

Abstract
Rationale CC16 (club cell secretory protein) is a pneumoprotein produced predominantly by pulmonary club cells. Circulating CC16 is associated with protection from the inception and progression of the two most common obstructive lung diseases (asthma and chronic obstructive pulmonary disease).
Objectives Although exact mechanisms remain elusive, studies consistently suggest a causal role of CC16 in mediating antiinflammatory and antioxidant functions in the lung. We sought to determine any novel receptor systems that could participate in CC16’s role in obstructive lung diseases.
Methods Protein alignment of CC16 across species led to the discovery of a highly conserved sequence of amino acids, leucine–valine–aspartic acid (LVD), a known integrin-binding motif. Recombinant CC16 was generated with and without the putative integrin-binding site. A Mycoplasma pneumoniae mouse model and a fluorescent cellular adhesion assay were used to determine the impact of the LVD site regarding CC16 function during live infection and on cellular adhesion during inflammatory conditions.
Measurements and Main Results CC16 bound to integrin α4β1), also known as the adhesion molecule VLA-4 (very late antigen 4), dependent on the presence of the LVD integrin-binding motif. During infection, recombinant CC16 rescued lung function parameters both when administered to the lung and intravenously but only when the LVD integrin-binding site was intact; likewise, neutrophil recruitment during infection and leukocyte adhesion were both impacted by the loss of the LVD site.
Conclusions We discovered a novel receptor for CC16, VLA-4, which has important mechanistic implications for the role of CC16 in circulation as well as in the lung compartment.
Keywords: CCSP, CC16, VLA-4, integrins, leukocyte adhesion
At a Glance Commentary
Scientific Knowledge on the Subject
CC16 (club cell secretory protein) detected in circulation is associated with protection from the inception and progression of the two most common obstructive lung diseases (asthma and chronic obstructive pulmonary disease). Mechanisms of CC16 action within circulation remain an area of intense study.
What This Study Adds to the Field
Our studies describe a novel receptor for CC16, the integrin complex known as VLA-4 (very late antigen 4), on leukocytes. Loss of the binding site within CC16 results in worse outcomes during live Mycoplasma infection, including airway hyperresponsiveness. Our data bring to light an immediately applicable role for CC16 in the circulation—to limit the number of activated leukocytes from entering and further damaging the delicate lung tissue during infectious insults.
CC16 (club cell secretory protein; also known as CCSP, CC10, and uteroglobin) is a homodimeric pneumoprotein encoded by the SCGB1A1 gene. Although CC16 is produced predominantly by club cells and nonciliated epithelial cells in the distal airways, it diffuses into the bloodstream and is easily detectable in serum (1, 2). CC16 has been the topic of much study for its potential as a biological marker of lung epithelial cell injury, and recent studies by our group and others have shown decreased serum CC16 concentrations in patients with obstructive lung diseases, such as asthma (3) and chronic obstructive pulmonary disease (COPD) (4), and in individuals with lung function deficits in the general population (5). In multiple epidemiological cohorts, low CC16 predicted subsequent impaired lung function growth in childhood and accelerated lung function decline and incident airflow limitation, the hallmark of COPD, in adult life (4). These observations build on previous reports showing that CC16 concentrations were significantly associated with the rate of change in FEV1 among smokers and patients with COPD (6, 7).
In addition, we have recently shown that low CC16 correlates with lung function deficits and airway hyperresponsiveness (AHR) not only in human cohorts but also in naive CC16-deficient mice (8). In the Tucson Children’s Respiratory Study, participants in the lowest tertile of serum CC16 had significant deficits in their lung function and enhanced AHR to methacholine challenge from 11 years throughout young adult life (8). Furthermore, we determined that Cc16−/− mice had significant deficits in lung function, increased sensitivity and airway resistance to methacholine, and airway structural alterations in an unchallenged state (no infection) as compared with wild-type (WT) mice (8), implicating deficits of this protein in the pathophysiology of progressive airway damage.
Although the biological functions of CC16 have not been completely described, mounting evidence suggests that this protein has critical effects in mediating antiinflammatory and antioxidant functions within the lung and, by virtue of this activity, may protect against the development of obstructive lung diseases. During respiratory syncytial virus infection, CC16-deficient (Cc16−/−) mice have heightened inflammation, viral load, Th2 cytokines, and AHR compared with WT mice (9). Despite enhanced clearance in Cc16−/− mice, Pseudomonas aeruginosa infection also led to increased inflammation and neutrophils in CC16-deficient mice (10). Relative to asthma studies, Cc16−/− mice had greater responses when challenged in the ovalbumin model of allergic airways disease with increased neutrophilia and eosinophilia, mucus production, and AHR compared with WT mice (11). Taken together, one common feature of these studies is an increase in the number of immune (neutrophils and/or eosinophils) cells infiltrating the airways when CC16 was absent, hinting at a potentially shared mechanism common to CC16 deficiency and increased immune-cell recruitment into the lung.
Taken together, these observations suggest that CC16 is not merely a biomarker but may be implicated in the inception and progression of the disease process. However, there is still a void in our mechanistic understanding of how this protein plays such a key role in shaping the course of lung function in health and disease. The purpose of this current study was to search for any undiscovered receptors through which CC16 may also function in the context of regulating lung function. Novel CC16 receptors could help guide studies with the intent of developing new therapeutics aimed at harnessing the activity of CC16 for a protective effect from lung function deficits linked to obstructive lung diseases, such as asthma and COPD.
Methods
Recombinant CC16 Constructs and Protein Purification
DNA corresponding with CC16 residues 22–91 was amplified out of human complementary DNA and cloned into a ligation-independent cloning vector (pMCSG7) to generate a human recombinant CC16 (rCC16) protein without the leader sequence and an N-terminal 6× His tag (12). The sequence confirmed vector was then subjected to site-directed mutagenesis using Pfu DNA polymerase and overlapping primers encoding the mutation aspartic acid to alanine at amino acid position 67 at the center of the primer. The parent rCC16 vector was then mutated to generate rCC16 with a mutation in the leucine–valine–aspartic acid (LVD) domain (rCC16 D67A), and it was subsequently sequence confirmed (see the online supplement for detailed methods on purification).
Plate-Bound Assay for CC16 Binding
We chose to test α4β1 and α4β7 integrins for potential binding to rCC16 on the basis of its expression in the lung and known importance in asthma and lung function. Plate-binding assays were conducted with rCC16 and rCC16 D67A mutant. The His-tagged proteins were used to saturate nickel-coated plates at a concentration of 10 μM in 50 μl for 30 minutes. Wells were washed three times with TRIS-buffered saline. Recombinant human integrin subunit α4 (Abcam) was combined in equimolar ratios with β1 (Abcam) or β7 (Abcam) to make α4β1 or α4β7, respectively. Combined integrin heterodimers (100 nmol) were added to wells with either bound rCC16 or rCC16 D67A for 1 hour, after which the plate was washed three times with TRIS-buffered saline with 0.5% Tween-20. A fluorescent anti-α4 red fluorescent protein antibody was used to detect the CC16-bound integrin by relative fluorescent intensity using a standard plate reader (BioTek).
