

Abstract
Moloney Murine Leukemia Virus (M-MLV) proviral DNA is transcriptionally silenced in embryonic cells by a large repressor complex tethered to the provirus by two sequence-specific DNA binding proteins, ZFP809 and YY1. A central component of the complex is Trim28, a scaffold protein that regulates many target genes involved in cell cycle progression, DNA damage responses, and viral gene expression. The silencing activity of Trim28, and its interactions with corepressors are often regulated by post-translational modifications such as sumoylation and phosphorylation. We defined the interaction domains of Trim28 and YY1, and investigated the role of sumoylation and phosphorylation of Trim28 in mediating M-MLV silencing. The RBCC domain of Trim28 was sufficient for interaction with YY1, and acidic region 1 and zinc fingers of YY1 were necessary and sufficient for its interaction with Trim28. Additionally, we found that residue K779 was critical for Trim28-mediated silencing of M-MLV in embryonic cells.
Keywords: Transcriptional silencing, Moloney murine leukemia virus, Trim28, YY1, SUMO, Phosphorylation, Protein-protein interaction
1. Introduction
M-MLV is a prototypical retrovirus that replicates efficiently in mouse cells in culture, and induces a T-cell leukemia in mice by insertional activation of endogenous protooncogenes. While M-MLV can carry out a productive infection in most cell types, virus replication is potently restricted in embryonic stem (ES) cells and embryonic carcinoma (EC) cells (Teich et al., 1977). M-MLV entry, reverse transcription, and viral DNA integration into the host genome occurs normally (Stewart et al., 1982), but the replication cycle is blocked at the transcriptional level (Gautsch and Wilson, 1983; Niwa et al., 1983). Transcriptional silencing of M-MLV in embryonic cells is attributable in some part to low levels of transcriptional enhancer activity (Hilberg et al., 1987; Linney et al., 1984) and in large part to the repressive activity of a trans-acting silencing complex (Loh et al., 1990; Petersen et al., 1991). The repressor complex mediates proviral silencing by inducing chromatin modifications and by de novo methylation of the provirus DNA (Stewart et al., 1982; Niwa et al., 1983; Leung et al., 2011).
Recent studies have identified some key components of the trans-acting silencing complex. Trim28 (also known as TIF1β/KAP1) is a co-repressor for a large family of transcriptional repressors known as the Krüppel-associated box (KRAB) domain-containing zinc-finger proteins (ZFPs) (Wolf and Goff, 2007). KRAB-ZFPs bind to specific DNA sequences and recruit Trim28 by their KRAB domain (Wolfe et al., 2000). In turn, Trim28 serves as a scaffold for additional co-repressor proteins, including heterochromatin protein 1 (HP1), the nucleosome remodeler CHD3 of the NuRD complex, and the H3K9 m6ethyltransferase ESET, which collectively contribute to heterochromatin formation and histone modifications to maintain long-term gene silencing (Le Douarin et al., 1996; Matsui et al., 2010; Schultz et al., 2002, 2001; Wolf et al., 2008). Trim28 interacts with these co-repressor proteins using several of its various domains, including the ring B1-B2 coiled-coil (RBCC) domain, chromoshadow domain, plant homeodomain (PHD), and bromodomain. The KRAB-ZFPs interact with the RBCC domain, HP1 interacts with the chromoshadow domain, and CHD3 and ESET interact with the plant homeodomain (PHD) and bromodomain (Schultz et al., 2002, 2001; Friedman et al., 1996; Lechner et al., 2000). Thus, Trim28 is the central repressor factor in the M-MLV silencing complex, linking repressor functions to specific gene targets.
In the context of M-MLV silencing, Trim28 is directed to the 5′ long-terminal repeat (LTR) with the aid of two ZFPs, ZFP809 and Yin Yang 1 (YY1). ZFP809 is a KRAB-ZFP that specifically binds to the primer binding site (PBS), an 18-nucleotide sequence just downstream of the 5′ LTR of M-MLV and a major site of repression (Wolf and Goff, 2007, 2009; Barklis et al., 1986; Loh et al., 1987). Mutation of the PBS region or knockdown of ZFP809 significantly reduces M-MLV silencing in embryonic cells (Barklis et al., 1986; Wolf and Goff, 2009). YY1 is a transcription factor that binds to another known site for repression, the negative control region (NCR) located in the M-MLV LTR (Flanagan et al., 1992, 1989). Deletion of the NCR or YY1 knockdown reduces M-MLV silencing and heterochromatin modifications on the provirus (Schlesinger et al., 2013). Unlike ZFP809, YY1 does not contain a KRAB domain, but coimmunoprecipitation (ci-IP) experiments show that YY1 also binds Trim28. Together, ZFP809 and YY1 recruit the Trim28 silencing complex to the provirus, resulting in a potent transcriptional silencing of M-MLV in embryonic cells.
One unexplored aspect of the YY1-Trim28 silencing complex is the nature of the biochemical interaction of the two proteins. As YY1 does not contain a domain predicted to interact with Trim28, it is unknown how this interaction occurs. An intriguing finding demonstrated that the YY1-Trim28 interaction occurred in embryonic cells but not in differentiated cells, even though YY1 and Trim28 are expressed in both cell types (Schlesinger et al., 2013). This suggests that the YY1-Trim28 interaction could be differentially regulated at a post-translational level. A possible mechanism for regulating this interaction includes the presence of supporting co-factors or absence of inhibitory co-factors. Another possibility is that post-translational modifications regulate the YY1-Trim28 interaction. There are no strong candidates for possible co-factors involved in the interaction, but there are a number of studies indicating the importance of sumoylation and phosphorylation modifications for regulating Trim28 interactions and repressive activity in the context of the DNA damage response pathway, cell cycle progression, and viral latency (Goodarzi et al., 2011; Ivanov et al., 2007; Lee et al., 2007; Li et al., 2007; Rauwel et al., 2015; Ziv et al., 2006).
Sumoylation is the covalently conjugation of a small ubiquitin-like modifier (SUMO) to a substrate, and this modification has been implicated in the regulation of various cellular processes such as transcription, DNA repair, transport, subcellular localization, and cell division (Geiss-Friedlander and Melchior, 2007; Verger et al., 2003). The mammalian SUMO family consists of SUMO1, the founding member of the family, and its relatives SUMO2, SUMO3, and SUMO4. SUMO1 has only 50% sequence identity with SUMO2/3 and demonstrates distinct cellular functions from SUMO2/3. SUMO2 and SUMO3 are 97% identical and do not show apparent functional differences. SUMO4 expression is restricted to a few cell types (Geiss-Friedlander and Melchior, 2007; Bohren et al., 2004). Protein targets of SUMO conjugation generally contain the tetrapeptide consensus motif, Ψ-K-x-D/E, where Ψ is a hydrophobic residue, K is the lysine that is conjugated to SUMO, x is any amino acid, and D/E is an acidic residue. Before SUMOs can be ligated to their targets, they need to be cleaved by SUMO-specific proteases (SENPs) to expose diglycine residues at the C-terminus, producing the mature SUMOs. SUMO conjugation can be reversed by the same SENPs that process the SUMOs into its mature form, and most substrates undergo rapid cycles of conjugation and deconjugation.
Trim28 contains six lysine residues that can be conjugated by the SUMOs: K554, K575, K676, K750, K779, and K804. Of these six, K554, K779, and K804 are previously reported to be the most important sites for Trim28 repressive activity (Goodarzi et al., 2011; Ivanov et al., 2007; Lee et al., 2007; Mascle et al., 2007). In a yeast two-hybrid assay, the interaction of Trim28 with ESET was disrupted by a mutation at K676, and the Trim28 interaction with CHD3 and ESET was disrupted by a double mutation at K676 and K779 (Ivanov et al., 2007). In mammalian cells, mutation of K554, K779, and K804 to arginine resulted in chromatin relaxation and in transcriptional derepression of the p21 promoter and of a reporter gene in a Gal4-based system (Lee et al., 2007; Mascle et al., 2007). While most of the work on Trim28 sumoylation has focused on SUMO1, SUMO2 has been specifically implicated in Trim28-mediated viral silencing. A recent genome-wide siRNA screen for proviral silencing factors in embryonic cells identified SUMO2 and the SUMO2 conjugating enzymes, but not SUMO1 or SUMO3 (Yang et al., 2015). Interestingly, the PHD of Trim28 functions as an E3 ligase and contributes to its auto-sumoylation as well as to the sumoylation of other proteins (Ivanov et al., 2007; Liang et al., 2011).
Phosphorylation of Trim28 abrogates its repressive activity and reduces SUMO conjugation of Trim28 (Goodarzi et al., 2011; Li et al., 2007; Rauwel et al., 2015). Specifically, phosphorylation of two residues, S824 and S473, reduces Trim28 repressive activity by perturbing the interaction of Trim28 with CHD3 and HP1, respectively (Goodarzi et al., 2011; Chang et al., 2008). Trim28 is also involved in establishing human cytomegalovirus (HCMV) latency in hematopoietic stem cells, and phosphorylation of Trim28 at the S824 residue relieves latency (Rauwel et al., 2015). It is unknown if these modifications are critical for Trim28-mediated repression of M-MLV in embryonic cells.
Here, we mapped the domains involved in the YY1-Trim28 interaction. We found that recombinant YY1 and Trim28 do not detectably interact in vitro. Sumoylation and phosphorylation were not found to be necessary for the YY1-Trim28 interaction in vivo, but the K779 residue in Trim28 was necessary for the silencing of M-MLV. We did not find the K554, K804, S473, or S824 residues to be important for Trim28 repression of M-MLV, suggesting that there are significant differences between the Trim28-dependent repressive mechanisms occurring on M-MLV in embryonic cells and those involved in the DNA repair response, cell cycle progression, and HCMV latency (Goodarzi et al., 2011; Ivanov et al., 2007; Lee et al., 2007; Rauwel et al., 2015).
