

Abstract
Capicua (CIC) is a transcriptional repressor that counteracts activation of genes in response to receptor tyrosine kinase (RTK)/Ras/ERK signaling. Following activation of RTK, ERK enters the nucleus and serine-phosphorylates CIC, releasing it from its targets to permit gene expression. We recently showed that ERK triggers ubiquitin-mediated degradation of CIC in glioblastoma (GBM). In this study, we examined whether another important downstream effector of RTK/EGFR, the non-RTK c-Src, affects CIC repressor function in GBM. We found that c-Src binds and tyrosine-phosphorylates CIC on residue 1455 to promote nuclear export of CIC. On the other hand, CIC-mutant allele (CIC-Y1455F), that escapes c-Src–mediated tyrosine phosphorylation, remains localized to the nucleus and retains strong repressor function against CIC targets, the oncogenic transcription factors ETV1 and ETV5. Furthermore, we show that the orally available Src family kinase inhibitor, dasatinib, which prevents EGF-mediated tyrosine phosphorylation of CIC and attenuates elevated ETV1 and ETV5 levels, reduces viability of GBM cells and glioma stem cells (GSC), but not of their control cells with undetectable c-Src activity. In fact, GBM cells and GSC expressing the tyrosine-defective CIC mutant (Y1455F) lose sensitivity to dasatinib, further endorsing the effect of dasatinib on Src-mediated tyrosine phosphorylation of CIC. These findings elucidate important mechanisms of CIC regulation and provide the rationale to target c-Src alongside ERK pathway inhibitors as a way to fully restore CIC tumor suppressor function in neoplasms such as GBM.
Introduction
Capicua (CIC) is a high-mobility group (HMG)-box transcriptional repressor that counteracts activation of genes downstream of receptor tyrosine kinase (RTK) Ras/ERK signaling and was first described in Drosophila to be involved in EGFR-mediated developmental patterning and cell fate (1–4). The importance of CIC in mammalian cells emerged after the discovery of loss-of-function mutations in CIC in tumors, such as oligodendrogliomas (5, 6), and gene fusions of CIC with either DUX4 or FOXO4 in round cell sarcomas (7, 8). Subsequently, CIC mutations have been linked to other tumor types (9, 10) and connected to additional biological processes, such as lung development, liver homeostasis, autoimmunity, and neurobehavioral processes (11). The oncogenic transcription factors ETV1, ETV4, and ETV5 (12), which mediate cell proliferation, motility, and invasion downstream of Ras (13), are the best-characterized CIC targets in mammalian cells. While these findings validate the importance of CIC, the molecular mechanisms regulating CIC repressor function are not well defined, especially in mammalian cells. Posttranslational events on CIC, including ERK-mediated serine/threonine phosphorylation (1–3, 14–16) have been shown to promote its inactivation by either degradation or nuclear-to-cytoplasmic shuttling of CIC, preventing its ability to function as a transcriptional repressor. We recently showed that in glioblastoma (GBM), CIC is degraded because of ERK-mediated serine (S173) phosphorylation of CIC, which promotes binding of the E3 ligase PJA1 to initiate ubiquitin-mediated degradation of CIC (17). Given the importance of posttranslational modifications of CIC on its repressor and tumor suppressor function, we examine in this report the role of tyrosine phosphorylation on the function of CIC.
Materials and Methods
Cells
HEK293A, HEK293T, MEF, triple knockout Src/Yes/Fyn SYF(−/−) MEFs [referred to as MEF Src(−/−) throughout the article], U87, U251, U118, A172, T98G, and GL261 were obtained from ATCC. Normal human astrocytes (NHA) were described previously (18). Normal mouse astrocytes were purchased from ScienCell Research Laboratories. Cells were maintained in DMEM (Invitrogen) supplemented with 10% heat-inactivated FBS (Wisent) at 37°C in a humidified 5% CO2 atmosphere. Six glioma stem cell (GSC) cultures (GSC 8–18, GSC 7-2, GSC 7–11, GSC 28, and GSC 30) were derived from freshly operated tumor samples from patients with GBM at the University of Texas MD Anderson Cancer Center (Houston, TX) as per guidelines set by the institutional review board and described previously (17). Each patient provided written informed consent for tumor tissues and this study was conducted under protocol LAB03-0687, which was approved by the Institutional Review Board of the University of Texas MD Anderson Cancer Center (Houston, TX; ref. 19). GSCs were maintained as neurospheres in either defined DMEM/F12 media or neurobasal media (Gibco), respectively, in the presence of growth factors EGF (20 ng/mL), recombinant basic FGF (20 ng/mL; R&D Systems), and B27 growth supplement with vitamin A (1:50 working concentration; Life Technologies) as described previously (17). Endogenously HA-tagged CIC in HEK293 cells was described previously (17). Briefly, the following DNA constructs were transfected: pRNAT-H1.3(Hygro), pX459-CICend, and double stranded donor DNA, 5′-CCCCAGCCCTCCCCCCCACCCCCAGGTCCCTCCACAGCTGCCACAGGCAGGTACCCCTACGACGTGCCCGACTACGCCTGAGGGACCCCTGAGAAGATGCCAGGACTTATAGTACCCCCTCAGGACATGG. Cells were selected with hygromycin and monoclonal lines were screened. To generate GL261, U87, or GSC 7-2 cells that express control, FLAG-CIC(WT), or FLAG-CIC(Y1455F) the following pMXs-GW-FLAG-IRES-BsdR transfer plasmids, along with pUMVC (Addgene 8449) and pCMV-VSV-G (Addgene 8454) were used to generate retroviral supernatants as described previously (17). Cells were selected in blasticidin. All cell lines were routinely tested for Mycoplasma infection using the PlasmoTest Kit (InvivoGen). Cell lines were not specifically authenticated and were used within 15 passages.
Plasmids
CIC cDNA was a kind gift from Paul Scotting (University of Nottingham, Nottingham, England). The cDNA was prepared for Gateway system using a two-step PCR with primary gene specific primers (5′- CAAAAAAGCAGGCTCCACCATGTATTCGGCCCACAGGCCC-3′; 5′-CAAGAAAGCTGGGTTTCACCTGCCTGTGGCAGCTGTG-3′) and secondary AttB-specific primers (5′- GGGGACAAGTTTGTACAAAAAAGCAGGCTCCACC- 3′; 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTT-3′). Mutations were introduced using site-directed mutagenesis with KOD polymerase (Novagen, Merck). Primers used: R201W (5′- ATGGGCCGCCAGATGTGGTCCTTCTCCCGCTT-3′; 5′-AAGCGGCACCAGGCCCTGGTCCACCAGC-3′) and R1515H (5′- CGTGAGGTGCACCAGAAGATCATGCAGGCTGC- 3′; 5′-GATCTTCTGGTGCACCTCACGGATCTTCAACTG-3′). The Y1455F plasmids were generated using overlap extension and the following four primers: CIC-Eco47IIIF: 5′-CAG CAG AGCGCTTTGCTGAGTTGCCTG. CIC-Y1455F-R: 5′-GGGAGGAGTATGGCACCTTGTCAAACTCTAGCTCCCCAAGCACGTCC CIC-Y1455F-F: 5′-GGACGTGCTTGGGGAGCTAGAGTTTGACAAGGTGCCATACTCCTCCC. NotI-CIC-R: 5′-CTGCTG GCGGCCGC TCACCTGCCTGTGGCAGCTGTG. The fragment generated was digested with Eco47III and NotI and ligated into the parental plasmid that was digested with the same enzymes and gel purified. All variants were sequence verified, using standard Gateway sequencing primers and gene internal primers (5′-TGCCCTACCCAAGGAACGG-3′; 5′-CAGGCGCTACAGGAACTGACG-3′; 5′-CGCCTGCTTCCTCCTCAGC-3′; 5′-CCACACTTGGTGGCTGGACC-3′; 5′-TCAGTTTCTCCCGTGCAGGC-3′; 5′-GCACCCACCTCCTCAGCACC-3′; 5′-CAGAGACCTGGACTCCCACGG-3′; and 5′-CCCACCCTGCAGTCTCTGGC-3′). Sequence was compared with accession number NM_015125. Subsequently the CIC cDNA was subcloned into modified pMXs Vektors (FLAG-tag, Gateway-cassette, IRES, and BsdR) using the LR-reaction following the manufacturer’s protocol (Invitrogen) a generous gift from Dr. Stefan Pusch (Heidelberg University Hospital, German Cancer Center). pCMV5-HA-CIC (DU19108) and pcDNA5-FRT/TO-GFP-CIC (DU16689) were purchased from MRC-PPU University of Dundee (Dundee, Scotland). HA-ERK (8974), c-Src (42202), and c-Src (K295R Y527F) were purchased from Addgene. EGFR-GFP was a kind gift from Dr. Yi Wang (Radiation Biology and Health Branch, Chalk River ON, Canada).
Mass spectrometry
HEK293 cells transfected with an empty plasmid control, HA-CIC(WT) alone or HA-CIC(WT) and c-Src together were lysed and immunoprecipitated with anti-HA antibody. The immunoprecipitated protein was trypsin digested, followed by LC/MS-MS), was performed at the SPARC BioCentre mass spectrometry facility of the Hospital for Sick Children (Toronto, Ontario, Canada).