Murine Studies
All experiments were done in accordance with the University of Arizona on Institutional Animal Care and Use Committee–approved animal protocols. For the Mycoplasma pneumoniae (Mp) infection, age-matched WT and Cc16−/− male mice on a C57BL/6J background were aged ∼6–8 weeks at the time of infection, and lung function was assessed 3 days after infection as described previously (8). For the infection model, Mp was purchased from American Type Culture Collection (ATCC) (15531) and grown in SP4 broth (Remel) at 35°C until adherent, approximately four passages, as previously described (13). On the day of infection, a frozen stock of adherent Mp was washed by centrifuging at 6,000 rpm for 5 minutes and was resuspended in sterile saline for infection at a concentration of 1×108 Mp/50 μl inoculum. Mp was delivered via intranasal instillation while mice were under isoflurane anesthesia. Some mice received rCC16 (0.75 mg/kg body weight) treatment via oropharyngeal delivery or retroorbital delivery while under light isoflurane anesthesia 2 hours before Mp infection, whereas vehicle control mice received sterile saline.
Pulmonary Function Tests in Mouse Models
Mice were analyzed on the Flexivent system (SCIREQ Inc.), after which they were killed, the lungs were lavaged with phosphate-buffered saline (PBS) (0.1 mM ethylenediaminetetraacetic acid [EDTA]), and lung tissue was obtained for further analysis, as previous described (14). Please refer to the online supplement for detailed methods on processing and further assessment.
Western Blots
To determine CC16 expression levels in response to infection with Mp, BAL fluid (BALF) was collected from mice by gently flushing the lungs three times with 1 ml of sterile saline (0.1 mM EDTA). An equal volume of reduced-lavage fluid samples was loaded onto a polyacrylamide gel for each mouse, and Western blots were assessed with an antimouse CC10 (also known as CC16 or CCSP) antibody from Santa Cruz (T-18: sc-9772) that recognized the monomeric form after reducing conditions. Films were taken after horseradish peroxidase exposure, and densitometry was assessed by Image J software.
Collection and Assessment of BALF
BALF was collected by tracheal cannulation using 1.5 ml of PBS with 0.1 mM of EDTA. The total cells collected from each sample were counted using a Countess II FL Automated Cell Counter (Life Technologies). To analyze the cell differentiation for each sample, slides were prepared using a Cytospin 3 (Shandon). Each slide sample was stained with an Easy III Rapid Differential Staining Kit (Azer Scientific), and neutrophil subpopulations were totaled by using standard morphological criteria via light microscopy. From a subset of samples, cell-free lavage fluid was plated on pleuropneumonia-like organism agar and allowed to incubate for 2 weeks at 35°C to determine Mp colony-forming units (CFUs).
Leukocyte–Endothelium Cell-Adhesion Assay
A CytoSelect Leukocyte-Endothelium Adhesion Assay Kit (Cell BioLabs, Inc.) was purchased and performed according to the manufacturers’ protocol. In brief, a 48-well tissue culture plate was gelatinized and allowed to incubate for 15 minutes at 37°C. Plates were washed once with PBS before plating TeloHAEC endothelial cells (ATCC) to the wells, which were then incubated at 37°C (5% CO2) until they formed an adherent monolayer (typically 24 h). Endothelial cells were treated with 2 ng/ml of TNFα to initiate activation and were incubated at 37°C (5% CO2) for 2 hours. THP-1 monocyte cells (ATCC) were prepped with LeukoTracker and left to incubate at 37°C (5% CO2) for 60 minutes. After the TeloHAEC incubation with TNFα, the cells were washed and treated with either rCC16 (WT) or the rCC16 D67A mutant at various concentrations (0–25 μg/ml) for 30 minutes at 37°C (5% CO2). After the THP-1 incubation with LeukoTracker, the monocytes were washed and added to the TeloHAEC cells and incubated for 60 minutes. After the final incubation, each well was washed three times. On the final wash, pictures of each well were taken via fluorescent light microscopy on the EVOS M5000 system (ThermoFisher). Then the lysis buffer provided within the kit was added to each well. The plate was incubated for 5 minutes at room temperature with shaking. Each sample was transferred to a black-bottom 96-well plate and read with a fluorescence plate reader (BioTek Instruments) at 480 nm/520 nm.
Statistical Analysis
For experiments examining lung function, raw data were log-transformed, and differences between each respective dose were determined by student’s T test, according to previous publications (8, 14). For other analysis, Prism software was used to determine significance by either T test or one-way ANOVA for multiple comparisons as appropriate. For the leukocyte adhesion studies, a linear regression model was used to test differences at respective doses of rCC16 (WT) versus rCC16 (D67A).
Results
CC16 Conservation throughout Species and Discovery of the LVD Site
Upon examination of the human and mouse full-length CC16 protein sequence, we observed that both contain an LVD motif that is present at the C-terminal end of α helix 3, a highly conserved sequence domain near the C-terminus that was also conserved in a wide variety of mammals (Figure 1) (15). Although integrin binding is most commonly shown via the Arginine-Glycine–Aspartic Acid (RGD) motif, both Leucine–Aspartic Acid–Valine (LDV) and LVD motifs have been found to influence integrin binding (16–19). Integrins are obligate heterodimers having an α and β subunit, in which both are required for activation and binding to extracellular matrix components. Upon binding to a target ligand, integrins activate signal transduction pathways that mediate various cellular signals. As such, integrins have been implicated in a myriad of diseases, including asthma (20, 21).
Figure 1.
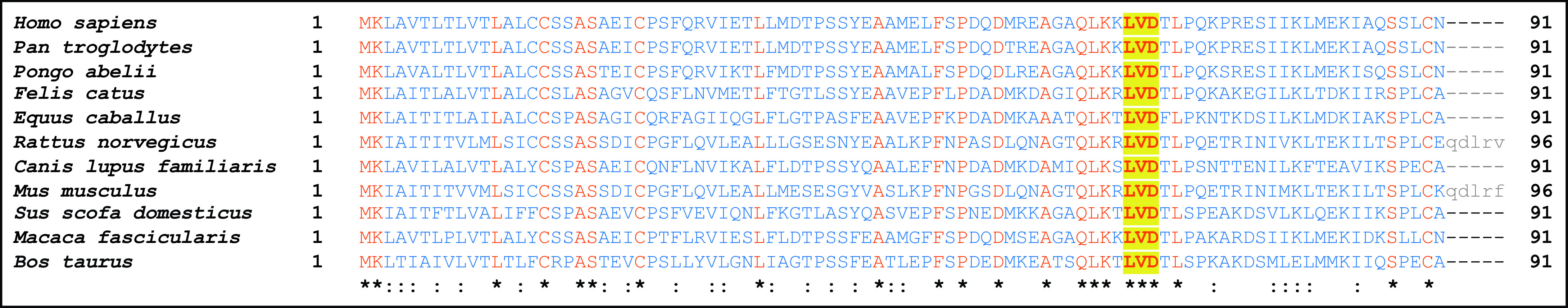
Conservation of leucine–valine–aspartic acid (LVD) sequence across mammalian species. Alignment was performed using the constraint-based multiple alignment tool on CC16 in different mammalian species. Protein residues that are identical in all species are shown in red and notated with an asterisk (*); the remaining residues are in blue. Residues that do not align (Rattus norvegicus and Mus musculus) are in gray. Furthermore, “:” represents highly conserved residues (by charge, size, and hydropreference [e.g., valine and alanine]). The conserved LVD site is bolded and highlighted in yellow. In order, Latin names correspond with human, chimpanzee, orangutan, cat, horse, rat, dog, mouse, pig, crab-eating macaque, and cow.
