fail to attach or cause SARS-COV-2 neutralization [66]. A possible explanation for such aberrant behavior may be the discrepancy of SARS-COV-2 RBDS with that of SARS-COV. Particularly, only 15 of the key residues contained in 33 amino acid in region between 460 and 492 essential for ACE2 binding in SARS-COV is conserved in SARS-COV-2 [67]. Nevertheless, a positive result of SARS-COV-2 pseudo-virus cross neutralization which has been possible in anti-sera of mouse produced against protein of SARS-COV suggests the presence of epitopes overlapping within these viruses [14]. COVID-19 patients have also revealed neutralization efficacy of anti-SARS-COV-2 spike RBD Abs. Thus, these RBD Abs mainly found in patients recovering from COVID-19 may extend temporary protection against reinfection [92]. In SARS-COV-2 disease, polyclonal responses are evident that mainly involves rapid class switching to IgG and sometimes to IgA isotypes. Moreover, somatic hypermutation in responding clones are also observed closely after onset of infection [68].
Complement Activation and Its Association with COV Infections
As seen from earlier reports, host-immune reaction is largely synchronized by complement system against CoV pathogenesis. Complement system is responsible for allowing detection by innate immunity and evoke corresponding protective response against foreign Ags [69]. With “dual edged sword” characteristics, complement system is essential for immune reaction. Stimulation of immune cells is known to be mediated by C3a and C5a (anaphylatoxins) leading to miscellaneous cytokine release. Cytolytic complement (terminal) C5b-9 and C3b, C5b fragments complex is generated by stimulated complement cascade [70]. Prostaglandin E2 (PGE2), leukotrienes and thromboxane B2 [71] are certain metabolites of arachidonic acid which are induced by these peptides that further recruits and activates eosinophils, neutrophils and monocytes provoking synthesis of wide range of cytokines along with pro-inflammatory mediators [72]. Inflammatory activities triggered by cytokines are coupled with fighting potential of innate immune response against virus. However, complement moderated incitement of host innate immunity should be finely adjusted since uncontrolled complement activation proves to be fatal and culminates into either inflammation or Disseminated intravascular coagulation (DIC), cell-death followed by immune-paralysis with subsequent multiple organ failure and even death.
Complement activation is dependent on either classical (CP), lectin (LP) or alternative (AP) pathway [73] (Fig. 4). LP involves binding of MBL (mannan-binding lectin, ficolins) with carbohydrate arrays belonging to mannan and N-acetylglucosamine residues located on either viral surface or pathogen infected cell-surface that in turn stimulates MBL-associated serine protease-2 (MASP-2), a MBL related protease acknowledged for direct initiation of complement system cascade [74] (Fig. 4). Recently, the N protein of SARS-COV-2 has been observed to activate MASP2 mediated LP [75] which may escalate microvascular injury and thrombosis in severely ill patients [76]. While AP gets stimulated by S proteins, advanced SARS-COV-2 infection may also trigger CP via C-reactive protein (CRP) or immune complexes [75].
Fig. 4.

Dysregulated complement cascade in SARS-COV-2 infection: SARS-COV-2 has been found to directly activate MASP2 and induce LP via N proteins. While S proteins activates the CP, AP is also stimulated in the advanced stages. Sequential aberrant activation of C3 compliment fraction promotes secretion of anaphylatoxins C3a and C5a. This anaphylatoxin along with C5B-9 may trigger potentiate cytokine storm, leukocyte activation and their infiltration in alveolar cells of lungs. Thereby dysregulated complement system further deteriorates the pathologic condition elevating the severity. CP classical pathway, AP alternative pathway, LP Lectin pathway, N protein nucleocapsid protein, S protein spike protein, MASP2: MBL Mannan-binding lectin serine protease 1, MBL Mannan-binding lectin, C3a/C5a complement activators C5/C3, MAC membrane attack complex, GM-CSF IL interleukins, TNF tumor necrosis factor, INF interferons
C5a (complement factor) recognized as the strongest peptide of the complement cascade is believed to play a major role in promoting inflammation during COVID-19 immunopathogenesis. Both C5a and C3a are known to induce secretion of proinflammatory cytokines like TNFα, IL-6, IL-1 from activated macrophages. The dexterity of COVID 19 infection is thus correlated with C5a concentration and macrophages with high C5a expression are detected in pulmonary lesions isolated from patient expired of COVID 19. C5b-9, the terminal product of complement system activated NF-κB, MCP1 and activator protein 1 (AP1) with subsequent liberation of IL-6 from smooth (vascular) muscle cells. Various evidences support the capability of C5 and C5b-9 in inculcating tissue damage in COVID-19 infection. Moreover, severe conditions have revealed histological accumulation of C4, MAC (membrane attack complex) which is interconnected with conditions like endotheliitis and microthrombi [77]. Thus, complement cascade may lead to procoagulant conditions and microvascular damage in advanced COVID 29 cases [78].
NOTCH Pathway: A Close Co-Ordination Between Adaptive and Innate Immunity
In addition to previous discussions, efficacy of innate as well as adaptive immunity is believed to be closely coordinated by NOTCH pathway. In COVID-19 affected patients, lack of balance between Th1 and Th2 cells evokes cytokine storm that brings about myocardial and pulmonary injury [79]. Polarization of macrophage (M1) is critically induced by Dll4/NOTCH pathway as both in-vitro and in-vivo data typically correlated NOTCH inhibition with direct reduction in inflammation [80]. IFN-γ modulates direct association of NOTCH1 and IL-6 promoter in macrophages thus causing positive regulation of IL-6 synthesis [81]. This IL-6 then subsequently augment NOTCH ligand (Dll4) expression that finally causes NOTCH signal supplementation with establishment of a loop of positive feedback thereby promoting further IL-6 generation [82] (Fig. 1). Alternatively, in Th1 cells, DII4 triggered Notch pathway enhances Th1/Th17 (proinflammatory) cytokines, while activated Th17 by IL-6 is suppressed by Jagged [80]. However, further extensive investigations considering NOTCH signaling and immune response in case of SARS-COV-2 are necessary.