2. Results
2.1. Recombinant YY1 and TRIM28 do not interact in vitro
In a previous study, we demonstrated that YY1 mediates the silencing of M-MLV in embryonic cells by recruiting Trim28 to the negative control region (NCR) in the 5′LTR of the provirus (Schlesinger et al., 2013). Although YY1 does not contain a domain predicted to interact with Trim28, we found that YY1 coimmunoprecipitates (co-IPs) with Trim28 in nuclear lysates of F9 embryonic cells but not of the differentiated NIH3T3 cells. These results prompted us to explore the nature of the YY1-Trim28 interaction and investigate possible mechanisms for regulating the YY1-Trim28 interaction and silencing activity. To test for a direct interaction between YY1 and Trim28, we conducted binding experiments using recombinant proteins. Trim28 has been demonstrated to interact directly with HP1, so this interaction was used as a positive control (Le Douarin et al., 1996). We expressed recombinant His6-tagged YY1, His6-tagged Trim28, and His6-Flag-tagged HP1 in bacteria and purified the recombinant proteins by nickel affinity chromatography. rYY1 was incubated with anti-YY1 antibody or a control antibody, the antibodies were loaded onto protein A/G beads, and the beads were incubated with rTrim28, under conditions of neutral pH, low salt, and low detergent concentration. In parallel, rFlag-HP1 was incubated with anti-Flag beads, followed by the addition of rTrim28. Bound proteins were removed from the beads and analyzed by Western blot. We could not detect rTrim28 bound to rYY1 above background using this approach. In contrast, we readily detected rTrim28 bound to rFlag-HP1 (Fig. 1). We performed similar experiments with an alternative binding buffer (EMSA buffer, see methods) or longer incubation periods, but under no conditions could we detect an interaction between rYY1 and rTrim28. We also generated GST-tagged rTrim28 protein, loaded the protein onto glutathione beads, and incubated the bound rTrim28 protein with rYY1. Proteins were removed from the beads and analyzed by Western blot probed with anti-YY1 antibody. We could not detect significant binding of rYY1 to rTrim28 with this approach (data not shown).
Fig. 1. Interaction between rYY1 and rTrim28.
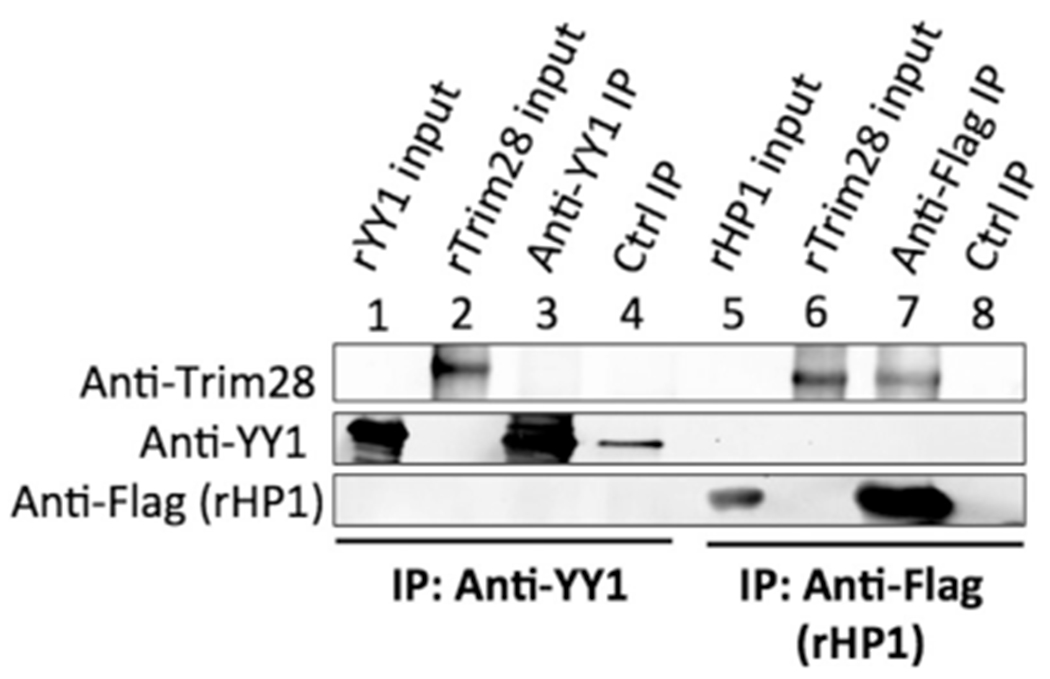
Lanes 1–4: rYY1 was incubated with anti-YY1 or control rabbit antiserum, and antibodies were then loaded onto protein A/G beads. The beads were incubated with rTrim28, then washed and analyzed by Western blot, probed with anti-Trim28, or anti-YY1 antibodies as indicated at left. Lanes 1 and 2: 10% of total input recombinant proteins. Lane 3: IP with anti-YY1 antibody. Lane 4: IP with control rabbit antibody. No rTrim28 was detected with anti-YY1 IP. Lanes 5–8: rFlag-HP1 was loaded onto anti-Flag beads. Beads were incubated with rTrim28, then washed and analyzed by Western blot, probed with anti-Trim28, or anti-Flag antibodies as indicated at left. Lanes 5 and 6: 10% of total input recombinant proteins. Lane 7: IP with anti-Flag antibody. Lane 8: IP with control rabbit antibody. rTrim28 was readily detected bound to rFlag-HP1.
One possible explanation for the failure to detect an interaction is that a portion of the YY1 antibody could have blocked the binding site for rTrim28. To test for this possibility, we conducted in vitro co-IPs with a different order of protein addition. rYY1 and rTrim28 were first incubated together, and rYY1 was subsequently immunoprecipitated using anti-YY1 antibody and protein A/G beads. Bound proteins were removed from the beads and analyzed by Western blot probing for Trim28. We again could not detect rTrim28 in the immunoprecipitates above background (data not shown). The inability of rYY1 and rTrim28 to co-IP in vitro suggests that their interaction in vivo may require protein modifications, or bridging cofactors.
2.2. Trim28 RBCC domain is necessary and sufficient for interaction with YY1
Further examination of the YY1-Trim28 interaction was carried out using immunoprecipitation from mammalian cell lysates. To determine the domains on Trim28 responsible for the YY1-Trim28 interaction, we conducted co-IP experiments with HA-tagged Trim28 mutants in F9 embryonic carcinoma cells, in which M-MLV silencing occurs. Engineering expression of Trim28 constructs, and depleting the endogenous wild-type Trim28, proved challenging. As F9 cells are difficult to transfect and only weakly recognize many promoters, all overexpression experiments were conducted by lentiviral transduction mediating expression of Trim28 and a drug resistance gene, driven by the EF1α promoter. Cells expressing exogenous Trim28 were first selected for stable expression of the drug resistance gene, followed by knockdown (KD) of endogenous Trim28 by shRNA. We found that endogenous Trim28 expression returned after prolonged culture, likely because Trim28 is critical for long-term embryonic cell function and survival (Cammas et al., 2000). Thus, Trim28 was knocked down immediately prior to using the cells in our various assays, and KD efficiency was confirmed by Western blot.
To determine which Trim28 domain(s) were necessary for its interaction with YY1, we expressed mutant forms of HA-tagged Trim28 with deletions of its major domains, generating Trim281–628 (with deletion of the PHD/Bromodomain), Trim28Δ441–522 (with deletion of the chromoshadow domain, denoted “PxVxL” for its pentapeptide sequence), and Trim28388–834 (with deletion of the RBCC domain) (Fig. 2A). An HA-Trim28WT cell line was also produced as a positive control. Cell lysates were incubated with specific antibodies, and bound proteins were isolated with protein A/G beads, displayed on SDS PAGE and transferred to Western blots, and probed for HA-Trim28 (Fig. 2B). All mutant constructs expressed well except for Trim28388–834, the construct missing the RBCC domain. Trim28WT, Trim281–628, and Trim28Δ441–522 were found to interact with endogenous YY1, indicating that the PHD/Bromodomain and chromoshadow domain are not necessary for Trim28 interaction with YY1. We did not detect HA-Trim28 or YY1 in any of the immunoprecipitates with the control antibody, demonstrating the specificity of the anti-YY1 antibody. To confirm the importance of the RBCC domain, we expressed a Trim28 mutant containing only the RBCC domain (Trim281–441) (Fig. 2A). Cell lysates were prepared, YY1 was immunoprecipitated, and bound proteins were examined by Western blot probing for HA-Trim28. The Trim281–441 mutant was detected with the immunoprecipitation of YY1 (Fig. 2B), demonstrating that the RBCC domain is sufficient for Trim28 interaction with YY1.
Fig. 2. Co-IP of YY1 with Trim28 mutants containing deletions of major domains.
