

Abstract
Systemic lupus erythematosus (SLE) is a prototypic autoimmune disease characterized by antinuclear antibodies (ANAs) that form immune complexes that mediate pathogenesis by tissue deposition or cytokine induction. Some ANAs bind DNA or associated nucleosome proteins, whereas other ANAs bind protein components of complexes of RNA and RNA-binding proteins (RBPs). Levels of anti-DNA antibodies can fluctuate widely, unlike those of anti-RBP antibodies, which tend to be stable. Because anti-DNA antibody levels can reflect disease activity, repeat testing is common; by contrast, a single anti-RBP antibody determination is thought to suffice for clinical purposes. Experience from clinical trials of novel therapies has provided a new perspective on ANA expression during disease, as many patients with SLE are ANA negative at screening despite previously testing positive. Because trial results suggest that patients who are ANA negative might not respond to certain agents, screening strategies now involve ANA and anti-DNA antibody testing to identify patients with so-called ‘active, autoantibody-positive SLE’. Evidence suggests that ANA responses can decrease over time because of the natural history of disease or the effects of therapy. Together, these findings suggest that, during established disease, more regular serological testing could illuminate changes relevant to pathogenesis and disease status.
Systemic lupus erythematosus (SLE) is a prototypic autoimmune disease, the immunological hallmark of which is the production of antinuclear antibodies (ANAs)1,2. These antibodies bind to nucleic acids (DNA or RNA), proteins and complexes of DNA or RNA with proteins. Although ANA production is not unique to SLE, the pattern of autoantibodies expressed by patients with this disease is highly characteristic, enabling the use of serology for screening, classification, diagnosis, prognosis and staging3–5. Furthermore, data from trials of new immunomodulatory therapies have raised the possibility that ANA-positive patients might respond differently from ANA-negative patients to certain agents, an effect possibly related to the important role of ANAs in cytokine production and tissue inflammation and damage6–8.
Testing for ANAs has been central to the evaluation of SLE for over 60 years4 and has spurred the development of many new assays (such as addressable laser bead immunoassays and peptide arrays)4,9,10 on the basis of advances in laboratory science. These new assays are well suited for hospital or commercial laboratories because of their high-throughput capacity; however, they differ from older assays in the properties of the antigens (for example, purified or molecularly cloned proteins compared with proteins in tissue extracts), the platform used for ANA detection and sensitivity11,12. As a result, the information provided by current serological tests could differ from that expected on the basis of classic literature or standard wisdom, and assay variability is now recognized as a major issue.
Coupled with an explosion of research into molecular and cellular events in SLE, the technological advances in antibody testing provide the basis for a new understanding of the role of ANAs in the pathogenesis of SLE and the use of these antibodies as clinical biomarkers. In this Review, we consider salient aspects of ANAs as biomarkers and advance the following ideas: that ANAs can promote both acute and chronic disease manifestations of SLE; that ANAs that mediate pathogenesis in SLE might not be specific for this disease; that ANA expression can vary over time following diagnosis; and that ANA expression reflects underlying disturbances in B cell populations that can be influenced by genetics, race and ethnicity. The changing perspective on ANAs as biomarkers is also an important theme of this Review, as we focus on the role of serology to not only classify patients, but also to stage and subset their disease for the determination of immunosuppressive therapy.
Controversies in ANA testing for SLE
As ANA testing has long been a mainstay of patient evaluation for a rheumatic disease, the properties of the assays used should be well known to clinicians and investigators. Nevertheless, studies from the past few years have prompted a rethinking of ANA assays, especially as new assay formats have been increasingly used. In addition, trials of many new agents for the treatment of SLE are ongoing, and this trial setting has revealed aspects of ANA expression not apparent with serological testing in the ordinary clinical setting.
Changing uses of ANAs as biomarkers
Awareness of the vagaries of serological testing for SLE has been stimulated most directly by experience gained from clinical trials of new therapeutics for SLE. Although ANA expression has long been viewed as essentially universal in SLE and, indeed, a defining feature, studies have indicated that as many as 30% of patients with SLE who are screened for a clinical trial for a new therapy are ANA negative6–8. Furthermore, as ANA-positive patients can have increased treatment responses to certain biological agents compared with ANA-negative patients, a new nosological entity known as ‘active, autoantibody-positive SLE’ has been created in the absence of strong evidence13. In this context, ANAs are being used as theranostic markers to identify a putatively treatment-responsive subset of patients with SLE.
The high prevalence of ANA negativity in patient cohorts enrolled in SLE clinical trials has been surprising in view of the longstanding idea that ANA-negative SLE is very rare, if it exists at all14. Data from clinical trials, therefore, suggest either limitations in the ANA assays that were not previously recognized or that some patients transition to a serologically negative state; both are possible. As efforts to harmonize and standardize the ANA assays used in clinical trials (as well as in routine testing) have been limited, considerable uncertainty exists around assay selection, as well as in the interpretation of any changes in the serological status of patients revealed during screening for clinical trials.
The publication in 2019 of SLE classification criteria developed by the ACR and EULAR has provided another impetus to rethink the serology of SLE15–17. Importantly, in these criteria, a positive ANA test is required for a patient to be classified as having SLE. In previously published criteria, such as those from the ACR or Systemic Lupus International Cooperating Clinics (SLICC), a positive ANA test is just one of several criteria that could count towards classification18,19. In the final version of the 2019 criteria, ANA positivity is defined as an immunofluorescence assay (IFA) at a titre of 1:80 or greater on HEp-2 cells or a solid-phase assay of at least equivalent performance16,17; however, the meaning of ‘equivalent performance’ has not yet been specified.
The repositioning of the ANA test signifies a fundamental shift in the conceptualization of SLE as a disease and the ANA test as a laboratory assay; the repositioning raises an apparent contradiction. Whereas the clinical trial setting has suggested a substantial prevalence of seronegativity among patients with established SLE, the 2019 SLE classification criteria posit ANA positivity as an essential feature of SLE. Reconciling these ideas represents a major priority and challenge, given that a change of a patient to a seronegative state could prevent classification as SLE, especially if the results of any prior ANA tests are not known.
Laboratory tests for ANAs
The performance characteristics of assays used for ANA determination have been reviewed in depth elsewhere3–5,11–13; therefore, issues around ANA testing are discussed only briefly, as serological assays are the basis for understanding the role of ANAs in pathogenesis.
The term ANA signifies that the target autoantigen is a nuclear constituent, whether that be a nucleic acid, protein or protein–nucleic acid complex. As such, nuclear localization represents the defining feature of these antigens, and the IFA the basis of this determination. As described in detail elsewhere3–5,11–13, current IFA assays use a long-term cell line called HEp-2, which can be fixed to a glass slide as the antigen source. Following incubation of the slide with the test fluid (serum, plasma or tissue culture fluid), antibody binding is assessed by microscopy using an immunofluorescent anti-immunoglobulin reagent. Data output includes an assessment of positivity, staining pattern and titre.
Although many IFA kits with HEp-2 cells are available, these kits can lead to divergent results in terms of titre and pattern when used with samples from the same patient20,21. These differences probably result from test variables, such as the effects of culture conditions on the display of nuclear antigens by the cells, the conditions for fixation and the properties of the anti-immunoglobulin detection reagents. Moreover, for IFA determinations, visual inspection and computerized analysis might both not be reproducible across laboratories22,23, although computerized results still require visual inspection and interpretation at present.
In addition to the IFA, ANA assays can use other immunochemical approaches such as enzyme-linked immunosorbent assay (ELISA), multiplex and line immunoassay formats (Box 1). The antigens used in these assays can vary in source and composition; depending on the assay, antigens can be mixtures of proteins and DNA, as well as cell extracts. DNA can be purified from natural sources without regard for species of origin, or synthetic DNA molecules can be used24. Similarly, assays for antibodies that recognize individual proteins can use synthetic peptides, cloned proteins or purified molecules from cells. Given the differences between these assays, variability is not unexpected. As such, a complete serological evaluation might require the use of more than one assay platform. Importantly, anyone ordering an ANA test should be aware of the specific assay that will be used and, ideally, its specificity and sensitivity4,13.
Box 1 |. High-throughput ANA testing platforms.
Antinuclear antibody (ANA) detection assays that can be automated for high-throughput use include enzyme-linked immunosorbent assay (ELISA), multiplex assays such as addressable laser bead immunoassay (ALBIA), line immunoassay and microarray assays4,9,10.
ELISA
ELISA is a solid-phase platform in which an antigen preparation is adhered to the wells of a plastic microtitre plate. A dilution of serum or plasma is added, and the amount of bound antibody is detected by an enzyme-conjugated anti-immunoglobulin reagent followed by an enzyme reaction; the reaction produces a product, the colour of which can be measured spectrophotometrically. The antigen in an ELISA can be a pure nucleic acid (natural or synthetic), a pure protein (cloned or purified) or a peptide. Variability between ELISAs (as well as between ELISA and indirect immunofluorescence assays) can occur because of the differences in the properties of the antigens (such as the use of a peptide fragment or a full-length protein). In addition, antigen mixes can be used for an ‘ANA ELISA’, although the composition of the mix will determine the array of ANAs that can be detected.
Multiplex assays
A multiplex assay involves the simultaneous detection of several different ANAs. ALBIA is a popular format for a multiplex assay in which purified antigens are bound to beads with different fluorescent properties. Antibody binding occurs in a similar manner to an ELISA, and results are analysed in terms of the amounts of antibody bound to each antigen bead; the differences in the fluorescent properties of the different beads enables the analysis of antigen-specific responses. As these assays involve a limited number of antigens, they can only detect some ANAs. As such, the results of an ALBIA do not represent an actual determination of ANA status because ANAs that recognize many antigens are not measured; nevertheless, ALBIAs are often called ANA tests or ANA screens. A line immunoassay is another type of multiplex assay, although in this instance antibodies bind to parallel lines of a limited number of antigen specificities on a strip and are measured by immunoblotting. A microarray assay is also a solid phase, multiplex assay that involves a very large number of proteins or nucleic acids bound to a matrix; because of the number of features of such arrays (hundreds or more), the detection occurs with a microscopic system.
In the absence of a direct comparison between kits, the results from one assay cannot be used to infer the results of another. Indeed, when the results of ANA testing with a particular kit or assay format are unexpected (usually a negative result when there is a high pre-test probability for SLE or another autoantibody-associated rheumatic disease), re-testing with another kit of the same type or the use of a different type of platform can help to resolve uncertainty. In this regard, the usual problem with ANA assays is a high prevalence of false-positive results in otherwise healthy individuals4. The high prevalence of negative results in patients with established SLE is a more recently recognized finding7,21.
In contrast to classic ANAs such as anti-DNA or anti-Sm antibodies, some SLE-associated autoantibodies bind to molecules in the cytoplasm25,26. For example, anti-ribosomal P (RibP) antibodies bind to a set of phosphoproteins associated with the ribosome. Formally, such antibodies are not considered to be ANAs because the target molecules are located in the cytoplasm, and laboratories can report sera with cytoplasmic staining as being ANA negative. Although anti-RibP antibodies are highly associated with SLE and could be readily designated as classification criteria, the 2019 SLE classification criteria require ANA positivity, thus preventing the classification of patients who solely have anti-RibP antibodies as having SLE if the term ANA is interpreted narrowly — an example of the nomenclature challenge. When reporting the results of tests in which antibodies produce cytoplasmic staining (such as with anti-RibP antibodies), designation as a positive ANA test can cause less confusion and ambiguity than reporting the results as a negative ANA. In this regard, the term anti-cellular (or anti-HEp-2) antibodies might be preferable to ANA, as use of HEp-2 cells enables the detection of binding to any antigen in the cell; however, changing the traditional nomenclature is likely to be difficult.
