

Abstract
The synthesis and characterisation of two novel self‐assembled amphiphiles (SSAs) SQS‐1 and SQS‐2 are reported. Both compounds, based on the squaramide motif, were fully soluble in a range of solvents and were shown to undergo self‐assembly through a range of physical techniques. Self‐assembly was shown to favour the formation of crystalline domains on the nanoscale but also fibrillar film formation, as suggested by SEM analysis. Moreover, both SQS‐1 and SQS‐2 were capable of anion recognition in DMSO solution as demonstrated using 1H NMR and UV/Vis absorption spectroscopy, but displayed lower binding affinities for various anions when compared against other squaramide based receptors. In more competitive solvent mixtures SQS‐1 gave rise to a colourimetric response in the presence of HPO4 2− that was clearly visible to the naked eye. We anticipate that the observed response is due to the basic nature of the HPO4 2− anion when compared against other biologically relevant anions.
Keywords: anion recognition, colorimetric sensors, self-assembly, squaramides, supramolecular chemistry
Sticky squaramides: Two squaramide‐based self‐assembled amphiphiles (SSAs) are reported that are capable of colorimetric anion recognition in both organic and aqueous solution. Although these SSAs displayed lower binding affinities for anions than other squaramide‐based receptors, in competitive aqueous mixtures, they are fully soluble and capable of selective recognition of HPO4 2− in comparison with other biologically relevant anions.

Introduction
Self‐assembly in molecular systems has inspired a new generation of supramolecular materials with applications ranging from solid‐state luminescent materials[1, 2, 3] to molecular recognition and binding.[4, 5, 6] Among the developments, self‐associating amphiphiles (SSAs) are emerging as a new motif in the design of supramolecular macromolecular materials.[7] Self‐assembly in these materials is brought about through a combination of weak non‐covalent interaction such as hydrogen‐bonds, halogen‐bonds, hydrophobic, electrostatic interactions, van der Waals interactions, etc., resulting in the overall formation of hierarchical materials.[8] SSAs have garnered considerable interest in the development of hierarchical nanostructures, biomolecular mimics, carrier molecules for drug delivery and molecular sensing.[9, 10] Recent work by Hiscock and co‐workers has explored the self‐associative and physicochemical properties for several low molecular weight SSAs.[11, 12] These urea‐sulfonate based SSAs contain a number of hydrogen bond donating (HBD) and accepting (HBA) functionalities that allow self‐association via hydrogen bond formation. Indeed, the combination of different HBD and HBA functionalities within these SSAs result in a variety of competitive, self‐associative hydrogen bonding modes forming ‘frustrated’ hydrogen bonded nanostructures.[13, 14] Importantly, such urea‐sulfonate based SSA compounds appear to be promising antimicrobial agents and also show application as selective cell membrane coordination/disruption systems.[15, 16, 17, 18, 19] Related work has also shown that di‐aryl ureas can form extended hydrogen bonding and π‐π interactions in the solid state, to produce supramolecular gels with antimicrobial properties against Staphylococcus aureus and Escherichia coli.[20] Our interest in SSAs stems from our recent report on squaramide‐naphthalimide conjugates that display self‐association characteristics.[21] Using this approach we were able to effectively disrupt self‐association bringing about a disaggregation induced emission response in the presence of halide anions. In particular, we discovered that the squaramide‐naphthalimide conjugates exhibited distinct responses with each halide, in the case of Br− resulting in a dramatic 500–600 % fluorescence enhancement. Through this work, we expected that disruption of self‐assembly in SSAs may also be exploited for the sensing of anionic analytes. It is now well established that anions play a significant role in both human health and environmental pollution with phosphate anions being particularly prevalent owing to their essential role in metabolism, energy transduction and their use in agricultural fertilizers and pesticides.[22, 23, 24, 25] While several phosphate sensors have been developed,[26, 27, 28, 29] the large majority of these have solely operated in organic solvents, and those that achieve selective anion recognition in aqueous solution remain scarce. Towards this end, Gunnlaugsson et al. developed naphthalimide thiourea based receptors, which also self‐assembled into highly ordered hydrogen bonded honeycomb structures, as effective colorimetric sensing probes for H2PO4 −. In aqueous solution a stark colour change from yellow to red was observed; a result of receptor deprotonation.[30, 31] However, the selectivity of these receptors was not optimal where AcO− and F− also exhibited a marked effect. More recently, the same group has also exploited meta‐phenylene bis(phenylurea) receptors as a supramolecular construct that used a phosphate anionic core to template a triple‐stranded helicate,[32] and in the development of urea tripodal systems, that can in the presence of anions such as H2PO4 − or SO4 2− result in the formation of SSA capsules, clusters and 2D nano‐structures.[33, 34, 35] Similarly, Caltagirone et al. synthesised diindolylureas that showed high affinity for oxoanions. These receptors showed particular selectivity for H2PO4 − in water/DMSO‐d6 binding with high affinity (Ka>104 M−1), however, no spectroscopic changes were reported.[36] Jolliffe and co‐workers have reported the synthesis of deltamides that display higher affinity for H2PO4 − than other oxoanions or halides.[37] Based on these results, we reasoned that exploiting SSAs as an anion recognition motif may provide advantages over previously reported strategies given the ability to modulate supramolecular assembly/disassembly.