Luciferase assay
Cells were transfected in triplicate with pGL3-ETV5 plasmid, which contains four consensus CIC octameric motifs, and pRL-SV40 (Promega) Renilla luciferase control as previously described (17). Cells were transfected with the indicated plasmids and lysates were assayed for luciferase activity. A Dual-Luciferase Reporter Assay System (Promega) was used, and luminescence was measured using a GloMax 20/20 Luminometer (Promega). Relative light units from firefly luciferase were normalized against Renilla luciferase values.
Immunoprecipitation, oligonucleotide pull-down assay, and immunoblotting
Immunoprecipitation and Western blotting were performed as described previously (17). Cells were harvested in EBC lysis buffer (50 mmol/L Tris, pH 8, 120 mmol/L NaCl, and 0.5% NP-40) and supplemented with protease inhibitors (Roche). Specifically, lysates were immunoprecipitated using the indicated antibodies along with protein A-Sepharose (Repligen). Bound proteins were washed five times in NETN buffer (20 mmol/L Tris, pH 8, 100 mmol/L NaCl, 1 mmol/L EDTA, and 0.5% NP-40), eluted by boiling in sample buffer and resolved by SDS-PAGE. Proteins were electro-transferred onto polyvinylidene difluoride membrane (Bio-Rad), blocked, and probed with the indicated antibodies. For experiments in which the nuclear or cytoplasmic fractions were prepared the Nuclear Extraction Kit (Cayman Chemicals) was used according to the manufacturer’s instruction and immunoblotting was performed as stated above. For denaturing immunoprecipitations used to detect ubiquitylation lysates were supplemented with 1% SDS and boiled for 5 minutes to denature proteins and disrupt interactions. The boiled lysates were diluted so the final concentration of SDS was less than 0.1% and immunoprecipitation was performed. Oligonucleotide pull-down assay was performed as described previously (17). The following oligos were annealed ETV5-WT octameric repeat: 5′-Biotin-CGCGTTTTTTATGAATGAAAAACGTCCTTA and 5′-TAAGGACGTTTTTCATTCATAAAAAACGCG or for ETV5-Mutant octameric repeat the following oligos were annealed: 5′-Biotin-CGCGTTTTTTATTAAAAGGAAACGTCCTTA and 5′-TAAGGACGTTTCCTTTTAATAAAAAACGCGC. Annealed oligonucleotides (1 μg) was bound to streptavidin agarose (Thermo Fisher Scientific) and mixed with lysate for 2 hours. Bound proteins were resolved as indicated above.
Antibodies
The following antibodies were obtained from Cell Signaling Technology: HA (C29F4) (1:6,000), c-Src (2108), pSrc(416) (2101), Lamin(A/C) (2032), pERK (4370), β-actin (8H10D10) (1:20,000), and α-tubulin (2144) (1:5,000). pERK (sc-7383 and sc-16982-R) and GFP (sc-9996) (1:6,000) were obtained from Santa Cruz Biotechnology. Phospho Ser/Thr (ab17464), capicua (ab123822), and ETV1 (ab81086) were obtained from Abcam. The following antibodies were purchased from Millipore: capicua (ABN446), capicua (MABN449), EGFR, and pTYR. FLAG-M2 (F1804), β-actin (A5316) (1:10,000), vinculin (V9264) (1:30,000), ETV5 (WH0002119M2), and polyclonal ERK (M5670) antibodies (1:5,000) were obtained from Sigma. All antibodies were utilized at a 1:1,000 dilution unless otherwise specified.
Chemicals and reagents
PD98509 (P215) and dasatinib were obtained from Sigma. EGF was purchased from Gibco. Streptavidin-agarose resin was obtained from Thermo Fisher Scientific (20349).
Cell proliferation
Equal numbers of cells were plated in triplicate in 96-well plates and cellular proliferation was assessed using Alamar Blue proliferation assay as per the manufacturer’s instructions (Invitrogen) or 5-bromodeoxyuridine (5-BrdU) cell proliferation assay as per the manufacturer’s instructions (BioVision) as described previously (17). Trypan blue exclusion assay was performed by directly counting cells plated in triplicate using the Beckman Coulter Vi-CELL (12-Sample Carousel) Cell Viability Analyzer (Beckman Coulter) as described previously (17). For drug sensitivity assays, 5,000 cells were seeded in 96-well plates and 24 hours later the cells were treated with increasing concentrations of drugs and Alamar Blue assay was performed.
Patient-derived tumor samples
Patient resected samples were obtained from Toronto Western Brain Tumor Bank and processed in accordance with a University Health Network Research Ethics Board–approved protocol.
Xenograft GBM model
All animal procedures were carried out according to animal user protocols approved ethically by the Institutional Animal Care Committee under the guidelines of the Canadian Council on Animal Care and the University Health Network Research Ethics Board as described previously (17). Briefly, NOD/SCID (male 8-week-old) were anaesthetized using 0.5 mg/g of intraperitoneal injection of 20 mg/mL Avertin (Sigma-Aldrich) and 5 mg/kg of the presurgical analgesic Anafen 1 mg/mL (Ketoprofen) was administered subcutaneously. TearGel (Novartis) was applied to the eyes to prevent corneal dehydration and abrasion. Once a toe pinch no longer elicited a response, the scalp was cleaned and hair removed, and a midline incision was made from the ears to the eyes. Underlying periosteum was frozen with 2% Lidocaine (Bimeda MTC) and removed with scissors. Mice were placed on a digital stereotaxic frame and from bregma the cortex coordinates were identified (X:1.6, Y:1, Z:0.6). A high-speed dental drill with a 0.7-mm adaptor (Fine Science Tools) was used to bore a whole through the skull. A total of 1 × 105 U87 cells resuspended in 10 mL of sterile 1× PBS were injected 3 mm deep through the hole using a 10-mL 30-G Hamilton microsyringe, over a 1-minute period. Mice were sutured and returned to a fresh sterile heated cage to recover and supplemented with 0.3 mg/ml enrofloxacin in the drinking water (Baytril Bayer/CDMV, catalog no.102207).
The Cancer Genome Atlas data analysis
mRNA dataset for GBM (n = 143) and normal patients (n = 5) were downloaded from the UCSC Cancer Genomics Browser (https://genome-cancer.ucsc.edu). The gene expression datasets (RSEM normalized count) were quantified using the Illumina Hiseq _RNASeqV2 by the UCSC Cancer Browser team and log2 transformed. We evaluated IDH wild-type GBM only and did not include any IDH-mutant GBM.
Ingenuity pathway analysis
To perform pathway analysis, we initially performed differential expression analysis between GBM versus normal patients. We then selected significantly differentially expressed genes (n = 6,668 genes) that were passed from a criteria (FDR < 0.05, fold change > 2 or fold change < −2) based on the changes in expression between GBM versus normal patients. Ingenuity pathway analysis spring release (March 2018) of differentially expressed genes in GBM versus normal patients identified several significantly contributed pathways.
Statistical analysis
All experiments were performed at least in triplicate with mean and SEM reported. Log-rank statistics were performed on survival curves. For direct comparisons, an unpaired Student t test was carried out. Significance was defined as *, P < 0.05 unless specifically stated in figure legends.
Results
CIC is tyrosyl-phosphorylated in response to EGF stimulation
While earlier work investigated phosphorylation of serine and threonine residues on CIC in response to ERK downstream of EGFR activation (17), tyrosine phosphorylation is an equally important posttranslational modification that regulates protein function. We asked whether CIC is posttranslationally modified via tyrosine phosphorylation in response to EGFR activation. HEK293 cells were transfected with plasmids encoding HA-tagged CIC and EGFR and cell lysates were immunoprecipitated using an anti-HA antibody. The HA-CIC immunoprecipitate reacted with anti-pSer/Thr antibody, as expected, but also exhibited marked tyrosyl phosphorylation in response to signaling in EGFR-activated cells (Fig. 1A). We confirmed this finding for mouse embryonic fibroblasts (MEF) cotransfected with HA-CIC and EGFR (Fig. 1B). We next asked whether CIC tyrosyl phosphorylation can be detected in an endogenous setting (in the absence of overexpression). CIC was tyrosyl-phosphorylated upon EGF treatment of HEK293 cells in which a C-terminal HA-epitope tag was genetically engineered onto endogenous CIC using CRISPR/Cas9 (Fig. 1C). Likewise, endogenous CIC in NHA was found to be tyrosyl-phosphorylated in response to EGF treatment (Fig. 1D). As expected, EGF treatment triggered derepression of CIC target ETV5 under these conditions (Fig. 1D). These results suggest that CIC can be posttranslationally modified by tyrosine phosphorylation in response to signaling downstream of EGFR.
Figure 1.