Inflammatory Immunopathogenesis
A malfunctioning immune system provoking cytokine storm that is potentially involved with inflammation of lungs. Lethal cases of both SARS and MERS-COV have depicted consistent increment in neutrophil infiltration together with monocytes and macrophages [83]. Also, overexpression of IFN1 with myeloid cells influx are the main causes of pulmonary dysfunction which have negative impact on disease outcome. As per speculation, delay in IFN1 production fails to contain virus in early stage of both SARS and MERS-COV disease that conferred in above-mentioned inflammatory cells infiltration. As the immune (innate) cells build-up, it has destructive role in body of affected host causing pneumonia or ARDS and analogous lung immunopathology [84]. COVID-19 patients in ICU were specifically reported to show intense plasma levels of IP-10, IL-10, IL-2, IL-7, Granulocyte colony-stimulating factor (G-CSF), Tumor necrosis factor (TNF), MCP1, and MIP1α which are associated with cytokine release syndrome [51]. Studies also revealed that with the progress of pathogenesis CRP and IL-6 concentration were elevated especially in patients who failed to survive with respect to COVID-19 survivors. Higher IL-6 concentration is often associated with severity of disease [85]. In case of SARS-COV infection, the negative feedback mechanism regulator of IL-6, Suppressor of cytokine signaling 3 (SOC3) is present in much higher levels suggesting extensive stimulation of IL-6 [86]. In fatal rather than milder COVID 19 afflicted patients, population of FCN1 + macrophage (from inflammatory monocyte) has also been reported to be intensively present in aspirated bronchoalveolar fluid [87] along with peripheral blood showing elevated CD14 + CD16 + monocytes (inflammatory) percentage [88]. These cells then participate in the secretion of MIP1α, MCP1 and IP-10 that contributes in advancement of the cytokine storm as discussed earlier. Moreover, rise in TNF levels along with other proinflammatory cytokines often cause septic shock and leads to tissue degeneration in diversified organs like liver, heart (leading to myocardial failure), kidney in addition to the lethal respiratory failure that may culminate into multi-organ failure in the critically ill patients. Lymph node necrosis in deceased patients with signs of splenic atrophy also indicates the prominent catastrophic outcome caused by unregulated immune response [89]. In this context, during viral attack, immune (innate) response favored liberation of cytokines results in neuroendocrine system stimulation which is involved in impairment of immune reaction mediated by various peptides as well as glucocorticoid release. So, this concept opens a new window of exploration in which the relation between SARS-COV-2 and inflammatory neuroendocrine (pathological) mediators release should be thoroughly investigated along with its role in imparting ARDS or other respiratory fatalities [89]. Apart from lung cells, SARS-COV has already been reported to attack other cellular targets as its presence was observed in DCs derived from monocytes [90], T-lymphocyte cells and also macrophages. While direct lymphocyte assassination may be the contributing factor behind the lymphopenia occurring in patients, immune cells (macrophages/ monocytes) attacked by pathogens cause abnormalities in cytokines generation [91]. However, the proximity with which SARS-COV-2 infects these cells still remains poorly defined.
The pulmonary cell damage caused by direct viral attack may also be aggravated by unchecked and intensive flux of inflammatory cells that result in protease secretion together with Reactive oxygen species (ROS) in high proportion. This condition steers not only diffuse alveolar tissue degradation but also causes alveolar cell desquamation, pulmonary edema along with hyaline membrane production [92] leading to constrained gaseous exchange capability of lungs which explain the breathing difficulties and decreased oxygen concentration in blood characterizing ARDS that accounts for 70% death in severe SARS-COV-2 infection. Such conditions also make the lungs impuissant to secondary infections [93]. During SARS-COV pathogenesis, there are defects and dysregulation in formation of hyaluronan (HA) which is in turn concurrent with ARDS [94] because of its high potency of water absorption (× 1000) over molecular weight. IL-1, TNF (inflammatory, cytokines) which are present in increased proportion in COVID-19 patients’ lungs are involved in dominant induction of HA-synthase-2 (HSA2) in fibroblasts, CD31 positive endothelium, EpCAM positive pulmonary alveolar (epithelial) cells [95]. Thereby modification of HA production may be advantageous in to relieve patients suffering from ARDS in COVID-19.
Inflammation Caused by SARS-COV-Induced ACE2 Downregulation and Shedding
The role of ACE2 in mediating lung damage in SARS-COV-2 has been widely reported. Downregulation of ACE2 by S-protein [96] along with active (catalytically) ectodomain of ACE2 shedding are related as characteristic features of the infection [97]. Disintegrin in association with metalloprotease-17/TNFα-cleavage enzyme (ADAM-17/TACE) accelerates cleaving of ACE2 with subsequent shedding in epithelial tissue of airway cells after cellular entry of virus to produce soluble, active (enzymatically) sACE2 that is released in extramembrane region [98]. ACE2 in general accounts for inhibiting the detrimental outcome of Angiotensin II (ANGII) binding to Angiotensin 1 receptor (AT1R) which results in aggravated inflammation, vasoconstriction and thrombosis. ACE2 deficiency in SARS-COV-2 thus results in ANGII accumulation in serum [99] and its unopposed effects through ACE2/ANGII/ATIR axis. ANGII via AT1R is known for stimulating inflammatory responses through activation of NF-κB which increases transcription of IL-1/6, TNFα, IL-1β, adhesive molecules [E and P selectin, Intracellular adhesion factor (ICAM), Vascular cell adhesion molecule 1 (VCAM)], various chemokines and proinflammatory factors as well [100]. In case of SARS-COV-2such functions of ANGII has been consistent. Moreover, elevated TGFβ expression as seen in COVID-19 patients by ANGII may trigger the formation of Th17 cells which in turn produces GM-CSF, IL-21, IL-17 etc. and leads to further release of diverse proinflammatory cytokines and chemokines [101]. Augmented ANGII/ATIR activation causes upregulation of ADAM17 and subsequent IL-6-sIL-6R complex trans-signaling drives STAT3 activation in pulmonary epithelial cells. This simultaneous activation of STAT3 along with NF-κB hyperactivates NF-κB stimulation mechanism via IL-6 amplifier (AMP) in lung which instigates cytokine storm followed by ARDS in severely in COVID 19 patients [102]. Recent preclinical and clinical studies have also demonstrated that attachment of ANGII to ATIR facilitates further internalization of ACE2 via ERK1/2 and MAPK signaling thereby suppressing ACE2 [103]. Thus, ACE2 repression by SARS-COV-2 leads to hyperinflammatory state due to dysregulated ACE/ANGII/AT1R axis and its maladjusted balance with ACE2/MasR axis in addition to upregulated complement system (C5a and C5b-9) components [104] (Fig. 5). Alveolar injury, high vascular penetrability of lungs, amplified lung-edema, conglomerated neutrophils and compromised pulmonary functions [105] reported in the infected conditions are consistent outcome of the reduced variant of ACE2.