We postulated that CC16 could bind to host integrins and, because of the functionality of the integrin family, that this integrin-binding site within CC16 may mediate a major and yet-to-be-identified role of this protein in the circulation as well as in the lung. We tested two combinations of integrins on the basis of their expression in the lung, known importance in asthma and lung function, and ability to bind to the LVD motif, α4β1 (VLA-4 [very late antigen-4]), and α4β7 (LPAM [lymphocyte Peyer patch adhesion molecule]) for CC16 binding (20, 22–26). VLA-4 is unique among the integrins because it is the only heterodimer to have been shown to both mediate cell–cell interactions and cell–extracellular matrix interactions (27, 28). The two known natural ligands for VLA-4 are fibronectin and VCAM-1, which mediate innate immune-cell adhesion (27) and are involved in asthma and lung remodeling (29–31). LPAM is expressed on lymphocytes and targets them to gut-associated lymphoid tissues. It does this through binding to MAdCAM (mucosal addressin cell-adhesion molecule) (26).
Generation and Testing of rCC16 with and without the LVD Site for Integrin Binding
We constructed rCC16 with (WT rCC16) and without (rCC16 D67A) the LVD motif intact. Through a solid-phase binding assay, we observed that rCC16 bound to VLA-4 in an LVD integrin motif–dependent manner (Figure 2A). No binding of rCC16 was detectable to α4β7 (data not shown), implying that the interaction with α4β1 is specific and/or that the binding of rCC16 with α4β7 had a significantly lower affinity than the detection threshold of this assay. These data suggest, for the first time, that CC16 potentially controls VLA-4 activation by limiting the binding of leukocytes expressing α4β1 to VCAM-1.
Figure 2.

Binding of recombinant CC16 (club cell secretory protein) (rCC16) to α4β1 integrin complex and relative abundance of CC16 during Mycoplasma pneumoniae (Mp) infection. (A) Plate-binding assays were conducted with human recombinant CC16 with (WT CC16) and without the leucine–valine–aspartic acid sequence (CC16 D67A) that were generated with a histidine tag. The His-tagged rCC16 protein was used to saturate nickel-coated plates and recombinant human integrin subunit α4 was combined with β1 to make the α4β1 integrin complex. Combined integrins were added to the plate-bound rCC16 for 1 hour, after which the plate was washed, and a fluorescent anti-α4 antibody was used to detect the CC16-bound integrin by relative fluorescent intensity. The CC16 to the anti-α4 antibody control was subtracted from each sample to give a final relative binding. Binding assays were conducted (n = 3 replicates) and averaged; **P < 0.01. (B) BAL fluid was examined for CC16 concentrations in nontreated and Mp-infected mice after 4 and 24 hours by Western blot and densitometry analysis. n = 3–5 mice/group; *P < 0.05 compared with nontreated control mice by one-way ANOVA for multiple comparisons. WT = wild-type.
In Vivo Pulmonary Function Tests of rCC16 Activity on an Mp Model of Infection
To test the rCC16 and CC16 D67A mutant in vivo, we established an infection model using a common asthma- and COPD-exacerbating agent, Mp. We observed that, in an acute infection model with Mp, CC16 concentrations detected in the BALF were dramatically elevated as early as 4 hours after infection and persisting at 24 hours after infection (Figure 2B). This result suggested that CC16 may be an important initial mediator that is upregulated in response to Mp infection.
When evaluating the effect of CC16 on lung function using an Mp infection model, we found that Mp-infected mice lacking CC16 (Cc16−/−) had significantly enhanced AHR during methacholine challenge compared with Mp-infected WT mice (Figure 3A; solid lines) and their respective saline control mice (Figure 3A; dotted lines) . At the 30 (P = 0.0037) and 100 (P = 0.05) mg/ml doses of methacholine, we observed significantly increased total airway resistance in infected Cc16−/− mice (Figure 3A; black line) compared with the infected WT mice (Figure 3A; gray line).
Figure 3.
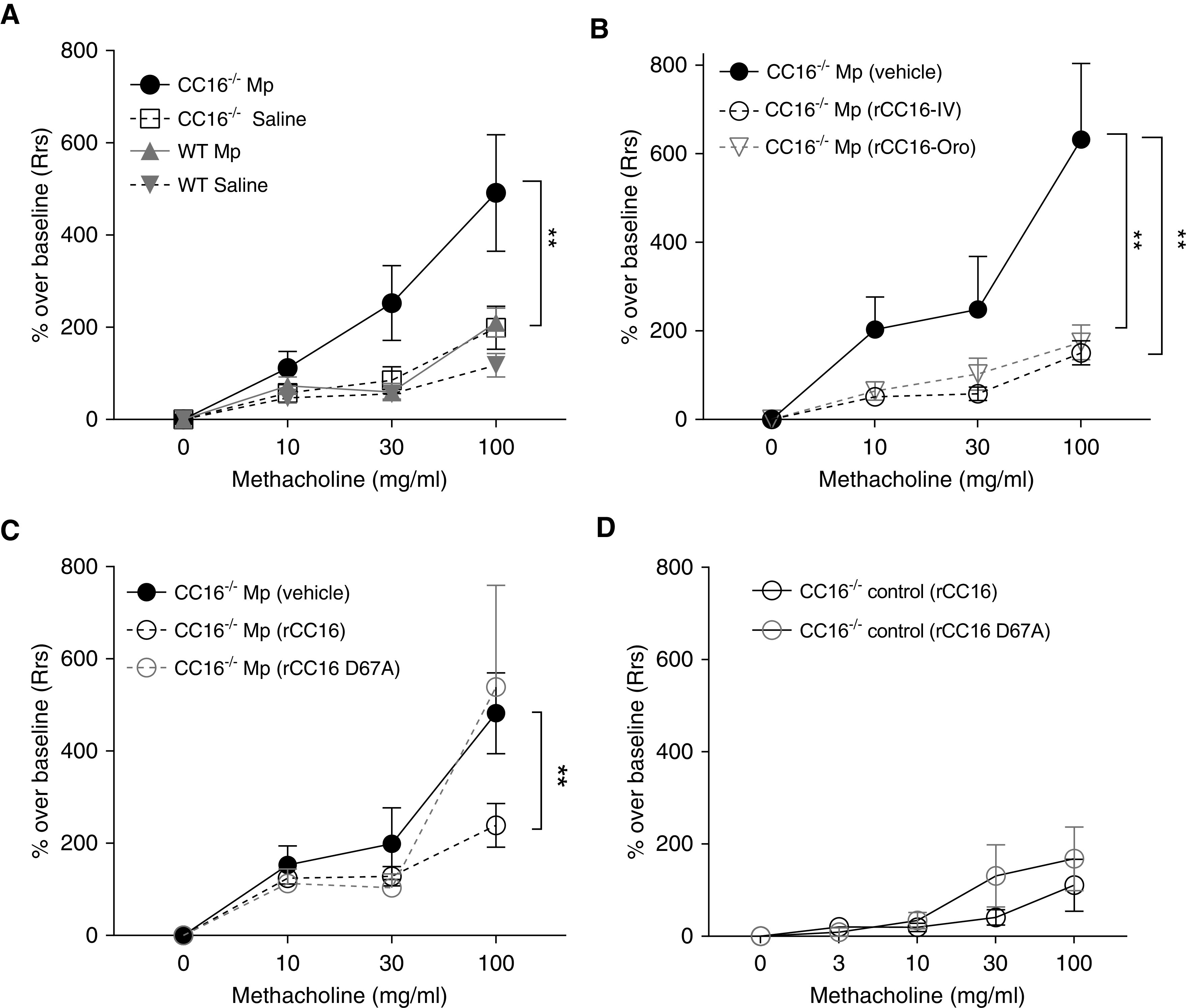
The impact of CC16 (club cell secretory protein) on airway hyperresponsiveness to methacholine challenge. (A) Wild-type (WT) and Cc16−/− mice were infected with Mycoplasma pneumoniae (Mp) for 3 days, after which their airway resistance during methacholine challenge was determined on the Flexivent machine. n = 10 WT mice/group; n = 15 Cc16−/− mice/group. (B) Recombinant CC16 (rCC16) given 2 hours before infection attenuated Mp-infected airway hyperresponsiveness in Cc16−/− mice whether given via oropharyngeal (Oro) or i.v. routes; n = 9 Mp only; n = 6 Oro; n = 6 i.v. (C) The D67A mutation alters the activity of i.v. rCC16 to reduce airway hyperresponsiveness in Mp-infected CC16−/− mice during Mp infection. n = 10–15 mice/group. (D) No differences in airway hyperresponsiveness were observed in Cc16−/− vehicle control mice given either rCC16 or rCC16 D67A; n = 4 mice/group; **P < 0.01 at the respective dose indicated. Rrs = respiratory system resistance.