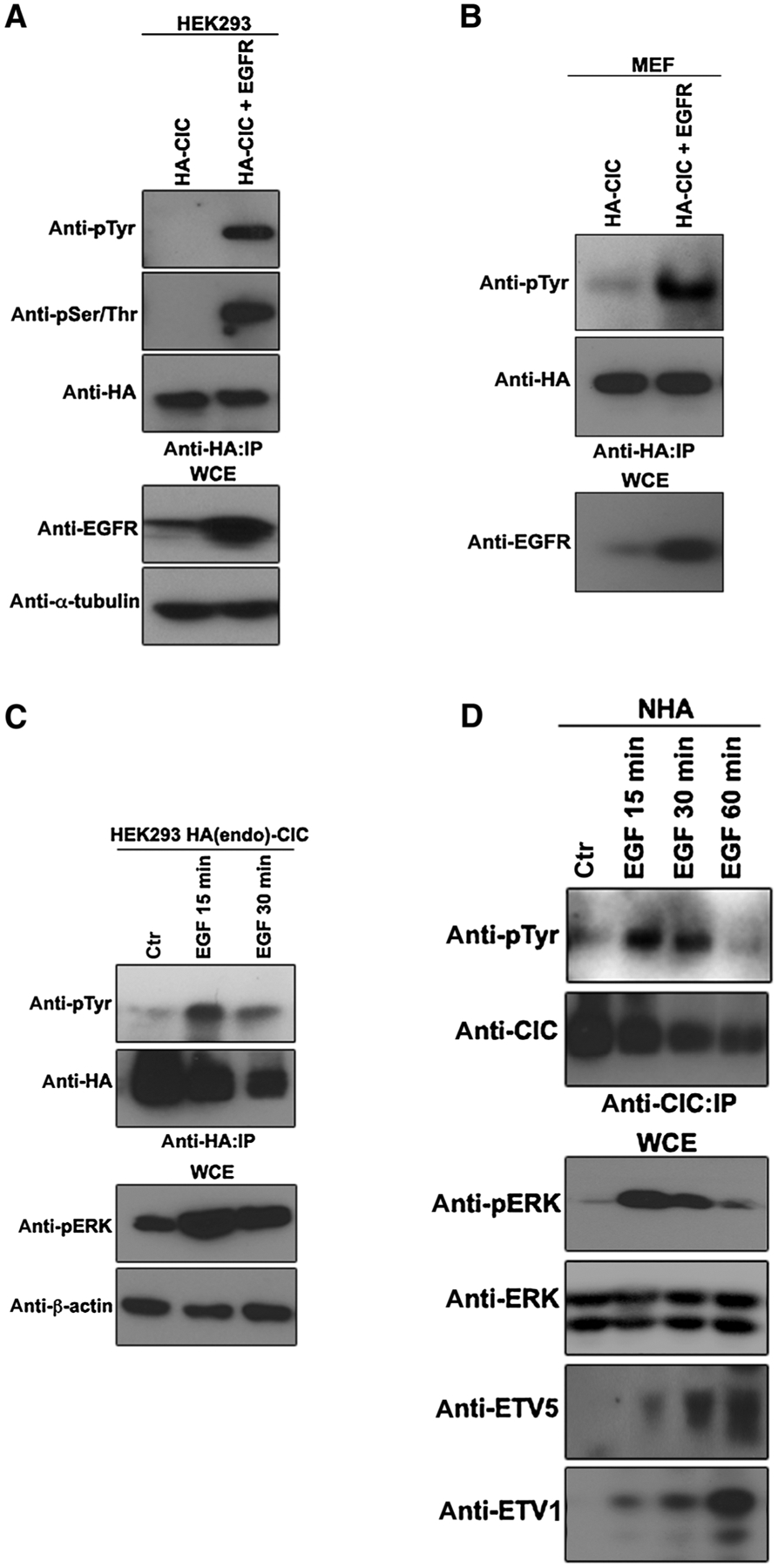
EGFR activation promotes tyrosine-phosphorylation of CIC. A, HEK293 cells transfected with the indicated plasmids were lysed and immunoprecipitated (IP) with anti-HA antibody followed by immunoblotting with indicated antibodies. B, MEFs transfected with the indicated plasmids were lysed and immunoprecipitated with anti-HA antibody followed by immunoblotting with indicated antibodies. C, HEK293 cells endogenously tagged with HA-CIC, treated with or without EGF at indicated time points were lysed, and immunoprecipitated with anti-HA antibody followed by immunoblotting with indicated antibodies. D, NHA treated with or without EGF at indicated time points were lysed and immunoprecipitated with anti-CIC antibody followed by immunoblotting with indicated antibodies. WCE, whole-cell extract. Ctr, control.
c-Src binds to and tyrosine-phosphorylates CIC
c-Src is another important kinase that is activated in response to EGFR signaling. c-Src tyrosine-phosphorylates its targets both in the cytoplasm and in the nucleus. To explore the potential of c-Src to tyrosine-phosphorylate CIC, HEK293 cells were engineered to overexpress c-Src, which was able to bind and promote the tyrosyl phosphorylation of endogenous CIC (Fig. 2A). A kinase-dead c-Src (K295R, Y527F) mutant was deficient in both of these abilities (Fig. 2A). We recently demonstrated that in GBM, endogenous CIC is serine-phosphorylated and cleared by activated ERK (17). To examine whether activated c-Src tyrosine-phosphorylates CIC, we stably expressed FLAG-CIC(WT) in U87 cells and cotransfected kinase activate and inactive c-Src. Like HEK293 cells, only kinase active c-Src–bound and tyrosine-phosphorylated CIC in U87 cells (Fig. 2B). Furthermore, we found that dasatinib, a well-established inhibitor of c-Src kinase activity, prevented EGF-mediated tyrosyl phosphorylation of CIC (Fig. 2C), further substantiating that EGFR utilizes c-Src to tyrosine-phosphorylate CIC. Importantly, we show that endogenous CIC is tyrosine phosphorylated in response to EGF treatment correlating with c-Src binding in NHA cells, while dasatinib pretreatment ameliorated this effect (Fig. 2D). To confirm that EGFR utilizes c-Src to tyrosine-phosphorylate CIC we obtained Src-knockout MEFs [MEF Src(−/−)] and showed that endogenous CIC was not tyrosine phosphorylated in response to EGF treatment in these cells lacking Src kinase activity (Fig. 2E). To localize the region within CIC that c-Src recognizes, we utilized two CIC fragments one comprising the N-terminal HMG-box DNA-binding domain CIC(1–450) and the other comprising the C-terminal C1 repressor domain CIC (450-End). Following cotransfection with c-Src into HEK293 cells, only CIC(1–450) fragment showed an interaction with c-Src (Fig. 2F), but surprisingly, was not tyrosyl phosphorylated (Fig. 2G). Specific hotspot mutations of CIC, CIC(R201W) and CIC(R1515H) found in human gliomas, which fail to bind to DNA (20) bound strongly to c-Src and underwent tyrosyl phosphorylation (Fig. 2H), suggesting that phosphorylation of CIC is not dependent on its ability to bind to the DNA. Collectively, these findings show that c-Src binds and tyrosine-phosphorylates CIC in response to EGFR activation.
Figure 2.
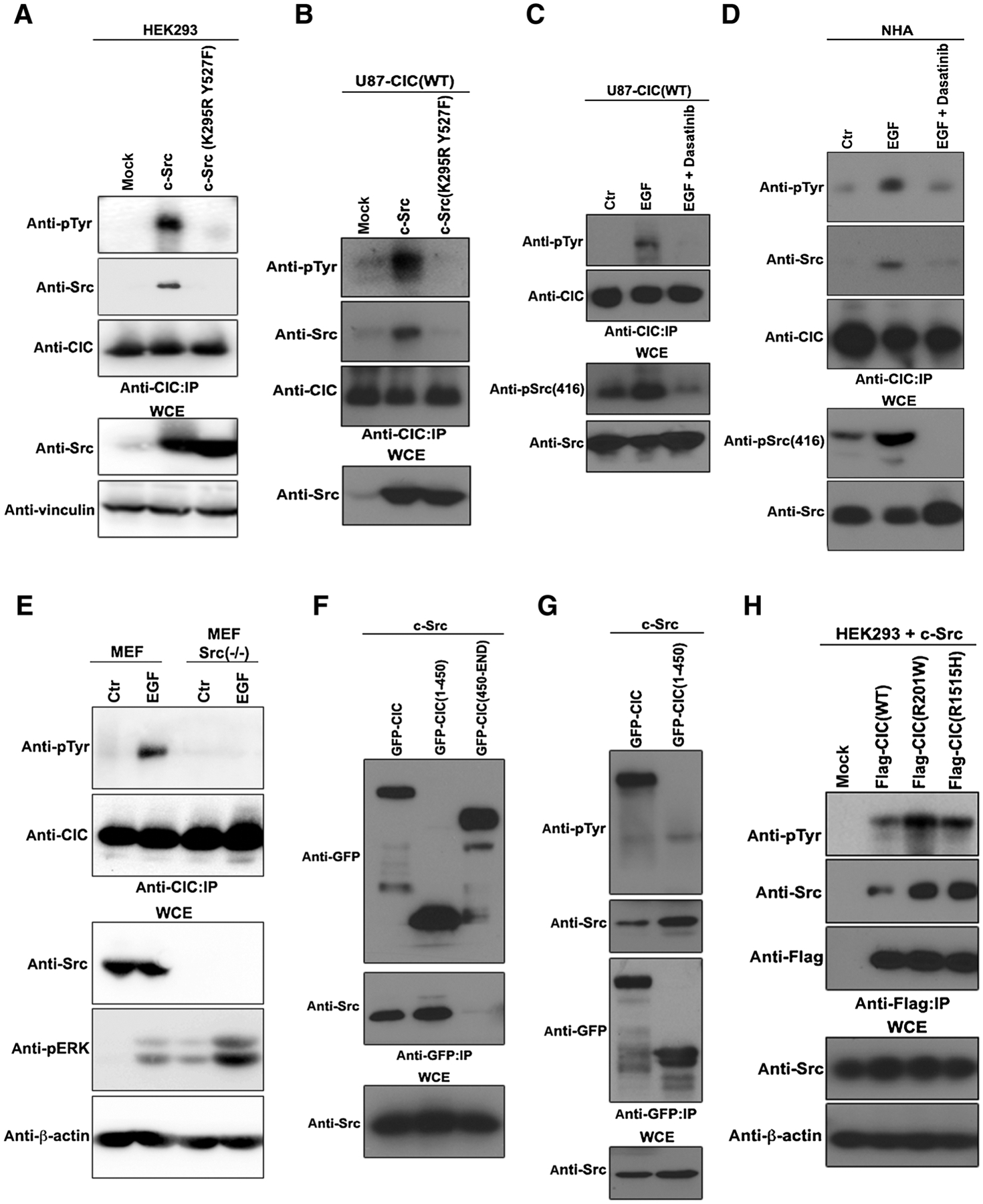
c-Src binds and tyrosine-phosphorylates CIC in response to EGFR activation. A, HEK293 cells transfected with the indicated plasmids were lysed and immunoprecipitated (IP) with anti-CIC antibody followed by immunoblotting with indicated antibodies. B, U87 stably expressing FLAG-CIC(WT) cells transfected with the indicated plasmids were lysed and immunoprecipitated with anti-CIC antibody followed by immunoblotting with indicated antibodies. C, U87 stably expressing FLAG-CIC(WT) cells were pretreated with or without 100 nmol/L of dasatinib for 1 hour prior to treatment with or without EGF were lysed and immunoprecipitated with anti-CIC antibody followed by immunoblotting with indicated antibodies. D, NHAs pretreated with or without 100 nmol/L of dasatinib for 1 hour prior to treatment with or without EGF were lysed and immunoprecipitated with anti-CIC antibody followed by immunoblotting with indicated antibodies. E, MEFs or Src(−/−) MEFs were treated with or without EGF and equal amounts of lysates were immunoprecipitated with anti-pTyr antibody and immunoblotted with the indicated antibodies. F, HEK293 cells transfected with the indicated plasmids were lysed and immunoprecipitated with anti-GFP antibody followed by immunoblotting with indicated antibodies. G, HEK293 cells transfected with the indicated plasmids were lysed and immunoprecipitated with anti-CIC antibody followed by immunoblotting with indicated antibodies. H, HEK293 cells transfected with the indicated plasmids were lysed and immunoprecipitated with anti-FLAG antibody followed by immunoblotting with indicated antibodies. WCE: whole-cell extract.