To improve hydrogen donor ability and potentially the self‐assembly properties of SSAs we sought to employ the squaramide motif in our design.[38] Squaramides display stronger hydrogen bond donor and acceptor abilities when compared with ureas and thioureas and are also known to display high levels of binding affinity for various anions. Furthermore, due to the restricted rotation about the C−N bond, double secondary squaramides exist predominantly as the extended anti/anti conformer, which allows the simultaneous participation of the two NH groups in the formation of hydrogen bonding interactions. This often giving rise to higher order structures in both solution and the solid state.[39, 40] We have reported several classes of squaramide based receptors for a variety of applications such as anion recognition, anion sensing and anion transport,[41, 42, 43, 44, 45, 46, 47, 48] and furthermore Costa and co‐workers and Kieltyka and co‐workers have exploited squaramides to create a wide variety of self‐assembled systems,[49, 50, 51, 52, 53, 54, 55] thus it was an obvious design decision. Herein, we report the design and synthesis of two novel squaramide based SSAs, an evaluation of their self‐assembly behaviour and their ability to recognise various anionic species in both organic and aqueous solutions.
Results and Discussion
The two squaramide based SSA structures SQS‐1 and SQS‐2 were designed to possess an aryl (UV‐Vis absorption) reporting group, possessing an electron withdrawing substituent, and both HBD and HBA functionalities within a single structure. Both compounds were synthesised by the reaction of two different substituted aniline derivatives with diethyl squarate (Scheme 1), followed by the addition of aminomethanesulfonate to give both SQS‐1 and SQS‐2 in moderate yields of 55–60 % as their NBu4 + salts. Both compounds were fully characterised as outlined in the experimental section, with the 1H NMR of SQS‐1 being shown in Figure 2 below (c.f. Supporting Information for all experimental data). Squaramides suffer routinely with poor solubility, however, the design of these HBD and HBA mixed systems, facilitated both SQS‐1 and SQS‐2 to be fully soluble in organic polar solvents such as DMSO, MeCN as well as in DCM and gratifyingly, also in highly competitive solvents such as H2O, MeOH and EtOH. Crystals suitable for single crystal X‐ray diffraction analysis were obtained for both SQS‐1 and SQS‐2 by recrystallization from a concentrated DMSO solution, which allowed for the evaluation of their solid‐state behaviour; the resulting structures shown in Figure 1.
Scheme 1.

Synthesis of SQS‐1 and SQS‐2 by the reaction of the appropriate mono substituted squarates with aminomethanesulfonate.
Figure 2.

(a) The NMR spectra of SQS‐2 in DMSO‐d6 (0.5 % H2O) at various concentrations (1–100 mM). (b) The changes in the various chemical shifts as a function of concentration.
Figure 1.

(a) Structure of SQS‐1 and (b) SQS‐2 as the NBu4 salt with heteroatom labelling scheme. Cations and selected hydrogen atoms are omitted for clarity. (c) Interaction of adjacent dimers in the structure of SQS‐1 showing the π‐π stacking between conjugated units. Deposition Numbers 2043909 (for SQS‐1) and 2087917 (for SQS‐2) contain(s) the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
The initial evidence for the self‐association of SQS‐1 was observed from X‐ray crystallography, with the diffraction data solved and refined in the monoclinic space group P21/n. The asymmetric unit contains one SQS‐1 anion and NBu4 + counterion, with no solvation or other guest species. As shown in Figure 1(a), two molecules of SQS‐1 associate into a centrosymmetric dimer through multiple hydrogen bonding interactions. The squaramide N−H groups (N1 and N2) act as hydrogen bond donors to sulfonate oxygen atom O1, with D⋅⋅⋅A distances of 2.8118(10) and 2.7761(10) Å and D−H⋅⋅⋅O angles of 156.2(14) and 167.0(14)°, respectively, and this interaction is reciprocated in each dimeric unit. These dimers further associate with one another through parallel face‐to‐face π‐π interactions which encompass one face of the entire conjugated portion of the molecule, at a mean interplanar distance of 3.28 Å, as is evident from Figure 1(c). Additional intermolecular interactions mostly take the form of C−H⋅⋅⋅O contacts from the α‐methylene groups of the NBu4 counterions; these interactions are shown in Figure S29 (See Supporting Information).
The structural model of SQS‐2 was refined in the monoclinic space group P21/c. The local structure of SQS‐2 is broadly similar to that of SQS‐1, with one anion in the asymmetric unit alongside a tetrabutylammonium cation, and no solvation or other guest species. An equivalent centrosymmetric dimer is observed in this structure where a sulfonate oxygen atom accepts chelating hydrogen bonds from the adjacent molecule (Figure 1(b),). The D⋅⋅⋅A distances of 2.865(3) and 2.804(3) Å for N1 and N2, respectively, are marginally longer on average than the equivalent distances in SQS‐1. The D−H⋅⋅⋅A angles of 148.85(15) and 164.93(14)°, respectively, are also smaller than those observed for SQS‐1, suggesting a less beneficial environment for dimerization in this species compared to the nitro analogue. The steric bulk of the trifluoromethyl groups (which also display minor crystallographic disorder) may also play a role in hindering π‐π stacking interactions in the structure of SQS‐2; while minor contacts are observed between the squaramide backbones, the aromatic surfaces of the phenyl rings are capped on both faces by weak C−H⋅⋅⋅π contacts from the tetrabutylammonium cations. Directional C−H⋅⋅⋅O contacts are also observed between the tetrabutylammonium cations and the squaramide and (non‐hydrogen bonded) sulfonate oxygen atoms, which are represented in Figure S30 (Supporting Information).
Molecular self‐assembly in solution was also observed; this behaviour being probed initially by 1H NMR spectroscopy where a dilution study was conducted at different concentrations of SQS‐1 and SQS‐2 in d6 ‐DMSO (1 mM to 100 mM) solution. Changes in the chemical shifts of various resonances were observed with the NH, CH2 and aromatic protons all showing some modulations as a function of concentration. For example, as shown in Figure 2 the NHa signal of SQS‐2 underwent a significant upfield shift (Δδ=0.33 ppm) upon dilution with the CH2, aromatic protons and NHb showing more gradual changes. This indicates that the chemical environment of all protons is perturbed to some extent as the molecules undergo self‐assembly at elevated concentrations. Similar behaviour was also observed for SQS‐1; the results of which are shown in Figures S12 and S13 (See Supporting Information).