The role of ANAs in disease
In addition to serving as markers of disease, ANAs can have a direct role in the clinical manifestations of SLE, although this role depends upon the specificity and amount of antibody present. Furthermore, identification of any disease role requires the use of an assay that allows specific measurement of that antibody. In considering the role of ANAs in disease, the properties of the target antigen are also important determinants of pathogenicity because, in some instances, the target antigens are the actual inducers of inflammation, and ANAs serve as conduits to the site of action.
Types of ANAs
ANAs can be characterized by their antigenic target, disease association or putative role in pathogenesis. In terms of antigenic targets, the most notable ANAs in SLE bind to either nucleosome components or RNA-binding proteins (RBPs). As previously discussed, the broad term ANA is also used for certain antibodies that recognize cytoplasmic proteins such as RibP, which might be present primarily in cytoplasm25,26. Nevertheless, as a group, ANAs that bind to nuclear or cytoplasmic autoantigens have some common features and patterns of expression irrespective of their precise cellular localization. Box 2 lists the properties of antigens that are the targets of autoantibodies in SLE.
Box 2 |. Properties of target antigens in systemic lupus erythematosus.
Are present in either the nucleus or the cytoplasm
Have a role in gene expression
Have conserved structures that are present widely in different mammalian species
Are complexes of protein and nucleic acids
Have a structural resemblance to bacterial or viral molecules
Display multiple epitopes
Have immunologically active nucleic acid components
Are cleaved and degraded intracellularly
Are released extracellularly during cell activation or cell death
Can be degraded by extracellular nucleases
Are present in extracellular vesicles or particles
ANAs that bind nucleosomes.
Nucleosomes are the form in which DNA is found in the cell nucleus and consist of DNA wrapped around a core octamer of histone proteins; the DNA between nucleosomes is called linker DNA. In SLE, ANAs can bind to DNA, histones or DNA–histone complexes24,27–29. Antibodies that bind to DNA–histone complexes are called anti-nucleosome or anti-chromatin antibodies. In general, antibodies that recognize nucleosomal antigens (such as anti-DNA and anti-histone antibodies) are expressed together or linked. Linkage is the potential consequence of epitope spreading; in epitope spreading, multiple nucleosome components, including antigenic structures comprising both histones and DNA, are recognized by the immune system following an initial antibody response to one component, possibly by molecular mimicry with a foreign antigen.
Of the anti-nucleosome antibodies, anti-DNA antibodies are the most distinctive; antibodies that recognize double-stranded DNA (dsDNA) are highly specific for the diagnosis (or classification) of SLE24,30. Anti-DNA antibodies can bind single-stranded DNA as well as dsDNA, and most antibodies bind to antigenic determinants present on both types of DNA24,31,32. Although most assays use dsDNA as an antigen, the term anti-DNA antibody can be used to encompass autoantibodies that recognize a broader spectrum of antigenic determinants present on the DNA molecule. Interestingly, some anti-DNA antibodies seem to recognize unusual DNA structures rather than the classic B conformation of dsDNA; the presence of these alternative forms of DNA (such as Z-DNA) depends on the sequence of the DNA as well as the salt concentrations used in the antigen-binding step of the assay31,32.
In contrast to other ANAs (and many antibodies that recognize foreign antigens), levels of anti-dsDNA antibodies can vary widely over time, especially in patients with active nephritis, and can essentially disappear during treatment, only to return during a flare33–36. As a result of the association between anti-DNA antibodies and disease activity, testing for these autoantibodies is often performed repeatedly during patient monitoring. Box 3 discusses the challenges around anti-DNA antibody testing. Testing for autoantibodies that recognize histones is much less commonly performed than evaluation of either ANAs or anti-DNA antibodies; however, some ANA assays use chromatin or nucleosomes as the target antigen. Assays that use chromatin as the test antigen can detect antibodies that recognize DNA–histone complexes as well as DNA or histones. Given the nature of chromatin, which contains DNA and histones, the actual antigen recognized might not be known unless another assay (such as an anti-DNA antibody assay) is also performed. Interest in testing for anti-histone antibodies relates to their presence in drug-induced lupus. Although antibodies that recognize certain histone proteins occur in drug-induced disease, anti-DNA antibodies are not present in this condition, whereas in SLE, both anti-DNA and anti-histone antibodies can occur together37,38.
Box 3 |. Anti-DNA antibody assays.
Although anti-DNA antibodies are an important biomarker, testing for these antibodies is problematic because of uncertainty around several important questions24,29,30. Is free or naked DNA the relevant antigen? Is anti-DNA antibody binding dependent on DNA sequence or structure? Are only high-avidity antibodies pathogenic?
In the absence of decisive answers to these questions, current assays make use of a variety of different DNA sources, ranging from mammalian DNA to bacterial plasmids to synthetic DNA. For some solid-phase anti-DNA antibody assays (such as enzyme-linked immunosorbent assays), a DNA-binding protein can be used to increase the concentration of DNA bound to the plastic plate; however, the effect of these proteins on DNA structure is unknown. Although the use of nucleosomes as antigens could provide a relevant form of DNA for antibody assays, nucleosomes can be difficult to prepare in a standardized and reproducible manner when purified from cells or constructed in vitro from histones or DNA28,29. In addition, although DNA in the double-stranded B conformation seems to be the target of most antibodies that recognize double-stranded DNA, alternative DNA structures can form depending on the sequence of the DNA and the salt concentration31,32. These conformations might exist only transiently in the cell, but can be represented in a stable form by synthetic or modified DNA. Notably, in the Crithidia luciliae immunofluorescence assay, the DNA source is a segment of double-stranded DNA that is present in an organelle called the kinetoplast24. Because the DNA is in a closed circle, it might be in a particular conformation that can be recognized by only some anti-DNA antibodies.
Studies comparing the results from different anti-DNA antibody assay kits have indicated striking differences between kits, with up to a twofold difference in the number of positive results in a population of patients with systemic lupus erythematosus35,57. The effects of these differences are important because the ability to detect anti-DNA antibodies can affect diagnosis, classification, assessment of disease activity and eligibility for clinical trials. Indeed, the use of the term ‘active, autoantibody positive systemic lupus erythematosus’ depends on the sensitivity of the assay used for these determinations, as well as the detection of various types of anti-DNA antibodies; however, rigorous studies on this issue are lacking8,13. For the clinician, understanding assay variability is important in interpreting discrepant results of anti-DNA antibody tests for patients who are seen by different providers or different health-care systems.
ANAs that bind RBPs and RBP-containing complexes.
Similar to anti-nucleosome antibodies, anti-RBP antibodies bind to complexes of proteins and nucleic acids, in this case, those that contain RNA39. In general, anti-RBP antibodies bind to the protein components of such complexes rather than to the associated RNA. As a group, antibodies that recognize RNA–protein complexes are sometimes termed anti-ENA antibodies because of the use of extractable nuclear antigen (ENA) as the antigen source in certain assays. ENA is a tissue extract that contains nuclear material along with other cellular components.
One class of anti-RBP antibodies recognizes protein components of small nuclear ribonucleoproteins (snRNPs) in complex with U-series RNA molecules; of these, anti-Sm antibodies bind primarily to B, B’ and D proteins, whereas anti-RNP antibodies bind to A, C and 70 K proteins. Because of the association of the U-series RNA molecules with proteins, anti-Sm antibodies can interact with U1, U2, U4 and U5 RNA snRNPs, and anti-RNP antibodies, which can be designated as anti-U1RNP antibodies, only react with the U1 snRNP39. In addition to anti-Sm and anti-RNP antibodies, the term anti-RBP antibody also covers anti-SSA/Ro and anti-SSB/La antibodies. Importantly, two types of anti-Ro antibodies exist: antibodies that recognize Ro60 bind to the protein component of a complex with small RNA molecules, whereas antibodies that recognize Ro52 bind to a protein called TRIM21 (a ubiquitin ligase molecule that does not bind RNA). Some assays allow measurement of antibodies that recognize both Ro60 and Ro52, enabling this distinction to be made40. Of the anti-RBP antibodies, only anti-Sm antibodies serve as a marker for SLE classification16–19; the other antibodies in this group, although common in SLE, also occur in other diseases. The expression of anti-RBP antibodies can also be linked; anti-Sm and anti-RNP antibodies are commonly expressed together, as are anti-SSA/Ro and anti-SSB/La antibodies41,42.
Disease associations of ANAs
Anti-DNA and anti-Sm antibodies occur primarily (and possibly exclusively) in SLE, which has led to their inclusion in SLE classification criteria16–19. By contrast, although anti-RibP antibodies are also specific for SLE, they are not part of the classification criteria and certain anti-RBP antibodies, which are common in patients with SLE, have a wider distribution among diseases. Thus, antibodies that recognize the Ro60 antigen occur in primary Sjögren syndrome (pSS) and systemic sclerosis (SSc), as well as in SLE40. Similarly, anti-RNP antibodies are also a feature of mixed connective tissue disease, a condition characterized by signs and symptoms of various rheumatic conditions, including SLE, rheumatoid arthritis and SSc. In mixed connective tissue disease, the titres of anti-RNP antibodies can be very high43,44. However, although not specific for SLE, anti-RNP and anti-SSA/Ro60 antibodies are nevertheless important biomarkers for constructing clinical and serological profiles of patients with this disease. In fact, anti-RNP antibodies are one of the most commonly expressed types of ANA in SLE, whereas anti-SSA/Ro60 is often present during pre-autoimmunity, a stage of disease when full-blown signs and symptoms have not yet developed despite increases in both ANA and cytokine expression45,46.
In the course of SLE, anti-nucleosome and anti-RBP antibodies can be expressed independently, a feature that also occurs in mouse models of this disease; in this instance, independence refers to the amount, time course and presence of ANAs in different patients or animal models34,47,48. These expression patterns imply differences in induction mechanisms, as well as in production by various B cell populations, and can also reflect the influence of race and ethnicity in patients. Thus, data suggest that patients with African ancestry are more likely to express anti-Sm and anti-RNP antibodies than those with European ancestry49. In view of the important immunological effects of anti-RBP antibodies, the severe disease manifestations that are commonly observed in patients with African ancestry (such as nephritis) might relate to their overall serological pattern, which comprises antibodies that recognize nucleosomes, as well as anti-RBP antibodies that are specific for SLE (such as anti-Sm antibodies) and those that can be expressed in other diseases (such as anti-RNP antibodies)50–53.
In evaluating the role of ANAs in such disease manifestations, it is important to consider that most sera contain multiple specificities of ANA, often in widely variable amounts. The presence of multiple ANAs in a serum sample can complicate the interpretation of IFA results, as the ability to detect certain binding patterns depends on the relative titres of the individual antibodies (Box 4). Furthermore, the failure to perform an end-point titre for the IFA or to use a quantitative assay such as an ELISA for a specific antigen limits the assessment of the serological profile of the patient and appreciation of the potential effect of ANAs on disease; as the amount of an ANA present can alter its effect on pathogenesis, qualitative determinations represent an incomplete picture.
Box 4 |. Issues with the detection of ANAs by IFA.
