

Abstract
The synthesis of the invariant natural killer (iNK) T cell agonist β-mannosylceramide along with a series of fatty amide analogues is reported. Of the six β-glycosylation protocols investigated, the sulfoxide methodology developed by Crich and co-workers proved to be the most effective where the reaction of a mannosyl sulfoxide and phytosphingosine derivative gave a key glycolipid intermediate as a 95 : 5 mixture of β- to α-anomers in high yield. A series of mannosyl ceramides were evaluated for their ability to activate D32.D3 NKT cells and induce antitumour activity.
Introduction
As part of a programme developing vaccines based on α-galactosylceramide (α-GalCer, 1) and antigenic peptides, we have developed syntheses of the parent glycolipid and its analogues that allow the preparation of therapeutic vaccine conjugates.2–4 α-GalCer, a synthetic compound discovered through SAR studies on a class of glycolipid originally isolated from a marine sponge,5,6 has been shown to be a functional CD1d-dependent ligand which activates iNKT cells to produce large amounts of cytokines, mainly IFN-γ and IL-4.7,8 This induces iNKT cell-dependent activation of antigen presenting cells (APCs), mediated by CD40-CD40L interactions.9 This, when co-administered with protein or peptide antigen, licences the APCs to stimulate antigen-specific T cell responses. Since the discovery of α-GalCer, a plethora of analogues has been reported leading to new compounds which could potentiate an anti-tumour immune response.10–12 Within this, O’Konek et al.13,14 have found that a related glycolipid, β-mannosylceramide (β-ManCer, 2), activates human and murine iNKT cells and shows anti-tumour activity in mice albeit via a different mechanism from α-GalCer. Here, activation of APCs was dependent on NOS and TNF-α. More recently, Terabe et al. demonstrated anti-CD1d-α-GalCer monoclonal antibodies can directly recognized β-ManCer-CD1d complexes and inhibit iNKT cell stimulation by β-ManCer.15 This result led the authors to postulate that β-ManCer can adopt a structural arrangement similar to α-GalCer within the CD1d presenting molecule, a feat that eludes other β-linked glycosylceramides. A potential problem encountered with stimulation of APCs by α-GalCer is that it induces iNKT cells to become hypo-responsive and unable to produce the same response on re-stimulation. In contrast, β-ManCer does not induce this anergic effect exemplifying its potential as an adjuvant for peptide-based subunit vaccines. Berzofsky et al.1 reported the first synthesis of β-ManCer where the key step is the stannylene acetal mediated β-selective glycosylation of acetal 3 and phytosphingosine derivative 4 giving key intermediate 5 (Scheme 1).
Scheme 1.
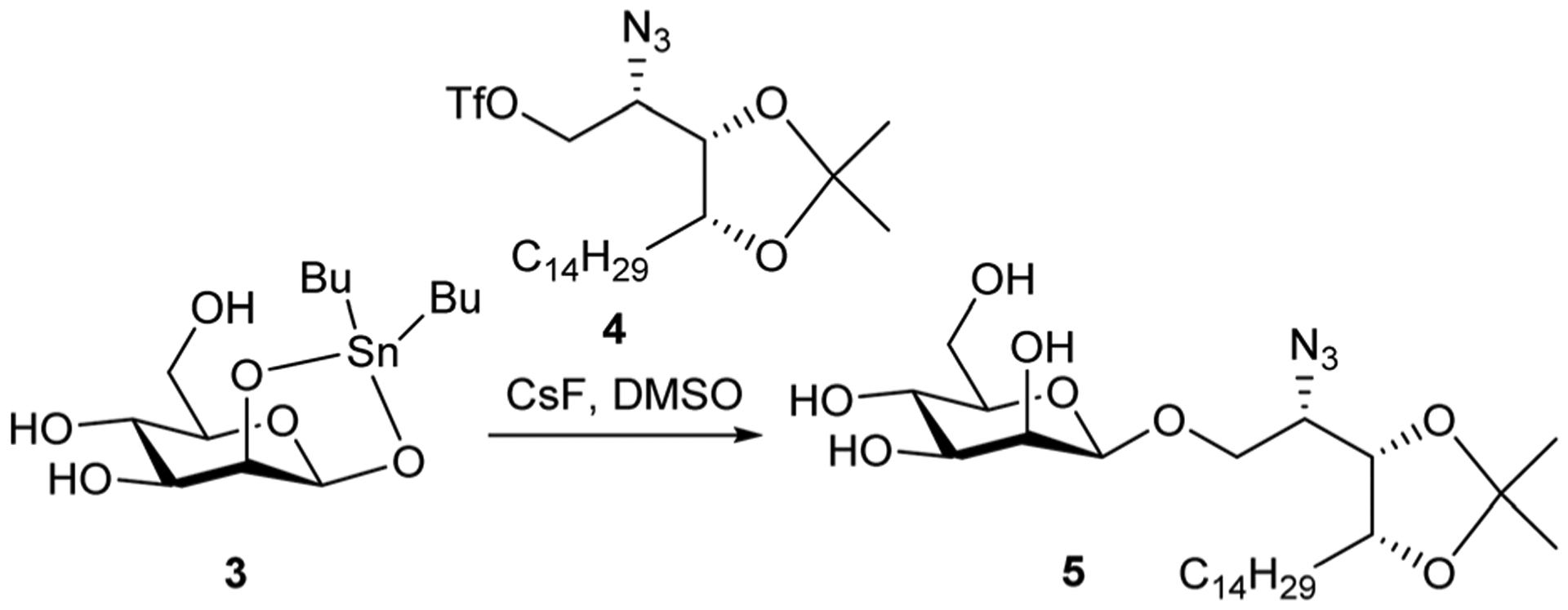
The key glycosylation step in Berzofsky et al.1 synthesis of β-ManCer 2.
Subsequent acid hydrolysis of the isopropylidene protecting group, H2S reduction of the azide and N-acylation with hexacosanyl chloride gave β-ManCer. Although highly effective, we proposed to investigate alternative methods which would allow us to prepare large quantities highly pure β-ManCer for biological studies. These strategies include reactions where tin reagents were avoided, or alternatively, employing stannyl-mediated chemistry with a suitably protected mannosyl donor which would prevent alkylation at O-3 on the sugar unit.16
Results and discussion
Due to the anomeric effect and an inability to invoke O-2 acyl participation, the 1,2-cis-β bond of β-mannosides is viewed as one of the more difficult glycosidic linkages to construct.17,18 With this in mind, we decided to explore a variety of methods for the synthesis of β-ManCer and its N-acyl analogues. Our preliminary approach (Method 1, Scheme 2) used the TMSOTf promoted reaction of mannosyl trichloroacetimidate 6a19 with protected phytosphingosine 720,21 which gave glycoside 8 in 70% yield as an inseparable 85 : 15 mixture of β- and α-anomers based upon integration of the benzylidene proton signals in the 1H NMR spectrum.
Scheme 2.
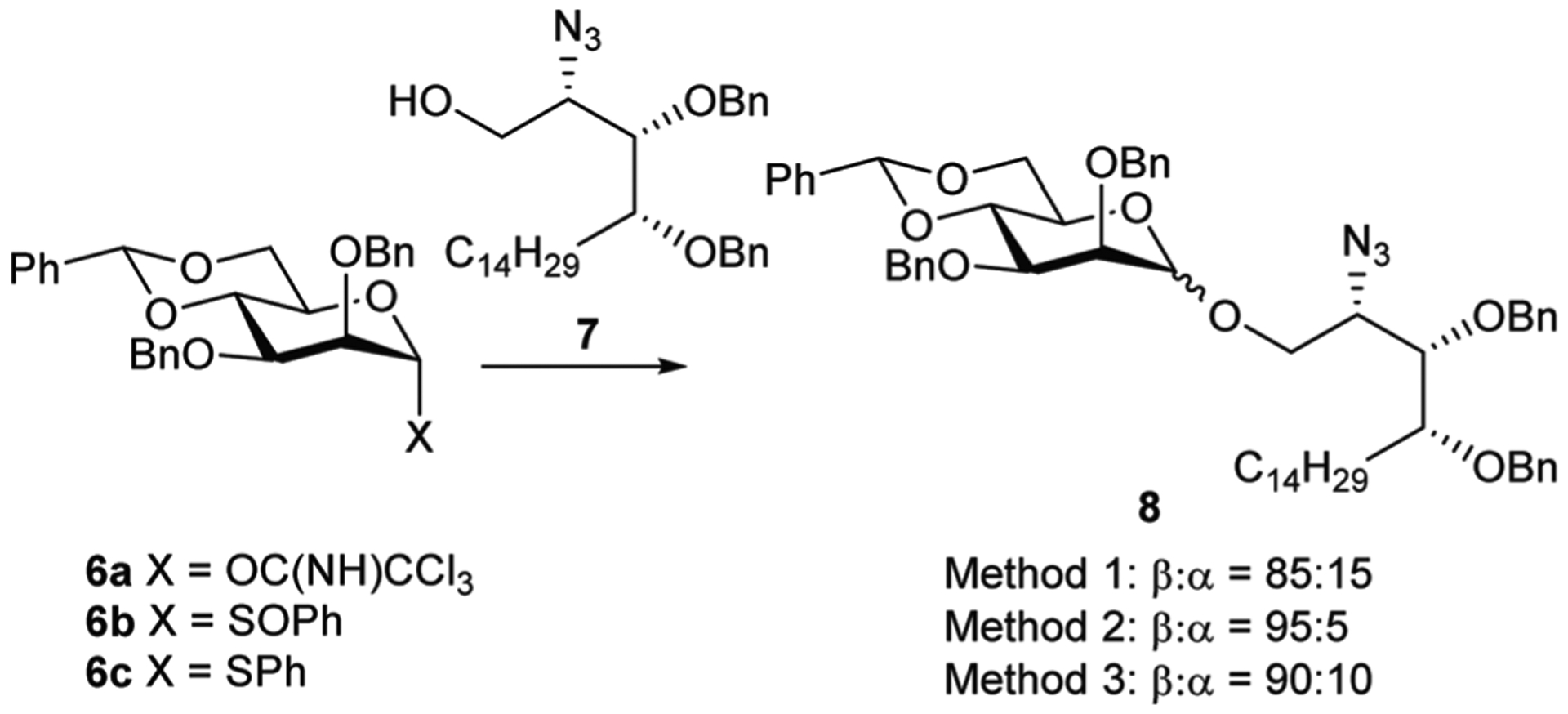
Reagents and conditions; Method 1: 7, TMSOTf, CH2Cl2, 4 Å MS, 0 °C; Method 2: (i) DTBMP, Tf2O, 4 Å MS, CH2Cl2, −78 °C, 15 min; (ii) 7, −78 °C → rt; Method 3: (i) DTBMP, Ph2SO, Tf2O, 4 Å MS, CH2Cl2, −78 °C, 25 min; (ii) 7, −78 °C → rt.