Hiscock and co‐workers have detailed how analogous urea based SSAs are capable of forming larger order nanostructures in aqueous media.[16, 56] With this in mind, we performed both DLS analysis of SQS‐1 in solution, as well as dropcasting samples of SQS‐1 and SQS‐2 from different solvent mixtures and investigating their morphological properties with scanning electron microscopy (SEM). The fact that the crystallographic results indicated that the HBD and HBA functionalities of these compounds resulted in dimer formation and further association due to favourable π‐π interactions, we wished to investigate the formation of higher order self‐assemblies.
In our hands, the DLS measurements indicated that SQS‐1 did not form spherical aggregates on the nanoscale under the conditions studied (25 μM–250 μM concentration) in solvents such as DMSO, EtOH or their mixtures with water. From our analysis at higher concentrations (250 μM), it was evident that a small number of larger particles were formed in DMSO as is evident from Figure 3, an indication that there was no homogeneous assembly in solution but instead the compound assembled abruptly forming a small number of very large particles that undergo sedimentation, and not diffusion.
Figure 3.
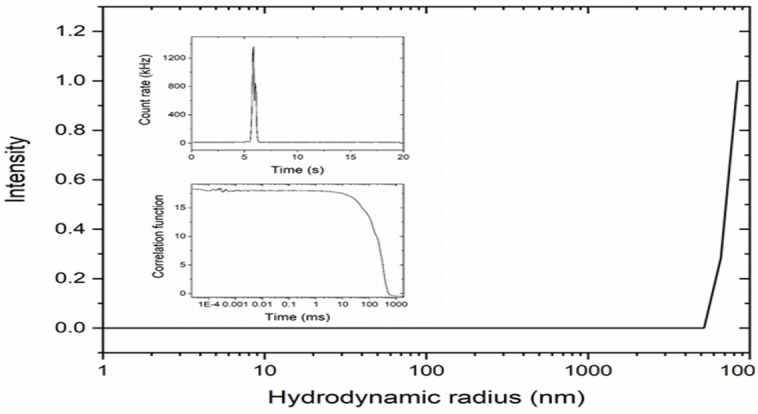
Hydrodynamic radius distribution of a 250 μM sample of SQS‐1 in DMSO. Inserts show the count rate over time and the correlation function.
Similarly, measurements in EtOH and EtOH:water mixtures suggested that particles arise from self‐assembly formation that results in crystallisation/nucleation of SQS‐1. These particles were observed at early time points in the measurements, and indicated that they formed immediately and subsequently underwent rapid sedimentation. Indeed, optically birefringent needle‐like crystals were observed by optical microscopy and suggest that the particles observed were crystals of SQS‐1. This result is particularly significant given the results reported by Hiscock et al., where analogous urea based molecules form discrete nanoscale particles under similar conditions[11, 12, 13] and it emphasises the role that the squaramide plays in dictating the self‐assembly properties of such SSAs.
To further probe any possible self‐assembly behaviour we also investigated, using SEM, the morphological features of both SQS‐1 and SQS‐2 in various solvents upon dropcasting the samples onto silica support using 1 μM stock solutions of each structure. The samples were allowed to dry in air and under vacuum prior to imaging. In DMSO, both compounds showed the formation of compact films (See Supporting Information; Figure S15 and S16). Similarly, in CH3CN, the formation of films was also observed, however, more fibrillar networks were particularly visible in the case of SQS‐1 (Figure 4(a)). Furthermore, the presence of microcrystalline domains was also observed (Figure 4(b)) in CH3CN. When recorded in EtOH, similar features were seen i. e. both the fibrous and microcrystalline materials were clearly visible. Imaging of SQS‐2 in CH3CN demonstrated the formation of more condensed films (Figure 4(c)), as well as the formation of crystalline structures within the film (Figure 4(d)). In EtOH, the formation of the microcrystalline structures was even more apparent for SQS‐2. We also investigated these features when both samples were dropcast using H2O and mixtures of DMSO:H2O solutions. On both occasions, the formation of self‐assemblies was observed, consisting of aggregated particles for SQS‐1 of ca. 200–400 nm, while in the case of SQS‐2, the aggregates exhibited a more rod‐like morphology (See Supporting Information). These results clearly demonstrate that both SQS‐1 and SQS‐2 undergo self‐assembly formation, however, their behaviours differ somewhat; an indication of the role the substituents play in modulating the hydrogen bonding properties and consequently on the assembly properties of these structures in different solvents.
Figure 4.

SEM images obtained from CH3CN solutions of SQS‐1 (top, (a) and (b)) and SQS‐2 (bottom, (c) and (d)). It is possible to appreciate the formation of condensed films containing microcrystalline domains.