We next examined the effect of rCC16 delivery before Mp infection in Cc16−/− mice, as those mice had significantly heightened AHR compared with WT mice. Because CC16 is produced in the lung but also detected at high concentrations in the circulation and because integrin binding may confer activity in the circulation, we tested delivery by both oropharyngeal (Oro) and retroorbital injection (i.v.). Our data demonstrate that delivery of WT rCC16 to an animal model with CC16 deficiency is sufficient to restore lung function (Figure 3B; lower total airway resistance = reduction in AHR) whether delivered directly to the lungs by Oro or indirectly through intravenous methods. We did not detect differences in the delivery of saline (vehicle) by either Oro or intravenous methods (not shown). We also tested our rCC16 in parallel with rCC16 purchased from R&D systems and found no differences regarding the source of CC16 in reducing Mp-induced AHR in Cc16−/− mice (data not shown).
As shown in Figure 3B, WT rCC16 was sufficient to restore lung function when given into circulation (i.v.). Discovery of the new VLA-4 integrin-binding site and successful rescue of the AHR phenotype when CC16 was given intravenously provided further evidence that CC16 could have an important mechanistic function in the bloodstream in addition to the previously noted functions in the lung. On the basis of the ability of rCC16 to bind to α4β1 in an LVD motif–dependent manner, we tested both WT rCC16 and rCC16 D67A mutant (both i.v.) in the Mp model of infection in Cc16−/− mice. Whereas WT rCC16 was able to rescue Mp-induced AHR, resulting in a lower overall resistance (Figure 3C), rCC16 D67A resulted in loss of activity and was unable to attenuate AHR, similar to the vehicle control. Cc16−/− control mice that were noninfected had no differences in AHR to the two rCC16 preparations (Figure 3D).
Assessment of rCC16 Activity on Mp-induced Muc5AC Expression and Cellular Recruitment
Because changes in AHR could be impacted by mucus production in the airways during live Mp infection, we assessed whether the LVD-binding site within CC16 had an impact on Muc5AC expression in the lung tissue of the infected mice. There were no differences in Muc5AC expression between the Mp-infected mice, whether they received vehicle, rCC16 (WT), or rCC16 (D67A) (Figure E1 in the online supplement).
The identification of an LVD integrin-binding site within CC16 suggests that this protein could exert its function at least in part by binding to integrins on immune cells both in circulation and within the pulmonary space. Indeed, in a subset of animals for which we measured pulmonary function (described above), we also assessed the LVD site on neutrophil influx into the lung. At 72 hours after infection, we only observed neutrophils and macrophages by differential staining of lavage fluid; no eosinophils or lymphocytes were detected. In line with AHR measures, mice that received the WT rCC16 had a significantly lower percentage of neutrophils in the lung lavage fluids compared with Mp-infected mice that received either vehicle or the rCC16 D67A mutant (Figure 4A). Immunofluorescent staining of tissue sections was also assessed for a subset of samples for a common leukocyte marker (CD45) and for a marker typically associated with mature T cells, B cells, and natural killer cells (CD2). Indeed, we observed robust staining for both CD45 and CD2 in lung sections from Mp-infected mice that were treated with vehicle. Overall, the lungs from the Mp-infected group that were given rCC16 (WT) had very little to no detectable staining for these markers; conversely, the lungs from Mp-infected mice given rCC16 (D67A) had robust and similar staining to the vehicle-treated lungs (Figure 4B).
