

Abstract
Objective
To confirm the efficacy and safety of intraarticular (IA) injection of diclofenac covalently linked to hyaluronic acid (diclofenac etalhyaluronate [DF‐HA]; ONO‐5704/SI‐613) in patients with knee osteoarthritis (OA).
Methods
In a phase III multicenter, randomized, double‐blind, placebo‐controlled trial, eligible subjects ages 40–75 years with symptomatic knee OA (Kellgren/Lawrence score of 2 or 3) were randomly assigned to receive IA injections of DF‐HA 30 mg or placebo (citric acid–sodium citrate buffered solution; 1:1) once every 4 weeks for 20 weeks (a total of 6 injections). Subjects were followed up for 24 weeks. The primary end point was the mean change from baseline to 12 weeks in Western Ontario and McMaster Universities Osteoarthritis Index version 3.1 (WOMAC) pain subscale scores, measured on a 100‐mm visual analog scale. Safety was evaluated by adverse event monitoring.
Results
All 440 subjects received investigational products (220 received placebo and 220 received DF‐HA). The full analysis set and safety population comprised 438 subjects (220 in the placebo group and 218 in the DF‐HA group) and 440 subjects, respectively. At 12 weeks, subjects receiving DF‐HA showed significant improvement from baseline in the WOMAC pain subscale score (–23.2 mm) compared to subjects receiving placebo ( −17.1 mm), with a difference of −6.1 mm (95% confidence interval −9.4, −2.8; P < 0.001). The difference between groups was significant as early as week 1, and a difference was maintained for 24 weeks, although the difference at week 24 was not significant. Anaphylactic reactions were observed in 2 subjects receiving DF‐HA.
Conclusion
Our findings indicate that treatment with DF‐HA results in significant improvement in the WOMAC pain subscale score compared to placebo over 12 weeks. Anaphylactic reactions were observed, and further safety evaluation is needed.
INTRODUCTION
Osteoarthritis (OA) is the most common degenerative joint disease, occurring frequently in the elderly (1). Current disease management mainly focuses on the treatment of symptoms (pain relief) to improve joint function and quality of life until the late stages of arthritis leading to knee replacement (2). The selection of knee OA treatment depends on disease severity, comorbidity, and patient preference (1, 3, 4, 5). Main treatment options include conservative therapies such as exercise therapy, physiotherapy, and pharmacotherapy. Pharmacotherapies include oral or topical nonsteroidal antiinflammatory drugs (NSAIDs), selective cyclooxygenase 2 (COX‐2) inhibitors, and intraarticular (IA) injection of glucocorticoids or hyaluronic acid (HA). NSAIDs are most commonly used for the symptoms of knee OA (6); however, upper and lower gastrointestinal (GI) tract disorders, adverse cardiovascular (CV) reactions, and renal dysfunction are associated with the long‐term use of oral NSAIDs (7, 8, 9, 10). Therefore, lower dosing of oral NSAIDs for a shorter period is recommended in patients at risk of these adverse events (1, 4). In addition, topical NSAIDs are recommended for patients with knee OA who have these comorbid conditions (4).
Although IA injection of glucocorticoids or HA is used for the conservative treatment of knee OA, these agents are a subject of controversy. IA glucocorticoids should be used for short‐term pain relief because of their short‐lived efficacy and safety issues (1, 11, 12, 13). The benefits of HA have been reported in multiple studies, although there is conflicting data. Clinical and nonclinical reports suggest that IA HA has antiinflammatory and analgesic effects, suppresses joint cartilage degeneration, and normalizes synovial fluid. HA also functions as a lubricant due to its viscoelasticity (14, 15). Nevertheless, treatment of knee OA with IA HA has not yet been established, and most guidelines do not currently recommend the use of IA HA based on poor evidence of its benefits (1, 3, 5).