With a view to further probing the self‐assembly process, the absorption properties of SQS‐1 and SQS‐2 were also investigated using UV/Vis absorption spectroscopy to examine if the self‐assembly behaviour could be monitored. SQS‐1 was highly coloured in DMSO (0.5 % H2O) solution and showed strong absorption maxima at 290 nm and 409 nm, respectively. In the presence of base (0.5 % Et3N in DMSO) absorption was observed at 538 nm, indicative of partial deprotonation of SQS‐1. Structurally analogous 4‐nitrophenyl squaramides are known to undergo deprotonation in DMSO.[57] We expected the addition of a protic solvent such as water to protonate the receptor and, indeed, as the proportion of water was increased a reduction in the band at 538 nm was observed with a concomitant hypsochromic shift of the absorption at 409 nm by approx. 20 nm (See Supporting Information; Figure S9). A dilution study in DMSO/H2O (99.5:0.5, v/v) demonstrated that bands at 285 nm and 409 nm increased linearly as a function of concentration (5–60 μM) for SQS‐1, obeying the Beer‐Lambert Law, and giving extinction coefficients of ca. 21,300 M−1 cm−1 and 25,500 M−1 cm−1 at 285 nm and 409 nm, respectively. Similar behaviour was observed for SQS‐2 with extinction coefficients of 19,000 M−1 cm−1 and 23,500 M−1 cm−1 at 285 nm and 345 nm, respectively over the same concentration range (See Supporting Information; Figures S10 and S11). This behaviour also suggests that the self‐assembly behaviour of these compounds either cannot be observed easily by UV/Vis spectroscopy at these concentrations or occurs only at higher concentrations.
As the UV/vis absorption properties of such squaramide receptors are known to be sensitive to anionic species,[48, 58, 59] their absorption properties upon addition of various anions were monitored in DMSO/H2O (99.5 : 0.5, v/v) solution (See Supporting Information; Figures S20–S23). Upon addition of anions, such as Br−, I−, Cl−, ClO4 − and NO3 −, as their TBA salt stock solutions, to SQS‐1 no colorimetric changes were observed. In contrast, more significant changes were seen in the absorption spectra upon addition of F−, AcO−, BzO−, H2PO4 − and SO4 2− to a solution of SQS‐1. Furthermore, these spectroscopic changes were clearly visible to the naked eye, as the solution was observed to change colour from yellow to red/purple. Similar changes were observed in the absorption spectrum where additions of anions such as F−, AcO−, SO4 2−, BzO− and H2PO4 − resulted in an increase in absorption at 538 nm and a concomitant decrease in the band at 409 nm. Similarly, the band at 290 nm showed a large hypochromism with a concomitant increase in the absorbance centered at 320 nm. Such behaviour indicates that these anions are capable of deprotonating SQS‐1 in DMSO solution. In contrast to these results, the absorption spectrum remained unchanged in the presence of Cl−, Br−, ClO4 − and I− at this concentration (4 mM).
More detailed titrations were performed for SQS‐1 in the presence of F− and SO4 2−. Addition of F− up to a concentration of 240 μM gave rise to an 89 % decrease at 409 nm and a large increase at 538 nm (Figure 5 (a)). In the presence of sulfate, similar changes were observed, however larger quantities of the anion were required to effect to exhibit the same degree of spectroscopic response. The changes observed for F− were accompanied with three clear isosbestic points appearing at 304 nm, 358 nm and 455 nm, respectively. Plotting the changes at the two main wavelengths, 409, and 538 nm, as a function of anion equivalents, is shown in Figure 5(b); the results demonstrating that only minor changes were observed within the addition of one equivalent of F−. However, between the addition of 1→3 equiv. significant changes are observed. It is possible that this is due to the initial hydrogen bonding of the F− anion with one or both of the N−H protons, followed by a full proton transfer, and hence, a deprotonation of one of the squaramide protons, the one adjacent to the nitrobenzene ring being the most likely candidate. This would result in the formation of the di‐anionic species as well as in the formation of HF. However, it is well documented that for thiourea and squaramide receptors, such protonation is often associated with the presence of a second F− anion, which results in the formation of HF2 −, and this is likely to account for the dramatic changes seen in the absorption spectra.[30, 48, 57, 60, 61]
Figure 5.

(a) Changes observed in the absorption spectrum of SQS‐1 (20 μM) upon addition of TBAF (0–240 μM) in 0.5 % H2O in DMSO solution. (b) Absorbance changes observed at 409 nm and 538 nm as a function of F− concentration.
To gain further structural insight into the interaction of SQS‐1 with these anions 1H NMR measurements were also carried out. Qualitative measurements were undertaken using a screening experiment in which 30 equiv. of several anions (AcO−, BnO−, H2PO4 −, SO4 2−, NO3 −, ClO4 −, F−, Cl−, Br−, and I− as their tetrabutylammonium salts) were added to the receptor in solution (5 mM in 0.5 % H2O in DMSO‐d6) (See Supporting Information; Figure S17). These results showed significant changes that suggested that AcO−, BzO−, H2PO4 −, SO4 2− and F− give rise to deprotonation of the receptor, in agreement with the UV/Vis results outlined above. In the case of F−, deprotonation was confirmed by the appearance of a triplet at 16.1 ppm suggesting HF2 − formation and supports the conclusions drawn from the absorption titration studies above. Conversely, Cl− and Br− addition suggested H‐bonding interactions due to significant downfield shifts of the NH signals associated with the squaramide. Other anions such as I−, ClO− and NO3 − did not appear to show any significant hydrogen bonding interactions or cause de‐protonation.
More detailed titrations were undertaken with SO4 2−, Cl− and Br−. From this data, Cl− resulted in the largest spectroscopic changes (See Supporting Information; Figures S18 and S19). Figure 6 shows the changes observed in the 1H NMR spectrum of SQS‐1 upon the addition of Cl− (30 equiv.). A gradual downfield shift of the NH signal (Ha) from 10.4 to 11.6 ppm was observed upon increasing Cl− concentration. When the resulting titration data was fitted to a 1 : 1 binding model using the open access BindFit software programme[62, 63, 64] it gave a low apparent stability constant of K=14.0 M−1 (±1.1 %). The changes suggest some degree of H‐bonding interaction between the receptor and the anion, however, the binding affinity for Cl− appears to be significantly lower than that reported for other squaramide based receptors in DMSO.[43, 46, 65, 66, 67, 68] This may be due to the proximity of the negatively charged sulfonate functionality to the squaramide binding site which results in electrostatic repulsion and prevents strong association with anionic species or it may suggest that self‐assembly of the squaramide subunits is competing with the anion recognition.