Given the reduced selectivity of this approach and the extra synthetic steps required to make the trichloroacetimidate donor, the corresponding sulfoxide methodology developed by Crich and co-workers was investigated (Method 2).22–24 Triflic anhydride mediated reaction of sulfoxide 6b25 and acceptor 721 gave 8 with a 95 : 5 β to α anomeric ratio in a 71% yield. Modification of the latter approach used diphenylsulfide bis (trifluoromethane-sulfonate), generated from diphenyl sulfoxide and Tf2O, as an activator of thiophenyl mannoside 6c (Method 3).26 Preactivation of 6c and subsequent addition of acceptor 7 gave 8 with a 90 : 10 β to α anomeric ratio in a 61% yield.
An alternative intramolecular aglycone delivery (IAD) approach pioneered by Ogawa and Ito27–29 was also investigated. After initially investigating a mannosyl substrate which included a p-methoxybenzyl group at O-2, the best results were obtained using 2-O-Nap-protected thiomannoside 930 that gave acetal 10 on treatment with DDQ and reaction with 7 (Scheme 3, Method 4). Activation of the thiophenyl group with diphenyl sulfoxide and Tf2O gave mannoside 11 in 45% yield over the two steps with high β-selectivity. However, due to its low yield and the difficultly encountered separating the target product from the various naphthyl by-products this method was not further investigated.
Scheme 3.
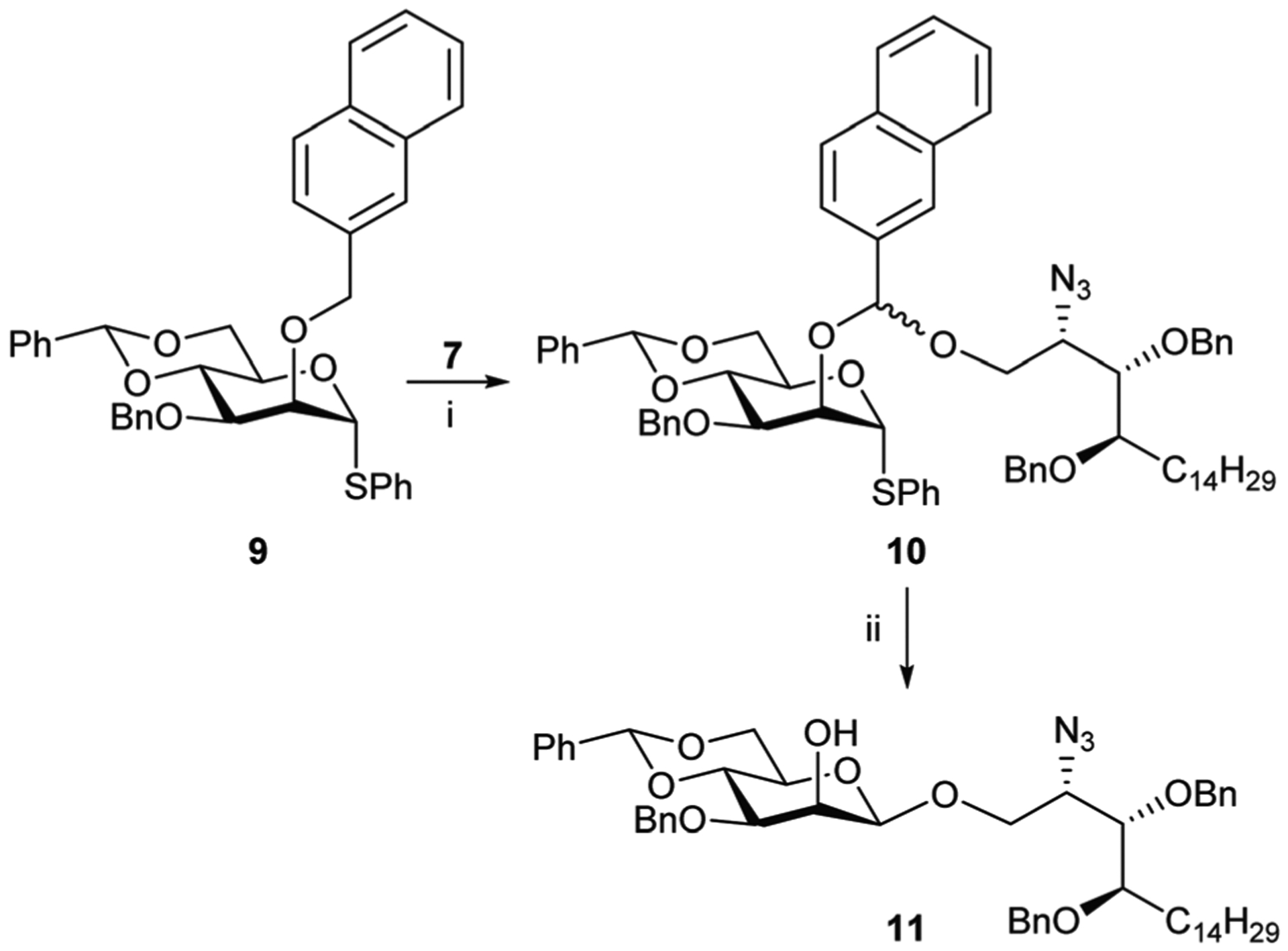
Reagents and conditions; Method 4: (i) (a) 7, anhyd. CH2Cl2, 4 Å powdered MS, rt, 40 min; (b) DDQ, rt, 3 h; (ii) (a) Ph2SO, DTBMP, 4 Å powdered MS, anhydrous ClCH2CH2Cl, 40 min; (b) Tf2O, −35 °C, 15 min; (c) −35 °C to rt, o/n, 45%.
The last direct approach (Method 5) used a stanylene-acetal mediated glycosylation (Scheme 4).16,31 Akin to the original synthesis of 2, employing benzyl protecting groups at O-3, −4, and −6 of mannose would ensure glycosylation exclusively at O-1 as well as providing an intermediate that would easily be purified from the resulting tin by-products by conventional chromatography on silica-gel. Accordingly, the reaction of 3,4,6-tri-O-benzyl-α-D-mannopyranose (12)32,33 with dibutyltin oxide in toluene under Dean–Stark conditions, followed by caesium fluoride assisted alkylation of the anomeric oxygen with phytosphingosine triflate 4, gave 13 with complete β-selectivity, in 62% yield.
Scheme 4.
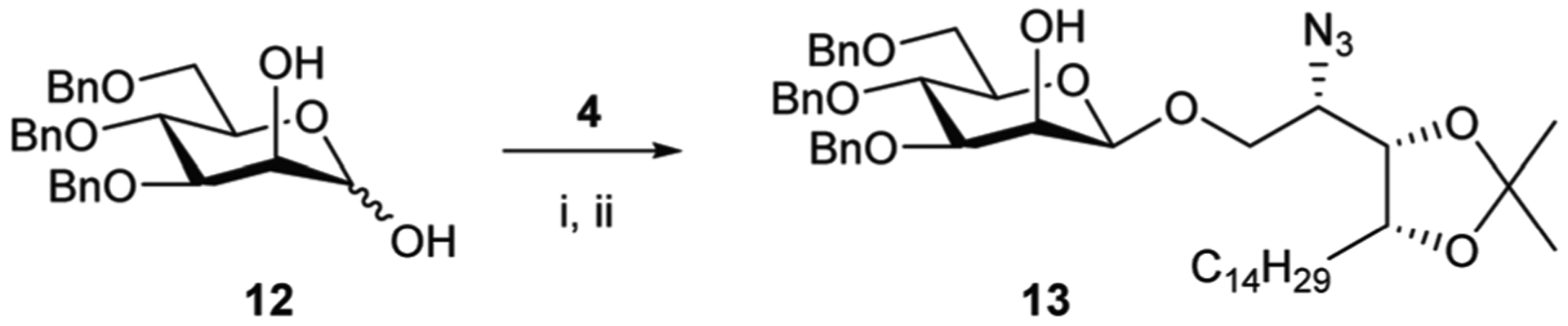
Reagents and conditions; Method 5: (i) Bu2SnO, tol, reflux, 3 h; (ii) (a) CsF, DMF, rt, 30 min; (b) 4, rt, o/n, 62%.