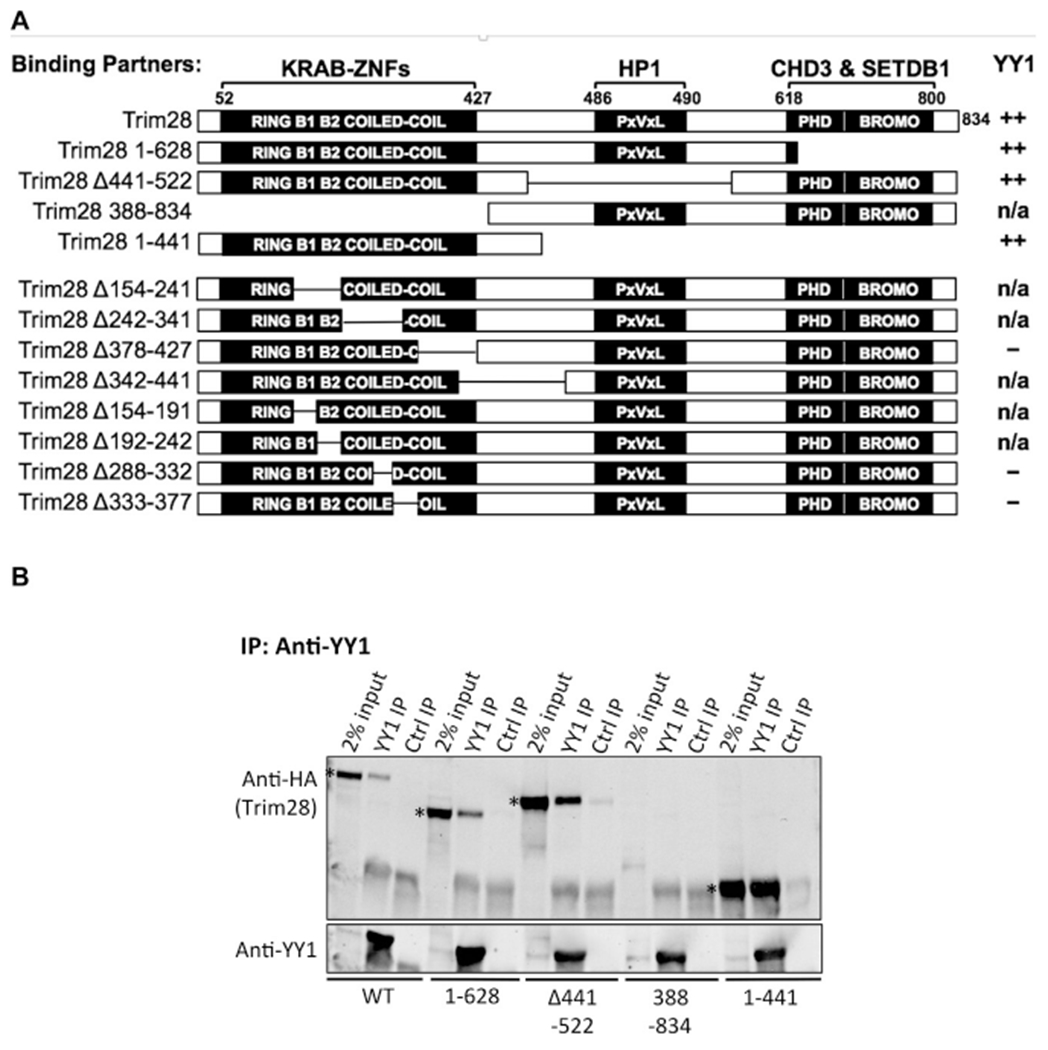
(A) Schematic of the Trim28 wild-type and mutant proteins. All mutants were HA-tagged at the N-terminus. Domains known to interact with other repressor partners are noted above the domain. The extent of interaction of Trim28 mutants with endogenous YY1 is indicated in the right column. A strong interaction is denoted by “++”, no interaction is indicated by “–”, and mutants that displayed poor expression are noted by n/a. (B) HA-Trim28 constructs were overexpressed in F9 cells, endogenous YY1 was immunoprecipitated with anti-YY1 antibody, and bound proteins were examined by Western blot probed with anti-HA antibody. For each mutant, groups of three lanes are shown: 2% of the total protein input to the IPs, YY1 IP, and control IP. Examples of positive and a poorly expressed mutant protein are shown. The bands corresponding to the Trim28 mutants positive for binding are marked in the input lanes by an asterisk.
We attempted to identify smaller domains of Trim28 that would be sufficient for YY1 interaction by making additional deletions in the RBCC domain (constructs Trim28Δ154–241, Trim28Δ242–341, Trim28Δ378–427, Trim28Δ342–441) (Fig. 2A). Lysates were prepared, incubated with YY1 on beads, and bound proteins were examined by Western blot. Of these mutants, only Trim28Δ378–427 expressed well, as indicated in the 2% input lanes for each mutant cell line. We did not detect an interaction between YY1 and Trim28Δ378–427, while control Trim28WT expressed well and interacted with YY1 (Supplementary Fig. 1, upper panel). To determine if smaller deletions in the RBCC could allow for Trim28 expression, we tested Trim28 constructs with 50 amino acid deletions (Trim28Δ154–191, Trim28Δ192–242, Trim28Δ288–332, Trim28Δ333–377) but these mutants also showed low or no expression (Supplementary Fig. 1, lower panel). These experiments suggest that an intact RBCC domain is important for Trim28 expression and that deletions in this region may be disruptive to its structure. The smallest region of Trim28 we could define as sufficient for the interaction with YY1 was the RBCC domain.
2.3. YY1 acidic region 1 and zinc fingers are necessary for interaction with Trim28
To determine the domains on YY1 necessary for interaction with Trim28, a panel of myc-tagged YY1 mutants were expressed in F9 cells and tested for their interaction with endogenous Trim28. The major domains of YY1, starting from the N-terminus, are the acidic region 1 (AR1), acidic region 2 (AR2), GA-rich region (GA), and the four zinc fingers (ZNFs). We were unable to produce a construct with a deleted AR1 region, but constructs with deletions of each of the other regions were made: YY1Δ54–144 (with deletion of the AR2), YY1Δ151–209 (with deletion of the GA), YY1Δ177–273 (with deletion of the spacer), and YY11–295 (with deletion of the ZNFs) (Fig. 3A). The empty vector was used as a negative control, and a YY1 wild-type construct (YY1WT) was used as a positive control. F9 cells were transduced with the empty vector, or the YY1WT or mutant YY1 vectors, and were selected for stable expression of the drug resistant gene. Lysates were prepared and exogenous YY1 was immunoprecipitated using anti-myc beads. Bound proteins were removed from the beads and analyzed by Western blot probed with an anti-Trim28 antibody (Fig. 3B, upper panel). Immunoprecipitation of myc-YY1, in the YY1WT cell lysate, demonstrated that Trim28 interacted with exogenous YY1. The low amount of Trim28 detected in the immunoprecipitates from the empty vector cell lysates reports the background Trim28 levels. The intensities of the various Trim28 bands were quantified, and signals above empty vector control were calculated relative to that seen with the YY1WT (Supplementary Table 1). Significant levels of Trim28 interacted with YY1 lacking the AR2 domain (YY1Δ54–144), indicating that this domain is not necessary for the interaction. However, only background levels of Trim28 levels were bound to myc-YY1Δ151–209, myc-YY1Δ177–273, and myc-YY11–295. We constructed additional mutants containing only the N-terminal domains (YY11–209), only the C-terminal domains (YY1Δ142–414 and YY1198–414), or both terminal domains (YY1Δ54–209, YY1Δ54–295, and YY1Δ85–144, Δ177–273) of YY1 (Fig. 3A). F9 cells expressing these mutants were generated and tested for binding Trim28 as before (Fig. 3B, lower panel). Mutants containing only the N-terminal domains (YY11–209) or only the C-terminal domains (YY1142–414, YY1198–414) did not interact with endogenous Trim28. On the other hand, YY1 mutants containing both terminal domains (YY1Δ54–209, YY1Δ54–295, and YY1Δ85–144, Δ177–273) did bind Trim 28. Of these mutants, YY1Δ54–295 was the mutant with the fewest domains that still successfully interacted with Trim28. These results demonstrate that the AR2, GA, and spacer are dispensable and that the combination of AR1 and ZNF domains are necessary and sufficient for YY1 interaction with Trim28.
Fig. 3. Co-IP of Trim28 and YY1 mutants.
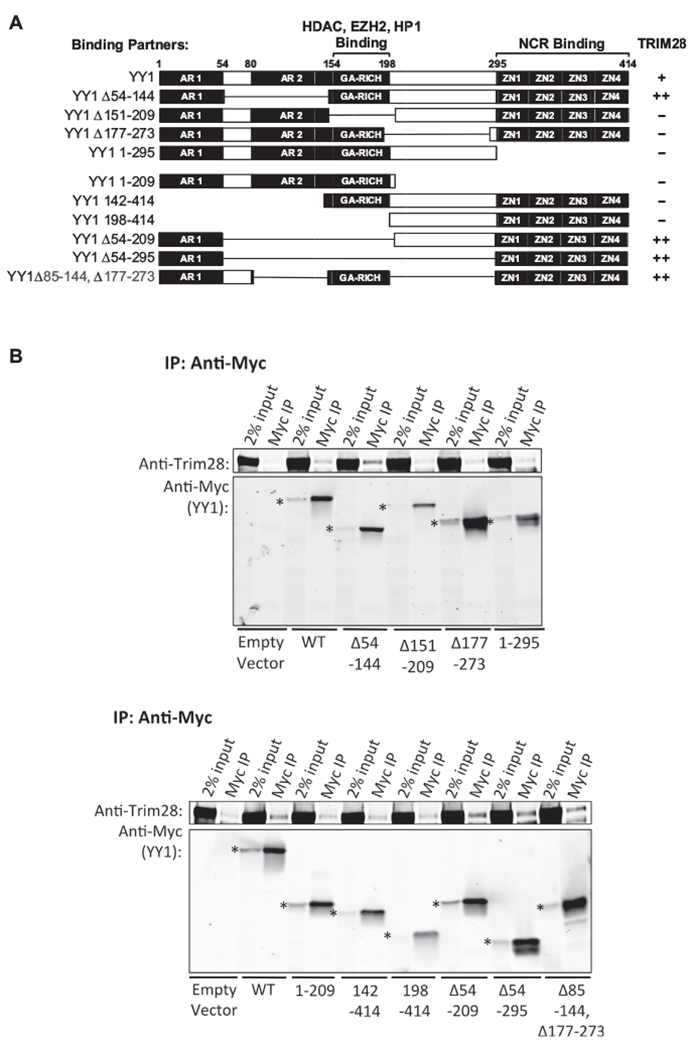
(A) Schematic of YY1 wild-type and mutant proteins. All mutants were myc-tagged at the N-terminus. Domains known to interact with other repressor partners are noted above the domain. Extent of interaction of YY1 mutants with endogenous Trim28 is indicated in the right column. A strong interaction is noted by “++”, a weak interaction is noted by “+”, and no interaction is indicated by “–”. (B) Upper panel: Myc-YY1 constructs were overexpressed in F9 cells, and cell lysates were prepared for co-IP experiments. Myc-YY1 proteins were immunoprecipitated with anti-myc beads, and bound proteins were examined by Western blot probed with an anti-Trim28 antibody. The band running below the Trim28 bands in the Myc IP lanes represent residual anti-myc antibody. The bands corresponding to the YY1 mutants are marked at the left of the input lanes by an asterisk. Mutants analyzed on this blot include the four mutants in which the AR2, GA, spacer, and ZNFs regions were independently deleted. Lower panel: Mutants analyzed on this blot include the mutants in which two or more regions were deleted.
We noted that the interaction between exogenous YY1WT and Trim28 was weaker relative to the interaction we detected between endogenous YY1 and Trim28. One possibility is that the myc-tag at the N-terminus interfered with YY1WT interaction with Trim28. Moving the myc-tag to the C-terminus of YY1WT did not improve its interaction with Trim28 (data not shown), making this possibility less likely. Another possibility is that there is a limiting level of endogenous Trim28 available to interact with exogenous YY1, because all available Trim28 molecules are already interacting with endogenous binding partners. To test this possibility, we expressed exogenous YY1WT with the KD of endogenous YY1 or with the overexpression of Trim28, but these conditions also did not improve the interaction.
