

Abstract
Diabetic peripheral neuropathy (DPN) is a complication of diabetes whose pathophysiology is linked to altered mitochondrial bioenergetics (mtBE). KU-596 is a small molecule neurotherapeutic that reverses symptoms of DPN, improves sensory neuron mtBE, and decreases the pro-oxidant protein, thioredoxin-interacting protein (Txnip) in a heat shock protein 70 (Hsp70)-dependent manner. However, the mechanism by which KU-596 improves mtBE and the role of Txnip in drug efficacy remains unknown. Mitophagy is a quality-control mechanism that selectively targets damaged mitochondria for degradation. The goal of this study was to determine if KU-596 therapy improved DPN, mtBE, and mitophagy in an Hsp70- and Txnip-dependent manner. Mito-QC (MQC) mice express a mitochondrially targeted mCherry-GFP fusion protein that enables visualizing mitophagy. Diabetic MQC, MQC × Hsp70 knockout (KO), and MQC × Txnip KO mice developed sensory and nerve conduction dysfunctions consistent with the onset of DPN. KU-596 therapy improved these measures, and this was dependent on Hsp70 but not Txnip. In MQC mice, diabetes decreased mtBE and increased mitophagy and KU-596 treatment reversed these effects. In contrast, KU-596 was unable to improve mtBE and decrease mitophagy in MQC × Hsp70 and MQC × Txnip KO mice. These data suggest that Txnip is not necessary for the development of the sensory symptoms and mitochondrial dysfunction induced by diabetes. KU-596 therapy may improve mitochondrial tolerance to diabetic stress to decrease mitophagic clearance in an Hsp70- and Txnip-dependent manner.
Keywords: C-terminal Hsp90 modulator, diabetic peripheral neuropathy, molecular chaperones, neuroprotection, novologues, sensory neuron
Graphical Abstract
INTRODUCTION
Diabetic peripheral neuropathy (DPN) develops in about 50–60% of patients with Type 1 or Type 2 diabetes.1 Although numerous pathologic mechanisms contribute to the onset of DPN, ameliorating mitochondrial dysfunction and the production of reactive oxygen species are emerging as viable approaches for its therapeutic management.2–4
Novologues are proprietary small molecule neurotherapeutics5 whose chemical biology is directed at modulating the activity and expression of molecular chaperones, such as heat shock protein 90 (Hsp90) and Hsp70. Over the last 12 years, we have published robust and rigorous data showing that novologues improve metabolic and clinical indices of DPN.6–11 KU-596 is a clinically advanced novologue (ChemSpider ID: 28665905) that binds to the C-terminal of Hsp90 to improve DPN in an Hsp70-dependent manner.6,12 Moreover, improvement in physiologic measures of DPN by KU-596 correlate with enhancing mitochondrial bioenergetics (mtBE) and decreasing oxidative stress.6,9 Although the efficacy of KU-596 in improving mtBE requires Hsp70, downstream mechanisms remain undefined.
Thioredoxin-interacting protein (Txnip) is a pro-oxidative stress13 and pro-inflammatory14 early response gene that is strongly induced by high glucose and whose expression is increased in diabetic sensory neurons.15,16,6 Importantly, transcriptomic analysis found that Txnip was among the genes with the greatest fold increase in sensory neurons obtained from diabetic WT and Hsp70 knockout (KO) mice.6 Though KU-596 therapy decreased Txnip expression in neurons from diabetic WT but not Hsp70 KO mice, whether Txnip contributed to the ability of KU-596 to improve mtBE and the sensory declines in DPN was unclear.
Txnip can modulate the cellular redox state by inhibiting mitochondrial thioredoxin-2 to increase mitochondrial oxidative stress and induce depolarization.17,18 Mitochondrial depolarization and fragmentation are key signals that promote mitophagy,19,20 a quality-control mechanism that selectively targets and eliminates damaged mitochondria.21,22 Though mitophagy serves to maintain the health and function of the mitochondrial network, the effects of modulating Txnip and mitophagy in the development of DPN remain unclear.
We examined the hypothesis that KU-596 improves mtBE and DPN by enhancing the rates of neuronal mitophagy in diabetic mice in a Txnip- and Hsp70-dependent manner. To study mitophagy in vivo, we used transgenic Mito-QC (MQC) mice, which have a global, constitutive knock-in of mCherry-GFP-FIS1101–152 that cause mitochondria to fluoresce both red and green under homeostatic conditions.23 During mitophagy, damaged mitochondria are eventually degraded in acidic mitolysosomes, which causes GFP to be quenched, resulting in only red fluorescence from mCherry. These red puncta within the cell are indicators of mitophagy and were assessed within the sensory neurons of lumbar DRG. Crossing the MQC mice with Hsp70 and Txnip KO mice allowed us to assess the role of these proteins in contributing to mitophagy and drug efficacy in DPN.
Diabetes induced a progressive sensory neuropathy, impaired mtBE, and increased mitophagy levels in MQC, MQC × Hsp70 KO, and MQC × Txnip KO mice. Daily in vivo treatment of KU-596 improved the sensory neuropathy, neuronal mtBE, and decreased mitophagy, but this efficacy was lost in MQC × Hsp70 KO mice. Though KU-596 therapy improved psychosensory and nerve conduction deficits in MQC × Txnip KO mice, the drug did not improve mtBE nor decrease mitophagy. Together, these results provide new insight into the role of Txnip and mitophagy in DPN and suggest that Txnip is downstream of Hsp70 in the mechanism of action of KU-596 to improve mtBE and decrease mitophagy in diabetic neurons.
RESULTS
KU-596 Therapy Improves DPN in MQC but not MQC × Hsp70 KO Mice.
We have shown that 8 weeks of KU-596 therapy reversed a pre-existing mechanical and thermal hypoalgesia, increased nerve conduction velocity, and improved deficits in mtBE that develop by 16 weeks of diabetes in C57Bl/6 mice.6,9 To assess the effect of diabetes and KU-596 treatment on mitophagy, male and female 6–8 week-old MQC mice were rendered diabetic with two intraperitoneal injections of 100 mg/kg streptozotocin (STZ) given on consecutive days. After 8 weeks of diabetes, mice were administered 1 mg/kg KU-596 or Captisol (drug vehicle) by daily oral gavage for an additional 8 weeks. Quantitative sensory testing was performed to monitor the progression of DPN, and mtBE and mitophagy were assessed in DRG sensory neurons after 16 weeks of diabetes. This intervention design provides meaningful therapeutic evidence of drug efficacy in ameliorating a pre-existing neuropathy, and a prevention trial (drug administration prior to developing DPN) was not performed.
Table 1 shows the weights and fasting blood glucose (FBG) of the MQC mice. As expected, the FBG of the diabetic MQC mice was significantly higher compared to that of nondiabetic mice. Curiously, the final FBG of both the nondiabetic and diabetic MQC mice treated with KU-596 was significantly decreased as compared to their respective controls. This was rather unexpected as previous work on the effect of KU-596 in C57Bl/6 mice, the genetic background of the MQC mice, had not yielded any effects of the drug on blood glucose levels in either control or diabetic mice. Moreover, Swiss Webster mice are an out-bred strain with a much broader genetic variability than C57Bl/6 mice, and though KU-596 improved DPN in this strain, this was also independent of any drug-induced change in FBG.7 The reason or mechanism underlying this discrepancy is unclear.
Table 1.
Effect of STZ and KU-596 on Weight, FBG, and HbA1c in MQC and MQC × Hsp70 KO Micea
| genotype | treatment | initial weight (g) | final weight (g) | final FBG (mg/dL) | HbA1c (%) |
|---|---|---|---|---|---|
| MQC | nondiabetic (7) | 19.6 ± 1.1 | 27.0 ± 1.7 | 154 ± 7 | nd |
| nondiabetic + KU-596 (8) | 19.0 ± 0.7 | 26.3 ± 1.4 | 142 ± 10^ | nd | |
| diabetic (15) | 19.6 ± 0.7 | 25.2 ± 1.0 | 261 ± 9* | nd | |
| diabetic + KU-596 (13) | 19.7 ± 0.6 | 24.9 ± 1.0 | 179 ± 9# | nd | |
| MQC × Hsp70 KO | nondiabetic (9) | 20.3 ± 1.3 | 26.6 ± 1.8 | 135 ± 5 | 4.4 ± 0.2 |
| nondiabetic + KU-596 (5) | 20.0 ± 1.3 | 25.4 ± 2.5 | 134 ± 11 | nd | |
| diabetic (10) | 21.1 ± 1.3 | 24.5 ± 1.2 | 311 ± 32* | 8.1 ± 1.4* | |
| diabetic + KU-596 (11) | 20.4 ± 1.4 | 24.8 ± 1.2 | 345 ± 38* | 7.2 ± 1.1* |
Animal number is indicated in parentheses.
, p < 0.05 vs nondiabetic,
, p < 0.05 versus nondiabetic,
, p < 0.05 versus diabetic.
nd, not determined.
Diabetic MQC mice developed a mechanical and thermal hypoalgesia by 8 weeks of diabetes (Figure 1A,B), and initiating therapy with KU-596 rapidly improved these sensory deficits. Drug treatment did not alter the paw withdrawal measurements in nondiabetic mice. Diabetic MQC mice also showed significantly slower motor nerve conduction velocity (MNCV) compared to nondiabetic control mice, and this was effectively improved by drug therapy (Figure 1C). These data suggest that KU-596 therapy can ameliorate the sensory hypoalgesia and electrophysiologic damage induced by diabetes, similar to that reported in diabetic C57Bl/6 mice.6
Figure 1.