Figure 6.

(a) Stack plot of 1H NMR spectra of SQS‐1 (5 mM) upon addition of TBACl (0–30 equiv.) in DMSO‐d6 at 25 °C. (b) Changes in the chemical shifts observed for the squaramide N−H and methylene protons as a function of increasing Cl− concentration, and the corresponding fit to a 1 : 1 binding model.
Given the observation that addition of water is capable of reprotonating these receptors in the presence of basic anions and their high degree of water solubility, we also sought to investigate if selectivity of spectroscopic response could be achieved by introducing a more competitive solvent. Consequently, the UV/Vis absorption measurements were carried out in DMSO/H2O (1 : 1, v/v) media where the changes in the absorption spectra of both SQS‐1 and SQS‐2 (20 μM) were monitored (See Supporting Information; Figures S24–S27). Under these conditions, two broad absorbance bands appeared for SQS‐1 at 290 nm and 392 nm, and at 285 nm and 345 nm for SQS‐2. Unfortunately, the addition of the sodium salts of anions such as I−, F−, Br−, AcO−, SO4 2−, Cl−, H2PO4 −, NO3 − and MoO4 2− resulted in minimal spectroscopic responses. However, addition of HPO4 2− gave rise to significant spectral changes. In the case of SQS‐1, a red shift from 290 nm to 310 nm, and from 390 nm to 455 nm was observed; this culminating in a visible colour change from yellow to orange‐red (Figure 7(a)). Given our previous observations on the deprotonation of 4‐nitro‐aniline containing squaramides[19] and the basicity of HPO4 2−, we suspected that these changes were once again likely to be due to deprotonation and not due to a H‐bonding interaction. Indeed, a pH titration was also performed in DMSO/H2O solution (1 : 1, v/v, 0.1 M TBAPF6) by addition of TBAOH across a pH range from 3.5–13.5 (Figure 7(b). During the measurements, SQS‐1 underwent a hypochromism at 410 nm band, which was accompanied by a concomitant hyperchromism at 510 nm. Concomitantly, an isosbestic point at 430 nm; mirroring the changes observed in the HPO4 2− titration and was also complemented with a similar colour change from yellow to orange‐red. A four parameter sigmoid curve was fitted through the data points using Sigma Plot (Systat Software Inc., Chicago, IL, USA) giving a pK a value of 11.47 in this solvent mixture.
Figure 7.

(a) The changes in the absorption as well as the visible colorimetric changes observed for SQS‐1 (20 μM) upon increasing concentration of HPO4 2− (0–20 mM) in DMSO/H2O (1 : 1, v/v) solution. (b) pH titration of SQS‐1 in DMSO/H2O (1 : 1) (0.1 M TBAPF6) showing the corresponding four parameter sigmoid curve fit to the data. The pH of the solution was increased by addition of TBAOH. The pK a value of 11.47 was determined by fitting a four parameter sigmoidal curves through the data points using Sigma Plot (Systat Software Inc., Chicago, IL, USA) with the point of inflexion corresponding to the pK a.
Conclusion
In conclusion, we have designed and synthesised two self‐associating amphiphiles based on the squaramide moiety SQS‐1 and SQS‐2. Importantly, both compounds are fully soluble in a range of both organic and aqueous solutions. Indeed, solubility is often a drawback with the use of squaramides and such an approach may be a useful strategy in increasing solubility of squaramide analogues. We have demonstrated their self‐association properties using X‐ray crystallography, 1H NMR measurements and through the use of SEM imaging. While DLS measurements suggest that these compounds do not form discrete spherical self‐assembly structures on the nanoscale in solution, the results seem to indicate that they rapidly form self‐assemblies at higher, mM concentrations, which ultimately results in crystallisation. Both SQS‐1 and SQS‐2 showed a non‐selective response to various anions where basic anions are capable of deprotonation while less basic anions such as Cl− are capable of H‐bonding in polar aprotic solvents. Such H‐bonding interactions do not appear to give rise to a significant spectroscopic response. Nevertheless, this interaction can be measured using 1H NMR. Finally, by increasing the competitiveness of the solvent medium, we have shown that both SQS‐1 and SQS‐2 undergo colormetric changes upon addition of HPO4 2− in an aqueous solution which is not observed for less basic anions. These changes are caused by the basicity of the HPO4 2− that results in a deprotonation of the squaramide receptor. Overall, these results further demonstrate the usefulness of squaramides as a scaffold for constructing self‐assembled materials and also as a useful moiety for anion recognition. Further work is on‐going in our laboratories where we seek to construct more complex molecular receptor structures that possess both increased sensitivity and selectivity for anions in competitive media, as well as exploring their materials properties further.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Dr. Lokesh Kumar Kumawat obtained his PhD in chemistry from IIT Roorkee, India in 2015. He was a UJ‐GES postdoctoral fellow from 2016 to 2017 at the University of Johannesburg, South Africa before being awarded an Irish Research Council Government of Ireland Postdoctoral Fellowship in 2018 at Maynooth University, Ireland under the mentorship of Dr. Rob Elmes. His research works are focused on the development of supramolecular chemistry, anion binding and sensing, and, recently, on the development of squaramide scaffolds for various applications.