2.4. Residue K779 of Trim28 is necessary for M-MLV silencing
SUMO modification of Trim28 was shown to be necessary for several Trim28 interactions (Goodarzi et al., 2011; Ivanov et al., 2007). To test whether sumoylation of Trim28 is important for the YY1-Trim28 interaction, we designed Trim28 constructs with mutations at the six lysine residues reported to be SUMO-modified (Fig. 4A). We mutated the lysine residues to arginine to eliminate the SUMO conjugation site while retaining the same positive charge. Each site was mutated independently (Trim28K554R, Trim28K575R, Trim28K676R, Trim28K750R, Trim28K779R, and Trim28K804R), or all six collectively (Trim286KR). F9 cells were transduced with the empty vector, or a Trim28WT or Trim28 mutant vector and selected for stable expression of the drug resistance gene. Subsequently, endogenous Trim28 was knocked down by shRNA. Lysates were prepared and analyzed by Western blot for Trim28 expression (Fig. 4B). We detected Trim28 in the untreated F9 cells (No KD), reflecting the basal levels of endogenous Trim28, and we detected only very low levels of Trim28 in the KD cells expressing the Trim28 shRNA and lacking the Trim28 cDNA (the empty vector lane). All the mutant proteins were expressed at high levels. YY1 was immunoprecipitated with an anti-YY1 antibody from cell lysates, and the levels of bound exogenous Trim28 were assessed by Western blot (Fig. 4C). Only background levels of Trim28 were detected when IPs were performed with non-specific antibodies (Ctrl IP lanes). Trim28 bound to YY1 in untreated F9 cell lysate (F9, YY1 IP lane) but not in the Trim28 KD cell lysate (EV, YY1 IP lane), demonstrating that endogenous Trim28 had been knocked down to levels such that any interaction with YY1 was not detectable. All mutant Trim28 proteins, including Trim286KR, were expressed at comparable levels and interacted with YY1 to similar degrees as Trim28WT, indicating that the six lysine residues examined on Trim28 are not necessary for the YY1-Trim28 interaction (YY1 IP lanes). In cases with high Trim28 expression, a faint band could be detected running below the major Trim28 band (the 2% input lanes for WT, K554R, K676R, K705R, K804R). The identity of this band is unknown.
Fig. 4. Mutations of major sumoylation sites on Trim28.
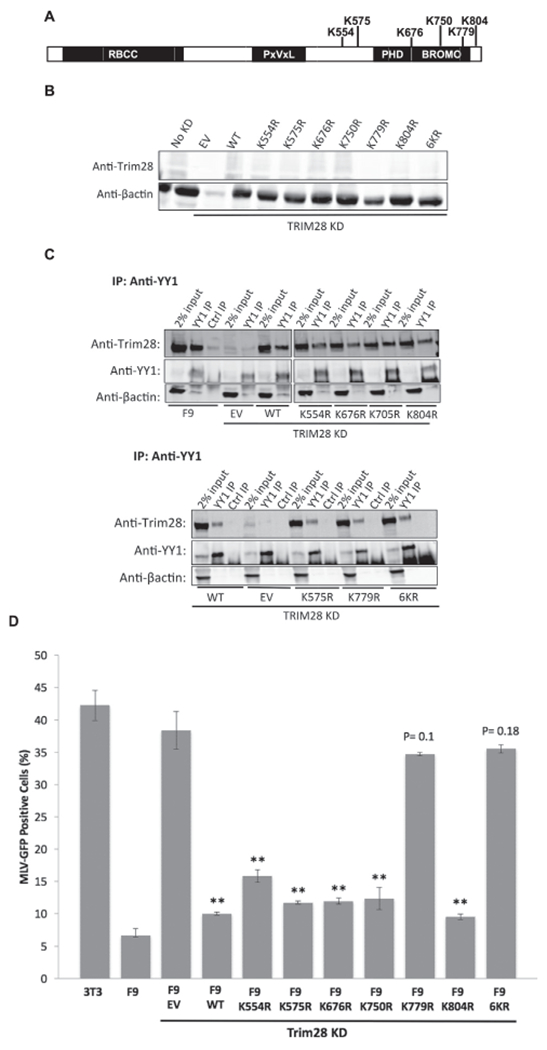
(A) Schematic of Trim28 protein with potentially sumoylated lysine residues indicated. (B) Mutant Trim28 was expressed and endogenous Trim28 was knocked down in F9 cells, and cell lysates were prepared and analyzed by Western blot. The “No KD” lane shows basal Trim28 levels in F9 cells. The empty vector (EV) lane shows the level of endogenous Trim28 expression in all KD lines, and the remaining lanes show expression levels of exogenous Trim28. (C) Western blots monitoring interaction of mutant Trim28 with YY1 by co-IP. YY1 was pulled-down with anti-YY1 antibody and the western blot was probed for Trim28. Co-IP results for four mutants are shown in the top panel and for the three remaining mutants on the bottom panel. (D) NIH3T3, untreated F9 cells, and Trim28 mutant F9 lines were infected with the M-MLV-GFP virus, and GFP expression was measured by flow cytometry. Uninfected cells were used to set our gates for counting “GFP-positive” expressing cells. NIH3T3 represent maximal M-MLV expression levels, and F9 cells represent maximal repression of M-MLV expression. Bars report the percentage of GFP-positive cells detected. Values represent the mean percentage of three biological replicates, and error bars represent the standard error of the mean. **denotes p < 0.01 significance of difference from EV control by student’s t-test. p values for samples with no significant difference from EV are indicated.
SUMO modifications of Trim28 have been reported as required for its transcriptional repressive activity (Goodarzi et al., 2011; Lee et al., 2007; Mascle et al., 2007). We asked if the sites of sumoylation were also required for silencing of M-MLV transcription in embryonic cell lines. To investigate this, we transduced Trim28 mutant lines with an M-MLV-GFP viral vector expressing the GFP reporter gene from M-MLV LTR, and containing both the NCR and the PBSpro sites for binding YY1 and ZFP809. Cells were transduced at a multiplicity of infection of approximately 1, and after two days the percentage of GFP-positive cells was determined by flow cytometry (Fig. 4D). Knockdown of endogenous Trim28 was confirmed at the time of M-MLV-GFP infection by Western blot, such that any M-MLV silencing detected could be attributed to the activity of the exogenous Trim28. The average percentage of GFP-positive cells across triplicates in NIH3T3 cells was 42%, representing maximal M-MLV expression, and that in the F9 cells was 6.7%, representing maximal M-MLV repression. F9 cells with Trim28 knocked down and carrying the empty vector control (EV) produced approximately the same percentage of GFP-positive cells (38%) as permissive NIH3T3 cells, underscoring the importance of Trim28 for M-MLV silencing. F9 cells expressing Trim28WT showed strong repression of M-MLV-GFP, demonstrating that exogenous Trim28 effectively rescued the repressive phenotype. Cells expressing five of the Trim28 mutants –Trim28K55R, Trim28K575R, Trim28K676R, Trim28K750R, Trim28K804R – displayed similar low percentages of GFP-positive cells as did Trim28WT cells, indicating that these lysine residues were not critical for M-MLV silencing. However, the repressive activity of the Trim28K779R and Trim286KR mutants were significantly compromised, yielding a high percentage of GFP-positive cells (34.7% and 35.5%, respectively). The fact that the Trim28K779R mutant showed the same loss of function as the Trim286KR mutant suggests that the K779R mutation provides most, if not all, of the derepression detected in the 6KR mutant. The importance of K779 for Trim28 repressive activity in this setting is consistent with what has been reported in previous studies of Trim28 silencing in other settings. In contrast, the finding that K554 and K804 are not critical for Trim28 silencing of M-MLV is distinct from findings for Trim28-mediated transcriptional silencing of other gene targets (Ivanov et al., 2007; Lee et al., 2007; Mascle et al., 2007).
Sumoylation is often important for intracellular localization of the modified protein, and thus the K779R mutation might block TRIM28 function by preventing proper nuclear localization. To test this possibility, we generated constructs expressing GFP-tagged versions of WT and K779R mutant TRIM28, and transfected NIH3T3 and F9 cells with these DNAs. Both GFP-WT TRIM28 and GFP-K779R TRIM28 were well expressed and were strictly nuclear in both cell types (Supplementary Fig. 2). We conclude that the K779R mutation did not act by causing mislocalization of TRIM28 but directly blocked its repressive function.
2.5. Trim28 is modified by SUMO2
The vast majority of studies on the effects of sumoylation modifications on Trim28 have involved the SUMO1 protein, but it is unknown if SUMO1, SUMO2/3, or all isoforms can mediate Trim28 repressive activity. A recent study implicating SUMO2 involvement in Trim28-mediated viral silencing (Yang et al., 2015) prompted us to investigate whether SUMO2 could be conjugated to Trim28 and if it conjugates the same lysine residues as those conjugated by SUMO1. To confirm that the six reported lysine residues on Trim28 were the only available sites for SUMO1 conjugation, we co-expressed HA-tagged Trim28WT or Trim286KR with Flag-tagged SUMO1 or SUMO1GG, a mutant form of SUMO1 that cannot be conjugated to any substrate, in 293T cells. Cell lysates were prepared using a lysis buffer that prevents SUMO deconjugation, HA-Trim28 was immunoprecipitated with anti-HA beads, and precipitated proteins were examined for SUMO1 modifications by Western blot probing for the Flag-tag. We detected a ladder of SUMO1-modified Trim28 products in the Trim28WT expressing cells but not in the Trim286KR expressing cells, demonstrating that Trim28 is modified by SUMO1 at one or more of the 6 lysine residues that were mutated in the 6KR mutant (Fig. 5A). As expected, SUMO1GG was not detected with the IP of Trim28WT or Trim286KR. These results confirmed that the six reported lysine residues on Trim28 encompass all available sites conjugated by SUMO1.
Fig. 5. SUMO conjugation of Trim28.
