

Abstract
Purpose:
Despite significant benefit for other cancer subtypes, immune checkpoint blockade (ICB) therapy has not yet been shown to significantly improve outcomes for men with castration-resistant prostate cancer (CRPC). Prior data have shown that DNA damage response (DDR) deficiency, via genetic alteration and/or pharmacologic induction using DDR inhibitors (DDRi), may improve ICB response in solid tumors in part due to induction of mitotic catastrophe and innate immune activation. Discerning the underlying mechanisms of this DDRi–ICB interaction in a prostate cancer–specific manner is vital to guide novel clinical trials and provide durable clinical responses for men with CRPC.
Experimental Design:
We treated prostate cancer cell lines with potent, specific inhibitors of ATR kinase, as well as with PARP inhibitor, olaparib. We performed analyses of cGAS–STING and DDR signaling in treated cells, and treated a syngeneic androgen-indifferent, prostate cancer model with combined ATR inhibition and anti–programmed death ligand 1 (anti–PD-L1), and performed single-cell RNA sequencing analysis in treated tumors.
Results:
ATR inhibitor (ATRi; BAY1895433) directly repressed ATR–CHK1 signaling, activated CDK1–SPOP axis, leading to destabilization of PD-L1 protein. These effects of ATRi are distinct from those of olaparib, and resulted in a cGAS–STING-initiated, IFN-β–mediated, autocrine, apoptotic response in CRPC. The combination of ATRi with anti–PD-L1 therapy resulted in robust innate immune activation and a synergistic, T-cell–dependent therapeutic response in our syngeneic mouse model.
Conclusions:
This work provides a molecular mechanistic rationale for combining ATR-targeted agents with immune checkpoint blockade for patients with CRPC. Multiple early-phase clinical trials of this combination are underway.
Translational Relevance.
We show that treatment of prostate cancer cells with potent, specific ATR inhibitors (ATRi) leads to robust activation of the cGAS–STING signaling pathway, growth inhibition, and cell death. We also demonstrate that ATRis repress ATR-Chk1 replication stress signaling, which results in activation of a CDK1–SPOP axis, PD-L1 degradation, and derepression of a cGAS–STING–IFN-β–IFNAR1-driven, autocrine, cytotoxic signaling pathway. ATRi and anti–PD-L1 combination treatment resulted in synergistic, antitumor activity in a syngeneic mouse androgen-indifferent, aggressive prostate cancer model. Single-cell RNA sequencing analysis of tumor tissue samples from this model demonstrated increased activation of cGAS–STING, IFN-β, and apoptotic signaling in combination versus ATRi single-agent treatment. Our findings provide a molecular mechanistic rationale for combining ATR-targeted agents with immune checkpoint blockade, leading to the development of active early-phase clinical trials and reveal mechanism-based opportunities for improving outcomes for men with advanced prostate cancer by combining immunotherapy and agents that induce mitotic catastrophe.
Introduction
Although androgen receptor (AR) biosynthesis and signaling inhibitors have significantly improved outcomes in patients with castration-resistant prostate cancer (CRPC), there is still a dearth of cytotoxic treatment options that provide durable responses. Clinical studies with immune checkpoint blockade (ICB), including anti–programmed death 1 (PD1)/ligand 1 (PD-L1) and anti–cytotoxic T-lymphocyte–associated protein 4 (CTLA-4), in patients with CRPC have shown relatively few responses, mostly limited to patients with mismatch repair deficiency/MSI-high tumors, and possibly other DNA damage response (DDR) defects, such as variants in CDK12 and/or BRCA2 (1–7). This lack of response to ICB in CRPC is thought to be due to multiple factors, including, but not limited to, relatively low mutational burden and lack of tumor-infiltrating immune cell populations. Thus, there is an urgent need for biomarker-driven, rational combinatorial treatment strategies to safely overcome this resistance in patients with CRPC.
Approximately 25% of men with metastatic CRPC are enriched for germline and/or somatic alterations in DDR genes (3, 8). Clinical trials of single-agent PARP inhibition (PARPi) have shown responses limited mostly to patients with deleterious BRCA2 variants, resulting in the recent FDA approval of two different PARP inhibitors, olaparib and rucaparib, for men with CRPC with certain variants, including BRCA2 (9, 10). Furthermore, preclinical studies of combination PARPi with ICB have shown the potential for additive benefit, even in BRCA1/2 wild-type cancer cells, whereby PARPi can induce immune activation through a variety of mechanisms not yet fully characterized (2, 11, 12). Early data from clinical trials combining olaparib with ICB agents have yielded mixed results, with benefit mostly seen in patients with DDR gene alterations (2, 13). DDR inhibitors (DDRi) have rapidly expanded, now including inhibitors of other pathways, including ATR kinase, which, along with ATM, serves as the key player in replication stress signaling (RSS; ref. 14–16). Although parallels between the known mechanism(s) of immune activation can be drawn between PARPis and ATRis, there are no known studies that compare and contrast these agents with regard to their mechanisms of action, impact on immune activation, and therapeutic opportunities for combination with ICB in immunocompetent models of prostate cancer. Previous reports have demonstrated that PARPis activate cGAS–STING signaling and induce PD-L1 protein expression through IRF3 activity (11, 17–19), yet the impact of ATRi on immune pathway activation is not clear. Furthermore, a recent report showed that ATRi treatment led to downregulation of PD-L1 in select cancer cell lines, although the underlying mechanism of this activity was not identified and synergy with anti–PD-1/L1 therapy not explored (20, 21). Because of these knowledge gaps, we sought to determine the resulting impact and the mechanism of action of ATR signaling inhibition on cytotoxicity, immune activation, PD-L1 protein regulation, and its capacity for safely improving the efficacy of ICB in prostate cancer, which can directly inform biomarker-driven ICB treatment strategies for patients with CRPC.
Materials and Methods
Cell lines and reagents
RM-9, RM-1, and RM-1-BM [generated from a bone tumor following repeated intracardiac injection of RM-1 cells (22), and obtained from Dr. Pamala J. Russell (Australian Prostate Cancer Research Centre and Queensland University of Technology, Queensland, AU)] mouse prostate cancer cell lines derived from ras+myc–induced mouse prostate cancer tumors (23–25) were maintained in DMEM cell culture medium supplemented with 10% FBS (Mediatech, Inc.). LNCaP C4–2b (C4–2b) cells (obtained from Dr. Gary E. Gallick, MD Anderson) and PC-3 cells (purchased from the ATCC) were maintained in RPMI-1640 cell culture medium. All cell lines were validated by short tandem repeat DNA fingerprinting in MD Anderson's Characterized Cell Line Core Facility, and tested for Mycoplasma (MycoAlert Mycoplasma Detection Kit, Lonza Group LTD.) prior to storage as Mycoplasma-negative frozen stock cells before use.
BAY1895344 (#S8666), VX-970 (#S7102), olaparib (KU-0059436) and bortezomib (PS-341, #S1013) were obtained from SelleckChem. Isotype control IgG and anti–PD-L1 [BE0101, clone 10F.9G2, anti-mouse CD8 (BP0061, clone 2.43) antibodies] antibodies were purchased from Bio X Cell. Anti-mouse and anti-human CD8–neutralizing antibodies were purchased from PBL Biomedical Laboratories.
Mammalian expression vector for CDK1 kinase, Cdc2-HA, and an expression vector for CDK1 kinase-dominant–negative inhibitor, Cdc2-DN-HA (Addgene plasmid # 1888; http://n2t.net/addgene:1888; RRID:Addgene_1888 and Addgene plasmid #1889; http://n2t.net/addgene:1889; RRID:Addgene_1889) were gifts from Sander van den Heuvel. pcDNA3.1 empty vector was purchased from Invitrogen. To generate pcDNA3.1-flag-SPOP, SPOP coding DNA fragment was amplified by PCR using reverse-transcribed cDNAs from NCI-H660 prostate cancer cells. The following are the primers for PCR: forward primer-5′-CTCGGATCCATGGCTGATTACAAGGATGACGACGATAAGGGTGATTACAAGGATGACGACGATAAGGGTGGTATGTCAAGGGTTCCAAGTCCTCC-3′ (2xFlag tag coding sequences are underlined, and BamHI restriction enzyme digestion site for clone is in italic) and reverse primer-5′-GTCTTGACCCTTTTAGGATTGCTTCAGGCGTTTGC-3′. The DNA fragment (amplified by PCR) was digested by BamHI, and ligated to pcDNA3.1 vector (linearized by BamHI/EcoRI and blunted at EcoRI site) using T4 DNA ligase. The plasmid construct was confirmed by DNA sequencing.
