

Abstract
Objective:
Cases of Clostridiodes difficile infection (CDI) diagnosed after hospital discharge account for a substantial proportion of new infections. It is unclear if post discharge infections originate from hospital-based transmission.
Design:
Retrospective cohort study.
Setting:
Tertiary care cancer center. Non outbreak setting.
Methods:
For all laboratory-identified cases of CDI in 2015– 2016, patients with post-discharge (PD) CDI within 8 weeks of their hospital stay were included in the study. Isolates from PD- CDI cases and their CDI positive unit-based contacts were first genotyped by MLST. Common strains were further examined by core genome sequencing (CGS) to evaluate transmission links.
Results:
Of 173 cases examined by MLST, 50 % of PD cases matched previous unit contacts. Next, 34 isolates, including 16 PD cases and their 18-unit contacts were examined by CGS. None were ≤3 SNVs apart. Seventy percent of PD cases had in-hospital antibiotic exposure before CDI onset in the community.
Conclusion:
Our study results suggest that symptomatic CDI cases are not a substantial source of transmission to PD cases. Frequent antibiotic exposure in post-discharge CDI cases is an important target for surveillance and stewardship efforts.
INTRODUCTION
Clostridiodes difficle (C. difficile) infection (CDI) is a leading cause of health-care associated (HA) infection in the United States(1).The incubation period of C. difficile can be highly variable, with infections diagnosed days to weeks after the initial exposure to C. difficile spores(2, 3). Surveillance studies estimate that 3 out of every 4 HA- CDIs have onset in the community(4); and up to 15 % occur within 4 weeks after a hospital stay (post-discharge cases )(5). Due to the latency between the initial acquisition and onset of diarrheal symptoms, the source of infection may remain obscure in the majority of HA- CDI cases.
The combined approach of temporospatial links and genetic relatedness has elucidated several aspects of hospital-based C. difficile transmission(3, 6). Amongst the various C. difficle genotyping techniques, whole or core genome sequencing (WGS or CGS) are highly discriminatory methods. Multilocus sequence typing (MLST) is a low-resolution method but offers the distinct advantage of low cost, simplified analysis, and high agreement with CGS for unrelated strains(7).
In a previous study by Eyre et al(8), 19 % of the CDI cases that were genetically related to a previous infection had evidence of hospital contact with the donor, including 13 % with ward-based contact and less than 1 % acquired infection indirectly from ward-based contamination (defined as possible environmental contamination persisting for four weeks after the first infectious patient had been discharged). Although the study linked post discharge cases with their previous hospital contacts, the number of exposures with all other CD cases and their time from discharge was not characterized in detail.
The National Healthcare Safety Network (NHSN) requires US hospitals to report post-discharge (PD) CDI events even though the contribution of hospital versus community-based transmission in PD cases is not known. We seek to determine if symptomatic cases of CDI serve as a source of infection to the same unit occupants who later develop CDI after hospital discharge. In this study, we apply a joint approach of MLST and CGS to isolates from PD-CDI cases and their previous unit based contacts.
METHODS
Study setting:
This study was performed at Memorial Sloan Kettering Cancer Center (MSK), a tertiary care cancer specialty hospital with 475 beds and approximately 25,000 annual admissions. For all laboratory-identified CDI’s in 2015 and 2016, patients with PD-CDI within eight weeks of hospital discharge were included in the study. CDI is nosocomial or community-based acquisition of infection; past and future CDI events among study participants were obtained from the Infection Control database (CKM, Canada). At our institution, clinical diagnostic testing for CDI is performed using the Cepheid GeneXpert PCR platform (one-step testing). A clinical case of CDI was defined by positive PCR result on unformed stool specimen tested by the Cepheid GeneXpert assay [Cepheid Xpert C. difficile Epi assayXpert®, Sunnyvale CA]. Screening of asymptomtic individuals for C. difficile was not done routinely in any patient population.
Community-acquired CDI cases were defined by onset in the community and without preceding hospitalization in 8 weeks. Hospital onset cases are defined as CDI diagnosed past 72 hours of inpatient admission. Putative donors were identified from the Infection control database using the following criteria:
Direct contact: C. difficile positive unit mates (overlapping stays) of post discharge cases.