The last approach (Method 6) utilized an indirect glycosylation strategy (Scheme 5). Here, the NIS/TfOH mediated glycosylation of thioglucoside 14 gave 15 which was deacetylated to give 16 in 67% over the two steps. A two-step inversion of stereochemistry at C-2 was achieved by oxidation, using Dess–Martin periodinane, and reduction of the resulting ulose with L-selectride providing mannoside 17 in 44% yield for the two steps after chromatographic purification. HPLC analysis of the crude reaction product showed the selectivity to be in the order of 99 : 1 of the manno- to gluco-configured glycosides.
Scheme 5.
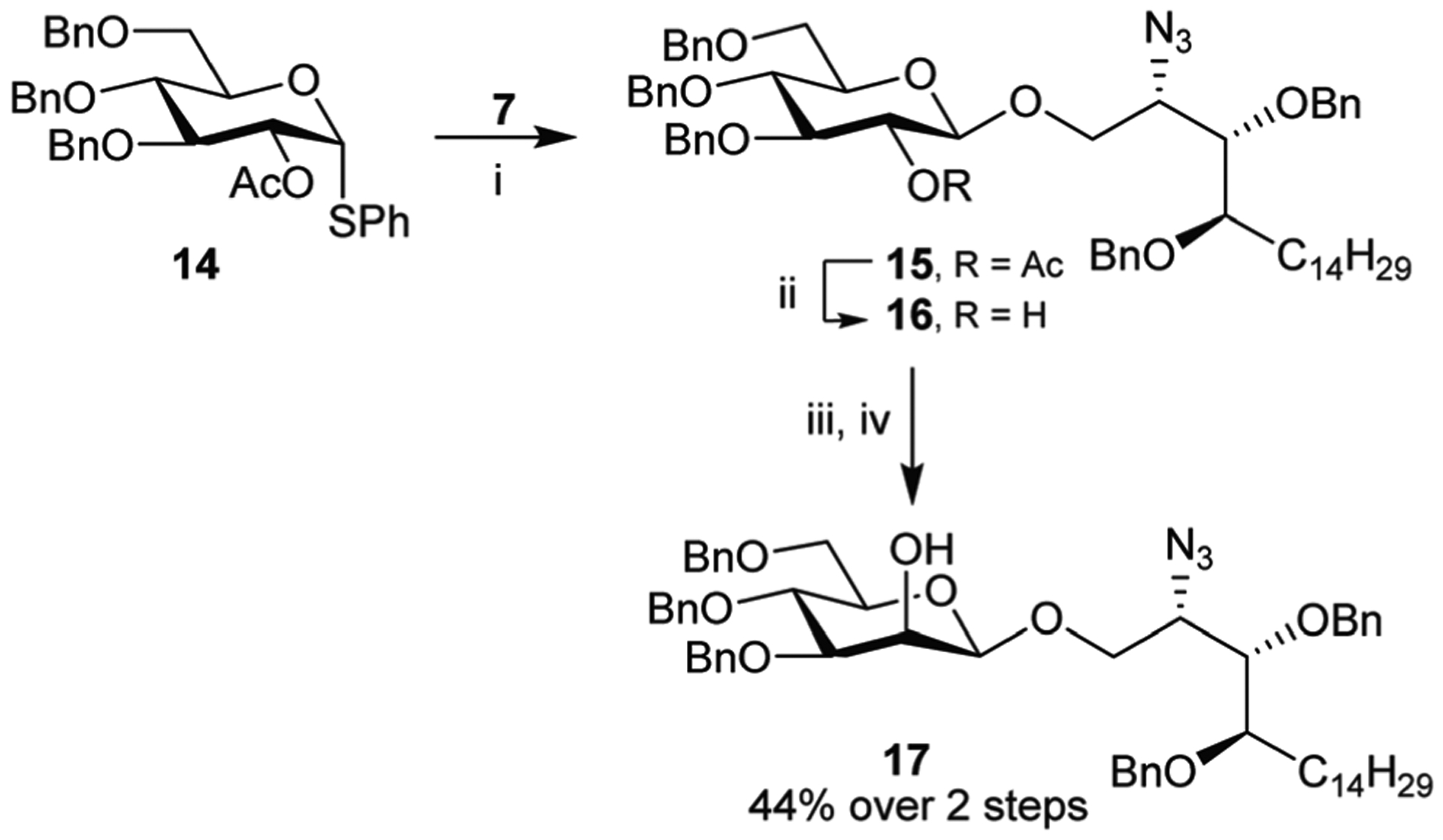
Reagents and conditions; Method 6: (i) (a) NIS, 4 Å MS, anhyd. CH2Cl2, −10 °C, 10 min; (b) 7, TfOH, −10 °C, 3 h, 70%. (ii) 2.5 M NaOMe, MeOH, rt, o/n, 96%; (iii) Dess–Martin periodinane, anhyd. CH2Cl2, rt, 16 h; (iv) L-selectride, anhyd. THF, −78 °C, 10 min, 44% over two steps.
Given the relative ease in preparing reactants 6b and 7, the high selectivity (β : α = 95 : 5) and good yield achieved using the sulfoxide methodology (Method 2), intermediate 8 was chosen to advance through to β-ManCer. Chemoselective reduction of the azide group of 8 was achieved on treatment with trimethylphosphine (Scheme 6). The crude product, amine 18, was acylated with cerotic acid to give a 95 : 5 mixture of 19-β and 19-α which, after column chromatography, provided 19-β in 81% yield over two steps. Global deprotection by hydrogenolysis over palladium on carbon in dichloromethane and methanol gave, after trituration of the product from water and precipitation from hot ethanol, β-ManCer 2 in 92% yield.
Scheme 6.
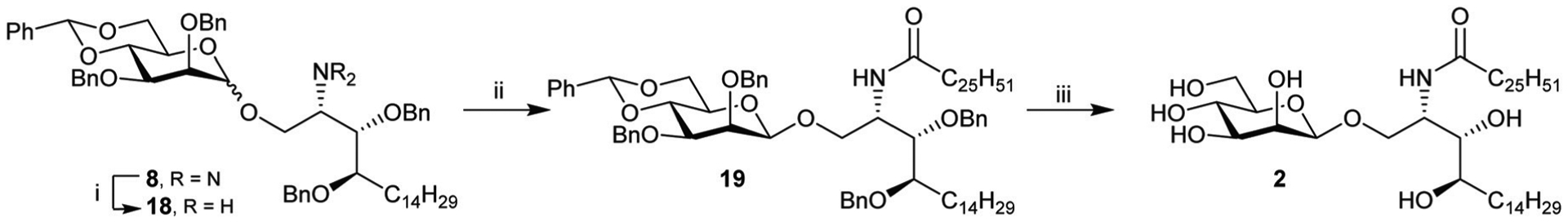
Reagents and conditions; (i) (a) Me3P (1 M in THF), anhyd. THF, 0 °C to rt, 5 h; (b) NaOH (2 M), o/n, quant. (ii) (a) EDCI·HCl, HOBt, C25H51COOH, 10 : 4 CH2Cl2 : DMF, rt, 30 min; (b) 18, DIPEA, anhyd. CH2Cl2, rt, o/n, 81% over two steps. (iii) Pd/C, CH2Cl2/MeOH H2, rt, o/n, 92%.
In order to assess whether traces of the α-ManCer 20 was present in β-ManCer 2 produced in this study, it was synthesised independently using the route shown in Scheme 7. The NIS/TfOH mediated glycosylation of thiomannoside 2134 and phytosphingosine 7 gave 22 that was subjected to global hydrogenolysis to provide amine 23 as its TFA salt after purification. N-Acylation with cerotic acid via the in situ formation of a carbonic anhydride gave a sample of 20. Comparison of the spectra of 2 and 20 clearly indicated that the former had been isolated as a single anomer (see ESI, Fig. SI-1† for 1H NMR spectral comparison).
Scheme 7.
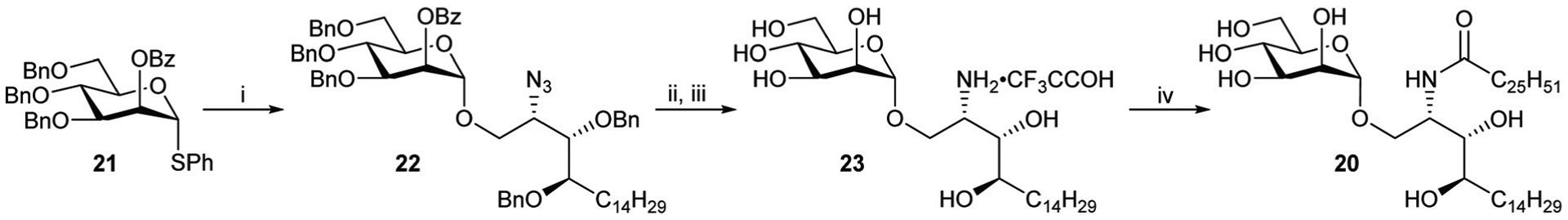
Reagents and conditions; (i) 7, TfOH, NIS, anhyd. CH2Cl2, 4 Å MS, −10 °C to rt, 74%; (ii) 2.5 M NaOMe, MeOH, rt, 3 h; (iii) Pd/C, H2, HCl, 1 : 1 CH2Cl2 : MeOH, o/n, 49% over two steps; (iv) (a) C25H51COOH, Et3N, isopropyl chloroformate (1 M in tol), anhyd. CH2Cl2, rt, 2 h; (c) 23, 1 : 0.2 CH2Cl2 : DMF, 0 °C to rt, o/n, 94%.