In pursuit of OA treatments, Seikagaku Corporation developed a novel compound, diclofenac etalhyaluronate (DF‐HA; ONO‐5704/SI‐613), which is DF covalently linked to HA. It is a high molecular weight fermented HA (600–1,200 kd) combining the purported advantages of IA HA injection and NSAIDs: antiinflammatory and analgesic effects, suppression of joint cartilage degeneration, and normalization of synovial fluid function (16, 17). Compared with oral DF therapy, IA injection of DF‐HA is thought to reduce systemic exposure to DF because the amount of DF released from DF‐HA is ~3.5 mg/dose. Indeed, in rabbits with antigen‐induced arthritis, systemic exposure to DF was much lower after a single effective IA dose of DF‐HA than after a single effective oral dose of diclofenac sodium (16). Furthermore, the sustained release of DF from DF‐HA after 1 injection into the joint tissue has potential analgesic effects lasting up to 28 days (16). The efficacy and safety of DF‐HA in patients with knee OA was confirmed in a previous trial (18), in which patients who received a total of 3 injections of DF‐HA, one every 4 weeks for 12 weeks, showed significant improvements in the Western Ontario and McMaster Universities Osteoarthritis Index 3.1 (WOMAC) (19) pain subscale score compared to patients who received placebo, with no major safety concerns.
We conducted a phase III trial to confirm the efficacy and safety of IA injection of DF‐HA 30 mg every 4 weeks into the knee joint cavities of patients with knee OA. The primary objective was to verify the superiority of DF‐HA over placebo by showing a greater change from baseline in the WOMAC pain subscale score over 12 weeks. The primary end point was the 12‐week mean change from baseline in WOMAC pain subscale score to verify the results of the phase II trial. The secondary objective was to evaluate the safety of DF‐HA when injected 6 times, once every 4 weeks, up to 24 weeks compared with placebo.
PATIENTS AND METHODS
Study design and characteristics of the subjects
A phase III multicenter, randomized, double‐blind, placebo‐controlled trial was performed at 50 sites in Japan. The study was conducted in accordance with the Declaration of Helsinki and the International Council for Harmonisation Guidelines for Good Clinical Practice and was approved by the institutional review boards at the central boards or at each local site. All subjects provided written informed consent.
Eligible subjects were ages 40–75 years and had knee OA (not secondary OA caused by trauma or another disease) with a Kellgren/Lawrence (K/L) grade of 2 or 3 diagnosed radiographically, pain for at least 1 year in the target knee, and a WOMAC pain subscale score and pain score on the 50‐foot walking test of 50–90 mm (on a 100‐mm visual analog scale [VAS]) in the target knee and ≤30 mm in the contralateral knee at the time of screening. Patients with lower extremity pain caused by other diseases; inflammation, infection, skin disease, or systemic disease at the target knee; body mass index (BMI) ≥35.0 kg/m2, which indicates a high risk of complications; hypersensitivity to DF, HA, or acetaminophen; aspirin‐induced asthma; surgical or invasive treatment within 1 year; or joint effusion removal within 7 days prior to screening were excluded.
Randomization and blinding
Subjects were allocated to receive either DF‐HA (30 mg/3 ml in a prefilled syringe; Seikagaku Corporation) or placebo (citric acid–sodium citrate buffered solution [3 ml in a prefilled syringe; Seikagaku Corporation]) at a 1:1 ratio, and randomization was performed using an interactive web response system. Balance between the treatment groups at each site was achieved using a minimization method (20). Groups were stratified by K/L grade, mean baseline WOMAC pain subscale score, and sex.
Both treatments were clear, colorless, and identical in appearance; however, the force needed to inject them differed because DF‐HA has a greater viscosity. To maintain blinding, an investigator other than the one who administered the injection performed all postinjection evaluations. Moreover, the investigator who administered the injection was prohibited from divulging information about the treatment to the investigators conducting the assessments or the subjects.
Procedures
Screening was conducted 1 week before randomization and on the day of randomization. The entire volume (3 ml) of study drug or placebo was injected IA into the target knee of each subject a total of 6 times, once every 4 weeks (at weeks 0, 4, 8, 12, 16, and 20). Although the administration method was not specified, IA injection was given by an orthopedist or a general physician who routinely performs IA injections, following the same procedure as in their normal clinical practice. The observation period began on the day of the first injection and ended at week 24. Efficacy and safety were assessed on the day of the first injection (week 0), and at weeks 1, 2, 4, 6, 8, 10, 12, 16, 20, and 24.
Analgesic agents (IA HA products, NSAIDs, glucocorticoids, opioid analgesics, and psychotherapeutic drugs), which are known to affect the underlying disease and its assessment, were prohibited from the specified washout period prior to the screening through the end of the study. Acetaminophen was provided for all subjects as a rescue medication from 7 days prior to screening through the end of the study and permitted at a dosage of up to 3,600 mg/day but was proscribed starting 2 days before each visit.