Fig. 5.

ACE2 downregulation and shedding in SARS-COV-2 infection mediates dysregulated ACE2/ANGII/ATIR axis. Virus mediated imbalance between ACE2/ANGII/ATIR and ACE2/ANG(1–7)/MasR axis results in stimulation of MAPK, NF-κB, TGFβ and complement cascade which mediates proinflammatory response. Moreover, ANGII further stimulates ADAM17 which causes shedding of ACE2 and directs formation of IL-6-Sil-6R complex which activates STAT3. This mechanism hyperactivates NF-κB signaling with results in IL-6 amplifier precipitating cytokine storm and leads to ARDS. ANGII angiotensin II, AT1R angiotensin 1 receptor, ACE2 angiotensin converting enzyme 2, STAT3 signal transducers and activators of transcription 3, TGFβ tumor growth factorβ, MAPK mitogen activated protein kinase, IL interleukins, VCAM vascular cell adhesion molecule 1, ICAM intercellular adhesion molecule 1, MCP1 macrophage inflammatory protein 1, NF-kB nuclear factor kappa light chain enhancer of activated B cells, IL-6-sIL-6R interleukin 6-soluble interleukin 6 receptor, GM-CSF granulocyte–macrophage colony-stimulating factor, Th T helper cells, ARDS acute respiratory disease syndrome, C5a/C3a complement activators 5a/3a, ADAM17 A disintegrin and metalloprotease 17
SARS-COV-2 Replication and Cellular Degradation Directed Inflammation
Earlier onset of rapidly extending viral replication may be the causative factor of substantial apoptosis of endothelial as well as epithelial cells along with vascular leakage that results in prompt emancipation of pluralistic chemokines and cytokines. As mentioned before, SARS-COV-2 pathology is integrated with pyroptosis causing ability in lymphocytes/macrophages [106]. Since peripheral blood lymphopenia has been perceived as a frequent phenomenon in SARS-COV-2 contaminated patients (82.1%), it also suggests the possible interlink with infiltrating pulmonary lymphocytes or apoptosis/pyroptosis evoked cellular damage [51]. Pyroptosis (Pyro: Fire; ptosis: falling) is a rapid inflammatory programmed cell assassination pathway which involves the inflammatory caspases 1,4,5. The host cell employs such proteases to contain different pathogenic infections. Following viral infection, activation of NLRP3 inflammosome stimulates caspase 1, which in turn cleaves gasdermin-D (GSDMD), a member of gasdermin family leading to perforation and permeability of cell membrane followed by cell lysis and death. This process of pyroptosis causes leakage of various molecules which triggers local cell reaction and inflammatory response [107]. After the translation of viral proteins in host cell, SARS-COV-2 involves formation ORF3a proteins which encodes calcium ion channel. Following this, its interaction with TRAF3 sequentially triggers NF-kB pathway transcription and effectuates pro-IL-IB gene transcription [108] on one hand and attune inflammasome complex (NLRP3-ACS-Caspase1) recruitment on the other hand (Supplementary Fig. 1). A secondary impulse including calcium ion ingression, caspase invigoration, ROS generation or even mitochondrial abrasion results in pro-IL-1B transmutation into IL-1B [109]. As IL-1B is liberated from macrophages, it may confer in cellular pyroptosis initiation leading to cytokine synthesis and release of proinflammatory constituents in bulk [110]. Apart from ORF3a, NLRP3 conciliated inflammosome pathway is also initialized by ORF8b protein which is longer in case of SARS-COV-2 (Supplementary Fig. 1). Intriguingly, these SARS-COV viruses also depict conservation of E-protein producing ion-channel which corresponds to cytokine hyperproduction via inflammasome (NLRP3) nexus [111]. Moreover, SARS-COV-2 is tracked to bind to GSDMD and thus inhibit its caspase mediated cleavage. Combination of all these mechanistic pathways succumbs to the cytokine storm contributing to typical COVID-19 respiratory suffering.
Inflammatory Responses Induced by Anti-Spike IgG (Anti-S-IgG) and Possible ADE
In some pathogenic cases, specific antibodies when binds to the surface proteins of virus, accelerates viral invasion into certain host cells thereby promoting infection. This condition known as ADE happens via two distinct molecular mechanism involving either enhanced infection or elevated activation of immune system. In the first case the NAbs or sub-neutralizing promotes FcɣRIIa (Fc gamma receptor IIa) directed viral endocytosis, increasing infection of monocytes and macrophages. This has been commonly reported in dengue virus infection. Second ADE mechanism reveals the capability of Fc mediated Nabs in initiating a powerful cascade of immune response that enhances respiratory distress. The immune complex formed by Nabs in conjugation with antigens leads to secretion of an array of cytokines (pro-inflammatory), activates complement system and also locally recruits immune cells within lungs. The consequent inflammation may effectuate obstruction of airways resulting in ARDS in severely ill COVID 19 patients [112].
Earlier in-vivo studies regarding SARS-COV demonstrated extensive lung tissue injury caused due to alteration in inflammatory reaction by anti-S viral protein (neutralizing) antibodies [113]. An array of studies on animal models clearly denoted that although virus was suppressed, the residing anti-S protein Ab during acute phase of SARS-COV infection can precipitate acute pulmonary deterioration that tends to prevail until the upcoming later phases. It is considered that occurrence of ADE of pathogen infection happens in individuals where early and trivial antiviral NAbs fail in complete neutralization of the virus. Rather the complexed virus-Nab binds to FcR (Fc receptor) that facilitates endocytosis of virus and target host cell infection. This cascade of events is finally linked with upsurge in overall viral replication and enhanced disease severity. And such events may also explain the probable reason for certain patient subpopulation developing early titres of NAbs being more prone to consistent inflammation, ARDS and demise while others can successfully surpass the inflammation and survive [114].