Biographical Information
Katrina (Kate) Jolliffe received her BSc (1993) and PhD (1997) from the University of New South Wales (Australia). She held positions at Twente University, The Netherlands; the University of Nottingham, UK and the Australian National University before taking up an Australian Research Council QEII fellowship at the University of Sydney in 2002. She currently holds the position of Payne‐Scott Professor at The University of Sydney. She is a Fellow of the Australian Academy of Science and has been awarded the Beckwith (2004), Biota (2006), Birch (2017), and H. G. Smith (2018) medals of the Royal Australian Chemical Institute. Her research interests are in the areas of supramolecular, peptide and organic chemistry, with a focus on the design and synthesis of functional molecules, such as molecular sensors capable of detecting anions in biological environments.

Biographical Information
Professor Thorri Gunnlaugsson holds a Personal Chair as Professor of Chemistry at the University of Dublin, Trinity College Dublin (TCD). Having obtained his PhD with Professor A. P. de Silva at Queen's University Belfast, he undertook his postdoctoral studies with Professor David Parker at Durham University. He was appointed to TCD in 1998 and has mentored close to 60 PhD students since then and collaborated with many postdoctoral fellows. He is the author of over 270 papers in the areas of supramolecular, materials and medicinal chemistries.

Biographical Information
Rob Elmes graduated with a B.A. Mod. (1st Class) in Medicinal Chemistry from Trinity College Dublin in 2007 before he was awarded an IRCSET Embark Scholarship to undertake his PhD under the supervision of Prof. Thorri Gunnlaugsson at TCD. After a short postdoctoral tenure at the Trinity Biomedical Sciences Institute in Dublin, Rob moved to The University of Sydney under the guidance of Prof. Kate Jolliffe where he was involved in the development of new platforms for the recognition, sensing and transport of biologically relevant anions. In late 2014, Rob returned to Ireland, taking up a position at Maynooth University where he is currently an Assistant Professor in Organic Chemistry within the Department of Chemistry and a Funded Investigator (FI) in the SSPC, The Science Foundation Ireland Research Centre for Pharmaceuticals. Rob's research interests lie in the fields of Supramolecular Chemistry and Chemical Biology where the group is trying to use supramolecular chemistry in applications such as bioconjugation methodologies, diagnostic tools and therapeutic agents.

Biographical Information
„I have had the benefit of superb mentorship and support through both my PhD (Thorri) and Postdoc (Kate) studies and I have tried to use this experience to foster a supportive and collaborative environment within my own research group. In my view, supportive mentorship is key to successful science and happy scientists and is perfectly exemplified by my own PhD supervisor's supervisor, the truly inspirational Prof. A.P. De Silva (pictured).“

Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
L.K.K. thanks the Irish Research Council for a Government of Ireland Postdoctoral Research Fellowship (GOIPD/2017/1091). L.E.B. thanks the Irish Research Council for a Government of Ireland Postgraduate Scholarship (GOIPG/2020/78). R.E. and T.G. acknowledge funding from Science Foundation Ireland (SFI), grant number 12/RC/2275/P2, which is co‐funded under the European Regional Development Fund. Science Foundation Ireland are also acknowledged for the funding of mass spectrometry facilities through the Opportunistic Infrastructure Fund (16/RI/3399). Open access funding provided by IReL.
L. K. Kumawat, C. Wynne, E. Cappello, P. Fisher, L. E. Brennan, A. Strofaldi, J. J. McManus, C. S. Hawes, K. A. Jolliffe, T. Gunnlaugsson, R. B. P. Elmes, ChemPlusChem 2021, 86, 1058.
References
- 1.Zhou B., Zhao Q., Tang L., Yan D., Chem. Commun. 2020, 56, 7698–7701. [DOI] [PubMed] [Google Scholar]
- 2.Yan D., Evans D. G., Mater. Horiz. 2014, 1, 46–57. [Google Scholar]
- 3.Lu B., Fang X., Yan D., ACS Appl. Mater. Interfaces 2020, 12, 31940–31951. [DOI] [PubMed] [Google Scholar]
- 4.Yan D., Chem. Eur. J. 2015, 21, 4880–4896. [DOI] [PubMed] [Google Scholar]
- 5.Yang Y., Fang X., Zhao S.-S., Bai F., Zhao Z., Wang K.-Z., Yan D., Chem. Commun. 2020, 56, 5267–5270. [DOI] [PubMed] [Google Scholar]
- 6.Li S., Lu B., Fang X., Yan D., Angew. Chem. Int. Ed. 2020, 59, 22623–22630; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 22812–22819. [Google Scholar]
- 7.Lombardo D., Kiselev M. A., Magazù S., Calandra P., Adv. Condens. Matter Phys. 2015, 2015, 151683. [Google Scholar]