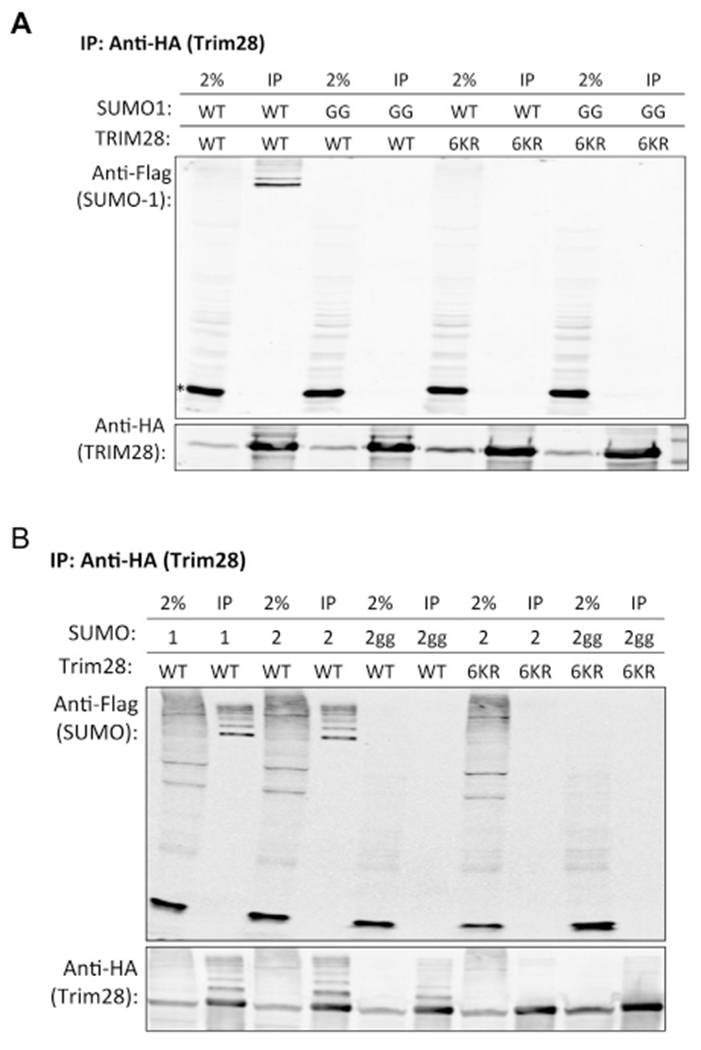
(A) Immunoprecipitation of HA-tagged Trim28 and probing for Flag-tagged SUMO1. Wild-type Flag-SUMO1 (WT) or Flag-SUMO1GG (GG) was stably co-expressed with wild-type HA-Trim28 (WT) or HA-Trim286KR (6KR) in 293T cells. The input lanes demonstrate the expression levels of Flag-SUMO1, Flag-SUMO1GG, HA-Trim28, or HA-Trim286KR. Trim28 was immunoprecipitated using anti-HA magnetic beads and bound proteins were analyzed by Western blot probed with anti-Flag antibodies to detect sumoylation levels onTrim28. The asterisk marks the position of the unconjugated monomeric SUMO1. (B) Immunoprecipitation of HA-tagged Trim28 and probing for Flag-tagged SUMO2. SUMO2 is indicated by “2” and SUMO2GG is indicated by “2gg, ” and immunoprecipitation of HA-Trim28 with SUMO1 was included as a control in the first two lanes.
To determine if SUMO2 can be conjugated to Trim28 and whether it conjugates to the same lysine residues as those conjugated by SUMO1, we co-expressed HA-Trim28WT and HA-Trim286KR with either Flag-SUMO2 or Flag-SUMO2GG in 293T cells and tested for SUMOylation as before. A ladder of SUMO2-modified Trim28WT products, similar to the ladder of SUMO1-modified Trim28WT products, was observed, while SUMO2 conjugation to Trim286KR was not detected (Fig. 5B). As expected, SUMO2GG was not detected on either Trim28 proteins. These results indicate that one or more of the six reported lysine residues on Trim28 are targets for both SUMO1 and SUMO2 conjugation, raising the possibility for M-MLV silencing to be mediated by SUMO1, SUMO2, and possibly, other modifications that can potentially occur at the K779 residue.
2.6. Major phosphorylation sites of Trim28 are not involved in the YY1-Trim28 interaction or derepression of M-MLV
The interaction of Trim28 with co-repressor proteins HP1 and CHD3 is disrupted by phosphorylation of S473 or S824 (Goodarzi et al., 2011; Ivanov et al., 2007; Chang et al., 2008; Bolderson et al., 2012). To test whether phosphorylation of these sites could also regulate the YY1-Trim28 interaction, we generated a panel of Trim28 phosphorylation mutants and tested them for binding to YY1. We constructed four Trim28 phosphorylation mutants: with an alanine substitution mutation at S473 (S473A) or S824 (S824A), or a phosphomimetic mutation at S473 (S473E) or S824 (S824D) (Fig. 6A). The alanine mutations rendered those sites unavailable for phosphorylation while the phosphomimetic mutations mimicked constitutively phosphorylated sites. The empty vector, Trim28WT, or a Trim28 mutant were expressed in F9 cells, followed by shRNA-mediated KD of endogenous Trim28 and preparation of lysates. Expression levels of mutant Trim28 constructs were analyzed by Western blots. We readily detected basal levels of endogenous Trim28 in untreated F9 cells (No KD), and only very low levels of Trim28 in the KD cells expressing the empty vector (EV). We detected high levels of exogenous Trim28 expression for all mutants except for Trim28S473A, which could not be tested further (Fig. 6B). YY1 was immunoprecipitated with an anti-YY1 antibody from cell lysates, and bound proteins were examined for the presence of exogenous Trim28 by Western blot. Trim28WT and all mutant Trim28 proteins interacted with YY1 to a similar extent (Fig. 6C), indicating that the S473E, S824A, and S824D mutations did not measurably impact the YY1-Trim28 interaction.
Fig. 6. Mutation of two major phosphorylation sites of Trim28.
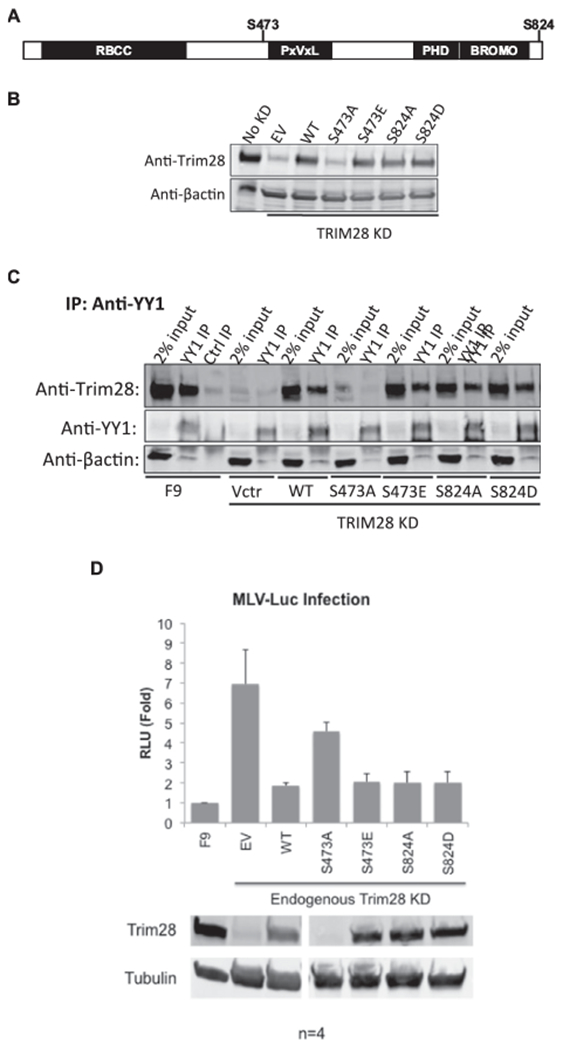
(A) Schematic of Trim28 protein with the major phosphorylation sites marked. Serine sites were mutated to alanine or to an acidic residue that mimics a constitutively phosphorylated residue. (B) Mutant Trim28 was expressed in F9 cells, endogenous Trim28 was knocked down, and cell lysates were prepared and analyzed by Western blot. The “No KD” lane shows basal Trim28 levels in F9 cells. The empty vector (EV) lane shows the level of endogenous Trim28 expression in all KD lines, and the remaining lanes show expression levels of exogenous Trim28. (C) Western blot monitoring interaction of mutant Trim28 with YY1 by co-IP. YY1 was immunoprecipitated with anti-YY1 antibody and the bound proteins were analyzed by western blot probed for Trim28. (D) Untreated F9 cells, and lines expressing empty vector (EV), wild-type Trim28 (WT), or Trim28 mutants, all with KD of endogenous Trim28, were infected with M-MLV-Luc virus, and luciferase activities were measured after 48 h. Relative light units (RLU) are values expressed relative to F9 control. Values indicate the mean of four biological replicates, and error bars represent the standard error of the mean. Lower panels: Lysates were analyzed by western blot with anti-Trim28 antibodies to monitor KD and expressed levels of Trim28. Anti-tubulin antibodies were used to report tubulin levels as a loading control.
Because phosphorylation modifications were also reported to interfere with Trim28 transcriptional repressive activity (Li et al., 2007; Rauwel et al., 2015), we investigated the importance of the phosphorylation sites for the silencing of GFP delivered on an M-MLV genome. Trim28 phosphorylation mutant cell lines were transduced with an M-MLV-Luc vector at a multiplicity of infection of approximately 1, and luciferase levels were measured after 48 h (Fig. 6D). All the cell lines expressing the Trim28 mutants displayed similar percentages of GFP-positive cells as those expressing Trim28WT, indicating that the mutant proteins were fully competent to suppress MLV genome expression. This finding suggests that these sites were not critical for regulating Trim28-mediated repression of M-MLV in embryonic cells. These results are distinct from those reported in studies of Trim28 regulation of HCMV latency and cell cycle progression (Li et al., 2007; Rauwel et al., 2015).
2.7. Trim28 could not be detected in YY1-DNA complexes
We made two efforts to detect Trim28 in association with a YY1-DNA complex. We first used an electromobility shift (EMSA) assay to probe protein complexes bound to the NCR DNA. We incubated F9 nuclear extracts with a biotinylated NCR or NCR mutant probe in which the YY1 binding site was scrambled. The lysates and probe mixture were displayed on a native gel, transferred to a membrane for Western blot, and examined for the migration of the biotinylated probe. The NCR probe, and not the NCR mutant probe, was shifted when incubated with F9 lysates, demonstrating the presence of proteins binding specifically to the NCR sequence (Supplementary Fig. 3A). Addition of anti-YY1 antibody resulted in large supershift of the NCR probe, indicating the presence of YY1 in this binding complex. However, addition of anti-Trim28 antibody did not result in a similar supershift. Secondly, we attempted to detect Trim28 binding to NCR DNA on beads. Biotinylated NCR or NCR mutant oligonucleotides were immobilized to streptavidincoated beads and incubated with F9 nuclear extract or with rYY1 as a positive control. Beads were washed and bound proteins were analyzed by Western blot. We found that rYY1 bound to NCR, but not NCR mutant oligonucleotide, and we also detected endogenous YY1 in the nuclear extract proteins bound to the NCR, but not the NCR mutant oligonucleotide. Low levels of Trim28 were also detected, but similar levels of Trim28 were detected in the proteins bound to both the NCR and NCR mutant oligonucleotides (Supplementary Fig. 3B), indicating that these are only background levels of Trim28 binding nonspecifically to the beads.