PicoGreen staining
PicoGreen staining was performed using Quant-iT Pico-Green dsDNA Reagent and Kits (Invitrogen) following the kit instruction manual as previously published (12). The slides were imaged using a Nikon microscope (TE2000-U) and a Leica SP5X laser scanning confocal microscope.
Protein immunoblot analysis
Whole-cell proteins were extracted by cell lysis buffer (NP-40 buffer or RIPA buffer, Cell Signaling Technology) with proteinase inhibitor cocktail (Roche) and phosphatase inhibitor cocktail (MilliporeSigma), and immunoblotting analysis was performed following a standard protocol.
RT-qPCR analysis
Total cell RNAs were extracted by TRizol reagent (cat #15596026, Invitrogen). The RNAs were reversely transcribed to cDNA using the high-capacity cDNA reverse transcription kit (Invitrogen). RT-qPCR was conducted using fast SYBR green master mix (Invitrogen), and the 2−ΔΔCt method was used to determine relative mRNA expressions compared with controls. The primer sequences used for RT-qPCR detection of genes' expression are listed in the Supplementary Table S3.
siRNA transfection
For siRNA transfection, cells were transfected with 20 nmol/L siRNA using the Lipofectamine RNAiMax transfection reagent (Invitrogen). The target sequences of siRNA used in the experiments are listed in the Supplementary Table S4.
Protein half-life assay (CHX chase assay)
Briefly, cycloheximide (CHX, MilliporeSigma) was added to the cell incubation at the final concentration of 100 μg/mL. The cells were lysed and collected at elapsed time points following the introduction of CHX incubation. The whole-cell lysates were separated by SDS-PAGE and analyzed for detection of specific proteins' expression by immunoblot assay with protein-specific antibodies. The blot signals were scanned, quantified, and plotted to display the time-elapsed presentation of proteins (compared with 0 time point) following CHX treatment to block the protein synthesis.
Transient transfection of expression vectors in mammalian cells
293T cells were transfected with expression vectors using Lipofectamine 2000 transfection reagent (Invitrogen) following the manufacturer's instructions. Forty-eight hours post-transfection, the cells were harvested and washed with PBS. Total cell lysates or total RNAs were prepared and used for co-immunoprecipitation (Co-IP), immunoblot, or RT-qPCR analysis for indicated experiments.
Co-IP analysis
Briefly, 293T cells were transfected with SPOP and CDK1 or empty vector (control) expression vectors. Forty hours after the transfection, 500 nmol/L (final concentration) PS-341 was added to incubation for another 8 hours. The cells were collected, washed by PBS, and lysed in NP-40 lysis buffer (Cell Signaling Technology) containing protease inhibitor cocktail (Roche) and phosphatase inhibitors (MilliporeSigma). Cell lysates were incubated with protein-specific antibodies and the immunocomplexes were collected by incubating the lysate-antibody mix with protein G Dynabeads at 4°C on a rotator. After extensive washing with NP-40 lysis buffer, the immunocomplexes were eluted from the beads by 1xSDS loading buffer and analyzed by immunoblotting.
MTS assay
Cells were plated in 96-well cell plates and treated as indicated. The viable cells were determined using CellTiter 96 AQueous One Solution Cell Proliferation Assay kit (Promega) following the manufacturer's instructions.
Flow cytometry
For cell cycle, cells were harvested after drug treatment, stained with propidium iodide (Thermo Fisher Scientific), and analyzed on a FACS Canto II flow cytometer (BD Biosciences). Quantitative data were obtained and analyzed using FlowJo software (TreeStar Inc.).
RM-1-BM syngeneic mouse prostate cancer model
Aliquots of 5 × 105 mouse RM-1-BM prostate cancer cells in 100 μL (1:1, PBS:Matrigel) were subcutaneously injected into the right flanks of 7-week-old C57BL/6N male mice (The Jackson Laboratory). Tumors were allowed to grow until they achieved a 30 to 50 mm3 volume before they were randomly distributed to receive one of the following treatments intraperitoneally for 18 days: vehicle control (water mixed with DMSO) for BAY1895344, vehicle control for olaparib [10% (2-Hydroxypropyl)-β-cyclodextrin] powder (MilliporeSigma) in PBS, isotype IgG control, anti–PD-L1 (Bio X Cell, 10 mg/kg/wk), BAY1895344 (40 mg/kg, every other day), olaparib (40 mg/kg/d, 5 days/week), or BAY1895344 combined with anti–PD-L1. For the BAY1895344 combination group, BAY1895344 was administered every other day, and initiated 2 days after the initial anti–PD-L1 injection. For the survival study, mice in each group were monitored until the tumor volume exceeded 1,000 mm3 or mice showed signs of being moribund.
For the anti-CD8 depletion study, tumor-bearing mice were divided into two groups, IgG or anti-CD8, and randomly selected to receive one of the five treatments: vehicle control, BAY1895344, anti–PD-L1 or the combination of BAY1895344 and anti–PD-L1 as described above. Each mouse received two intraperitoneal injections of either IgG (200 μg) or anti-CD8 (200 μg) before the five treatments were initiated, and then two injections/wk thereafter.
Tumor volume was measured every 3 days and calculated on the basis of (width × width × length)/2. Eighteen days after the initial treatment, the tumors were harvested. All animal experiments were conducted in accordance with accepted standards of humane animal care approved by MDACC IACUC.
Immunofluorescence analysis
Immunofluorescence analysis of cultured prostate cancer cells was performed using a primary antibody to cleaved caspase 7 (Asp 198; Cell Signaling Technology, #8438) following a standard protocol. Positively stained cells were counted using image analysis.
ELISA
Detection of IFN-β cytokine secreted by tumor cells was conducted by ELISA according to the manufacture's instruction (PBL Biomedical Laboratories). To determine IFN-β expression in vivo in treated tumor tissues, tissues were lysed by RIPA buffer (Cell Signaling Technology), and IFN-β ELISA assay was used to detect the IFN-β expression. For comparative analysis of treated tumor tissues, IFN-β values were determined by normalizing primary results to the total protein of tissue samples.
Single-cell RNA sequencing
Tumor tissues were recovered from eight RM-1-BM tumor-grafted mice (two for each treatment) at day 10 treated by vehicle controls (2), IgG (2), ATRi (BAY1895344; 2), or BAY1895344+anti–PD-L1 (2), respectively. For the isolation of single cells of each treatment tumor tissue sample, the tumors were finely minced and treated by trypsin at 37°C for 5 minutes. Then they were transferred into DMEM medium with 10% FBS and dissociated into single-cell suspension. The resulting single cells were washed in BSA-PBS twice and prepared to be picked as single cells using the Chromium Single Cell 3′ system (10X Genomics), according to the manufacturer's instructions. The single-cell RNA sequencing (scRNA-seq) data were pre-processed using the Cell Ranger pipeline (26). R packages Seurat, Monocle, and DESeq2 were used to assess the quality, normalize, cluster, and perform differential expression analysis on the single-cell data (27–29). Garnett software package was used to classify the cell types in the data (30). Pre-ranked gene set enrichment analysis (GSEA) was performed on the basis of test statistic obtained from differential expression among treatment groups using KEGG and other pathways of interest from MsigDB (31).
Statistical analysis
Data were presented as the mean ± SEM. T tests were used for analysis of MTS assay, RT-PCR, ELISA, tumor volume (growth), and tumor wet weights. Benjamini and Hochberg adjusted P values < 0.05 were considered statistically significant. The Wilcoxon rank-sum test was used for data with non-normal distributions in flow cytometry assay. Synergistic effects were evaluated by the Bliss independence model (32). Combined percentage of inhibition for the two treatments (x-ATRi, y-anti–PD-L1) was predicted (Exy, P). The difference between the observed combined percentage of inhibition from the two treatments (Exy, O) and the predicted combined percentage of inhibition was used to assess the combined effect of two treatments (synergistic if, Eab, O > Eab, P).