Outcome measures
The primary outcome measure was the WOMAC pain subscale score, measured on a 100‐mm VAS, and the primary end point was the mean change from baseline in WOMAC pain subscale score at 12 weeks. The secondary outcome measures were WOMAC index score (stiffness subscale score, physical function subscale score, and total score on a 100‐mm VAS), pain score on a 100‐mm VAS after a 50‐foot walking test, daily pain score on an 11‐point numerical rating scale as recorded in the subject’s diary, rates of responders and strict responders according to the Outcome Measures in Rheumatology (OMERACT)–Osteoarthritis Research Society International (OARSI) response criteria (21), patient global assessment, physician global assessment, Medical Outcomes Study Short Form 36 (SF‐36) health survey (22, 23, 24), EuroQol 5‐domain (EQ‐5D) (25), and acetaminophen consumption.
Safety was evaluated by monitoring treatment‐emergent adverse events (TEAEs) after the initial injection, based on the definitions listed in Supplementary Table 1, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41725/abstract. AEs that led to study drug withdrawal were considered important AEs, while those that occurred at the injection site, and were associated with GI disorders, CV disorders, renal dysfunction, anaphylactic reaction, or hypersensitivity were considered AEs of special interest.
Clinical laboratory tests (hematology, blood biochemistry, and urine test), measurement of vital signs (body temperature, blood pressure, and pulse rate), examination of the target knee (for joint effusion, swelling, redness, and warmth) to assess the injection site reaction, and radiography, including anteroposterior standing, lateral standing, and patellar axial radiographs to observe structural changes (worsening) in the target knee (osteophytes, joint space narrowing, osteosclerosis, or deformity of epiphysis) were conducted based on the protocol at preinjection and at 24 weeks or at the time of study discontinuation. To prevent variations in evaluation at each site, instructions for radiography were distributed (Supplementary Table 2, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41725/abstract). Imaging evaluation was performed by the investigator. Findings that were judged by the investigator to be unfavorable or unintended were considered AEs.
Statistical analysis
We estimated that a sample size of 220 subjects per group would be needed to attain at least 90% power to detect a significant difference between groups with a 2‐sided significance level of 5%, assuming that the difference between groups in the change from baseline in WOMAC pain subscale score was −7.5 mm, with an SD of 23 mm and a dropout rate of ~10% (18).
Efficacy analyses were conducted in the full analysis set, which included subjects who received ≥1 injection and were evaluated for efficacy. For the primary analysis, the mean change from baseline in the WOMAC pain subscale score over 12 weeks was analyzed using a mixed‐effects model for repeated measures (MMRM) at each time point from weeks 1 through 12. The model included treatment group, time point, treatment group–by–time point interaction, baseline WOMAC pain subscale score, K/L grade, sex, and pooled site as fixed effects. The covariance structure was assumed to be unstructured. The Kenward‐Roger method was used to calculate degrees of freedom. For the secondary analysis, the change from baseline in WOMAC pain subscale score at each time point of weeks 1 through 24 was compared between the groups using the same MMRM as used in the primary analysis. To evaluate the impact of missing data, a sensitivity analysis was conducted using the pattern‐mixture model approach to multiple imputation under the missing‐not‐at‐random assumption by creating control‐based pattern imputation. Mean changes from baseline in WOMAC pain subscale score over 12 weeks in the subgroups were analyzed using the same MMRM model without K/L grade, sex, and pooled site.
Secondary continuous outcomes expected for EQ‐5D and mean daily acetaminophen consumption were analyzed using the same MMRM as used for analyses of the primary outcome. The responder rate and strict responder rate were analyzed using a generalized estimating equation. The odds ratio over 12 weeks and at each time point were calculated using the model that included treatment group, time point, treatment group–by–time point interaction, baseline WOMAC pain subscale score, K/L grade, sex, and pooled site. The covariance structure was assumed to be unstructured. The change from baseline in the mean daily acetaminophen consumption at each time point was compared with that in the placebo group using Wilcoxon’s rank sum test.