3. Discussion
Trim28 is a key repressor protein in the complex that mediates transcriptional silencing of the M-MLV provirus in embryonic cells (Wolf and Goff, 2007; Rowe et al., 2010). Trim28 is ubiquitously expressed across all developmental stages, and its selectivity for embryonic cells is mediated by the regulated functionality of two zinc-finger proteins, ZFP809 and YY1, responsible for recruiting Trim28 to the provirus DNA. The full length ZFP809 protein is only expressed in embryonic cells, such that its PBS-dependent silencing is regulated by limitation of ZFP809 expression (Wolf and Goff, 2009). Our lab recently elucidated the mechanism for this, showing that ZFP809 protein is stable in embryonic cells but is ubiquitinated and rapidly degraded in differentiated cells (Wang and Goff, 2017). The selectivity of YY1 activity is achieved by a different mechanism. YY1 is expressed well in both embryonic and differentiated cells, but the YY1-Trim28 interaction is detected preferentially in embryonic cells (Schlesinger et al., 2013), suggesting that this interaction per se is regulated. The domains of both partners that would be involved in their interaction could not be predicted from earlier studies. Several functional domains of Trim28 – an RBCC domain, chromoshadow domain, PHD domain, and bromodomain – are known to interact with critical co-repressor proteins. The chromoshadow domain, also called the HP1 box, interacts with HP1 (Le Douarin et al., 1996), the PHD-Bromodomain interacts with CHD3 and ESET (Schultz et al., 2002, 2001), and the RBCC domain interacts with the repressive KRAB domain of KRAB-ZNFs, such as ZFP809 (Friedman et al., 1996; Wolf and Goff, 2009). Our findings demonstrated that the RBCC domain of Trim28 is sufficient for interaction with YY1, despite YY1 not containing a KRAB domain (Fig. 2B). The interaction of the RBCC domain of Trim28 with YY1 may involve other KRAB domain-containing proteins that serve as bridging proteins. This notion is consistent with our inability to detect a direct interaction between recombinant forms of YY1 and Trim28 (Fig. 1).
A peculiar feature of YY1 is that it can behave as a transcriptional activator or repressor depending on the context of its target DNA and the partners available for interaction (Shi et al., 1997). The repressor domains of YY1 have been mapped to the GA-rich region and the ZNFs, while the activating domains have been mapped to the N-terminal domains, AR1 and AR2 (Thomas and Seto, 1999). Thus far, it is known that the GA-rich region of YY1 is required for its interaction with several co-repressor proteins, including the histone deacetylase 1 (HDAC1), enhancer of zeste homolog 2 (EZH2), and the HP1 proteins (Morey et al., 2012; Yang et al., 1996). Our experiments showed that the interaction of YY1 with Trim28 depended on the AR1 and ZNFs (Fig. 3B). The involvement of the ZNFs is consistent with what is thought to be the repressive domains of YY1, but the requirement for the AR1 was unexpected (Bushmeyer et al., 1995). We consistently detected a weaker interaction between exogenous YY1WT and Trim28 relative to the interaction detected between endogenous YY1 and Trim28. Moving the myc-tag, knocking down endogenous YY1, or overexpressing Trim28 did not improve this interaction.
One possible complication in our studies is the presence of YY2, an isotype of YY1. YY2 cross-reacts with YY1 antibodies, binds to some of the same sequences that YY1 occupies, and is 86% identical to YY1 in its zinc fingers (Chen et al., 2010; Klar, 2010; Klar and Bode, 2005; Nguyen et al., 2004). Thus, what was previously believed to be the silencing activities of only YY1, may actually reflect the combined repressive activities of YY1 and YY2. It is also possible that YY2 interacts with Trim28 and is immunoprecipitated with the anti-YY1 antibody. However, YY2 does not contain the AR1 domain demonstrated to be necessary for Trim28 interaction, making this scenario less likely.
Several studies have demonstrated the importance of sumoylation and phosphorylation for the interaction of Trim28 with its partners and for its repressive activity (Goodarzi et al., 2011; Ivanov et al., 2007; Li et al., 2007; Rauwel et al., 2015; Chang et al., 2008; Bolderson et al., 2012). A study using a yeast-two hybrid assay previously demonstrated that the K779R mutation did not perturb binding of the Trim28 PHD/Bromo domain to CHD3 or ESET, but mutations at both K779 and K676 eliminated CHD3 and ESET binding (Ivanov et al., 2007). None of the sites tested in our studies were found to be responsible for regulating the YY1-Trim28 interaction. Thus, the basis for the regulated interaction between these proteins remains unknown. Functional tests, however, showed that residue K779 was necessary for Trim28-mediated silencing of M-MLV (Fig. 4D). In addition to K779, previous studies have highlighted the importance of the K554 and K804 residues for the repressive activity of Trim28 in the DNA damage response pathway and in the regulation of cell cycle genes (Goodarzi et al., 2011; Lee et al., 2007; Mascle et al., 2007). In contrast, the K554 and K804 residues were not found to be critical for the silencing of M-MLV. It is still possible that these sites have redundant repressive functions, thereby requiring the combined mutation of multiple residues to disrupt silencing activity. Furthermore, whereas Trim28 phosphomimetic mutations at S473 or S824 abrogated Trim28 repressive activity in the context of HCMV latency, the DNA damage response, and the expression of cell cycle genes (Goodarzi et al., 2011; Rauwel et al., 2015; Chang et al., 2008), phosphomimetic mutations at these sites had no impact on Trim28 silencing of M-MLV (Fig. 6D). We note that Trim28S473A did not express well, suggesting that this mutation may affect protein folding or stability.
We also investigated which of the SUMO proteins can conjugate to Trim28 at the six lysine residues examined. Most studies addressed the role of SUMO1 in Trim28-mediated repression, and specifically tested for activity in differentiated cells and in the context of the DNA damage or cell cycle pathways. Very few studies have explored the role of Trim28 modification by SUMO2. A recent genomic-wide siRNA screen showed that SUMO2 and its associated sumoylation factors are necessary for silencing of endogenous retroviruses in embryonic cells (Yang et al., 2015). Moreover, this study demonstrates that the sets of endogenous retroviral genes upregulated after Trim28 KD and SUMO2 KD greatly overlap, implicating the two proteins in a co-dependent repressive function. We showed that both SUMO1 and SUMO2 can be conjugated to Trim28 and that mutation of the six lysine residues prevented conjugation by both SUMO1 and SUMO2 (Fig. 5A and B). It is currently unknown if only one SUMO, either of the SUMOs, or possibly another modification regulates Trim28 silencing activity at the K779 residue.
In conclusion, we here mapped the interacting domains on Trim28 and YY1 and demonstrated the necessity of residue K779 on Trim28 for mediating M-MLV silencing. We demonstrated that the RBCC domain of Trim28 is sufficient for its interaction with YY1 and that the AR1 and the zinc fingers of YY1 were necessary and sufficient for its interaction with Trim28. The K779 residue on Trim28 was shown to be necessary for its repression of M-MLV, but the other known sumoylation and phosphorylation sites were dispensable. Thus, our results suggest that silencing of M-MLV in embryonic cells utilizes a Trim28-mediated silencing mechanism different from that involved in DNA damage, cell cycle progression, and HCMV latency. Further studies aimed at elucidating the consequences of Trim28 modifications at the various residues in different pathways and cell types will be important for developing a more comprehensive understanding of how Trim28 silencing activity is regulated.
4. Materials and methods
4.1. Cell culture and RNA interference
F9, PCC4, and NIH3T3 cells were cultured in DMEM media with 10% FBS, 100U/mL penicillin, 0.05 mM streptomycin, and 2 mM l-glutamine. E14 cells were cultured in DMEM media with 15% FBS, 20 mM HEPES, 0.1 mM nonessential amino acids, 0.1 mM 2-mercaptoethanol, 100 U/mL penicillin, 0.05 mM streptomycin, 2 mM l-glutamine, and leukemia inhibitory factor was added fresh at the time of culture. Plates were coated with 0.1% gelatin prior to plating ES/EC cells. RNAi KD was performed with pLKO.1 shRNA (Sigma or GE Dharmacon). The target sequence used for mTrim28 KD and mYY1 KD are listed below:
mYY1 shRNA: CGACGGTTGTAATAAGAAGTT
mTRIM28 shRNA: CCGCATGTTCAAACAGTTCAA
4.2. Plasmid construction
Mutant and wild-type Trim28 and YY1 cDNAs were cloned into the pLVX-EF1a-IRES vector containing the neomycin, hygromycin, or puromycin drug resistance gene (Clontech). Silent mutations at the YY1 and Trim28 shRNA target were created to prevent the knockdown of the exogenous protein. SUMO and phosphorylation mutant Trim28 were created using GENEART Site-Directed Mutagenesis System (Thermo Fisher Scientific). Trim28, YY1, and HP1 were cloned into the pQE80L vector (Qiagen) for the generation of recombinant protein.
To construct WT or K779R GFP-TRIM28 plasmids, overlapping PCR was performed using pEGFP-N1 (Clontech) and pLVX-Eif1a-WT-TRIM28 with primers 5′-AAATTTCCGAATTCATGGTGAGCAAGGGCGAGG-3′, 5′-GCAGTCGCTGCCGCCGAGGCCGCCATCTTGTACAGCTCGTCCATGCCG-3′, 5′-CGGCATGGACGAGCTGTACAAGATGGCGGCCTCGGCGGCAGCGACTGC-3′, and 5′-AGGGGCTCATGCTTGTGCACATTACAGTAGACTGTTCGC-3′. The rsulting PCR fragment was inserted into pLVX-EF1a-WT-TRIM28 or pLVX-K779R-Trim29 at EcoRI and XhoI restriction sites.
