

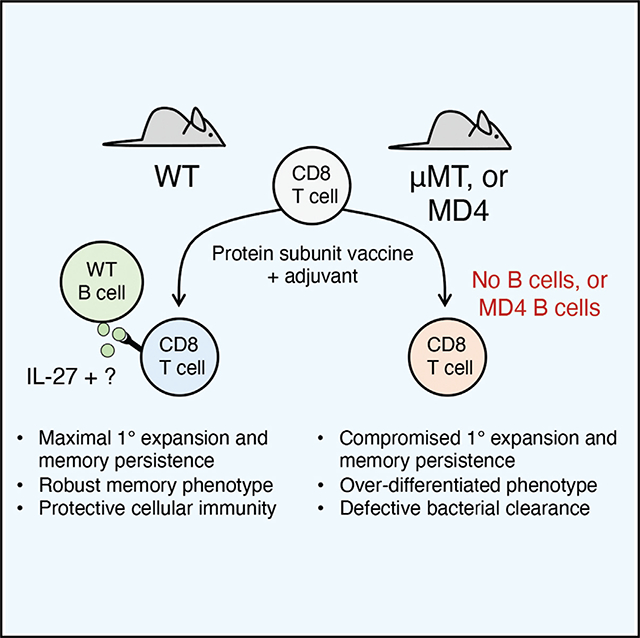
SUMMARY
The relationship between B cells and CD4 T cells has been carefully studied, revealing a collaborative effort in which B cells promote the activation, differentiation, and expansion of CD4 T cells while the so-called “helper” cells provide signals to B cells, influencing their class switching and fate. Interactions between B cells and CD8 T cells are not as well studied, although CD8 T cells exhibit an accelerated contraction after certain infections in B-cell-deficient mice. Here, we find that B cells significantly enhance primary CD8 T cell responses after vaccination. Moreover, memory CD8 numbers and function are impaired in B-cell-deficient animals, leading to increased susceptibility to bacterial challenge. We also show that interleukin-27 production by B cells contributes to their impact on primary, but not memory, CD8 responses. Better understanding of the interactions between CD8 T cells and B cells may aid in the design of more effective future vaccine strategies.
In brief
Generating cytotoxic CD8 T cell responses with vaccines can greatly improve their efficacy, but inducing adequate numbers of these cells can be challenging. Klarquist et al. reveal that the magnitude, persistence, and function of CD8 T cell vaccine responses depend on B cells.
Graphical abstract
INTRODUCTION
Studies in mice utilizing experimental infectious agents have helped outline the mechanisms driving T cell immunity. Given the similarly robust cellular responses they elicit, it is often assumed that protein subunit vaccine formulations function via the factors that govern responses to infections. In contrast to these assumptions, numerous mechanisms of adjuvant-elicited CD8 T cell immunity that are not shared by their infection-elicited T cell counterparts have been identified. We recently documented the amount of interleukin (IL)-27 produced by conventional type 1 dendritic cells (cDC1s) as an innate correlate of CD8 T cell protective memory after adjuvant administration (Kilgore et al., 2018, 2020). This correlation was sufficiently robust so as to facilitate the predictive stratification of adjuvants for their CD8 T-cell-eliciting potential (Kilgore et al., 2020). The predictive value of this relationship fit well with the dependency of adjuvant-elicited, but not infection-elicited, CD4 and CD8 T cell responses on T-cell-intrinsic IL-27 signaling (Klarquist et al., 2018; Pennock et al., 2014). Indeed, maximal T cell expansion to adjuvant administration requires multiple cytokines (IL-27, IL-15) and transcription factors (Tbet, Eomesodermin [Eomes]) (Klarquist et al., 2018) that are not required by T cells responding to infectious challenges (Banerjee et al., 2010; Becker et al., 2002; Intlekofer et al., 2008; Liu et al., 2014; Mayer et al., 2008; Obar et al., 2004; Wehrens et al., 2018; Wherry et al., 2002). After vaccine adjuvant delivery, these cytokines are necessary for enabling a unique metabolic program in subunit vaccine-elicited T cells (Klarquist et al., 2018). Whereas T cells responding to infection depend on glycolysis to fuel clonal expansion, mitochondrial function is overwhelmingly favored during adjuvant-elicited clonal expansion, and the proliferating T cells are largely indifferent to glycolytic blockade. Thus, there are major differences in the factors relied upon by infection-elicited and adjuvant-elicited CD8 T cell responses to support clonal expansion, survival, and memory formation.
Whereas IL-15 and IL-27 are produced by cDC1s (Kilgore et al., 2018; Sosinowski et al., 2013), other antigen (Ag)-presenting cells (APCs) including B cells, also have the capacity to make one or both cytokines. Indeed, one member of the IL-27 heterodimer, Ebi3 (Epstein-Barr virus [EBV] induced gene 3), was originally isolated from EBV-infected B cells, and B cells produce IL-15 and IL-27 upon stimulation with Toll-like receptor (TLR) ligands or CD40L (Schneider et al., 2011; Yan et al., 2020). Furthermore, B cell expression of IL-27 has been linked to CD8 T cell recovery after lymphoablation (Ayasoufi et al., 2019), and T cell memory in IL-27Rα−/− hosts is functionally compromised (Pennock et al., 2014). Similarly, IL-15 production by B cells has been observed in mouse models of multiple sclerosis (MS) and systemic lupus erythematosus (SLE), as well as in human MS patients (Ma et al., 2014; Schneider et al., 2011). In addition to cytokine production, B cells may also be competent for cross-presentation to CD8 T cells. Although still controversial (Keller et al., 2009), multiple groups have demonstrated B cell cross-presentation to CD8 T cells using a variety of model systems (Castiglioni et al., 2005; Hon et al., 2005; Ke and Kapp, 1996; Robson et al., 2008; Zanetti et al.,2004). CD8 T cells are not classically thought to traffic into or reside within B cell zones, legitimately calling into question the in vivo applicability of in vitro demonstrations of B cell cross-presentation. However, a large portion of CD8 T cells actually do migrate into the marginal and B cell zones of the spleen within the first 3 days after lymphocytic choriomeningitis virus (LCMV) challenge (Hu et al., 2011; Jung et al., 2010), and a substantial proportion of memory CD8 T cells can be found within the B cell zone at late time points after Listeria monocytogenes (LM) challenge (Khanna et al., 2007). These data suggest that there may be ample opportunity for CD8 T-cell:B-cell interactions over the course of adaptive immune generation.
Previous work in various infection models has broadly documented an indifference of primary CD8 T cell responses to the presence of B cells, including LM (Shen et al., 2003), influenza (León et al., 2014), LCMV (Asano and Ahmed, 1996; Homann et al., 1998), vaccinia virus (Bründler et al., 1996), and vesicular stomatitis virus (Bründler et al., 1996). Several of these studies detailed an accelerated contraction that yielded either low (Shen et al., 2003) or normal (Asano and Ahmed, 1996; León et al., 2014) numbers of Ag-specific memory CD8 T cells; however, this finding was not universal, with B cells dispensable for CD8 T cell memory in other studies (Bründler et al., 1996; Di Rosa and Matzinger, 1996; Whitmire et al., 2009). Indeed, in some model systems, CD8 T cells actually limit the B cell response as a result of Ag-mediated recognition and killing of Ag-specific B cells (Barnaba et al., 1990). These data do not necessarily contradict, but rather underscore, the idea that immune processes learned from one infectious model do not define immunity at large. In one study, antibodies were found to play a role in capturing and stabilizing Ag after influenza infection, thereby enhancing Ag presentation by DCs (León et al., 2014). However, in all other examples where B cells influenced CD8 T cell contraction (Asano and Ahmed, 1996; Bründler et al., 1996; Homann et al., 1998; Shen et al., 2003), underlying mechanisms have been left largely unexplained.
In exploring a potential role for B cells in the generation of CD8 T cell responses, we found that B cells impact both the primary and memory Ag-specific CD8 T cell responses to subunit vaccines. These results were independent of CD4 T cells or antibody production and were instead due to B-cell-intrinsic factors that were absent in mice deficient in B cells or with a fixed B cell receptor (BCR). Collectively, our results challenge the conventional paradigm of how B cells influence cellular immunity and suggest that some form of CD8 T cell:B cell collaboration is critical for elicitation of maximal CD8 T cell responses after subunit vaccination.
RESULTS
B cells augment primary CD8 T cell responses to subunit vaccination
After adjuvanted subunit immunization, total B cell numbers increase as much as 4-fold within 4 days, in a manner dependent on the adjuvant. The largest increase seen was predictably with a vaccine that utilizes whole ovalbumin protein (OVA) with poly(I:C) and agonistic CD40 antibody (combined-adjuvant), but all single adjuvants evaluated also induced substantial B cell expansion after vaccination (Figure 1A). The considerable size of the B cell pool at these early time points led us to investigate whether B cells contributed to the CD8 T cell response. To explore this, we compared the CD8 T cell response to combined-adjuvant immunization in C57BL/6 wild-type (WT) and μMT−/− B-cell-deficient mice. To mitigate any possible confounding effects on the T cell repertoire of having no B cells present during T cell development, 200 T cell receptor (TCR) transgenic OT1 T cells were transferred 1 day before immunization, a number known to respond to immunological challenge but not completely outcompete an endogenous CD8 T cell response (Badovinac et al., 2007). The number and frequency of endogenous and transferred Ag-specific CD8 T cells were significantly reduced in B-cell-deficient mice at the peak of the response, 7 days after combined-adjuvant immunization (Figures 1B and 1 C). This was not simply due to the lower number of total CD8 T cells typically found in μMT−/− mice, because (1) the total number of transferred OT1 T cells was significantly lower, and (2) endogenous tetramer+ cells were significantly reduced as a percentage of total CD8 T cells in μMT−/− mice compared to WT controls (Figure 1D).
Figure 1. B cells promote primary CD8 T cell responses to subunit vaccines.

(A) Total splenic B220+CD19+ B cells from WT C57BL/6 mice 0–7 days after i.v. administration of combined-adjuvant vaccine (anti-CD40, poly(I:C), and OVA).
(B–D) 200 CD45.1+ OT1 T cells were transferred into WT or μMT−/− mice 1 day before combined-adjuvant vaccination. Spleens were harvested 7 days later. (B) Representative tetramer and CD45.1 staining. (C) Total OT1 cells (left) and tetramer+ endogenous CD8 T cells (right). (D) Tetramer as a percentage of endogenous CD8 T cells.
(E) Representative immunofluorescent staining of 2 naive mice per group with DAPI (blue), CD3 (yellow), B220 (magenta), and CD169 (green). Scale bar represents 50 μm for all images.
(F) Total splenic B220+CD19+ B cells from WT or MD4 mice 0–7 days after combined-adjuvant vaccine.
(G and H) Total splenic tetramer+ CD8 T cells (top) or tetramer+ cells as a percentage of CD8 T cells (bottom) were measured 7 days after subunit vaccination or infection. (G) Ag-specific CD8 T cell responses after i.v. (left) or s.c. (right) administration of combined-adjuvant + OVA vaccine. (H) Responses after i.v. subunit immunization with the single adjuvants poly(I:C) (left) or Pam3Cys (right).
(I) Responses after i.p. administration of OVA emulsified in alum.
(J) Responses after infection with LM-OVA.
Data shown are means ± SEM; n ≥ 3 mice per group, representative of ≥ 2 experiments, except for (F) and (H), which are from one experiment each. Significance was defined by two-tailed, unpaired Student’s t tests, where *p < 0.05, **p < 0.01, and ***p < 0.001.
See also Figure S2.
A trivial explanation for these results might be that the complete absence of B cells indirectly impacted T cell priming simply because of the disruption of secondary lymphoid tissue architecture (Ngo et al., 2001). Therefore, we used the MD4 transgenic mouse, in which >90% of the B cells have a fixed specificity toward hen egg lysozyme (HEL) (Goodnow et al., 1988). Unlike the μMT−/− mice, the splenic architecture in MD4 mice is relatively normal (Figure 1E), and the MD4 mouse has a normal frequency of CD4 T cells, cDC1s, and Tregs (Figure S1). Compared to WT, MD4 mice have slightly fewer CD8 T cells and B cells overall (Figure S1) but exhibited a similar early expansion and contraction of total B cells after combined-adjuvant subunit vaccination compared to WT controls (Figure 1F). As in the B-cell-deficient host, CD8 T cell responses to combined-adjuvant subunit vaccination in the MD4 mice were substantially reduced. This was true for combined-adjuvant immunization administered intravenously (i.v.) or subcutaneously (s.c.) (Figure 1G), as well as for single adjuvants delivered as soluble (Figure 1H) or alum-precipitated (Figure 1I) formulations. Again, the CD8 T cell response was reduced in both numbers (Figures 1G–1I, top) and frequency (Figures 1G–1I, bottom). In contrast to these various forms of subunit vaccination, there was no difference in the CD8 T cell response between MD4 and WT controls after infection with OVA-expressing LM (LM-OVA), (Figure 1J), in line with previous reports of LM infections in μMT−/− mice (Shen et al., 2003). Finally, the quantitative difference in primary CD8 T cell responses between WT and MD4 mice was independent of Ag dose (Figure S2A) and was not restricted to the use of OVA; we obtained similar data when immunizing with herpes simplex virus glycoprotein B (HSVgB) or severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike receptor-binding domain (RBD) proteins (Figures S2B–S2D). CD8 T cell responses to SARS-CoV-2 RBD were analyzed by intracellular cytokine staining after restimulation with the peptide VVLSFELL, a peptide within the RBD known to elicit CD8 T cell responses to SARS-CoV (Zhao et al., 2010), and conserved in SARS-CoV-2 (Davenport et al., 2021). Collectively, we concluded that B cells influence the CD8 T cell response to subunit vaccination, but not infection, and that this influence is not limited to a specific adjuvant formulation, Ag, or route of administration.