Results
ATR inhibition induces cytoplasmic DNA and cGAS–STING activation
We used prostate cancer cell lines derived from ras+myc–induced mouse prostate tumors [RM-9 (ARhigh), RM-1 (ARlow; refs. 33, 34), and a highly metastatic, ARlow RM-1 derivative, RM-1-BM (22, 35); Supplementary Fig. S1] and C4–2b and PC-3 human prostate cancer cells to analyze the effects of ATRi (BAY1895344 or VX-970) or PARPi (olaparib) on cell proliferation, DNA damage, and activation of cGAS–STING signaling. Previous publications and sequencing analysis data from our laboratory and others have shown that these human and mouse prostate cancer cell lines are wild-type for canonical DDR genes, including BRCA1, BRCA2, and ATM genes (Supplementary Table S1). ATRis and olaparib treatments both resulted in significant suppression of cell proliferation (MTS assay), with ATRis demonstrating marginally greater activity than olaparib, reflecting different potencies in this assay, particularly in human prostate cancer cells (Fig. 1A–D). Analysis of cell cycle with flow cytometry revealed marked differences in the effects of ATRi (BAY1895344) compared with olaparib, with ATRi demonstrating reduced cell accumulation in G2–M, and olaparib showing increased accumulation in G2–M. Increased accumulation of prostate cancer cells in subG1 (indicating apoptosis) was generally observed for ATRis compared with olaparib for all cell lines (Fig. 1E and F). Quantitative analysis of cytoplasmic double-strand DNA (dsDNA) indicated that ATRis or olaparib generated significantly higher levels of this cGAS–STING activator compared with controls at the concentrations used (Fig. 1G–I; Supplementary Fig. S2). Immunoblotting (IB) analysis was performed to analyze the cGAS–STING downstream signaling in mouse prostate cancer cells. The results showed marked activation of this pathway, including induction of p-TBK1 and p-IRF3 following treatment with ATRis or olaparib (Fig. 1J; Supplementary Table S2). Markedly increased levels of IRF3-induced cytokines CCL5 and CCL10 following ATRi or olaparib treatment were also apparent (2–10-fold, P < 0.05; Fig. 1K–N). Importantly, siRNA knockdown of cGAS–STING signaling components (cGAS, STING, TBK1, or IRF3) led to suppression of CCL5 and CXCL10 gene expression following ATRi treatment (P < 0.05; Fig. 1O and P; Supplementary Fig. S3, Supplementary Tables S3 and S4).
Figure 1.
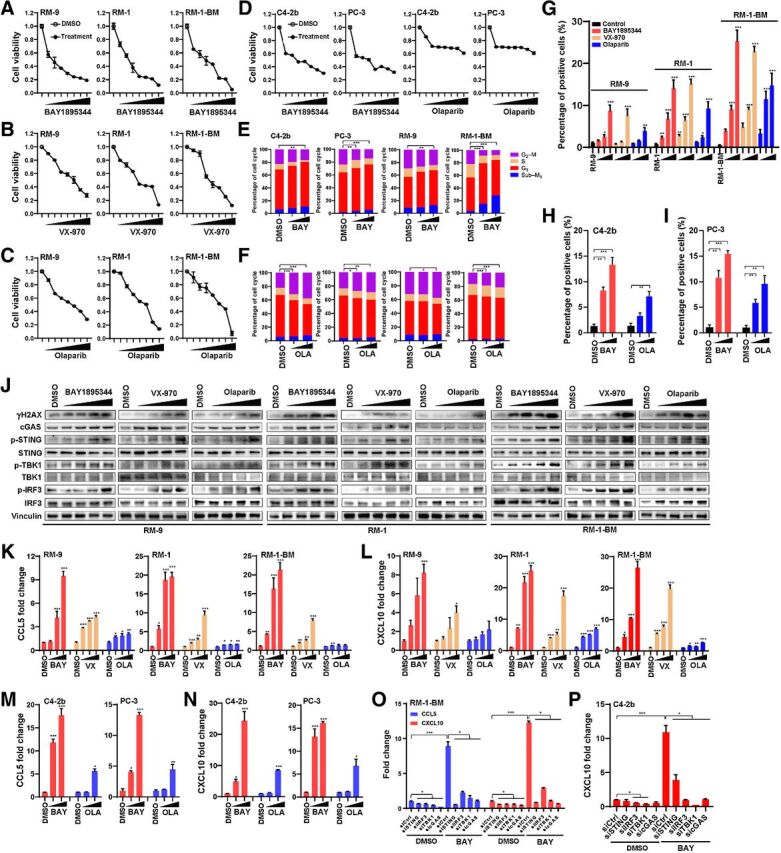
ATRi demonstrates marked cytotoxicity and cGAS–STING activation in prostate cancer models. A–D, MTS assay of human (C4–2b and PC-3) and mouse (RM-9, RM-1, and RM-1-BM) prostate cancer cell lines treated with ATRi (BAY1895344 and VX-970) or olaparib (OLA). Concentrations range from 0.125 to 8 μmol/L as labeled in the figures (x-axis). Specific concentrations within this range (0.125, 0.25, 0.5, 1, 2, 4, and 8 μmol/L) were used for other experiments as indicated. Viable cells are represented as the fold change relative to DMSO (vehicle control) treatment. E and F, Flow cytometry cell-cycle analyses of BAY1895344-treated (10 and 100 nmol/L, in E) and olaparib-treated (250 and 500 nmol/L, in F) C4–2b, PC-3, RM-9, and RM-1-BM prostate cancer cell lines. G–I, Quantitative analysis of PicoGreen staining in prostate cancer cells treated with DMSO (vehicle control), BAY1895344 (concentrations were 0.5, 1, and 2 μmol/L), VX-970 (concentrations were 0.75, 1.5, and 3 μmol/L), or olaparib (concentrations were 1.2, 2.5, and 5 μmol/L) for 48 hours. Data represent mean ± SEM of three independent experiments. J, Immunoblots of select proteins in prostate cancer cells treated with increasing doses of BAY1895344, VX-970, or olaparib (concentrations ranging from 0.125 to 2 μmol/L). K–N, RT-qPCR analysis of CCL5 and CXCL10 in prostate cancer cells treated with DMSO (vehicle control), BAY1895344, VX-970, or olaparib (concentrations ranging from 0.5 to 2 μmol/L). O and P, RT-qPCR analysis of CCL5 and CXCL10 in RM-1-BM or C4–2b prostate cancer cells in response to BAY1895344 (2 μmol/L) or DMSO (vehicle control) following knockdown of cGAS, STING, TBK1, or IRF3 gene expression in vitro via transfection with control siRNA or gene-specific siRNA (sicGAS, siSTING, siTBK1, or siIRF3). n.s., not significant; * P < 0.05; **, P < 0.01; ***, P < 0.001.
ATR inhibition suppresses PD-L1 protein expression
A recent study demonstrated that olaparib can upregulate PD-L1 protein levels through cGAS–STING signaling and IRF3-mediated transcriptional activation in small cell lung cancer cells (11), yet recent studies have shown that ATRi treatment leads to downregulation of PD-L1 protein in various cell line models (20, 21). To determine the mechanisms of ATRi regulation of PD-L1, we first analyzed expression of PD-L1 following ATRi (BAY1895344). We found that ATRi treatment led to upregulation of PD-L1 mRNA in RM-9 and RM-1-BM cells that was suppressed by cGAS–STING pathway siRNAs (Fig. 2A). Congruent with the results of published studies in other tumor types, olaparib treatment increased PD-L1 protein expression in RM-9 and RM-1-BM prostate cancer cells, as revealed by IB analysis. In contrast, ATRi or ATR siRNA treatment led to downregulation of PD-L1 protein expression (Fig. 2B; Supplementary Fig. S4A and S4B), but ATRi treatment induced the expression of PD-L1 mRNA (Fig. 2A). Further studies demonstrated that an ATRi- and proteasomal degradation–dependent mechanism led to suppression of PD-L1 protein levels (Fig. 2C), which resulted in a significant reduction in PD-L1 protein half-life in ATRi- but not olaparib-treated prostate cancer cells (Fig. 2D and E). These results indicate opposing regulatory pathways for ATRi and PARPi with regard to PD-L1 protein levels.
Figure 2.