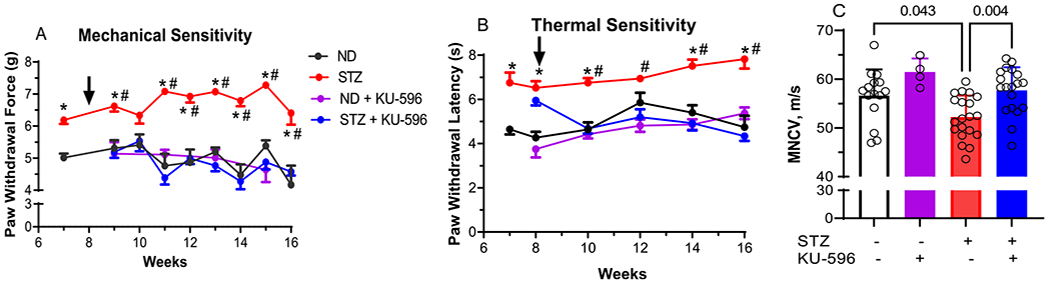
KU-596 treatment improves sensory function and MNCV in diabetic MQC mice. (A) Mechanical and (B) thermal sensitivity were assessed at the indicated weeks, and the arrow indicates the initiation of daily drug therapy. ND, nondiabetic. Results are mean ± SEM, and number of animals per group is shown in (C). *, p < 0.05 versus time-matched ND mice. #, p < 0.05 versus time-matched STZ + KU-596. (C) MNCV, symbols indicate responses from individual animals, and the results are mean ± SD.
As we have previously shown that many of the in vivo effects of KU-596 on improving DPN are Hsp70-dependent,6,9 MQC × Hsp70 KO mice were generated and the absence of Hsp70 expression was verified by genotyping and immunoblot analysis (Figure S1A,B). MQC × Hsp70 KO mice were rendered diabetic, and FBG levels and HbA1c at 16 weeks were consistent with prolonged diabetes that was unaffected by KU-596 therapy (Table 1). MQC × Hsp70 KO mice also developed a significant sensory hypoalgesia (Figure 2A,B) and deficits in MNCV (Figure 2C), but these were unaffected by 8 weeks of novologue therapy.
Figure 2.
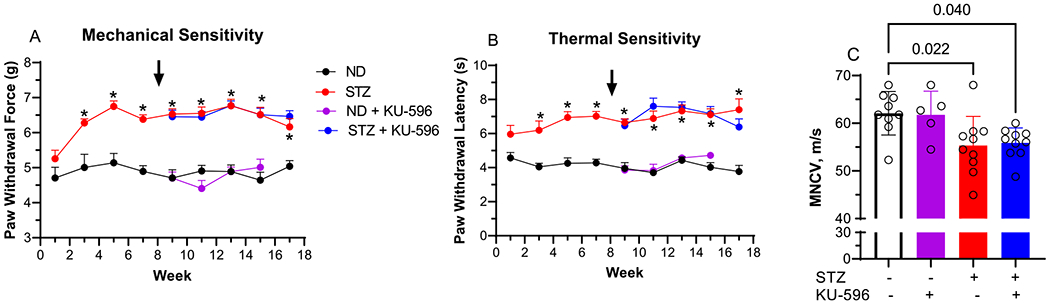
KU-596 treatment does not improve sensory function and MNCV in diabetic MQC × Hsp70 KO mice. (A) Mechanical and (B) thermal sensitivity were assessed at the indicated weeks, and the arrow indicates the initiation of daily drug therapy. ND, nondiabetic. Results are mean ± SEM, and number of animals per group is shown in (C). *, p < 0.05 versus time-matched ND mice. (C) MNCV, symbols indicate response from individual animals, and the results are mean ± SD.
KU-596 Therapy Improves Sensory Neuron mtBE and Decreases Mitophagy in an Hsp70-Dependent Manner.
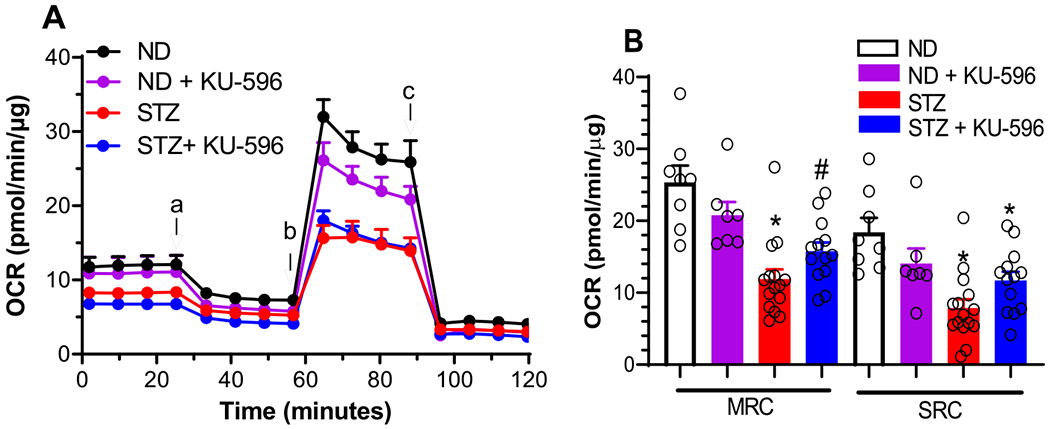
To determine the effect of diabetes and KU-596 on mtBE, mitochondrial respiration was assessed ex vivo using sensory neurons isolated from the lumbar DRG of nondiabetic and diabetic MQC mice treated with vehicle or KU-596 for 8 weeks. Nondiabetic and diabetic MQC × Hsp70 KO mice were treated similarly to determine the role of Hsp70 in drug efficacy.
Cellular respiration was determined using a Seahorse analyzer, which measures the total oxygen consumption rate (OCR) in intact neurons. The contribution of mitochondrial and nonmitochondrial components to overall respiration was assessed by the addition of respiratory chain poisons. The first four rate measures in Figure 3A reflect basal OCR, and no significant difference was observed between the groups of MQC mice. The addition of the ATP synthase inhibitor, oligomycin, decreased respiration, and the magnitude of this effect reflects the contribution of ATP-coupled respiration to basal respiration. No significant difference emerged, indicating that the efficiency of oxygen consumption to ATP production is similar between the cells. This ratio is called the coupling efficiency, and its value was not significantly affected by diabetes or drug treatment (data not shown).
Figure 3.
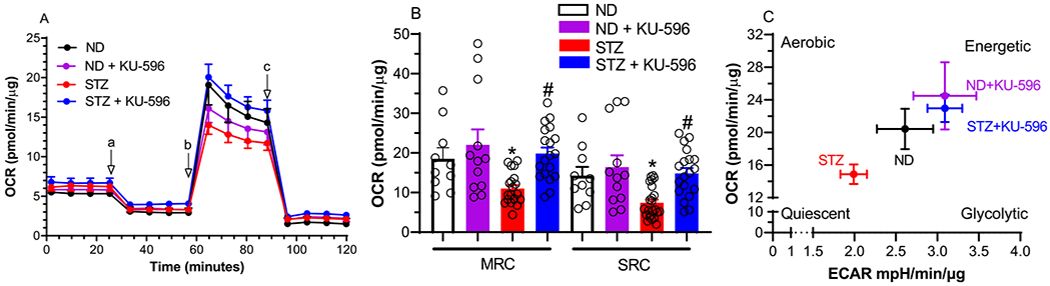
KU-596 improves mtBE in ex vivo MQC diabetic neurons. (A) Basal OCR was measured in sensory neurons prior to the addition of oligomycin (a), FCCP (b), and rotenone/antimycin A (c). ND, nondiabetic. (B) Effect of treatments on MRC and SRC. *, p = 0.047 and 0.039 versus ND-MRC and ND-SRC, respectively; #, p < 0.001 and 0.004 versus STZ-MRC and STZ-SRC, respectively. Symbols are responses from pooled neurons in each well that were isolated from lumbar DRG of 3–6 mice per group. (C) Drug treatment increased energy metabolism in diabetic neurons.
Since the rate of maximal electron transfer through the electron transport chain (ETC) is limited by the proton and electrochemical gradient across the inner mitochondrial membrane; the disruption of this gradient by the protonophore FCCP promotes maximal respiratory capacity (MRC). A decrease in MRC supports that diabetes has promoted mitochondrial dysfunction.24 Diabetic sensory neurons from the MQC mice showed a significant decline in MRC that was improved by KU-596 therapy (Figure 3A,B)
Spare respiratory capacity (SRC) reflects the bioenergetic reserve that is available for cells to respond to additional energy demands and is the difference between MRC and basal respiration.24 SRC was also significantly impaired in diabetic neurons, and this was reversed by KU-596 therapy (Figure 3A,B). The increase in MRC and SRC following drug therapy suggests that modulating molecular chaperones with KU-596 is improving the overall energetic profile of the neurons. This was visualized by plotting the OCR versus the rate of glycolysis, which is measured indirectly by assessing the extracellular acidification rate (ECAR) due to the production of lactate from pyruvate. The bioenergetic profile has four quadrants that reflect the ratio of aerobic to glycolytic metabolism and energy availability. As shown in Figure 3C, control neurons had an energetic profile of relatively equal rates of oxidative phosphorylation versus glycolysis. Diabetes decreased aerobic metabolism pushing the cells toward a more quiescent energy profile. However, 8 weeks of novologue treatment improved the overall energetic profile, indicating that KU-596 therapy improved mtBE in the diabetic MQC sensory neurons.