Although the exact technique of governing inflammation along with pulmonary tissue injury by anti-S (neutralizing) Ab remains vaguely understood, S-IgG presence preceding clearance of virus may propose altering mechanistic polarization of macrophages (alveolar) in critically infected macaques. Besides, anti-S-IgG accelerates cumulation of monocytes and macrophages (proinflammatory factors) along with IL-8 and MCP-1 production. Such pro-inflammatory feedback reactions are supposed to occur when viral-anti-S-IgG complex binds FcR expressed on surface of either macrophages or monocytes [113]. Another probability that warrants further validation is the activation of classical complement pathway by such anti-S-IgG complex that may subsequently effectuate cellular destruction. Involvement of Antibody-dependent cell-mediated cytotoxicity (ADCC) is also considered. Thus, this phenomenon indicates the role of anti-S-IgG complex interacting complex with FcR in augmenting and accelerating replication of virus and inflammatory outcomes in host patients’ lungs [114].
Assuming mutual similarities regarding inflammatory reaction in SARS-COV and SARS-COV-2, the outbreak of inflammation may be therefore categorized into two phases, primary response that becomes apparent at early stage after the viral infection followed by secondary reactions that are characterized by NAbs presence.
Conclusion
Conclusively current observations imply the adaptability of coronaviruses for manipulation and evasion of immune scrutinization as well as suppression of host immune reaction that may probably contribute to their extended incubation period by absconding detection by human immune system during early infection phase. Belonging to Betacoronavirus genus, mechanism of COVID 19 immune circumvention play an important role and may potentially be identical with SARS and MERS-CoV. This host immune escape mechanism thus largely relies on (a) Repression of immune (innate) responses, especially mediated by blocking the prompt expression of IFN-1 and also retarding virus identification and signaling primarily by hindering phosphorylation of STAT-1. Viral proteins such as membrane (M) or nonstructural (NS) proteins (e.g., NS4a, NS4b, NS15) participate as chief molecules in modulation host immune feedback [115] (b) Evading adaptive immunity by truncating MHC I/II mediated antigen presentation orchestrated by the virus by infecting macrophage/Dendritic cells (DCs) which significantly depleted active T cells [116] (c) immune system exhaustion through extensive and detained IFN1 generation via plasmacytoid dendritic cells (pDCs) supervise the third defensive technique maneuvering fatal lung immunopathology and (d) “cytokine storm” that potentially debilitate immune system of host due to IFN-1-directed apoptosis of T-cells. In addition to these, a recent study has proposed a hypothesis that depletion of cellular adenosine triphosphate (c-ATP) may confer into possible immune cell dysfunction as ATP is known to be involved in advocating IFN production, IFN signaling as well as preventing cytokine storm. Keeping these in mind, along with further exploration and investigation of molecular host-virus interactions will assist to enlighten the escape routes from the fatal COVID-19 and accelerate the development of immunoregulation strategies and also effective COVID-19 interventions (supplementary Fig. 2).
Supplementary Information
Below is the link to the electronic supplementary material.
Author Contributions
SPP had the idea for the article. PB and SPP performed the literature research. PB and PS wrote the manuscript. SPP, PB and PS critically revised the work. All authors read and approved the final manuscript.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or non-profit sectors.
Declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Novel Coronavirus (2019) Situation report‐2 [Published online ahead of print January 21, 2020]. https://www.who.int/docs/default‐source/coronaviruse/situationreports/20200122‐sitrep‐2‐2019‐ncov.pdf
- 2.Li X, Geng M, Peng Y, Meng L, Lu S. Molecular immune pathogenesis and diagnosis of COVID-19. J Pharm Anal. 2020;10:102–108. doi: 10.1016/j.jpha.2020.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sarzi-Puttini P, Giorgi V, Sirotti S, Marotto D, Ardizzone S, Rizzardini G, Antinori S, Galli M. COVID-19, cytokines and immunosuppression: what can we learn from severe acute respiratory syndrome? Clin Exp Rheumatol. 2020;38(2):337–342. [PubMed] [Google Scholar]
- 5.Fehr AR, Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol Biol. 2015;1282:1–23. doi: 10.1007/978-1-4939-2438-7_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gorbalenya A, Baker S, Baric R, et al. Coronaviridae study group of the international committee on taxonomy of viruses the species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. 2020;5(4):536. doi: 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS-CoV-2. Nat Med. 2020;26(4):450–452. doi: 10.1038/s41591-020-0820-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li G, FanY LY, Han T, Li Z, Zhou P, et al. Coronavirus infections and immune responses. J Med Virol. 2020;92(4):424–432. doi: 10.1002/jmv.25685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xiao X, Chakraborti S, Dimitrov AS, Gramatikoff K, Dimitrov DS. The SARS-CoV S glycoprotein: expression and functional characterization. Biochem Biophys Res Commun. 2003;312(4):1159–1164. doi: 10.1016/j.bbrc.2003.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu S, Xiao G, Chen Y, He Y, Niu J, He Y, Niu J, Escalante CR, et al. Interaction between heptad repeat 1 and 2 regions in spike protein of SARS-associated coronavirus: implications for virus fusogenic mechanism and identification of fusion inhibitors. Lancet. 2004;363(9413):938–947. doi: 10.1016/S0140-6736(04)15788-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Masters PS. The molecular biology of coronaviruses. Adv Virus Res. 2006 doi: 10.1016/S0065-3527(08)60286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim Y, Jedrzejczak R, Maltseva NI, Wilamowski M, Endres M, Godzik A, et al. Crystal structure of Nsp15 endoribonuclease NendoU from SARS-CoV-2. Protein Sci. 2020;29(7):1596–1605. doi: 10.1002/pro.3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coutard B, Valle C, de Lamballerie X, Canard B, Seidah NG, Decroly E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res. 2020;176:104742. doi: 10.1016/j.antiviral.2020.104742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181(2):281–292. doi: 10.1128/MCB.00116-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qiu H, Tang X, Ma J, Shaverdashvili K, Zhang K, Bedogni B. Notch1 autoactivation via transcriptional regulation of furin, which sustains Notch1 signaling by processing Notch1-activating proteases ADAM10 and membrane type 1 matrix metalloproteinase. Mol Cell Biol. 2015;35:3622–3632. doi: 10.1128/MCB.00116-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmieder RE. Mechanisms for the clinical benefits of angiotensin II receptor blockers. Am J Hypertension. 2005;18(5):720–730. doi: 10.1016/j.amjhyper.2004.11.032. [DOI] [PubMed] [Google Scholar]