Indirect contact: Previously discharged occupants from the same unit in the 12 weeks before admission of PD case.
For study purposes, the six most frequent strains at MSK are included: ST 1, 2, 3, 8, 11, and 42. First, MLST types of PD cases and their direct or indirect contacts were compared. For indirect contacts, the time interval was determined by admission date of PD case and, any CDI case on the same unit within 4 weeks, 4–8 weeks, 8–12 weeks of this date. Next, confirmatory genetic analysis with core genome sequencing (CGS) was conducted on a subset of MLST identical PD cases and their donors. Based on a plausibly higher likelihood of transmission, the analysis was restricted to unit mates with direct overlap or most proximal hospitalization to the PD case (≤ 4 weeks). Pearson Chi-Square tests are applied to assess differences in proportions between groups.
Laboratory methods:
Stools were thawed and ethanol shocked with a 1:4 dilution in 100% ethanol for at least 1 hour. After incubation and centrifugation, samples were inoculated on selective media for the detection of CD and incubated anaerobically. Growth confirmation of a single colony by PRO disk was performed with the remaining portion of the colony subbed to a blood agar plate for isolation. After a 48-hour anaerobic incubation, samples were submitted for core genome sequencing.
MLST was done as previously described(7). CGS was performed using established methods. Briefly, genomic DNA (gDNA) was extracted from C. difficile isolates using the QIAmp DNA Mini-Kit bacterial suspension protocol with some modifications as previously described(9). Libraries were prepared using Nextera XT reagents (Illumina, San Diego, CA) per manufacturers’ instructions. Sequencing was performed on the Illumina MiSeq platform to generate 150 base paired-end reads. Bioinformatic analysis was performed by an in-house pipeline using publicly available tools. FASTQ files were demultiplexed and adaptor trimmed using the in-house pipeline. Quality of the sequences was evaluated and reads with a Q30 score of less than 30 for more than 50% of the bases were filtered out using FASTX. A range of kmer and coverage values was used with Velvet assembler to generate multiple assemblies for each isolate. Each assembly was evaluated, and the best assembly was used based on the highest N50 value. Genes were predicted on draft assemblies using prodigal (10). Genes were assigned functions using RPS-BLAST (11) and CDD database (12). PGAP pipeline was used for pan-genome analysis and building an SNP-based phylogeny tree. PGAP pipeline can perform five analytic functions, including cluster analysis of functional genes, pan-genome profile analysis, genetic variation analysis of functional genes, species evolution analysis, and function enrichment analysis of gene clusters (13).
Isolates were considered isogenic if they differed by ≤2 core genome SNVs (isolates collected <124 days apart) or ≤3 core genome SNVs (isolates collected 124–364 days apart), based on analyses of C. difficile evolutionary rate established by previous studies (8, 14).
This study was reviewed and appoved be instiutional IRB and granted HIPAA waiver of authorization.
RESULTS
During the two-year study period, 1,365 new CDI cases were diagnosed, 542 community- acquired (40%), 440 hospital-onset (32%), and 383 (28%) post-discharge cases. Among these, 1,112 (82%) were successfully genotyped by MLST. Five hundred and twenty-nine (47.6%) met incusion criteria based on the strain type.
For the 383 PD cases, the median age was 58 years, 180 (47%) were females. The median time to CDI diagnosis from discharge was 19 days, with 162 PD cases diagnosed within 48 hours of readmission. Seventy percent (n=267) of patients received antibiotics during the index hospitalization. The most common antibiotic was beta-lactam- lactamase inhibitor in 135 (51%) patients, fluoroquinolones in 69 (26%), and 3rd or 4th generation cephalosporins in 87 (36%) patients. The median length of antibiotic therapy was four days.