Responder analyses were performed post hoc to interpret clinically meaningful treatment benefits of group differences (26). These analyses were conducted using the percentages of subjects with an improvement in WOMAC pain subscale score from baseline of >20%, >30%, or >50%, who were considered to have clinically meaningful pain reduction, and subjects whose WOMAC pain subscale score reached <40 mm, <30 mm, or <20 mm based on different levels of response on ratings of treatment satisfaction at 1, 4, 12, and 24 weeks.
TEAEs and other safety outcomes were summarized in the safety population, which included subjects who received treatment at least once. The incidences of TEAEs were calculated for each treatment group. TEAEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA) version 21.1. TEAEs associated with GI disorders, CV disorders, renal dysfunction, anaphylactic reaction, or hypersensitivity were categorized using a standardized MedDRA query.
SAS software, version 9.4 (SAS Institute) was used for all statistical analyses. P values less than 0.05 (2‐sided) were considered significant.
RESULTS
Between March 24, 2017 and July 30, 2018, 539 subjects were screened (Figure 1). Of the 539 subjects, 440 were determined to be eligible for the study, randomized, and received treatment (220 received placebo and 220 received DF‐HA). The safety population comprised 440 subjects (220 in the placebo group and 220 in the DF‐HA group), and the full analysis set comprised 438 subjects (220 in the placebo group and 218 in the DF‐HA group), which was the safety population minus 2 subjects who had no efficacy evaluation data. Demographic and other baseline characteristics were similarly distributed between the treatment groups (Table 1).
Figure 1.
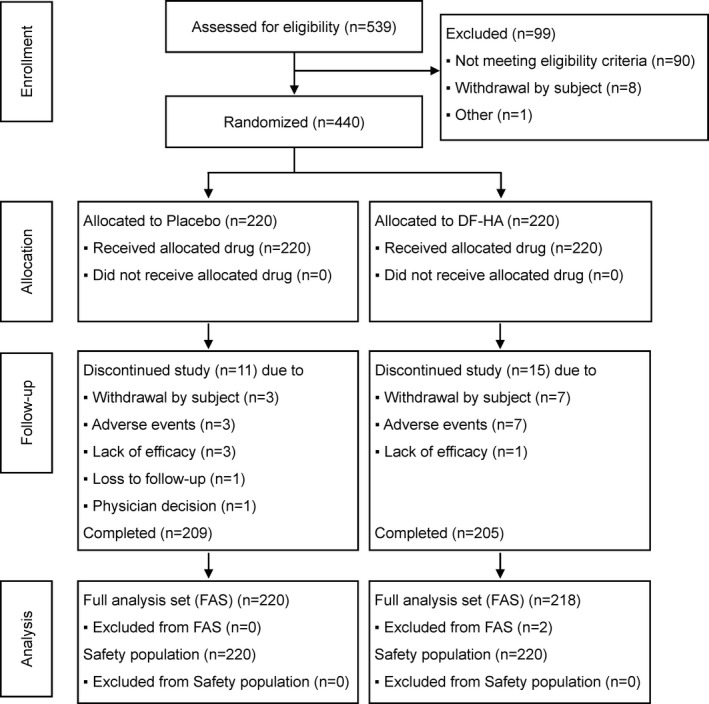
Disposition of the study subjects. Details are given according to the Consolidated Standards of Reporting Trials (CONSORT) statement for reporting randomized controlled trials. DF‐HA = diclofenac etalhyaluronate.
Table 1.