- 17.Shi Y, Wang Y, Shao C, Huang J, Gan J, Huang X, et al. COVID-19 infection: the perspectives on immune responses. Cell Death Differ. 2020;27:1451–1454. doi: 10.1038/s41418-020-0530-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pobezinskaya YL, Kim YS, Choksi S. The function of TRADD in signaling through tumor necrosis factor receptor 1 and TRIFdependent Toll-like receptors. Nat Immunol. 2008;9(9):1047–1054. doi: 10.1038/ni.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu J, Wu P, Gao F, Qi J, Kawana-Tachikawa A, Xie J, et al. Novel immunodominant peptide presentation strategy: a featured HLA-A* 2402-restricted cytotoxic T-lymphocyte epitope stabilized by intrachain hydrogen bonds from severe acute respiratory syndrome coronavirus nucleocapsid protein. J Virol. 2010;84(22):11849–11857. doi: 10.1128/JVI.01464-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen YM, Liang SY, Shih YP, Chen CY, Lee YM, Chang L, et al. Epidemiological and genetic correlates of severe acute respiratory syndrome coronavirus infection in the hospital with the highest nosocomial infection rate in Taiwan in 2003. J Clin Microbiol. 2006;44(2):359–365. doi: 10.1128/JCM.44.2.359-365.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang SF, Chen KH, Chen M, Li WY, Chen YJ, Tsao CH, et al. Human-leukocyte antigen class I Cw 1502 and class II DR 0301 genotypes are associated with resistance to severe acute respiratory syndrome (SARS) infection. Viral Immunol. 2011;24(5):421–426. doi: 10.1089/vim.2011.0024. [DOI] [PubMed] [Google Scholar]
- 22.Tu X, Chong WP, Zhai Y, Zhang H, Zhang F, Wang S, et al. Functional polymorphisms of the CCL2 and MBL genes cumulatively increase susceptibility to severe acute respiratory syndrome coronavirus infection. J Infect. 2015;71(1):101–109. doi: 10.1016/j.jinf.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hajeer AH, Balkhy H, Johani S, Yousef MZ, Arabi Y. Association of human leukocyte antigen class II alleles with severe middle east respiratory syndrome-coronavirus infection. Ann Thorac Med. 2016;11(3):211. doi: 10.4103/1817-1737.185756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giamarellos-bourboulis EJ, Netea MG, Rovina N, Koulouris N, Gogos C, Koutsoukou A, et al. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe. 2020;27:1–9. doi: 10.1016/j.chom.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tomita Y, Ikeda T, Sato R, Sakagami T. Association between HLA gene polymorphisms and mortality of COVID-19: an insilico analysis. Immunity Inflammat Dis. 2020 doi: 10.1002/iid3.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang BX, Fish EN. Global virus outbreaks: interferons as 1st responders. Semin Immunol. 2019;43:101300. doi: 10.1016/j.smim.2019.101300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fensterl V, Chattopadhyay S, Sen GC. No love lost between viruses and interferons. Annu Rev Virol. 2015;2(1):549–572. doi: 10.1146/annurev-virology-100114-055249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sa Ribero M, Jouvenet N, Dreux M, Nisole S. Interplay between SARS-CoV-2 and the type I interferon response. PLoS Pathog. 2020;16(7):e1008737. doi: 10.1371/journal.ppat.1008737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loo YM, Gale M., Jr Immune signaling by RIG-I-like receptors. Immunity. 2011;34:680–692. doi: 10.1016/j.immuni.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ziegler CGK, Allon SJ, Nyquist SK, Mbano I, Miao VN, Cao Y, et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is enriched in specific cell subsets across tissues. Cell. 2020;181(5):1016–1035. doi: 10.1016/j.cell.2020.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patil S, Fribourg M, Ge Y, Batish M, Tyagi S, Hayot F, et al. Single-cell analysis shows that paracrine signaling by first responder cells shapes the interferon-b response to viral infection. Sci Signal. 2015;8(363):16. doi: 10.1126/scisignal.2005728. [DOI] [PubMed] [Google Scholar]
- 32.Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Møller R. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181:1036–1045.e9. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arunachalam PS, Wimmers F, Mok CKP, Perera RAPM, Scott M, et al. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science. 2020;369:1210–1220. doi: 10.1126/science.abc6261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schulte-Schrepping J, Reusch N, Paclik D, Babler K, Schlickeiser S, et al. Deutsche COVID-19 OMICS initiative (DeCOI). Severe COVID-19 is marked by a dysregulated myeloid cell compartment. Cell. 2020;182:1419–1440.e23. doi: 10.1016/j.cell.2020.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lucas C, Wong P, Klein J, Castro TBR, Silva J, Sundaram M, et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature. 2020;584:463–469. doi: 10.1038/s41586-020-2588-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Israelow B, Song E, Mao T, Lu P, Meir A, Liu F, et al. Mouse model of SARS-CoV-2 reveals inflammatory role of type I interferon signaling. J Exp Med. 2020;217(12):e20201241. doi: 10.1084/jem.20201241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Siu KL, Chan CP, Kok KH, Jin DY. Suppression of innate antiviral response by severe acute respiratory syndrome coronavirus M protein is mediated through the first transmembrane domain. Cell Mol Immunol. 2014;11(2):141–149. doi: 10.1038/cmi.2013.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kindler E, Thiel V, Weber F. Interaction of SARS and MERS coronaviruses with the antiviral interferon response. Adv Virus Res. 2016;96:219–243. doi: 10.1016/bs.aivir.2016.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frieman M, Yount B, Heise M, Kopecky-Bromberg SA, Palese P, Baric RS. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J Virol. 2007;81:9812–9824. doi: 10.1128/JVI.01012-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schultze JL, Aschenbrenner AC. COVID-19 and the human innate immune system. Cell. 2021;184:1–22. doi: 10.1016/j.cell.2021.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Villadangos JA, Schnorrer P. Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo. Nat Rev Immunol. 2007;7(7):543–555. doi: 10.1038/nri2103. [DOI] [PubMed] [Google Scholar]