Figure 4.
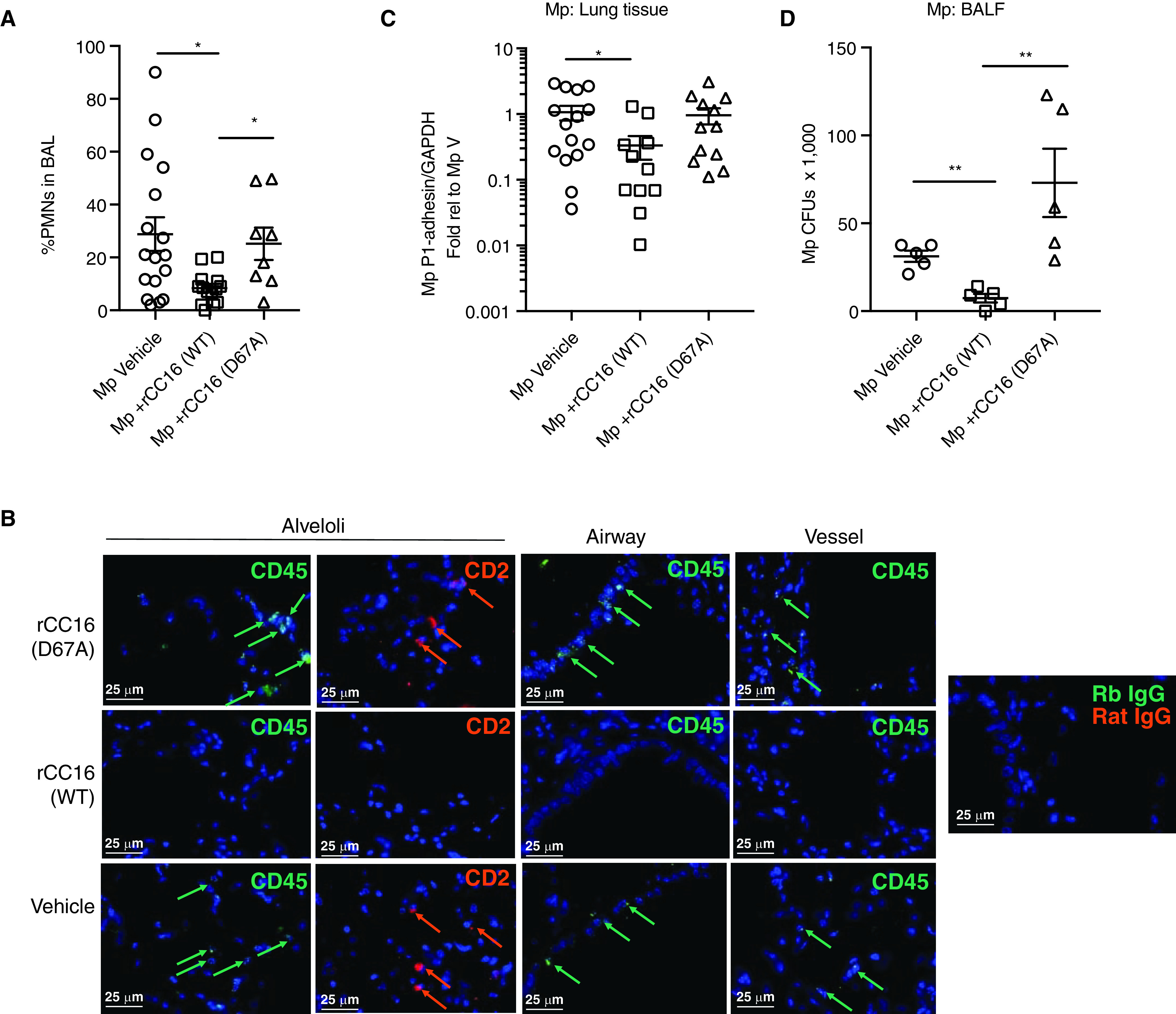
Inflammatory cell assessment and pathogen burden based on CC16 (club cell secretory protein) treatment. (A) A subset of Cc16−/− mice that were assessed for pulmonary function tests were also assessed for the presence of neutrophils in the lavage fluid by differential staining. CC16-deficient mice that received recombinant CC16 (rCC16) (WT) had a significantly lower percentage of neutrophils (PMNS) versus those that received no CC16 or rCC16 (D67A). (B) Representative (n = 4/treatment group) immunofluorescence staining for CD45 and CD2 in alveoli, airways, and vessels in the specific treatment groups. Arrows indicate positively stained cells relative to negative antibody controls (Rb IgG and rat IgG). Scale bars, 25 μm. (C) Assessment of Mycoplasma pneumoniae (Mp) burden in lung tissue by RT-PCR for Mp-specific P1-adhesin gene relative to GAPDH. Data shown as fold relative to Mp vehicle. (D) Assessment of Mp CFUs present in cell-free BAL at time of harvest. *P < 0.05 and **P < 0.01 by one-way ANOVA with Kruskal-Wallis test for multiple comparisons. BALF = BAL fluid; CFU = colony-forming unit; PMNS = polymorphonuclear leukocytes; V = vehicle; WT = wild-type.
Assessment of Mp Pathogen Burden in Association with LVD-Binding Site within CC16
Because the LVD site within CC16 appears to be important in mediating neutrophil recruitment during Mp infection, we sought to determine whether the pathogen burden would be impacted by the presence or absence of this site. Lung samples were assessed for pathogen burden by RT-PCR for the Mp-specific P1-adhesin gene, and a subset of those samples were further assessed for Mp CFUs present in BAL at the time of harvest. Somewhat surprisingly, the mice that received rCC16 (WT) had significantly less Mp detected in the lung tissue and in the BAL compared with the mice that received vehicle only (Figures 4C and 4D). The mice that received rCC16 (D67A) had Mp burden in both the tissue and CFUs in the BAL, which was similar to those that received vehicle only.
Assessment of rCC16 on Leukocyte Adhesion
The loss of activity in rCC16 when the integrin-binding site was mutated in the context of our infectious insult model suggests that this binding site is of vital importance in ameliorating loss of lung function and reducing AHR, which could in part be attributed to attenuated inflammatory cell recruitment into the lung compartment. To further explore this idea, we performed a leukocyte adhesion assay using the following human cell lines: THP-1 cells for leukocytes, as they are commonly used in studies for VLA-4 activity (32), and TeloHAEC for endothelial cells. rCC16 (WT) dose-dependently inhibited the adhesion of fluorescent THP-1 cells to activated endothelial cells, whereas rCC16 D67A mutant displayed significantly less activity in preventing cell adhesion (Figures 5A and 5B).
Figure 5.
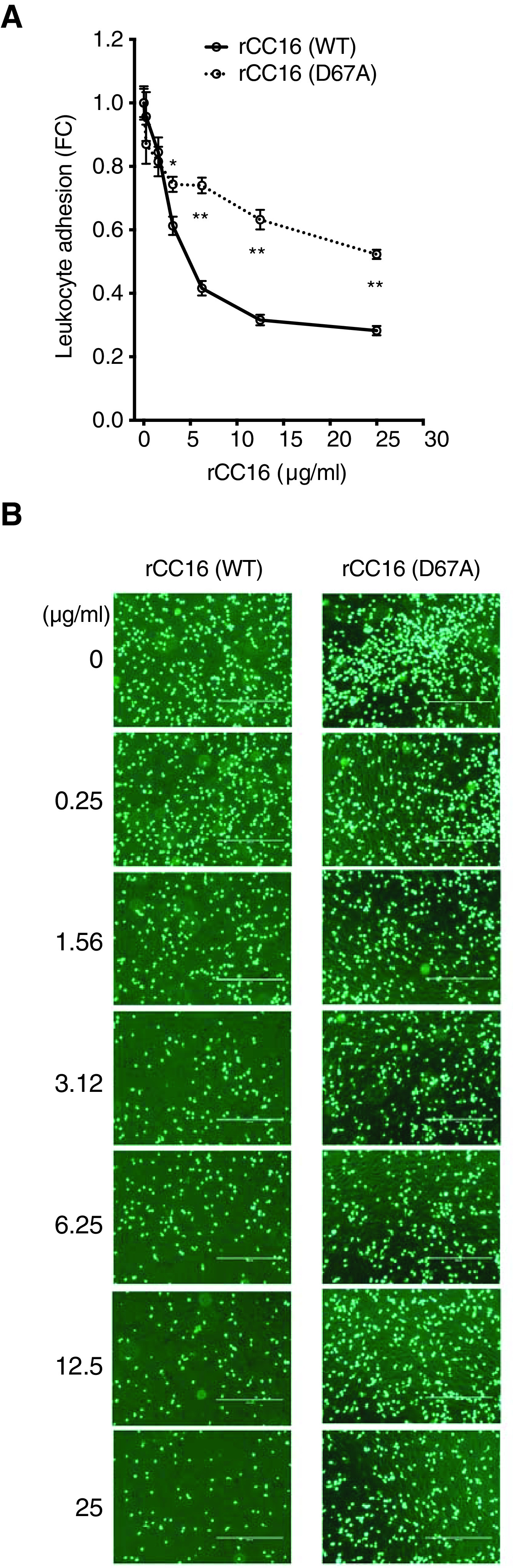
The VLA-4 (very late antigen 4) binding sequence impacts leukocyte adhesion. (A) recombinant CC16 (club cell secretory protein) (rCC16) (WT) dose-dependently inhibited fluorescent THP-1 adhesion to human endothelial cells after TNF-α activation, whereas D67A was significantly less active. Fluorescence was determined at 480 nm/520 nm wavelength, and data were plotted as the FC reduction from the non–CC16-treated values. N = minimum of 4 replicates/2 repeat experiments. By linear regression, *P < 0.05 and **P < 0.001. (B) Representative pictures of the various rCC16 doses of adhered fluorescent THP-1 cells to human endothelial cells. Scale bars, 400 μm. FC = fold change; WT = wild-type.