- 8.Wang C., Wang Z., Zhang X., Acc. Chem. Res. 2012, 45, 608–618. [DOI] [PubMed] [Google Scholar]
- 9.Yu G., Jie K., Huang F., Chem. Rev. 2015, 115, 7240–7303. [DOI] [PubMed] [Google Scholar]
- 10.Calatrava-Pérez E., Acherman S., Stricker L., McManus G., Delente J., Lynes A. D., Henwood A. F., Lovitt J. I., Hawes C. S., Byrne K., Schmitt W., Kotova O., Gunnlaugsson T., Scanlan E. M., Org. Biomol. Chem. 2020, 18, 3475–3480. [DOI] [PubMed] [Google Scholar]
- 11.White L. J., Tyuleva S. N., Wilson B., Shepherd H. J., Ng K. K. L., Holder S. J., Clark E. R., Hiscock J. R., Chem. Eur. J. 2018, 24, 7761–7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.White L. J., Wells N. J., Blackholly L. R., Shepherd H. J., Wilson B., Bustone G. P., Runacres T. J., Hiscock J. R., Chem. Sci. 2017, 8, 7620–7630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blackholly L. R., Shepherd H. J., Hiscock J. R., CrystEngComm 2016, 18, 7021–7028. [Google Scholar]
- 14.Gumbs T. L., White L. J., Wells N. J., Shepherd H. J., Hiscock J. R., Supramol. Chem. 2018, 30, 286–295. [Google Scholar]
- 15.Ng K. K. L., Dimitrovski M., Boles J. E., Ellaby R. J., White L. J., Hiscock J. R., Supramol. Chem. 2020, 1–11. [Google Scholar]
- 16.Tyuleva S. N., Allen N., White L. J., Pépés A., Shepherd H. J., Saines P. J., Ellaby R. J., Mulvihill D. P., Hiscock J. R., Chem. Commun. 2019, 55, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.White L. J., Boles J. E., Allen N., Alesbrook L. S., Sutton J. M., Hind C. K., Hilton K. L. F., Blackholly L. R., Ellaby R. J., Williams G. T., Mulvihill D. P., Hiscock J. R., J. Mater. Chem. B 2020, 8, 4694–4700. [DOI] [PubMed] [Google Scholar]
- 18.Townshend G., Thompson G. S., White L. J., Hiscock J. R., Ortega-Roldan J. L., Chem. Commun. 2020, 56, 4015–4018. [DOI] [PubMed] [Google Scholar]
- 19.Allen N., White L. J., Boles J. E., Williams G. T., Chu D. F., Ellaby R. J., Shepherd H. J., Ng K. K. L., Blackholly L. R., Wilson B., Mulvihill D. P., Hiscock J. R., ChemMedChem 2020, 15, 2193–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pandurangan K., Kitchen J. A., Blasco S., Paradisi F., Gunnlaugsson T., Chem. Commun. 2014, 50, 10819–10822. [DOI] [PubMed] [Google Scholar]
- 21.Kumawat L. K., Abogunrin A. A., Kickham M., Pardeshi J., Fenelon O., Schroeder M., Elmes R. B. P., Front. Chem. 2019, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Finger T. E., Danilova V., Barrows J., Bartel D. L., Vigers A. J., Stone L., Hellekant G., Kinnamon S. C., Science 2005, 310, 1495–1499. [DOI] [PubMed] [Google Scholar]
- 23.Silveira M. L., Vendramini J. M. B., Sollenberger L. E., Int. J. Agron. 2010, 2010, 517603. [Google Scholar]
- 24.El Kateb A., Stalder C., Rüggeberg A., Neururer C., Spangenberg J. E., Spezzaferri S., PLoS One 2018, 13, e0197731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berndt T., Kumar R., Annu. Rev. Physiol. 2007, 69, 341–359. [DOI] [PubMed] [Google Scholar]
- 26.Busschaert N., Caltagirone C., Van Rossom W., Gale P. A., Chem. Rev. 2015, 115, 8038–8155. [DOI] [PubMed] [Google Scholar]
- 27.Sarwar M., Leichner J., Naja G. M., Li C.-Z., Microsyst. Nanoeng. 2019, 5, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu B., Wang H., Yang D., Tan R., Zhao R. R., Xu R., Zhou Z. J., Zhang J. F., Zhou Y., Dyes Pigm. 2016, 133, 127–131. [Google Scholar]
- 29.Hewitt S. H., Ali R., Mailhot R., Antonen C. R., Dodson C. A., Butler S. J., Chem. Sci. 2019, 10, 5373–5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gunnlaugsson T., Glynn M., Tocci G. M., Kruger P. E., Pfeffer F. M., Coord. Chem. Rev. 2006, 250, 3094–3117. [Google Scholar]
- 31.Gunnlaugsson T., Kruger P. E., Jensen P., Tierney J., Ali H. D. P., Hussey G. M., J. Org. Chem. 2005, 70, 10875–10878. [DOI] [PubMed] [Google Scholar]
- 32.Gillen D. M., Hawes C. S., Gunnlaugsson T., J. Org. Chem. 2018, 83, 10398–10408. [DOI] [PubMed] [Google Scholar]
- 33.Pandurangan K., Kitchen J. A., Blasco S., Boyle E. M., Fitzpatrick B., Feeney M., Kruger P. E., Gunnlaugsson T., Angew. Chem. Int. Ed. 2015, 54, 4566–4570; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 4649–4653. [Google Scholar]
- 34.Aletti A. B., Miljkovic A., Toma L., Bruno R., Armentano D., Gunnlaugsson T., J. Org. Chem. 2019, 84, 4221–4228. [DOI] [PubMed] [Google Scholar]
- 35.Aletti A. B., Blasco S., Aramballi S. J., Kruger P. E., Gunnlaugsson T., Chem. 2019, 5, 2617–2629. [Google Scholar]
- 36.Kim J.-I., Juwarker H., Liu X., Lah M. S., Jeong K.-S., Chem. Commun. 2010, 46, 764–766. [DOI] [PubMed] [Google Scholar]