Relatedness among PD cases and prior unit contacts by low resolution MLST
Based on the study inclusion criteria, 147/383 (38.4%) cases were due to selected endemic strains (Figure1). The breakdown by sequence type (ST) is as follows: ST2=46; ST42=39; ST8 =26; ST11=15; ST1=12; ST3=9. Among the 147 PD cases, 69 (47%) had contact with an isogenic MLST case [ 43 indirect; 8 direct, and 18 cases with both] (Figure 2). Table 1 shows the genotypic relationship by type of contact between patients.Only 8.3 % (125/ 1507) of the PD cases and their unit contacts had similar MLST strains. No differences were found when examined by the type of contact (direct vs. indirect; p= 0.574) or by the time interval for those with indirect contact (p= 0.343) (Table 1).
Figure 1.
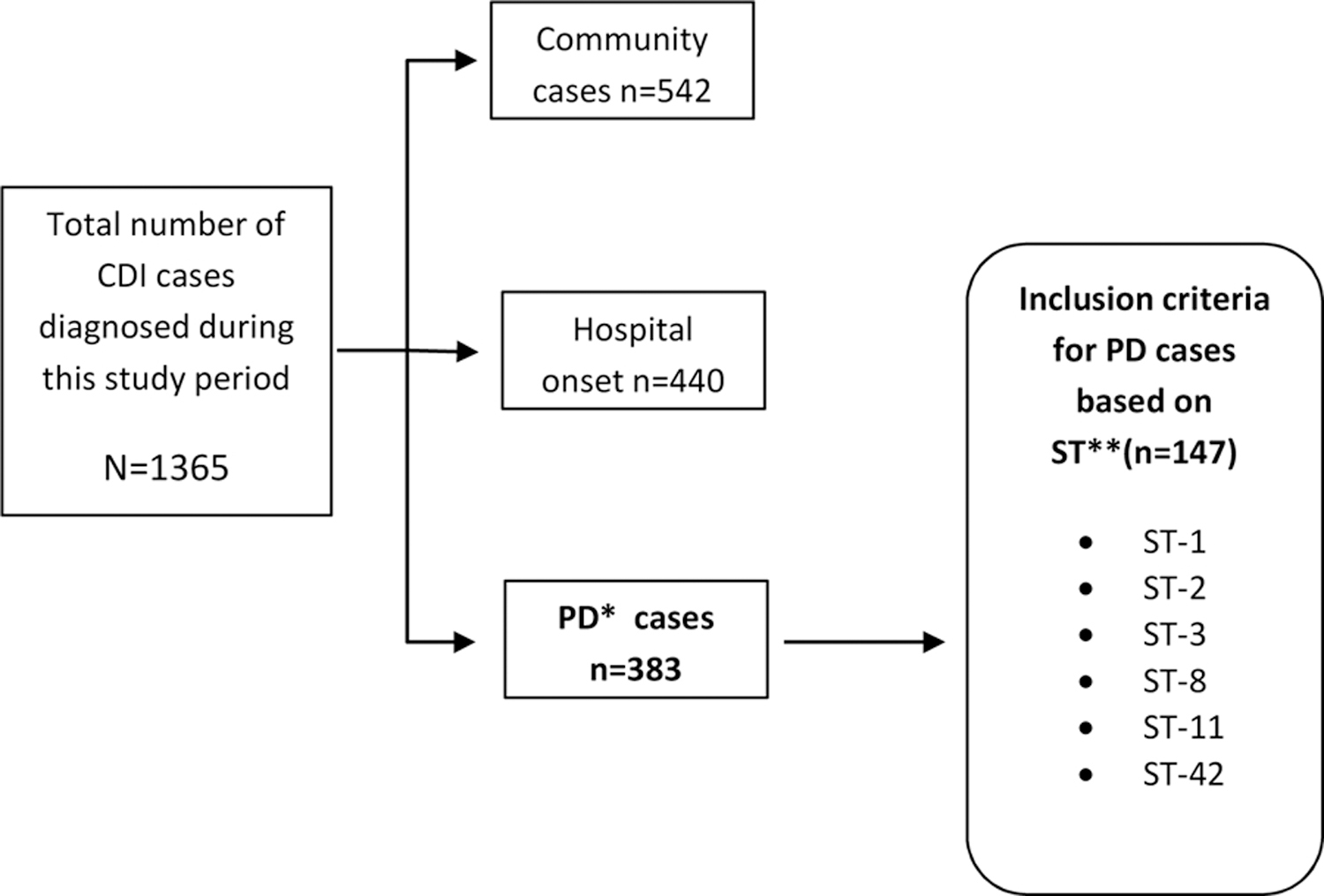
Inclusion citeria for Postdischarge (PD) C. difficile cases during the two year study period.