Residue Y1455 of CIC is phosphorylated by c-Src
To examine the tyrosine residue(s) phosphorylated by c-Src, we conducted mass spectrometry of CIC-enriched HEK293 lysates cotransfected with c-Src, which revealed tyrosine 1455 as the potential phosphorylation site (Fig. 3A), consistent with our findings showing that the 450-end region of CIC was tyrosine-phosphorylated (Fig. 2G). A cross-species alignment of CIC protein shows that Y1455 is a conserved residue and that aspartic acid (D) or glutamic acid (E) residue immediately follows a tyrosine (Y) residue, and in humans is consistent with a candidate YD c-Src phosphorylation motif (ref. 21; Fig. 3B). To validate that c-Src phosphorylates tyrosine 1455 on CIC, HEK293 cells were cotransfected with c-Src and a plasmid encoding a mutant [tyrosine (Y) to phenylalanine (F) at residue 1455] to prevent Y1455 tyrosine phosphorylation. The Y1455F allele showed markedly reduced phosphorylation of CIC compared with wild-type CIC (Fig. 3C). To examine whether CIC is tyrosine phosphorylated on the same residue in GBM cells we stably expressed FLAG-CIC(WT) and FLAG-CIC(Y1455F) in patient-derived GSCs that are otherwise devoid of CIC (17). Similar to HEK293 cells, wild-type, but not tyrosine-defective CIC-Y1455F mutant was tyrosine phosphorylated in response to EGF treatment (Fig. 3D). Notably, Y1455 is located near the C1 repressor domain, which is essential for the recognition and binding of the N-terminal HMG-box to target DNA, as well as nuclear localization signal (NLS) and predicted nuclear export signals (NES) of CIC (Fig. 3E) implicating the potential functional implication of this phosphorylation event on CIC function. These results show that tyrosine 1455 is phosphorylated on CIC in response to EGF treatment or c-Src expression.
Figure 3.
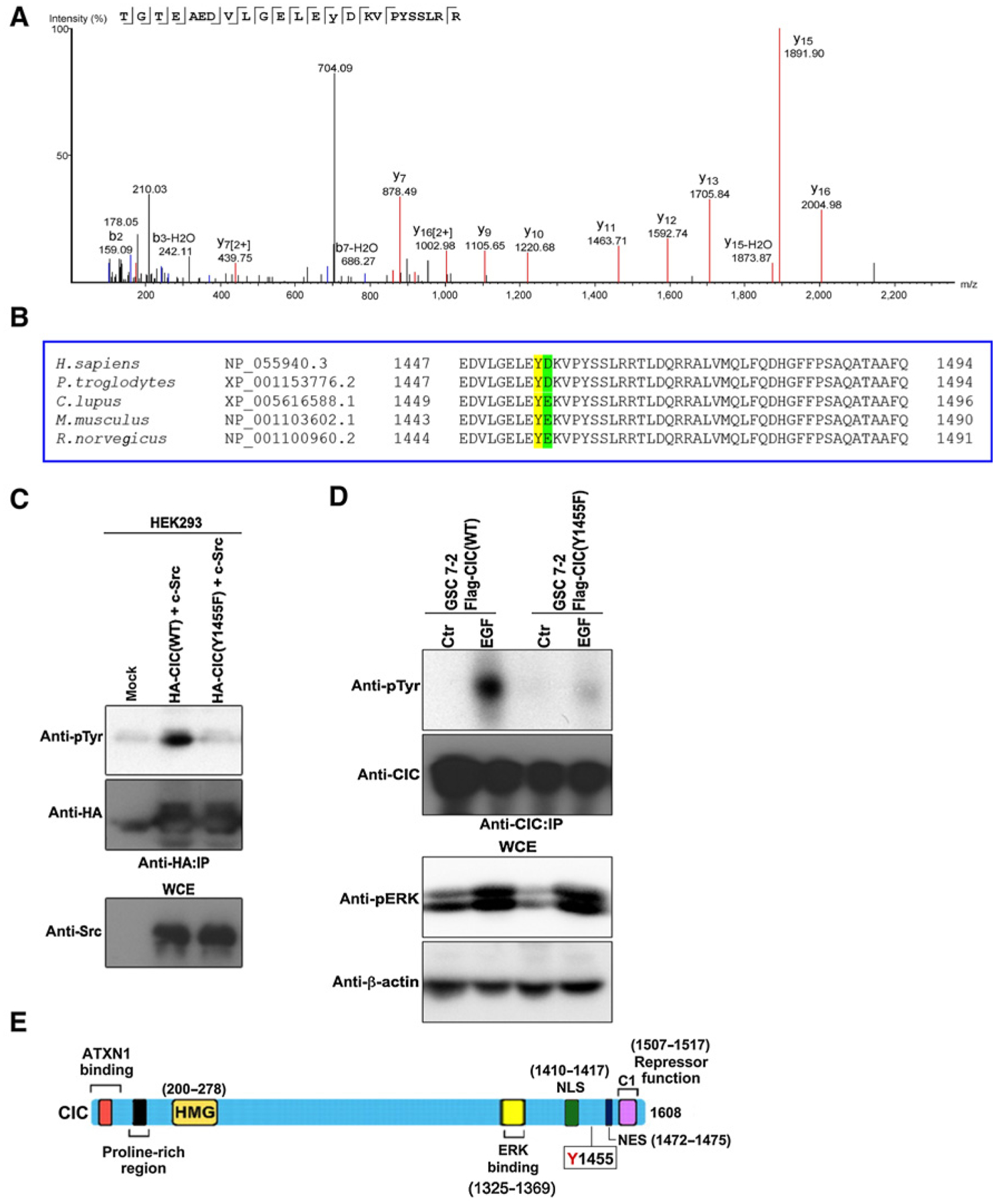
c-Src phosphorylates CIC on tyrosine 1455 residue. A, Mass spectrometry analysis identifies tyrosine phosphorylation on residue 1455 of CIC from HEK293 lysates cotransfected with HA-CIC and c-Src. CIC was immunoprecipitated (IP) using HA antibody, trypsin digest of the sample, followed by LC/MS-MS analysis was performed at the SPARC BioCentre mass spectrometry facility of the Hospital for Sick Children (Toronto, Ontario, Canada). Shown is an annotated representative CID spectrum for phosphorylated peptide containing Y1455. β ions are indicated in blue, and γ ions are indicated in red. B, A multiple sequence alignment was performed using Clustal Omega (29). CIC tyrosine (Y) 1455 residues are conserved across species and are in yellow. Shown in blue are aspartic acid (D) residues or glutamic acid (E) that immediately follows a Tyr (Y) residue, making a candidate c-Src phosphorylation motif (21). C, HEK293 cells transfected with the indicated plasmids were lysed and immunoprecipitated with anti-HA antibody followed by immunoblotting with indicated antibodies. D, GSC 7–2 stably expressing either FLAG-CIC(WT or Y1455F) cells were treated with or without EGF, lysed, and immunoprecipitated with anti-HA antibody followed by immunoblotting with indicated antibodies. E, Diagram depicting functional domains of CIC with the tyrosine (Y) 1455 residue. Ataxin 1 (ATXN1)-binding domain, DNA-binding HMG box domain, NLS, and the predicted NES that were generated using the publically available NES prediction server, NESbase (24). Figure adapted with permission from Journal of Cell Science (12). Ctr, control.