In the case of α-GalCer, it has been shown that modification of the N-acyl chain can produce potent analogues that activate iNKT cells. Of the many variants, 7DW8–535 and C3436 reported by Wong and co-workers, and C20:237 by Yu et al. are the most promising. 7DW8–5 and C34 have a stronger binding affinity for CD1d biasing the cytokine release profile towards a TH1 response. Conversely, C20:2 induces a TH2 bias triggering IL-4 production without inducing an INF-γ response. To understand if a similar trend could be observed for β-ManCer, its corresponding N-acyl analogues were targeted with the hope that a more potent, less anergic version of 2 could be discovered. Towards this, the trimethylphosphine mediated reduction of azide 8 and subsequent acylation with 2438,39 and 2540 provided, after column chromatography, amides 26 and 27 in 70% and 67% yields (respectively) with the latter isolated as a 95 : 5 mixture of β- and α-anomers. Hydrogenolysis of 26 provided 7DW8–5 analogue 28 in 83% yield. Similarly, 27 gave C34 analogue 29 as a 97 : 3 mixture of anomers. Acetylation allowed separation of the β-peracetate 30 from the corresponding α-anomer which upon deacetylation provided β−29.
To complete the library of these glycolipids β-mannosylphytosphingosine (β-ManPhs) and C20:2 analogues were also prepared (Schemes 9 and 10). Reduction of the azide group of 8 (β : α = 95 : 5) to amine 18, followed by reaction with Boc-anhydride, gave the corresponding mixture of β- and α-anomers of 31 that were separated by column chromatography to give β−31 in 78% yield over the two steps. Hydrogenolysis and then treatment with TFA afforded β-ManPhs (32) in 77% yield as its TFA salt (Scheme 8).
Scheme 9.
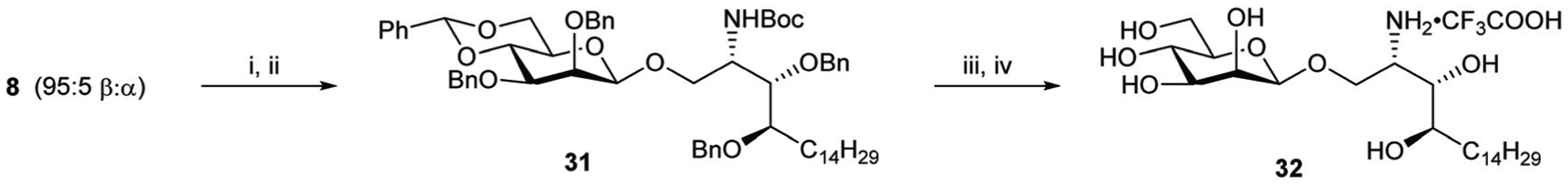
Reagents and conditions; (i) (a) Me3P (1 M in THF), anhyd. THF, 0 °C to rt, 5 h; (b) NaOH (2 M), o/n; (ii) Boc2O, DIPEA, DMF, 50 °C, o/n, 31 (β-only) 78% over two steps; (iii) Pd/C, H2, 1 : 1 CH2Cl2 : MeOH, rt, o/n; (iv) TFA, CH2Cl2, rt, o/n, 77% over two steps.
Scheme 10.
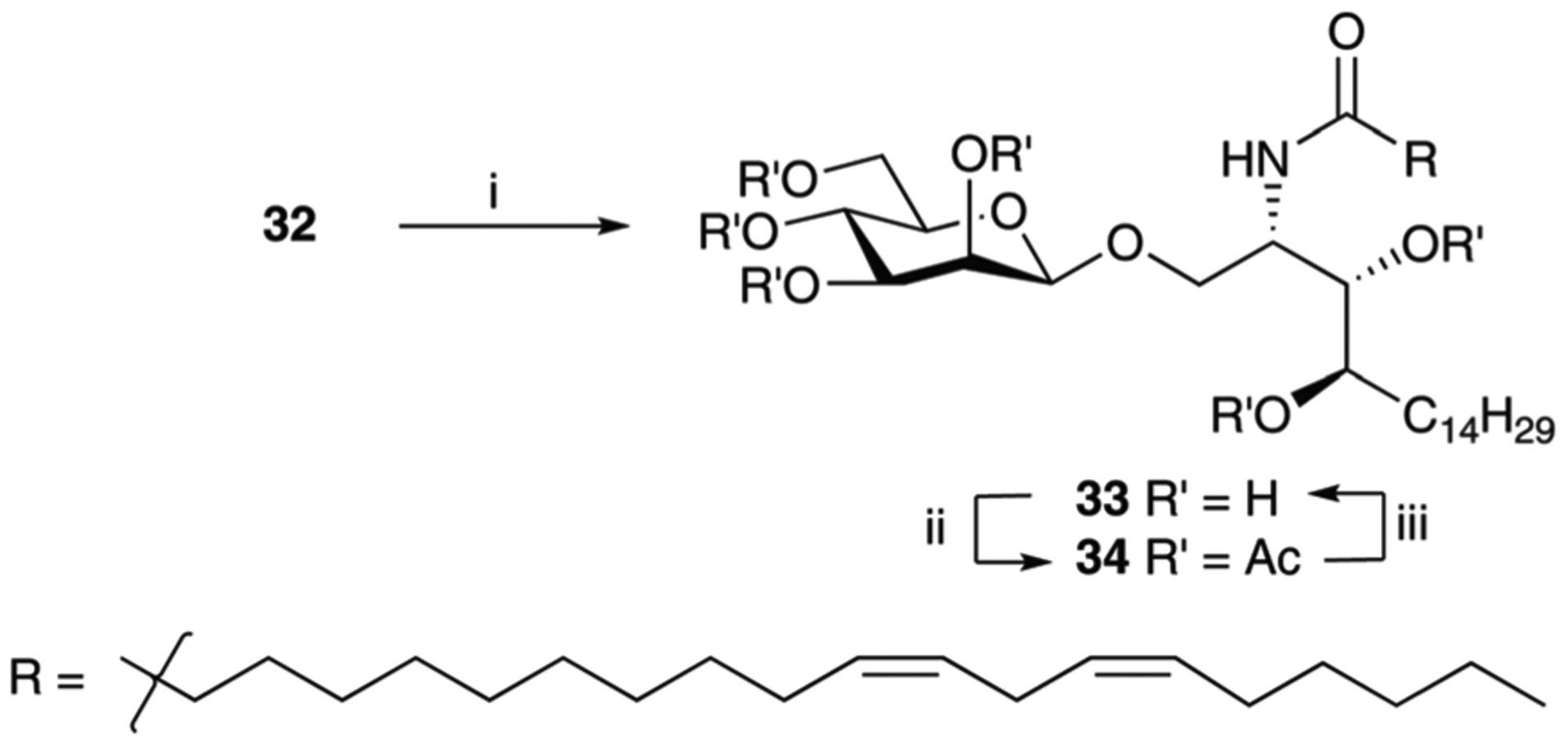
Reagents and conditions; (i) (a) elicosadienoic acid, Et3N, isopropyl chloroformate (1 M in tol), anhyd. CH2Cl2, rt, 50 min; (b) rt to 0 °C; (c) 32, DMF, 0 °C to rt, 3 h; (ii) Ac2O, DMAP, o/n, 86%; (iii) 2.5 M NaOMe, MeOH, rt, 3 h, 88%.
Scheme 8.