CDK1-SPOP-PD-L1 signaling is selectively activated by ATRi. A, RT-qPCR analysis showing that ATRi BAY1895344 (2 μmol/L) transcriptionally activates expression of PD-L1 mRNA through the cGAS–STING pathway. RM-9 and RM-1-BM prostate cancer cells were transfected with control siRNAs or gene-specific siRNAs (sicGAS, siSTING, siTBK1, or siIRF3), and cells were treated with DMSO or BAY1895344 (2 μmol/L) for 48 hours before total RNAs were extracted for RT-qPCR analysis of PD-L1 mRNA expression. B, Immunoblot analysis to show PD-L1 protein expression in RM-9 or RM-1-BM cells following BAY1895344 or olaparib (OLA) treatment (2 μmol/L for 24 and 48 hours). C, Immunoblot analysis of protein expression of PD-L1 and phosphorylated ATR in human (C4–2b and PC-3) and mouse (RM-9 and RM-1-BM) prostate cancer cells treated with BAY1895344 (2 μmol/L for 24 hours), or concomitant treatment with BAY1895344 and the proteasome inhibitor bortezomib (PS-341, 0.5 μmol/L for 8 hours). D and E, Cycloheximide (CHX)-chase protein half-life analysis to examine the degradation of PD-L1 protein affected by BAY1895344 and olaparib in RM-1-BM prostate cancer cells. RM-1-BM cells were pretreated with DMSO (vehicle control), BAY1895344 (2 μmol/L), or olaparib (2 μmol/L) for 36 hours before treatment with CHX. Immunoblot analysis was used to determine the expression of PD-L1 protein expression and the autoradiographic bands were scanned, quantified by densitometer, and normalized (to the internal reference protein blot signals, vinculin protein, and plotted; in E). F, Immunoblot analysis to show dose-dependent CDK1 activation (dephosphorylation of p-Y15-CDK1 by ATRi BAY1895344 in doses ranging from 0.125 to 2 μmol/L) compared with CDK1 inhibition (increase in p-Y15-CDK1 by olaparib) resulting from inhibition of the cell-cycle kinase signaling cascade ATR-CHK1-CDC25C/WEE1-CDK1 in human (C4–2b and PC-3) and mouse (RM-1-BM) prostate cancer cells.
To determine the origins of these opposing PD-L1 protein regulatory pathways implicated in ATRi and olaparib treatment, we analyzed the effects of these agents on RSS, with a focus on regulation of the G2–M checkpoint (36, 37). As expected, IB analysis showed that ATRi suppressed p-ATR/total ATR, and p-Chk1 (S317)/total Chk1 in mouse (RM-1-BM) and human (C4–2b and PC-3) prostate cancer cells. In contrast, olaparib treatment led to ATR-Chk1 activation (Fig. 2F). Analysis of ATRi- versus olaparib-regulated ATR-Chk1 activities were extended to specific downstream Chk1 target proteins that regulate G2–M, including CDC25C, WEE1, and CDK1. ATRi treatment led to activation of CDC25C [reduced p-CDC25C (S216)/total CDC25C] and inhibition of WEE1 [reduced p-WEE1 (S642)/total WEE1], whereas olaparib suppressed CDC25C and activated WEE1 (Fig. 2F). Importantly, ATRi activation of CDC25C and repression of WEE1 was associated with dephosphorylation of CDK1 (p-Y15), and its activation (see below), whereas olaparib suppression of CDC25C and activation of WEE1 was associated with inhibitory phosphorylation of CDK1 (p-Y15) and its repression (Fig. 2F). These results provide an underlying mechanism for possible superior cytotoxicity of ATRi against BRCA–wild-type prostate cancer cells compared with olaparib, in light of recent findings that premature, unregulated activation of CDK1 can lead to lethal mitotic events (38, 39).
ATR inhibition suppresses PD-L1 through the CDK1–SPOP axis
A recent report showed that cyclin D–mediated activation of CDK4 regulates PD-L1 levels through phosphorylation-mediated stabilization and upregulation of SPOP, a substrate-binding adaptor protein of SPOP/Cullin3 E3 ubiquitin ligase complex that binds to and promotes ubiquitination of PD-L1 for degradation through the ubiquitination proteomic pathway (40). Interestingly, we found that the phosphorylation site in SPOP that is recognized by CDK4 is also a CDK1 consensus sequence (Supplementary Fig. S4C), raising the possibility that SPOP may be differentially regulated by ATRi or PARPi through CDK1-mediated phosphorylation. Indeed, our IB analysis showed that activated CDK1 increased total SPOP, and suppressed PD-L1, whereas olaparib, which suppresses CDK1 activity, reduced total SPOP and upregulated PD-L1 protein (Fig. 2F). Our results further suggest that the observed tumor cell–intrinsic innate immune pathway induced by DDR-targeting agents is modulated by the SPOP E3 ubiquitin ligase complex, which is differentially regulated by ATRi or PARPi.
To determine whether ATRi-induced signaling targets PD-L1 through CDK1 activation of SPOP, we initially co-transfected FLAG-tagged SPOP together with HA-tagged CDK1 or HA-tagged–dominant negative inhibitor of CDK1 kinase activity (CDK1-DN) in 293T cells and showed that increased expression of CDK1 led to upregulation of SPOP, whereas overexpression of CDK1-DN (41) effectively destabilized and downregulated SPOP levels in a dose-dependent manner (Fig. 3A). Additional experiments using this system demonstrated that CDK1 significantly increased the half-life of SPOP from approximately 30 to more than 120 minutes in 293T cells (Fig. 3B and C). Using the co-IP assay, we observed that CDK1 and SPOP directly interact within the same complex pulled down by protein-specific antibody recognizing SPOP or CDK1 (Fig. 3D). To examine the regulatory mechanisms controlling ATRi–CDK1–SPOP–PD-L1 interactions, we transfected C4–2b and RM-1-BM cells with CDK1 siRNA and analyzed SPOP and PD-L1 levels by IB. The results supported our hypothesis (Fig. 3E), showing that CDK1 siRNA led to downregulation of SPOP expression, but upregulation of PD-L1, and that transfection of SPOP siRNA led to upregulation of PD-L1 protein expression, but did not alter PD-L1 mRNA levels (Fig. 3F; Supplementary Fig. S4D). Furthermore, transfection of SPOP siRNA or non-silencing control siRNA (siCtrl) followed by ATRi (BAY1895344) treatment demonstrated that SPOP siRNA “rescued” PD-L1 protein expression levels reduced by ATRi (Fig. 3G). In summary, these observations demonstrate that the suppression of PD-L1 protein in prostate cancer cells via ATRi is dependent on activation of the CDK1–SPOP axis.
Figure 3.

The CDK1–SPOP axis regulates PD-L1 protein levels. A, Immunoblot analysis to demonstrate that dose-dependent overexpression of HA-tagged CDK1 stabilizes SPOP and that overexpression of dominant negative inhibitor of the kinase activity of CDK1 (HA-tagged CDK1-DN) led to destabilization of SPOP expression in 293T cells. Specifically, 293T cells were cotransfected with Flag-tagged SPOP together with HA-tagged CDK1 or HA-tagged CDK1-DN expression vectors. After 48 hours, cells were lysed and immunoblot analysis was performed to analyze the expression of SPOP (anti-Flag). B, Protein half-life analysis for CDK1-targeted SPOP. 293T cells were cotransfected with expression vectors for Flag-tagged SPOP and empty vector (non-overexpression control) or HA-tagged CDK1. Thirty-six hours after transfection, CHX-chase assay was conducted, followed by immunoblot analysis of cell lysates. C, Autoradiographic bands from the immunoblots were scanned, quantified by densitometer, normalized (to the internal reference protein blot signals, vinculin protein), and plotted to show the increased half-life of SPOP with CDK1 overexpression (from ∼30 to over 60 minutes). D, Co-IP analysis to examine the protein–protein interaction of SPOP and CDK1 in vitro. 293T cells were cotransfected by SPOP, CDK1, or empty vector control. E, Knockdown of CDK1 with siRNA (siCDK1) downregulates SPOP expression and upregulates PD-L1 protein in C4–2b and RM-1-BM prostate cancer cells. F, Immunoblot analysis of lysates from RM-1-BM and C4–2b cells previously transfected with SPOP siRNA (siSPOP), showing that SPOP knockdown effectively stabilizes PD-L1 in these prostate cancer cells. G, Immunoblot analysis of PD-L1 protein expression in cell lysates from RM-1-BM and C4–2b cells pre-transfected with control siRNA (siCtrl, siRNA non-silencing transfection control) or siSPOP followed by treatment with BAY1895344 (0.5 and 2 μmol/L) showed that SPOP knockdown in C4–2b and RM-1-BM cells effectively prevents the degradation of PD-L1 caused by BAY1895344. n.s., not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
IFN-β–driven autocrine, apoptotic signaling following ATR or PARP inhibition
A previous report showed that cell-intrinsic PD-L1 signaling protects cancer cells from IFN-β–mediated cytotoxicity and accelerates tumor progression by inhibiting the IFNAR1-JAK1–promoted phosphorylation of STAT3 at Y705 (42). On the basis of this report and our own results, we hypothesized that ATRi-mediated downregulation of PD-L1 may lead to enhanced tumor cell–intrinsic, pro-apoptotic IFN-β signaling. To test this hypothesis, we initially analyzed intrinsic IFN-β expression in human (C4–2b, PC-3) and mouse (RM-9, RM-1-BM) prostate cancer cells in response to ATRi (BAY1895344) or olaparib in vitro using RT-qPCR. The results indicated that IFN-β mRNA expression was induced by ATRi or olaparib compared with controls, at the indicated concentrations (Fig. 4A). In line with this observation, higher levels of secreted IFN-β protein were also detected (by ELISA assay) in conditioned culture media from ATRi- or olaparib-treated cancer cells, at the indicated concentrations (Fig. 4B). Analysis of the effects of ATRi or olaparib in the presence or absence of exogenously added IFN-β on the viability of human and mouse prostate cancer cells demonstrated significantly greater ATR-induced cytotoxicity in the presence of exogenously added IFN-β, whereas this was not the case with olaparib at the indicated concentrations. Importantly, IFN-β–induced cytotoxicity was neutralized by antibody depletion of IFN-β under ATRi or olaparib treatment conditions (Fig. 4C). We further analyzed the effects of ATRi (BAY1895344) or olaparib on constitutive IFNAR1 signaling to determine the potential effects of differentially expressed IFN-β on these treatments in vitro. We found that ATRi treatment specifically led to increased levels of IFNAR1, p-JAK1/tJAK1, p-STAT3 (Y705)/tSTAT3, and cleaved caspases 3 and 7 in RM-1-BM and C4–2b prostate cancer cells (Fig. 4D). Although olaparib treatment led to induction of IFNAR1-JAK1 signaling, the effects (JAK1-STAT3 signal transduction to phosphorylate STAT3 at Y705) were largely blunted compared with ATRi treatment. These results are similar to those from a recent study that demonstrated a cancer cell intrinsic IFNAR1–JAK1–STAT3 (p-Y705)–caspase 7–dependent cytotoxic pathway induced by deletion of a specific intracytoplasmic domain of PD-L1 (42). In addition, multiple studies previously showed that caspase 7 mediates IFN-β–induced apoptosis in cancer cells (42–44). RT-qPCR analysis of cGAS mRNA levels indicated that ATRi (BAY1895344) or olaparib treatment produced a dose-dependent increase in cGAS mRNA expression in RM-1-BM and/or C4–2b prostate cancer cells, at the indicated concentrations (Fig. 4E).