Y1455 phosphorylation is not required for CIC’s repressor function
Residue 1455 is near the functional C1 domain, which mediates CIC’s DNA-binding ability (20) and we therefore asked whether c-Src–mediated tyrosine phosphorylation at this residue influences CIC’s repressor function on its targets, including the oncogenic transcription factors ETV1, ETV4, and ETV5 (12, 13). To test this, we first examined the role of tyrosine phosphorylation of CIC on its repressor function. Using the Y1455F phosphorylation-defective CIC mutant we show that it retains the ability to repress luciferase activity through the octameric consensus motif (TGAATGAA) of ETV5, similar to levels of wild-type CIC (Fig. 4A). Consistent with this finding, we found that cells stably expressing FLAG-CIC(Y1455F) suppressed ETV5 and ETV1 protein levels and reduced cellular proliferation similar to wild-type controls. We note that this result is in contrast to cells that express the disease-associated mutation in residue 1515 (R1515H) or 201 (R201W; Fig. 4B and C). The finding that the Y1455F CIC allele retained its transcriptional repressive activity was confirmed in additional cell lines, including patient-derived GSCs (Fig. 4D–G). Overall, tyrosine phosphorylation at residue 1455 did not affect CIC’s repressor activity and effect on proliferation in GBM cells. These findings suggest that c-Src–mediated tyrosine phosphorylation negatively regulates CIC repressor function by promoting release of CIC from its target genes in a manner similar to ERK-mediated serine/threonine phosphorylation. We then used a biotinylated DNA pull-down assay that encodes a portion of the ETV5 promoter containing the octameric CIC-binding motif (TGAATGAA). With this assay, we show that overexpression of c-Src promotes the release of both wild-type CIC and the Y1455F phosphorylation–defective mutant from the ETV5 DNA octamer (Fig. 4H), in a manner that was rescued by pretreatment with the specific ERK inhibitor PD98509 (Fig. 4I). These findings indicate that c-Src–induced tyrosine 1455 phosphorylation of CIC does not mediate the release of DNA-bound CIC.
Figure 4.
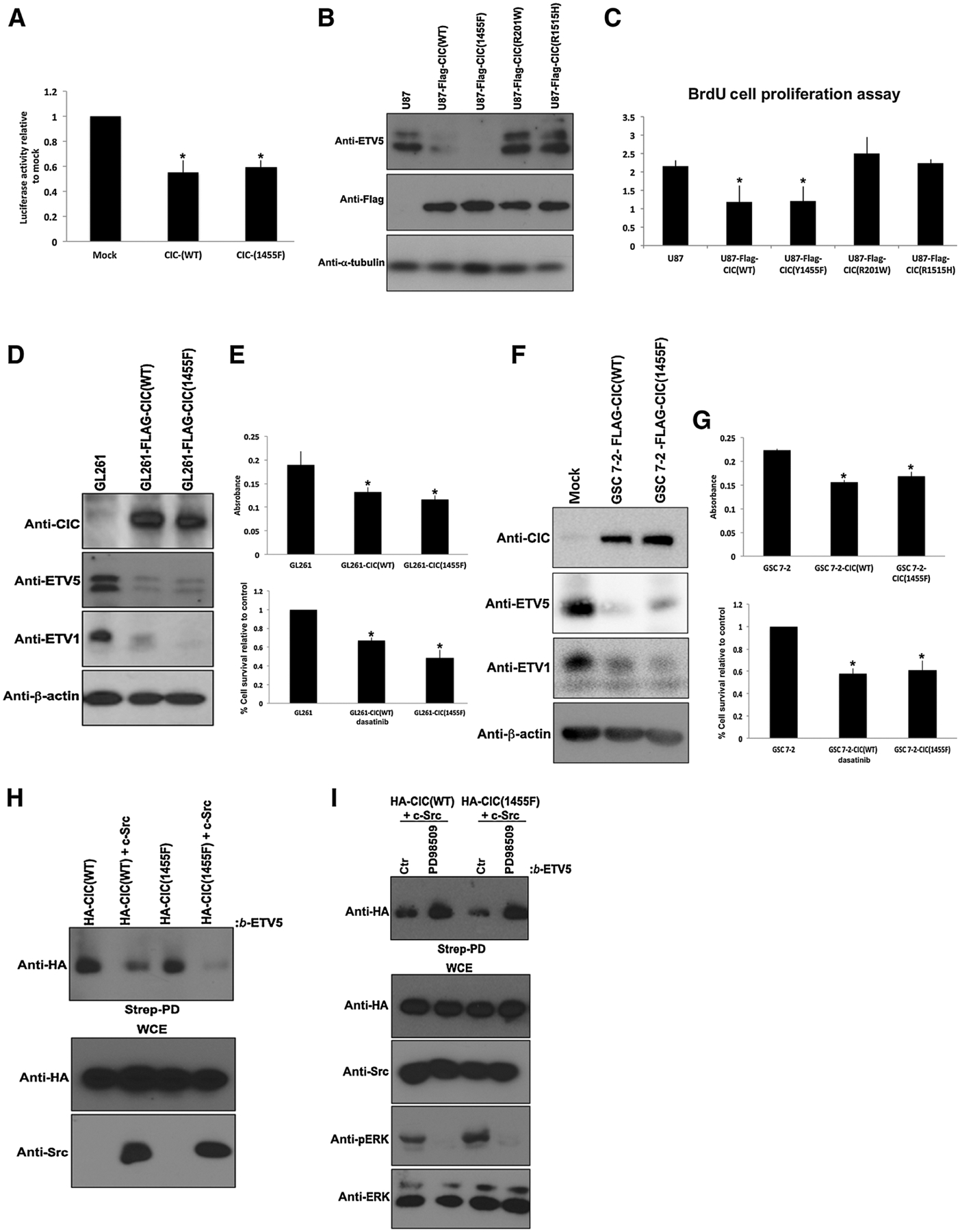
Phosphorylation-defective CIC-Y1455F mutant retains strong repressor function. A, pGL3–ETV5 was cotransfected with empty vector (mock), HA-CIC(WT), or HA-CIC(Y1455F)-mutant plasmid into HEK293 cells, and luciferase expression was measured. The bar graph depicts the relative levels of luciferase activity normalized to empty vector control. The data shown are the mean ± SEM from luciferase assays performed in triplicate. *, P < 0.05 Student t test compared with mock. B, U87 control or stably expressing FLAG-CIC(WT, Y1455F, R201W, or R1515H) cells were lysed and immunoblotted with indicated antibodies. C, Equal number of U87 control or stably expressing FLAG-CIC(WT, Y1455F, R201W, or R1515H) cells were plated and cell proliferation was assessed by BrdU incorporation assay. Data represent mean ± SEM of three independent experiments performed in octuplet. *, P < 0.05 Student t test compared with control. D, GL261 cells stably expressing control, CIC-FLAG(WT), or CIC-FLAG(Y1455F) plasmids were lysed and immunoblotted with indicated antibodies. E, Equal number of GL261 cells stably expressing either the control, CIC-FLAG(WT), or CIC-FLAG(Y1455F) plasmids were plated and Alamar blue (top) or trypan blue exclusion (bottom) assay was performed. Data represent mean ± SEM of three independent experiments performed in octuplet. *, P < 0.05 Student t test compared with GL261 control. F, GSC 7-2 stably expressing either the control, CIC-FLAG(WT), or CIC-FLAG(Y1455F) plasmids were lysed and immunoblotted with indicated antibodies. G, Equal number of GSC 7-2 stably expressing either the control, CIC-FLAG(WT), or CIC-FLAG(Y1455F) plasmids were plated and Alamar blue (top) or trypan blue exclusion (bottom) assay was performed. Data represent mean ± SEM of three independent experiments performed in octuplet. *, P < 0.05 Student t test compared with GSC 7-2 control. H, HEK293 cells transfected with indicated plasmids were lysed and an equivalent amount of lysate was incubated with Streptavidin agarose bound to biotinylated ETV5 oligonucleotides octameric motif, and bound proteins were detected by immunoblotting. I, HEK293 cells transfected with indicated plasmids were either pretreated with or without the ERK inhibitor, PD98509, lysed, and an equivalent amount of lysate was incubated with Streptavidin agarose bound to biotinylated ETV5 oligonucleotides octameric motif, and bound proteins were detected by immunoblotting. Ctr, control; strep-PD, Streptavidin pull-down; WCE, whole-cell extract.