Discussion
In this study, we sought to better understand what role CC16 plays on a mechanistic level in protecting from lung function deficits in the hopes of advancing future novel CC16-based therapeutics for asthma and COPD. The importance of our discovery of an LVD integrin-binding site within CC16 is of high relevance given the potentially new mechanism through which CC16 can participate by binding to integrins in both the circulation and within the pulmonary space.
Integrins are obligate heterodimers and have an α and β subunit, in which both are required for activation and binding to extracellular matrix components. Upon binding a target ligand, integrins activate signal transduction pathways that mediate cellular signals, including the regulation of cell cycle, organization of intracellular cytoskeleton, and movement of new receptors to the cell membrane. As such, they have been implicated in a myriad of diseases, including asthma (20, 21). We tested two combinations of integrins on the basis their expression in the lung and known importance in asthma and lung function (α4β1 and α4β7) for CC16 binding (22–24). On the basis of the presence of the LVD sequence and phenotypes associated with CC16 protection during inflammation, we suspected that CC16 would be able to bind to α4β1 (VLA-4) (20, 25). Indeed, we found rCC16 to bind to VLA-4 but not to α4β7, which was dependent on the LVD sequence. This binding is of relative importance to lung inflammation, as VLA-4 is known to bind to VCAM-1, which mediates innate immune-cell adhesion (27). Although we were able to show that rCC16 inhibited cellular adhesion ex vivo in our human endothelial cell/leukocyte model and neutrophil recruitment in a live infection mouse model, future studies are needed to determine whether CC16 competes for VLA-4 binding with VCAM-1.
Furthermore, VLA-4 is unique among the integrins because it is the only heterodimer to have been shown to both mediate cell–cell interactions and cell–extracellular matrix interactions (27, 28). Another known natural ligand for VLA-4 is fibronectin, which is involved in asthma and lung remodeling (29–31). As we have previously shown naive Cc16−/− mice to have enhanced remodeling factors, which likely impacts lung function (8), further assessment of the interaction or, more likely, the competition of CC16 with fibronectin through VLA-4 interactions will also be vital. Adding a level of complexity, CC16 has been shown to bind with high affinity to fibronectin (independent of the LVD site) and prevents its monomeric self-aggregation (33).
We would like to point out that the dampening effect of rCC16 on Mp-induced AHR is entirely consistent with other animal models of lung injury in which rCC16 was administered either intravenously or by intratracheal instillation and improved mechanical lung function (9, 34–40). In addition, these results are consistent with previous studies demonstrating the essential nature of D67 to overall CC16 function, including phospholipase-A2 inhibition (41). An unexpected finding was that although rCC16 (WT) led to a reduction in neutrophils in the BAL and CD45 + leukocytes in the lung tissue, those mice also had less Mp detected in both BAL and lung tissue compared with infected mice that received vehicle or rCC16 (D67A). This suggests that some other relevant antimicrobial host responses are being mediated by rCC16 and should be further explored in future studies.
We also acknowledge that several additional receptors and multiple mechanisms of action for CC16 have previously been identified (reviewed in Reference 42). Among those previously described mechanisms for CC16 that could also impact phenotypes similar to what we are reporting are phospholipase A2 inhibition (43), accelerated airway epithelial repair (44), and scavenging of reactive oxygen species (34). CC16 is a ligand for ALX (lipoxin receptor A4) and inhibits SSA (serum amyloid-A)-driven inflammation (45). In addition, CC16 has also been reported to influence the migration of certain cell populations via interaction with the N-formyl-Met-Leu-Phe receptor on neutrophils and eosinophils (46) and the lipocalin-1 receptor on cancer cells (47). Nevertheless, our observations underscore the idea that CC16 impacts several basic inflammatory, injury, and repair pathways and processes common to many different types of pulmonary insults and that interaction with VLA-4 represents yet another mechanism contributing to the activity of this pleotropic protein.
With all that is known relative to CC16 receptors and modes of action, the majority of therapeutic delivery mechanisms for CC16 in the clinic have previously only targeted the lung (48, 49). Based on the studies presented herein, the circulation should not be neglected in future drug testing for optimal activity of CC16. The results of our study may have important implications for future personalized strategies to prevent and treat obstructive lung diseases, including COPD and asthma.
COPD has now risen to the third leading cause of death in the United States, and asthma remains a highly prevalent chronic condition, particularly in childhood (50, 51). Longitudinal studies have demonstrated that these conditions may frequently coexist and up to 50% of COPD cases develop the disease through a trajectory of low lung function from childhood into adult life, for which asthma is the most important risk factor (52). Thus, understanding the role that endogenous proteins such as CC16 may have in the onset and progression of these conditions and the mechanisms by which they affect disease risk is of paramount importance.
Our data also bring to light an immediately applicable role for CC16 in circulation—to limit the number of activated leukocytes from entering and further damaging the delicate lung tissue during infectious insults. Future trials should evaluate not only the potential of rCC16 therapeutics but also consider our results for the possible design of targeted small-molecule therapeutics or peptidomimetics associated with the LVD-binding site to be delivered to individuals with or at risk of obstructive and inflammatory lung diseases.
Acknowledgment
The authors thank Stephanie Delgado and Renata Vallecillo for technical assistance and Samuel Packard for statistical advice. They also thank Natalie Iannuzo and Michael Tomchaney for assistance with immunofluorescent staining and blinded assessment of tissue sections by microscopy.
Footnotes
F.D.M. is Deputy Editor of AJRCCM. His participation complies with American Thoracic Society requirements for recusal from review and decisions for authored works.
Supported by NIH grants HL142769, AI135108.