Figure 4.

Interaction between PD-L1 and tumor cell–intrinsic IFN-β cytotoxic signaling. A, RT-qPCR of IFN-β expression in cells treated with DMSO vehicle control, BAY1895344 (BAY; 0.5 and 2 μmol/L), or olaparib (OLA; 0.5 and 2 μmol/L) in human and mouse prostate cancer cells. B, ELISA assay to detect the IFN-β protein secreted by C4–2b and RM-1-BM cancer cells following treatment with BAY1895344 (2 and 4 μmol/L) or olaparib (5 and 10 μmol/L) in vitro (36 hours). n.s., not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001. C, MTS assay of human prostate cancer cells (top) and mouse prostate cancer cells (bottom) treated with BAY1895344 or olaparib in the absence or presence of mouse or human recombinant IFN-β (100 U/mL). Separate prostate cancer cell cultures treated with BAY1895344 (2 μmol/L) or olaparib (5 μmol/L) in the presence of recombinant IFN-β were also treated with IFN-β–neutralizing antibody. After these treatments, viable cells were determined by MTS assays. n.s., not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001. D, Immunoblot analysis showed greater induction of IFN-β–induced cytotoxic signaling through IFN-β-JAK1-STAT3–caspase 7 cascade following treatment with BAY1895344 than olaparib (42). E, BAY1895344 treatment (0.5 and 2 μmol/L) led to a dose-dependent induction of cGAS mRNA (RT-PCR) in C4–2b and RM-1-BM prostate cancer cells, whereas olaparib treatment (0.5 and 2 μmol/L) led to increased cGAS mRNA in C4–2b to a lesser extent than ATRi and did not induce cGAS mRNA in RM-1-BM.
PD-L1 knockdown sustained STAT3 proapoptotic signaling following PARP inhibition
To demonstrate the role of PD-L1 in regulation of IFNAR1 signaling in prostate cancer cells, we transfected RM-1-BM or C4–2b prostate cancer cells with PD-L1 siRNA before olaparib treatment and analyzed selected IFNAR1 signaling activities using IB (Fig. 5A). In addition, we assessed the impact of the PD-L1 siRNA on cGAS mRNA expression by RT-qPCR and levels of cleaved caspase 7 by immunofluorescence analysis in olaparib-treated cells (Fig. 5B–D, respectively). The results showed that PD-L1 siRNA treatment led to increased cGAS mRNA levels and sustained robust IFNAR1–JAK1–STAT3–cleaved caspase 7 signaling in these cell lines following olaparib treatment. These results are consistent with a previous report demonstrating that cGAS is an IFN-stimulated gene (ISG) that contains IFN-sensitive response elements in its promoter region (45).
Figure 5.

PD-L1 knockdown sustains IFNAR1–JAK1–STAT3–cleaved caspase 7 signaling following olaparib treatment. A, Immunoblotting analysis shows that PD-L1 knockdown by siRNA effectively sustains the JAK1–STAT3–caspase 7 apoptotic signaling cascade, including p-STAT3, p-STAT1, and cleaved caspase 7, following olaparib (2 μmol/L) treatment in C4–2b and RM-1-BM cells. Immunoblotting also demonstrated that sequential siPD-L1 and olaparib treatment maintained induction of cGAS protein levels in these cells. B, RT-qPCR analysis demonstrated that siPD-L1 treatment led to increased cGAS mRNA levels following olaparib treatment in C4–2b and RM-1-BM cells. C, Immunostaining analysis of cleaved caspase 7 (Asp198) also showed that siPD-L1 resulted in significant upregulation of caspase 7 following olaparib in C4–2b and RM-1-BM cells. D, Image analysis demonstrated that the numbers of caspase 7–positive cells were significantly higher in C4–2b and RM-1-BM cells that were treated with siPD-L1 before olaparib treatment compared with olaparib treatment alone. n.s., not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
ATR inhibition leads to greater induction of innate immune pathway activation and prolonged survival in a syngeneic prostate cancer model
Having used comparative analysis of ATRi and olaparib activities in vitro to clarify intrinsic cytotoxic mechanisms induced by specifically ATRi in prostate cancer cells, we next focused on ATRi therapy activities using a syngeneic prostate cancer model. Analysis of single-agent ATRi (BAY1895344), anti–PD-L1, or ATRi and anti–PD-L1 combination treatment showed that ATRi together with anti–PD-L1 was safe and resulted in significantly greater inhibition of tumor growth than either single agent in an RM-1-BM syngeneic prostatic cancer model (Fig. 6A and B). The combination treatment revealed a synergistic interaction between ATRi and anti–PD-L1 by Bliss independence analysis (Fig. 6B; Supplementary Fig. S5A), and IB analysis of tumor tissues showed that ATRi suppressed PD-L1 protein levels compared with vehicle control–treated tumors (Supplementary Fig. S5B). To determine the role of specific host immune cells in these treatment responses, we used anti-CD8 antibody to generate mice deficient in CD8+ T cells and treated RM-1-BM tumors with ATRi or ATRi+anti–PD-L1. Compared with IgG antibody, which we used as a control for anti-CD8, we found that CD8+ T-cell depletion via anti-CD8 substantially abrogated, but did not entirely eliminate, the therapy effect in all treatment groups, demonstrating that CD8+ T cells were key effectors of growth suppression in the RM-1-BM tumors treated with ATRi and anti–PD-L1, but also that T-cell–independent mechanisms contributed to the therapeutic response (Fig. 6C). T-cell–dependent therapeutic effects of PARPi + anti–PD-L1 have been previously shown for PARPi in small cell lung cancer and triple-negative breast cancer mouse models (11, 17). To determine the relative levels and downstream effects of IFN-β in the tumor microenvironment, we analyzed IFN-β, CCL5, and CXCL10 mRNA (RT-qPCR) and IFN-β protein (ELISA) in tumor tissues from all treatment conditions. The results showed significant differences between ATRi and ATRi+anti–PD-L1 treatment for the specified cytokine mRNAs (Fig. 6D) and IFN-β protein (Fig. 6E). In a survival study, we examined the treatment response durability of ATRi as a single agent or together with anti–PD-L1 in the RM-1-BM model in vivo, which demonstrated significantly increased survival of tumor-engrafted animals for ATRi+anti–PD-L1 compared with ATRi or anti–PD-L1 single-agent treatment (Fig. 6F).
Figure 6.