Baseline characteristics of the subjects with knee OA*
|
Placebo (n = 220) |
DF‐HA 30 mg (n = 218) |
|
|---|---|---|
| Age, years | 62.4 ± 8.1 | 63.3 ± 8.7 |
| Sex, no, (%) male/female | 75 (34.1)/145 (65.9) | 75 (34.4)/143 (65.6) |
| BMI, kg/m2 | 25.61 ± 3.95 | 25.46 ± 3.75 |
| Duration of current pain, weeks | 268.3 ± 257.8 | 252.4 ± 262.0 |
| K/L grade, no. (%) | ||
| 2 | 124 (56.4) | 122 (56.0) |
| 3 | 96 (43.6) | 96 (44.0) |
| WOMAC pain score, mm† | 65.2 ± 7.6 | 64.9 ± 7.9 |
| WOMAC pain score category, no. (%)† | ||
| <70 mm | 163 (74.1) | 162 (74.3) |
| ≥70 mm | 57 (25.9) | 56 (25.7) |
| WOMAC stiffness score, mm† | 57.5 ± 21.4 | 56.6 ± 20.9 |
| WOMAC physical function score, mm† | 61.3 ± 12.8 | 59.8 ± 12.8 |
| WOMAC total score, mm† | 61.8 ± 11.0 | 60.6 ± 11.3 |
| 50‐foot walking test pain score, mm† | 68.0 ± 7.6 | 68.0 ± 8.5 |
| Daily pain score | 6.69 ± 1.20‡ | 6.60 ± 1.27 |
| Patient global assessment score, mm† | 70.1 ± 12.7 | 69.7 ± 12.9 |
| Physician global assessment score, mm† | 62.5 ± 14.1 | 62.9 ± 14.0 |
| SF‐36 summary score | ||
| MCS score | 55.8 ± 8.3 | 55.4 ± 8.1 |
| RCS score | 45.1 ± 13.0 | 47.2 ± 13.1 |
| PCS score | 25.8 ± 10.5 | 25.9 ± 11.6 |
| EQ‐5D | ||
| QOL score | 0.6778 ± 0.1354 | 0.6906 ± 0.1254 |
| VAS score | 66.3 ± 16.2 | 67.0 ± 16.3 |
| Daily acetaminophen dosage, mg/day | 220.5 ± 390.0 | 230.2 ± 399.5 |
Except where indicated otherwise, values are the mean ± SD. OA = osteoarthritis; DF‐HA = diclofenac etalhyaluronate; BMI = body mass index; K/L = Kellgren/Lawrence; WOMAC = Western Ontario and McMaster Universities Osteoarthritis Index; SF‐36 = Short Form 36; MCS = mental component summary; RCS = social role component summary; PCS = physical component summary; EQ‐5D = EuroQol 5‐domain; QOL = quality of life.
On a 100‐mm visual analog scale (VAS).
Data were available for 219 patients.
The least squares mean (LSM) change from baseline in the WOMAC pain subscale score at 12 weeks was −17.1 mm for placebo and −23.2 mm for DF‐HA (Table 2). The difference in LSM between the groups was −6.1 mm (95% confidence interval [95% CI] −9.4, −2.8; P < 0.001), which demonstrates the superiority of DF‐HA over placebo. Results of the MMRM sensitivity analysis using placebo multiple imputation were similar to the results of the primary analysis without imputation of missing data. Figure 2 shows forest plots of the difference in LSM between the groups and the 95% CI for all subjects and for subgroups of subjects. The change from baseline in WOMAC pain subscale score over 12 weeks was greater in all DF‐HA subgroups than in all placebo subgroups. The between‐group differences in LSM were similar for all subgroups.
Table 2.
Primary and secondary outcomes at 12 weeks*
| LSM change from baseline (95% CI) |
Difference (95% CI) |
P | ||
|---|---|---|---|---|
| Placebo | DF‐HA | |||
| Primary outcome | ||||
| WOMAC pain score, mm† | ||||
| Primary analysis | −17.1 (−19.8, −14.4) | −23.2 (−25.9, −20.4) | −6.1 (−9.4, −2.8) | <0.001 |
| Sensitivity analysis | −17.2 (−19.9, −14.4) | −23.2 (−25.9, −20.4) | −6.0 (−9.3, −2.7) | <0.001 |
| Secondary outcomes | ||||
| WOMAC stiffness score, mm† | −13.3 (−16.1, −10.5) | −17.9 (−20.7, −15.1) | −4.6 (−8.0, −1.2) | 0.008 |
| WOMAC physical function score, mm† | −13.2 (−15.8, −10.5) | −18.9 (−21.5, −16.2) | −5.7 (−8.9, −2.5) | <0.001 |
| Total score | −14.0 (−16.6, −11.4) | −19.6 (−22.2, −16.9) | −5.6 (−8.7, −2.4) | <0.001 |
| 50‐foot walking test pain score, mm† | −21.3 (−24.3, −18.2) | −28.1 (−31.1, −25.1) | −6.8 (−10.5, −3.2) | <0.001 |
| Mean daily pain score | −1.20 (−1.41, −0.99) | −1.76 (−1.97, −1.55) | −0.56 (−0.82, −0.31) | <0.001 |
| Patient global assessment score, mm† | −18.1 (−20.7, −15.4) | −24.6 (−27.2, −22.0) | −6.5 (−9.7, −3.3) | <0.001 |
| Physician global assessment score, mm† | −16.4 (−18.6, −14.2) | −20.9 (−23.1, −18.7) | −4.5 (−7.2, −1.9) | <0.001 |
| SF‐36 summary score | ||||
| MCS | −0.1 (−1.0, 0.7) | 0.5 (−0.3, 1.4) | 0.6 (−0.4, 1.7) | 0.209 |
| RCS | 2.5 (1.2, 3.8) | 2.1 (0.8, 3.4) | −0.4 (−2.0, 1.1) | 0.588 |
| PCS | 4.3 (3.0, 5.6) | 5.9 (4.5, 7.2) | 1.6 (0.0, 3.2) | 0.049 |
| OMERACT‐OARSI response | ||||
| Responder‡ | −0.11 (−0.36, 0.14)§ | 0.41 (0.15, 0.67)§ | 1.69 (1.24, 2.30)¶ | <0.001 |
| Strict responder# | −1.26 (−1.56, −0.97)§ | −0.71 (−0.99, −0.43)§ | 1.74 (1.25, 2.43)¶ | 0.001 |
OMERACT‐OARSI = Outcome Measures in Rheumatology–Osteoarthritis Research Society International (see Table 1 for other definitions).