Author Contributions: M.D.L.J., A.E.C., F.P., C.E.R., M.K., F.D.M., S.G., and J.G.L. participated in project inception and experimental design. M.D.L.J., U.S.Y., S.V.M., K.J.A., M.W., and J.G.L. performed the experiments. M.D.L.J., U.S.Y., A.L.P., S.G., and J.G.L. participated in writing the manuscript; all authors reviewed the manuscript.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202006-2576OC on December 16, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Broeckaert F, Clippe A, Knoops B, Hermans C, Bernard A. Clara cell secretory protein (CC16): features as a peripheral lung biomarker. Ann N Y Acad Sci . 2000;923:68–77. doi: 10.1111/j.1749-6632.2000.tb05520.x. [DOI] [PubMed] [Google Scholar]
- 2. Lakind JS, Holgate ST, Ownby DR, Mansur AH, Helms PJ, Pyatt D, et al. A critical review of the use of Clara cell secretory protein (CC16) as a biomarker of acute or chronic pulmonary effects. Biomarkers . 2007;12:445–467. doi: 10.1080/13547500701359327. [DOI] [PubMed] [Google Scholar]
- 3. Shijubo N, Itoh Y, Yamaguchi T, Sugaya F, Hirasawa M, Yamada T, et al. Serum levels of Clara cell 10-kDa protein are decreased in patients with asthma. Lung . 1999;177:45–52. doi: 10.1007/pl00007626. [DOI] [PubMed] [Google Scholar]
- 4. Guerra S, Halonen M, Vasquez MM, Spangenberg A, Stern DA, Morgan WJ, et al. Relation between circulating CC16 concentrations, lung function, and development of chronic obstructive pulmonary disease across the lifespan: a prospective study. Lancet Respir Med . 2015;3:613–620. doi: 10.1016/S2213-2600(15)00196-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rava M, Tares L, Lavi I, Barreiro E, Zock JP, Ferrer A, et al. Serum levels of Clara cell secretory protein, asthma, and lung function in the adult general population. J Allergy Clin Immunol . 2013;132:230–232. doi: 10.1016/j.jaci.2013.01.023. [DOI] [PubMed] [Google Scholar]
- 6. Vestbo J, Edwards LD, Scanlon PD, Yates JC, Agusti A, Bakke P, et al. ECLIPSE Investigators. Changes in forced expiratory volume in 1 second over time in COPD. N Engl J Med . 2011;365:1184–1192. doi: 10.1056/NEJMoa1105482. [DOI] [PubMed] [Google Scholar]
- 7. Petersen H, Leng S, Belinsky SA, Miller BE, Tal-Singer R, Owen CA, et al. Low plasma CC16 levels in smokers are associated with a higher risk for chronic bronchitis. Eur Respir J . 2015;46:1501–1503. doi: 10.1183/13993003.00682-2015. [DOI] [PubMed] [Google Scholar]
- 8. Zhai J, Insel M, Addison KJ, Stern DA, Pederson W, Dy A, et al. Club cell secretory protein deficiency leads to altered lung function. Am J Respir Crit Care Med . 2019;199:302–312. doi: 10.1164/rccm.201807-1345OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang SZ, Rosenberger CL, Bao YX, Stark JM, Harrod KS. Clara cell secretory protein modulates lung inflammatory and immune responses to respiratory syncytial virus infection. J Immunol . 2003;171:1051–1060. doi: 10.4049/jimmunol.171.2.1051. [DOI] [PubMed] [Google Scholar]
- 10. Hayashida S, Harrod KS, Whitsett JA. Regulation and function of CCSP during pulmonary Pseudomonas aeruginosa infection in vivo. Am J Physiol Lung Cell Mol Physiol . 2000;279:L452–L459. doi: 10.1152/ajplung.2000.279.3.L452. [DOI] [PubMed] [Google Scholar]
- 11. Wang SZ, Rosenberger CL, Espindola TM, Barrett EG, Tesfaigzi Y, Bice DE, et al. CCSP modulates airway dysfunction and host responses in an Ova-challenged mouse model. Am J Physiol Lung Cell Mol Physiol . 2001;281:L1303–L1311. doi: 10.1152/ajplung.2001.281.5.L1303. [DOI] [PubMed] [Google Scholar]
- 12. Stols L, Gu M, Dieckman L, Raffen R, Collart FR, Donnelly MI. A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr Purif . 2002;25:8–15. doi: 10.1006/prep.2001.1603. [DOI] [PubMed] [Google Scholar]
- 13. Ledford JG, Lo B, Kislan MM, Thomas JM, Evans K, Cain DW, et al. Surfactant protein-A inhibits mycoplasma-induced dendritic cell maturation through regulation of HMGB-1 cytokine activity. J Immunol . 2010;185:3884–3894. doi: 10.4049/jimmunol.1000387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Addison KJ, Morse J, Robichaud A, Daines MO, Ledford JG. A novel in vivo system to test bronchodilators. J Infect Pulm Dis . 2017;3 doi: 10.16966/2470-3176.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Papadopoulos JS, Agarwala R. COBALT: constraint-based alignment tool for multiple protein sequences. Bioinformatics . 2007;23:1073–1079. doi: 10.1093/bioinformatics/btm076. [DOI] [PubMed] [Google Scholar]
- 16. Clements JM, Newham P, Shepherd M, Gilbert R, Dudgeon TJ, Needham LA, et al. Identification of a key integrin-binding sequence in VCAM-1 homologous to the LDV active site in fibronectin. J Cell Sci . 1994;107:2127–2135. doi: 10.1242/jcs.107.8.2127. [DOI] [PubMed] [Google Scholar]
- 17. Tselepis VH, Green LJ, Humphries MJ. An RGD to LDV motif conversion within the disintegrin kistrin generates an integrin antagonist that retains potency but exhibits altered receptor specificity: evidence for a functional equivalence of acidic integrin-binding motifs. J Biol Chem . 1997;272:21341–21348. doi: 10.1074/jbc.272.34.21341. [DOI] [PubMed] [Google Scholar]
- 18. Chigaev A, Wu Y, Williams DB, Smagley Y, Sklar LA. Discovery of very late antigen-4 (VLA-4, alpha4beta1 integrin) allosteric antagonists. J Biol Chem . 2011;286:5455–5463. doi: 10.1074/jbc.M110.162636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chouhan BS, Käpylä J, Denessiouk K, Denesyuk A, Heino J, Johnson MS. Early chordate origin of the vertebrate integrin αI domains. PLoS One . 2014;9:e112064. doi: 10.1371/journal.pone.0112064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Humphries MJ. Integrin structure. Biochem Soc Trans . 2000;28:311–339. [PubMed] [Google Scholar]
- 21. Hynes RO, Zhao Q. The evolution of cell adhesion. J Cell Biol . 2000;150:F89–F96. doi: 10.1083/jcb.150.2.f89. [DOI] [PubMed] [Google Scholar]
- 22. Johansson MW, Mosher DF. Integrin activation States and eosinophil recruitment in asthma. Front Pharmacol . 2013;4:33. doi: 10.3389/fphar.2013.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Metzger WJ. Therapeutic approaches to asthma based on VLA-4 integrin and its counter receptors. Springer Semin Immunopathol . 1995;16:467–478. doi: 10.1007/BF00196101. [DOI] [PubMed] [Google Scholar]
- 24. Oshima T, Jordan P, Grisham MB, Alexander JS, Jennings M, Sasaki M, et al. TNF-alpha induced endothelial MAdCAM-1 expression is regulated by exogenous, not endogenous nitric oxide. BMC Gastroenterol . 2001;1:5. doi: 10.1186/1471-230X-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Humphries JD, Byron A, Humphries MJ. Integrin ligands at a glance. J Cell Sci . 2006;119:3901–3903. doi: 10.1242/jcs.03098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Petrovic A, Alpdogan O, Willis LM, Eng JM, Greenberg AS, Kappel BJ, et al. LPAM (alpha 4 beta 7 integrin) is an important homing integrin on alloreactive T cells in the development of intestinal graft-versus-host disease. Blood . 2004;103:1542–1547. doi: 10.1182/blood-2003-03-0957. [DOI] [PubMed] [Google Scholar]