Primary CD8 T cell responses to subunit vaccines are not dependent on Ag-antibody (Ag-Ab) complexes or CD4 T cell responses
Since the presence of B cells specific for an irrelevant Ag did not rescue the CD8 T cell response, it was possible that the production of Ag-specific antibody was required for maximal primary CD8 T cell responses to vaccines. Indeed, the formation of Ag-specific Ag-Ab complexes have been shown to facilitate CD8 T cell immunity by enhancing DC Ag capture, retention, and presentation (León et al., 2014). Therefore, we investigated whether mice deficient in the capacity to make Ag-Ab complexes, formed either by secreted natural immunoglobulin (Ig)M or Ag-specific IgG, would also show reduced CD8 response to vaccination. However, no defects in CD8 T cell primary cell numbers were observed after vaccination of secreted IgM-deficient mice (sIgM−) or activation-induced cytidine deaminase-knockout (AID−/−) hosts (Figures 2A and 2B), effectively ruling out these possibilities. We did not specifically test a possible role for secreted, circulating IgD, although this seems highly unlikely, given that detectable sIgD largely depends on class-switched IgD+IgM− cells (Chen et al., 2009; Choi et al., 2017), a process that requires AID (Choi et al., 2017) and was therefore tested in our AID−/− experiments. We also eliminated the possibility that defective CD4 T cell responses were responsible for the diminished primary response in both μMT−/− and MD4 mice by depleting CD4 T cells 2–5 days before immunization (Figure S3). CD4 depletion did not significantly change the Ag-specific CD8 T cell numbers at day 7 (Figure 2C), consistent with our published data showing no role for CD4 T cells in the primary expansion of CD8 T cells to subunit vaccination (Ahonen et al., 2004; Edwards et al., 2013).
Figure 2. Primary CD8 T cell responses are not dependent on Ag-Ab complexes or CD4 T cell responses.
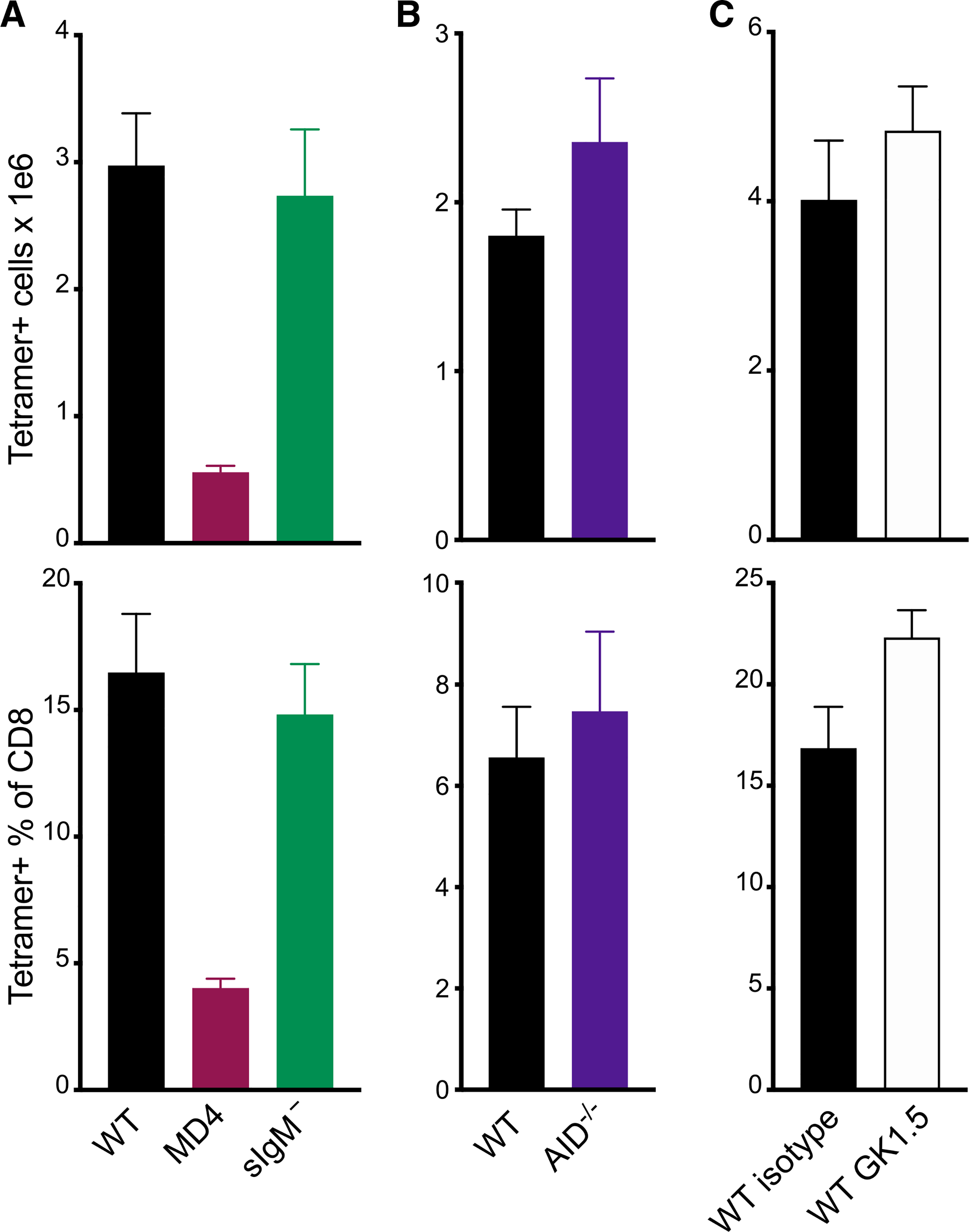
(A–C) Mice were immunized with the combined-adjuvant vaccine. Total splenic tetramer+ CD8 T cells (top) or tetramer+ cells as a percentage of CD8 T cells (bottom) were measured 7 days after subunit vaccination in: (A) WT, MD4, or sIgM− mice; (B) WT or AID−/− mice; or (C) WT mice treated with control antibodies (isotype) or CD4-depleting antibodies (GK1.5). Data shown are means ± SEM; 3–5 mice per group, representative of 2 experiments.
See also Figure S3.
Cytokine and transcription factor analyses indicate that B cells promote vaccine-elicited CD8 T cell memory skewing
To determine whether there were any differences in the quality of the response, we performed ex vivo cytokine restimulation assays at the peak of the response to combined-adjuvant immunization. In line with our tetramer data, there was a significant difference in the frequency of cytokine-producing, Ag-specific CD8 T cells between WT and MD4 mice (Figures 3A–3C). Additionally, more detailed analyses revealed striking differences in the quality of the response. Although there were no differences observed in the percentage of cells producing 2 or more cytokines (Figures 3D and 3E), a significantly higher percentage of cytokine+ cells in WT mice produced IL-2, compared to those in MD4 mice (Figure 3F). Moreover, WT cells produced higher levels of interferon gamma (IFNγ) and IL-2 on a per-cell basis (Figure 3G). These data were potentially highly significant, given that IL-2 is uniquely capable of driving T cell memory development (Feau et al., 2011; Sarkar et al., 2008; Williams et al., 2006), and suggested a skewing of the response toward effectors in the MD4 host. Consistent with this, Ag-specific T cells in MD4 mice substantially increased their percentage of granzyme B+CD127− T cells (Figures 3H and 3I). In contrast, CD8 T cells in WT mice skewed more toward a memory phenotype, with a reduced percentage of granzyme B+ cells (Figure 3I) that also produced less granzyme B on a per-cell basis (Figure 3J).
Figure 3. Cytokine and granzyme production by effector cells demonstrates greater terminal differentiation in mice with a fixed B cell repertoire.
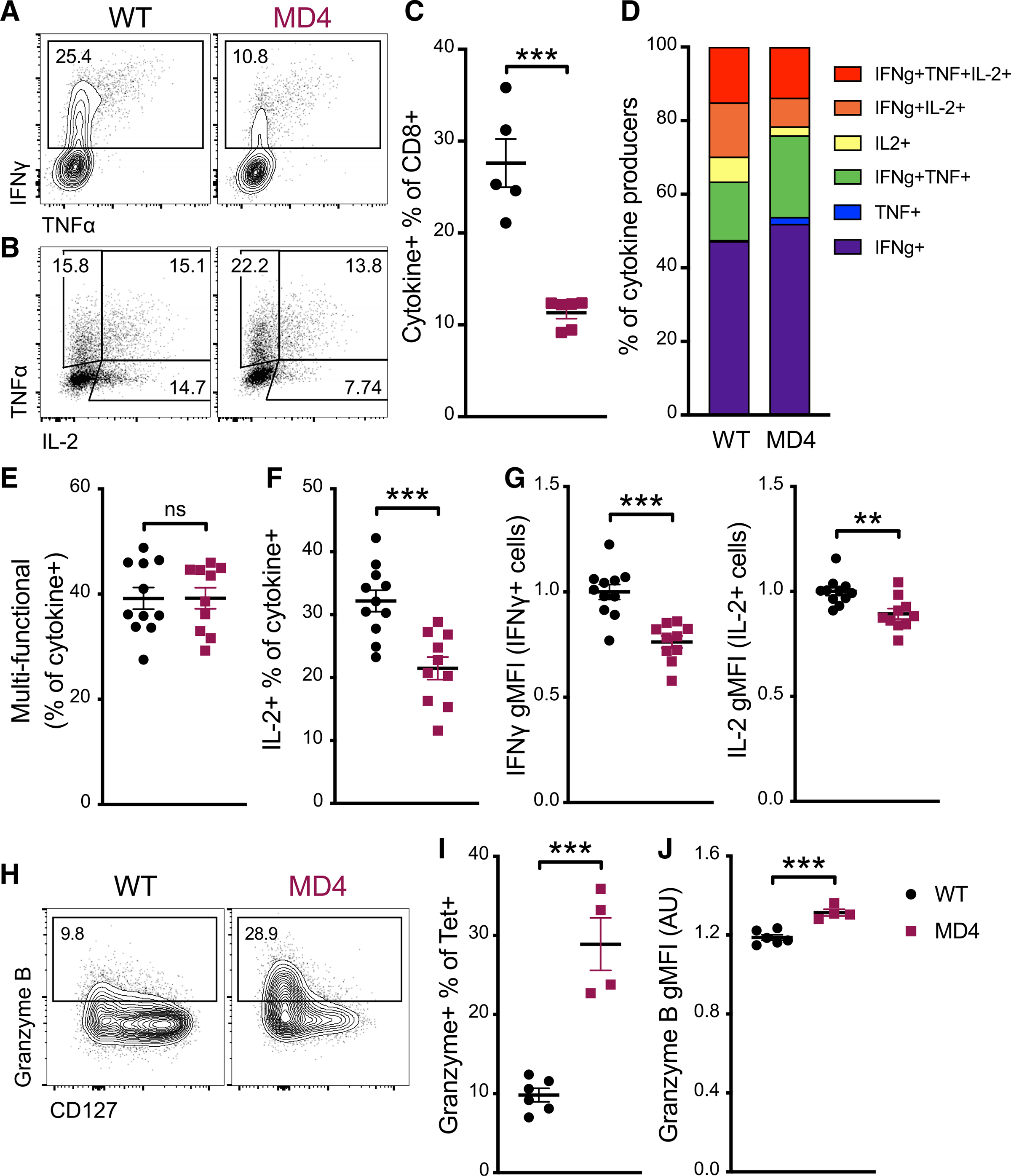
(A–G) Seven days after immunization with the combined-adjuvant vaccine, splenocytes were incubated with SIINFEKL peptide ex vivo and assessed for cytokine production. (A) Representative flow cytometry plots of total CD8 T cells and (B) IFNγ+ CD8 T cells, showing gating for IFNγ, TNF-α, and IL-2. (C) The percentage of CD8 T cells positive for one or more of the cytokines IFNγ, TNF-α, and IL-2. (D) The distribution of single-, double-, and triple-positive cells within all cytokine-producing CD8 T cells. (E) Multi-functional CD8 T cells, defined as producing more than one cytokine, as a percentage of all cytokine-producing CD8 T cells. (F) The percentage of cytokine-producing cells making IL-2. (G) The gMFI for IFNγ (left) and IL-2 (right) of the respective cytokine+ cells.