- 42.Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. 2013;31:563–604. doi: 10.1146/annurev-immunol-020711-074950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rao VUS, Arakeri G, Subash A, Rao J, Jadhav S, Suhail Sayeed M, et al. COVID-19: loss of bridging between innate and adaptive immunity? Med Hypotheses. 2020;144:109861. doi: 10.1016/j.mehy.2020.109861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Law HKW, Cheung CY, Ng HY, Sia SF, Chan YO, Luk W, et al. Chemokine up-regulation in SARS-coronavirus-infected, monocyte-derived human dendritic cells. Blood. 2005;106(7):2366–2374. doi: 10.1182/blood-2004-10-4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tseng C-TK, Perrone LA, Zhu H, Makino S, Peters CJ. Severe acute respiratory syndrome and the innate immune responses: modulation of effector cell function without productive infection. J Immunol. 2005;174:7977. doi: 10.4049/jimmunol.174.12.7977. [DOI] [PubMed] [Google Scholar]
- 46.Cervantes-Barragan L, Züst R, Weber F, Spiegel M, Lang KS, Akira S, et al. Control of coronavirus infection through plasmacytoid dendritic-cell– derived type I interferon. Blood. 2006;109:1131–1137. doi: 10.1182/blood-2006-05-023770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bertram S, Heurich A, Lavender H, Gierer S, Danisch S, Perin P, et al. Influenza and SARS-coronavirus activating proteases TMPRSS2 and HAT are expressed at multiple sites in human respiratory and gastrointestinal tracts. PLoS ONE. 2012;7(4):e35876. doi: 10.1371/journal.pone.0035876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sanchez-Cerrillo I, Landete P, Aldave B, Sanchez-Alonso S, Azofra AS, Marcos-Jimenez A, et al. Differential redistribution of activated monocyte and dendritic cell subsets to the lung associates with severity of COVID-19. MedRxiv. 2020 doi: 10.1101/2020.05.13.20100925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Borges RC, Hohmann MS, Borghi SM. Dendritic cells in COVID-19 immunopathogenesis: insights for a possible role in determining disease outcome. Int Rev Immunol. 2021;40(1–2):108–125. doi: 10.1080/08830185.2020.1844195. [DOI] [PubMed] [Google Scholar]
- 50.Yang D, Chu H, Hou Y, Chai Y, Shuai H, Lee AC-Y, et al. Attenuated interferon and proinflammatory response in SARS-CoV-2- infected human dendritic cells is associated with viral antagonism of STAT1 phosphorylation. J Infect Dis. 2020;222(5):734–745. doi: 10.1093/infdis/jiaa356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cui W, Fan Y, Wu W, Zhang F, Wang JY, Ni AP. Expression of lymphocytes and lymphocyte subsets in patients with severe acute respiratory syndrome. Clin Infect Dis. 2003;37(6):857–859. doi: 10.1086/378587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pascal KE, Coleman CM, Mujica AO, Kamat V, Badithe A, Fairhurst J, et al. Pre-and postexposure efficacy of fully human antibodies against Spike protein in a novel humanized mouse model of MERS-CoV infection. Proc Nat Acad Sci. 2015;112(28):8738–8743. doi: 10.1128/JVI.01505-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Channappanavar R, Fett C, Zhao J, Meyerholz DK, Perlman S. Virus specific memory CD8 T cells provide substantial protection from lethal severe acute respiratory syndrome coronavirus infection. J Virol. 2014;88(19):11034–11044. doi: 10.1128/JVI.01505-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang Y, Xiong Z, Zhang S, Yan Y, Nguyen J, Ng B, et al. Bcl-xL inhibits T-cell apoptosis induced by expression of SARS coronavirus E protein in the absence of growth factors. Biochem J. 2005;392(1):135–143. doi: 10.1042/BJ20050698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mubarak A, Alturaiki W, Hemida MG. Middle east respiratory syndrome coronavirus (MERS-CoV): infection, immunological response, and vaccine development. J Immunl Res. 2019 doi: 10.1155/2019/6491738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen J, Lau YF, Lamirande EW, Paddock CD, Bartlett JH, Zaki SR, et al. Cellular immune responses to severe acute respiratory syndrome coronavirus (SARS-CoV) infection in senescent BALB/c mice: CD4+ T cells are important in control of SARS-CoV infection. J Virol. 2010;84(3):1289–1301. doi: 10.1128/JVI.01281-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dutzan N, Abusleme L. T helper 17 cells as pathogenic drivers of periodontitis. Adv Exp Med Biol. 2019;1197:107–117. doi: 10.1007/978-3-030-28524-1_9. [DOI] [PubMed] [Google Scholar]
- 59.Li CK, Wu H, Yan H, Ma S, Wang L, Zhang M, et al. T cell responses to whole SARS coronavirus in humans. J Immunol. 2008;181(8):5490–5500. doi: 10.4049/jimmunol.181.8.5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shin HS, Kim Y, Kim G, Lee JY, Jeong I, Joh JS, et al. Immune responses to middle east respiratory syndrome coronavirus during the acute and convalescent phases of human infection. Clin Infect Dis. 2019;68(6):984–992. doi: 10.3389/fimmu.2020.00827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX. Clinical characteristics of coronavirus disease 2019 in China. N Eng J Med. 2020;382(18):1708–1720. doi: 10.1056/NEJMoa2002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19) Front Immunol. 2020;11:827. doi: 10.3389/fimmu.2020.00827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Laing AG, Lorenc A, Del Barrio IDM, Das A, Fish M, Monin L (2020) A consensus Covid-19 immune signature combines immuno-protection with discrete sepsis-like traits associated with poor prognosis. medRxiv.