Despite the MQC × Hsp70 KO mice being more diabetic than their MQC counterparts (Table 1), diabetes blunted but did not significantly decrease MRC and SRC versus the nondiabetic neurons (Figure 4), Interestingly, KU-596 significantly increased MRC and SRC in neurons from nondiabetic MQC × Hsp70 KO mice. In contrast, the drug did not improve MRC and SRC in neurons from diabetic MQC × Hsp70 KO mice. We have previously observed that nondiabetic Hsp70 KO neurons treated with KU-596 had a higher MRC and SRC than vehicle-treated nondiabetic neurons.9 Thus, the drug may have some Hsp70-independent effects to improve mtBE in nondiabetic neurons, but improving mtBE in diabetic neurons requires Hsp70.9
Figure 4.
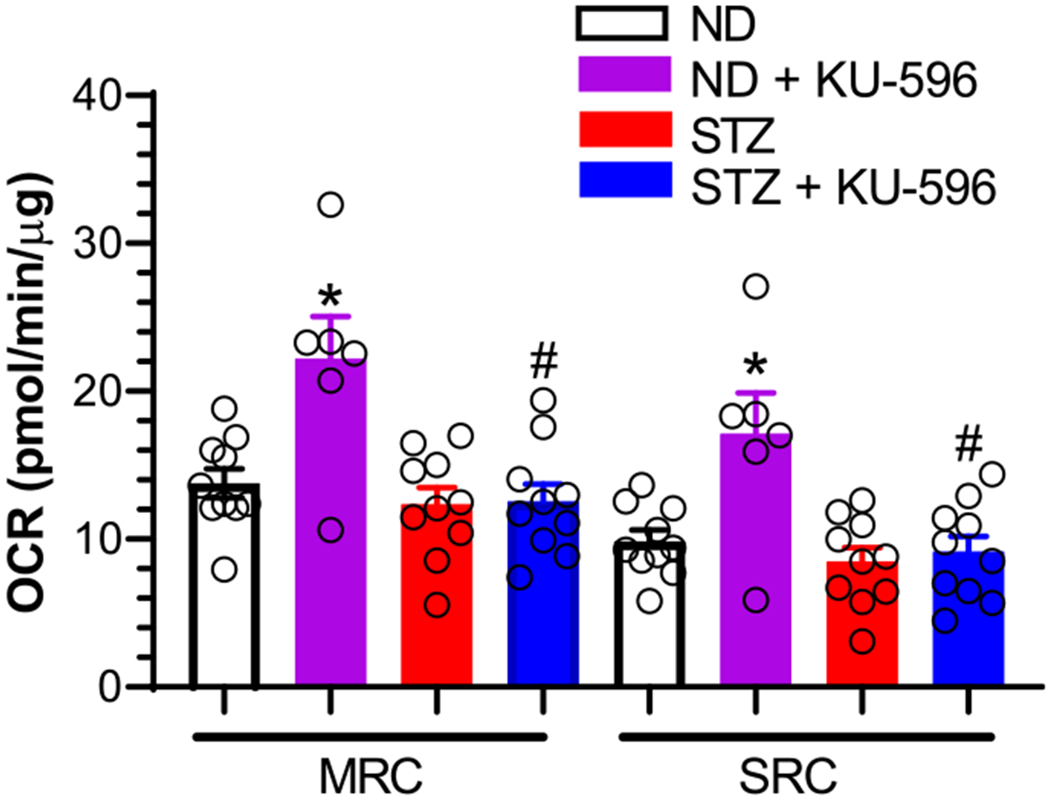
Hsp70 is necessary for KU-596 to improve mtBE in ex vivo diabetic neurons. Effect of treatments on MRC and SRC. ND, nondiabetic. *, p < 0.003 and 0.004 versus ND-MRC and ND-SRC, respectively; #, p < 0.001 and 0.002 versus MRC and SRC from ND + KU-596, respectively. Symbols are responses from pooled neurons in each well that were isolated from lumbar DRG of 3–6 mice per group.
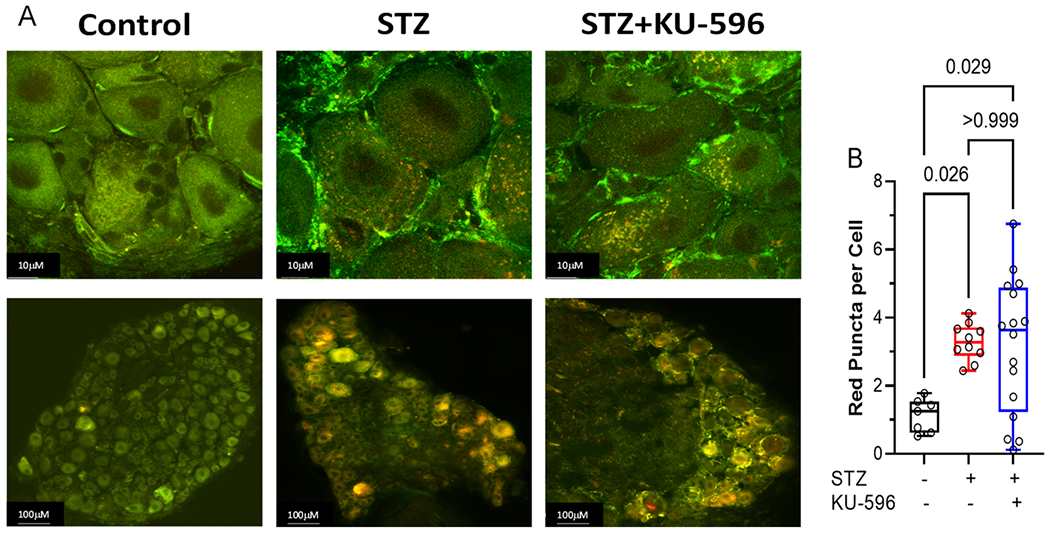
To determine the role of mitophagy in diabetic sensory neurons, L4–L6 lumbar DRGs were isolated from diabetic MQC mice and mitophagy, visualized, and quantified as described in the Methods. Nondiabetic sensory neurons had a low level of basal mitophagy, and this was significantly increased after 16 weeks of diabetes as shown by an increase in the number of red puncta per cell (Figure 5A,B). Mitophagy was similarly increased after 16 weeks of diabetes in the MQC × Hsp70 KO mice (Figure 5C,D). This data suggests that elevated mitophagy may be an adaptive response to clear damaged mitochondria and that increased mitophagy in the diabetic sensory neurons occurs in an Hsp70-independent manner.
Figure 5.
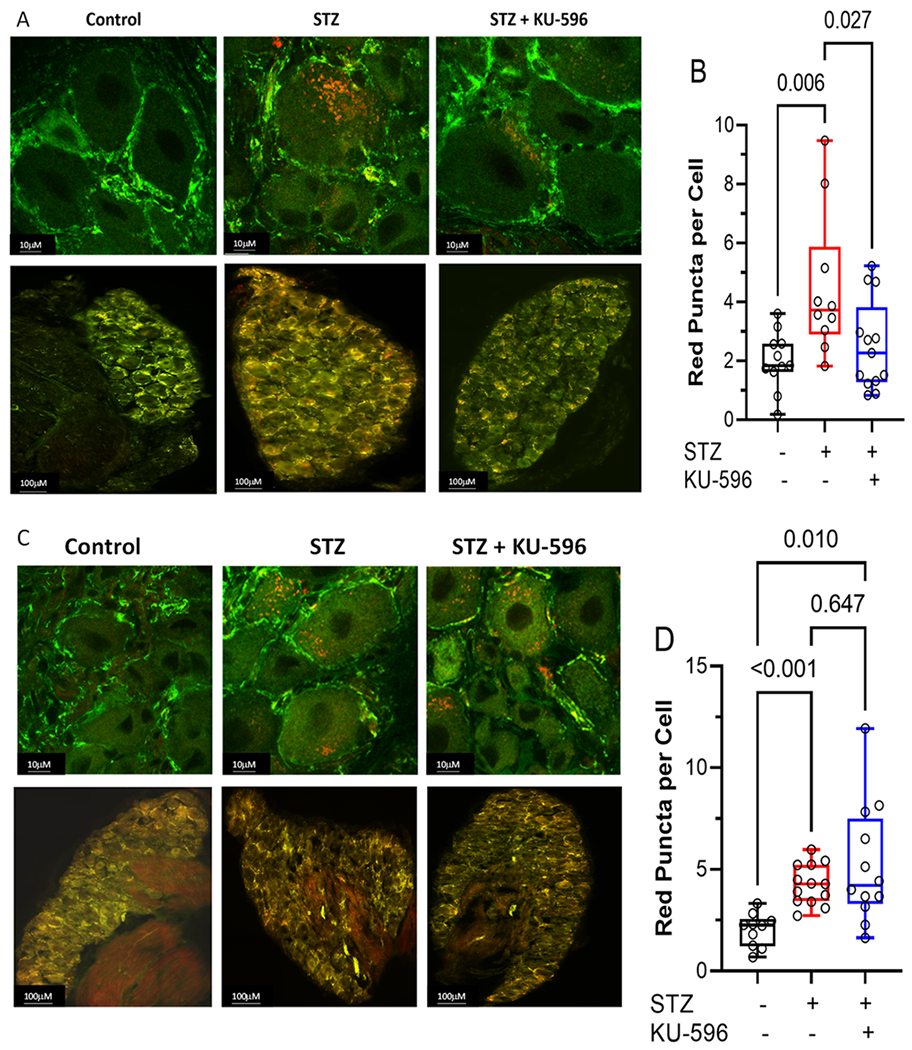
KU-596 decreased mitophagy in (A and B) MQC but not (C and D) MQC × Hsp70 diabetic neurons. (A and C) Top panels show representative images of sensory neurons and bottom panels show the entire DRG. (B and D) Symbols represent average puncta per cell from one animal. The number of cells counted per ganglia ranged from 50 to 100. Box, 25th, median, and 75th percentiles; whiskers, minimum, and maximum.