(H and I) Ag-specific CD8 T cells were identified by tetramer-staining splenocytes 7 days after mice received combined-adjuvant vaccine. (H) Representative plots displaying CD127 and granzyme B staining of tetramer+ cells. (I) Granzyme-B-positive cells as a percentage of tetramer+ CD8 T cells.
(J) The gMFI of granzyme-B-positive tetramer+ cells.
Data shown are means ± SEM; n ≥ 3 mice per group, representative of 2 experiments. Significance was defined by two-tailed, unpaired Student’s t tests, where *p < 0.05, **p < 0.01, and ***p < 0.001.
These results were highly informative, given that the primary goal of prophylactic vaccination is the development of long-lasting, protective immunity. Therefore, we examined more closely the memory/terminal effector phenotypes of CD8 T cells 7 days after vaccination in WT and MD4 mice, including their expression of transcription factors known to influence these cell-fate decisions. Eomes, although dispensable for the primary response to infectious challenge, is critical to the primary response to subunit vaccination (Klarquist et al., 2018). Endogenous CD8 T cells responding in the MD4 host had reduced levels of Eomes compared to those in the WT (Figures 4A and 4B). The biological impact of this was apparent in the reduction in CD122 (IL-2Rβ) expression, a shared component of the heterotrimeric IL-15 and IL-2 receptors that Eomes is known to regulate (Intlekofer et al., 2005). Both of these factors are downstream of IL-27, which is essential for promoting the necessary metabolic support for the CD8 T cell responses to subunit vaccination (Klarquist et al., 2018). Indeed, the CD8 T cells in the MD4 phenocopied (EomeslowCD122low) vaccine responses generated in the absence of IL-27 (Klarquist et al., 2018; Pennock et al., 2014). Additionally, the percentage of cells maintaining high TCF1, which correlates with the self-renewal capacity of pre-memory T cell populations (Im et al., 2016; Kratchmarov et al., 2018; Lin et al., 2016), was significantly reduced in Ag-specific CD8 T cells from MD4 mice. Reciprocally, those T cells exhibited higher IRF4, which can inhibit TCF1 and impair memory development (Man et al., 2017) (Figures 4A and 4B). These results, along with the IL-2 and granzyme B data noted earlier, suggested that CD8 T cells from the MD4 host would display increased phenotypic characteristics of effector cells (high KLRG1 and/or low CD127) and decreased characteristics of memory precursors (high CD127). Indeed, and in contrast to previous reports on infection in B-cell-deficient hosts (León et al., 2014), we observed significant changes in the memory precursor versus terminally differentiated effector phenotype of the cells after immunization. In fact, the entire defect in the MD4 primary response was limited to a loss of CD127high cells (Figures 4C and 4D). Taken together, the cytokine profile, transcriptional program, and surface protein phenotype of Ag-specific CD8 T cells in MD4 animals demonstrated a skewing away from the memory phenotype that usually predominates subunit vaccination.
Figure 4. B cells promote CD8 T cell memory programming following subunit vaccination.
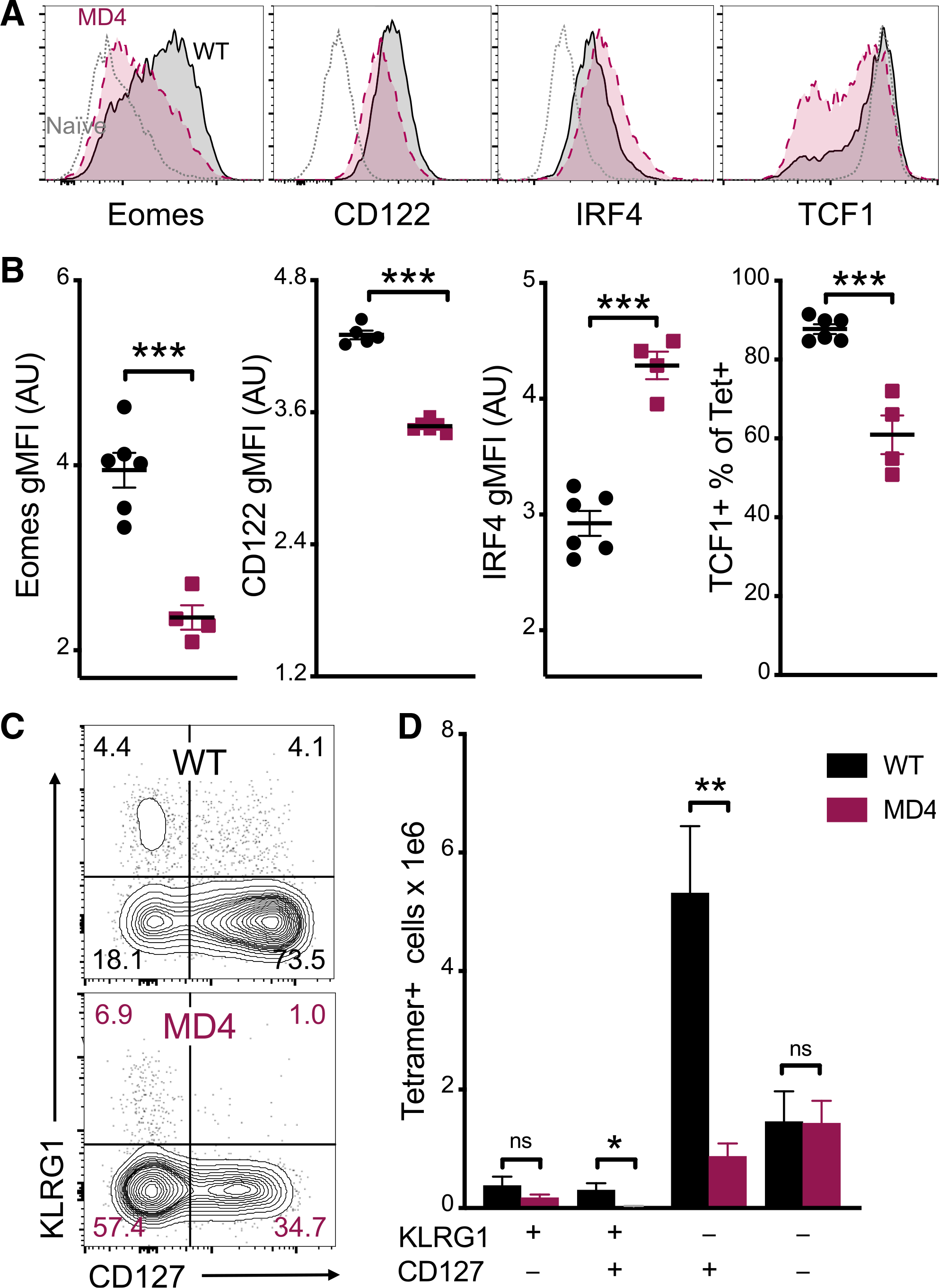
(A) Representative histograms for Eomes, CD122, IRF4, and TCF1 staining of tetramer+ cells from WT (black) or MD4 (maroon) mice 7 days after combined-adjuvant vaccination or for CD44− naive control CD8 T cells (gray).
(B) The gMFI of tetramer+ cells for Eomes, CD122, and IRF4, where the x axis intersects the y axis at the average gMFI value for CD44− naive controls, and the percentage of tetramer+ cells that stained positive for TCF1.
(C and D) Representative KLRG1 versus CD127 staining (C) and numbers of tetramer+ splenocytes in the respective quadrants (D), 7 days after immunization.
Data shown are means ± SEM; n : mice per group, representative of 3 experiments. Significance was defined by two-tailed, unpaired Student’s t tests, where *p < 0.05, **p < 0.01, and ***p < 0.001.
B cells, not CD4 T cells, are critical to the persistence and function of CD8 T cell memory after subunit vaccination
To determine the long-term consequences of the functional and phenotypic differences we observed earlier, we tracked Ag-specific CD8 T cell memory for 12 weeks after immunization. To control for the requirement of CD4 help during priming, we depleted CD4 T cells 2–5 days before vaccination (Figure S3). As seen in the spleens 7 days after immunization (Figure 2), CD4 T cell depletion actually resulted in an overall increase in tetramer+ CD8 T cell frequency at all time points (Figure 5A). This increase was independent of a polyclonal B cell repertoire, as it also occurred in the MD4 host (Figure 5A). However, circulating Ag-specific CD8 T cell survival to memory was dramatically reduced in MD4 mice compared to WT controls in both the intact and CD4-depleted conditions. Analysis of these results by a two-way ANOVA yielded a significant interaction effect (p < 0.001), indicating that the MD4 defect in CD8 memory was exaggerated compared to what would have been predicted simply by the reduced primary response; that is, the slope of the decay in Ag-specific CD8 T cells was significantly increased in MD4 mice compared to that in WT mice. Analysis of the Ag-specific CD8 T cell responses in WT and MD4 mice depleted of CD4 T cells before vaccination also resulted in a statistically significant interaction effect, indicating that the accelerated decay of CD8 T cells in the MD4 host was not CD4 dependent. Moreover, there was no significant interaction effect when comparing the decay of memory T cells over time between isotype-control-treated and CD4-depleted WT mice; thus, while initially depleting CD4 T cells changed the peak magnitude of the CD8 T cell response, it had no impact on the persistence of memory CD8 T cells. This memory T cell defect was also not dependent on the use of the combined-adjuvant, as MD4 mice also had significantly reduced splenic tetramer+ cell numbers after vaccination with single adjuvants (Figure 5B). Furthermore, within the splenic memory CD8 T cells, there were significantly lower proportions of CD62L+ central memory cells at 3 months post-vaccination, and fewer memory CD8 T cells expressed TCF1, which regulates the differentiation and longevity of CD8 memory (Figure 5C). Thus, we conclude that B cells influence the maintenance of CD8 T cell memory after vaccination in a manner independent of CD4 T cells.
Figure 5. Defective persistence of CD8 T cell memory cells in the absence of a normal B cell repertoire.
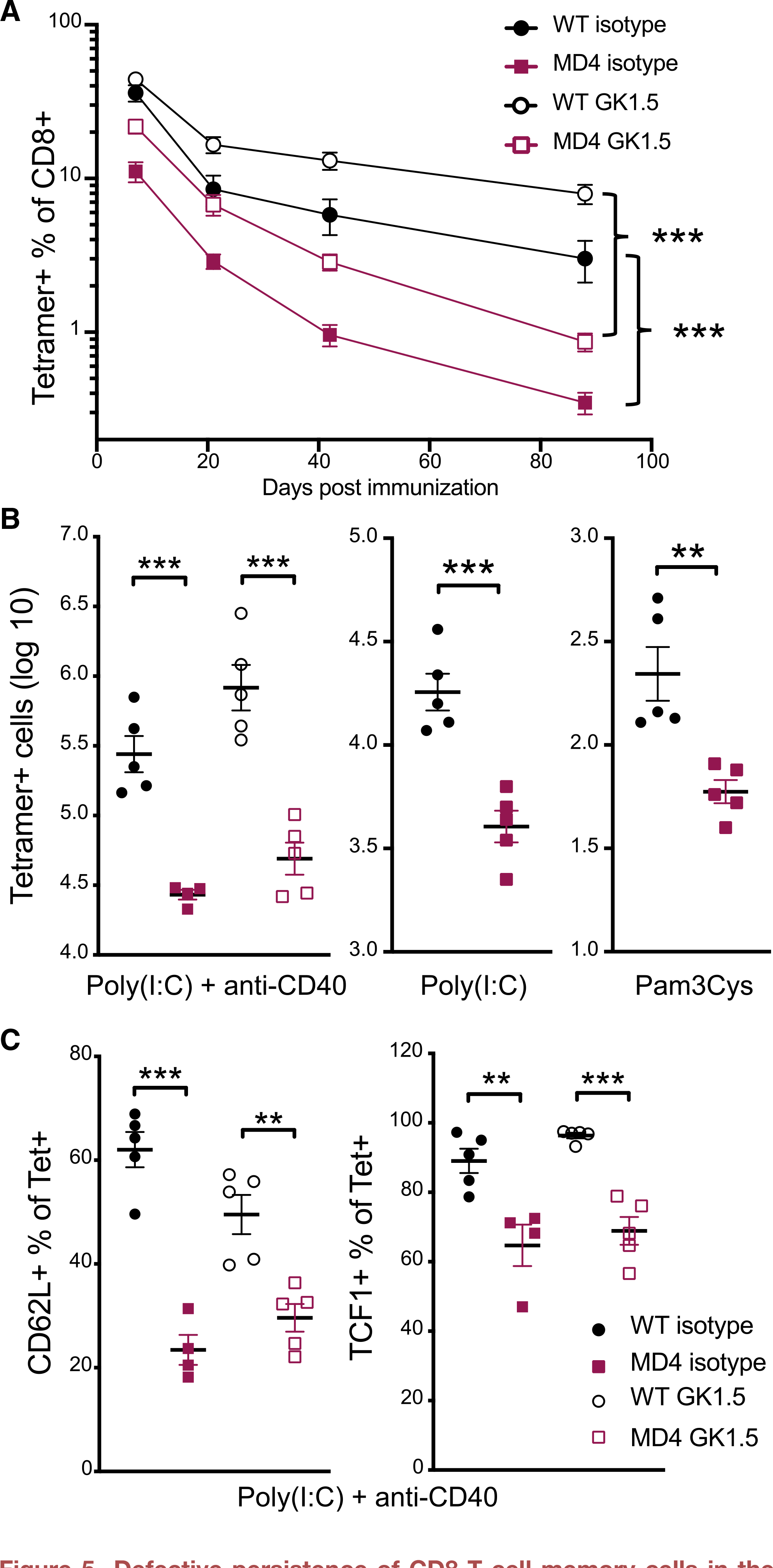
(A) Endogenous tetramer+ cells as a percentage of CD8 T cells in the blood over time after combined-adjuvant immunization of CD4-depleted mice (GK1.5) or controls (isotype).