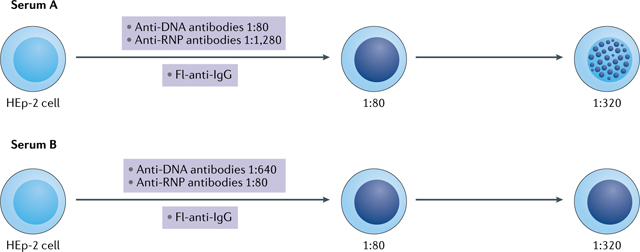
One of the issues with antinuclear antibody (ANA) testing by the indirect immunofluorescence assay (IFA) relates to the interpretation of results from sera that contain more than one, or even multiple, ANAs that produce different patterns of staining4. In this situation, the ability to detect more than one ANA depends on the relative titre of each ANA present. As an example, the figure shows the IFA patterns of two sera that both contain anti-DNA and anti-ribonucleoprotein (RNP) antibodies. In the top panel (serum A), the titre of the anti-DNA antibodies is 1:80, whereas the titre of anti-RNP antibodies is 1:1,280. In this hypothetical example, the laboratory assesses titres only to a dilution of 1:320. Anti-DNA and related anti-nucleosome antibodies can lead to homogeneous staining, whereas anti-RNP antibodies produce a speckled pattern. At 1:80, the homogeneous staining obscures the speckled staining, whereas at 1:320, the speckles become visible because the contribution of anti-DNA antibodies is no longer apparent. As such, the laboratory report would indicate ‘homogeneous 1:80 and speckles at 1:320’. In the bottom panel (serum B), anti-DNA antibodies are present at higher titres than anti-RNP antibodies. As such, the presence of speckles is obscured at both the 1:80 and 1:320 dilutions. For both sera, a titre of 1:320 is reported because the titration stops at a dilution of 1:320, which is more than sufficient to establish positivity. The much higher amount of anti-RNP antibodies in serum A is not detected because an end-point titre was not performed; the end point is determined when fluorescence is no longer visible. The use of assays that detect antibodies to specific nuclear antigens (such as multiplex assays) can complement the analysis by IFA and enable the better determination of a serum that contains multiple ANA specificities. Fl-anti-IgG, fluorescein-labelled anti-immunoglobulin antibodies.
In general, current serological assay formats provide information about autoantigen binding (often as either a positive or negative result for anti-RBP or anti-ENA antibodies) although, with many of the technologies available, immunoglobulin isotypes could be readily measured. Multiplex assays such as addressable laser bead immunoassays (Box 1) enable the assessment of a limited number of autoantibodies, including those that occur either commonly or have utility for diagnosis or classification. However, although the term ANA assay can be used for such multiplex platforms, they typically do not detect antibodies that recognize many well-defined nuclear antigens that can occur in SLE or other conditions, albeit at low frequencies. Indeed, the antigens in these assays represent the most common and characteristic specificities for conditions that include SLE, pSS, myositis and SSc. A full evaluation of serological status might, therefore, require more than one approach, with a multiplex assay providing information for screening for the most common ANAs in different diseases (including pSS, SSc and SLE)4,13.
Current assays usually do not characterize immunochemical features of antibodies such as avidity, cross-reactivity or effector function (including complement fixation or Fc receptor binding). However, although no longer commonly performed because of availability and concerns about radioactivity, the Farr assay for anti-DNA antibody determination is an exception, as it does provide information relevant to avidity54–56; the high salt conditions required for precipitation by ammonium sulfate in the Farr assay favour the detection of high-avidity antibodies. Anti-DNA antibody results obtained by the Farr assay often differ from those of an ELISA or multiplex assay, the latter of which can detect a broader array of anti-DNA antibodies, including less avid antibodies57,58. The limited information provided by conventional serology is one factor preventing the clear-cut association of laboratory findings with clinical events, although the importance of avidity for pathogenicity, although possible, has not been rigorously proven.
The pathogenicity of ANAs
The expression of an ANA can be categorized as pathological insofar as antibodies of that specificity are found in patients with disease and are not found in otherwise healthy individuals; as such, the production of these antibodies signifies a pathological disturbance in the mechanisms that should prevent B cell and T cell reactivity to self-antigens. However, the aberrant expression of a particular ANA does not imply a causal role in disease manifestations in terms of signs and symptoms or organ damage; these manifestations include tissue inflammation as well as cellular dysfunction and death. In contrast to ANAs that are present in the blood in a disease, ANAs that actually promote or augment disease are considered pathogenic59. Figure 1 summarizes the mechanisms by which two main types of ANAs (anti-DNA and anti-RBP antibodies) can promote disease in SLE.
Fig. 1 |. Anti-DNA and anti-RBP antibodies in systemic lupus erythematosus.
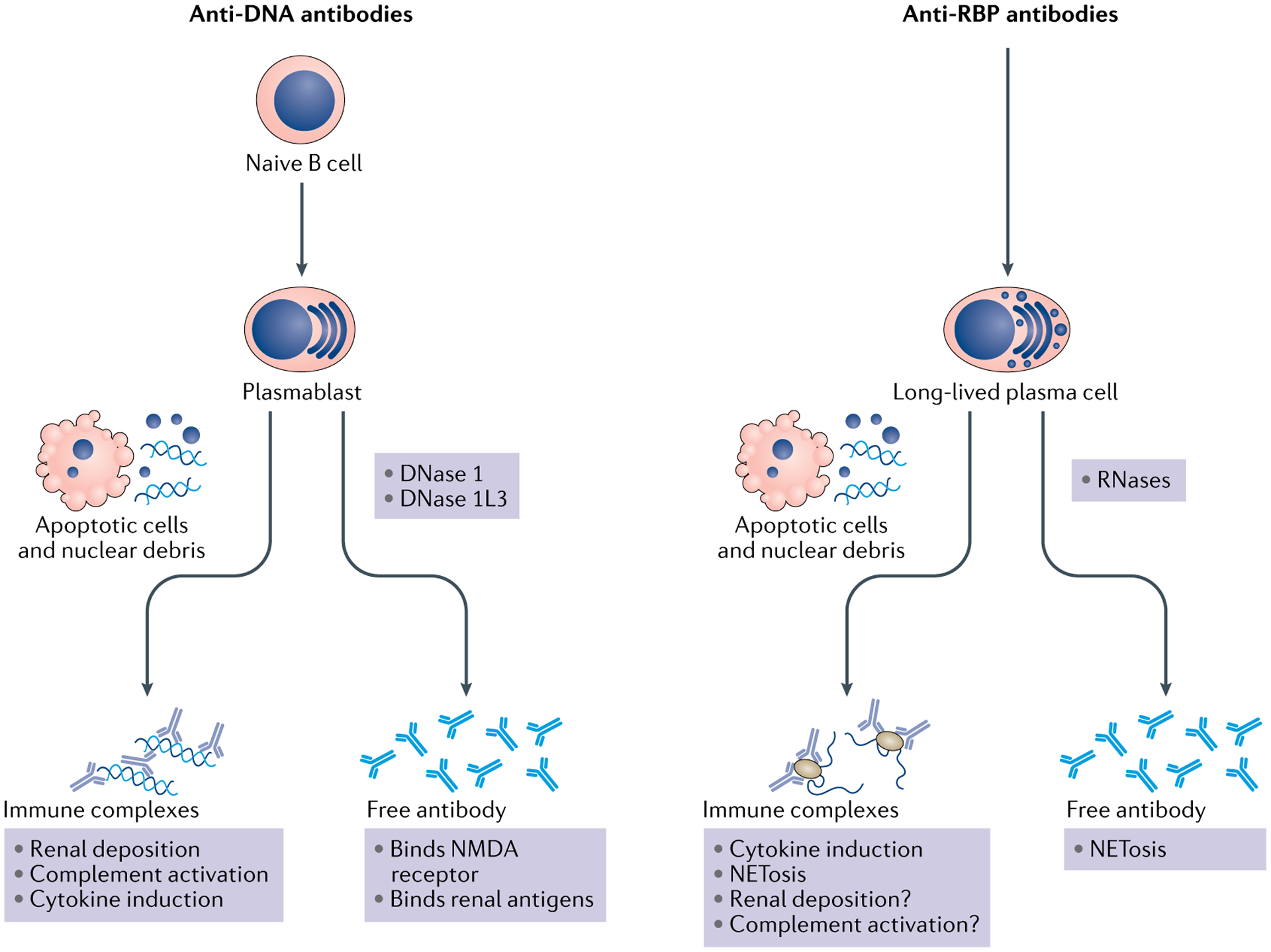
Anti-DNA and anti-RNA binding protein (RBP) antibodies can arise from different B cell populations. This difference can account for the pattern of expression during the course of systemic lupus erythematosus and the apparent relationship to disease activity. Whereas titres of anti-RBP antibodies are consistent over time because they are produced by long-lived plasma cells, anti-DNA antibody titres can vary markedly because they arise from naive B cells that transition to plasmablasts, which can be short-lived. Once secreted, both anti-DNA and anti-RBP antibodies can mediate pathogenesis either as free antibodies or as components of immune complexes, although in some instances, the actual role of these antibodies is uncertain. Cells undergoing apoptosis are considered to be the most common source of nuclear antigens, which can be released in a free form or as constituents of microparticles. Extracellular nuclear molecules can also arise from cells undergoing other forms of cell death (such as necroptosis or necrosis) or from neutrophils releasing neutrophil extracellular traps, which can occur with cell death (known as NETosis) or without. The amount of this material that is extracellular is determined by the clearance of dead and dying cells, as well as by the digestion of released nucleic acids by nucleases. Anti-DNA antibodies can also contribute to neuropsychological manifestations by mediating neuronal cell death by cross-reactive binding to the N-methyl-d-aspartate (NMDA) receptor on neurons; activation of microglial cells can subsequently affect neurons by dendritic pruning. DNase 1L3, DNase 1 like 3.
Immune complex formation between autoantigens and ANAs.
Immune complex formation is a major mechanism by which ANAs mediate inflammation in SLE, either by deposition in the tissue or by promoting cytokine production by cells of the innate immune system. An immune complex is a molecular structure formed by the binding of an antibody to its cognate antigen. Depending on the size of the antigen and the number of different antibodies that recognize sites on the antigen, an immune complex can vary in mass, solubility and biological activity. For the formation of immune complexes, a source of autoantigen is essential. In SLE, nuclear self-antigens can arise from cell death, and both excessive cell death and impairment of the clearance mechanisms can boost the amount of extracellular nuclear material available60,61. For DNA, deficiency of nucleases such as DNase 1 and DNase 1-like 3 (1L3) can increase the amount of self-antigen in the blood that can form immune complexes62–65. Disturbances in RNase activity might have a similar role for RBP antigens66,67. Alternatively, more stable forms of RNA, such as microRNAs, could serve as antigens in immune complexes. Finally, nucleic acids in the circulation can exist in a particulate form as microvesicles or microparticles; the amount of DNA in a free or particle form is determined by nucleases68–70.
Given the crucial role of self-antigen in the formation of immune complexes, it is, therefore, possible that the amount of self-antigen available for complex formation could determine the pathogenicity of an ANA. Thus, an ANA might be present without clinical consequences during the pre-autoimmune stage of SLE or in other clinical conditions because adequate concentrations of the cognate antigen are not present in the blood or tissue. The measurement of self-antigen is not commonly performed, although assays for both nucleosomes and DNA are commercially available. Direct measurement of the total amount of immune complexes is also not often performed because the added value of these determinations in the clinical setting is not clear71–73. By contrast, the determination of complement components (such as complement proteins C3 and C4 or erythrocyte-bound complement components) is widely used to infer the presence of biologically active immune complexes74,75. Box 5 discusses issues related to assays used to determine immune complex concentrations.