Though KU-596 improved mtBE, whether this was related to an increase or decrease in mitophagy in the diabetic neurons was unclear. Treating diabetic MQC mice with KU-596 for 8 weeks promoted a significant decline in the overall number of red puncta per cell. However, the drug was unable to decrease the level of mitophagy in MQC × Hsp70 KO mice. These data contrasted with our initial hypothesis that KU-596 may improve mtBE by increasing the mitophagic removal of damaged organelles, in an Hsp70-dependent manner. However, the decrease in mitophagy is clearly Hsp70-dependent.
Txnip is not Necessary to Develop DPN.
We have previously reported that KU-596 therapy dramatically decreased Txnip mRNA expression in diabetic sensory neurons in an Hsp70-dependent manner.6 However, it was unclear whether Txnip contributed to the drug-induced improvements in mtBE and sensory deficits. To examine the role of Txnip in the onset of DPN and drug efficacy, we generated MQC × Txnip KO mice. The absence of Txnip expression was validated by genotyping (Figure S2A) and immunoblot analysis of tissue and MEFs exposed to hyperglycemia (Figure S2B,C).
Txnip deficiency has been reported to protect against diabetes, and the MQC × Txnip KO mice often required up to four injections with STZ to be rendered diabetic. However, after 16 weeks of diabetes, FBG and HbA1c levels were consistent with those of the other MQC strains, indicating a prolonged diabetes (Table 2). Indeed, diabetic MQC × Txnip KO mice developed a significant mechanical and thermal hypoalgesia as well as MNCV deficits that were similar to the other MQC strains (Figure 6A–C). Thus, it does not seem that Txnip is necessary to develop these measure measures of DPN despite its elevated expression in diabetic DRG.6,15,26
Table 2.
Effect of STZ and KU-596 on Weight, FBG, and HbA1c in MQC × Txnip KO Micea
| treatment | final weight (g) | final FBG (mg/dL) | HbA1c (%) |
|---|---|---|---|
| nondiabetic + vehicle (11) | 29.3 ± 1.1 | 124 ± 8 | 4.5 ± 0.3 |
| nondiabetic + KU-596 (10) | 29.1 ± 1.4 | 130 ± 5 | 4.2 ± 0.1 |
| diabetic + vehicle (16) | 26.7 ± 0.8 | 284 ± 24* | 7.3 ± 2.1 |
| diabetic + KU-596 (17) | 26.8 ± 0.8 | 242 ± 33* | 8.3 ± 0.9* |
Animal number is indicated in parentheses.
, p < 0.05 vs nondiabetic.
Figure 6.
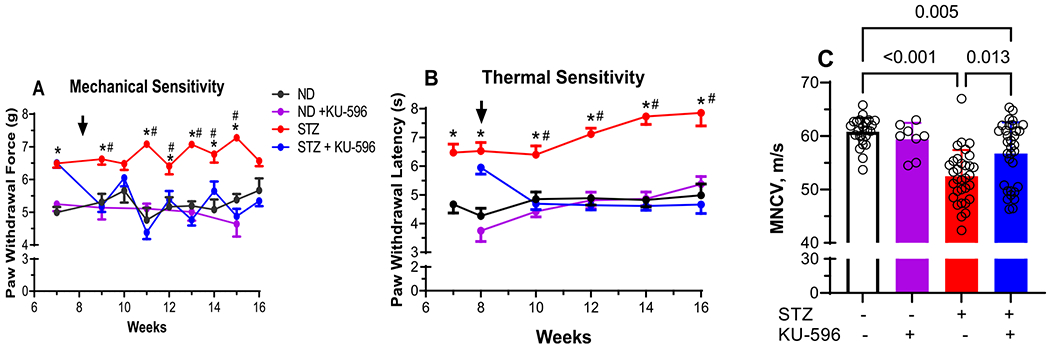
KU-596 improves sensory function and MNCV in diabetic MQC × Txnip KO mice. (A) Mechanical and (B) thermal sensitivity were assessed at the indicated weeks, and the arrow indicates the initiation of daily drug therapy. Results are mean ± SEM, and number of animals per group is shown in (C). ND, nondiabetic. *, p < 0.05 versus time-matched ND mice; #, p < 0.05 versus time-matched STZ + KU-596. (C) MNCV, symbols indicate responses from individual animals, and the results are mean ± SD.
After 8 weeks of diabetes, the mice were treated daily with 1 mg/kg KU-596 for an additional 8 weeks. Compared to the untreated diabetic MQC × Txnip KO mice, KU-596 attenuated the sensory hypoalgesia and significantly improved the MNCV deficit. However, in contrast to the MQC mice, MNCV remained significantly lower than nondiabetic mice. These data indicate that KU-596 therapy can ameliorate the sensory hypoalgesia and partially recover the electrophysiologic damage induced by diabetes via largely Txnip-independent mechanisms.
Txnip is not Necessary to Develop Deficits in mtBE and to Increase Mitophagy in Diabetic Sensory Neurons.
Though Txnip has been linked to affecting mitochondrial function, its role in diabetic sensory neurons remains poorly defined. To determine if the deletion of Txnip affected mtBE and the response to KU-596, OCR was assessed in sensory neurons obtained from nondiabetic and diabetic MQC × Txnip KO mice with and without 8 weeks of KU-596 treatment. Compared to nondiabetic neurons, neurons isolated from diabetic MQC × Txnip KO mice showed a significantly diminished MRC and SRC (Figure 7A,B) and an increase in mitophagy (Figure 8A,B). These results are similar to the effect of 16 weeks of diabetes in the MQC mice and indicate that Txnip is not required for the diabetes-induced deficit in mtBE or increase in mitophagy. In contrast, 8 weeks of KU-596 therapy were unable to improve MRC and SRC and decrease mitophagy in the MQC × Txnip KO neurons.
Figure 7.
KU-596 does not improves mtBE in ex vivo MQC × Txnip KO diabetic neurons. (A) Basal OCR was measured in sensory neurons prior to the addition of oligomycin (a), FCCP (b), and rotenone/antimycin A (c). ND, nondiabetic. (B) Effect of treatments on MRC and SRC. *, p < 0.001 vs ND-MRC or ND-SRC; #, p = 0.002 versus ND-MRC. Symbols are responses from pooled neurons in each well that were isolated from the lumbar DRG of 3–6 mice per group.
Figure 8.
KU-596 does not decrease mitophagy in MQC × Txnip KO Mice. (A) Top panels show representative images of sensory neurons, and bottom panels show entire DRG. (B) Symbols represent average puncta per cell from one animal. The number of cells counted per ganglia ranged from 50 to 100. Box, 25th, median, and 75th percentiles; whiskers, minimum, and maximum.
DISCUSSION
Mitophagy is emerging as a major mechanism involved in the development of diabetic nephropathy and cardiomyopathy, as well as neurodegenerative diseases.27–30 Using MQC mice to visualize and quantify mitophagy, we provide novel evidence that mitophagy is increased in diabetic sensory neurons. Additionally, though prior work has shown that Txnip expression is increased in diabetic neurons; its contribution to the onset of DPN has not been assessed. Our use of the MQC × Txnip KO mice also provides novel support that Txnip is not necessary for the development of a sensory hypoalgesia, a decline in MNCV, decreased mtBE, nor the increase in mitophagy.
Diabetic MQC and MQC × Hsp70 KO mice exhibited a progressive onset of a mechanical/thermal hypoalgesia and a diminished MNCV, indicative of the presence of a distal peripheral neuropathy. Thus, the expression of the mitochondrially targeted MQC transgene or loss of Hsp70 did not interfere with the development of these indices of DPN. On the contrary, we were unable to detect a significant decline in sensory NCV (SNCV) in these mice (data not shown). Since it is hard to rationalize that the global expression of the MQC transgene would preferentially impact SNCV but not MNCV, it is unclear why SNCV was unaffected in these mice. Treating the diabetic MQC mice with KU-596 improved the psychosensory and MNCV deficits, and this is consistent with prior results that the drug can improve DPN in models of Type 16,8 and Type 2 diabetes7 without improving FBG. However, given the unexpected decreased in FBG in drug-treated nondiabetic and diabetic MQC mice, it is possible that the improvements in hypoalgesia and MNCV were partially due to a reduction in blood glucose. When the MQC × Hsp70 KO mice were treated with KU-596, neither the measures of DPN nor blood glucose were affected. This supports that the observed improvements in hypoalgesia, MNCV, and FBG in the MQC mice by KU-596 were Hsp70-dependent.