(B) The absolute number of tetramer+ CD8 T cells 3–5 months after combined-adjuvant (left) or single-adjuvant subunit vaccinations (middle and right).
(C) The percentage of tetramer+ splenocytes positive for CD62L (left) or TCF1 (right) 3 months after combined-adjuvant vaccination.
Data shown are means ± SEM; n ≥ 3 mice per group, representative of ≥ 2 experiments for (A) and one experiment each for (B) and (C). Significance was defined by 2-way ANOVA interaction effects (A) or by two-tailed, unpaired Student’s t tests (B and C), where *p < 0.05, **p < 0.01, and ***p < 0.001.
See also Figure S3.
The data thus far indicates increased effector differentiation, decreased memory fate commitment, and accelerated loss of memory in the CD8 T cells from immunized MD4 hosts. Our next goal was to determine whether these factors ultimately result in decreased memory T cell function. Using an in vivo killing assay, we found that CD8 T cell memory function was indeed compromised on a per-cell basis in MD4 mice. Transferring differentially CTV-labeled targets bearing control or vaccine-specific Ag into WT or MD4 mice 3 months post-immunization revealed reduced killing of the Ag-pulsed targets in MD4 hosts (Figure 6A). This defect in the MD4 CD8 T cells remained even when accounting for the differences in memory T cell frequency between WT and MD4 hosts (Figure 6B), indicating reduced lytic function of memory CD8 T cells in the MD4 host on a per-cell basis. Interestingly, this was unaffected by CD4 depletion (Figure 6B), once again identifying this as a B-cell-dependent phenomenon.
Figure 6. Dysfunctional CD8 memory in the absence of a normal B cell repertoire.

(A and B) Mice that were either depleted of CD4 T cells 5 days before vaccination or kept intact received 500 OT1 cells immediately before combined-adjuvant vaccination. The function of resting memory cells was assessed by an in vivo killing assay 3 months later. SIINFEKL-loaded splenocytes and HSVgB-peptide-loaded splenocytes were labeled with low and high levels of CTV, respectively; mixed; and injected intravenously into mice. Two hours later, splenic target and effector numbers were quantified. (A) Representative raw data are shown with staggered histograms. (B) Data are plotted as percentage of total SIINFEKL-loaded targets killed versus total number of effector cells.
(C–N) Separate WT and MD4 mice were administered the i.v. combined-adjuvant subunit vaccine. After 3 months, they were challenged with 105 colony-forming units (CFUs) of LM-OVA and analyzed 4 days later. (C) LM CFUs in the liver of mice 4 days post-challenge. (D) Number of splenic tetramer+ cells 4 days post-challenge. (E) Fold secondary expansion of Ag-specific CD8 T cells in the blood as determined by dividing the frequency of tetramer+ (out of a total CD8 T cells) 3 months after immunization, i.e., pre-challenge, by the frequency of tetramer+ cells 4 days post-challenge. (F) LM CFUs from (C) were correlated with Ag-specific T cell numbers in (B), and analysis of the MD4 data yielded a significant linear regression with an R2 of 0.74. (G–I) Representative flow cytometry plots of memory CD8 T cells from WT and MD4 4 days post-LM challenge. (G) IFNγ and TNF-α intracellular cytokine staining of CD8 T cells after 5-h stimulation with SIINFEKL (OVA257–264) peptide. (H) TNF-α and IL-2 staining of IFNγ+ T cells from (E). (I) Granzyme B staining directly ex vivo 4 days post-challenge, showing tetramer+ CD8 T cells (black) overlayed with CD44− naive control cells (gray). (J) IFNγ+ as a percentage of CD8 T cells after 5-h incubation with SIINFEKL (left) or an irrelevant control peptide (HSV gB498–505, right). (K) Multi-functional cells—double- or triple-positive for IFNγ, TNF-α, and/or IL-2—as a percentage of total cytokine producers. (L) IFNγ and (M) TNF-α gMFI of respective cytokine+ cells. (N) Granzyme B gMFI of tetramer+granzyme+ CD8 T cells.
Data shown are means ± SEM; n ≥ 3 mice per group, representative of 2 experiments. Significance was defined by two-tailed, unpaired Student’s t tests, where *p < 0.05, **p < 0.01, and ***p < 0.001.
These results described the functional capacity of resting memory T cells 3 months post-vaccination. To assess the capacity of these cells for a secondary response, the mice were challenged with a lethal dose of LM-OVA at this same time point post-vaccination. Four days after LM-OVA challenge, the frequency and function of Ag-specific T cells and bacterial load were determined in the spleen and liver, respectively. Consistent with the decreased per-cell killing observed in the resting memory T cells (Figure 6B), 9/10 MD4 mice were unable to clear the infection, whereas WT mice had no detectable bacterial burden (Figure 6C). Unlike previous results in the context of infection-elicited immunity (Shen et al., 2003), bacterial challenge yielded fewer secondary effector CD8 T cells post-challenge in MD4 mice (Figure 6D), although MD4 mice exhibited a largely unimpaired secondary expansion compared to WT controls (Figure 6E). Secondary responses were unaffected in sIgM− and AID−/− mice (Figure S4A), demonstrating that capture and stabilization of Ag (León et al., 2014) by secreted natural IgM or Ag-specific IgG also does not play a role in the development of CD8 memory after subunit vaccination. Notably, μMT−/− mice exhibited poor CD8 T cell memory persistence after immunization, similar to MD4 mice, although their secondary expansion was significantly impaired, in stark contrast with previous data (Shen et al., 2003). Those same factors in sIgM− and AID−/− mice mirrored that of WT mice (Figures S4A–S4E). Since antibodies do not naturally afford protection against LM (Matsuzaki et al., 1999; Shen et al., 2003), the lack of naive, Ag-specific B cells in μMT−/− and MD4 mice is not a confounding variable when assessing bacterial load. In MD4 mice, bacterial titers negatively correlated with numbers of secondary effectors (Figure 6F). Notably, several mice had high bacterial titers despite similar numbers of secondary effectors to WT mice, again indicating a deficit in effector function on a per-cell basis. This could not be accounted for by reductions in cytokine or granzyme production, as, somewhat counterintuitively, T cells from MD4 mice were highly polyfunctional and produced more IFNγ, tumor necrosis factor alpha (TNF-α), and granzyme B on a per-cell basis than their WT counterparts in ex vivo restimulation assays (Figures 6G–6N). However, the ongoing bacterial load in these mice (Figure 6C) and the propensity of these CD8 T cells toward effector differentiation (Figure 3) collectively suggest an elevated degree of terminal effector differentiation. The reduced capacity of the MD4 T cells for persistence into memory (Figure 5) further indicates a reduced capacity for self-renewal, a critical factor for T-cell-mediated host protection (Wherry et al., 2003). Substantial cytokine production was detected in control wells lacking cognate peptide (Figure 6J), evidence in further support of over-differentiation driven by continued antigenic stimulation in vivo. Taken together, these results indicate that B cells, not CD4 T cells, influence the magnitude of the primary CD8 T cell response to vaccination; its transition to, and persistence as a memory T cell pool; and its capacity to enact host-protective functions.
B cells can cross-present Ag to CD8 T cells, but B cell Ag presentation is not required for maximal CD8 responses to subunit vaccines
The data thus far suggest that a restricted B cell repertoire is detrimental to the generation and maintenance of vaccine-elicited CD8 T cell responses. Immunofluorescence evaluation of spleen sections after immunization revealed that, as B cells expand early after vaccination, a high percentage of Ag-specific T cells are found within B cell zones on days 2–4 post-vaccination (Figure S5). This localization of CD8 T cells into the B cell zone has been previously observed (Hu et al., 2011; Jung et al., 2010) and could conceivably facilitate direct CD8 T cell:B cell interactions at these early time points.
Given previous conflicting reports on whether B cells can cross-present Ag (Castiglioni et al., 2005; Hon et al., 2005; Ke and Kapp, 1996; Keller et al., 2009; Robson et al., 2008; Zanetti et al., 2004), we began by assessing their abilities in an in vitro experimental system. We compared the capacity of highly purified B cells from WT or MD4 spleens (Figure S6A) to cross-present Ag to proliferation-dye-labeled OT1 T cells. APCs were cultured with fast protein liquid chromatography (FPLC)-purified whole protein HEL-OVA conjugates or OVA. As positive controls, we included WT splenocytes enriched for DCs as well as SIINFEKL-peptide-pulsed DCs/B cells. Analysis of OT1 proliferation 2–3 days later demonstrated that, whereas DCs induced OT1 proliferation in all conditions, WT B cells only did so in the presence of peptide, and MD4 B cells did so only in the context of HEL-OVA conjugates or peptide (Figure 7A). These results confirm published data that B cells can indeed cross-present Ag to CD8 T cells in vitro and that they do so in a BCR-restricted manner.
Figure 7. B cell production of IL-27, not cross-presentation, contributes to CD8 T cell vaccine responses.
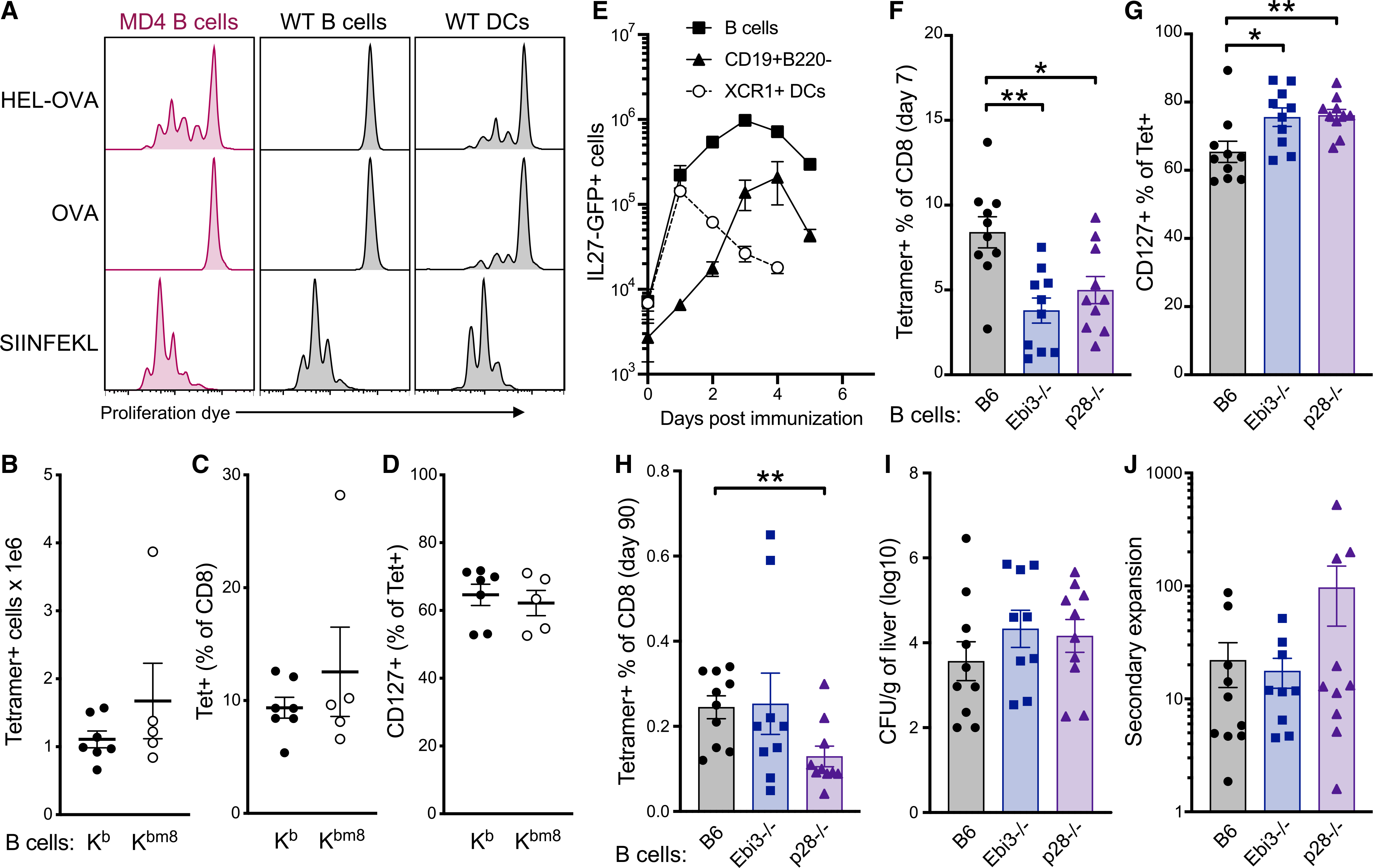
(A) B cells or splenocytes enriched for DCs were pre-incubated with protein or peptide for 1 h and then washed thoroughly before combining with proliferation dye-labeled OT1s. OT1 proliferation was determined by proliferation dye dilution 72 h after incubation with the indicated cell type.