Box 5 |. Immune complex detection assays.
Owing to the important role of immune complexes in the pathogenesis of systemic lupus erythematosus (SLE), the assay of immune complex concentrations could potentially provide an important biomarker that does not depend on either the specificity of the antibody or the antigen in the complex. Many assays of this kind have, therefore, been developed on the basis of several different immunochemical principles71–74. These assays include those that determine the presence and amounts of immune complexes on the basis of physical properties (such as size, precipitation in the cold and precipitation with polyethylene glycol), as well as assays for the binding of immune complexes to complement protein C1q in either the fluid or solid phase; the binding of complement protein C3 or C3 fragments within immune complexes to complement receptors on cells (usually the Raji B lymphoblastoid cell line, which has receptors for C3b, C3d, C1q and Fc); and the binding of immune complexes by rheumatoid factor. Rheumatoid factor has a binding preference for conformational determinants on the Fc portion of IgG that occur with antigen interaction. As with indirect immunofluorescence assays, many kits for measuring immune complexes have been developed, but their use for assessing either diagnosis or staging disease activity has been limited by variability and uncertainty concerning the most appropriate format for SLE71. Furthermore, as immune complexes occur in many inflammatory or infectious diseases, these assays are not specific for SLE. Compared with direct assay of immune complexes, indirect assays such as the measurement of complement proteins or the presence of complement fragments fixed to cells have proven more reliable and informative74. In the future, assay of IgG bound to microparticles might represent a new approach to the direct assay of immune complexes, although assays of this kind require flow cytometry68–70.
The role of immune complexes in lupus nephritis.
Of the mechanisms for the generation of tissue lesions in SLE, the formation and deposition of immune complexes in the kidney has received the most investigative interest76. Those ANAs that mediate nephritis are termed nephritogenic, of which anti-DNA antibodies are the most characteristic. DNA and anti-DNA antibodies can form immune complexes that localize to the kidney, where complement is activated (Fig. 2a). Several forms of laboratory and histopathological findings provide evidence that supports the pathogenic deposition of immune complexes in the kidney. The most compelling of these findings is the demonstration of immune complexes and their components (such as immunoglobulins and complement) in the kidney by light, electron and immunofluorescence microscopy1,2. Increased anti-DNA antibody levels and decreased amounts of complement component proteins C3 and C4 during active disease also support a pathogenic role for immune complexes; as immune complex formation and deposition can take place over time and precede clinical events, concentrations of immunoreactants might not be raised at the time of patient presentation. Indeed, autoantibody levels might be reduced because of tissue deposition77.
Fig. 2 |. Mechanisms of glomerulonephritis induction in systemic lupus erythematosus.
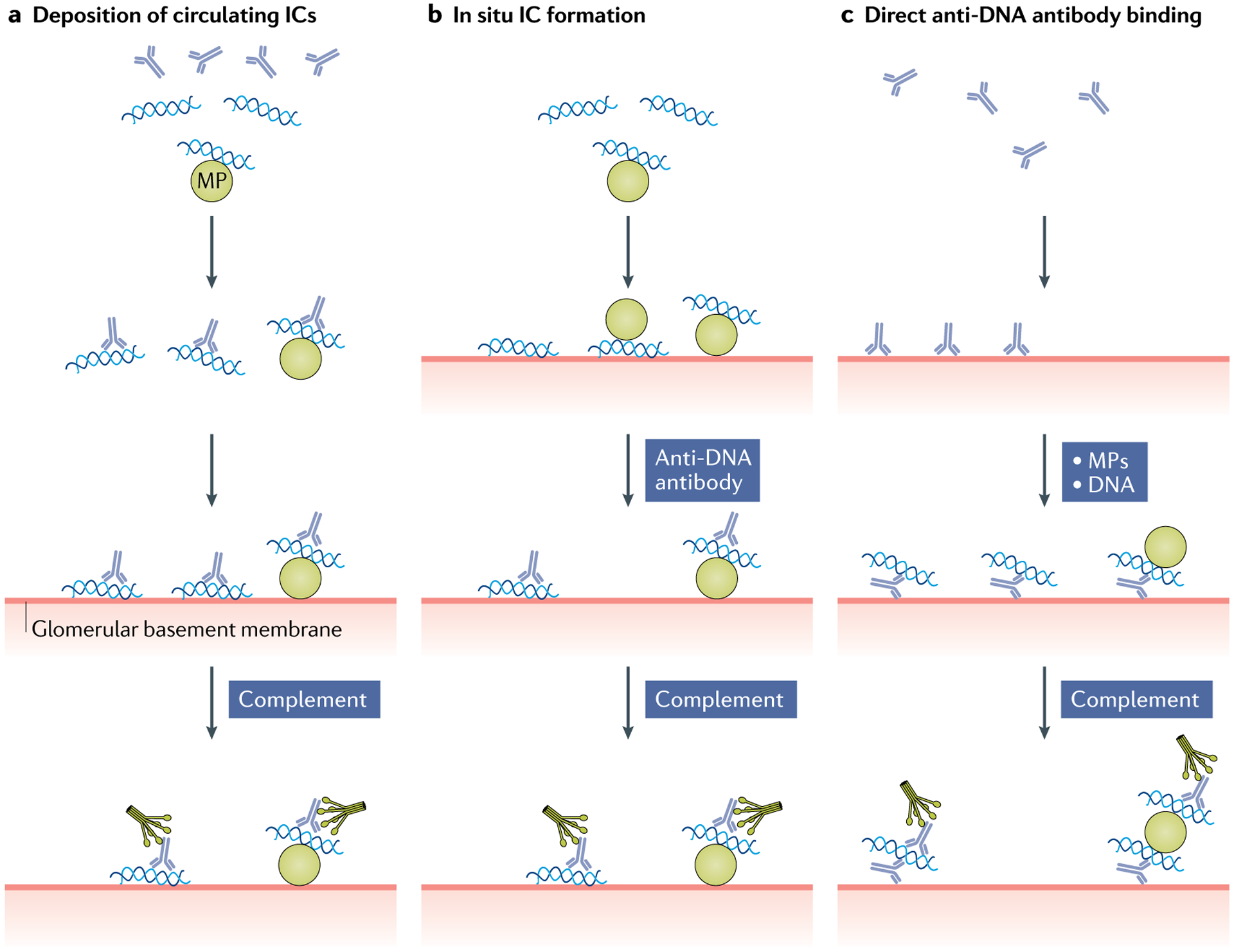
Anti-DNA antibodies can promote nephritis by several different mechanisms in systemic lupus erythematosus. a | Nephritogenic anti-DNA antibodies can form immune complexes (ICs) with circulating DNA, as well as with DNA on microparticles (MPs); these ICs can then be deposited on the glomerular basement membrane and activate complement to induce damage and recruit inflammatory cells. In this process, the charge of the antigen present might influence renal localization. b | During in situ IC formation, complexes can form with DNA or MPs that have bound to the glomerular basement membrane; such antigens are termed ‘planted’. These ICs can then activate complement. c | Anti-DNA antibodies can bind to fixed sites on the glomerular basement membrane, attracting circulating DNA in a free form or as MPs. The assembly of ICs in this mechanism requires that antibody bound to the glomerular basement membrane is, nevertheless, able to interact with circulating antigen to serve as a nidus for IC formation.
Although a role for anti-DNA antibodies in immune complex formation seems likely, it has been difficult to demonstrate circulating immune complexes using immunochemical techniques, perhaps because of issues such as assay sensitivity and time of sampling. As a result, alternative mechanisms for immune complex pathogenicity have been proposed. Thus, immune complexes might form in situ in the kidney (Fig. 2b); anti-DNA antibodies could interact with DNA that has arrived separately and has been attracted to the glomerular basement membrane on the basis of charge (either of the DNA itself or the associated histones in the nucleosome), known as ‘planted’24,30. DNA and nucleosomal antigens in the form of chromatin can also be generated by local cell death, which can increase in the kidney owing to a local decrease in DNase 1 (REFS78,79). Another mechanism for nephritogenicity involves the direct interaction of anti-DNA antibodies with the glomerular basement membrane, creating a nidus for subsequent local immune complex deposition (Fig. 2c). In this regard, as nuclear antigens can be components of microparticles, the immune complex might involve these large structures; similarly, a particle might serve as a planted antigen.
The role of immune complexes in cytokine induction.
In the pathogenesis of nephritis, cytokines released by cells such as monocytes and macrophages can promote inflammation. In addition to inducing complement activation in the kidney, immune complexes can induce the production of pro-inflammatory cytokines, most notably type I interferon. Anti-DNA antibodies can bind to DNA and effectively be internalized into innate immune cells, most prominently plasmacytoid dendritic cells80. Once inside the cell, DNA can interact with the endosomal Toll-like receptor (TLR), TLR9, as well as with other nucleic acid sensors in the cytoplasm. These sensors can either access DNA following its uptake as part of an immune complex, or they can respond to DNA that is aberrantly present in the cell as a result of infection by intracellular organisms, mitochondrial release of DNA from stressed or damaged mitochondria, or the formation of micro-nuclei in response to genotoxic agents or chromosomal instability81–83. Thus, nucleic acid sensors represent a type of internal host defence system, for which DNA in immune complexes is just one type of trigger.
Of these sensors, the cyclic GMP-AMP synthase (cGAS)–stimulator of interferon genes (STING) system mediates the production of type I interferon and other cytokines. Once bound by DNA, cGAS catalyses the formation of cyclic GMP-AMP that binds to STING. The outcome of these interactions is the stimulation of type I interferon production and its downstream effects84,85. This stimulation only occurs with certain anti-DNA antibodies; however, the properties of these antibodies that enable the formation of immune complexes that can stimulate type I interferon production are not known. Furthermore, it is not clear if the DNA–anti-DNA antibody immune complexes that are deposited in the kidney can induce cytokine production by plasmacytoid dendritic cells. At present, there is no term for pathogenic antibodies that induce cytokine production as opposed to renal disease.
Although initial studies on cytokine induction by immune complexes implicated anti-DNA antibodies as an important factor80, subsequent research has shown that immune complexes with anti-RBP antibodies can stimulate type I interferon production and might actually be the main promoters of this pathway49. The differing results of these studies probably reflect the demographics of the patient populations studied, as the increased expression of anti-RBP antibodies in patients with African ancestry can affect the assessment of pathogenicity86–88. In this mechanism, immune complexes containing RNA are ingested by phagocytes, in which they interact with internal RNA sensors in the endosome (such as TLR3 and TLR7) or in the cytoplasm81–83. In addition, cytoplasmic sensors of RNA, including the retinoic acid-inducible gene 1 and melanoma differentiation-associated protein 5 pathways, can lead to the production of type I interferon when stimulated by RNA that has entered cells in the form of immune complexes with anti-RBP antibodies, which provide a conduit for entry of RNA into the cell. Studies thus far suggest that anti-RBP antibodies of all known specificities can form immune complexes that have immunostimulatory activity; however, it is possible that, depending on the immunochemical properties of autoantibodies such as fine specificity or avidity, only certain anti-RBP antibodies can induce these responses.