Tyrosyl phosphorylation of CIC triggers nucleocytoplasmic shuttling
EGFR signaling triggers derepression of CIC from its target genes, which can be sustained by two mechanisms, protein degradation and nuclear export. We have previously demonstrated that ERK activation triggers nuclear degradation of CIC through the E3 ubiquitin ligase PJA1 (17). Because c-Src–mediated tyrosyl phosphorylation of CIC did not affect its DNA-binding ability (Fig. 4H and I), we hypothesized that c-Src mediates tyrosyl phosphorylation of CIC to influence nucleocytoplasmic shuttling. The close proximity of Y1455 to the putative NES of CIC is consistent with this as a possible mechanism (Fig. 3E). Ectopic expression of c-Src or EGFR, but not kinase dead c-Src(K295R, Y527F) triggered nuclear export of wild-type CIC (Fig. 5A and B), demonstrating that active c-Src is required for nucleocytoplasmic shuttling of CIC to occur. To confirm these findings in an endogenous setting, HEK293 cells endogenously tagged with HA-CIC were treated with EGF in the presence and absence of either the ERK inhibitor (PD98509) or Src kinase inhibitor (dasatinib). Nuclear CIC was significantly reduced in EGF-treated cells that were rescued in cells pretreated with PD98509 (Fig. 4C). This confirms our previous findings that ERK promotes degradation of nuclear CIC bound to the DNA (1). Importantly, we show for the first time that EGF treatment concordantly promotes cytoplasmic accumulation of CIC that can be rescued by pretreatment with dasatinib (Fig. 5C). Similarly, in MEFs lacking any Src kinase activity, CIC failed to accumulate in the cytoplasm following EGF treatment (Fig. 5D). These findings suggest that in addition to ERK-mediated degradation of nuclear CIC that is bound to the DNA (1), EGFR also employs c-Src to clear nuclear CIC to the cytoplasm. In fact, we show that the two disease-causing CIC mutants (R201W and R1515H) have a higher affinity to interact with c-Src, to be tyrosine phosphorylated (Fig. 2H) and are preferentially accumulated in the cytoplasm compare with wild-type CIC (Fig. 5E). Because these disease-causing CIC mutants fail to bind to their DNA target (17, 20), further supports our speculation that c-Src tyrosine-phosphorylates nuclear CIC (that is not bound to DNA) for export to cytoplasm. To formally assess the role of tyrosine 1455 phosphorylation and nuclear export, we first transfected HEK293 cells with plasmids encoding HA-CIC(WT) or HA-CIC(Y1455F) in combination with c-Src. Nuclear export of CIC(Y1455F) in response to c-Src expression was drastically reduced compared with the wild-type control (Fig. 5F). Similarly, EGF treatment failed to promote cytoplasmic accumulation in GSCs stably expressing FLAG-CIC(Y1455F) compared with FLAG-CIC(WT)-expressing cells (Fig. 5G). These results suggest that c-Src–mediated tyrosyl phosphorylation of CIC on residue 1455 preferentially promotes nuclear export of CIC that is not bound to its target DNA (Fig. 5H), thereby preventing CIC from its function as a transcriptional repressor.
Figure 5.
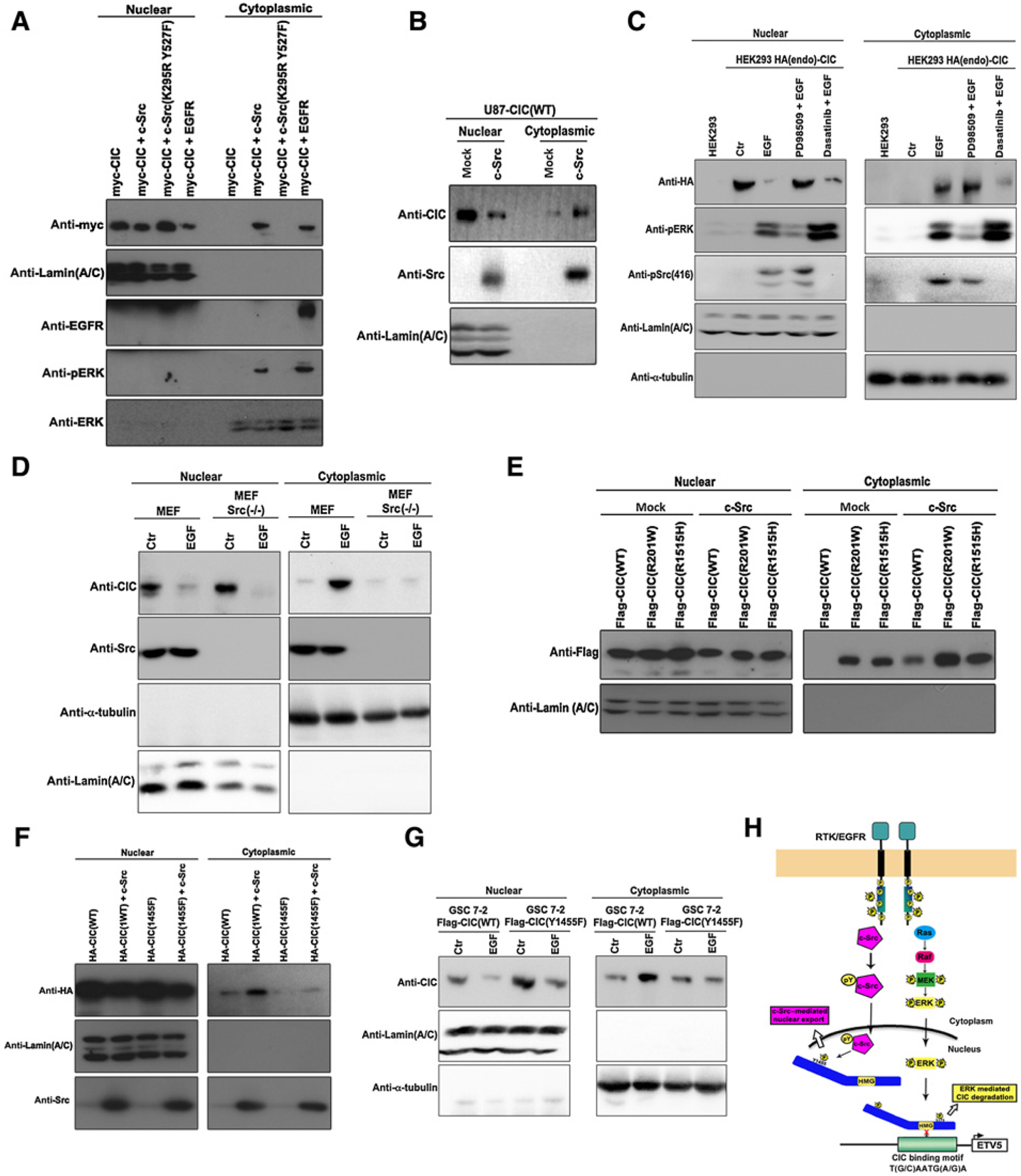
c-Src–mediated tyrosine 1455 phosphorylation regulates CIC nuclear export. A, HEK293 cells transfected with indicated plasmids were lysed and nuclear or cytoplasmic fractions were obtained and immunoblotted with indicated antibodies. B, U87 cells stably expressing CIC-FLAG(WT) transfected with indicated plasmids were lysed and nuclear or cytoplasmic fractions were obtained and immunoblotted with indicated antibodies. C, HEK293 parental cells or endogenously tagged with HA-CIC pretreated for 1 hour with either PD98509 or dasatinib prior to treatment with or without EGF were lysed and nuclear or cytoplasmic fractions were obtained and immunoblotted with indicated antibodies. D, MEFs or Src(−/−) MEFs treated with or without EGF were lysed and nuclear or cytoplasmic fractions obtained and immunoblotted with indicated antibodies. E and F, HEK293 cells transfected with indicated plasmids were lysed and nuclear or cytoplasmic fractions were obtained and immunoblotted with indicated antibodies. G, GSC 7-2 stably expressing FLAG-CIC(WT or Y1455F) treated with or without EGF were lysed and nuclear or cytoplasmic fractions were obtained and immunoblotted with indicated antibodies. H, Diagram depicting two parallel mechanism that regulate CIC function upon EGFR activation, the c-Src–mediated phosphorylation of tyrosine 1455 residue of nuclear CIC that is not DNA bound and ERK-mediated serine phosphorylation and degradation of DNA-bound CIC. Ctr, control.
Dasatinib affects the viability of GBM cells and not their normal counterparts
RTK signaling is a feature of GBM and promotes activation of other important signaling pathways including c-Src. To test whether c-Src is concordantly active in GBM, we assessed phosphorylation of c-Src on tyrosine 416 residue, which is an established marker of c-Src kinase activity (22). All five GBM cell lines test (U87, U251, A172, U118, and T98G) displayed a greater proportion of active c-Src compared with control NHA cells (Fig. 6A). Intracranial xenografts of U87 cells in mice revealed active c-Src in the xenograft tissue, but not in the normal mouse brain tissue (Fig. 6B). Compared with control neural stem cells (NSC), five GSC cultures displayed increase in active c-Src (Fig. 6C). A similar result was observed in 17 of 17 newly diagnosed GBM human tumors tested compared with four samples derived from nonneoplastic brain tissue (Fig. 6D). As additional evidence, mouse glioma GL261 cells also had elevated pSrc(416) level compared with normal mouse astrocytes (Fig. 6E). To validate the relevance of c-Src activation in GBM, we compared differentially expressed genes in GBM versus nonneoplastic brain and found c-Src activation as one of the top pathways upregulated in GBM in The Cancer Genome Atlas data (Supplementary Fig. S1A).