(B–D) Bone marrow chimeras were generated by reconstituting H-2Kb/Kbm8CD45.1/2 F1 mice with a mixture of μMT−/−CD45.1/2 + WTCD45.2/2, or H-2Kbm8CD45.2/2 bone marrow. Chimeric mice were immunized 10 weeks after reconstitution and analyzed for Ag-specific CD8 T cell responses 7 days later. (B) Total splenic tetramer+ CD8 T cells. (C) Tetramer+ cells as a percentage of CD8 T cells. (D) CD127+ cells as a percentage of tetramer+ cells.
(E) The percentage of cells producing IL-27 was tracked over time in B cells (CD19+B220+), CD19+B220− cells, and XCR1+ DCs (B220−CD11chighXCR1+) using IL-27p28-GFP reporter mice after combined-adjuvant immunization.
(F–J) Bone marrow chimeras were generated by reconstituting WTCD45.1/2 mice with a mixture of μMT−/−CD45.1/2 + WTCD45.2/2, Ebi3−/−CD45.2/2, or p28−/−CD45.2/2 bone marrow. Chimeric mice were immunized 10 weeks after reconstitution. (F) Tetramer+ cells as a percentage of grafted CD8 T cells in the blood 7 days after vaccination. (G) CD127+ cells as a percentage of tetramer+ cells on day 7. (H) Tetramer+ cells as a percentage of grafted CD8 T cells 3 months after vaccination. (I) Secondary expansion of Ag-specific CD8 T cells in the blood defined as tetramer+ percentage of CD8 (pre-challenge) divided by tetramer+ percentage of CD8 (post-challenge). (J) LM CFUs in the liver of mice 4 days post-challenge.
Data in (A) are representative of 3 experiments. Data shown in (B)–(D) and (F)–(J) are means ± SEM from 2 combined experiments. Data shown in (E) are means ± SEM; n ≥ 3 mice per group, from 3 experiments combined. Significance was defined by two-tailed, unpaired Student’s t tests, where *p < 0.05, **p < 0.01, and ***p < 0.001.
See also Figure S6.
However, whether Ag presentation to CD8 T cells by B cells occurs in vivo and is necessary for B cell promotion of CD8 T cell responses to vaccines remained open questions. To test these, bone marrow chimeras were generated so that the B cell compartment was selectively limited in its ability to present the SIINFEKL peptide. Bone marrow from μMT−/− mice was mixed with bone marrow from either H-2Kbm8 mice (that express a mutant form of H-2Kb that cannot present the SIINFEKL peptide to CD8 T cells) or WT H-2Kb mice, at a ratio of 9:1. This ratio ensures that although all B cells express either the mutant or WT H-2Kb allele, ~90% of all other cells express WT H-2Kb derived from the μMT−/− background. H-2Kb 3 H-2Kbm8 F1 mice were used as recipients to ensure acceptance of bone marrow grafts. Ten weeks after lethal irradiation and bone marrow reconstitution, mice were vaccinated with combined-adjuvant + OVA, and the T cell responses were measured at the peak of the response, 7 days later. The chimeric mice mounted similar Ag-specific CD8 T cell responses regardless of whether B cells were (Kb) or were not (Kbm8) capable of presenting cognate SIINFEKL peptide (Figures 7B and 7C). The CD127high memory phenotype was also equal between these groups (Figure 7D), consistent with the conclusion that Ag presentation by B cells is not responsible for the promotion of CD8 T cell responses after immunization.
IL-27 produced by B cells contributes to the primary CD8 T cell vaccine response
As noted earlier, CD8 T cell responses to adjuvanted subunit vaccination are uniquely dependent on IL-27 (Klarquist et al., 2018; Pennock et al., 2014), which is not required for the primary CD8 T cell response to infections (Pennock et al., 2014; Wehrens et al., 2018) or, in some cases, even inhibits the response (Liu et al., 2014). We have characterized the IL-27 production by DC subsets after vaccination and its unique capacity to predict the magnitude of protective CD8 T cell memory. Given the importance of IL-27 to the vaccine-elicited response, it seemed possible that the influence of B cells on the CD8 T cells might be through their production of this cytokine. We used our recently described IL-27p28-GFP reporter mouse (Kilgore et al., 2018) to examine B cell production of IL-27, specifically over the time frame in which we had observed Ag-specific CD8 T cells within the B cell zone early after vaccination (Figure S5). Although the percentage of B cells producing IL-27p28 was considerably lower than that seen in XCR1+ cDCs (Figures S6B and S6C), a significant number of GFP+ B cells were observed 15 days after combined-adjuvant vaccination (Figure 7E). We again utilized the bone marrow from μMT−/− mice in mixed bone marrow chimeras, this time in combination with bone marrow derived from Ebi3−/−, IL-27p28−/−, and WT controls. We used the 9:1 ratio in favor of μMT−/−, ensuring that all B cells were the desired cytokine-deficient phenotype, whereas the majority of non-B cells were WT (Figure S6D). In contrast to the chimeras in which B cells were deficient in Ag presentation, selective B cell deficiency in either of the heterodimeric IL-27 subunits significantly compromised the primary vaccine-elicited CD8 T cell response (Figure 7F). The Ebi3 subunit can be shared with either IL-35 and the newly described IL-39, whereas p28, either alone or in concert with cytokine-like factor 1 (CLF), can exert signaling as IL-30. Thus, the fact that we observed similar results in mice with B cells deficient in either p28 or Ebi3 effectively identifies B cell production of IL-27 as the cytokine necessary for maximal vaccine-elicited CD8 responses. Intriguingly, however, this defect in CD8 T cell expansion was not associated with the characteristic skewing away from memory phenotype seen in the MD4 hosts. Indeed, significantly more of the Ag-specific CD8 T cells from the B-cell-selective IL-27-deficient bone marrow chimeras were CD127high, although similar percentages of tetramer+ cells were seen 3 months post-vaccination (Figures 7G and 7H). Furthermore, bacterial challenge of chimeric mice resulted in similar bacterial burden and secondary expansion (Figures 7I and 7J). Thus, although the production of IL-27 by B cells does not fully account for the effect of B cells on CD8 memory, their production of this cytokine promotes the primary CD8 T cell response to subunit vaccines.
DISCUSSION
The data presented here establish that B cells promote the primary and memory responses of CD8 T cells to subunit vaccines. Our data suggest that perhaps the role of B cells may have been underappreciated in earlier studies of infection that demonstrated accelerated CD8 T cell contraction in B-cell-deficient mice (Asano and Ahmed, 1996; Bründler et al., 1996; Homann et al., 1998). Given the numerous differences cited earlier between vaccine- and infection-elicited CD8 T cell responses, it is likely that the participation of B cells in CD8 T cell immunity also has functions unique to subunit vaccination. Our documented role for B cell IL-27 production is consistent with this prediction, as IL-27 clearly has a role in vaccine-elicited cellular immunity not shared with T cells responding to infectious challenge. Although we have yet to fully appreciate the reciprocal nature of this relationship, recent reports that describe the production of IL-21 by CD8 T cells in the context of influenza virus infection (Yang et al., 2016) and experimental autoimmune disease (Valentine et al., 2018) suggest that it needs reconsideration. In these studies, CD8 T cells were also found to colocalize with B cells and, in particular, with germinal center B cells, where they promoted class switching and the production of antibodies. With the data we present here, these studies collectively raise the question as to whether a B cell-CD8 T cell bidirectional collaboration has been similarly underappreciated.
The function of B cells as APCs presenting Ag to CD4 T cells and promoting their activation, differentiation, and expansion is well documented (Constant, 1999; Molnarfi et al., 2013). Although B cells are clearly capable of Ag cross-presentation to CD8 T cells in vitro, we found that this was not the mechanism by which they help CD8 T cell responses to vaccines in vivo. Similarly, we found that B cells do not indirectly impact the magnitude of the CD8 T cell response via CD4 T cells. Indeed, antibody depletion of CD4 T cells before vaccination actually increased the primary and memory CD8 responses, indicating a more direct action of B cells, independent of CD4 cells, in augmenting CD8 T cell responses. CD4 T cells seem instead to play an inhibitory role, possibly through the action of Tregs or by competing for interactions with APCs. We previously showed that memory but not primary CD8 cell numbers were reduced in CD4-deficient, major histocompatibility complex class II knockout (MHCII−/−) mice (Edwards et al., 2013). In the context of the data presented here, the poor memory survival in our MHCII−/− mouse studies is more likely to have been caused by B cell dysfunction rather than deriving from a lack of CD4 T cells. Indeed, MHCII−/− mice have no germinal centers (Madsen et al., 1999), and even B-cell-specific MHCII deletion prevents activated B cell differentiation and proliferation (Giles et al., 2015). This fits well with the data we provide here, where B cells were critical for maintaining CD8 memory after vaccination. The role of CD4 T cells in suppressing CD8 T cell responses to subunit vaccines remains an open question.
Our observed defect in the CD8 T cell responses to vaccination in mice with a limited or absent B cell repertoire has numerous practical implications. Rituximab, an anti-CD20, B-cell-depleting therapy, has been used since 1997, when it was first approved by the Food and Drug Administration to treat non-Hodgkin’s lymphoma. It has since been approved for additional diseases, and its off-label use has increased in recent years to more than half of patients receiving the drug (Delate et al., 2020). As almost all of the uses of the various anti-CD20 monoclonal antibody treatments involve prolonged B cell depletion, our data from μMT hosts are directly relevant. Consistent with this, B cell depletion in a mouse model of immune thrombocytopenia (ITP) showed reduced CD8 T cell proliferation in response to TCR stimulation (Guo et al., 2016). This is additionally applicable to disease settings in which limiting the function of CD8 T cells by depleting B cells may be beneficial. Rituximab use in patients with antineutrophil cytoplasmic antibody-associated vasculitis (AAV) alters the CD8 T cell memory compartment, reducing the frequency of effector memory cells re-expressing CD45RA (TEMRA) (Néel et al., 2019). B cells isolated from these patients increased the cytokine output of CD8 T cells in ex vivo co-cultures, providing evidence for a direct, positive effect of B cells on CD8 T cells in this disease setting, where CD8 T cells are pathogenic. A contemporary implication of our data is that of next-generation vaccines deployed to combat emerging viruses, such as SARS-CoV-2. Necessary to consider are the negative consequences of B cell deficiency in the generation of the T cell responses, which might otherwise help compensate for impaired humoral immunity.
The data presented here should also inform our understanding of common variable immunodeficiency (CVID), a heterogenous group of disorders characterized by hypogammaglobulinemia of unknown cause. Multiple CD8 T cell abnormalities have been documented in CVID (Wong and Huissoon, 2016), including decreased numbers of naive CD8 T cells (Bateman et al., 2012; Giovannetti et al., 2007), an increase in markers of exhaustion (Klocperk et al., 2020; Paquin-Proulx et al., 2013), dysregulated cytokine production (Giovannetti et al., 2007; North et al., 1998; Thon et al., 1997), and impaired proliferation after TCR engagement (Fischer et al., 1994; Jaffe et al., 1993). The reasons for these CD8-related sequelae have not been described, but our data suggest that it is reasonable to postulate a causal relationship between the B cell and CD8 T cell dysfunctions. Consistent with what we observed for T cells in MD4 compared to WT mice 7 days after immunization and after bacteria challenge, CD8 T cells from patients with CVID (Klocperk et al., 2020) exhibit higher granzyme B than controls. This is similarly true of CD8 T cells after B cell depletion in an experimental model for ITP (Guo et al., 2016). Further studies to uncover the B cell deficiencies in CVID may aid in better understanding the accompanying cellular defects and, in so doing, provide new avenues for therapeutic intervention for the frequent infections experienced by these patients.