On a 100‐mm visual analog scale.
Responders were defined as subjects with ≥20% improvement from baseline and an absolute change of ≥10 mm in ≥2 of the following 3 measures: WOMAC pain subscale score, WOMAC physical function subscale score, and/or patient global assessment score.
Values are the log odds (95% confidence interval [95% CI]).
Values are the odds ratio (95% CI).
Strict responders were defined as subjects with ≥50% improvement from baseline and absolute change of ≥20 mm in the WOMAC pain subscale score or WOMAC physical function subscale score.
Figure 2.
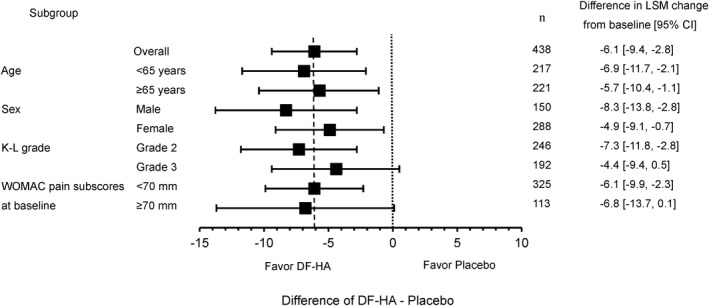
Forest plot showing the change from baseline over 12 weeks in Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) pain subscale score in the full analysis set (overall) and in the indicated subgroups of subjects with knee osteoarthritis receiving diclofenac etalhyaluronate (DF‐HA) or placebo. Values are the least squares mean (LSM) change from baseline (95% confidence interval [95% CI]). Values were estimated using a mixed‐effects model for repeated measures. K/L = Kellgren/Lawrence.
Table 2 also shows the results of the secondary outcomes over 12 weeks. Significant differences were observed for all outcomes except for the mental component summary score and social role component summary score of the SF‐36. OMERACT‐OARSI responder analysis indicated that the responder and strict responder rates at week 12 were 63.7% for placebo versus 76.7% for DF‐HA and 41.0% for placebo versus 54.3% for DF‐HA, respectively (Supplementary Table 3, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41725/abstract). The odds ratios for achieving a response or a strict response at week 12 with DF‐HA versus placebo were 1.69 (95% CI 1.24, 2.30; P < 0.001) and 1.74 (95% CI 1.25, 2.43; P = 0.001), respectively (Table 2).
Figure 3 shows the time course of the WOMAC pain subscale score, WOMAC function subscale score, and patient global assessment score, which improved after each DF‐HA injection. The between‐group difference was significant beginning at week 1, and a difference was maintained for 24 weeks, although the difference at week 24 was not significant. The between‐group differences in LSM in WOMAC pain subscale score were −7.2 mm at week 1 (95% CI −10.5, −3.9; P < 0.001), −6.2 mm at week 4 (95% CI −9.9, −2.5; P = 0.001), −6.1 mm at week 12 (95% CI −10.3, −2.0; P = 0.004), and −4.0 mm at week 24 (95% CI −8.6, 0.5; P = 0.082) (Supplementary Table 4, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41725/abstract). Similar effects were observed for the secondary outcomes (Supplementary Tables 4–10, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41725/abstract).