For the bioenergetic analyses, lumbar sensory neurons were isolated from diabetic animals as they provide the cell bodies for the axons that are the primary targets in diabetic neuropathy.2 Using the ex vivo cell culture model of nondiabetic and diabetic sensory neurons allows for a more accurate reflection of the in vivo impact of chronic diabetes and KU-596 therapy on mtBE versus treating neurons in vitro with an acute exposure to hyperglycemia and drug.31,32 Sensory neurons from diabetic MQC mice showed decreased MRC and SRC, which is consistent with a decline in overall mitochondrial bioenergetic capacity. As observed previously, mitochondrial OCR was improved by KU-596 in an Hsp70-dependent manner, but the mechanism by which Hsp70 improves mtBE is unclear.
An increase in oxidative damage is thought to contribute to mitochondrial dysfunction in diabetic neurons, and using electron paramagnetic resonance spectroscopy, we observed an increase in mitochondrial superoxide production in diabetic neurons that was diminished with KU-596 treatment. However, the ability of KU-596 to improve mtBE was not solely dependent on decreasing mitochondrial superoxide.9 Oxidative stress and mitophagy are interrelated processes implicated in a wide range of pathological conditions involving high energy demanding tissues. However, the effect of diabetes on mitophagy in sensory neurons is poorly characterized.
It is well-established that prolonged diabetes can lead to the depolarization of mitochondria,2 which is a classic signal to promote mitophagy.33 Though mitophagy was decreased in sciatic nerve of eight week diabetic rats,34 16 weeks of diabetes led to an increase in mitophagy as indicated by the rise in the number of red puncta in the sensory neurons, and this correlated with an decrease in mtBE. One interpretation of this data is that mitophagy is not impaired at this time in the diabetic neurons, and the increase reflects an active and adaptive response to remove damaged mitochondria. However, this was clearly insufficient to prevent the decline in mtBE that was seen in the diabetic MQC mice. On the contrary, mitophagy is a very dynamic process, and since our assessment occurred at only a single time point, it is possible that degradation in mitolysosomes is impaired, leading to an accumulation of the red puncta. It would not be surprising if mitophagy may eventually decrease since chronic diabetes may exhaust the capacity of this autophagic removal and lead to an increase in mitochondrial fragmentation and more severe deficits in mtBE.1
Since a primary goal of the current work was to assess whether KU-596 may improve mtBE by affecting mitophagy, it was necessary to identify a time point when mitophagic pathways were active, and our preliminary data suggested that mitophagy was maximal by 16 weeks of diabetes. Therefore, our intervention design of initiating KU-596 therapy after 8 weeks of diabetes allowed us to assess if the drug may impact mitophagy.
KU-596 therapy improved mtBE in neurons derived from the diabetic MQC mice, and this correlated with a decrease in the level of red puncta in the sensory neurons. It is possible that KU-596 may help neurons tolerate diabetic stress by decreasing oxidative damage to improve overall mitochondrial integrity,6,9 thereby decreasing the need to clear organelles that have undergone damage by chronic hyperglycemia. As mentioned above, however, our assessment at a single time point does not rule out the possibility that the drug may be increasing the rate of mitophagic flux, which would manifest as a decrease in mitophagy when measured at a single time point. Nonetheless, the effects of KU-596 on mtBE and mitophagy were Hsp70-dependent, and the quantification of the red puncta indicated 2.7- and 2.1-fold increases in sensory neuron mitophagy after 16 weeks of diabetes in the MQC and MQC × Hsp70 KO mice, respectively. These data suggest that the lack of Hsp70 has little impact on the extent of sensory neuron mitophagy despite evidence in muscle that Hsp70 is essential for the interaction of parkin, a cytosolic E3-ubiquitin ligase,35 to clear damaged mitochondria.36 It is possible that the translocation of parkin to the mitochondrial membrane is not as dependent on Hsp70 in DRG compared to muscle or that parkin-independent pathways may be more involved in regulating mitophagy in sensory neurons versus muscle.
The homeostasis of glucose regulation, cellular redox, and mitochondria function have been linked to Txnip, and its expression is increased in diabetic animals.15,37 Since it is unclear whether Txnip affects mtBE, mitophagy, and the development of DPN, the MQC × Txnip KO mice allowed us to assess the role of this protein in the onset of DPN and drug efficacy. Txnip deficiency has been reported to protect against diabetes and compared to the MQC and MQC × Hsp70 KO mouse lines; rendering the MQC × Txnip KO mice diabetic sometimes required 3–4 injections of STZ over a two week period. However, once the mice were rendered diabetic, the kinetics and magnitude of the mechanical and thermal hypoalgesia that evolved was comparable with those of the diabetic MQC mice. The decline in MNCV and mtBE and the increase in mitophagy was also notably similar to the diabetic MQC mice. Thus, despite the reported increase in Txnip expression in diabetic sensory neurons and its role in increasing oxidative stress,15,37 it does not seem necessary to promote these physiologic and biochemical indices of DPN. Surprisingly, the improvement in sensory function with KU-596 therapy was Txnip-independent, but the improvement in mtBE and reduction in mitophagy was Txnip-dependent.
Although Txnip gene expression was significantly upregulated in STZ-induced diabetic rats, this was unaffected by decreasing oxidative stress with lipoic acid or a p38 MAP kinase inhibitor. However, insulin treatment and the partial correction of blood glucose did prevent the diabetes-induced increase in Txnip expression.15 These data suggest that Txnip expression does not require oxidative stress to be induced in diabetic tissue, but its increase may still promote oxidative stress.18 Since we did not measure markers of oxidative stress, it is unclear if the onset of DPN in the MQC × Txnip KO mice was associated with changes in superoxide levels as we previously reported.9 KU-596 therapy can reduce oxidative stress, and this was associated with an improvement in sensory function and decreased Txnip expression.6 Though KU-596 did improve MNCV in the diabetic MQC × Txnip KO mice, it remained significantly below that of nondiabetic control mice. Thus, it is possible that a drug-induced decrease in oxidative stress may contribute to the improvement in MNCV and occur in a Txnip-independent manner. This data could further imply that either a longer duration of KU-596 treatment or a higher concentration may further improve MNCV in diabetic MQC × Txnip KO mice.
The absence of Txnip did not prevent diabetic stress from causing mitochondrial dysfunction since mtBE were significantly decreased in diabetic MQC × Txnip KO mice. Txnip deficiency in heart caused a repression of mitochondrial respiration and a switch toward enhanced anaerobic glycolysis.38,39 Since we did not observe a significant difference between OCR in nondiabetic MQC versus MQC × Txnip KO mice or an increase in glycolysis, the absence of Txnip is not repressing mitochondrial respiration and switching metabolism toward glycolysis in the sensory neurons. Other studies have also reported that Txnip deficiency causes defects in mitochondrial metabolism.38,39 The metabolic profiling of Txnip null mice has shown that oxidative insufficiencies were due to deficits in enzymes required for catabolism of amino acids, ketones, lactate, and enzymes of the TCA cycle.39 This suggests that the decrease in mtBE may be due to hyperglycemic stress damaging the ETC but also a substrate deficit due to impaired TCA cycle functioning resulting from Txnip deficiency. Though the deletion of Txnip causes defects in mitochondrial metabolism, it may play a tissue specific role in determining intracellular bioenergetic flux.38,39
The lack of improvement in MRC and SRC with KU-596 therapy implies that ameliorating mtBE deficits in diabetic neurons is Txnip-dependent. Since KU-596 did not down-regulate Txnip expression in diabetic Hsp70 KO mice,6 a drug-induced decrease in Txnip seems to be downstream of Hsp70 induction and is critical to improving mtBE.
Diabetic MQC × Txnip KO mice also exhibited elevated levels of mitophagy, whereas the inhibition of Txnip reduced mitophagy in diabetic kidney proximal tubules.20,40 This may be due to observed differences in the high level of basal mitophagy in proximal tubule epithelia relative to the low basal level observed in sensory neurons.23,41 Although mitophagy increased independent of the presence of Txnip, KU-596 required Txnip to reduce mitophagy. As discussed above, a drug-induced decrease in Txnip seems to be downstream of Hsp70 induction and is critical to improving mitophagy as well as mtBE. However, the mechanism by which KU-596 decreases Txnip expression is unclear. KU-596 is thought to work by disrupting the association of heat shock factor 1 with Hsp90 to activate a heat shock response and increase chaperone expression. Paradoxically, overexpressing heat shock factor 1 upregulates Txnip expression and could be prevented by deleting heat shock elements within the Txnip gene promoter.16 Since Txnip can interact with Hsp90 and Hsp70,42 an increase in Hsp70 could target Txnip to increase its degradation and counterbalance its pro-oxidant effect.43
CONCLUSIONS
In summary, diabetic MQC, MQC × Hsp70 KO, and MQC × Txnip KO mice developed sensory and electrophysiologic deficits consistent with the onset of DPN. Eight weeks of KU-596 therapy improved these deficits in an Hsp70-dependent manner. In contrast, the ability of KU-596 to improve sensory and electrophysiologic function was largely independent of Txnip.