Last, it is worth noting that these studies have unveiled an unexpected role for B cell production of IL-27 in vaccine-elicited CD8 T cell immunity. Despite the fact that that Ebi3 was originally discovered in B cells, there are surprisingly few reports on IL-27 in B cells and its influence on adaptive immunity, humoral or cellular. Moreover, while canonically pleotropic, the influence of IL-27 on cellular immunity, outside of subunit vaccination, is overwhelmingly suppressive, so that any T-cell-centric elimination of IL-27 signaling typically results in elevated lymphoproliferation and its related pathology. In addition to the findings we report here, we and our colleagues recently documented the extent of B-cell-produced IL-27 in virus-infected and TLR-agonist-treated mice (Yan et al., 2020). In these studies, IL-27 was highly expressed in mice treated with a range of TLR agonists often used as adjuvants. Further, in mice with a B-cell-specific deletion of IL-27, there was substantially reduced vaccinia-specific IgG as compared to WT virus challenge mice. The immune defects in vaccinia-challenged mice with IL-27-deficient B cells were substantial enough so as to significantly delay virus clearance. Thus, the importance of IL-27 in the normal functions of B cells responding to adjuvant and virus challenges is also underappreciated. Collectively, our results reveal functions for B cell production of IL-27 in vaccine-elicited CD8 T cell immunity. Future studies will be necessary to determine the specific B cells involved in producing IL-27 and when and exactly how this IL-27 shapes primary CD8 T cell responses, particularly in the context of DC-produced IL-27. It remains to be determined whether new avenues of therapeutic application for this cytokine are possible and in what clinical areas these data will find their most influential application. Finally, unraveling the additional mechanism(s) by which B cells contribute to CD8 T cell memory responses to vaccination remains the subject of ongoing investigation, which will hopefully yield further avenues for improving vaccine-elicited immunity.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ross Kedl (Ross.Kedl@cuanschutz.edu).
Materials availability
This study did not generate unique reagents.
Data and code availability
Microscopy data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All experiments involving mice were conducted following protocols approved by the University of Colorado Institutional Animal Care and Use Committee (IACUC) according to guidelines provided by the Association for Assessment and Accreditation of Laboratory Animal Care. WT (C57BL/6J), congenic CD45.1 B6 (B6.SJL-Ptprca Pepcb/BoyJ), OVA-specific TCR-transgenic OT-1 (C57BL/6-Tg(TcraTcrb)1100Mjb/J), μMT−/− (B6.129S2-Ighmtm1Cgn/J) and MD4 (C57BL/6-Tg(IghelMD4)4Ccg/J) mice were originally obtained from the Jackson Laboratory and subsequently bred inhouse at the University of Colorado. MD4 mice were bred as hemizygous MD4 x C57BL/6J, and hemizygous MD4 mice were compared to WT littermate controls. Secreted-IgM-deficient (Boes et al., 1998), AID−/− (Muramatsu et al., 2000), and H-2Kbm8 (B6(C)-H2-Kbm8/LrpJ) mice were provided by Drs. Claudia Jakubzick, Jing H. Wang, and Larry Pease, respectively. IL-27p28-GFP mice were generated by our lab as previously described (Kilgore et al., 2018) and used as hemizygotes. IL-27p28−/− mice, originally provided by Janssen Research & Development (Spring House, PA), and Ebi3−/− mice (B6.129X1-Ebi3tm1Rsb/J), from Jackson laboratory, were bred in house the University of Pennsylvania. Experiments were performed in 6–10-week-old male and female mice.
METHOD DETAILS
Immunizations and infections
Mice were immunized via tail-vein injection (I.V.), subcutaneous injection in the ear (S.C.), or intraperitoneal injection (I.P.) with the indicated adjuvant(s) with 150 μg (only 25 μg in the case of SC immunization) of whole chicken OVA (Sigma) or 100 μg of SARS-CoV-2 spike protein receptor binding domain (RBD) protein plus adjuvant. OVA (Sigma) and HSVgB (produced inhouse by baculovirus expression) proteins were detoxified by phase separation (Aida and Pabst, 1990) and were lipopolysaccharide-free as determined by a limulus assay. SARS-CoV-2 spike RBD protein (GenBank: MT380724.1) was generated by transfection of HEK293 T cells with a His-tagged vector (a gift from F. Krammer, Icahn School of Medicine at Mount Sinai, New York, NY) (Amanat et al., 2020) and subsequent purification over an ATKA nickel column (Cytiva) by the University of Colorado Cell Technologies Shared Resource. The following adjuvant doses were used for immunizations: 40 μg poly(I:C) (5 μg for S.C., Invivogen), 40 μg αCD40 (5 μg for S.C., clone FGK4.5, BioXCell), 40 μg Pam3Cys, or 100 μL Alu-Gel-S (1.3% aluminum hydroxide, SERVA Electrophoresis GmbH). Vaccines were made immediately prior to immunization with the exception of alum; OVA was emulsified in alum by gentle rocking at 4C overnight. Unless otherwise specified, mice were immunized I.V. with combined-adjuvant subunit vaccine (anti-CD40, poly(I:C), and OVA). Mice were infected with 2,000 CFU of OVA-expressing Listeria monocytogenes (LM-OVA) or challenged with 1–2 × 105 CFU of LM-OVA by I.V. injection. Four days after challenge, spleens, blood, and liver were harvested from euthanized mice. Livers were massed then processed in 5 mL of 0.02% NP40 using sterile plastic tissue homogenizers (Miltenyi). Bacterial counts were performed after incubating multiple dilutions of liver homogenates for 48 hours at 37C on BHI agar plates with 5 μg/ml erythromycin (Sigma).
Bone marrow chimeras
Recipient BoyJ mice (CD45.1/1) were lethally irradiated with 2 × 600 rads 3 hours apart and reconstituted 18 hours later with T cell-depleted bone marrow from sex-matched μMT−/− (CD45.1/2) and either C57BL/6, H-2Kbm8, IL-27p28−/−, or Ebi3−/− (CD45.2/2) bone marrow at a 9:1 ratio. T cells were depleted using CD3 (clone 145–2C11), CD4 (GK1.5), CD8 (53.67) and CD90.2 (30-H12) APC-conjugated antibodies and anti-APC magnetic beads (BioLegend). At 10 weeks post-reconstitution, mice were immunized as described above.
Flow cytometry
Spleen single cell suspensions or whole blood (collected by tail vein puncture into 5mM EDTA in HBSS), as indicated, were subjected to ACK red blood cell lysis and counted using a Vi-Cell automated cell counter (Beckman Coulter). Cells were then incubated with αCD16/32 (clone 2.4G2; hybridoma supernatant) and tetramer stained as previously described at 37C for 30 minutes, or in the case of OT-1 transfer, cells were stained for CD45.1 (clone A20; BioLegend) at room temperature (RT) for 10 minutes. Kb-SIINFEKL tetramers were provided by the NIH Tetramer Core. Following tetramer staining, surface antibodies were added for 10 min. at 37C, including CD3e (145–2C11), B220 (RA3–6B2), CD8a (53–6.7), CD44 (IM7; Tonbo), CD62L (MEL-14), CD122 (TM-β1), CD127 (A7R34; Tonbo), and KLRG1 (2F1/KLRG1) (all from BioLegend, unless otherwise noted). For transcription factor analysis, cells were surface stained, then fixed and permeabilized using Tonbo Foxp3 fixation/permeabilization buffers (Tonbo) and stained for Eomes (Dan11mag; eBioscience), IRF4 (3E4; eBioscience), and TCF1 (C63D9; Cell Signaling Technology). Intracellular cytokine staining was performed after a 5-hour stimulation with either 1 μg/ml peptide of SIINFEKL for OVA immunizations, or VVLSFELL for SARS-CoV-2 RBD immunizations (ChinaPeptides) and 3 μg/ml brefeldin A. After fixation and permeabilization, CD8 T cells were stained for intracellular IFNγ (XMG1.2), TNFα (MP6-XT22), and IL-2 (JES6–5H4); granzyme B (GB11; all from BioLegend) was stained directly ex vivo and analyzed on tetramer-positive cells. Flow cytometry data were acquired on a four-laser (405, 488, 561, 638 nm) CytoFLEX S flow cytometer (Beckman Coulter) and analysis was performed using FlowJo (version 10.7.1; BD Biosciences).
OT1 adoptive transfers and CD4-depletion
CD8 T cells were magnetically purified (BioLegend) from OT1 mice to > 95% purity. Cells were counted and 200–500 (as indicated) OT1s were transferred to recipient mice by tail vein injection 1 day prior to immunization. CD4 T cell-depletion was achieved by IV injection of 50 μg αCD4 antibody (clone GK1.5, BioXCell) 2–5 days prior to immunization.
Immunofluorescence
Whole spleens were frozen in OCT compound at −80C until cutting 7 μm sections with a cryostat followed by acetone fixation. Sections were dried, then rehydrated with PBS. Non-specific staining was blocked using PBS + 10% FBS and 2.5% anti-CD16/32 (2.4G2) hybridoma supernatant, and an Avidin/Biotin blocking system (BioLegend). Sections were then stained using CD45.1-PE (clone A20; BioLegend), CD169-AlexaFluor488 (3D6.112; BioLegend), B220-DyLight650 (RA3–6B2; made inhouse with hybridoma), F480-AlexaFluor488 (BM8; BioLegend), CD3e-biotin (145–2C11; BioLegend) followed by streptavidin-AlexaFluor750 (ThermoFisher), and/or IgM-Cy5 (JacksonImmunoResearch), followed by incubation with 300nM DAPI (ThermoFisher) and coverslipping with ProLong Gold anti-fade reagent (ThermoFisher). Images were captured by whole-slide scanning with a Vectra Polaris (Akoya Biosciences) and analyzed using inForm software (Akoya).
In vitro cross-presentation assay
B cells were highly purified by staining with CD43-APC (clone S11) and CD11c-APC (clone N418; BioLegend) followed by anti-APC bead separation over magnetic LS columns (Miltenyi), where the supernatant consisted of ~99% B220+CD19+ B cells. DCs were enriched to ~30% by CD11c-APC staining and magnetic bead separation. CD8 T cells from OT1 mice were enriched to > 95% as described above and stained with 2 μM CellTrace Violet proliferation dye (ThermoFisher). HEL-OVA conjugates were created by glutaraldehyde crosslinking essentially as previously described (Allen et al., 2007). Whole OVA and HEL (Sigma) were combined at a 1:1 molar ratio in KPO4 (pH 7.2) with 0.023% glutaraldehyde (Sigma). Crude protein conjugates were dialyzed overnight with 20kD dialysis cassettes (ThermoFisher) in PBS. Precipitates were removed by 0.2 μm filtration. Conjugated HEL-OVA was concentrated with 30kD MWCO spin concentrators (Sigma), then purified by FPLC (AKTA). B cells or DCs were incubated with 10 μg/ml FPLC-purified HEL-OVA, OVA, or SIINFEKL peptide and 10 μg/ml anti-CD40 for 1 h at 37C, then washed 3 times with RPMI 1640 containing 10% FBS, 10 mM HEPES, 0.1 mM β-ME, 0.1 mM non-essential amino acids, 0.1 mM sodium pyruvate, 2 mM L-glutamine and penicillin-streptomycin (complete media). B cells or DCs were plated at 100,000 cells/well together with 50,000 dye-labeled OT1 cells/well and incubated for 48–72 hours in 200 μL complete media.
In vivo killing assay
Splenocytes were peptide-pulsed for 1 h in complete media with 1 μg/ml of OVA257–264 (target cells) or HSVgB498–505 (control cells), washed, then labeled with 0.05 μM or 0.5 μM CellTrace Violet (ThermoFisher) dye, respectively. Recipient WT or MD4 mice were immunized with anti-CD40, poly(I:C), and OVA 1 day after adoptive transfer of 500 OT1 T cells. After 3 months, 8.5 million target splenocytes combined with 1.5 million control splenocytes were transferred into mice by tail vein injection. Mice were euthanized after 2 h and the ratio of target:control was used to determine the percent killing compared to the same ratio observed for non-immunized control mice. Percent killing was plotted against the total number of splenic Ag-specific CD8 T cells as determined by tetramer staining.