- 27. Pulido R, Campanero MR, García-Pardo A, Sánchez-Madrid F. Structure-function analysis of the human integrin VLA-4 (alpha 4/beta 1): correlation of proteolytic alpha 4 peptides with alpha 4 epitopes and sites of ligand interaction. FEBS Lett . 1991;294:121–124. doi: 10.1016/0014-5793(91)81356-d. [DOI] [PubMed] [Google Scholar]
- 28. Pulido R, Elices MJ, Campanero MR, Osborn L, Schiffer S, García-Pardo A, et al. Functional evidence for three distinct and independently inhibitable adhesion activities mediated by the human integrin VLA-4: correlation with distinct alpha 4 epitopes. J Biol Chem . 1991;266:10241–10245. [PubMed] [Google Scholar]
- 29. Hocking DC. Fibronectin matrix deposition and cell contractility: implications for airway remodeling in asthma. Chest . 2002;122(Suppl):275S–278S. doi: 10.1378/chest.122.6_suppl.275s. [DOI] [PubMed] [Google Scholar]
- 30. Januskevicius A, Gosens R, Sakalauskas R, Vaitkiene S, Janulaityte I, Halayko AJ, et al. Suppression of eosinophil integrins prevents remodeling of airway smooth muscle in asthma. Front Physiol . 2017;7:680. doi: 10.3389/fphys.2016.00680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kong DH, Kim YK, Kim MR, Jang JH, Lee S. Emerging roles of vascular cell adhesion molecule-1 (VCAM-1) in immunological disorders and cancer. Int J Mol Sci . 2018;19:1057. doi: 10.3390/ijms19041057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McGilvray ID, Lu Z, Bitar R, Dackiw AP, Davreux CJ, Rotstein OD. VLA-4 integrin cross-linking on human monocytic THP-1 cells induces tissue factor expression by a mechanism involving mitogen-activated protein kinase. J Biol Chem . 1997;272:10287–10294. doi: 10.1074/jbc.272.15.10287. [DOI] [PubMed] [Google Scholar]
- 33. Zhang Z, Kundu GC, Yuan CJ, Ward JM, Lee EJ, DeMayo F, et al. Severe fibronectin-deposit renal glomerular disease in mice lacking uteroglobin. Science . 1997;276:1408–1412. doi: 10.1126/science.276.5317.1408. [DOI] [PubMed] [Google Scholar]
- 34. Lopez E, Fujiwara O, Nelson C, Winn ME, Clayton RS, Cox RA, et al. Club cell protein, CC10, attenuates acute respiratory distress syndrome induced by smoke inhalation. Shock . 2020;53:317–326. doi: 10.1097/SHK.0000000000001365. [DOI] [PubMed] [Google Scholar]
- 35. Cai Y, Winn ME, Zehmer JK, Gillette WK, Lubkowski JT, Pilon AL, et al. Preclinical evaluation of human secretoglobin 3A2 in mouse models of lung development and fibrosis. Am J Physiol Lung Cell Mol Physiol . 2014;306:L10–L22. doi: 10.1152/ajplung.00037.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Miller TL, Shashikant BN, Melby JM, Pilon AL, Shaffer TH, Wolfson MR. Recombinant human Clara cell secretory protein in acute lung injury of the rabbit: effect of route of administration. Pediatr Crit Care Med . 2005;6:698–706. doi: 10.1097/01.pcc.0000165565.96773.08. [DOI] [PubMed] [Google Scholar]
- 37. Shashikant BN, Miller TL, Welch RW, Pilon AL, Shaffer TH, Wolfson MR. Dose response to rhCC10-augmented surfactant therapy in a lamb model of infant respiratory distress syndrome: physiological, inflammatory, and kinetic profiles. J Appl Physiol (1985) . 2005;99:2204–2211. doi: 10.1152/japplphysiol.00246.2005. [DOI] [PubMed] [Google Scholar]
- 38. Hung CH, Chen LC, Zhang Z, Chowdhury B, Lee WL, Plunkett B, et al. Regulation of TH2 responses by the pulmonary Clara cell secretory 10-kd protein. J Allergy Clin Immunol . 2004;114:664–670. doi: 10.1016/j.jaci.2004.05.042. [DOI] [PubMed] [Google Scholar]
- 39. Chandra S, Davis JM, Drexler S, Kowalewska J, Chester D, Koo HC, et al. Safety and efficacy of intratracheal recombinant human Clara cell protein in a newborn piglet model of acute lung injury. Pediatr Res . 2003;54:509–515. doi: 10.1203/01.PDR.0000081300.49749.87. [DOI] [PubMed] [Google Scholar]
- 40. Nosratabadi AR, Ljungman AG, Lindahl M, Welch R, Pilon A, Tagesson C. Clara cell 10-KDA protein inhibits endotoxin-induced airway contraction in isolated perfused rat lungs. Exp Lung Res . 2003;29:455–473. doi: 10.1080/01902140303778. [DOI] [PubMed] [Google Scholar]
- 41. Chowdhury B, Mantile-Selvaggi G, Miele L, Cordella-Miele E, Zhang Z, Mukherjee AB. Lys 43 and Asp 46 in alpha-helix 3 of uteroglobin are essential for its phospholipase A2 inhibitory activity. Biochem Biophys Res Commun . 2002;295:877–883. doi: 10.1016/s0006-291x(02)00767-2. [DOI] [PubMed] [Google Scholar]
- 42. Mukherjee AB, Zhang Z, Chilton BS. Uteroglobin: a steroid-inducible immunomodulatory protein that founded the Secretoglobin superfamily. Endocr Rev . 2007;28:707–725. doi: 10.1210/er.2007-0018. [DOI] [PubMed] [Google Scholar]
- 43. Mantile G, Miele L, Cordella-Miele E, Singh G, Katyal SL, Mukherjee AB. Human Clara cell 10-kDa protein is the counterpart of rabbit uteroglobin. J Biol Chem . 1993;268:20343–20351. [PubMed] [Google Scholar]
- 44. Bustos ML, Mura M, Hwang D, Ludkovski O, Wong AP, Keating A, et al. Depletion of bone marrow CCSP-expressing cells delays airway regeneration. Mol Ther . 2015;23:561–569. doi: 10.1038/mt.2014.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Antico G, Aloman M, Lakota K, Miele L, Fiore S, Sodin-Semrl S. Uteroglobin, a possible ligand of the lipoxin receptor inhibits serum amyloid A-driven inflammation. Mediators Inflamm . 2014;2014:876395. doi: 10.1155/2014/876395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Johansson S, Andersson K, Wennergren G, Wennerås C, Rudin A. CC16 inhibits the migration of eosinophils towards the formyl peptide fMLF but not towards PGD2. Inflammation . 2009;32:65–69. doi: 10.1007/s10753-008-9103-1. [DOI] [PubMed] [Google Scholar]
- 47. Zhang Z, Kim SJ, Chowdhury B, Wang J, Lee YC, Tsai PC, et al. Interaction of uteroglobin with lipocalin-1 receptor suppresses cancer cell motility and invasion. Gene . 2006;369:66–71. doi: 10.1016/j.gene.2005.10.027. [DOI] [PubMed] [Google Scholar]
- 48. Levine CR, Gewolb IH, Allen K, Welch RW, Melby JM, Pollack S, et al. The safety, pharmacokinetics, and anti-inflammatory effects of intratracheal recombinant human Clara cell protein in premature infants with respiratory distress syndrome. Pediatr Res . 2005;58:15–21. doi: 10.1203/01.PDR.0000156371.89952.35. [DOI] [PubMed] [Google Scholar]
- 49. Davis JM, Pilon AL, Shenberger J, Breeze JL, Terrin N, Mazela J, et al. The role of recombinant human CC10 in the prevention of chronic pulmonary insufficiency of prematurity. Pediatr Res . 2019;86:254–260. doi: 10.1038/s41390-019-0419-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.National Center for Health Statistics. National health interview survey. Atlanta, GA: Centers for Disease Control and Prevention; 2017. [Google Scholar]
- 51. Mutti A, Corradi M, Goldoni M, Vettori MV, Bernard A, Apostoli P. Exhaled metallic elements and serum pneumoproteins in asymptomatic smokers and patients with COPD or asthma. Chest . 2006;129:1288–1297. doi: 10.1378/chest.129.5.1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Melén E, Guerra S. Recent advances in understanding lung function development. F1000 Res . 2017;6:726. doi: 10.12688/f1000research.11185.1. [DOI] [PMC free article] [PubMed] [Google Scholar]