Sixteen weeks of diabetes was sufficient to decrease mitochondrial bioenergetics and increase mitophagy in sensory neurons, and this was ameliorated by 8 weeks of novologue therapy in an Hsp70- and Txnip-dependent manner. Thus, Txnip would seem to be a critical downstream target for the Hsp70-dependent improvement of mitochondrial function and mitophagy by KU-596. However, the ability of KU-596 to improve sensory function in diabetic MQC × Txnip KO mice suggests that the recovery of sensory deficits in DPN may not be tightly linked to improving neuronal mtBE and decreasing mitophagy.
METHODS
Animals.
Mito-QC (MQC) mice globally express a constitutive knock-in of mCherry-GFP-FIS1101–152 in the mouse Rosa26 locus on a C57BL/6 background mice and were a gift from Dr. Ian Ganley (University of Dundee).23 Homozygous male and female MQC mice were used in this study. Hsp70 KO mice on a C57Bl/6 background were initially obtained from the Mutant Mouse Resource and Research Center, and MQC × Hsp70 KO mice were generated by interbreeding the two strains. The verification of the genetic background was performed by PCR-based genotyping of genomic DNA obtained from an ear punch. The primer sets and PCR conditions for the MQC transgene23 and Hsp70 deletion44 were as previously described.
Txnip KO mice were generously provided by Dr. Richard Lee (Harvard University), and MQC × Txnip KO mice were generated by interbreeding the two strains. The PCR-based genotyping of genomic DNA for the Txnip deletion used 5′-CTTCACCCCCCTAGAGTGAT-3′ (WT forward, 128 bp amplicon), 5′-TTTCGTTTGGGTTTTCAAGC-3′ (KO forward, 240 bp amplicon), and 5′-CCCAGAGCACTTTCTTGGAC-3′ (common reverse) as described.45 Experimental animals were maintained on a 12 h light/dark cycle with ad libitum access to water and 7022 NIH-07 rodent chow (5.2% fat). All animal procedures were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee and in compliance with standards and regulations for the care and use of laboratory rodents set by the National Institutes of Health.
Induction of Diabetes and Randomization.
Following a 6 h fast, male and female 6–8 week old mice were rendered diabetic with an intraperitoneal (IP) injection of 100 mg/kg streptozotocin (STZ) (Enzo Life Science, #ALX-380-010) freshly prepared in 50 mM sodium citrate, pH 4.5, and given on two consecutive days. Control mice received only the vehicle. One week after the second injection, mice were fasted for 6 h, blood was obtained from the tail, and mice with fasting blood glucose (FBG) ≥ 250 mg/dL were deemed diabetic. If necessary, mice were given a second STZ regimen and FBG was assessed 1 week later. Animals not rendered diabetic after four STZ injections were removed from the study. Less than 5% of STZ-treated mice from all the genotypes required exclusion. No diabetic mice included in the study required veterinary evaluation and recommendation for euthanization due to excessive dehydration or ketoacidosis over the 16 week study.
Prior to all studies, equal numbers of male and female mice were randomized to each of the four treatment groups on the basis of their ear tag number. After 8 weeks, nondiabetic mice were then divided in their pre-assigned group and given drug vehicle or KU-596 (see below). Similarly, after 8 weeks of diabetes, STZ-treated mice were divided into their pre-assigned groups and received either drug vehicle or KU-596 for 8 weeks. At study termination, blood glycated hemoglobin (HbA1c) was measured using an A1c Now+ kit.
Synthesis and Administration of KU-596.
KU-596, N-(2-(5-(((3R,4S,5R)-3,4-dihydroxy-5-methoxy-6,6-dimethyltetra hydro-2Hpyran-2-yl)oxy)-3′-fluoro-[1,1′-biphenyl]-2-yl)ethyl)-acetamide, was synthesized, and the structural purity (>95%) was verified as described previously (Kusuma et al., 2012)5. KU-596 was solubilized at a concentration of 5 mg/mL in 50 mM research grade Captisol (β-cyclodextrin sulfobutylethers; CyDex Pharmaceuticals, Lenexa, KS) dissolved in water. KU-596 (1 mg/kg) was administered by daily oral gavage in an adjusted volume of 0.2 mL for up to 8 weeks. Controls received a similar volume of drug vehicle only. No mice of any genotype exhibited adverse drug effects that necessitated veterinary evaluation and a recommendation for removal from the study.
Behavioral Testing.
To monitor the development of DPN, thermal and mechanical sensitivity tests were performed after the induction of diabetes. Thermal sensitivity was assessed using a Hargreaves Analgesiometer (Stoelting Inc.). An increasing temperature stimulus to the mouse’s hind paw was applied, and the animal’s paw withdraw latency was automatically recorded after lifting or flinching of the paw. Mechanical sensitivity was assessed using a dynamic plantar aesthesiometer (Stoelting Inc.), fitted with a stiff monofilament. An increasing force (up to 8g) was applied to the mouse’s hind paw, and the paw withdraw force was automatically recorded. Responses from each animal were assessed four times on alternate feet with a least a 5 min interval between measures and averaged. Sensory measures were assessed 18–24 h after the prior drug dose and before administration of the next daily dose.
Nerve Conduction Velocity Assessment.
Approximately 24 h after the final drug dose, animals were deeply anesthetized with a ketamine/xylazine cocktail prior to assessing motor nerve conduction velocity (MNCV). MNCV measurements were performed using a TECA Synergy N2-EMG monitoring system as previously described.46 The body temperature of the anesthetized mice was monitored with a rectal probe and maintained at 37 °C using a heating pad. Limb temperature was monitored with a subcutaneous sensor and maintained at 36–37 °C with the aid of a heat lamp. The sciatic nerve was stimulated proximally at the sciatic notch and distally at the ankle via bipolar electrodes with a supramaximal stimulus (9.9 mA) of 0.05 ms duration with low and high filter settings of 3 and 10 kHz. MNCV (in meters/second) was calculated by measuring the latencies from the stimulus artifact to the onset of the negative M-wave deflection of the compound muscle action potentials recorded from the first interosseous muscle and dividing by the distance (in millimeters) between the electrodes.
SNCV was recorded in the digital nerve to the second toe by stimulating with a square-wave pulse of 0.05 ms duration using the smallest intensity current that resulted in a maximal amplitude response. The sensory nerve action potential was recorded behind the medial malleolus, and the maximal SNCV was calculated by measuring the latency to the peak of the initial negative deflection divided by the distance between the electrodes.
Immunoblot Analysis and Antibodies.
Mouse tissues were homogenized on ice using a glass tissue homogenizer in 250 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1% NP-40, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 1 mM β-glycerophosphate, and 1 mM Na3VO4 containing complete protease inhibitors (ThermoFisher, Pittsburgh, PA). Lysates were clarified by centrifugation at 10 000g for 10 min at 4 °C. Supernatants were removed, frozen, and stored at −20 °C. Samples contained 20–30 μg of denatured protein lysate; the proteins were resolved on 10% SDS-PAGE gels and electrotransfered onto nitrocellulose. Equal loading was evaluated by staining membranes with 0.5% Ponceau-S. After blocking in 5% dry milk in PBST (phosphate-buffered saline with 0.1% Tween-20), membranes were incubated with primary antibodies overnight at 4 °C in 5% dry milk in PBST. The membranes were washed 3× with PBST and incubated with secondary antibodies for 2 h at room temperature. Membranes were washed 3× with PBST, and bands were visualized with ECL reagent and exposure to film or imaging on a BioRad ChemiDoc System. Primary antibodies used were GFP (Abcam, ab13970), LC3A/B (Cell Signaling, D3U4C), Hsp70 (Enzo Life Sciences, CF92F3A-5), and Txnip Clone JY2 monoclonal antibody (Medical and Biological Laboratories).
Sensory Neuron Isolation from Adult DRG.
Mouse spines were dissected and L4–L6 lumbar DRGs were collected into a 60 mm tissue culture dish containing Hamm’s F10 media and 10% fetal bovine serum (FBS). The isolated ganglia were trimmed, placed in a 35 mm dish containing 2 mL of F10 media without serum, and digested with 1 mL of 1.25% collagenase at 37 °C for 45 min. The collagenase was aspirated, 1 mL of 2% trypsin was added, and the tissue was incubated at 37°C for another 30 min. Fresh F10 media was added; the ganglia were transferred to a 15 mL conical tube and centrifuged at 1000g for 5 min at 4 °C. The supernatant was discarded, and the pellet was resuspended in 4 mL of F10 media and triturated using a fire polished glass pipet. The cell suspension was layered onto 10 mL of a sterile and freshly prepared, isosmotic Percoll solution and centrifuged at 800g for 20 min at room temperature. The supernatant was aspirated, and the cell pellet was resuspended in F10 media containing N2 supplement (4250 μL of F10 media, 50 mg of transferrin powder, 100 μL of 2 μM progesterone solution, 500 μL of 10 mM putrescine solution, 150 μL of 3 μM sodium selenite, 50 mg BSA) (N2/F10 = 1:100). The isolated adult sensory neurons were seeded at a density of 5 × 103 cells/well into a pre-coated 96-well plate. Plates were prepared by adding 100 μL of dl-polyornithine (0.5 mg/mL) per well and incubating at 37 °C for 5–6 h. After aspiration of the solution, the wells were coated with 100 μL of 2 μg/mL laminin for 1 h, excess liquid was removed, and the plates were air-dried. The neurons were maintained overnight and used the following day to assess mitochondrial respiration.
Mouse embryonic fibroblasts (MEFs) were isolated by the digestion of decapitated and eviscerated e15–e18 embryonic mice using a MEF isolation kit from ThermoScientific per the manufacturer’s protocol.