QUANTIFICATION AND STATISTICAL ANALYSIS
GraphPad Prism (version 9.1.2, GraphPad) was used for all statistical analyses. Figure legends detail the number of experimental replicates and n-values. Unless noted, data shown are means ± SEM. Significance was defined by a two-tailed, unpaired Student’s t tests unless otherwise specified, where *p < 0.05, **p < 0.01, and ***p < 0.001.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| APC anti-mouse CD45.1 | BioLegend | Cat# 110714 |
| Brilliant Violet 421 anti-mouse CD3e | BioLegend | Cat# 155617 |
| PerCP anti-mouse B220 | BioLegend | Cat# 103234 |
| Brilliant Violet 605 anti-mouse CD8a | BioLegend | Cat# 100744 |
| redFluor 710 anti-Human/Mouse CD44 | Tonbo Biosciences | Cat# 80–0441-U100 |
| FITC anti-mouse CD62L | BioLegend | Cat# 104406 |
| FITC anti-mouse CD122 (IL-2Rβ) | BioLegend | Cat# 123208 |
| PE anti-Mouse CD127 (IL-7Ra) | Tonbo Biosciences | Cat# 50–1271–U100 |
| PE/Cyanine7 anti-mouse/human KLRG1 | BioLegend | Cat# 138416 |
| Foxp3 / Transcription Factor Staining Buffer Kit | Tonbo Biosciences | Cat# TNB-0607-KIT |
| PE/Cyanine7 anti-mouse/human Eomes | ThermoFisher Scientific | Cat# 25–4875–82 |
| PE anti-mouse/human IRF4 | ThermoFisher Scientific | Cat# 12–9858–82 |
| Alexa Fluor 488 anti-mouse/human TCF1 | Cell Signaling | Cat# 6444S |
| APC anti-mouse IFN-γ | BioLegend | Cat# 505810 |
| PE anti-mouse IL-2 | BioLegend | Cat# 503808 |
| PE/Cyanine 7 anti-mouse TNF-α | BioLegend | Cat# 506324 |
| FITC anti-human/mouse Granzyme B | BioLegend | Cat# 372206 |
| MojoSort Mouse anti-APC Nanobeads | BioLegend | Cat# 480072 |
| InVivomAb anti-mouse CD4 (GK1.5) | BioXCell | Cat# BE0003–1 |
| PE anti-mouse CD45.1 | BioLegend | Cat# 110708 |
| Alexa Fluor® 488 anti-mouse CD169 (Siglec-1) | BioLegend | Cat# 142419 |
| Alexa Fluor® 488 anti-mouse F4/80 | BioLegend | Cat# 123120 |
| Biotin anti-mouse CD3ε | BioLegend | Cat# 100304 |
| Cy5 AffiniPure Goat Anti-Mouse IgM, μ chain specific | Jackson ImmunoResearch | Cat# 115–175-075 |
| APC anti-mouse CD43 | BioLegend | Cat# 143208 |
| APC anti-mouse CD11c | BioLegend | Cat# 117310 |
| InVivoMAb anti-mouse CD40 (FGK4.5) | BioXCell | Cat# BE0016–2 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Streptavidin, Alexa Fluor 750 conjugate | ThermoFisher Scientific | Cat# S21384 |
| DAPI (4’,6-Diamidino-2-Phenylindole, Dihydrochloride) | ThermoFisher Scientific | Cat# D1306 |
| ProLong Gold Antifade Mountant | ThermoFisher Scientific | Cat# P36934 |
| CellTrace Violet Cell Proliferation Kit | ThermoFisher Scientific | Cat# C34557 |
| Albumin from chicken egg white (ovalbumin) | Millipore Sigma | Cat# A5503 |
| Avidin/Biotin Blocking System (Previously Covance catalog# SIG-31126) | BioLegend | Cat# 927301 |
| Alu-Gel-S | Serva | Cat# 12261.01 |
| Ghost Dye Red 780 | Tonbo | Cat# 13–0865-T500 |
|
| ||
| Critical commercial assays | ||
|
| ||
| DyLight 650 Antibody Labeling Kit | ThermoFisher Scientific | Cat# 84535 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: C57BL/6J | The Jackson Laboratory | Cat# 000664 |
| Mouse: B6 CD45.1 | The Jackson Laboratory | Cat# 002014 |
| Mouse: OT-1 | The Jackson Laboratory | Cat# 003831 |
| Mouse: μMT−/− | The Jackson Laboratory | Cat# 002288 |
| Mouse: MD4 | The Jackson Laboratory | Cat# 002595 |
| Mouse: sIgM− | Boes et al., 1998 | N/A |
| Mouse: AID−/− | Muramatsu et al., 2000 | N/A |
| Mouse: H-2Kbm8 | The Jackson Laboratory | Cat# 023029 |
| Mouse: IL-27p28-GFP | Kilgore et al., 2018 | N/A |
| Mouse: IL-27p28−/− | Janssen Research and Development | N/A |
| Mouse: Ebi3−/− | The Jackson Laboratory | Cat# 008691 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Poly(I:C) HMW (long synthetic analog of dsRNA) | Invivogen | Cat# tlrl-pic-5 |
|
| ||
| Recombinant DNA | ||
|
| ||
| SARS-CoV-2 spike vector | Amanat et al., 2020 | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Prism version 9.1.2 | GraphPad | N/A |
| inForm version 2.5.1 | Akoya Biosciences | N/A |
| FlowJo version 10.7.1 | BD Biosciences | N/A |
Highlights.
Primary and memory CD8 T responses to vaccines are hampered in B-cell-deficient mice
B cells support a self-renewal program in vaccine-elicited CD8 T cells
IL-27 from B cells drives the magnitude of the primary CD8 T cell vaccine response
ACKNOWLEDGMENTS
We would like to thank Divij Mathew, Raul Torres, Roberta Pelanda, and John Cambier for helpful discussions. We thank Jing Wang, Claudia Jakubzick, and Larry Pease for providing mouse strains necessary for these studies. We thank Cody Rester and Trevor Blain for technical assistance. We also thank Tem Morrison, Lori Sherman, and the CU Cancer Center Cell Technologies Shared Resource for producing SARS-CoV-2 spike-derived RBD protein. This work was supported by National Institute of Allergy and Infectious Diseases training grant AI074491 (to J.K.) and grants AI066121, AI148919, and AI126899 (to R.M.K.).
Footnotes
DECLARATION OF INTERESTS
R.M.K. is a founder of ImmuRx Inc., a vaccine company for which intellectual property is based on the combined TLR agonist/anti-CD40 immunization platform. All other authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109591.
REFERENCES
- Ahonen CL, Doxsee CL, McGurran SM, Riter TR, Wade WF, Barth RJ, Vasilakos JP, Noelle RJ, and Kedl RM (2004). Combined TLR and CD40 triggering induces potent CD8+ T cell expansion with variable dependence on type I IFN. J. Exp. Med 199, 775–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aida Y, and Pabst MJ (1990). Removal of endotoxin from protein solutions by phase separation using Triton X-114. J. Immunol. Methods 132, 191–195. [DOI] [PubMed] [Google Scholar]
- Allen CDC, Okada T, Tang HL, and Cyster JG (2007). Imaging of germinal center selection events during affinity maturation. Science 315, 528–531. [DOI] [PubMed] [Google Scholar]
- Amanat F, Stadlbauer D, Strohmeier S, Nguyen THO, Chromikova V, McMahon M, Jiang K, Arunkumar GA, Jurczyszak D, Polanco J, et al. (2020). A serological assay to detect SARS-CoV-2 seroconversion in humans. Nat. Med 26, 1033–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano MS, and Ahmed R (1996). CD8 T cell memory in B cell-deficient mice. J. Exp. Med 183, 2165–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayasoufi K, Zwick DB, Fan R, Hasgur S, Nicosia M, Gorbacheva V, Keslar KS, Min B, Fairchild RL, and Valujskikh A (2019). Interleukin-27 promotes CD8+ T cell reconstitution following antibody-mediated lymphoablation. JCI Insight 4, e125489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badovinac VP, Haring JS, and Harty JT (2007). Initial T cell receptor transgenic cell precursor frequency dictates critical aspects of the CD8(+) T cell response to infection. Immunity 26, 827–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A, Gordon SM, Intlekofer AM, Paley MA, Mooney EC, Lindsten T, Wherry EJ, and Reiner SL (2010). Cutting edge: The transcription factor eomesodermin enables CD8+ T cells to compete for the memory cell niche. J. Immunol 185, 4988–4992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnaba V, Franco A, Alberti A, Benvenuto R, and Balsano F (1990). Selective killing of hepatitis B envelope antigen-specific B cells by class I-restricted, exogenous antigen-specific T lymphocytes. Nature 345, 258–260. [DOI] [PubMed] [Google Scholar]
- Bateman EAL, Ayers L, Sadler R, Lucas M, Roberts C, Woods A, Packwood K, Burden J, Harrison D, Kaenzig N, et al. (2012). T cell phenotypes in patients with common variable immunodeficiency disorders: associations with clinical phenotypes in comparison with other groups with recurrent infections. Clin. Exp. Immunol 170, 202–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker TC, Wherry EJ, Boone D, Murali-Krishna K, Antia R, Ma A, and Ahmed R (2002). Interleukin 15 is required for proliferative renewal of virus-specific memory CD8 T cells. J. Exp. Med 195, 1541–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boes M, Esau C, Fischer MB, Schmidt T, Carroll M, and Chen J (1998). Enhanced B-1 cell development, but impaired IgG antibody responses in mice deficient in secreted IgM. J. Immunol. 160, 4776–4787. [PubMed] [Google Scholar]
- Bründler MA, Aichele P, Bachmann M, Kitamura D, Rajewsky K, and Zinkernagel RM (1996). Immunity to viruses in B cell-deficient mice: influence of antibodies on virus persistence and on T cell memory. Eur. J. Immunol 26, 2257–2262. [DOI] [PubMed] [Google Scholar]
- Castiglioni P, Gerloni M, Cortez-Gonzalez X, and Zanetti M (2005). CD8 T cell priming by B lymphocytes is CD4 help dependent. Eur. J. Immunol 35, 1360–1370. [DOI] [PubMed] [Google Scholar]
- Chen K, Xu W, Wilson M, He B, Miller NW, Bengtén E, Edholm ES, Santini PA, Rath P, Chiu A, et al. (2009). Immunoglobulin D enhances immune surveillance by activating antimicrobial, proinflammatory and B cell-stimulating programs in basophils. Nat. Immunol 10, 889–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, Wang K-W, Zhang D, Zhan X, Wang T, Bu C-H, Behrendt CL, Zeng M, Wang Y, Misawa T, et al. (2017). IgD class switching is initiated by microbiota and limited to mucosa-associated lymphoid tissue in mice. Proc. Natl. Acad. Sci. USA 114, E1196–E1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constant SL (1999). B lymphocytes as antigen-presenting cells for CD4+ T cell priming in vivo. J. Immunol 162, 5695–5703. [PubMed] [Google Scholar]
- Davenport BJ, Morrison TE, Kedl RM, and Klarquist J (2021). Conserved and Novel Mouse CD8 T Cell Epitopes within SARS-CoV-2 Spike Receptor Binding Domain Protein Identified following Subunit Vaccination. J. Immunol 206, 2503–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delate T, Hansen ML, Gutierrez AC, and Le KN (2020). Indications for rituximab use in an integrated health care delivery system. J. Manag. Care Spec. Pharm 26, 832–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Rosa F, and Matzinger P (1996). Long-lasting CD8 T cell memory in the absence of CD4 T cells or B cells. J. Exp. Med 183, 2153–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards LE, Haluszczak C, and Kedl RM (2013). Phenotype and function of protective, CD4-independent CD8 T cell memory. Immunol. Res 55, 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feau S, Arens R, Togher S, and Schoenberger SP (2011). Autocrine IL-2 is required for secondary population expansion of CD8(+) memory T cells. Nat. Immunol 12, 908–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer MB, Hauber I, Eggenbauer H, Thon V, Vogel E, Schaffer E, Lokaj J, Litzman J, Wolf HM, Mannhalter JW, et al. (1994). A defect in the early phase of T-cell receptor-mediated T-cell activation in patients with common variable immunodeficiency. Blood 84, 4234–4241. [PubMed] [Google Scholar]
- Giles JR, Kashgarian M, Koni PA, and Shlomchik MJ (2015). B Cell-Specific MHC Class II Deletion Reveals Multiple Nonredundant Roles for B Cell Antigen Presentation in Murine Lupus. J. Immunol 195, 2571–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannetti A, Pierdominici M, Mazzetta F, Marziali M, Renzi C, Mileo AM, De Felice M, Mora B, Esposito A, Carello R, et al. (2007). Unravelling the complexity of T cell abnormalities in common variable immunodeficiency. J. Immunol 178, 3932–3943. [DOI] [PubMed] [Google Scholar]
- Goodnow CC, Crosbie J, Adelstein S, Lavoie TB, Smith-Gill SJ, Brink RA, Pritchard-Briscoe H, Wotherspoon JS, Loblay RH, Raphael K, et al. (1988). Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature 334, 676–682. [DOI] [PubMed] [Google Scholar]
- Guo L, Kapur R, Aslam R, Speck ER, Zufferey A, Zhao Y, Kim M, Lazarus AH, Ni H, and Semple JW (2016). CD20+ B-cell depletion therapy suppresses murine CD8+ T-cell-mediated immune thrombocytopenia. Blood 127, 735–738. [DOI] [PubMed] [Google Scholar]