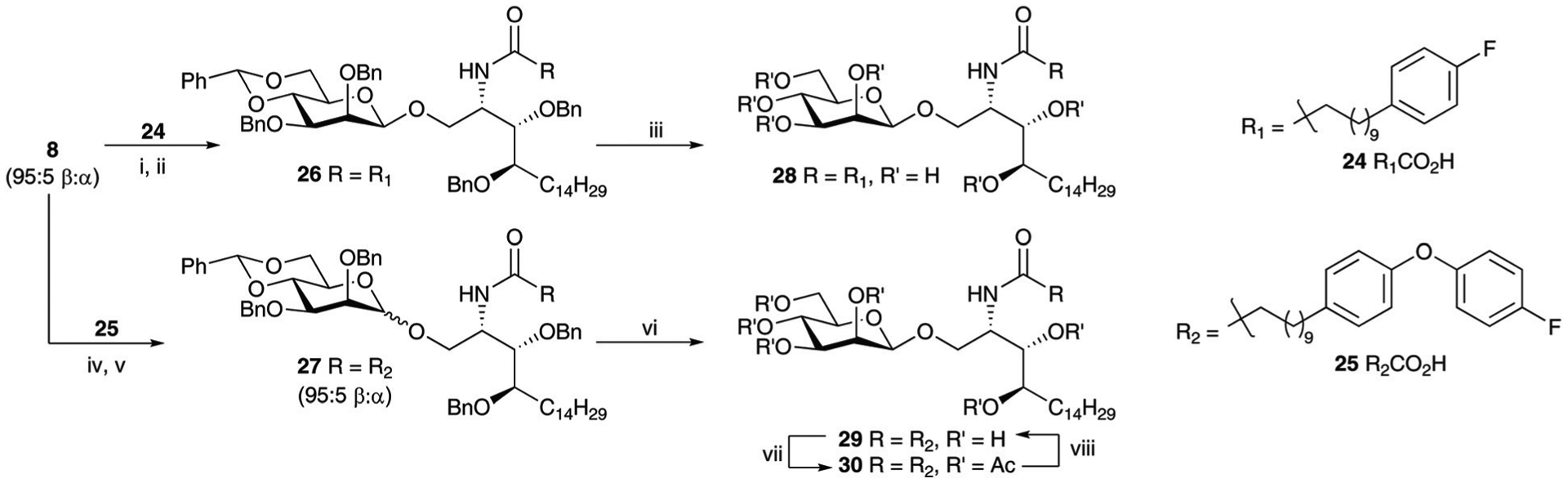
Reagents and conditions; (i) (a) Me3P (1 M in THF), anhyd. THF, 0 °C to rt, 5 h; (b) NaOH (2 M), o/n, 18 quant; (ii) (a) EDCI·HCl, HOBt, 10 : 4 CH2Cl2 : DMF, rt, 30 min; then 18, DIPEA, anhyd. CH2Cl2, rt, o/n; β−26 70% over two steps; (iii) Pd/C, H2, rt, o/n; 28 83% (β-only); (iv) (a) Me3P (1 M in THF), anhyd. THF, 0 °C to rt, 5 h; (b) NaOH (2 M), o/n, 18 quant; (v) (a) EDCI·HCl, HOBt, 10 : 4 CH2Cl2 : DMF, rt, 30 min; then 18, DIPEA, anhyd. CH2Cl2, rt, o/n; 27 – α : β 5 : 95, 65% over two steps; (vi) Pd/C, H2, rt, o/n; 29 94% (mixture of anomers); (vii) Ac2O, py, rt, o/n β−30 74%; (viii) 2.5 M NaOMe, MeOH, rt, 3 h.
Acylation of 32 with cis-11,14-elicosadienoic acid and subsequent acetylation to aid in purification gave per-acetate 34 in 86% yield. Deacetylation was effected on treatment with a catalytic amount of sodium methoxide in methanol to give 33 in 88% yield after precipitation from hot ethanol.
β-ManCer (2), its N-acyl analogues (28, 29 and 33) and α-ManCer (20) were then tested for their ability to activate the DN32.D3 NKT cell hybridoma (Fig. 1). Following well-established protocols and using α-GalCer as a positive control, both β- and α-ManCer induced IL-2 secretion. As expected,13 β-ManCer was less potent than α-GalCer and α-ManCer was at least one log less potent than β-ManCer. Interestingly, we were unable to detect any NKT cell activation when the analogues 28, 29 and 33 where tested in this model system. This finding supports that obtained with the α-carba-GalCer series of compounds whereby the increased potency of certain fatty acyl analogues in the α-GalCer series did not transfer to the corresponding C-glycoside version.41
Fig. 1.
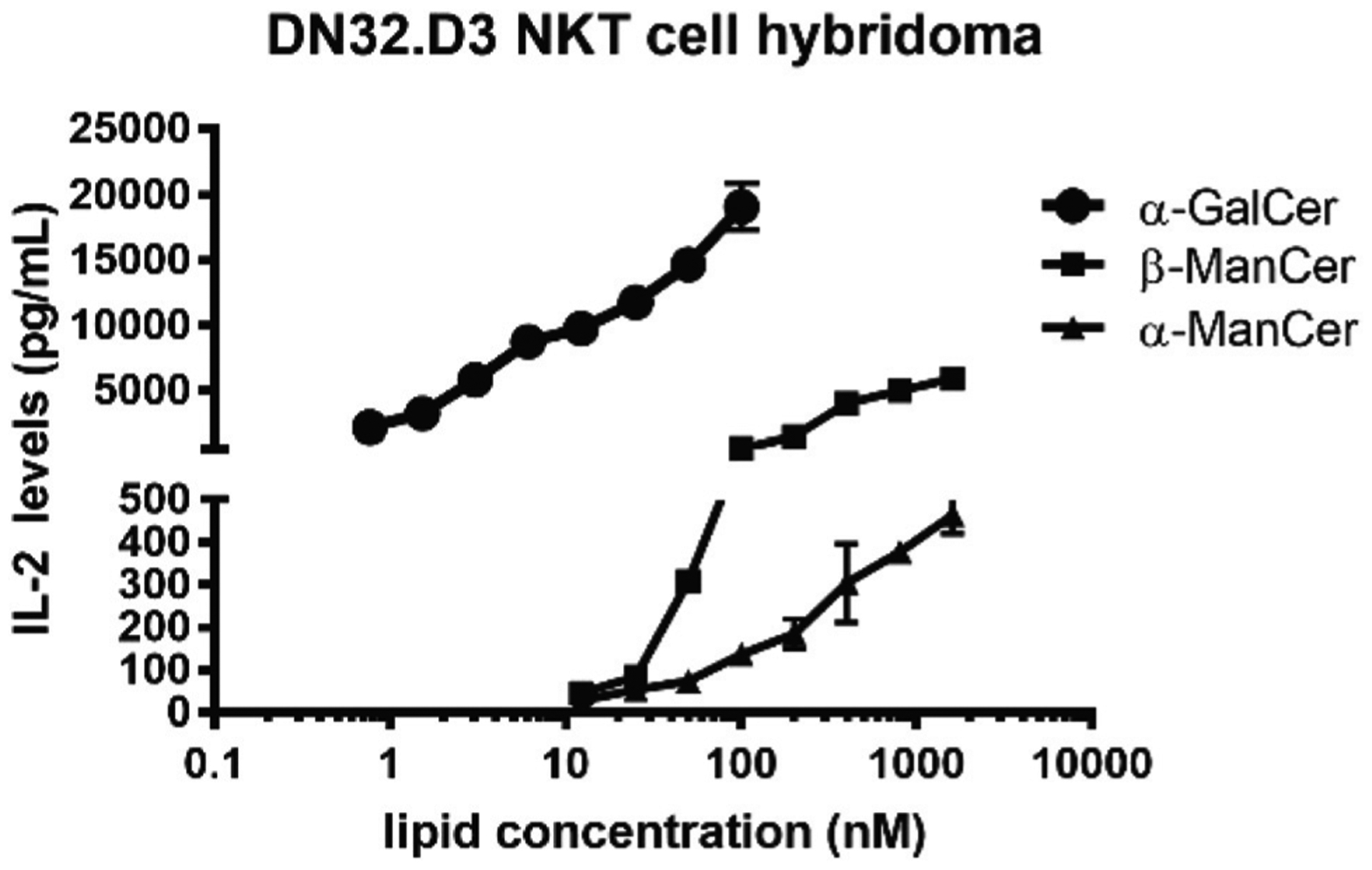
Dose dependent response of DN32.D3 hybridoma against α-GalCer 1, β-ManCer 2, and α-ManCer 20 on cytokine secretion of IL-2. DN32.D3 were stimulated with T cell-depleted spleen cells together with α-GalCer at concentrations ranging from 100 nM to 0.781 nM, β-ManCer or α-ManCer at concentrations ranging from 1600 nM to 12.25 nM. Supernatants from the culture were collected after 18 h, and the concentrations of IL-2 were measured by ELISA. Each condition was tested in triplicate. Data presented are representative of two independent experiments.
As previously reported β-ManCer (2) could protect against lung metastasis but, mirroring the inactivity observed in the in vitro model, none of the N-acyl analogues tested showed any protection against tumour growth (Fig. 2). α-ManCer (20) showed some antitumour activity in this model when administered at a two logs higher dose than required for β-ManCer (ESI, Fig. SI-2†).
Fig. 2.