- 37.Zwicker V. E., Yuen K. K. Y., Smith D. G., Ho J., Qin L., Turner P., Jolliffe K. A., Chem. Eur. J. 2018, 24, 1140–1150. [DOI] [PubMed] [Google Scholar]
- 38.Marchetti L. A., Kumawat L. K., Mao N., Stephens J. C., Elmes R. B. P., Chem. 2019, 6, 1398–1485. [Google Scholar]
- 39.Bujosa S., Castellanos E., Frontera A., Rotger C., Costa A., Soberats B., Org. Biomol. Chem. 2020, 18, 888–894. [DOI] [PubMed] [Google Scholar]
- 40.Ramos J., Arufe S., Martin H., Rooney D., Elmes R. B. P., Erxleben A., Moreira R., Velasco-Torrijos T., Soft Matter. 2020, 16, 7916–7926. [DOI] [PubMed] [Google Scholar]
- 41.Qin L., Vervuurt S. J. N., Elmes R. B. P., Berry S. N., Proschogo N., Jolliffe K. A., Chem. Sci. 2020, 11, 201–207. [Google Scholar]
- 42.Qin L., Wright J. R., Lane J. D., Berry S. N., Elmes R. B. P., Jolliffe K. A., Chem. Commun. 2019, 55, 12312–12315. [DOI] [PubMed] [Google Scholar]
- 43.Qin L., Hartley A., Turner P., Elmes R. B. P., Jolliffe K. A., Chem. Sci. 2016, 7, 4563–4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Elmes R. B. P., Jolliffe K. A., Supramol. Chem. 2015, 27, 321–328. [Google Scholar]
- 45.Elmes R. B. P., Busschaert N., Czech D. D., Gale P. A., Jolliffe K. A., Chem. Commun. 2015, 51, 10107–10110. [DOI] [PubMed] [Google Scholar]
- 46.Elmes R. B. P., Jolliffe K. A., Supramol. Chem. 2014, 27, 321–328. [Google Scholar]
- 47.Busschaert N., Elmes R. B. P., Czech D. D., Wu X., Kirby I. L., Peck E. M., Hendzel K. D., Shaw S. K., Chan B., Smith B. D., Jolliffe K. A., Gale P. A., Chem. Sci. 2014, 5, 3617–3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Elmes R. B. P., Turner P., Jolliffe K. A., Org. Lett. 2013, 15, 5638–5641. [DOI] [PubMed] [Google Scholar]
- 49.Pina M. N., Rotger C., Soberats B., Ballester P., Deya P. M., Costa A., Chem. Commun. 2007, 963–965. [DOI] [PubMed] [Google Scholar]
- 50.Rotger C., Soberats B., Quiñonero D., Frontera A., Ballester P., Benet-Buchholz J., Deyà P. M., Costa A., Eur. J. Org. Chem. 2008, 1864–1868. [Google Scholar]
- 51.Soberats B., Martínez L., Sanna E., Sampedro A., Rotger C., Costa A., Eur. J. Org. Chem. 2012, 18, 7533–7542. [DOI] [PubMed] [Google Scholar]
- 52.López C., Ximenis M., Orvay F., Rotger C., Costa A., Chem. Eur. J. 2017, 23, 7590–7594. [DOI] [PubMed] [Google Scholar]
- 53.Saez Talens V., Makurat D. M. M., Liu T., Dai W., Guibert C., Noteborn W. E. M., Voets I. K., Kieltyka R. E., Polym. Chem. 2019, 10, 3146–3153. [Google Scholar]
- 54.Tong C., Liu T., Saez Talens V., Noteborn W. E. M., Sharp T. H., Hendrix M. M. R. M., Voets I. K., Mummery C. L., Orlova V. V., Kieltyka R. E., Biomacromolecules 2018, 19, 1091–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Talens V. S., Englebienne P., Trinh T. T., Noteborn W. E. M., Voets I. K., Kieltyka R. E., Angew. Chem. Int. Ed. 2015, 54, 10502–10506; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10648–10652. [Google Scholar]
- 56.Hiscock J. R., Bustone G. P., Wilson B., Belsey K. E., Blackholly L. R., Soft Matter 2016, 12, 4221–4228. [DOI] [PubMed] [Google Scholar]
- 57.Amendola V., Bergamaschi G., Boiocchi M., Fabbrizzi L., Milani M., Chem. Eur. J. 2010, 16, 4368–4380. [DOI] [PubMed] [Google Scholar]
- 58.Rostami A., Colin A., Li X. Y., Chudzinski M. G., Lough A. J., Taylor M. S., J. Org. Chem. 2010, 75, 3983–3992. [DOI] [PubMed] [Google Scholar]
- 59.Jagleniec D., Siennicka S., Dobrzycki Ł., Karbarz M., Romański J., Inorg. Chem. 2018, 57, 12941–12952. [DOI] [PubMed] [Google Scholar]
- 60.Boiocchi M., Del Boca L., Gómez D. E., Fabbrizzi L., Licchelli M., Monzani E., J. Am. Chem. Soc. 2004, 126, 16507–16514. [DOI] [PubMed] [Google Scholar]
- 61.Duke R. M., Gunnlaugsson T., Tet. Lett. 2007, 48, 8043–8047. [Google Scholar]
- 62.Thordarson P., Chem. Soc. Rev. 2011, 40, 1305–1323. [DOI] [PubMed] [Google Scholar]
- 63.Lowe A. J., Pfeffer F. M., Thordarson P., Supramol. Chem. 2012, 24, 585–594. [Google Scholar]
- 64.Brynn Hibbert D., Thordarson P., Chem. Commun. 2016, 52, 12792–12805. [DOI] [PubMed] [Google Scholar]
- 65.Elmes R. B. P., Yuen K., Jolliffe K. A., Chem. Eur. J. 2014, 20, 7373–7380. [DOI] [PubMed] [Google Scholar]
- 66.Edwards S. J., Valkenier H., Busschaert N., Gale P. A., Davis A. P., Angew. Chem. Int. Ed. 2015, 54, 4592–4596; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 4675–4679. [Google Scholar]
- 67.Berry S. N., Soto-Cerrato V., Howe E. N. W., Clarke H. J., Mistry I., Tavassoli A., Chang Y.-T., Perez-Tomas R., Gale P. A., Chem. Sci. 2016, 7, 5069–5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Prohens R., Portell A., Font-Bardia M., Bauza A., Frontera A., Chem. Commun. 2018, 54, 1841–1844. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information