Notably, although signalling via TLR7 and TLR9 superficially seems similar, the induction of pro-inflammatory cytokines and pathological outcomes can be quite different89. In mouse models of SLE, TLR7 engagement seems to exacerbate disease, whereas TLR9 engagement seems to suppress disease; nevertheless, both TLR7 and TLR9 can bias the production of autoantibodies that recognize RNA and DNA, respectively. The differences in clinical and serological outcomes following TLR7 and TLR9 engagement probably reflect the various effects of TLR stimulation during B cell development and activation90,91.
Other pathogenic roles of ANAs.
As well as renal deposition and cytokine induction, ANAs, either alone or in the form of immune complexes, can induce neutrophil extracellular trap (NET) formation92,93. NETs are complex structures that can be released from neutrophils as a defence against bacteria and fungi by entrapping and killing them. A NET comprises high molecular weight DNA that is decorated with enzymes such as myeloperoxidase that are usually contained within cytoplasmic granules. Although NETs have an important role in host defence, they can also damage tissues, especially the endothelium. Thus, the stimulation of NET formation represents another mechanism by which certain ANAs, including anti-RNP antibodies, can promote SLE pathogenesis94,95.
Promoting disease by another mechanism, certain anti-DNA antibodies can cross-react with neuronal antigens, which might contribute to central nervous system manifestations in SLE in both mouse models and patients96,97. As one example, anti-DNA antibodies that cross-react with the N-methyl-d-aspartate receptor in the brain can induce excitotoxic cell death; these antibodies can also contribute to microglial activation that kills neurons and mediates synaptic pruning96,97. Cross-reactivity between certain ANAs and constituents of the gut microbiota has also been reported, suggesting the possibility that some ANAs arise from gut dysbiosis98,99. The presence of gut organisms that are bound to antibodies in patients with SLE suggests that ANAs can affect the composition of the microbiota and its influence on the immune system. Given the number of organisms in the gut, it will be important to determine those that can serve as a target for autoreactivity and the mechanisms by which disturbances in the immune system alter the microbiome98,99.
Changing attitudes to ANA testing
ANAs represent one of the most venerable biomarkers in rheumatology if not all of medicine; nevertheless, the field remains in flux. Although new technologies enable an improved breadth and depth of serological testing, the interpretation of findings from existing technologies is often uncertain. Furthermore, the observations that the presence of ANAs can influence treatment responses suggest that these antibodies have a profound influence on the course of SLE in a way not previously appreciated or understood.
Current uses for ANA testing
As suggested in the previous sections, ANAs have the ability to induce a wide variety of immune disturbances and clinical manifestations in SLE (and other diseases). Indeed, one antibody could potentially induce such disparate events as cytokine induction, immune complex deposition in the kidney and the stimulation of NET formation; however, the time frame of these different pathogenic events can vary, occurring both acutely and chronically. Importantly, a pathogenic antibody might not be disease specific, as pathogenicity can occur with antibodies that are not relevant for classification or even considered to be characteristic of SLE. Anti-RNP and anti-SSA/Ro60 antibodies are two prominent examples of such pathogenic antibodies that are not specific for SLE40,49,87.
A variety of approaches have been used to determine the contribution of a given antibody specificity to either overall disease activity or to the occurrence of a particular manifestation (such as dermatitis or nephritis). Analogous to Koch’s postulates for infection, a clinical condition can be assumed to be autoimmune in aetiology on the basis of certain criteria. For B cell-mediated conditions such as SLE, these criteria include the presence of an autoantibody, documentation of the autoantibody in the pathological lesion and demonstration that the autoantibody can cause tissue pathology. To evaluate these criteria, techniques such as trans-placental transmission of clinical disease, adoptive transfer into experimental animals or in vitro effects on cellular function can be used. Supportive evidence for autoimmunity can also involve an appropriate animal model, clinical benefit from immunosuppressive therapy, association with other evidence of autoimmunity and lack of evidence of other causes of the condition100.
ANAs as indicators of disease activity.
Although serology in SLE is inherently complicated, determining the role of different ANAs in individual patients is, nevertheless, important, as these antibodies can provide valuable biomarker information for staging, as well as assessing prognosis and disease activity. In this regard, the utility of ANA testing can vary depending on the context in which it is used. For example, anti-RBP antibodies (other than anti-Sm antibodies) are not useful for SLE classification, but it seems likely that all members of this group might promote type I interferon production when in the form of immune complexes49.
Among the many ANAs expressed by patients with SLE, anti-RNP antibodies seem to be a strong determinant of the presence of an interferon signature, as data suggest that the increased expression of anti-RNP antibodies in patients with SLE with African ancestry increases the likelihood of their having an interferon signature49. Patients with different ancestral backgrounds might also differ in the simultaneous expression of multiple ANAs. For example, the increased expression of anti-RBP antibodies might lead to a more diverse pattern of ANA expression in patients with SLE who have African ancestry compared with those who have European ancestry.
Although ANAs were discovered more than 60 years ago, the role of these antibodies in specific SLE manifestations, as well as in overall disease activity, still remains uncertain. Indeed, among the many ANA specificities identified, only anti-DNA antibodies have the status of a marker of disease activity, as exemplified by their inclusion in activity measures such as the SLE Disease Activity Index (SLEDAI)101,102. Notably, however, the FDA does not consider anti-DNA antibodies to be either biomarkers or surrogate markers for SLE. The inclusion of anti-DNA antibodies as a measure of disease activity in the SLEDAI relates, in part, to its highly variable expression over time. During a flare, levels of anti-DNA antibodies can rise sharply in association with decreases in complement proteins; levels can also decrease and essentially disappear after therapy with glucocorticoids or other immunosuppressive agents33–36. Because disease flares characterize the course of SLE, the dramatic rises and falls of anti-DNA antibodies strengthen the idea that these autoantibodies contribute to disease.
The pattern of expression of anti-DNA antibodies differs from that of anti-RBP antibodies, which are generally expressed at a more consistent level than anti-DNA antibodies and show relatively little variation in the blood over time34,103. However, it is important to recognize that data on the longitudinal expression of anti-RBP antibodies are limited compared with data for anti-DNA antibodies. In this regard, even though current technologies readily enable quantitative determinations of essentially all ANAs, monitoring of ANA titres for the assessment of disease activity has primarily focused on anti-DNA antibodies, leaving a considerable gap in the literature.
ANA variation as an indicator of pathogenesis.
The differences between anti-DNA and anti-RBP antibody expression suggest that these antibodies represent fundamentally different B cell responses (Fig. 1). In view of their often persistent expression, anti-RBP antibodies seem to arise from long-lived plasma cells in the bone marrow that are no longer proliferating and are, therefore, less susceptible to immunosuppressive therapy. By contrast, anti-DNA antibodies seem to arise from newly generated plasmablasts that require proliferation for their differentiation or maintenance, and some evidence suggests that these cells might arise from naive or pre-naive B cells104–107. As such, the elimination of anti-RBP antibodies might require different types of therapy from those required to attenuate anti-DNA antibodies. In fact, the persistence of anti-RBP antibody responses despite treatment with conventional immunosuppressive therapy might be one of the reasons why these therapies have limited efficacy in some patients with SLE34. Notably, some therapies, such as belimumab, might be able to reduce titres of anti-RBP antibodies108, although determining such an effect requires a quantitative assay or at least a determination of seroconversion from positive to negative. Interestingly, during treatment with belimumab, the reduction in anti-DNA antibody titres exceeds that of anti-RBP antibodies, providing additional evidence that these autoantibodies arise from distinct mechanisms108.
In their duration, anti-RBP antibody responses resemble those of antibodies that develop following immunization or infection, especially chronic viral infections; these antibodies can persist for years to decades109. Given that existing data suggest that anti-RBP antibody responses are usually long-lived, repeat testing is usually deemed unnecessary in routine care after initial serological assessment and is not commonly performed. In addition, many assays for anti-RBP antibodies are qualitative (the results are provided in terms of either positive or negative) and do not provide information about the potential effect of quantitative variations or other immunochemical features on pathogenicity; with qualitative assays, even a large change in an antibody response and corresponding change in the amounts of immune complexes can be missed, as this loss of antibodies to complex formation would be below the detection limits of any assay.
In general, in SLE, the approach to determining the pathogenicity of an ANA for a clinical manifestation is to use association studies in which the expression of an ANA and a manifestation are compared. This approach has yielded evidence of associations between anti-DNA antibodies and nephritis, and anti-SSA/Ro antibodies and subacute cutaneous lupus and neonatal lupus syndrome40. Studies of this kind have also suggested an association between anti-RibP antibodies and central nervous system manifestations, although this relationship is unclear because of the variety of nervous system manifestations in SLE and the variability in test results owing to different assay formats or antigen preparations25,26.
Efforts to meet the aforementioned criteria for an autoimmune disease more completely have occurred essentially only with anti-DNA antibodies and nephritis, although cross-sectional studies have examined the relationship between serological findings and kidney disease; studies on eluates from glomeruli have also shown the presence of antibodies of a number of specificities in the kidney110–113. The relationship between many clini cal manifestations and particular ANAs remains uncertain and speculative, and the extent to which autoantibodies mediate many important features of SLE (including pleuritis and pericarditis), has not been determined. Thus, the use of anti-DNA antibodies and complement testing in the general assessment of disease activity, as well as activity in organs other than the kidney, is uncertain.
A new perspective on ANA testing
Despite many uncertainties in the field, we believe that the role of ANAs in manifestations of SLE is, in fact, substantial and determinative of outcome, but that current approaches for serological testing are inadequate. We also believe that available assay platforms can be modified and expanded to provide more robust serological information than is currently available. We therefore advocate for considerable changes to current serological strategies, which we believe can provide a much deeper and clearer picture of the role of ANAs in SLE.
At the heart of this re-evaluation and re-conceptualization of the role of ANAs in SLE pathogenesis are the following ideas: ANAs should be considered as potentially pathogenic whether or not they are specific for SLE or useful for diagnosis and classification; quantitative assays should be used for ANA determinations; the frequency of anti-RBP antibody assessment should be increased; analytic methods should be developed to explore the role of ANAs as an ensemble rather than separate entities; immune system changes during established disease should be defined; studies on ANA expression should be integrated with studies on B cell phenotype and functional properties; and ANA assays should be validated for various biomarker uses.
Consider all ANAs as potentially pathogenic.
In studies on the pathogenesis in SLE, there has been a tendency to focus on anti-DNA antibodies. Thus, for an animal model to achieve status as a model for the study of SLE, anti-DNA antibody expression is required24; few animal models have been designed to elucidate or even consider the contribution of anti-RBP antibodies to immunological events. With the focus on anti-DNA antibodies, there has been limited attention given to ANAs that are neither specific for SLE nor vary substantially in expression over time. For example, anti-SSA/Ro60 antibodies are among the first autoantibodies produced by patients with SLE, and evidence exists that these antibodies can form immune complexes and be deposited in the kidney112,114–116. In comparison with anti-DNA antibodies, few studies have addressed the nature of the complexes formed by anti-SSA/Ro60 antibodies. As a result, most studies using monoclonal antibodies of either murine or human origin to explore pathogenicity bind to DNA, although such studies for anti-SSA/Ro60 antibodies or other ANAs could be very informative.
Use quantitative assays for ANA determinations.
Although results are often provided as just positive or negative, current ELISA and multiplex assays enable the quantitative assessment of all ANAs. Theoretically, a greater amount of antibody should lead to greater inflammation or damage; similarly, antibody avidity or fine specificity would be expected to contribute to pathogenicity. Therefore, some of the difficulties in relating ANA responses to disease manifestations might result from insufficient quantification and immunochemical characterization.