- Homann D, Tishon A, Berger DP, Weigle WO, von Herrath MG, and Oldstone MBA (1998). Evidence for an underlying CD4 helper and CD8 T-cell defect in B-cell-deficient mice: failure to clear persistent virus infection after adoptive immunotherapy with virus-specific memory cells from muMT/muMT mice. J. Virol 72, 9208–9216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon H, Oran A, Brocker T, and Jacob J (2005). B lymphocytes participate in cross-presentation of antigen following gene gun vaccination. J. Immunol 174, 5233–5242. [DOI] [PubMed] [Google Scholar]
- Hu JK, Kagari T, Clingan JM, and Matloubian M (2011). Expression of chemokine receptor CXCR3 on T cells affects the balance between effector and memory CD8 T-cell generation. Proc. Natl. Acad. Sci. USA 108, E118–E127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, et al. (2016). Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, Mullen AC, Gasink CR, Kaech SM, Miller JD, et al. (2005). Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat. Immunol 6, 1236–1244. [DOI] [PubMed] [Google Scholar]
- Intlekofer AM, Banerjee A, Takemoto N, Gordon SM, Dejong CS, Shin H, Hunter CA, Wherry EJ, and Reiner SL (2008). Anomalous Type 17 Response to Viral Infection by CD8+ T Cell Lacking T-bet and Eomesodermin. Science 321, 408–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe JS, Strober W, and Sneller MC (1993). Functional abnormalities of CD8+ T cells define a unique subset of patients with common variable immunodeficiency. Blood 82, 192–201. [PubMed] [Google Scholar]
- Jung YW, Rutishauser RL, Joshi NS, Haberman AM, and Kaech SM (2010). Differential localization of effector and memory CD8 T cell subsets in lymphoid organs during acute viral infection. J. Immunol. 185, 5315–5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Y, and Kapp JA (1996). Exogenous antigens gain access to the major histocompatibility complex class I processing pathway in B cells by receptor-mediated uptake. J. Exp. Med 184, 1179–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller SA, von Allmen CE, Hinton HJ, Bauer M, Muntwiler S, Dietmeier K, Saudan P, and Bachmann MF (2009). Follicular and marginal zone B cells fail to cross-present MHC class I-restricted epitopes derived from viral particles. J. Immunol 182, 6261–6266. [DOI] [PubMed] [Google Scholar]
- Khanna KM, McNamara JT, and Lefrançois L (2007). In situ imaging of the endogenous CD8 T cell response to infection. Science 318, 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilgore AM, Welsh S, Cheney EE, Chitrakar A, Blain TJ, Kedl BJ, Hunter CA, Pennock ND, and Kedl RM (2018). IL-27p28 Production by XCR1+ Dendritic Cells and Monocytes Effectively Predicts Adjuvant-Elicited CD8+ T Cell Responses. Immunohorizons 2, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilgore AM, Pennock ND, and Kedl RM (2020). cDC1 IL-27p28 Production Predicts Vaccine-Elicited CD8+ T Cell Memory and Protective Immunity. J. Immunol 204, 510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klarquist J, Chitrakar A, Pennock ND, Kilgore AM, Blain T, Zheng C, Danhorn T, Walton K, Jiang L, Sun J, et al. (2018). Clonal expansion of vaccine-elicited T cells is independent of aerobic glycolysis. Sci. Immunol 3, 9822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klocperk A, Unger S, Friedmann D, Seidl M, Zoldan K, Pfeiffer J, Hausmann O, Benes V, Andrieux G, Boettler T, et al. (2020). Exhausted phenotype of follicular CD8 T cells in CVID. J. Allergy Clin. Immunol 146, 912–915.e13. [DOI] [PubMed] [Google Scholar]
- Kratchmarov R, Magun AM, and Reiner SL (2018). TCF1 expression marks self-renewing human CD8+ T cells. Blood Adv 2, 1685–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- León B, Ballesteros-Tato A, Randall TD, and Lund FE (2014). Prolonged antigen presentation by immune complex-binding dendritic cells programs the proliferative capacity of memory CD8 T cells. J. Exp. Med 211, 1637–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin WW, Nish SA, Yen B, Chen Y-H, Adams WC, Kratchmarov R, Rothman NJ, Bhandoola A, Xue H-H, and Reiner SL (2016). CD8+ T Lymphocyte Self-Renewal during Effector Cell Determination. Cell Rep 17, 1773–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu FDM, Kenngott EE, Schröter MF, Kühl A, Jennrich S, Watzlawick R, Hoffmann U, Wolff T, Norley S, Scheffold A, et al. (2014). Timed action of IL-27 protects from immunopathology while preserving defense in influenza. PLoS Pathog 10, e1004110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma N, Xing C, Xiao H, He Y, Han G, Chen G, Hou C, Marrero B, Wang Y, Zhang S, et al. (2014). BAFF suppresses IL-15 expression in B cells. J. Immunol 192, 4192–4201. [DOI] [PubMed] [Google Scholar]
- Madsen L, Labrecque N, Engberg J, Dierich A, Svejgaard A, Benoist C, Mathis D, and Fugger L (1999). Mice lacking all conventional MHC class II genes. Proc. Natl. Acad. Sci. USA 96, 10338–10343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man K, Gabriel SS, Liao Y, Gloury R, Preston S, Henstridge DC, Pellegrini M, Zehn D, Berberich-Siebelt F, Febbraio MA, et al. (2017). Transcription Factor IRF4 Promotes CD8+ T Cell Exhaustion and Limits the Development of Memory-like T Cells during Chronic Infection. Immunity 47, 1129–1141.e5. [DOI] [PubMed] [Google Scholar]
- Matsuzaki G, Vordermeier HM, Hashimoto A, Nomoto K, and Ivanyi J (1999). The role of B cells in the establishment of T cell response in mice infected with an intracellular bacteria, Listeria monocytogenes. Cell. Immunol 194, 178–185. [DOI] [PubMed] [Google Scholar]
- Mayer KD, Mohrs K, Reiley W, Wittmer S, Kohlmeier JE, Pearl JE, Cooper AM, Johnson LL, Woodland DL, and Mohrs M (2008). Cutting edge: T-bet and IL-27R are critical for in vivo IFN-gamma production by CD8 T cells during infection. J. Immunol 180, 693–697. [DOI] [PubMed] [Google Scholar]
- Molnarfi N, Schulze-Topphoff U, Weber MS, Patarroyo JC, Prod’-homme T, Varrin-Doyer M, Shetty A, Linington C, Slavin AJ, Hidalgo J, et al. (2013). MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. J. Exp. Med 210, 2921–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, and Honjo T (2000). Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 102, 553–563. [DOI] [PubMed] [Google Scholar]
- Néel A, Bucchia M, Néel M, Tilly G, Caristan A, Yap M, Rimbert M, Bruneau S, Cadoux M, Agard C, et al. (2019). Dampening of CD8+ T Cell Response by B Cell Depletion Therapy in Antineutrophil Cytoplasmic Antibody-Associated Vasculitis. Arthritis Rheumatol 71, 641–650. [DOI] [PubMed] [Google Scholar]
- Ngo VN, Cornall RJ, and Cyster JG (2001). Splenic T zone development is B cell dependent. J. Exp. Med. 194, 1649–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North ME, Webster ADB, and Farrant J (1998). Primary defect in CD8+ lymphocytes in the antibody deficiency disease (common variable immunodeficiency): abnormalities in intracellular production of interferon-gamma (IFN-gamma) in CD28+ (‘cytotoxic’) and CD28- (‘suppressor’) CD8+ subsets. Clin. Exp. Immunol 111, 70–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obar JJ, Crist SG, Leung EK, and Usherwood EJ (2004). IL-15-independent proliferative renewal of memory CD8+ T cells in latent gammaherpesvirus infection. J. Immunol 173, 2705–2714. [DOI] [PubMed] [Google Scholar]
- Paquin-Proulx D, Santos BAN, Carvalho KI, Toledo-Barros M, Barreto de Oliveira AK, Kokron CM, Kalil J, Moll M, Kallas EG, and Sandberg JK (2013). IVIg immune reconstitution treatment alleviates the state of persistent immune activation and suppressed CD4 T cell counts in CVID. PLoS ONE 8, e75199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennock ND, Gapin L, and Kedl RM (2014). IL-27 is required for shaping the magnitude, affinity distribution, and memory of T cells responding to subunit immunization. Proc. Natl. Acad. Sci. USA 111, 16472–16477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson NC, Donachie AM, and Mowat AMI (2008). Simultaneous presentation and cross-presentation of immune-stimulating complex-associated cognate antigen by antigen-specific B cells. Eur. J. Immunol 38, 1238–1246. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Kalia V, Haining WN, Konieczny BT, Subramaniam S, and Ahmed R (2008). Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J. Exp. Med 205, 625–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider R, Mohebiany AN, Ifergan I, Beauseigle D, Duquette P, Prat A, and Arbour N (2011). B cell-derived IL-15 enhances CD8 T cell cytotoxicity and is increased in multiple sclerosis patients. J. Immunol 187, 4119–4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Whitmire JK, Fan X, Shedlock DJ, Kaech SM, and Ahmed R (2003). A specific role for B cells in the generation of CD8 T cell memory by recombinant Listeria monocytogenes. J. Immunol 170, 1443–1451. [DOI] [PubMed] [Google Scholar]
- Sosinowski T, White JT, Cross EW, Haluszczak C, Marrack P, Gapin L, and Kedl RM (2013). CD8α+ dendritic cell trans presentation of IL-15 to naive CD8+ T cells produces antigen-inexperienced T cells in the periphery with memory phenotype and function. J. Immunol 190, 1936–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thon V, Wolf HM, Sasgary M, Litzman J, Samstag A, Hauber I, Lokaj J, and Eibl MM (1997). Defective integration of activating signals derived from the T cell receptor (TCR) and costimulatory molecules in both CD4+ and CD8+ T lymphocytes of common variable immunodeficiency (CVID) patients. Clin. Exp. Immunol 110, 174–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentine KM, Davini D, Lawrence TJ, Mullins GN, Manansala M, AlKuhlani M, Pinney JM, Davis JK, Beaudin AE, Sindi SS, et al. (2018). CD8 Follicular T Cells Promote B Cell Antibody Class Switch in Autoimmune Disease. J. Immunol 201, 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrens EJ, Wong KA, Gupta A, Khan A, Benedict CA, and Zuniga EI (2018). IL-27 regulates the number, function and cytotoxic program of antiviral CD4 T cells and promotes cytomegalovirus persistence. PLoS ONE 13, e0201249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, Becker TC, Boone D, Kaja MK, Ma A, and Ahmed R (2002). Homeostatic proliferation but not the generation of virus specific memory CD8 T cells is impaired in the absence of IL-15 or IL-15Ralpha. Adv. Exp. Med. Biol 512, 165–175. [DOI] [PubMed] [Google Scholar]
- Wherry EJ, Teichgräber V, Becker TC, Masopust D, Kaech SM, Antia R, von Andrian UH, and Ahmed R (2003). Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat. Immunol 4, 225–234. [DOI] [PubMed] [Google Scholar]
- Whitmire JK, Asano MS, Kaech SM, Sarkar S, Hannum LG, Shlomchik MJ, and Ahmed R (2009). Requirement of B cells for generating CD4+ T cell memory. J. Immunol 182, 1868–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MA, Tyznik AJ, and Bevan MJ (2006). Interleukin-2 signals during priming are required for secondary expansion of CD8+ memory T cells. Nature 441, 890–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong GK, and Huissoon AP (2016). T-cell abnormalities in common variable immunodeficiency: the hidden defect. J. Clin. Pathol 69, 672–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Wang R, Wang J, Wu S, Fernandez M, Rivera C, Cervantes C, Moroney J, Li X-D, Zhang N, et al. (2020). BATF3-dependent induction of IL-27 in B cells bridges the innate and adaptive stages of the antibody response. BioRxiv. [Google Scholar]
- Yang R, Masters AR, Fortner KA, Champagne DP, Yanguas-Casás N, Silberger DJ, Weaver CT, Haynes L, and Rincon M (2016). IL-6 promotes the differentiation of a subset of naive CD8+ T cells into IL-21-producing B helper CD8+ T cells. J. Exp. Med 213, 2281–2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanetti M, Castiglioni P, Rizzi M, Wheeler M, and Gerloni M (2004). B lymphocytes as antigen-presenting cell-based genetic vaccines. Immunol. Rev 199, 264–278. [DOI] [PubMed] [Google Scholar]
- Zhao J, Zhao J, and Perlman S (2010). T cell responses are required for protection from clinical disease and for virus clearance in severe acute respiratory syndrome coronavirus-infected mice. J. Virol 84, 9318–9325. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Microscopy data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.