4.3. Virus production
Viruses were produced in 293T cells. 3.5 × 106 293T cells were plated in a 10 cm plate. The following day, 293T cells were transfected with 8 μg of a retroviral transfer plasmid, 4 μg of a vector contain the VSV-G envelope gene, and 4 μg of a plasmid expressing the gag and pol genes. MLV-GFP viruses were produced by co-transfection of 293T cells with pNCA-GFP, pCMV-intron, and pMD.G DNAs, as previously described (Lim et al., 2002). Lentiviruses were produced by co-transfection of 293T cells with pLVX-EF1a-IRES or pLKO.1 vectors with pCMVΔr8.2 and pMD.G DNAs, as previously described (Schlesinger et al., 2013). Transfections were carried out using Polyethylenimine (PEI). Virions were harvested 48 h and 72 h after transfection, and cells were transduced with virus for 3 h with 8 μg/mL of polybrene.
4.4. GFP expression from MLV vector
F9 cells were plated at 2 × 105 cells per well of a 6-well dish and transduced with virus preparations containing an MLV vector expressing GFP (pNCA-GFP) at an MOI of 1. At 48 h after infection, cells were trypsinized, washed with D-PBS, and re-suspended in D-PBS supplemented with 2% FBS. Cells were analyzed by flow cytometry on the Guava flow cytometer (EMD Millipore) and analyzed with FlowJo software (TreeStar).
4.5. Recombinant protein expression and purification
BL21 cells (NEB) were transformed with pQE80L vectors expressing recombinant proteins. Bacterial cultures were grown with ampicillin and 1 mM IPTG for 4 h. Cells were harvested and lysed with Buffer A (6 M GuHCl, 100 mM NaH2PO4, 10 mM Tris-Cl, 5 mM B-mercaptoethanol, 10 mM imidazole, pH 8.0) for 15 min. Lysates were clarified by centrifugation at 16.1 RCF for 1 h. The lysates were mixed with pre-washed Ni-NTA Agarose beads (Qiagen) for 30 min and then loaded onto a centrifuge column (Pierce). Beads were washed 4 times with buffer A and recombinant protein was eluted with elution buffer (Buffer A with 100 mM imidazole). Recombinant protein was dialyzed into folding buffer (Golebiowski et al., 2011) or PBS using the Slide-a-Lyzer Dialysis Cassette (Pierce).
4.6. Coimmunoprecipitation
Cells were grown to confluence in 10-cm dishes and collected and washed in ice cold D-PBS for each immunoprecipitation reaction. Ice-cold 0.1% NP40 lysis buffer (0.1% NP40, 250 mM NaCl, 20 mM Na3PO4, pH 7.0, 30 mM Na4P2O7, 5 mM EDTA, 10 mM NaF) with 1 × complete protease inhibitor (Roche) was added to the pellet at 2 × the cell pellet volume. Cells were lysed on ice for 30 min and the lysates were clarified by centrifugation for 15 min at 14,000 RPM at 4 °C. The nuclear extracts were diluted to 400 μl total volume in lysis buffer, per IP reaction. 2% of total lysate volume was saved for input lanes, and remaining lysate was incubated with 4 μg anti-YY1 antibody (sc281, Santa Cruz Biotechnology) or rabbit control antibody (sc-2027, Santa Cruz Biotechnology) for 16 h in 4 °C. Prewashed protein A/G dynabeads (Thermo Fisher Scientific) were added to the lysates and incubated for 1 h in 4 °C. For anti-myc IP experiments, lysates were incubated with 20 μl of pre-washed anti-myc beads (Pierce) for 1 h in 4 °C. For in vitro co-IP experiments, recombinant proteins were incubated with antibody or beads in 200 μl of 0.1% NP40 lysis buffer with 1 × complete protease inhibitor (Roche). 10 μl of prewashed anti-Flag beads (M8823, Sigma) were used for the IP of Flag-rHP1 protein. Beads were washed 3 × with 0.1% NP40 lysis buffer and bound proteins were eluted and analyzed by Western blot.
4.7. Lysate preparation and immunoprecipitation of sumoylated substrates
Flag-tagged SUMO proteins were co-expressed with HA-tagged Trim28 in 293 T cells. Cell lysates were prepared in SDS lysis buffer (5% SDS, 30% glycerol, 0.15 M Tris-HCl, pH 6.8), diluted 1:4 with 0.5% NP40/PBS, added to pre-washed anti-HA magnetic beads (Pierce), and incubated overnight at 4 °C in rotation. Beads were washed with 0.5% NP40/PBS three times and bound proteins were removed with 1 × SDS sample buffer and boiling for 5 min at 95 °C. Co-IP of proteins was analyzed by Western blot.
4.8. Antibodies
Antibodies used for Western blots were as follows: anti-Trim28 20C1 (ab22553, Abcam), anti-YY1 C-20 (sc281, Santa Cruz Biotechnology), anti-HA.11 (901515, BioLegend), anti-myc 71D10 (2278, Cell Signaling Technology), anti-myc 9E10 (sc-40, Santa Cruz Biotechnology), anti-Flag M2 (F3165, Sigma-Aldrich), pS824-Trim28 (ab70369), anti-Oct3/4 (H-134, Santa Cruz Biotechnology), and anti-β-actin (A1978, Sigma). Antibodies used for co-IP experiments are as follows: anti-YY1 C-20 (sc281, Santa Cruz Biotechnology), rabbit control antibody (sc-2027, Santa Cruz Biotechnology). Antibodies used for EMSA shifts were as follows: anti-YY1 C-20 (sc281, Santa Cruz Biotechnology), anti-Trim28 20C1 (ab22553, Abcam), and rabbit control antibody (sc-2027, Santa Cruz Biotechnology).
4.9. EMSA
Nuclear extracts were prepared as previously described (Wolf and Goff, 2007). Double-stranded DNA probes were end labeled with biotin and used for EMSA reactions with the LightShift Chemiluminescent EMSA Kit (cat. #20148; Thermo Scientific) according to the manufacturer’s instructions. For the detection of supershifts, antibody was added 30 min prior to adding the probe. Protein complexes were analyzed by a 5% native polyacrylamide gels and transferred to 0.45 μM pore nitrocellulose membrane for 40 min at 380 mAmps in 4 °C. Membranes were immediately crosslinked using the UV Crosslinker (Fischer Scientific) for 1 min prior to blocking. The biotinylated oligonucleotides were purchased from IDT and their sequences (plus strand) are listed here:
NCR: 5′AGCTTAAGTAACGCCATTTTGCAAGGCA3′
NCRm: 5′AGCTTAAGTAATACGGCTATGCAAGGCA3′
4.10. Protein binding to DNA beads
50 μl of pre-washed streptavidin coated dynabeads (ThermoFisher) were incubated with 50 pmols of annealed biotinylated oligonucleotide in 200 μl binding buffer (1 M NaCl, 5 mM Tris, 1 mM EDTA) for 10 min. The same biotinylated oligonucleotides sequences used for EMSA reactions were used here. Beads were washed twice and resuspended with 200 μl EMSA buffer (10 mM Tris, 50 mM KCl, 2.5% glycerol, 5 mM MgCl2, and 0.05% NP40). 2 μg poly(dI: dC) (Sigma) and 2 μg rYY1 protein or F9 nuclear lysate was added to the beads and incubated for 30 min. Beads were washed 3 times with EMSA buffer (100 mM KCl, 5% glycerol, 10 mM MgCl2, 0.1% NP40, and 10 mM Tris, pH 7.5) and the bound proteins were eluted and analyzed by SDS-Page and Western blot.
Supplementary Material
Acknowledgements
This work was supported by a grant from the NCI (R01CA 30488). A.L. was supported by a training grant from the NCI (T32 CA 009503). S.P.G. is an Investigator of the Howard Hughes Medical Institute. These studies used the resources of the Cancer Center Flow Core Facility funded in part through Center Grant P30CA013696.