As an approach to exploring pathogenicity, assays that measure the ability of antibodies to fix complement can be very informative. Although assessment of complement fixation is an old approach, it is, nevertheless, highly relevant to conditions mediated by immune complexes. Characterizing the ability of anti-RBP antibodies to fix complement, for example, could help researchers to understand the reduced complement concentrations that are often encountered in patients in the absence of anti-DNA antibodies or nephritis (so-called serological negative, clinically negative disease).
Increase the frequency of anti-RBP antibody assessment.
Although many studies indicate that anti-RBP antibody titres vary little over time (consistent with antibody production by long-lived plasma cells), other data indicate that anti-RBP antibody titres can vary considerably in some patients and correlate with disease activity43,117–121. The infrequent assessment of anti-RBP antibody titres that currently takes place during the course of SLE following an initial evaluation is a reflection of reliance on older and incomplete data. Over a time frame of years, gains and losses of specific ANAs from the anti-RBP antibody group can occur, providing a rationale for periodic retesting of patients122. Therefore, it seems important to test the full array of ANAs at the time of a flare, rather than relying on a test result obtained months or even years before a clinical event. Periodic serological retesting seems well justified in view of data showing that some immunosuppressive drugs in widespread use, such as mycophenolate mofetil, might decrease plasma cell populations (the presumed source of anti-RBP antibodies)123–127.
Develop analytic methods to explore the role of ANAs as an ensemble.
Studies suggest that anti-DNA and anti-RBP antibodies can both form immune complexes that promote type I interferon production49,80,86–88. To elucidate determinants of type I interferon expression, one study simply summed the titres of individual ANAs and demonstrated that the magnitude of type I interferon induction in an in vitro system related to the total amount of ANA present87. Similarly, if autoantibodies other than anti-DNA antibodies (such as anti-Sm antibodies) can cause nephritis, assessing these responses in tandem might be more illuminating than considering the effects of anti-DNA antibodies alone51.
Studies on the role of different ANAs in SLE clearly indicate redundancy in pathogenic mechanisms (such as nephritis and type I interferon stimulation). An intriguing question relates to the possibility of synergy, which could occur if, for example, immune complexes could simultaneously stimulate TLR and non-TLR sensors for both DNA and RNA. This issue might be especially relevant for patients with African ancestry because of their frequent expression of anti-RBP antibodies49.
Thus, it seems reasonable to characterize patients with SLE into serologically defined subsets depending on their positivity for anti-DNA and anti-RBP antibodies: anti-DNA antibody-positive, anti-RBP antibody-negative; anti-DNA antibody-negative, anti-RBP antibody-positive; double positive; and double negative. Analysing treatment responses in these serologically defined subsets should provide a valuable perspective for assessing treatment efficacy. Studies using autoantibodies to novel autoantigens involved in signalling pathways provide support for this type of approach and the value to characterizing autoantibody clusters as elements in pathogenesis128.
Define immune system changes during established disease.
As previously described, studies related to clinical trials for new therapies for SLE have indicated that a substantial percentage (up to ~30%) of patients with established SLE who are screened for trial entry are ANA negative8,129. Although such seronegativity might be a result of the performance characteristics of assays, at least some ANA responses seem to wane over time and diminish appreciably in a stage of disease that can be termed post-autoimmunity130; this stage occurs subsequent to the diagnosis of disease, just as pre-autoimmunity occurs prior to diagnosis. During post-autoimmunity, reduction of ANA responses might reflect the natural history of the disease as well as the effects of therapy. Hydroxychloroquine, a mainstay of SLE therapy, might modulate the overall balance of the immune system via effects on TLR and other signalling pathways such as cGAS–STING, thereby indirectly affecting antibody production131. In addition, rituximab, belimumab, cyclophosphamide and mycophenolate mofetil can all affect B cell function, although belimumab and mycophenolate mofetil can have direct effects on plasma cells and glucocorticoids can affect IgG levels123–127. However, the extent to which these actions can alter ANA production, especially by long-lived plasma cells, remains to be proven, and the expression of new ANAs can, nevertheless, occur following diagnosis, with the process of epitope spreading continuing despite therapy.
Integrate studies on ANA expression and B cells.
Elegant studies have established the unique properties of B cells from patients with SLE, delineating the effects of disturbances in checkpoints on the antibody repertoire132–136. Furthermore, these studies have documented the role of specific B cell populations in the generation of ANA responses, as well as the role of extrafollicular pathways for autoantibody generation as opposed to the perhaps expected role of germinal cell reactions. An important next step will be to determine how these disturbances lead to the production of specific ANAs and the immunochemical properties that underlie pathogenicity, for example, during the formation of immune complexes. The rules for antibody recognition of antigen and the generation of a mature antibody response have mostly been derived from studies on immunization models in animals. By comparison, studies in humans have been limited and have involved only select antigens such as influenza virus and HIV137,138. Studies in humans can also involve adjuvants that differ from those used in many animal studies. Therefore, comparison of the responses to foreign antigens in patients with SLE and otherwise healthy individuals can be informative, as disturbances that promote ANA responses might also affect the specificity and avidity of antibodies elicited to a virus139.
Validate ANA assays for various biomarker uses.
Current ANA assays have been validated primarily for screening patients in routine patient care. Although these assays perform reasonably well in the setting of SLE, assays for the purpose of classification or assessment of disease activity might have different performance characteristics from those for routine clinical testing4. Experience gained from using ANA assays to determine eligibility for clinical trials highlights the uncertainty in current assay approaches8. Until assays are developed and standardized for the unique setting of SLE, it will be difficult to answer fundamental questions about this disease, such as whether SLE is always ANA positive, if classification changes with serological status and if ANA negativity at the time of classification (rather than at an unknown time in the past) precludes the classification of SLE. Given that classification is the foundation of research into both mechanisms of SLE and its management, we believe that a detailed and comprehensive examination of available ANA assays is essential for future progress.
Conclusions
The expression of autoantibodies to nuclear molecules is an important immunological feature of SLE that provides biomarkers for elucidating pathogenesis as well as facilitating diagnosis and treatment. Although these responses have long been the focus of investigation, new ideas and new technologies promise novel insights into the origin of ANAs, as well as their contributions to tissue inflammation and damage. We believe that these studies would benefit if greater attention were paid to the full gamut of ANAs expressed in SLE, whether specific for SLE or not, and from the development of both qualitative and quantitative assays to provide better markers of disease activity and better approaches to therapy.
Key points.
Antinuclear antibodies (ANAs) bind DNA, RNA and complexes of nucleic acids and protein.
In addition to ANAs, anti-DNA and anti-Sm antibodies are part of the classification criteria for systemic lupus erythematosus (SLE).
Anti-DNA antibodies and antibodies that recognize RNA-binding proteins show distinct patterns of expression that relate to their origin from different B cell populations.
ANAs can mediate events in the pathogenesis of SLE as either free antibodies or as immune complexes.
Amounts of ANAs in patients with SLE can change over time as a result of the natural history of the disease or the effects of immunosuppressive agents.
Rescreening of ANA levels after disease onset could provide important information about disease mechanisms and disease status.
Acknowledgements
The work of D.S.P. is supported by a Veterans Administration Merit Review grant and by a National Institutes for Health grant (1R01AR073935). The work of P.E.L. is supported by the RILITE Foundation.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Kaul A et al. Systemic lupus erythematosus. Nat. Rev. Dis. Prim 2, 16039 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Tsokos GC, Lo MS, Costa Reis P & Sullivan KE New insights into the immunopathogenesis of systemic lupus erythematosus. Nat. Rev. Rheumatol 12, 716–730 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Agmon-Levin N et al. International recommendations for the assessment of autoantibodies to cellular antigens referred to as anti-nuclear antibodies. Ann. Rheum. Dis 73, 17–23 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Pisetsky DS Antinuclear antibody testing: misunderstood or misbegotten? Nat. Rev. Rheumatol 13, 495–502 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Damoiseaux J et al. Clinical relevance of HEp-2 indirect immunofluorescent patterns: the International Consensus on ANA patterns (ICAP) perspective. Ann. Rheum. Dis 78, 879–889 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wallace DJ et al. A phase II, randomized, double-blind, placebo-controlled, dose-ranging study of belimumab in patients with active systemic lupus erythematosus. Arthritis Rheum. 61, 1168–1178 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Furie R et al. A phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum. 63, 3918–3930 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pisetsky DS, Rovin BH & Lipsky PE New perspectives in rheumatology: biomarkers as entry criteria for clinical trials of new therapies for systemic lupus erythematosus: the example of antinuclear antibodies and anti-DNA. Arthritis Rheumatol. 69, 487–493 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Hueber W, Utz PJ, Steinman L & Robinson WH Autoantibody profiling for the study and treatment of autoimmune disease. Arthritis Res. 4, 290–295 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li QZ et al. Protein array autoantibody profiles for insights into systemic lupus erythematosus and incomplete lupus syndromes. Clin. Exp. Immunol 147, 60–70 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deng X et al. Utility of antinuclear antibody screening by various methods in a clinical laboratory patient cohort. J. Appl. Lab. Med 1, 36–46 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Olsen NJ, Choi MY & Fritzler MJ Emerging technologies in autoantibody testing for rheumatic diseases. Arthritis Res. Ther 19, 172 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pisetsky DS, Bossuyt X & Meroni PL ANA as an entry criterion for the classification of SLE. Autoimmun. Rev 18, 102400 (2019). [DOI] [PubMed] [Google Scholar]
- 14.Choi MY et al. Antinuclear antibody-negative systemic lupus erythematosus in an international inception cohort. Arthritis Care Res. 71, 893–902 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leuchten N et al. Performance of antinuclear antibodies for classifying systemic lupus erythematosus: a systematic literature review and meta-regression of diagnostic data. Arthritis Care Res. 70, 428–438 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Aringer M et al. 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Arthritis Rheumatol. 71, 1400–1412 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aringer M et al. 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Ann. Rheum. Dis 78, 1151–1159 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Tan EM et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 25, 1271–1277 (1982). [DOI] [PubMed] [Google Scholar]