*PD, post discharge cases
**ST; sequence types
Figure 2.
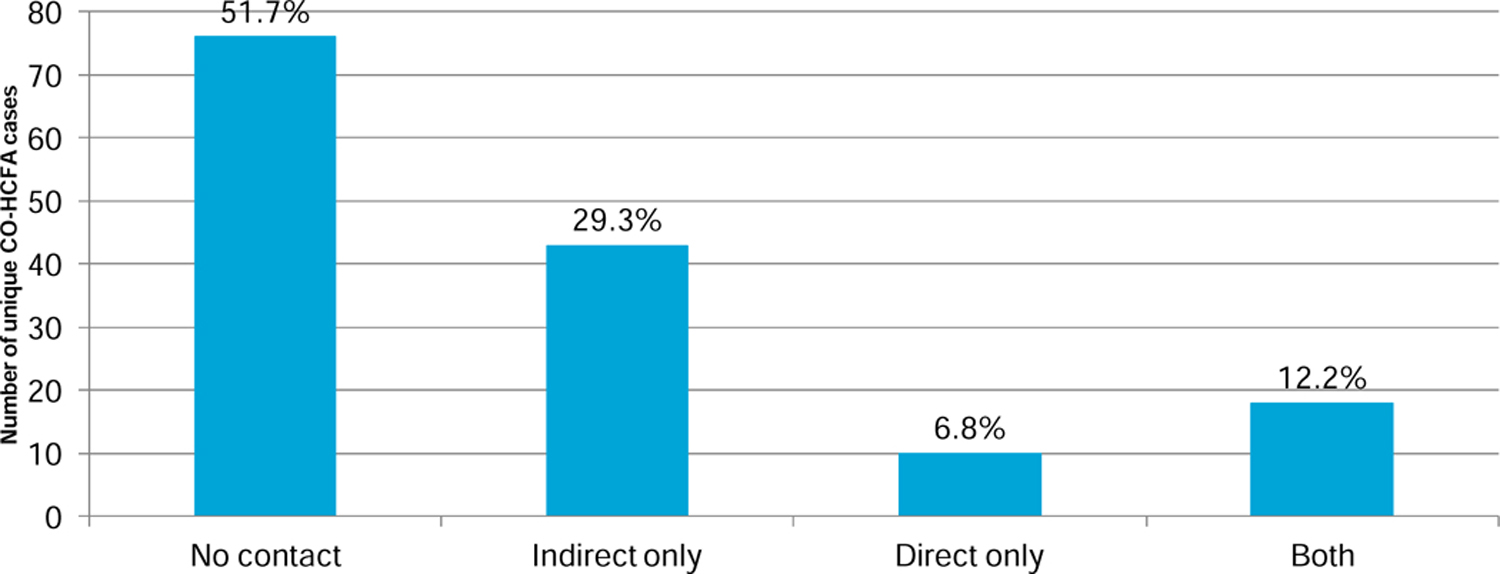
Type of exposure for 147 PD cases and previous CDI cases on the same unit with shared MLST strain type
Table 1.
Total number of CDI positive unit contacts by time and MLST concordant status for all PD cases (n=147)
| Exposure | PD cases with prior CDI contact ^ | Total previous contacts with CDI | Contacts with same ST type (Presumed transmission) | 95% CI | Overall PD cases with same ST contact¶ |
|---|---|---|---|---|---|
| Indirect overlap * | |||||
| <4 weeks | 121 | 389 | 30, 7.7% | (5.2–10.8%) | 27 |
| 4–8 weeks | 115 | 324 | 32, 9.8% | (6.9–13.7%) | 20 |
| 8–12 weeks | 103 | 340 | 30, 8.8% | (6.0–12.4%) | 20 |
| Direct Overlap | 117 | 454 | 33, 7.2% | (5.1–10.1%) | 26 |
| Total | 1507 | 125, 8.3% | (7.0–9.8%) |
Interval between discharge of previous unit contact that is CDI positive and date of admission of CO-HCFA case.