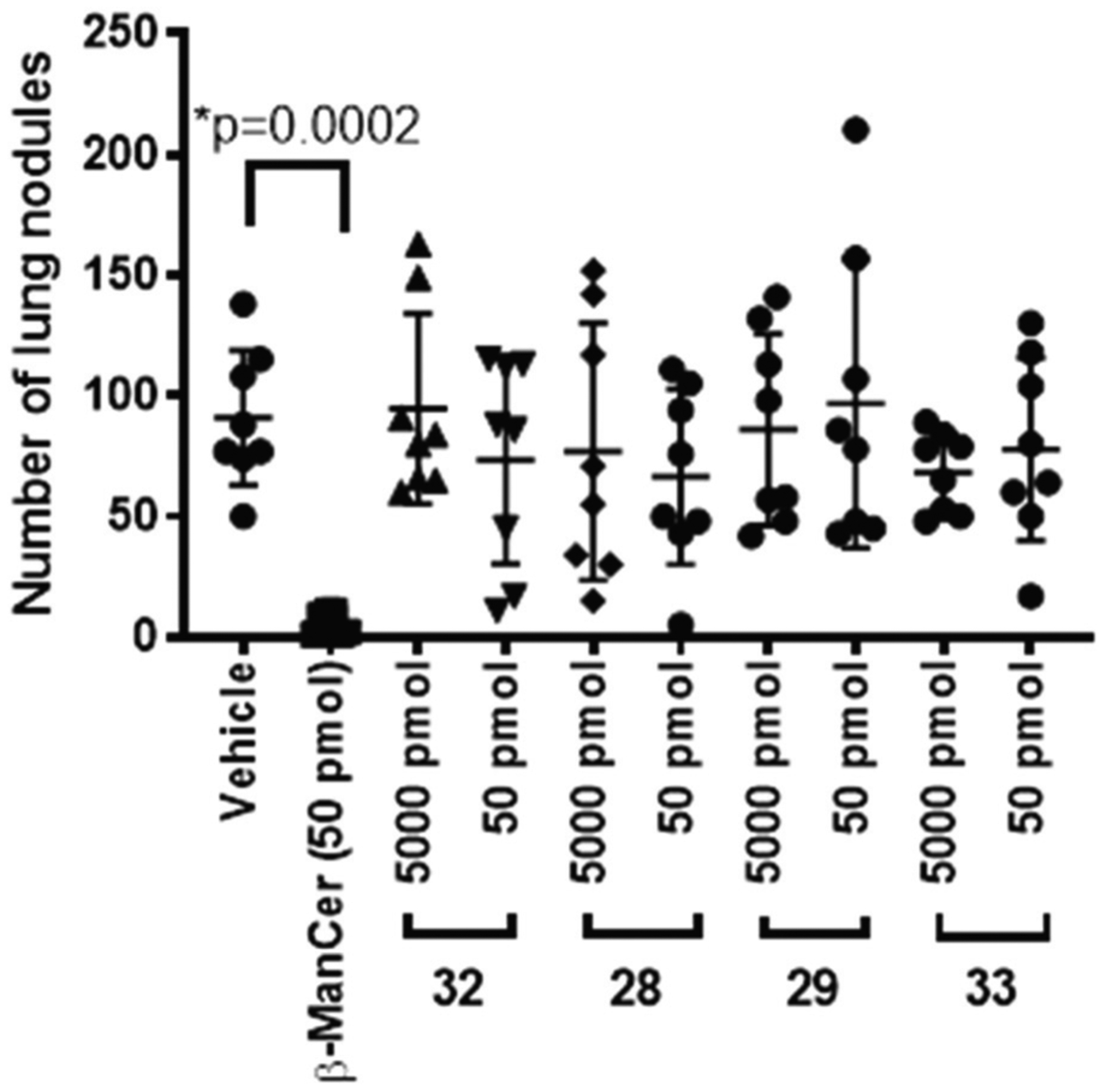
Effects of 32 (β-mannosylphytospingosine), 28 (7DW8–5 version), 29 (C34 version) and 33 (C20:2 version) compared to vehicle (PBS/TWEEN20) and β-ManCer at 5000 and 50 pmol. (N = 8 mice/group) *p < 0.05 compared with vehicle control (Mann–Whitney).
Conclusion
In conclusion we have scoped a number of glycosylation protocols for the synthesis of β-ManCer (2) and found that the most expedient approach utilised a triflic anhydride mediated β-glycosylation of sulfoxide 6b and phytosphingosine 7 giving intermediate 8 as a 95 : 5 mixture β- and α-anomers. This was then used to synthesise β-ManCer and a series of β-mannosyl fatty amide analogues which mirror the immunologically active α-GalCer variants 7DW8–5, C34 and C20:2 (28, 29 and 33, respectively). To complete the library, β-ManPhs (32) and α-ManCer (20) were also synthesised. Of the compounds prepared here, only β-ManCer and α-ManCer were able to activate DN32.D3 NKT cells and induce antitumour activity. Beyond substantiating that α-GalCer is significantly more potent than β-ManCer at activating NKT cells, our results indicate that structural variations that alter immunomodulatory activity of the α-GalCer analogues 7DW8–5, C34 and C20:2 is not transferrable to the β-ManCer series and that the C-26 lipid of β-ManCer remains optimal for NKT cell activation.
Experimental
General experimental are reported in ESI.†
Experimental for key glycosylation reactions
1-((2′S,3′S,4′R)-2′-Azido-3′,4′-di-O-benzyl-1′-octadecanyl) 2,3-di-O-benzyl-4,6-O-benzylidene-β-D-mannopyranoside (8).
Method 1:
To a stirred solution of alcohol 7 (260 mg, 0.496 mmol) and trichloroacetimidate 6a (1.00 g, 1.687 mmol) in dry CH2Cl2 (30 mL) with powdered 4 Å sieves and cooled to 0 °C was added TMSOTf (12 μL, 0.0615 mmol). The reaction mixture was allowed to warm to rt over 1 h before being quenched by the addition of Et3N (0.2 mL), filtered through Celite and the solvent removed. Purification of the residue by column chromatography (PE to 8 : 1 PE : EtOAc) gave the title compound 8 as an inseparable mixture of anomers (α : β = 15 : 85, 331 mg, 70%) as a colourless oil. Rf = 0.50 (5 : 1 PE : EtOAc); ṽmax (cm−1) 2920, 2850, 2127, 2100, 1453, 1375, 1215, 1184, 1093, 1074, 745; 1H NMR (500 MHz, CDCl3) for β-anomer δH inter alia 7.49 (4H, ddt, J = 17.4, 5.8, 1.6 Hz), 7.42–7.35 (3H, m), 7.35–7.21 (18H, m), 5.61 (1H, s), 4.99 (1H, d, J = 12.1 Hz), 4.85 (1H, d, J = 12.1 Hz), 4.69 (1H, d, J = 12.6 Hz), 4.67 (1H, s), 4.67 (1H, s), 4.60 (1H, d, J = 12.5 Hz), 4.56 (1H, d, J = 11.6 Hz), 4.53 (1H, d, J = 11.5 Hz), 4.38 (1H, d, J = 0.9 Hz, H-1), 4.24 (1H, dd, J = 10.4, 4.8 Hz), 4.22–4.11 (2H, m), 3.94 (1H, dd, J = 3.1, 0.8 Hz), 3.88 (1H, t, J = 10.3 Hz), 3.79–3.72 (2H, m), 3.69 (1H, td, J = 6.5, 3.0 Hz), 3.64 (1H, dt, J = 7.7, 3.9 Hz), 3.55 (1H, dd, J = 9.9, 3.1 Hz), 3.23 (1H, td, J = 9.7, 4.9 Hz), 1.70 (1H, dddd, J = 14.5, 10.4, 7.5, 4.9 Hz), 1.62–1.51 (1H, m), 1.48–1.36 (1H, m), 1.32–1.24 (23H, m), 0.88 (3H, t, J = 7.1 Hz); 1H NMR (126 MHz, CDCl3) δC inter alia 138.70, 138.49, 138.43, 138.15, 137.71, 129.01, 128.58, 128.52, 128.47, 128.45, 128.33, 128.28, 128.24, 128.21, 128.06, 128.02, 127.97, 127.84, 127.72, 127.69, 127.62, 126.21, 102.26 (d, 1JCH = 155.29 Hz, C-1), 101.56, 79.30, 79.11, 78.68, 77.81, 76.41, 75.18, 74.08, 72.41, 72.10, 69.63, 68.68, 67.80, 62.00, 32.08, 30.05, 29.95, 29.86, 29.84, 29.82, 29.80, 29.78, 29.52, 25.53, 22.85, 14.28; HRMS (ESI-pos): calcd for C59H75N3O8Na [M + Na]+ m/z 976.5446 found m/z 976.5441; Anal. Calcd for C59H75N3O8: C, 74.26; H, 7.92; N, 4.40. Found C, 74.03; H, 7.97; N, 4.38.
Method 2:
A mixture of sulfoxide 6b (445 mg, 0.800 mmol), DTBMP (422 mg, 2.012 mmol), and activated 4 Å molecular sieves (100 mg) was stirred in dry CH2Cl2 (10 mL) for 1 h at rt before it was cooled to −78 °C. Tf2O (150 μL, 0.888 mmol) was added and after being stirred at the same temperature for 10 min, a solution of alcohol 7 (503 mg, 0.961 mmol) in dry CH2Cl2 (4 mL) was added dropwise. The resulting mixture was allowed to warm to −50 °C over 3 h before being quenched with the addition of saturated aqueous NaHCO3. After being warmed to rt, the mixture was filtered and the filtrate was extracted with CH2Cl2, the combined organic layers were washed with saturated aqueous NaHCO3 and brine. Removal of the solvent and purification of the residue by column chromatography (PE to 8 : 1 PE : EtOAc) gave the title compound 8 as an inseparable mixture of anomers (α : β = 5 : 95, 505 mg, 66%) as a colourless oil.