Assessment of Mitochondrial Bioenergetics (mtBE).
Sensory neuron mtBE were analyzed using a Seahorse XF96 analyzer (Seahorse Biosciences). The day before the assay, the cartridge was hydrated overnight as per the manufacturer’s recommendation and the various respiratory chain poisons placed in designated ports. The growth medium was aspirated from the sensory neurons and replaced with 180 μL of bicarbonate and phenol free DMEM containing 1 mM glucose, 1 mM pyruvate, and 2 mM glutamine. The plate was incubated at 37 °C without CO2 for 1 h. The plate was introduced into the Seahorse analyzer, where the program used a 3 min mix cycle to oxygenate the medium followed by a 4 min measurement of the oxygen consumption rate (OCR). The initial four OCRs provide a measure of basal respiration prior to the determining ATP-linked respiration with 1.5 μg/mL oligomycin (Sigma-Aldrich, #75351), an inhibitor of the ATP synthase complex. Any remaining OCR after oligomycin injection is from uncoupled respiration (proton leak) that is not linked to ATP synthesis. The injection of a protonophore, 5 μM of carbonyl cyanide-p-trifluoromethoxy-phenyl-hydrazon (FCCP) (Sigma-Aldrich, #C2920) causes a depolarization of the inner mitochondrial membrane (IMM) membrane potential and allows the electron transport chain (ETC) to function at its maximal rate. Lastly, complex I and III inhibitors, 0.5 μM rotenone + 0.5 μM antimycin A (Sigma #R8875, #A8674), are added to shut down the ETC allowing for the calculation of nonmitochondrial respiration. Maximal respiratory capacity (MRC) was calculated as the difference between the OCR induced by FCCP and oligomycin. Spare respiratory capacity (SRC) was calculated by subtracting basal respiration from the MRC. Bioenergetic parameters were normalized to total protein per well (BCA protein assay) after subtracting background and nonmitochondrial respiration. Wave 2.6.0 was used to view all oxygen consumption rates, and measurements were exported into the Seahorse XF Cell Mito Stress Test Report Generator software.
Immunohistochemistry.
Adult mice were terminally anesthetized, and whole DRG were excised and processed via immersion fixation in sterile-filtered 3.7% (wt/vol) paraformaldehyde in 200 mM Hepes, pH 7.0 at 4 °C. Fixed tissues were washed extensively in 1× PBS at 4 °C, before cryoprotection in filtered 30% sucrose in PBS at 4 °C. Cryoprotected tissues were oriented in Peel-A-Way molds (Pelco), embedded in OCT (Sakura Tissue-Tek), and slowly frozen on dry ice. Frozen tissues were sectioned on a CM3050S cryostat (Leica Biosystems) and mounted directly to Superfrost plus microscope slides. Tissues were washed with filtered PBS, and a cover glass was attached with Vectashield hard setting antifade mounting media.
Preliminary and zoomed in images were taken using a Leica laser scanning confocal upright microscope fitted with a 60× objective. Images used for quantification were imaged using a 3i Olympus Inverted Epifluorescence microscope fitted with a 60× objective. These images were quantified using a custom Matlab script developed by Dr. Erika Nolte, and measurements were placed in GraphPad Prism 8 software for statistical analysis.
The custom Matlab script was designed to identify green, yellow, and red pixels. Images are imported into a folder associated with the Matlab script. These images are processed through the script where green and yellow pixels allow for the identification of individual cells. The script then counts the red puncta within those identified cells. Once this process is complete, the software outputs a processed image that shows the cells and puncta that were identified along with a run-log that lists the image name and the number of red puncta and cells that were identified.
To validate that the red puncta are true indicators of mitophagy and not background fluorescence or foreign particles, LC3 was used as a biomarker for mitophagy. LC3 facilitates autophagosome formation by recruiting the ubiquitin-tagged damaged mitochondria.25 Nondiabetic neurons had a very low basal fluorescence for MQC and LC3 that was strongly increased in sensory neurons of diabetic MQC mice (Figure S3). The colocalization of mCherry and LC3 within the diabetic sensory neurons is evident by the presence of purple fluorescence and supports that the red puncta are an indicator of mitophagy.
Statistical Analysis.
All statistical analysis was performed using GraphPad Prism 9.0.2 software. Ordinary one-way and two way-ANOVAs were conducted for between-group comparison of parametric data. Post hoc analysis was applied to determine differences between treatments using Tukey’s test. Nonparametric data was analyzed using a Kruksal–Wallis test followed by a Dunn’s test for the post hoc analysis. All data sets were examined for statistical outliers using the Rout method and Q = 1%. This led to the removal of one statistical outlier from the STZ and STZ + KU-596 groups in Figure 3B prior to the one-way ANOVA and Tukey’s multiple comparison. All data are presented as mean ± SEM unless indicated otherwise.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the National Institute of Diabetes, Digestive and Kidney Diseases [DK095911] and the National Institute of Neurologic Diseases [NS114355] to R.T.D., [NS075311] to B.S.J.B. and R.T.D., and the National Cancer Institute [CA120458] to B.S.J.B.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.1c00340.
Figures of genotyping data, lack of Hsp70 and Txnip expression, Ponceau S staining image, immunoblot image, and increased red puncta in diabetic sensory neurons (PDF)
The authors declare the following competing financial interest(s): Drs. Blagg and Dobrowsky are named inventors on US and foreign patents claiming KU-596 and related compounds, which are among a series of patents and patent applications licensed to Reata Pharmaceuticals by the University of Kansas.
Complete contact information is available at: https://pubs.acs.org/10.1021/acschemneuro.1c00340
Contributor Information
Yssa A. Rodriguez, Department of Pharmacology and Toxicology, University of Kansas, Lawrence, Kansas 66045, United States
Sukmanjit Kaur, Department of Pharmacology and Toxicology, University of Kansas, Lawrence, Kansas 66045, United States.
Erika Nolte, Department of Pharmacology and Toxicology, University of Kansas, Lawrence, Kansas 66045, United States.
Zhang Zheng, Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46556, United States.
Brian S. J. Blagg, Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46556, United States.
Rick T. Dobrowsky, Department of Pharmacology and Toxicology, University of Kansas, Lawrence, Kansas 66045, United States.
REFERENCES
- (1).Feldman EL, Nave KA, Jensen TS, and Bennett DL (2017) New Horizons in Diabetic Neuropathy: Mechanisms, Bioenergetics, and Pain. Neuron 93 (6), 1296–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Akude E, Zherebitskaya E, Chowdhury SK, Smith DR, Dobrowsky RT, and Fernyhough P (2011) Diminished superoxide generation is associated with respiratory chain dysfunction and changes in the mitochondrial proteome of sensory neurons from diabetic rats. Diabetes 60 (1), 288–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Fernyhough P, Roy Chowdhury SK, and Schmidt RE (2010) Mitochondrial stress and the pathogenesis of diabetic neuropathy. Expert Rev. Endocrinol. Metab. 5 (1), 39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Chowdhury SK, Smith DR, and Fernyhough P (2013) The role of aberrant mitochondrial bioenergetics in diabetic neuropathy. Neurobiol. Dis. 51, 56–65. [DOI] [PubMed] [Google Scholar]
- (5).Kusuma BR, Zhang L, Sundstrom T, Peterson LB, Dobrowsky RT, and Blagg BSJ (2012) Synthesis and Evaluation of Novologues as C-Terminal Hsp90 Inhibitors with Cytoprotective Activity against Sensory Neuron Glucotoxicity. J. Med. Chem. 55, 5797–5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Ma J, Pan P, Anyika M, Blagg BS, and Dobrowsky RT (2015) Modulating Molecular Chaperones Improves Mitochondrial Bioenergetics and Decreases the Inflammatory Transcriptome in Diabetic Sensory Neurons. ACS Chem. Neurosci. 6 (9), 1637–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Ma J, Farmer KL, Pan P, Urban MJ, Zhao H, Blagg BS, and Dobrowsky RT (2014) Heat shock protein 70 is necessary to improve mitochondrial bioenergetics and reverse diabetic sensory neuropathy following KU-32 therapy. J. Pharmacol. Exp. Ther. 348 (2), 281–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Urban MJ, Pan P, Farmer KL, Zhao H, Blagg BS, and Dobrowsky RT (2012) Modulating Molecular Chaperones Improves Sensory Fiber Recovery and Mitochondrial Function in Diabetic Peripheral Neuropathy. Exp. Neurol. 235, 388–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).You Z, Zhang Z, Blagg BSJ, and Dobrowsky RT (2019) KU-596 decreases mitochondrial superoxide and improves bioenergetics following downregulation of manganese superoxide dismutase in diabetic sensory neurons. Exp. Neurol. 313, 88–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zhang L, Zhao H, Blagg BS, and Dobrowsky RT (2012) C-terminal heat shock protein 90 inhibitor decreases hyperglycemia-induced oxidative stress and improves mitochondrial bioenergetics in sensory neurons. J. Proteome Res. 11 (4), 2581–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Urban MJ, Li C, Yu C, Lu Y, Krise JM, McIntosh MP, Rajewski RA, Blagg BS, and Dobrowsky RT (2010) Inhibiting heat-shock protein 90 reverses sensory hypoalgesia in diabetic mice. ASN Neuro 2 (4), No. e00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zhang X, Li C, Fowler SC, Zhang Z, Blagg BSJ, and Dobrowsky RT (2018) Targeting Heat Shock Protein 70 to Ameliorate c-Jun Expression and Improve Demyelinating Neuropathy. ACS Chem. Neurosci. 9 (2), 381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Devi TS, Somayajulu M, Kowluru RA, and Singh LP (2017) TXNIP regulates mitophagy in retinal Muller cells under high-glucose conditions: implications for diabetic retinopathy. Cell Death Dis. 8 (5), No. e2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Gokulakrishnan K, Mohanavalli KT, Monickaraj F, Mohan V, and Balasubramanyam M (2009) Subclinical inflammation/oxidation as revealed by altered gene expression profiles in subjects with impaired glucose tolerance and Type 2 diabetes patients. Mol. Cell. Biochem. 324 (1–2), 173–81. [DOI] [PubMed] [Google Scholar]