- 64.Long QX, Liu BZ, Deng HJ, Wu GC, Deng K, Chen YK, et al. Antibody responses to SARS-CoV-2 in patients with COVID-19. Nat Med. 2020;26:845–848. doi: 10.1038/s41591-020-0897-1. [DOI] [PubMed] [Google Scholar]
- 65.Wang B, Wang L, Kong X, Geng J, Xiao D, Ma C, et al. Longterm coexistence of SARS-CoV-2 with antibody response in COVID-19 patients. J Med Virol. 2020;92:1684–1689. doi: 10.1002/jmv.25946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang C, Li W, Drabek D, Okba NM, van Haperen R, Osterhaus AD, et al. A human monoclonal antibody blocking SARS-CoV-2 infection. Nat Commun. 2020;11(1):1–6. doi: 10.1038/s41467-020-16256-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li F, Li W, Farzan M, Harrison SC. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. 2005;309(5742):1864–1868. doi: 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- 68.Nielsen SC, Yang F, Hoh RA, Jackson KJ, Roeltgen K, Lee JY, et al. B cell clonal expansion and convergent antibody responses to SARS-CoV-2. Cell Host Microbe. 2020;28:516–525. doi: 10.1016/j.chom.2020.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Baker S, Kessler E, Darville-Bowleg L, Merchant M. Different mechanisms of serum complement activation in the plasma of common (Chelydraserpentina) and alligator (Macrochelystemminckii) snapping turtles. PLoS ONE. 2019;14(6):0217626. doi: 10.1371/journal.pone.0217626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cole DS, Morgan BP. Beyond lysis: how complement influences cell fate. Clin Sci. 2003;104(5):455–466. doi: 10.1042/CS20020362. [DOI] [PubMed] [Google Scholar]
- 71.Imagawa DK, Osifchin NE, Paznekas WA, Shin ML, Mayer MM. Consequences of cell membrane attack by complement: release of arachidonate and formation of inflammatory derivatives. Proc Nat Acad Sci. 1983;80(21):6647–6651. doi: 10.1093/cvr/cvn277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sánchez-Galán E, Gómez-Hernández A, Vidal C, Martín-Ventura JL, Blanco-Colio LM, et al. Leukotriene B4 enhances the activity of nuclear factor-κB pathway through BLT1 and BLT2 receptors in atherosclerosis. Cardiovasc Res. 2009;81(1):216–225. doi: 10.1093/cvr/cvn277. [DOI] [PubMed] [Google Scholar]
- 73.Wills-Karp M. Complement activation pathways: a bridge between innate and adaptive immune responses in asthma. Proc Am Thorac Soc. 2007;4(3):247–251. doi: 10.1513/pats.200704-046AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wallis R. Interactions between mannose-binding lectin and MASPs during complement activation by the lectin pathway. Immunobiol. 2007;212(4–5):289–299. doi: 10.1016/j.imbio.2006.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gao T, Hu M, Zhang X, Li H, Zhu L, Liu H, et al. Highly pathogenic coronavirus N protein aggravates lung injury by MASP-2- mediated complement over-activation. Infect Dis. 2020 doi: 10.1101/2020.03.29.20041962. [DOI] [Google Scholar]
- 76.Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res. 2020;220:1–3. doi: 10.1016/j.trsl.2020.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Carvelli J, Demaria O, Vély F, Batista L, Benmansour NC, Fares J, et al. Association of COVID-19 inflammation with activation of the C5a–C5aR1 axis. Nature. 2020;588:146–150. doi: 10.1038/s41586-020-2600-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Benmansour NC, Carvelli J, Vivier E. Complement cascade in severe forms of COVID-19: recent advances in therapy. Eur J Immunol. 2021;19:1–8. doi: 10.1002/eji.202048959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Radojcic V, Maillard I. Notch signaling and alloreactivity. Transplantation. 2016;100(12):2593. doi: 10.1097/TP.0000000000001468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vieceli Dalla Sega F, Fortini F, Aquila G, Campo G, Vaccarezza M, Rizzo P. Notch signaling regulates immune responses in atherosclerosis. Front Immunol. 2019;10:1130. doi: 10.3389/fimmu.2019.01130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wongchana W, Palaga T. Direct regulation of interleukin-6 expression by Notch signaling in macrophages. Cell Mol Immunol. 2012;9(2):155–162. doi: 10.1038/cmi.2011.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hildebrand D, Uhle F, Sahin D, Krauser U, Weigand MA, Heeg K. The interplay of notch signaling and STAT3 in TLR-activated human primary monocytes. Front Cell Infect Microbiol. 2018;8:241. doi: 10.3389/fcimb.2018.00241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zumla A, Hui DS, Perlman S. Middle east respiratory syndrome. Lancet. 2015;386(9997):995–1007. doi: 10.1016/S0140-6736(15)60454-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Channappanavar R, Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin Immunopathol. 2017;39:529–539. doi: 10.1007/s00281-017-0629-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang Y, Li J, Zhan Y, Wu L, Yu X, Zhang W, et al. Analysis of serum cytokines in patients with severe acute respiratory syndrome. Infect Immun. 2004;72(8):4410–4445. doi: 10.1128/IAI.72.8.4410-4415.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Okabayashi T, Uhle F, Yokota SI, Iki S, Indoh T, Yokosawa N, et al. Cytokine regulation in SARS coronavirus infection compared to other respiratory virus infections. J Med Virol. 2006;78(4):417–424. doi: 10.1002/jmv.20556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med. 2020;26(6):842–844. doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- 88.Zhou Y, Fu B, Zheng X, Wang D, Zhao C, et al. Pathogenic T-cells and inflammatory monocytes incite inflammatory storms in severe COVID-19 patients. Nat Sci Rev. 2020;7(6):998–1002. doi: 10.1093/nsr/nwaa041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cao X. COVID-19: immunopathology and its implications for therapy. Nat Rev Immunol. 2020;20(5):269–270. doi: 10.1038/s41577-020-0308-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Law HK, Cheung CY, Ng HY, Sia SF, Chan YO, Luk W, et al. Chemokine up-regulation in sars-coronavirus–infected, monocyte-derived human dendritic cells. Blood. 2005;106:2366–2374. doi: 10.1182/blood-2004-10-4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tseng CT, Perrone LA, Zhu H, Makino S, Peters CJ. Severe acute respiratory syndrome and the innate immune responses: modulation of effector cell function without productive infection. J Immunol. 2005;174(12):7977–7985. doi: 10.4049/jimmunol.174.12.7977. [DOI] [PubMed] [Google Scholar]