Method 3:
Thioglycoside 6c (100 mg, 0.185 mmol), diphenyl sulfoxide (62 mg, 0.308 mmol), DMAP (104 mg, 0.496 mmol), and activated powdered 4 Å molecular sieves (100 mg) was stirred in anhydrous CH2Cl2 (2.5 mL) for 40 min at rt under Ar before being cooled to −60 °C. Tf2O (42 μL, 0.249 mmol) was added and after being stirred at the same temperature for 20 min a solution of alcohol 7 (105 mg, 0.200 mmol) in anhydrous CH2Cl2 (2 mL) was added dropwise. The reaction was stirred at −78 °C for an additional 2.5 h before being warmed slowly to rt where it was quenched on addition of Et3N. The mixture was filtered through Celite and the filter cake washed with CH2Cl2. The filtrate was washed with saturated aqueous NH4Cl, dried filtered and concentrated in vacuo to afford the residue which was purified by flash column chromatography (PE to 5 : 1 PE/EtOAc) to give the title compound 8 as an inseparable mixture of anomers (α : β = 1 : 9, 107 mg, 61%) as a colourless oil.
1-((2′S,3′S,4′R)-2′-Azido-3′,4′-di-O-benzyl-1′-octadecanyl) 3-O-benzyl-4,6-O-benzylidene-β-D-mannopyranoside (11).
Method 4:
To a mixture of IAD donor 9 (102 mg, 0.173 mmol) and alcohol 7 (113 mg, 0.217 mmol) in anhydrous CH2Cl2 (5 mL) was added freshly activated 4 Å powdered molecular sieves (100 mg). The mixture was stirred at rt for 40 min under Ar. Then DDQ (53 mg, 0.233 mmol) was added, the resulting mixture was stirred for 3 h at rt under Ar. The reaction was quenched with aqueous ascorbic acid buffer (0.7% ascorbic acid, 1.3% citric acid, 0.9% NaOH), and filtered through Celite. The filtrate was extracted with CH2Cl2 and washed with saturated aqueous NaHCO3 and brine. The combined organic layers were dried, filtered and concentrated in vacuo to give the crude mixed acetal 10 that was used directly in the next step. Mixed acetal 10 (102 mg, 0.092 mmol), diphenylsulfoxide (28 mg, 0.133 mmol), DTBMP (50 mg, 0.239 mmol) and freshly activated 4 Å powdered molecular sieves (100 mg) in anhydrous DCE (5 mL) was stirred at rt under Ar for 40 min prior to being cooled to −35 °C. Tf2O (20 μL, 0.117 mmol) was added and the mixture was allowed to stir at the same temperature for 15 min, before being warmed slowly to rt. The reaction was stirred overnight and then quenched with the addition of a few drops of Et3N, before being filtered through Celite. Aqueous NH4Cl was added and the mixture extracted into CH2Cl2. The organic layer was washed with brine, dried filtered and concentrated in vacuo. The residue was purified by column chromatography (PE to 5 : 1 PE/EtOAc) to afford the title compound 11 (36 mg, 0.042 mmol, 45%) as a slightly impure colourless oil. 1H NMR (400 MHz, CDCl3) δH 7.53–7.27 (20H, m), 5.60 (1H, s), 4.86 (1H, d, J = 12.4 Hz), 4.79 (1H, d, J = 12.4 Hz), 4.69 (1H, d, J = 11.4 Hz), 4.65 (1H, d, J = 11.3 Hz), 4.56 (2H, s), 4.38 (1H, d, J = 1.1 Hz, H-1), 4.26 (1H, dd, J = 10.5, 4.9 Hz), 4.21–4.07 (3H, m), 3.89–3.78 (3H, m), 3.67–3.61 (2H, m), 3.59 (1H, dd, J = 9.5, 3.3 Hz), 3.24 (1H, td, J = 9.8, 4.9 Hz), 2.46 (1H, bs), 1.75–1.50 (4H, m), 1.32–1.22 (22H, br s), 0.92–0.84 (3H, m); 13C NMR (101 MHz, CDCl3) δC 138.41, 138.12, 137.99, 137.58, 131.19, 129.46, 129.09, 128.58, 128.54, 128.54, 128.38, 128.33, 128.22, 128.09, 128.04, 128.02, 127.97, 127.88, 126.18, 124.94, 101.67, 100.32 (d, 1JCH = 162.89 Hz, C-1), 79.43, 79.12, 78.41, 76.61, 73.87, 72.60, 72.19, 69.93, 69.84, 68.69, 67.09, 61.94, 32.08, 30.12, 29.95, 29.86, 29.85, 29.83, 29.82, 29.78, 29.76, 29.52, 25.34, 22.84, 14.28; HRMS (ESI-pos): calcd for C52H69O8N3Na [M + Na]+ m/z 886.4977 found m/z 886.5003.
1-((2′S,3′S,4′R)-2′-Azido-3′,4′-O-isopropylidene-1′-octadecanyl) 3,4,6-tri-O-benzyl-β-D-mannopyranoside (13).
3,4,6-Tri-O-D-mannose (12) (706 mg, 1.567 mmol) and dibutyl tin oxide (550 mg, 2.209 mmol) were dried together under high vacuum. The flask was backfilled with argon and toluene (15 mL) was added. The reaction was refluxed at 135 °C for 3 h before the resulting yellow/orange solution was allowed to cool to rt and the solvent removed in vacuo to give the intermediate dibutyl tin acetal as a yellow oil. The residue was placed under high vacuum for 30 min then under argon. Cesium fluoride (340 mg, 2.238 mmol) was added and anhydrous DMF (6 mL) was cannulated into the mixture. Allowed to stir at rt for 30 min before the addition of triflate 4 (1020 mg, 1.979 mmol) as a solution in DMF (1 mL). The reaction was stirred overnight at rt under argon before being concentrated in vacuo. The residue was re-dissolved in EtOAc and washed with saturated aqueous KF solution. The organic layer was concentrated in vacuo and the residue purified by flash column chromatography (PE to 3 : 1 PE/EtOAc) to afford the title compound 13 (793 mg, 0.972 mmol, 62%) as a colourless oil. δH (500 MHz, CDCl3): 7.45–7.27 (15H, m), 7.21 (2H, dd, J = 7.7, 1.9 Hz), 4.89 (1H, d, J = 10.9 Hz), 4.78 (1H, d, J = 11.9 Hz), 4.67 (1H, d, J = 11.9 Hz), 4.63 (1H, d, J = 12.2 Hz), 4.57 (1H, d, J = 10.3 Hz), 4.55 (1H, d, J = 9.7 Hz), 4.50 (1H, d, J = 1.1 Hz), 4.23–4.08 (3H, m), 3.99 (1H, dd, J = 10.6, 2.8 Hz), 3.91 (1H, t, J = 9.2 Hz), 3.86 (1H, dd, J = 9.6, 5.5 Hz), 3.78 (1H, dd, J = 10.9, 2.3 Hz), 3.73 (1H, dd, J = 10.9, 5.2 Hz), 3.64–3.55 (2H, m), 3.45 (1H, ddd, J = 9.6, 5.2, 2.3 Hz), 1.80 (1H, br s), 1.69–1.47 (2H, m), 1.39 (3H, s), 1.37–1.31 (2H, m), 1.30 (3H, s), 1.34–1.22 (22H, m), 0.88 (3H, t, J = 6.9 Hz); δC(126 MHz, CDCl3): 138.44, 138.36, 137.98, 128.61, 128.52, 128.49, 128.23, 128.06, 127.97, 127.92, 127.88, 127.70, 108.50, 99.73 (d, 1JCH = 157.87 Hz, C-1), 81.39, 77.94, 75.76, 75.71, 75.27, 74.25, 73.64, 71.53, 70.66, 69.29, 68.21, 59.83, 32.08, 29.85, 29.81, 29.77, 29.75, 29.71, 29.57, 29.51, 28.33, 26.59, 25.80, 22.85, 14.28; HRMS (ESI-pos): calcd for C48H69O8N3Na [M + Na]+ m/z 838.4977 found m/z 838.4970.
1-((2′S,3′S,4′R)-2′-Azido-3′,4′-di-O-benzyl-1′-octadecanyl) 2-O-acetyl-3,4,6-tri-O-benzyl-β-D-glucopyranoside (15).
Triflic acid (15 μL, 0.170 mmol) was added to a mixture of thioglycoside 14 (240 mg, 0.458 mmol), alcohol 7 (323 mg, 0.552 mmol), and NIS (190 mg, 0.802 mmol) in anhydrous CH2Cl2 (5 mL) containing pre-activated 4 Å MS at −10 °C under argon. When TLC indicated completion (ca. 3 h), the reaction was diluted with CH2Cl2 and filtered through Celite. The filtrate was washed with NaHCO3 and concentrated in vacuo to afford the residue which was purified by column chromatography (PE to 8 : 1 PE/EtOAc) to afford the title compound 15 (258 mg, 0.258 mmol, 70%) as a white solid. δH (500 MHz, CDCl3): 7.44–7.27 (23H, m), 7.23–7.10 (2H, m), 5.06 (1H, t, J = 8.6 Hz), 4.80 (2H, dd, J = 11.1, 6.1 Hz), 4.69 (3H, dd, J = 11.0, 5.5 Hz), 4.59 (3H, dd, J = 17.9, 11.4 Hz), 4.51 (2H, dd, J = 11.9, 5.6 Hz), 4.40 (1H, d, J = 7.9 Hz), 4.16 (1H, dd, J = 10.6, 6.2 Hz), 3.83 (1H, dd, J = 10.6, 3.3 Hz), 3.78–3.59 (7H, m), 3.46 (1H, dd, J = 9.6, 3.3 Hz), 1.96 (3H, s), 1.68 (1H, dtd, J = 13.8, 9.0, 4.8 Hz), 1.54 (1H, tdt, J = 14.2, 9.3, 4.7 Hz), 1.43 (1H, dq, J = 10.6, 5.3 Hz), 1.33–1.22 (23H, m), 0.89 (3H, t, J = 6.8 Hz); δC (126 MHz, CDCl3): 169.58, 138.59, 138.33, 138.32, 138.23, 138.04, 128.57, 128.51, 128.47, 128.27, 128.16, 128.05, 128.00, 127.92, 127.86, 127.83, 127.74, 127.72, 100.77 (C-1), 83.08, 79.48, 78.63, 78.06, 75.44, 75.20, 75.16, 74.12, 73.63, 73.13, 72.06, 68.82, 68.75, 61.55, 32.08, 30.01, 29.91, 29.86, 29.83, 29.82, 29.79, 29.77, 29.52, 25.61, 22.85, 21.14, 14.28; HRMS (ESI-pos): calcd for C61H79O9N3Na [M + Na]+ m/z 1020.5709, found m/z 1020.5736.