Represents 147 PD cases - not mutually exclusive by exposure categories
Represents 69 PD cases with concordant contact- not mutually exclusive by exposure categories
Core genome sequence (CGS) comparison of PD cases and same unit contacts
CGS was done to compare further the PD cases and their putative donors with similar MLST types. Fifty-one PD cases were eligible to be included, 16 (31%) could be successfully sequenced and analyzed due to various reasons (lost PD or donor sample, no growth in culture, sequencing failure). The analyzable cohort of 34 cases included the 16 PD infections (designated recipients “R” in Figure 3), and their 18 unit-based contacts (designated donors “D” in Figure 3). Each PD case had at least one matched donor, and two had one additional donor each. For these 18 donors, 10 were overlapping stays with the PD case, and eight were on the same unit within four weeks before admission of the PD case. Pairwise SNV comparison color-coded by PD case [R] and the same unit donor [D] is shown in Figure 3. None of these 18 contacts had ≤ 3 SNV differences from PD cases. None of the common ST types examined by CGS had ≤2 SNP differences regardless of the epidemiologic link.
Figure 3.

Pairwise SNV comparison between spatially linked MLST concordant PD cases and putative donors. Pairs are color coded for comparison. PD cases labelled as recipients and previous contacts as donors.
DISCUSSION
Our study results suggest that symptomatic CDI cases and environmental contamination from these cases are not the source of infection for unit mates who develop CDI after discharge from the hospital. Half of the PD cases in our study had a putative source by low resolution genotyping (MLST). However, no transmission was detected when CGS examined a subset of the cases with the closest epidemiological link. Antibiotic exposure during the index hospitalization was common.
Several recent population studies suggest a rise in community-onset CDI rates. According to a population-based report in Olmstead county, Minnesota two third of CDI cases have onset in the community. Approximately half of these cases were hospitalized within 12 weeks prior to CDI diagnosis(15). Pinzon et al’s study from the VHA found that 9.1% of ~ 20,000 CDI episodes between 2011–2014 occurred within 4 weeks of discharge from a hospital(16). Further, antibiotic use was highest in this group (50 %) compared to an overall 40% exposure among all other CDI cases. Despite this shift in epidemiology, genotyping has not been applied to examine the role of hospital-based acquisition in PD CDI cases.
The lack of any transmission by CGS and high antibiotic use in PD-CDI cases suggests that C. difficle infections may start outside the hospital, and emphasis on antibiotic stewardship efforts is more likely to be effective in preventing PD-CDI.
There are several limitations in our study: isolates examined are restricted to unit-based contacts, not hospital-wide or outpatient contacts. Despite a robust two-step approach, our conclusions are based on successful core genome sequencing of ~ a third of the eligible cohort, including 10 pairs with direct overlap and the highest spatial probability of transmission. Recent studies applying WGS show that hospital-based CDI transmission is more likely to originate from cases then carriers(6). The role of environmental reservoir is not ascertained – only contamination that could have occurred from symptomatic cases in a predefined time interval. ST-1 cases were not isolated among the donor- recipient pairs examined by CGS (17)(Figure 3). Cases were defined based on symptoms of diarrhea and positive PCR. In a cancer population, C. difficile carriage rates are high, and diarrhea due to non-infectious causes is frequently encountered. This is a well recognized limitation of current assays and introduces the possibility of overestimating carriers as CDI cases. Recent studies applying WGS show that hospital-based CDI transmission is more likely to originate from cases then carriers(6)
In summary, our study results suggest that symptomatic CDI cases are not a substantial source of transmission to PD cases. Frequent antibiotic exposure in post-discharge CDI cases is an important target for surveillance and stewardship efforts.
Acknowledgements and funding source :
The authors acknowledge support from the MSK Cancer Center Support Grant/Core Grant (P30 CA008748).
The authors wish to acknowledge Natalie Chow for assistance with sequencing.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
This work has been presented in part at the poster session of ID week on October 6, 2017.