- 92.Tian S, Hu W, Niu L, Liu H, Xu H, Xiao SY. Pulmonary pathology of early phase 2019 novel coronavirus (COVID-19) pneumonia in two patients with lung cancer. J Thorac Oncol. 2020;15(5):700–704. doi: 10.1016/j.jtho.2020.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395:507–513. doi: 10.1016/S0140-6736(20)30211-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest. 2020;130(5):2620–2629. doi: 10.1172/JCI137244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bell TJ, Brand OJ, Morgan DJ, Salek-Ardakani S, Jagger C, Fujimori T, et al. Defective lung function following influenza virus is due to prolonged, reversible hyaluronan synthesis. Matrix Biol. 2019;80:14–28. doi: 10.1016/j.matbio.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Glowacka I, Bertram S, Herzog P, Pfefferle S, Steffen I, Muench MO, et al. Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63. J Virol. 2010;84(2):1198–1205. doi: 10.1128/JVI.01248-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jia HP, Look DC, Tan P, Shi L, Hickey M, Gakhar L, et al. Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am J Physiol Lung Cell Mol Physiol. 2009;297(1):84–96. doi: 10.1152/ajplung.00071.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lambert DW, Yarski M, Warner FJ, Thornhill P, Parkin ET, Smith AI, et al. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2) J Biol Chem. 2005;280(34):30113–30119. doi: 10.1074/jbc.M505111200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liu Y, Yang Y, Zhang C, Huang F, Wang F, Yuan J, et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci. 2020;63(3):364–374. doi: 10.1007/s11427-020-1643-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kim JM, Heo HS, Ha YM, Ye BH, Lee EK, Choi YJ, et al. Mechanism of Ang II involvement in activation of NF-κB through phosphorylation of p65 during aging. Age. 2012;34(1):11–25. doi: 10.1007/s11357-011-9207-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fang C, Stavrou E, Schmaier AA, Grobe N, Morris M, Chen A, et al. Angiotensin 1–7 and Mas decrease thrombosis in Bdkrb2–/– mice by increasing NO and prostacyclin to reduce platelet spreading and glycoprotein VI activation. Blood J Am Soc Hematol. 2013;121(15):3023–3032. doi: 10.1182/blood-2012-09-459156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hirano T, Murakami M. COVID-19: a new virus, but a familiar receptor and cytokine release syndrome. Immunity. 2020;52(5):731–733. doi: 10.1016/j.immuni.2020.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Xie X, Chen J, Wang X, Zhang F, Liu Y. Age- and gender-related difference of ACE2 expression in rat lung. Life Sci. 2006;78:2166–2171. doi: 10.1016/j.lfs.2005.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mahmudpour M, Roozbeh J, Keshavarz M, Farrokhi S, Nabipour I. COVID-19 cytokine storm: the anger of inflammation. Cytokine. 2020;133:155151. doi: 10.1016/j.cyto.2020.155151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nat Commun. 2005;436:112–116. doi: 10.1038/nature03712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yang M (2020) Cell pyroptosis, a potential pathogenic mechanism of 2019-nCoV infection. SSRN 3527420 10.2139/ssrn.3527420
- 107.Man SM, Karki R, Kanneganti TD. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev. 2017;277:61–75. doi: 10.1111/imr.12534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Siu KL, Yuen KS, Castano-Rodriguez C, Ye ZW, Yeung ML, Fung SY, et al. Severe acute respiratory syndrome Coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019;33(8):8865–8877. doi: 10.1096/fj.201802418R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Vellingiri B, Jayaramayya K, Iyer M, Narayanasamy A, Govindasamy V, Giridharan B, et al. COVID-19: a promising cure for the global panic. Sci Total Environ. 2020 doi: 10.1016/j.scitotenv.2020.138277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chen IY, Moriyama M, Chang MF, Ichinohe T. Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Front Microbiol. 2019 doi: 10.3389/fmicb.2019.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nieto-Torres JL, Verdiá-Báguena C, Jimenez-Guardeño JM, Regla-Nava JA, Castaño-Rodriguez C, Fernandez-Delgado R, et al. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology. 2015;485:330–339. doi: 10.1016/j.virol.2015.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lee WS, Wheatley AK, Kent SJ, DeKosky BJ. Antibody-dependent enhancement and SARS-CoV-2 vaccines and therapies. Nat Microbiol. 2020;5(10):1185–1191. doi: 10.1038/s41564-020-00789-5. [DOI] [PubMed] [Google Scholar]
- 113.Liu L, Wei Q, Lin Q, Fang J, Wang H, Kwok H, et al. Anti-spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS-CoV infection. JCI Insight. 2019 doi: 10.1172/jci.insight.123158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fu Y, Cheng Y, Wu Y. Understanding SARS-CoV-2-mediated inflammatory responses: from mechanisms to potential therapeutic tools. Virol Sin. 2020;35(3):266–271. doi: 10.1007/s12250-020-00207-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Faure E, Poissy J, Goffard A, Fournier C, Kipnis E, et al. Distinct immune response in two MERS-CoV-infected patients: can we go from bench to bedside? PLoS ONE. 2014;9(2):88716. doi: 10.1371/journal.pone.0088716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shokri S, Mahmoudvand S, Taherkhani R, Farshadpour F. Modulation of the immune response by middle east respiratory syndrome coronavirus. J Cell Physiol. 2019;234:2143–2151. doi: 10.1002/jcp.27155. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.