Supplementary Material
Acknowledgements
This work was financially supported by the New Zealand Ministry of Business Innovation and Employment (RTVU1603), and SAR thanks the University of Otago for financial support for a PhD stipend.
Footnotes
Electronic supplementary information (ESI) available: Experimental procedures, supporting figures, NMR spectra and HPLC chromatograms. See DOI: 10.1039/d0ob00223b
Conflicts of interest
M. T. and J. A. B. are co-inventors on US Patent 8 835 613 issued September 16, 2014 and subsequent US divisional applications as well as Canadian Patent 2792754, issued November 12, 2019, entitled “Beta-Mannosylceramide and Stimulation of NKT cell Anti-tumor Immunity”. The original patent applications were filed prior to the research described in this publication.
Notes and references
- 1.Berzofsky JA, O’Konek JJ, Terabe M, Illarionov P and Besra GS, US Pat., US 8835613 B2 20140916, 2014.
- 2.Anderson RJ, Compton BJ, Tang C.-w., Authier-Hall A, Hayman CM, Swinerd GW, Kowalczyk R, Harris P, Brimble MA, Larsen DS, Gasser O, Weinkove R, Hermans IF and Painter GF, Chem. Sci, 2015, 6, 5120–5127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson RJ, Tang C.-w., Daniels NJ, Compton BJ, Hayman CM, Johnston KA, Knight DA, Gasser O, Poyntz HC, Ferguson PM, Larsen DS, Ronchese F, Painter GF and Hermans IF, Nat. Chem. Biol, 2014, 10, 943–949. [DOI] [PubMed] [Google Scholar]
- 4.Compton BJ, Tang C.-w., Johnston KA, Osmond TL, Hayman CM, Larsen DS, Hermans IF and Painter GF, Org. Lett, 2015, 17, 5954–5957. [DOI] [PubMed] [Google Scholar]
- 5.Natori T, Koezuka Y and Higa T, Tetrahedron Lett, 1993, 34, 5591–5592. [Google Scholar]
- 6.Tan RX and Chen JH, Nat. Prod. Rep, 2003, 20, 509–534. [DOI] [PubMed] [Google Scholar]
- 7.Togashi Y, Chamoto K, Wakita D, Tsutsumi N, Iwakura Y, Matsubara N, Kitamura H and Nishimura T, Cancer Sci, 2007, 98, 721–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fujii S, Shimizu K, Smith C, Bonifaz L and Steinman RM, J. Exp. Med, 2003, 198, 267–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hermans IF, Silk JD, Gileadi U, Salio M, Mathew B, Ritter G, Schmidt R, Harris AL, Old L and Cerundolo V, J. Immunol, 2003, 171, 5140–5147. [DOI] [PubMed] [Google Scholar]
- 10.Laurent X, Bertin B, Renault N, Farce A, Speca S, Milhomme O, Millet R, Desreumaux P, Henon E and Chavatte P, J. Med. Chem, 2014, 57, 5489–5508. [DOI] [PubMed] [Google Scholar]
- 11.Tashiro T and Mori K, Stud. Nat. Prod. Chem, 2014, 42, 1–31. [Google Scholar]
- 12.Tashiro T, Biosci., Biotechnol., Biochem, 2012, 76, 1055–1067. [DOI] [PubMed] [Google Scholar]
- 13.O’Konek JJ, Illarionov P, Khursigara DS, Ambrosino E, Izhak L, Castillo II BF, Raju R, Khalili M, Kim H-Y, Howell AR, Besra GS, Porcelli SA, Berzofsky JA and Terabe M, J. Clin. Invest, 2011, 121, 683–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Konek JJ, Kato S, Takao S, Izhak L, Xia Z, Illarionov P, Besra GS, Terabe M and Berzofsky JA, Clin. Cancer Res, 2013, 19, 4404–4411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clark K, Yau J, Bloom A, Wang J, Venzon DJ, Suzuki M, Pasquet L, Compton BJ, Cardell SL, Porcelli SA, Painter GF, Zajonc DM, Berzofsky JA and Terabe M, Front. Immunol, 2019, 10, 2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hodosi G and Kovac P, Carbohydr. Res, 1998, 308, 63–75. [DOI] [PubMed] [Google Scholar]
- 17.Modern Methods in Carbohydrate Synthesis, ed. Khan SH and O’Neill RA, 1996, p. 251. [Google Scholar]
- 18.Nigudkar SS and Demchenko AV, Chem. Sci, 2015, 6, 2687–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roy S and Roy N, J. Carbohydr. Chem, 2003, 22, 521– 535. [Google Scholar]
- 20.Xia C, Schuemann J, Emmanuel R, Zhang Y, Chen W, Zhang W, De Libero G and Wang PG, J. Med. Chem, 2007, 50, 3489–3496. [DOI] [PubMed] [Google Scholar]
- 21.Costantino V, Fattorusso E, Imperatore C and Mangoni A, Tetrahedron, 2002, 58, 369–375. [Google Scholar]
- 22.Crich D, Banerjee A, Li W and Yao Q, J. Carbohydr. Chem, 2005, 24, 415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crich D and Smith M, Org. Lett, 2000, 2, 4067–4069. [DOI] [PubMed] [Google Scholar]
- 24.Crich D and Smith M, J. Am. Chem. Soc, 2001, 123, 9015–9020. [DOI] [PubMed] [Google Scholar]
- 25.Crich D and Sun S, Tetrahedron, 1998, 54, 8321–8348. [Google Scholar]
- 26.Codee JDC, Litjens REJN, den Heeten R, Overkleeft HS, van Boom JH and van der Marel GA, Org. Lett, 2003, 5, 1519–1522. [DOI] [PubMed] [Google Scholar]
- 27.Ishiwata A, Lee YJ and Ito Y, Org. Biomol. Chem, 2010, 8, 3596–3608. [DOI] [PubMed] [Google Scholar]
- 28.Ishiwata A, Munemura Y and Ito Y, Eur. J. Org. Chem, 2008, 4250–4263, DOI: 10.1002/ejoc.200800249. [DOI] [Google Scholar]
- 29.Ito Y and Ogawa T, Angew. Chem, 1994, 106, 1843–1845, (see also Angew. Chem., Int. Ed. Engl., 1994, 1833(1817), 1765–1847). [Google Scholar]
- 30.Adero PO, Jarois DR and Crich D, Carbohydr. Res, 2017, 449, 11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shaffer KJ and Taylor CM, Org. Lett, 2006, 8, 3959–3962. [DOI] [PubMed] [Google Scholar]
- 32.Norberg O, Wu B, Thota N, Ge J-T, Fauquet G, Saur A-K, Aastrup T, Dong H, Yan M and Ramstroem O, Carbohydr. Res, 2017, 452, 35–42. [DOI] [PubMed] [Google Scholar]
- 33. See ESI† for experimental details for the synthesis of 12.
- 34.Heuckendorff M, Poulsen LT, Hedberg C and Jensen HH, Org. Biomol. Chem, 2018, 16, 2277–2288. [DOI] [PubMed] [Google Scholar]
- 35.Li X, Fujio M, Imamura M, Wu D, Vasan S, Wong C-H, Ho DD and Tsuji M, Proc. Natl. Acad. Sci. U. S. A, 2010, 107, 13010–13015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin K-H, Liang J-J, Huang W-I, Lin-Chu S-Y, Su C-Y, Lee Y-L, Jan J-T, Lin Y-L, Cheng Y-SE and Wong C-H, Antimicrob. Agents Chemother, 2010, 54, 4129–4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu KOA, Im JS, Molano A, Dutronc Y, Illarionov PA, Forestier C, Fujiwara N, Arias I, Miyake S, Yamamura T, Chang Y-T, Besra GS and Porcelli SA, Proc. Natl. Acad. Sci. U. S. A, 2005, 102, 3383–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu Z and Bittman R, Org. Lett, 2012, 14, 620–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Panza L, Compostella F and Imperio D, Carbohydr. Res, 2019, 472, 50–57. [DOI] [PubMed] [Google Scholar]
- 40.Liang P-H, PCT Int. Appl, WO 2012094540 A2, 2012.
- 41.Altiti AS, Ma X, Zhang L, Ban Y, Franck RW and Mootoo DR, Carbohydr. Res, 2017, 443–444, 73–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.