- 19.Petri M et al. Derivation and validation of the systemic lupus international collaborating clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 64, 2677–2686 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chevrier M, Jordan J, Schreiter J & Benson J Comparitive analysis of anti-nuclear antibody testing using blinded replicate samples reveals variability between commercial testing laboratories [abstract]. Arthritis Rheumatol. 68, 2809 (2016). [Google Scholar]
- 21.Pisetsky DS, Spencer DM, Lipsky PE & Rovin BH Assay variation in the detection of antinuclear antibodies in the sera of patients with established SLE. Ann. Rheum. Dis 77, 911–913 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Binder SR, Genovese MC, Merrill JT, Morris RI & Metzger AL Computer-assisted pattern recognition of autoantibody results. Clin. Diagn. Lab. Immunol 12, 1353–1357 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park Y et al. Automated versus conventional microscopic interpretation of antinuclear antibody indirect immunofluorescence test. Ann. Clin. Lab. Sci 49, 127–133 (2019). [PubMed] [Google Scholar]
- 24.Pisetsky DS Anti-DNA antibodies — quintessential biomarkers of SLE. Nat. Rev. Rheumatol 12, 102–110 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Viana VT, Durcan L, Bonfa E & Elkon KB Ribosomal P antibody: 30 years on the road. Lupus 26, 453–462 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Choi MY, FitzPatrick RD, Buhler K, Mahler M & Fritzler MJ A review and meta-analysis of anti-ribosomal P autoantibodies in systemic lupus erythematosus. Autoimmun. Rev 19, 102463 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Ghiggeri GM et al. An update on antibodies to necleosome components as biomarkers of sistemic lupus erythematosus and of lupus flares. Int. J. Mol. Sci 20, 5799 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mehra S & Fritzler MJ The spectrum of anti-chromatin/nucleosome autoantibodies: independent and interdependent biomarkers of disease. J. Immunol. Res 2014, 368274 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rekvig OP, van der Vlag J & Seredkina N Review: antinucleosome antibodies: a critical reflection on their specificities and diagnostic impact. Arthritis Rheumatol. 66, 1061–1069 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Rekvig OP The anti-DNA antibody: origin and impact, dogmas and controversies. Nat. Rev. Rheumatol 11, 530–540 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Stollar BD Antibodies to DNA. Crit. Rev. Biochem 20, 1–36 (1986). [DOI] [PubMed] [Google Scholar]
- 32.Jang YJ & Stollar BD Anti-DNA antibodies: aspects of structure and pathogenicity. Cell. Mol. Life Sci 60, 309–320 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schur PH & Sandson J Immunologic factors and clinical activity in systemic lupus erythematosus. N. Engl. J. Med 278, 533–538 (1968). [DOI] [PubMed] [Google Scholar]
- 34.McCarty GA, Rice JR, Bembe ML & Pisetsky DS Independent expression of autoantibodies in systemic lupus erythematosus. J. Rheumatol 9, 691–695 (1982). [PubMed] [Google Scholar]
- 35.Ward MM, Pisetsky DS & Christenson VD Antidouble stranded DNA antibody assays in systemic lupus erythematosus: correlations of longitudinal antibody measurements. J. Rheumatol 16, 609–613 (1989). [PubMed] [Google Scholar]
- 36.ter Borg EJ, Horst G, Hummel EJ, Limburg PC & Kallenberg CG Measurement of increases in anti-double-stranded DNA antibody levels as a predictor of disease exacerbation in systemic lupus erythematosus. A long-term, prospective study. Arthritis Rheum. 33, 634–643 (1990). [DOI] [PubMed] [Google Scholar]
- 37.Portanova JP, Arndt RE, Tan EM & Kotzin BL Anti-histone antibodies in idiopathic and drug-induced lupus recognize distinct intrahistone regions. J. Immunol 138, 446–451 (1987). [PubMed] [Google Scholar]
- 38.Vaglio A et al. Drug-induced lupus: traditional and new concepts. Autoimmun. Rev 17, 912–918 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Migliorini P, Baldini C, Rocchi V & Bombardieri S Anti-Sm and anti-RNP antibodies. Autoimmunity 38, 47–54 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Schulte-Pelkum J, Fritzler M & Mahler M Latest update on the Ro/SS-A autoantibody system. Autoimmun. Rev 8, 632–637 (2009). [DOI] [PubMed] [Google Scholar]
- 41.To CH & Petri M Is antibody clustering predictive of clinical subsets and damage in systemic lupus erythematosus? Arthritis Rheum. 52, 4003–4010 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Ching KH et al. Two major autoantibody clusters in systemic lupus erythematosus. PLoS One 7, e32001 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.St Clair EW et al. Expression of autoantibodies to recombinant (U1) RNP-associated 70K antigen in systemic lupus erythematosus. Clin. Immunol. Immunopathol 54, 266–280 (1990). [DOI] [PubMed] [Google Scholar]
- 44.Sharp G The origin of mixed connective tissue disease: a stimulus for autoimmune disease research. Lupus 18, 1031–1032 (2009). [DOI] [PubMed] [Google Scholar]
- 45.Arbuckle MR et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N. Engl. J. Med 349, 1526–1533 (2003). [DOI] [PubMed] [Google Scholar]
- 46.Heinlen LD et al. 60 kD Ro and nRNP A frequently initiate human lupus autoimmunity. PLoS One 5, e9599 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pisetsky DS, McCarty GA & Peters DV Mechanisms of autoantibody production in autoimmune MRL mice. J. Exp. Med 152, 1302–1310 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eisenberg RA, Craven SY, Warren RW & Cohen PL Stochastic control of anti-Sm autoantibodies in MRL/Mp-lpr/lpr mice. J. Clin. Invest 80, 691–697 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Catalina MD et al. Patient ancestry significantly contributes to molecular heterogeneity of systemic lupus erythematosus. JCI Insight 110, 102359 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tikly M, Burgin S, Mohanlal P, Bellingan A & George J Autoantibodies in black South Africans with systemic lupus erythematosus: spectrum and clinical associations. Clin. Rheumatol 15, 143–147 (1996). [DOI] [PubMed] [Google Scholar]
- 51.Alba P et al. Anti-dsDNA, anti-Sm antibodies, and the lupus anticoagulant: significant factors associated with lupus nephritis. Ann. Rheum. Dis 62, 556–560 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ko K, Koldobskaya Y, Rosenzweig E & Niewold TB Activation of the interferon pathway is dependent upon autoantibodies in African-American SLE patients, but not in European-American SLE patients. Front. Immunol 4, 309 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Elbagir S et al. Sudanese and Swedish patients with systemic lupus erythematosus: immunological and clinical comparisons. Rheumatology 59, 968–978 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Riley RL, Addis DJ & Taylor RP Stability of DNA/anti-DNA complexes. II. Salt lability and avidity. J. Immunol 124, 1–7 (1980). [PubMed] [Google Scholar]
- 55.Smeenk R, van der Lelij G & Aarden L Avidity of antibodies to dsDNA: comparison of IFT on Crithidia luciliae, Farr assay, and PEG assay. J. Immunol 128, 73–78 (1982). [PubMed] [Google Scholar]
- 56.Smeenk RJ, Van Rooijen A & Swaak TJ Dissociation studies of DNA/anti-DNA complexes in relation to anti-DNA avidity. J. Immunol. Methods 109, 27–35 (1988). [DOI] [PubMed] [Google Scholar]
- 57.Haugbro K, Nossent JC, Winkler T, Figenschau Y & Rekvig OP Anti-dsDNA antibodies and disease classification in antinuclear antibody positive patients: the role of analytical diversity. Ann. Rheum. Dis 63, 386–394 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Compagno M et al. Clinical phenotype associations with various types of anti-dsDNA antibodies in patients with recent onset of rheumatic symptoms. Results from a multicentre observational study. Lupus Sci. Med 1, e000007 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pisetsky DS Antinuclear antibodies in rheumatic disease: a proposal for a function-based classification. Scand. J. Immunol 76, 223–228 (2012). [DOI] [PubMed] [Google Scholar]
- 60.Mahajan A, Herrmann M & Munoz LE Clearance deficiency and cell death pathways: a model for the pathogenesis of SLE. Front. Immunol 7, 35 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Elkon KB Review: cell death, nucleic acids, and immunity: inflammation beyond the grave. Arthritis Rheumatol. 70, 805–816 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Al-Mayouf SM et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat. Genet 43, 1186–1188 (2011). [DOI] [PubMed] [Google Scholar]
- 63.Sisirak V et al. Digestion of chromatin in apoptotic cell microparticles prevents autoimmunity. Cell 166, 88–101 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Soni C & Reizis B DNA as a self-antigen: nature and regulation. Curr. Opin. Immunol 55, 31–37 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weisenburger T et al. Epistatic interactions between mutations of deoxyribonuclease 1-like 3 and the inhibitory Fc gamma receptor IIB result in very early and massive autoantibodies against double-stranded DNA. Front. Immunol 9, 1551 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Crow YJ & Manel N Aicardi-Goutieres syndrome and the type I interferonopathies. Nat. Rev. Immunol 15, 429–440 (2015). [DOI] [PubMed] [Google Scholar]
- 67.Uggenti C, Lepelley A & Crow YJ Self-awareness: nucleic acid-driven inflammation and the type I interferonopathies. Annu. Rev. Immunol 37, 247–267 (2019). [DOI] [PubMed] [Google Scholar]
- 68.Nielsen CT et al. Increased IgG on cell-derived plasma microparticles in systemic lupus erythematosus is associated with autoantibodies and complement activation. Arthritis Rheum. 64, 1227–1236 (2012). [DOI] [PubMed] [Google Scholar]
- 69.Zirngibl M et al. Loading of nuclear autoantigens prototypically recognized by systemic lupus erythematosus sera into late apoptotic vesicles requires intact microtubules and myosin light chain kinase activity. Clin. Exp. Immunol 179, 39–49 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mobarrez F et al. Microparticles in the blood of patients with SLE: size, content of mitochondria and role in circulating immune complexes. J. Autoimmun 102, 142–149 (2019). [DOI] [PubMed] [Google Scholar]
- 71.Abrass CK, Nies KM, Louie JS, Border WA & Glassock RJ Correlation and predictive accuracy of circulating immune complexes with disease activity in patients with systemic lupus erythematosus. Arthritis Rheum. 23, 273–282 (1980). [DOI] [PubMed] [Google Scholar]
- 72.Lock RJ & Unsworth DJ Measurement of immune complexes is not useful in routine clinical practice. Ann. Clin. Biochem 37, 253–261 (2000). [DOI] [PubMed] [Google Scholar]
- 73.Wener MH Tests for circulating immune complexes. Methods Mol. Biol 1134, 47–57 (2014). [DOI] [PubMed] [Google Scholar]
- 74.Putterman C et al. Cell-bound complement activation products in systemic lupus erythematosus: comparison with anti-double-stranded DNA and standard complement measurements. Lupus Sci. Med 1, e000056 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Trouw LA, Pickering MC & Blom AM The complement system as a potential therapeutic target in rheumatic disease. Nat. Rev. Rheumatol 13, 538–547 (2017). [DOI] [PubMed] [Google Scholar]
- 76.Nowling TK & Gilkeson GS Mechanisms of tissue injury in lupus nephritis. Arthritis Res. Ther 13, 250 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ho A, Magder LS, Barr SG & Petri M Decreases in anti-double-stranded DNA levels are associated with concurrent flares in patients with systemic lupus erythematosus. Arthritis Rheum. 44, 2342–2349 (2001). [DOI] [PubMed] [Google Scholar]