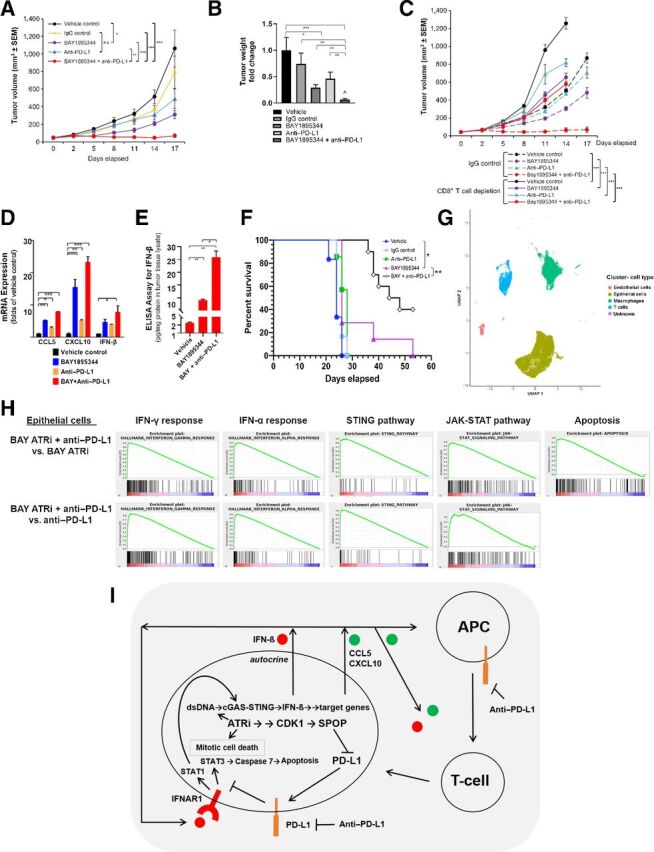
ATRi is synergistic with anti–PD-L1 in a syngeneic mouse prostate cancer model. A, RM-1-BM subcutaneous tumor volume (growth), and (B) wet weight (fold-change relative to vehicle control) at the study termination of vehicle control, IgG, BAY1895344, anti–PD-L1, and BAY1895344+anti–PD-L1 treatment. C, Tumor volume during vehicle control, BAY1895344, anti–PD-L1, and BAY1895344+anti–PD-L1 treatment, administered together with IgG or anti-CD8. D, RT-qPCR analysis of IFN-β, CCL5, and CXCL10 mRNA from tumor samples following treatments described previously in A and B. E, ELISA analysis of IFN-β from tumor samples following treatments described previously in A and B. F, Survival curve of mice treated with vehicle control, IgG, anti–PD-L1, BAY1895344, or BAY1895344+anti–PD-L1. *, P < 0.05; **, P < 0.01; ***, P < 0.001. P values determined by the Mantel–Cox test. ⁁, indicates that the combination treatment revealed a synergistic interaction between ATRi and anti–PD-L1 by Bliss independence analysis. G, Selective markers were used to define cell types in the data using Garnett, which include endothelial cells, epithelial (predominantly tumor) cells, macrophages, and T cells. H, GSEA for BAY1895344+anti–PD-L1 treatment versus BAY1895344 or anti–PD-L1 single-agent treatment in epithelial cell cluster. I, Conceptual mechanistic model for ATRi-mediated intrinsic cytotoxicity driven by activation of the CDK1–SPOP axis, suppression of PD-L1 levels through SPOP destabilization, and derepression of intrinsic IFN-β–driven apoptosis. Degradation of PD-L1 through the CDK1–SPOP axis activity leads to derepression of an IFN-β-IFNAR1-driven, STAT3–caspase 7 autocrine, cytotoxic signaling pathway (42). ATRi-mediated induction of cGAS mRNA through IFN-β–IFNAR1 signaling (45) completes a feed-forward loop.
We further analyzed the impact of ATR inhibition on the immune microenvironment, stroma, and tumor cells using single scRNA-seq (day 10 following treatment, see Fig. 6A). Epithelial (cancer cell)-, T-cell–, macrophage-, and endothelial cell–specific markers were used to identify cell types after clustering the single-cell data (Fig. 6G; Supplementary Fig. S6 and Supplementary Table S5). GSEA in the epithelial cell cluster demonstrated enrichment of IFN-γ signaling, IFN-β signaling, STING signaling, and JAK–STAT signaling pathways in ATRi+anti–PD-L1–treated cells compared with ATRi- or anti–PD-L1–treated cells (Fig. 6H). The apoptotic signaling pathway also illustrated a trend of enrichment in ATRi+anti–PD-L1–treated cells compared with ATRi-treated cells.
Discussion
The results of our study uncover a novel ATRi-induced, IFN-β–IFNAR1-driven, autocrine, cytotoxic signaling pathway in prostate cancer cells that underlies synergistic ATRi+anti–PD-L1 therapeutic response. ATRi can promote superior therapeutic effects in prostate cancer cells through abrogation of the ATR–Chk1–CDK1-regulated G2–M cell-cycle checkpoint, which leads to cell death and activation of cGAS–STING signaling (Fig. 6I). This mechanism differs from PARPi, which activates the ATR–Chk1–CDK1 cell-cycle checkpoint (G2–M arrest) through unrepaired DNA damage, but also leads to cell death and, in surviving cancer cells, activation of cGAS–STING signaling. The differences in the mechanisms of action for ATRi and PARPi demonstrated by our results point to distinctive properties of agents that specifically target RSS. These differences must be considered when selecting drug candidates for combination therapy approaches using DDRi-based therapies. ATRi- or PARPi-induced cGAS–STING activation results in increased IFN-β and ISG (CCL5 and CXCL10) expression. However, cGAS–STING output and IFN-β response are sustained and greater in ATRi-treated than in PARPi-treated prostate cancer cells (Figs. 1 and 4). Importantly, we demonstrate that, in opposition to PARPi, ATRi-induced abrogation of ATR–Chk1 signaling and activation of CDK1 results in activation of the CDK1–SPOP axis, which leads to destabilization and degradation of PD-L1 in prostate cancer cells. We also show that ATRi in the RM-1-BM syngeneic prostate cancer model leads to reduced PD-L1 protein levels (Supplementary Fig. S5B). Notably, previous studies have shown that Chk1 inhibition is similar to PARPi, in that both treatments lead to induction of PD-L1 in a syngeneic mouse small cell lung cancer model (11). Degradation of PD-L1 via the CDK1–SPOP axis leads to derepression of an IFN-β–IFNAR1-driven, STAT3–caspase 7 autocrine, cytotoxic signaling pathway (Figs. 2 and 4; ref. 42). We further demonstrate significant transcriptional induction of cGAS expression mediated by ATRi, reflecting a previously described IFN-β–IFNAR1 signaling activity that identified cGAS as an ISG (45), mechanistically constituting a feed-forward loop (Fig. 5). Together with the continued generation of immune-stimulatory cytoplasmic DNA, this signaling pathway sustains cGAS–STING activation in tumor cells that escape cell death, and induces intrinsic IFN-β expression and production of downstream ISG products. These ISG products, including CCL5 and CXCL10, in turn, stimulate cGAS–STING pathway activities in specific immune cells that comprise the tumor microenvironment (11, 17, 46–48). Analysis of epithelial tumor cells using scRNA-seq provided mechanistic insight into the synergistic effects of ATRi and anti–PD-L1 combination therapy in the RM-1-BM syngeneic prostatic cancer model. In particular, GSEA analysis demonstrated significantly greater induction of IFN-β signaling and the STING pathway, and apoptotic signaling in ATRi+anti–PD-L1–treated prostate cancer cells compared with single-agent treatment(s) in vivo (Fig. 6H). Increased IFN-β production within the tumor microenvironment certainly contributed to the marked increase in IFN-β signaling in tumor epithelial cells induced by ATRi and anti–PD-L1 combination therapy (Fig. 6D and E). In addition, although CD8+ T-cell depletion via anti-CD8 demonstrated that CD8+ T cells were key effectors of growth suppression in the RM-1-BM tumors treated with ATRi and anti–PD-L1, our results showed that T-cell–independent mechanisms contributed to the therapeutic response (Fig. 6C). Taken together, these results are consistent with a novel, intrinsic, IFN-β–driven, proapoptotic signaling mechanism, regulated by ATRi-mediated PD-L1 destablization, which can potentially augment T-cell–mediated tumor cell death induced by ATRi and anti–PD-L1 combination therapy in prostate cancer (Fig. 6I).