Figure 6.
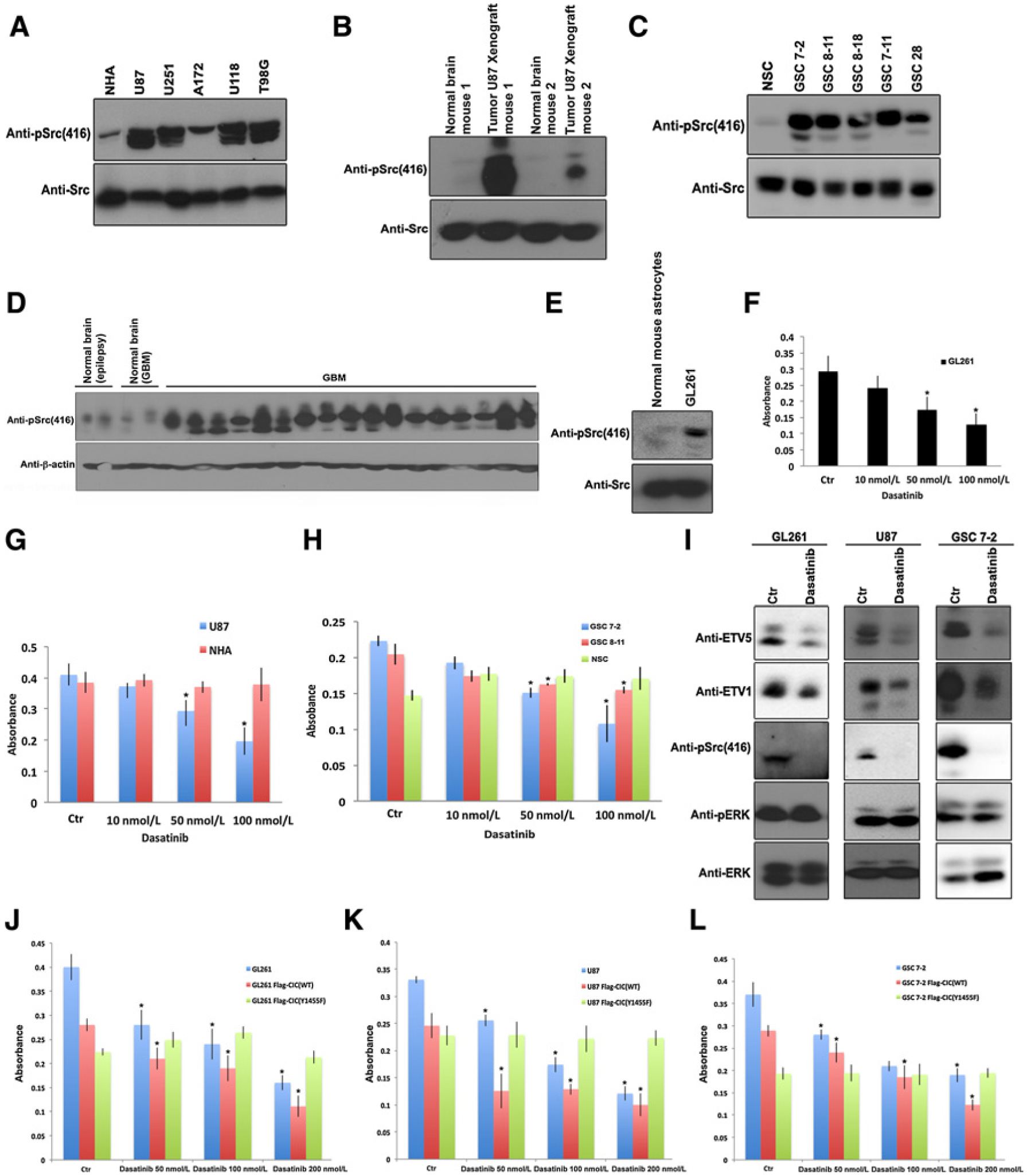
Dasatinib affects viability of GBM cells with elevated c-Src activity but not normal cells with negligible Src activity. A, Human-derived GBM cell lines or NHAs were lysed and protein lysates were immunoblotted with indicated antibodies. B, Tumors or unaffected normal brains obtained from intracranial U87 xenograft from two different mice were lysed and protein lysates were immunoblotted with indicated antibodies. C, GSCs or NSCs were lysed and protein lysates were immunoblotted with indicated antibodies. D, Human operative GBM samples or normal derived brains (NB) were lysed and immunoblotted with indicated antibodies. E, GL261 cells or normal mouse astrocytes were lysed and protein lysates were immunoblotted with indicated antibodies. F, Equal number of GL261 cells were plated and treated with or without indicated concentrations of dasatinib and cell viability was assessed by an Alamar blue assay. Data represent mean ± SEM of three independent experiments performed in octuplet. *, P < 0.05 Student t test compared with control. G, Equal number of U87 or NHAs treated with or without indicated concentrations of dasatinib was plated and cell viability was assessed by an Alamar blue assay. Data represent mean ± SEM of three independent experiments performed in octuplet. *, P < 0.05 Student t test compared with control. H, Equal number of GSC 7-2, GSC 8–11, or NSCs treated with or without indicated concentrations of dasatinib were plated and cell viability was assessed by an Alamar blue assay. Data represent mean ± SEM of three independent experiments performed in octuplet. *, P < 0.05 Student t test compared with control. I, GL261, U87, or GSC 7-2 treated with or without dasatinib 100 nmol/L were lysed and immunoblotted with indicated antibodies. GL261 (J), U87 (K), or GSC 7-2 (L) control or stably expressing FLAG-CIC(WT or Y1455F) treated with or without indicated concentrations of dasatinib were plated and cell viability was assessed by an Alamar blue assay. Data represent mean ± SEM of three independent experiments performed in octuplet. *, P < 0.05 Student t test compared with respective controls.
Given that the c-Src pathway is active in GBM, we tested the effect of increasing concentrations of highly specific Src kinase inhibitor, dasatinib, using clinically relevant doses and lower, on viability of GBM cells versus their normal counterparts. We show that concentrations of dasatinib that are below the clinically relevant dose (100 nmol/L) significantly affect viability of GL261 (Fig. 6F), U87 (Fig. 6G), and GSC-7-2 (Fig. 6H). Notably, dasatinib at 100 nmol/L did not affect viability of normal human astrocytes (NHA) (Supplementary Fig. S1B), NHA cells (Fig. 6G), or NSC (Fig. 6H). NSC did not show a reduction in viability even at doses as high as 750 nmol/L (Supplementary Fig. S1C), further substantiating that dasatinib affects only the viability of cells with active c-Src such as in GBM. Importantly, we found that dasatinib treatment reduces activation of c-Src, but not of ERK, in GL261, U87, and GSC 7-2 cells (Fig. 6I). Consistent with our data showing an interaction of CIC and c-Src, we also found that dasatinib treatment correlated with reduced levels of the key CIC targets ETV1 and ETV5 in these cells (Fig. 6I). To demonstrate that the specificity of dasatinib on cell viability is through c-Src–mediated tyrosine-phosphorylation of CIC we assessed the effect of dasatinib in GL261, U87, and GSC stably expressing FLAG-CIC(WT) or FLAG-CIC(Y1455F). These results demonstrate that dasatinib had no significant effect on the viability of GL261 (Fig. 6J), U87 (Fig. 6K), or GSC 7-2 (Fig. 6L) cells expressing Flag-CIC(Y1455F), compared with cells expressing wild-type CIC. Similar results were observed in U87 cells expressing CIC (R201W or R1515H) mutants (Supplementary Fig. S1D). These findings further support the specificity of dasatinib treatment on c-Src inhibition that concordantly affects CIC repressor function and the expression of its ETV targets in GBM cells.
Discussion
In this article, we explored the regulation of CIC in a series of experiments and validated its relevance in GBMs to further elucidate its biological role in this tumor type. CIC is a transcriptional repressor that inhibits activation of genes downstream of RTK/Ras/ERK signaling. This well-conserved signaling pathway controls many different biological processes and is commonly activated in cancer. We recently found that in the highly malignant and aggressive GBM, which is characterized by hyperactive RTK signaling, CIC protein levels are undetectable due to ERK-induced serine phosphorylation and proteasomal degradation of CIC (17). We extend these findings by showing that the non-RTK c-Src regulates CIC repressor function by tyrosine-phosphorylating and exporting CIC to the cytoplasm. Our findings suggest that activation of EGFR employs two parallel regulatory mechanisms to ensure CIC inactivation and derepression of EGFR-responsive genes. First, EGFR activation leads to ERK-mediated serine-phosphorylation that induces degradation of DNA-bound CIC (17). Second, EGFR activates c-Src, which in turn results in tyrosine-phosphorylation of nuclear CIC that is not DNA bound, promoting its nuclear export (Fig. 5H). Accordingly, it is likely that c-Src–mediated tyrosine-phosphorylation of CIC ensures that free, unbound nuclear CIC is rapidly exported to the cytoplasm and is inaccessible for additional binding to its target DNA following degradation of formerly DNA-bound CIC.