Footnotes
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.virol.2018.01.012
References
- Barklis E, Mulligan RC, Jaenisch R, 1986. Chromosomal position or virus mutation permits retrovirus expression in embryonal carcinoma cells. Cell 47, 391–399. [DOI] [PubMed] [Google Scholar]
- Bohren KM, Nadkarni V, Song JH, Gabbay KH, Owerbach D, 2004. A M55V polymorphism in a novel SUMO gene (SUMO-4) differentially activates heat shock transcription factors and is associated with susceptibility to type I diabetes mellitus. J. Biol. Chem 279, 27233–27238. [DOI] [PubMed] [Google Scholar]
- Bolderson E, Savage KI, Mahen R, Pisupati V, Graham ME, Richard DJ, Robinson PJ, Venkitaraman AR, Khanna KK, 2012. Kruppel-associated Box (KRAB)-associated co-repressor (KAP-1) Ser-473 phosphorylation regulates heterochromatin protein 1beta (HP1-beta) mobilization and DNA repair in heterochromatin. J. Biol. Chem 287, 28122–28131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushmeyer S, Park K, Atchison ML, 1995. Characterization of functional domains within the multifunctional transcription factor, YY1. J. Biol. Chem 270, 30213–30220. [DOI] [PubMed] [Google Scholar]
- Cammas F, Mark M, Dolle P, Dierich A, Chambon P, Losson R, 2000. Mice lacking the transcriptional corepressor TIF1beta are defective in early postimplantation development. Development 127, 2955–2963. [DOI] [PubMed] [Google Scholar]
- Chang CW, Chou HY, Lin YS, Huang KH, Chang CJ, Hsu TC, Lee SC, 2008. Phosphorylation at Ser473 regulates heterochromatin protein 1 binding and corepressor function of TIF1beta/KAP1. BMC Mol. Biol 9, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Shioda T, Coser KR, Lynch MC, Yang C, Schmidt EV, 2010. Genome-wide analysis of YY2 versus YY1 target genes. Nucleic Acids Res. 38, 4011–4026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan JR, Krieg AM, Max EE, Khan AS, 1989. Negative control region at the 5′ end of murine leukemia virus long terminal repeats. Mol. Cell. Biol 9, 739–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan JR, Becker KG, Ennist DL, Gleason SL, Driggers PH, Levi BZ, Appella E, Ozato K, 1992. Cloning of a negative transcription factor that binds to the up-stream conserved region of Moloney murine leukemia virus. Mol. Cell. Biol. 12, 38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JR, Fredericks WJ, Jensen DE, Speicher DW, Huang XP, Neilson EG, Rauscher FJ 3rd, 1996. KAP-1, a novel corepressor for the highly conserved KRAB repression domain. Genes Dev. 10, 2067–2078. [DOI] [PubMed] [Google Scholar]
- Gautsch JW, Wilson MC, 1983. Delayed de novo methylation in teratocarcinoma suggests additional tissue-specific mechanisms for controlling gene expression. Nature 301, 32–37. [DOI] [PubMed] [Google Scholar]
- Geiss-Friedlander R, Melchior F, 2007. Concepts in sumoylation: a decade on. Nat. Rev. Mol. Cell Biol 8, 947–956. [DOI] [PubMed] [Google Scholar]
- Golebiowski FM, Gorecki A, Bonarek P, Dziedzicka-Wasylewska M, 2011. Efficient overexpression and purification of active full-length human transcription factor Yin Yang 1 in Escherichia coli. Protein Expr. Purif 77, 198–206. [DOI] [PubMed] [Google Scholar]
- Goodarzi AA, Kurka T, Jeggo PA, 2011. KAP-1 phosphorylation regulates CHD3 nucleosome remodeling during the DNA double-strand break response. Nat. Struct. Mol. Biol 18, 831–839. [DOI] [PubMed] [Google Scholar]
- Hilberg F, Stocking C, Ostertag W, Grez M, 1987. Functional analysis of a retroviral host-range mutant: altered long terminal repeat sequences allow expression in embryonal carcinoma cells. Proc. Natl. Acad. Sci. USA 84, 5232–5236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov AV, Peng H, Yurchenko V, Yap KL, Negorev DG, Schultz DC, Psulkowski E, Fredericks WJ, White DE, Maul GG, Sadofsky MJ, Zhou MM, Rauscher FJ 3rd, 2007. PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol. Cell 28, 823–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klar M, 2010. It is not necessarily YY1–the frequently forgotten Yin-Yang-2 transcription factor. Proc. Natl. Acad. Sci. USA 107E190; author reply E191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klar M, Bode J, 2005. Enhanceosome formation over the beta interferon promoter underlies a remote-control mechanism mediated by YY1 and YY2. Mol. Cell. Biol 25, 10159–10170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Douarin B, Nielsen AL, Garnier JM, Ichinose H, Jeanmougin F, Losson R, Chambon P, 1996. A possible involvement of TIF1 alpha and TIF1 beta in the epigenetic control of transcription by nuclear receptors. EMBO J. 15, 6701–6715. [PMC free article] [PubMed] [Google Scholar]
- Lechner MS, Begg GE, Speicher DW, Rauscher FJ 3rd, 2000. Molecular determinants for targeting heterochromatin protein 1-mediated gene silencing: direct chromoshadow domain-KAP-1 corepressor interaction is essential. Mol. Cell. Biol 20, 6449–6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YK, Thomas SN, Yang AJ, Ann DK, 2007. Doxorubicin down-regulates Kruppel-associated box domain-associated protein 1 sumoylation that relieves its transcription repression on p21WAF1/CIP1 in breast cancer MCF-7 cells. J. Biol. Chem 282, 1595–1606. [DOI] [PubMed] [Google Scholar]
- Leung DC, Dong KB, Maksakova IA, Goyal P, Appanah R, Lee S, Tachibana M, Shinkai Y, Lehnertz B, Mager DL, Rossi F, Lorincz MC, 2011. Lysine methyltransferase G9a is required for de novo DNA methylation and the establishment, but not the maintenance, of proviral silencing. Proc. Natl. Acad. Sci. USA 108, 5718–5723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Lee YK, Jeng JC, Yen Y, Schultz DC, Shih HM, Ann DK, 2007. Role for KAP1 serine 824 phosphorylation and sumoylation/desumoylation switch in regulating KAP1-mediated transcriptional repression. J. Biol. Chem 282, 36177–36189. [DOI] [PubMed] [Google Scholar]
- Liang Q, Deng H, Li X, Wu X, Tang Q, Chang TH, Peng H, Rauscher FJ 3rd, Ozato K, Zhu F, 2011. Tripartite motif-containing protein 28 is a small ubiquitin-related modifier E3 ligase and negative regulator of IFN regulatory factor 7. J. Immunol 187, 4754–4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim D, Orlova M, Goff SP, 2002. Mutations of the RNase H C helix of the Moloney murine leukemia virus reverse transcriptase reveal defects in polypurine tract recognition. J. Virol 76, 8360–8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linney E, Davis B, Overhauser J, Chao E, Fan H, 1984. Non-function of a Moloney murine leukaemia virus regulatory sequence in F9 embryonal carcinoma cells. Nature 308, 470–472. [DOI] [PubMed] [Google Scholar]
- Loh TP, Sievert LL, Scott RW, 1987. Proviral sequences that restrict retroviral expression in mouse embryonal carcinoma cells. Mol. Cell. Biol 7, 3775–3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh TP, Sievert LL, Scott RW, 1990. Evidence for a stem cell-specific repressor of Moloney murine leukemia virus expression in embryonal carcinoma cells. Mol. Cell. Biol 10, 4045–4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascle XH, Germain-Desprez D, Huynh P, Estephan P, Aubry M, 2007. Sumoylation of the transcriptional intermediary factor 1beta (TIF1beta), the Co-repressor of the KRAB Multifinger proteins, is required for its transcriptional activity and is modulated by the KRAB domain. J. Biol. Chem 282, 10190–10202. [DOI] [PubMed] [Google Scholar]
- Matsui T, Leung D, Miyashita H, Maksakova IA, Miyachi H, Kimura H, Tachibana M, Lorincz MC, Shinkai Y, 2010. Proviral silencing in embryonic stem cells requires the histone methyl transferase ESET. Nature 464, 927–931. [DOI] [PubMed] [Google Scholar]
- Morey L, Pascual G, Cozzuto L, Roma G, Wutz A, Benitah SA, Di Croce L, 2012. Nonoverlapping functions of the Polycomb group Cbx family of proteins in embryonic stem cells. Cell Stem Cell 10, 47–62. [DOI] [PubMed] [Google Scholar]
- Nguyen N, Zhang X, Olashaw N, Seto E, 2004. Molecular cloning and functional characterization of the transcription factor YY2. J. Biol. Chem 279, 25927–25934. [DOI] [PubMed] [Google Scholar]
- Niwa O, Yokota Y, Ishida H, Sugahara T, 1983. Independent mechanisms involved in suppression of the Moloney leukemia virus genome during differentiation of murine teratocarcinoma cells. Cell 32, 1105–1113. [DOI] [PubMed] [Google Scholar]
- Petersen R, Kempler G, Barklis E, 1991. A stem cell-specific silencer in the primer-binding site of a retrovirus. Mol. Cell. Biol 11, 1214–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauwel B, Jang SM, Cassano M, Kapopoulou A, Barde I, Trono D, 2015. Release of human cytomegalovirus from latency by a KAP1/TRIM28 phosphorylation switch. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe HM, Jakobsson J, Mesnard D, Rougemont J, Reynard S, Aktas T, Maillard PV, Layard-Liesching H, Verp S, Marquis J, Spitz F, Constam DB, Trono D, 2010. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 463, 237–240. [DOI] [PubMed] [Google Scholar]
- Schlesinger S, Lee AH, Wang GZ, Green L, Goff SP, 2013. Proviral silencing in embryonic cells is regulated by Yin Yang 1. Cell Rep. 4, 50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz DC, Friedman JR, Rauscher FJ 3rd, 2001. Targeting histone deacetylase complexes via KRAB-zinc finger proteins: the PHD and bromodomains of KAP-1 form a cooperative unit that recruits a novel isoform of the Mi-2alpha subunit of NuRD. Genes Dev. 15, 428–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ 3rd, 2002. SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyl transferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 16, 919–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lee JS, Galvin KM, 1997. Everything you have ever wanted to know about Yin Yang 1. Biochim. Biophys. Acta 1332, F49–66. [DOI] [PubMed] [Google Scholar]
- Stewart CL, Stuhlmann H,, Jahner D, Jaenisch R, 1982. De novo methylation, expression, and infectivity of retroviral genomes introduced into embryonal carcinoma cells. Proc. Natl. Acad. Sci. USA 79, 4098–4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teich NM, Weiss RA, Martin GR, Lowy DR, 1977. Virus infection of murine teratocarcinoma stem cell lines. Cell 12, 973–982. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Seto E, 1999. Unlocking the mechanisms of transcription factor YY1: are chromatin modifying enzymes the key? Gene 236, 197–208. [DOI] [PubMed] [Google Scholar]
- Verger A, Perdomo J, Crossley M, 2003. Modification with SUMO. A role in transcriptional regulation. EMBO Rep. 4, 137–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Goff SP, 2017. Differential control of retrovirus silencing in embryonic cells by proteasomal regulation of the ZFP809 retroviral repressor. Proc. Natl. Acad. Sci. USA 114, E922–E930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf D, Goff SP, 2007. TRIM28 mediates primer binding site-targeted silencing of murine leukemia virus in embryonic cells. Cell 131, 46–57. [DOI] [PubMed] [Google Scholar]
- Wolf D, Goff SP, 2009. Embryonic stem cells use ZFP809 to silence retroviral DNAs. Nature 458, 1201–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf D, Cammas F, Losson R, Goff SP, 2008. Primer binding site-dependent restriction of murine leukemia virus requires HP1 binding by TRIM28. J. Virol 82, 4675–4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe SA, Nekludova L, Pabo CO, 2000. DNA recognition by Cys2His2 zinc finger proteins. Annu. Rev. Biophys. Biomol. Struct 29, 183–212. [DOI] [PubMed] [Google Scholar]
- Yang BX, El Farran CA, Guo HC, Yu T, Fang HT, Wang HF, Schlesinger S, Seah YF, Goh GY, Neo SP, Li Y, Lorincz MC, Tergaonkar V, Lim TM, Chen L, Gunaratne J, Collins JJ, Goff SP, Daley GQ, Li H, Bard FA, Loh YH, 2015. Systematic identification of factors for provirus silencing in embryonic stem cells. Cell 163, 230–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WM, Inouye C, Zeng Y, Bearss D, Seto E, 1996. Transcriptional repression by YY1 is mediated by interaction with a mammalian homolog of the yeast global regulator RPD3. Proc. Natl. Acad. Sci. USA 93, 12845–12850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC, Lukas J, Bekker-Jensen S, Bartek J, Shiloh Y, 2006. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat. Cell Biol 8, 870–876. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.