In addition to identifying this ATRi-induced, IFN-β–IFNAR1-driven, autocrine, cytotoxic signaling pathway in prostate cancer, our study also raises important considerations and potential applications for combined DDR targeting and ICB for patients with prostate cancer and other malignancies. First, suppression of ATR replication stress response signaling and activation of the CDK1–SPOP axis will likely lead to the identification of other SPOP functions that are relevant to this therapeutic approach. These functions may reveal novel predictive biomarkers and new therapy targets that will expand the landscape of DDR targeting and combination therapies. Second, our results demonstrate a mechanistic interaction between DDR targeting and immune checkpoint suppression that can inform clinical trials involving these agents, including our early-phase clinical trials combining ATR inhibition and immune checkpoint blockade in patients with advanced malignancies, including prostate cancer (NCT04266912 and NCT04095273). Overall, our findings provide underlying molecular rationale for combining ATR-targeted agents with ICB, and reveals mechanism-based opportunities for improving outcomes of therapeutic strategies using DDRi and I–O agents for men with advanced prostate cancer.
Authors' Disclosures
P.G. Pilié reports grants from Prostate Cancer Foundation and ASCO Career Development Award during the conduct of the study, as well as a patent for Replication Stress Response Biomarker pending. T.A. Yap reports grants from Artios, Constellation, Cyteir, Eli Lilly, Forbius, GlaxoSmithKline, Genentech, Immune Sensor, Ipsen, Jounce, Karyopharm, Kyowa, Novartis, Ribon Therapeutics, Regeneron, Sanofi, Scholar Rock, Tesaro, and Vertex Pharmaceuticals; grants and personal fees from AstraZeneca, Bayer, Clovis, EMD Serono, F-Star, Merck, Pfizer, Repare, and Seattle Genetics; and personal fees from Almac, Aduro, Atrin, Axiom, Bristol Myers Squibb, Calithera, Cybrexa, Guidepoint, Ignyta, I-Mab, Janssen, Roche, Rubius, Schrodinger, Varian, and Zai Labs outside the submitted work. T.C. Thompson reports grants from Bayer Healthcare Pharmaceuticals, Inc. outside the submitted work. No disclosures were reported by the other authors.
Supplementary Material
Supplementary Table S1 shows the origin and selected gene alteration status of C4-2b, PC-3, RM-9, RM-1 and RM-1-BM prostate cancer cell line models. Supplementary Table S2 shows details of the antibodies used in the experiments. Supplementary Table S3 shows primer sequences for RT-qPCR analysis. Supplementary Table S4 shows siRNA sequences used in the experiments. Supplementary Table S5 shows quantification of cell types among treatments derived from single cell RNA sequencing data.
Supplementary Figure S1 shows androgen responsiveness of RM-9 and RM-1-BM prostate cancer cell lines including immunoblot analysis of AR and PD-L1 protein expression and response to increasing concentrations of enzalutamide (MTS analysis).
{kind=link}
Supplementary Figure S2 shows generation of cytosolic dsDNA (PicoGreen staining) in RM-9, RM-1 and RM-1-BM prostate cancer cells treated by BAY1895344, and BAY1895344- or olaparib-dose dependent activation of cGAS-STING signaling pathway in C4-2b and PC-3 prostate cancers (immunoblot analysis).
{kind=link}
Supplementary Figure S3 shows siRNA knockdown efficiencies of gene specific siRNA transfections in RM-1-BM prostate cancer cells (immunoblot analysis).
{kind=link}
Supplementary Figure S4 shows ATRi or ATR siRNA knockdown downregulates PD-L1 protein expression in RM-1-BM prostate cancer cells (immunoblot analysis), and SPOP siRNA transfection does not alter PD-L1 mRNA levels in RM-1-BM and C4-2b prostate cancer cells (suggesting SPOP post-transcriptional regulation of PD-L1 expression).
{kind=link}
Supplementary Figure S5 shows ATRi, anti-PD-L1 or ATRi and anti-PD-L1 effects on tumor weights, and the effects of ATRi on PD-L1 in tumor tissues in the RM-1-BM syngeneic prostate cancer tumor model.
{kind=link}
Supplementary Figure S6 shows expression level of markers used to annotate cell types from analysis of single cell RNA sequencing data.
{kind=link}
Acknowledgments
We acknowledge the editorial assistance of Sarah E. Townsend. This research was supported by MD Anderson NCI Prostate Cancer SPORE grant P50 CA140388, the NCI Cancer Center Support grant P30 CA16672, and a Young Investigator's Award from the Prostate Cancer Foundation (to P.G. Pilié).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Contributions
Z. Tang: Formal analysis, investigation, methodology, writing–original draft. P.G. Pilié: Conceptualization, data curation, formal analysis, writing–original draft, project administration, writing–review and editing. C. Geng: Conceptualization, data curation, formal analysis, supervision, investigation, methodology, writing–original draft, project administration, writing–review and editing. G.C. Manyam: Conceptualization, formal analysis, investigation, methodology, writing–original draft, writing–review and editing. G. Yang: Formal analysis, investigation, methodology, writing–original draft. S. Park: Formal analysis, supervision, investigation, methodology, writing–original draft, project administration. D. Wang: Investigation, methodology. S. Peng: Investigation, methodology. C. Wu: Investigation, methodology. G. Peng: Conceptualization, methodology. T.A. Yap: Conceptualization, writing–review and editing. P.G. Corn: Conceptualization, writing–review and editing. B.M. Broom: Conceptualization, formal analysis, supervision, funding acquisition, methodology, writing–original draft, project administration, writing–review and editing. T.C. Thompson: Conceptualization, data curation, supervision, funding acquisition, writing–original draft, project administration, writing–review and editing.
References
- 1. Beer TM, Kwon ED, Drake CG, Fizazi K, Logothetis C, Gravis G, et al. Randomized, double-blind, phase III trial of ipilimumab versus placebo in asymptomatic or minimally symptomatic patients with metastatic chemotherapy-naive castration-resistant prostate cancer. J Clin Oncol 2017;35:40–7. [DOI] [PubMed] [Google Scholar]
- 2. Karzai F, VanderWeele D, Madan RA, Owens H, Cordes LM, Hankin A, et al. Activity of durvalumab plus olaparib in metastatic castration-resistant prostate cancer in men with and without DNA damage repair mutations. J Immunother Cancer 2018;6:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015;162:454. [DOI] [PubMed] [Google Scholar]
- 4. Subudhi SK, Vence L, Zhao H, Blando J, Yadav SS, Xiong Q, et al. Neoantigen responses, immune correlates, and favorable outcomes after ipilimumab treatment of patients with prostate cancer. Sci Transl Med 2020;12:eaaz3577. [DOI] [PubMed] [Google Scholar]
- 5. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N Engl J Med 2012;366:2443–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wu YM, Cieslik M, Lonigro RJ, Vats P, Reimers MA, Cao X, et al. Inactivation of CDK12 delineates a distinct immunogenic class of advanced prostate cancer. Cell 2018;173:1770–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sharma P, Pachynski RK, Narayan V, Flechon A, Gravis G, Galsky MD, et al. Nivolumab plus ipilimumab for metastatic castration-resistant prostate cancer: preliminary analysis of patients in the CheckMate 650 trial. Cancer Cell 2020;38:489–99. [DOI] [PubMed] [Google Scholar]
- 8. Pritchard CC, Mateo J, Walsh MF, De Sarkar N, Abida W, Beltran H, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med 2016;375:443–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mateo J, Porta N, Bianchini D, McGovern U, Elliott T, Jones R, et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): a multicentre, open-label, randomised, phase 2 trial. Lancet Oncol 2020;21:162–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. de Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med 2020;382:2091–102. [DOI] [PubMed] [Google Scholar]
- 11. Sen T, Rodriguez BL, Chen L, Corte CMD, Morikawa N, Fujimoto J, et al. Targeting DNA damage response promotes antitumor immunity through sting-mediated T-cell activation in small-cell lung cancer. Cancer Discov 2019;9:646–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shen J, Zhao W, Ju Z, Wang L, Peng Y, Labrie M, et al. PARPi triggers the STING-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCAness. Cancer Res 2019;79:311–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yu EY, Wu H, Schloss C. KEYNOTE-365: phase 1b/2 trial of pembrolizumab combination therapy for metastatic castration-resistant prostate cancer (mCRPC). European Urology Supplements 2017;16:e360. [Google Scholar]