We found that c-Src specifically phosphorylates CIC at tyrosine 1455, which is located within the regulatory C-terminal region of the protein (Fig. 3E). In addition to its close proximity to the C1 repressor domain, which is essential for the binding of the N-terminal HMG-box to its target DNA, Y1455 is also close to the ERK-binding domain and the C-terminal NLS (Fig. 3E). Our work adds to the existing literature on the regulation of localization of CIC. Specifically, prior work suggests that ERK-mediated serine-phosphorylation of CIC prevents its nuclear import (23). Our study provides evidence that c-Src–mediated tyrosine phosphorylation also plays a role in the localization of CIC by promoting its nuclear export. Furthermore, the publicly available NES prediction server, NESbase, a database of proteins experimentally authenticated to identify leucine-rich NES (24), predicted one potential NES at the C-terminal region (1472–1475) of CIC (Fig. 3E). Although this requires further validation, the close proximity of the predicted NES to tyrosine 1455 reinforces our findings that c-Src–mediated tyrosine 1455 phosphorylation is important in the nuclear export of CIC, and is consistent with a role for c-Src–mediated nuclear export of other transcription factors including YAP1 (25), FOXO (26), and RUNX3 (27).
c-Src has several important functional domains that regulate its activity. For instance, c-Src typically recognizes its substrates using its SH3 domain and tyrosine phosphorylates its substrates using its protein kinase domain (22). Consistently, we found that while c-Src tyrosine phosphorylates CIC at its C-terminal end, it bound to CIC at its N-terminus (Fig. 2F and G). Because an N-terminal proline-rich region, close to the DNA-binding HMG box, has been identified (Fig. 3E), we speculate that c-Src recognizes this proline-rich region via its SH3 domain to initiate tyrosine 1455 phosphorylation. This interplay is analogous to binding of CIC to DNA, which occurs through the concerted effort of the C-terminal C1 repressor domain and the N-terminal HMG-box (20).
The activation of c-Src is observed in a large proportion of tumors from multiple organ sites (28), and in this study we characterize a role for c-Src in GBM. We show that GBM cells are highly sensitive to the c-Src inhibitor, dasatinib, even at low concentrations. Importantly, we show that dasatinib does not affect activation of ERK in GBM cells, but it results in derepression of ETV levels, which are known CIC targets. Results from our study demonstrate that in response to EGFR activation, two parallel mechanisms (ERK and c-Src activation) operate to negatively regulate CIC function. Because GBM is characterized with hyper-activated EGFR signaling, our work suggests that dual ERK and c-Src kinase inhibition may be required to fully restore CIC’s tumor suppressor function in GBM, and future studies will be aimed to address this.
Supplementary Material
Implications:
c-Src tyrosine-phosphorylates CIC exports to cytoplasm and inactivates its repressor function in GBM.
Acknowledgments
This work was supported by funding from the Canada Cancer Society Research Institute Innovation Grant and the MacFeeters Hamilton Neuro-Oncology Program.
Footnotes
Note: Supplementary data for this article are available at Molecular Cancer Research Online (http://mcr.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Jimenez G, Guichet A, Ephrussi A, Casanova J. Relief of gene repression by torso RTK signaling: role of capicua in Drosophila terminal and dorsoventral patterning. Genes Dev 2000;14:224–31. [PMC free article] [PubMed] [Google Scholar]
- 2.Roch F, Jimenez G, Casanova J. EGFR signalling inhibits Capicua-dependent repression during specification of Drosophila wing veins. Development 2002; 129:993–1002. [DOI] [PubMed] [Google Scholar]
- 3.Tseng A-SK, Tapon N, Kanda H, Cigizoglu S, Edelmann L, Pellock B, et al. Capicua regulates cell proliferation downstream of the receptor tyrosine kinase/ras signaling pathway. Curr Biol 2007;17:728–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Atkey MR. Capicua regulates follicle cell fate in the Drosophila ovary through repression of mirror. Development 2006;133:2115–23. [DOI] [PubMed] [Google Scholar]
- 5.Bettegowda C, Agrawal N, Jiao Y, Sausen M, Wood LD, Hruban RH, et al. Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science 2011;333:1453–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gleize V, Alentorn A, Connen de Kérillis L, Labussière M, Nadaradjane AA, Mundwiller E, et al. CIC inactivating mutations identify aggressive subset of 1p19q codeleted gliomas. Ann Neurol 2015;78:355–74. [DOI] [PubMed] [Google Scholar]
- 7.Sugita S, Arai Y, Tonooka A, Hama N, Totoki Y, Fujii T, et al. A novel CICFOXO4 gene fusion in undifferentiated small round cell sarcoma: a genetically distinct variant of Ewing-like sarcoma. Am J Surg Pathol 2014;38:1571–6. [DOI] [PubMed] [Google Scholar]
- 8.Antonescu CR, Owosho AA, Zhang L, Chen S, Deniz K, Huryn JM, et al. Sarcomas with CIC-rearrangements are a distinct pathologic entity with aggressive outcome: a clinicopathologic and molecular study of 115 cases. Am J Surg Pathol 2017;41:941–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu HC, Tan Q, Rousseaux MW, Wang W, Kim JY, Richman R, et al. Disruption of the ATXN1-CIC complex causes a spectrum of neurobehavioral phenotypes in mice and humans. Nat Genet 2017;49:527–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kalender Atak Z, Gianfelici V, Hulselmans G, De Keersmaecker K, Devasia AG, Geerdens E, et al. Comprehensive analysis of transcriptome variation uncovers known and novel driver events in T-cell acute lymphoblastic leukemia. PLos Genet 2013;9:e1003997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simón-Carrasco L, Jiménez G, Barbacid M, Drosten M. The Capicua tumor suppressor: a gatekeeper of Ras signaling in development and cancer. Cell Cycle 2018;17:702–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jimenez G, Shvartsman SY, Paroush Z. The Capicua repressor–a general sensor of RTK signaling in development and disease. J Cell Sci 2012;125:1383–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oh S, Shin S, Janknecht R. ETV1, 4 and 5: an oncogenic subfamily of ETS transcription factors. Biochim Biophys Acta 2012;1826:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goff DJ, Nilson LA, Morisato D. Establishment of dorsal-ventral polarity of the Drosophila egg requires capicua action in ovarian follicle cells. Development 2001;128:4553–62. [DOI] [PubMed] [Google Scholar]
- 15.Astigarraga S, Grossman R, Díaz-Delfín J, Caelles C, Paroush Z, Jiménez G. A MAPK docking site is critical for downregulation of Capicua by Torso and EGFR RTK signaling. EMBO J 2007;26:668–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ajuria L, Nieva C, Winkler C, Kuo D, Samper N, Andreu MJ, et al. Capicua DNA-binding sites are general response elements for RTK signaling in Drosophila. Development 2011;138:915–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bunda S, Heir P, Metcalf J, Li ASC, Agnihotri S, Pusch S, et al. CIC protein instability contributes to tumorigenesis in glioblastoma. Nat Commun 2019; 10:661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ding H, Roncari L, Shannon P, Wu X, Lau N, Karaskova J, et al. Astrocyte-specific expression of activated p21-ras results in malignant astrocytoma formation in a transgenic mouse model of human gliomas. Cancer Res 2001;61: 3826–36. [PubMed] [Google Scholar]
- 19.Wei J, Barr J, Kong L-Y, Wang Y, Wu A, Sharma AK, et al. Glioma-associated cancer-initiating cells induce immunosuppression. Clin Cancer Res 2010;16: 461–73. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Forés M, Simón-Carrasco L, Ajuria L, Samper N, González-Crespo S, Drosten M, et al. A new mode of DNA binding distinguishes Capicua from other HMG-box factors and explains its mutation patterns in cancer. PLos Genet 2017;13: e1006622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwartz D, Gygi SP. An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat Biotechnol 2005; 23:1391–8. [DOI] [PubMed] [Google Scholar]
- 22.Boggon TJ, Eck MJ. Structure and regulation of Src family kinases. Oncogene 2004;23:7918–27. [DOI] [PubMed] [Google Scholar]
- 23.Dissanayake K, Toth R, Blakey J, Olsson O, Campbell DG, Prescott AR, et al. ERK/p90(RSK)/14-3-3 signalling has an impact on expression of PEA3 Ets transcription factors via the transcriptional repressor capicua. Biochem J 2011; 433:515–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.la Cour T NESbase version 1.0: a database of nuclear export signals. Nucleic Acids Res 2003;31:393–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ege N, Dowbaj AM, Jiang M, Howell M, Hooper S, Foster C, et al. Quantitative analysis reveals that actin and Src-family kinases regulate nuclear YAP1 and its export. Cell Syst 2018;6:692–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bülow MH, Bülow TR, Hoch M, Pankratz MJ, Jünger MA. Src tyrosine kinase signaling antagonizes nuclear localization of FOXO and inhibits its transcription factor activity. Sci Rep 2014;4:4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goh Y-M, Cinghu S, Hong ETH, Lee Y-S, Kim J-H, Jang J-W, et al. Src kinase phosphorylates RUNX3 at tyrosine residues and localizes the protein in the cytoplasm. J Biol Chem 2010;285:10122–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dehm SM, Bonham K. SRC gene expression in human cancer: the role of transcriptional activation. Biochem Cell Biol 2004;82:263–74. [DOI] [PubMed] [Google Scholar]
- 29.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 2011;7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.