- (15).Price SA, Gardiner NJ, Duran-Jimenez B, Zeef LA, Obrosova IG, and Tomlinson DR (2006) Thioredoxin interacting protein is increased in sensory neurons in experimental diabetes. Brain Res. 1116 (1), 206–14. [DOI] [PubMed] [Google Scholar]
- (16).Tsubaki H, Tooyama I, and Walker DG (2020) Thioredoxin-Interacting Protein (TXNIP) with Focus on Brain and Neurodegenerative Diseases. Int. J. Mol. Sci. 21 (24), 9357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Chen J, Saxena G, Mungrue IN, Lusis AJ, and Shalev A (2008) Thioredoxin-interacting protein: a critical link between glucose toxicity and beta-cell apoptosis. Diabetes 57 (4), 938–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Minn AH, Hafele C, and Shalev A (2005) Thioredoxin-interacting protein is stimulated by glucose through a carbohydrate response element and induces beta-cell apoptosis. Endocrinology 146 (5), 2397–405. [DOI] [PubMed] [Google Scholar]
- (19).Singh LP, Devi TS, and Yumnamcha T (2017) The Role of Txnip in Mitophagy Dysregulation and Inflammasome Activation in Diabetic Retinopathy: A New Perspective. JOJ. Ophthalmol 4 (4), 555643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Huang C, Zhang Y, Kelly DJ, Tan CY, Gill A, Cheng D, Braet F, Park JS, Sue CM, Pollock CA, and Chen XM (2016) Thioredoxin interacting protein (TXNIP) regulates tubular autophagy and mitophagy in diabetic nephropathy through the mTOR signaling pathway. Sci. Rep. 6, 29196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Williams JA, Zhao K, Jin S, and Ding WX (2017) New methods for monitoring mitochondrial biogenesis and mitophagy in vitro and in vivo. Exp. Biol. Med. 242 (8), 781–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ni HM, Williams JA, and Ding WX (2015) Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 4, 6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).McWilliams TG, Prescott AR, Allen GF, Tamjar J, Munson MJ, Thomson C, Muqit MM, and Ganley IG (2016) mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J. Cell Biol. 214 (3), 333–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Brand MD, and Nicholls DG (2011) Assessing mitochondrial dysfunction in cells. Biochem. J. 435 (2), 297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Nguyen TN, Padman BS, Usher J, Oorschot V, Ramm G, and Lazarou M (2016) Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J. Cell Biol. 215 (6), 857–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Price SA, Zeef LA, Wardleworth L, Hayes A, and Tomlinson DR (2006) Identification of changes in gene expression in dorsal root ganglia in diabetic neuropathy: correlation with functional deficits. J. Neuropathol. Exp. Neurol. 65 (7), 722–32. [DOI] [PubMed] [Google Scholar]
- (27).Liu J, Liu W, Li R, and Yang H (2019) Mitophagy in Parkinson’s Disease: From Pathogenesis to Treatment. Cells 8 (7), 712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Lautrup S, Lou G, Aman Y, Nilsen H, Tao J, and Fang EF (2019) Microglial mitophagy mitigates neuroinflammation in Alzheimer’s disease. Neurochem. Int. 129, 104469. [DOI] [PubMed] [Google Scholar]
- (29).Saxena S, Mathur A, and Kakkar P (2019) Critical role of mitochondrial dysfunction and impaired mitophagy in diabetic nephropathy. J. Cell. Physiol. 234 (11), 19223–19236. [DOI] [PubMed] [Google Scholar]
- (30).Tong M, Saito T, Zhai P, Oka SI, Mizushima W, Nakamura M, Ikeda S, Shirakabe A, and Sadoshima J (2019) Mitophagy Is Essential for Maintaining Cardiac Function During High Fat Diet-Induced Diabetic Cardiomyopathy. Circ. Res. 124 (9), 1360–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Chowdhury SK, Zherebitskaya E, Smith DR, Akude E, Chattopadhyay S, Jolivalt CG, Calcutt NA, and Fernyhough P (2010) Mitochondrial respiratory chain dysfunction in dorsal root ganglia of streptozotocin-induced diabetic rats and its correction by insulin treatment. Diabetes 59 (4), 1082–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Zherebitskaya E, Akude E, Smith DR, and Fernyhough P (2009) Development of selective axonopathy in adult sensory neurons isolated from diabetic rats: role of glucose-induced oxidative stress. Diabetes 58 (6), 1356–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Killackey SA, Philpott DJ, and Girardin SE (2020) Mitophagy pathways in health and disease. J. Cell Biol. 219 (11), No. e202004029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Yerra VG, and Kumar A (2017) Adenosine mono-phosphate-activated protein kinase abates hyperglycaemia-induced neuronal injury in experimental models of diabetic neuropathy: Effects on mitochondrial biogenesis, autophagy and neuroinflammation. Mol. Neurobiol. 54 (3), 2301–2312. [DOI] [PubMed] [Google Scholar]
- (35).Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, and Youle RJ (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8 (1), No. e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Drew BG, Ribas V, Le JA, Henstridge DC, Phun J, Zhou Z, Soleymani T, Daraei P, Sitz D, Vergnes L, Wanagat J, Reue K, Febbraio MA, and Hevener AL (2014) HSP72 is a mitochondrial stress sensor critical for Parkin action, oxidative metabolism, and insulin sensitivity in skeletal muscle. Diabetes 63 (5), 1488–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Schulze PC, Yoshioka J, Takahashi T, He Z, King GL, and Lee RT (2004) Hyperglycemia promotes oxidative stress through inhibition of thioredoxin function by thioredoxin-interacting protein. J. Biol. Chem. 279 (29), 30369–74. [DOI] [PubMed] [Google Scholar]
- (38).Yoshioka J, Chutkow WA, Lee S, Kim JB, Yan J, Tian R, Lindsey ML, Feener EP, Seidman CE, Seidman JG, and Lee RT (2012) Deletion of thioredoxin-interacting protein in mice impairs mitochondrial function but protects the myocardium from ischemia-reperfusion injury. J. Clin. Invest. 122 (1), 267–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).DeBalsi KL, Wong KE, Koves TR, Slentz DH, Seiler SE, Wittmann AH, Ilkayeva OR, Stevens RD, Perry CG, Lark DS, Hui ST, Szweda L, Neufer PD, and Muoio DM (2014) Targeted metabolomics connects thioredoxin-interacting protein (TXNIP) to mitochondrial fuel selection and regulation of specific oxidoreductase enzymes in skeletal muscle. J. Biol. Chem. 289 (12), 8106–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Huang C, Lin MZ, Cheng D, Braet F, Pollock CA, and Chen XM (2014) Thioredoxin-interacting protein mediates dysfunction of tubular autophagy in diabetic kidneys through inhibiting autophagic flux. Lab. Invest. 94 (3), 309–20. [DOI] [PubMed] [Google Scholar]
- (41).McWilliams TG, Prescott AR, Montava-Garriga L, Ball G, Singh F, Barini E, Muqit MMK, Brooks SP, and Ganley IG (2018) Basal Mitophagy Occurs Independently of PINK1 in Mouse Tissues of High Metabolic Demand. Cell Metab. 27 (2), 439–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Hirata CL, Ito S, and Masutani H (2019) Thioredoxin interacting protein (Txnip) forms redox sensitive high molecular weight nucleoprotein complexes. Arch. Biochem. Biophys. 677, 108159. [DOI] [PubMed] [Google Scholar]
- (43).Cazzaniga A, Locatelli L, Castiglioni S, and Maier JAM (2019) The dynamic adaptation of primary human endothelial cells to simulated microgravity. FASEB J. 33 (5), 5957–5966. [DOI] [PubMed] [Google Scholar]
- (44).Li C, Ma J, Zhao H, Blagg BS, and Dobrowsky RT (2012) Induction of heat shock protein 70 (Hsp70) prevents neuregulin-induced demyelination by enhancing the proteasomal clearance of c-Jun. ASN Neuro 4 (7), No. 20120047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Dotimas JR, Lee AW, Schmider AB, Carroll SH, Shah A, Bilen J, Elliott KR, Myers RB, Soberman RJ, Yoshioka J, and Lee RT (2016) Diabetes regulates fructose absorption through thioredoxin-interacting protein. eLife 5, e18313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).McGuire JF, Rouen S, Siegfreid E, Wright DE, and Dobrowsky RT (2009) Caveolin-1 and Altered Neuregulin Signaling Contribute to the Pathophysiological Progression of Diabetic Peripheral Neuropathy. Diabetes 58, 2677–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.