- 14. Pilie PG, Tang C, Mills GB, Yap TA. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat Rev Clin Oncol 2019;16:81–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wengner AM, Siemeister G, Lucking U, Lefranc J, Wortmann L, Lienau P, et al. The novel ATR inhibitor BAY 1895344 is efficacious as monotherapy and combined with DNA damage-inducing or repair-compromising therapies in preclinical cancer models. Mol Cancer Ther 2020;19:26–38. [DOI] [PubMed] [Google Scholar]
- 16. Yap TA, O'Carrigan B, Penney MS, Lim JS, Brown JS, de Miguel Luken MJ, et al. Phase I trial of first-in-class ATR inhibitor M6620 (VX-970) as monotherapy or in combination with carboplatin in patients with advanced solid tumors. J Clin Oncol 2020;38:3195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pantelidou C, Sonzogni O, De Oliveria Taveira M, Mehta AK, Kothari A, Wang D, et al. PARP inhibitor efficacy depends on CD8(+) T-cell recruitment via intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast cancer. Cancer Discov 2019;9:722–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ding L, Kim HJ, Wang Q, Kearns M, Jiang T, Ohlson CE, et al. PARP inhibition elicits STING-dependent antitumor immunity in brca1-deficient ovarian cancer. Cell Rep 2018;25:2972–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pilie PG, Gay CM, Byers LA, O'Connor MJ, Yap TA. PARP inhibitors: extending benefit beyond BRCA-mutant cancers. Clin Cancer Res 2019;25:3759–71. [DOI] [PubMed] [Google Scholar]
- 20. Sun LL, Yang RY, Li CW, Chen MK, Shao B, Hsu JM, et al. Inhibition of ATR downregulates PD-L1 and sensitizes tumor cells to T-cell–mediated killing. Am J Cancer Res 2018;8:1307–16. [PMC free article] [PubMed] [Google Scholar]
- 21. Sato H, Niimi A, Yasuhara T, Permata TBM, Hagiwara Y, Isono M, et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat Commun 2017;8:1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Power CA, Pwint H, Chan J, Cho J, Yu Y, Walsh W, et al. A novel model of bone-metastatic prostate cancer in immunocompetent mice. Prostate 2009;69:1613–23. [DOI] [PubMed] [Google Scholar]
- 23. Thompson TC, Southgate J, Kitchener G, Land H. Multistage carcinogenesis induced by ras and myc oncogenes in a reconstituted organ. Cell 1989;56:917–30. [DOI] [PubMed] [Google Scholar]
- 24. Lu X, Park SH, Thompson TC, Lane DP. Ras-induced hyperplasia occurs with mutation of p53, but activated ras and myc together can induce carcinoma without p53 mutation. Cell 1992;70:153–61. [DOI] [PubMed] [Google Scholar]
- 25. Baley PA, Yoshida K, Qian W, Sehgal I, Thompson TC. Progression to androgen insensitivity in a novel in vitro mouse model for prostate cancer. J Steroid Biochem Mol Biol 1995;52:403–13. [DOI] [PubMed] [Google Scholar]
- 26. Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun 2017;8:14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 2014;32:381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM III, et al. Comprehensive integration of single-cell data. Cell 2019;177:1888–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pliner HA, Shendure J, Trapnell C. Supervised classification enables rapid annotation of cell atlases. Nat Methods 2019;16:983–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. BLISS CI. The toxicity of poisons applied JOINTLY1. Ann Appl Biol 1939;26:585–615. [Google Scholar]
- 33. Soki FN, Cho SW, Kim YW, Jones JD, Park SI, Koh AJ, et al. Bone marrow macrophages support prostate cancer growth in bone. Oncotarget 2015;6:35782–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Watanabe M, Yang G, Cao G, Tahir SA, Naruishi K, Tabata K, et al. Functional analysis of secreted caveolin-1 in mouse models of prostate cancer progression. Mol Cancer Res 2009;7:1446–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tse BW, Russell PJ, Lochner M, Forster I, Power CA. IL-18 inhibits growth of murine orthotopic prostate carcinomas via both adaptive and innate immune mechanisms. PLoS ONE 2011;6:e24241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Saldivar JC, Cortez D, Cimprich KA. The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol 2017;18:622–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lecona E, Fernandez-Capetillo O. Targeting ATR in cancer. Nat Rev Cancer 2018;18:586–95. [DOI] [PubMed] [Google Scholar]
- 38. Lemmens B, Hegarat N, Akopyan K, Sala-Gaston J, Bartek J, Hochegger H, et al. DNA replication determines timing of mitosis by restricting CDK1 and PLK1 activation. Mol Cell 2018;71:117–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Szmyd R, Niska-Blakie J, Diril MK, Renck Nunes P, Tzelepis K, Lacroix A, et al. Premature activation of Cdk1 leads to mitotic events in S phase and embryonic lethality. Oncogene 2019;38:998–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT, et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 2018;553:91–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. van den Heuvel S, Harlow E. Distinct roles for cyclin-dependent kinases in cell-cycle control. Science 1993;262:2050–4. [DOI] [PubMed] [Google Scholar]
- 42. Gato-Canas M, Zuazo M, Arasanz H, Ibanez-Vea M, Lorenzo L, Fernandez-Hinojal G, et al. PDL1 signals through conserved sequence motifs to overcome interferon-mediated cytotoxicity. Cell Rep 2017;20:1818–29. [DOI] [PubMed] [Google Scholar]
- 43. Sanceau J, Hiscott J, Delattre O, Wietzerbin J. IFN-beta induces serine phosphorylation of Stat-1 in Ewing's sarcoma cells and mediates apoptosis via induction of IRF-1 and activation of caspase-7. Oncogene 2000;19:3372–83. [DOI] [PubMed] [Google Scholar]
- 44. Saito R, Mizuno M, Hatano M, Kumabe T, Yoshimoto T, Yoshida J. Two different mechanisms of apoptosis resistance observed in interferon-beta induced apoptosis of human glioma cells. J Neurooncol 2004;67:273–80. [DOI] [PubMed] [Google Scholar]
- 45. Ma F, Li B, Liu SY, Iyer SS, Yu Y, Wu A, et al. Positive feedback regulation of type I IFN production by the IFN-inducible DNA sensor cGAS. J Immunol 2015;194:1545–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Francica BJ, Ghasemzadeh A, Desbien AL, Theodros D, Sivick KE, Reiner GL, et al. TNFalpha and radioresistant stromal cells are essential for therapeutic efficacy of cyclic dinucleotide STING agonists in nonimmunogenic tumors. Cancer Immunol Res 2018;6:422–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Iurescia S, Fioretti D, Rinaldi M. Targeting cytosolic nucleic acid-sensing pathways for cancer immunotherapies. Front Immunol 2018;9:711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bakhoum SF, Cantley LC. The multifaceted role of chromosomal instability in cancer and its microenvironment. Cell 2018;174:1347–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1 shows the origin and selected gene alteration status of C4-2b, PC-3, RM-9, RM-1 and RM-1-BM prostate cancer cell line models. Supplementary Table S2 shows details of the antibodies used in the experiments. Supplementary Table S3 shows primer sequences for RT-qPCR analysis. Supplementary Table S4 shows siRNA sequences used in the experiments. Supplementary Table S5 shows quantification of cell types among treatments derived from single cell RNA sequencing data.
Supplementary Figure S1 shows androgen responsiveness of RM-9 and RM-1-BM prostate cancer cell lines including immunoblot analysis of AR and PD-L1 protein expression and response to increasing concentrations of enzalutamide (MTS analysis).
Supplementary Figure S2 shows generation of cytosolic dsDNA (PicoGreen staining) in RM-9, RM-1 and RM-1-BM prostate cancer cells treated by BAY1895344, and BAY1895344- or olaparib-dose dependent activation of cGAS-STING signaling pathway in C4-2b and PC-3 prostate cancers (immunoblot analysis).
Supplementary Figure S3 shows siRNA knockdown efficiencies of gene specific siRNA transfections in RM-1-BM prostate cancer cells (immunoblot analysis).
Supplementary Figure S4 shows ATRi or ATR siRNA knockdown downregulates PD-L1 protein expression in RM-1-BM prostate cancer cells (immunoblot analysis), and SPOP siRNA transfection does not alter PD-L1 mRNA levels in RM-1-BM and C4-2b prostate cancer cells (suggesting SPOP post-transcriptional regulation of PD-L1 expression).
Supplementary Figure S5 shows ATRi, anti-PD-L1 or ATRi and anti-PD-L1 effects on tumor weights, and the effects of ATRi on PD-L1 in tumor tissues in the RM-1-BM syngeneic prostate cancer tumor model.
Supplementary Figure S6 shows expression level of markers used to annotate cell types from analysis of single cell RNA sequencing data.