No conflict of interest (all authors)
REFERENCES
- 1.Lessa FC, Winston LG, McDonald LC, Emerging Infections Program CdST. Burden of Clostridium difficile infection in the United States. N Engl J Med. 2015;372(24):2369–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Muto CA, Pokrywka M, Shutt K, Mendelsohn AB, Nouri K, Posey K, et al. A large outbreak of Clostridium difficile-associated disease with an unexpected proportion of deaths and colectomies at a teaching hospital following increased fluoroquinolone use. Infection control and hospital epidemiology : the official journal of the Society of Hospital Epidemiologists of America. 2005;26(3):273–80. [DOI] [PubMed] [Google Scholar]
- 3.Kamboj M, Sheahan A, Sun J, Taur Y, Robilotti E, Babady E, et al. Transmission of Clostridium difficile During Hospitalization for Allogeneic Stem Cell Transplant. Infection control and hospital epidemiology : the official journal of the Society of Hospital Epidemiologists of America. 2016;37(1):8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Centers for Disease C, Prevention. Vital signs: preventing Clostridium difficile infections. MMWR Morb Mortal Wkly Rep. 2012;61(9):157–62. [PubMed] [Google Scholar]
- 5.Kutty PK, Benoit SR, Woods CW, Sena AC, Naggie S, Frederick J, et al. Assessment of Clostridium difficile-associated disease surveillance definitions, North Carolina, 2005. Infect Control Hosp Epidemiol. 2008;29(3):197–202. [DOI] [PubMed] [Google Scholar]
- 6.Kong LY, Eyre DW, Corbeil J, Raymond F, Walker AS, Wilcox MH, et al. Clostridium difficile: Investigating Transmission Patterns Between Infected and Colonized Patients Using Whole Genome Sequencing. Clin Infect Dis. 2019;68(2):204–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Griffiths D, Fawley W, Kachrimanidou M, Bowden R, Crook DW, Fung R, et al. Multilocus sequence typing of Clostridium difficile. Journal of clinical microbiology. 2010;48(3):770–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eyre DW, Cule ML, Wilson DJ, Griffiths D, Vaughan A, O’Connor L, et al. Diverse sources of C. difficile infection identified on whole-genome sequencing. N Engl J Med. 2013;369(13):1195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sim JH, Anikst V, Lohith A, Pourmand N, Banaei N. Optimized Protocol for Simple Extraction of High-Quality Genomic DNA from Clostridium difficile for Whole-Genome Sequencing. J Clin Microbiol. 2015;53(7):2329–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McGinnis S, Madden TL. BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004;32(Web Server issue):W20–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marchler-Bauer A, Bo Y, Han L, He J, Lanczycki CJ, Lu S, et al. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017;45(D1):D200–D3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao Y, Wu J, Yang J, Sun S, Xiao J, Yu J. PGAP: pan-genomes analysis pipeline. Bioinformatics. 2012;28(3):416–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kociolek LK, Gerding DN, Espinosa RO, Patel SJ, Shulman ST, Ozer EA. Clostridium difficile Whole Genome Sequencing Reveals Limited Transmission Among Symptomatic Children: A Single-Center Analysis. Clin Infect Dis. 2018;67(2):229–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khanna S, Pardi DS, Aronson SL, Kammer PP, Orenstein R, St Sauver JL, et al. The epidemiology of community-acquired Clostridium difficile infection: a population-based study. Am J Gastroenterol. 2012;107(1):89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mora Pinzon MC, Buie R, Liou JI, Shirley DK, Evans CT, Ramanathan S, et al. Outcomes of Community and Healthcare-onset Clostridium difficile Infections. Clin Infect Dis. 2019;68(8):1343–50. [DOI] [PubMed] [Google Scholar]
- 17.Martin JSH, Eyre DW, Fawley WN, Griffiths D, Davies K, Mawer DPC, et al. Patient and Strain Characteristics Associated With Clostridium difficile Transmission and Adverse Outcomes. Clin Infect Dis. 2018;67(9):1379–87. [DOI] [PMC free article] [PubMed] [Google Scholar]