- 78.Zykova SN, Tveita AA & Rekvig OP Renal Dnase1 enzyme activity and protein expression is selectively shut down in murine and human membranoproliferative lupus nephritis. PLoS One 5, 0012096 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pedersen HL et al. Lupus nephritis: low urinary DNase I levels reflect loss of renal DNase I and may be utilized as a biomarker of disease progression. J. Pathol. Clin. Res 4, 193–203 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vallin H, Perers A, Alm GV & Ronnblom L Anti-double-stranded DNA antibodies and immunostimulatory plasmid DNA in combination mimic the endogenous IFN-alpha inducer in systemic lupus erythematosus. J. Immunol 163, 6306–6313 (1999). [PubMed] [Google Scholar]
- 81.Shrivastav M & Niewold TB Nucleic acid sensors and type I interferon production in systemic lupus erythematosus. Front. Immunol 4, 319 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sharma S, Fitzgerald KA, Cancro MP & Marshak-Rothstein A Nucleic acid-sensing receptors: rheostats of autoimmunity and autoinflammation. J. Immunol 195, 3507–3512 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Barrat FJ, Elkon KB & Fitzgerald KA Importance of nucleic acid recognition in inflammation and autoimmunity. Annu. Rev. Med 67, 323–336 (2016). [DOI] [PubMed] [Google Scholar]
- 84.Sun L, Wu J, Du F, Chen X & Chen ZJ Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li T & Chen ZJ The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med 215, 1287–1299 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lovgren T, Eloranta ML, Bave U, Alm GV & Ronnblom L Induction of interferon-alpha production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG. Arthritis Rheum. 50, 1861–1872 (2004). [DOI] [PubMed] [Google Scholar]
- 87.Kirou KA et al. Activation of the interferon-alpha pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum. 52, 1491–1503 (2005). [DOI] [PubMed] [Google Scholar]
- 88.Weckerle CE et al. Network analysis of associations between serum interferon-alpha activity, autoantibodies, and clinical features in systemic lupus erythematosus. Arthritis Rheum. 63, 1044–1053 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jackson SW et al. Opposing impact of B cell-intrinsic TLR7 and TLR9 signals on autoantibody repertoire and systemic inflammation. J. Immunol 192, 4525–4532 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Christensen SR et al. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity 25, 417–428 (2006). [DOI] [PubMed] [Google Scholar]
- 91.Tilstra JS et al. B cell-intrinsic TLR9 expression is protective in murine lupus. J. Clin. Invest 130, 3172–3187 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Garcia-Romo GS et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl Med 3, 73ra20 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.van Dam LS et al. Intrinsically distinct role of neutrophil extracellular trap formation in antineutrophil cytoplasmic antibody-associated vasculitis compared to systemic lupus erythematosus. Arthritis Rheumatol. 71, 2047–2058 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gupta S & Kaplan MJ The role of neutrophils and NETosis in autoimmune and renal diseases. Nat. Rev. Nephrol 12, 402–413 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Apel F, Zychlinsky A & Kenny EF The role of neutrophil extracellular traps in rheumatic diseases. Nat. Rev. Rheumatol 14, 467–475 (2018). [DOI] [PubMed] [Google Scholar]
- 96.Kowal C et al. Human lupus autoantibodies against NMDA receptors mediate cognitive impairment. Proc. Natl Acad. Sci. USA 103, 19854–19859 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nestor J et al. Lupus antibodies induce behavioral changes mediated by microglia and blocked by ACE inhibitors. J. Exp. Med 215, 2554–2566 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Azzouz D et al. Lupus nephritis is linked to disease-activity associated expansions and immunity to a gut commensal. Ann. Rheum. Dis 78, 947–956 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Silverman GJ, Azzouz DF & Alekseyenko AV Systemic lupus erythematosus and dysbiosis in the microbiome: cause or effect or both? Curr. Opin. Immunol 61, 80–85 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Diamond B & Lipsky PE in Harrison’s Principles of Internal Medicine 20th edn, Ch. 348 (eds Jameson JL, Fauci AS, Kasper DL, Hauser SL, Longo DL & Loscalzo J) 2510–2515 (McGraw-Hill Education; 2018). [Google Scholar]
- 101.Bombardier C, Gladman DD, Urowitz MB, Caron D & Chang CH Derivation of the SLEDAI. A disease activity index for lupus patients. The committee on prognosis studies in SLE. Arthritis Rheum. 35, 630–640 (1992). [DOI] [PubMed] [Google Scholar]
- 102.Gladman DD, Ibanez D & Urowitz MB Systemic lupus erythematosus disease activity index 2000. J. Rheumatol 29, 288–291 (2002). [PubMed] [Google Scholar]
- 103.St Clair EW, Burch JA Jr, Ward MM, Keene JD & Pisetsky DS Temporal correlation of antibody responses to different epitopes of the human La autoantigen. J. Clin. Invest 85, 515–521 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ferraccioli G & Houssiau FA Which B-cell subset should we target in lupus? Ann. Rheum. Dis 72, 1891–1892 (2013). [DOI] [PubMed] [Google Scholar]
- 105.Tipton CM et al. Diversity, cellular origin and autoreactivity of antibody-secreting cell population expansions in acute systemic lupus erythematosus. Nat. Immunol 16, 755–765 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hiepe F & Radbruch A Plasma cells as an innovative target in autoimmune disease with renal manifestations. Nat. Rev. Nephrol 12, 232–240 (2016). [DOI] [PubMed] [Google Scholar]
- 107.Tipton CM, Hom JR, Fucile CF, Rosenberg AF & Sanz I Understanding B-cell activation and autoantibody repertoire selection in systemic lupus erythematosus: a B-cell immunomics approach. Immunol. Rev 284, 120–131 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Stohl W et al. Belimumab reduces autoantibodies, normalizes low complement levels, and reduces select B cell populations in patients with systemic lupus erythematosus. Arthritis Rheum. 64, 2328–2337 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Amanna IJ, Carlson NE & Slifka MK Duration of humoral immunity to common viral and vaccine antigens. N. Engl. J. Med 357, 1903–1915 (2007). [DOI] [PubMed] [Google Scholar]
- 110.Mannik M, Merrill CE, Stamps LD & Wener MH Multiple autoantibodies form the glomerular immune deposits in patients with systemic lupus erythematosus. J. Rheumatol 30, 1495–1504 (2003). [PubMed] [Google Scholar]
- 111.Vilá LM et al. Clinical and prognostic value of autoantibodies in Puerto Ricans with systemic lupus erythematosus. Lupus 15, 892–898 (2006). [DOI] [PubMed] [Google Scholar]
- 112.Ahlin E et al. Autoantibodies associated with RNA are more enriched than anti-dsDNA antibodies in circulating immune complexes in SLE. Lupus 21, 586–595 (2012). [DOI] [PubMed] [Google Scholar]
- 113.Arroyo-Avila M et al. Clinical associations of anti-Smith antibodies in PROFILE: a multi-ethnic lupus cohort. Clin. Rheumatol 34, 1217–1223 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Maddison PJ & Reichlin M Deposition of antibodies to a soluble cytoplasmic antigen in the kidneys of patients with systemic lupus erythematosus. Arthritis Rheum. 22, 858–863 (1979). [DOI] [PubMed] [Google Scholar]
- 115.Venables PJ, Yi T, Woodrow DF, Moss J & Maini RN Relationship of precipitating antibodies to soluble cellular antigens and histologically defined renal lesions in systemic lupus erythematosus. Ann. Rheum. Dis 42, 17–22 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Simmons-O’Brien E et al. One hundred anti-Ro (SS-A) antibody positive patients: a 10-year follow-up. Medicine 74, 109–130 (1995). [DOI] [PubMed] [Google Scholar]
- 117.Barada FA Jr, Andrews BS, Davis JS IV & Taylor RP Antibodies to Sm in patients with systemic lupus erythematosus. Correlation of Sm antibody titers with disease activity and other laboratory parameters. Arthritis Rheum. 24, 1236–1244 (1981). [DOI] [PubMed] [Google Scholar]
- 118.Praprotnik S, Bozic B, Kveder T & Rozman B Fluctuation of anti-Ro/SS-A antibody levels in patients with systemic lupus erythematosus and Sjögren’s syndrome: a prospective study. Clin. Exp. Rheumatol 17, 63–68 (1999). [PubMed] [Google Scholar]
- 119.Tench CM & Isenberg DA The variation in anti-ENA characteristics between different ethnic populations with systemic lupus erythematosus over a 10-year period. Lupus 9, 374–376 (2000). [DOI] [PubMed] [Google Scholar]
- 120.Faria AC, Barcellos KS & Andrade LE Longitudinal fluctuation of antibodies to extractable nuclear antigens in systemic lupus erythematosus. J. Rheumatol 32, 1267–1272 (2005). [PubMed] [Google Scholar]
- 121.Ippolito A et al. Autoantibodies in systemic lupus erythematosus: comparison of historical and current assessment of seropositivity. Lupus 20, 250–255 (2011). [DOI] [PubMed] [Google Scholar]
- 122.Fisher DE, Reeves WH, Wisniewolski R, Lahita RG & Chiorazzi N Temporal shifts from Sm to ribonucleoprotein reactivity in systemic lupus erythematosus. Arthritis Rheum. 28, 1348–1355 (1985). [DOI] [PubMed] [Google Scholar]
- 123.Butler WT & Rossen RD Effects of corticosteroids on immunity in man. I. Decreased serum IgG concentration caused by 3 or 5 days of high doses of methylprednisolone. J. Clin. Invest 52, 2629–2640 (1973). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bijl M, Horst G, Bootsma H, Limburg PC & Kallenberg CG Mycophenolate mofetil prevents a clinical relapse in patients with systemic lupus erythematosus at risk. Ann. Rheum. Dis 62, 534–539 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ng KP et al. B cell depletion therapy in systemic lupus erythematosus: long-term follow-up and predictors of response. Ann. Rheum. Dis 66, 1259–1262 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tew GW et al. Baseline autoantibody profiles predict normalization of complement and anti-dsDNA autoantibody levels following rituximab treatment in systemic lupus erythematosus. Lupus 19, 146–157 (2010). [DOI] [PubMed] [Google Scholar]
- 127.Eickenberg S et al. Mycophenolic acid counteracts B cell proliferation and plasmablast formation in patients with systemic lupus erythematosus. Arthritis Res. Ther 14, R110 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lewis MJ et al. Autoantibodies targeting TLR and SMAD pathways define new subgroups in systemic lupus erythematosus. J. Autoimmun 91, 1–12 (2018). [DOI] [PubMed] [Google Scholar]
- 129.Pisetsky DS, Thompson DK, Wajdula J, Diehl A & Sridharan S Variability in antinuclear antibody testing to assess patient eligibility for clinical trials of novel treatments for systemic lupus erythematosus. Arthritis Rheumatol. 71, 1534–1538 (2019). [DOI] [PubMed] [Google Scholar]
- 130.Putterman C et al. The SLE-key test serological signature: new insights into the course of lupus. Rheumatology 57, 1632–1640 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.An J, Minie M, Sasaki T, Woodward JJ & Elkon KB Antimalarial drugs as immune modulators: new mechanisms for old drugs. Annu. Rev. Med 68, 317–330 (2017). [DOI] [PubMed] [Google Scholar]
- 132.Yurasov S et al. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J. Exp. Med 201, 703–711 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yurasov S et al. Persistent expression of autoantibodies in SLE patients in remission. J. Exp. Med 203, 2255–2261 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Dorner T & Lipsky PE B cells: depletion or functional modulation in rheumatic diseases. Curr. Opin. Rheumatol 26, 228–236 (2014). [DOI] [PubMed] [Google Scholar]
- 135.Suurmond J et al. Patterns of ANA+B cells for SLE patient stratification. JCI Insight 4, e127885 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Suurmond J et al. Loss of an IgG plasma cell checkpoint in patients with lupus. J. Allergy. Clin. Immunol 143, 1586–1597 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.McMichael AJ, Borrow P, Tomaras GD, Goonetilleke N & Haynes BF The immune response during acute HIV-1 infection: clues for vaccine development. Nat. Rev. Immunol 10, 11–23 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lee J et al. Molecular-level analysis of the serum antibody repertoire in young adults before and after seasonal influenza vaccination. Nat. Med 22, 1456–1464 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kaur K et al. High affinity antibodies against influenza characterize the plasmablast response in SLE patients after vaccination. PLoS One 10, e0125618 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]