Figure 3.
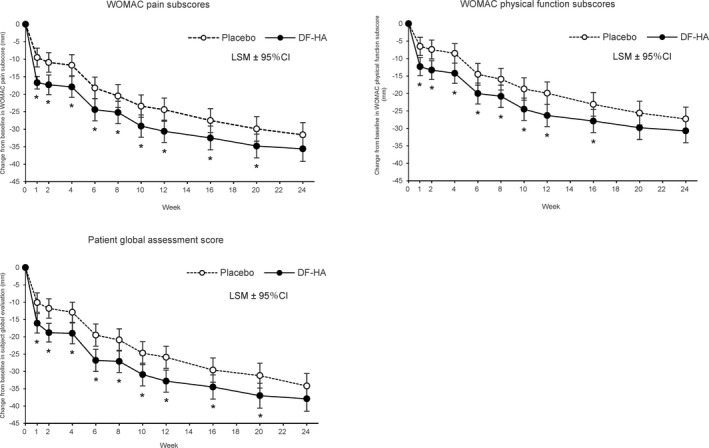
Time course of change from baseline in Western Ontario and McMaster Universities Osteoarthritis Index version 3.1 (WOMAC) pain subscale, WOMAC physical function subscale, and patient global assessment scores up to week 24 in the full analysis set of patients with knee OA receiving diclofenac etalhyaluronate (DF‐HA) or placebo. Values are the least squares mean (LSM) change from baseline with 95% confidence interval (95% CI). * = P < 0.05 versus placebo.
The OMERACT‐OARSI response rates were higher in the DF‐HA group than the placebo group at all time points until week 24. The response rates for placebo versus DF‐HA were 26.9% versus 45.0% at week 1 and 71.8% versus 81.1% at week 24, and the response rates in each group increased after injection (Supplementary Table 3). The decrease from baseline in mean daily acetaminophen consumption at each time point was greater in the DF‐HA group than in the placebo group, and the difference was significant at all time points through week 12 (Supplementary Table 11, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41725/abstract).
Post hoc responder analyses showed that the DF‐HA group had a higher proportion of subjects with improved pain than the placebo group at all cutoffs and time points in terms of the percentages of subjects with clinically meaningful pain reduction (improvement of >20%, >30%, or >50%), and subjects reporting different levels of response on ratings of treatment satisfaction (pain level of <40 mm, <30 mm, or <20 mm) (Supplementary Table 12, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41725/abstract). The proportions of subjects with improvement from baseline of >20%, >30%, and >50% at week 12 in the placebo group versus the DF‐HA group were 66.5% versus 78.1%, 56.1% versus 71.0%, and 38.2% versus 51.9%, respectively, and the proportions of subjects with a pain level of <40 mm, <30 mm, and <20 mm at week 12 in the placebo group versus the DF‐HA group were 49.5% versus 61.4%, 37.3% versus 48.6%, and 26.9% versus 38.1%, respectively (Supplementary Table 12, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41725/abstract).
Table 3 shows that the incidence of TEAEs was 126 (57.3%) in the placebo group and 134 (60.9%) in the DF‐HA group. No severe TEAEs occurred in either group. The incidence of serious TEAEs was 1 (0.5%) in the placebo group and 5 (2.3%) in the DF‐HA group. Serious TEAEs were nausea and vomiting in 1 subject in the placebo group and anaphylactic shock, anaphylactic reaction, autonomic seizure, unstable angina, and strabismus correction in 1 subject each in the DF‐HA group. All serious TEAEs were moderate in severity and were resolved with complete recovery except for unstable angina (resolving). Anaphylactic shock and anaphylactic reaction occurred on the first day of DF‐HA injection. The subject with anaphylactic shock was not hospitalized, and the subject with anaphylactic reaction was hospitalized. Anaphylactic shock and anaphylactic reaction were mitigated on the day the subjects received various antianaphylaxis therapies or medications and were resolved at 6 and 8 days after onset, respectively. Autonomic seizure was caused by pain during puncture and was judged to be a serious TEAE based on symptoms such as SpO2 decline. Emergency treatment was performed, and recovery was confirmed 2 hours after the onset. Unstable angina and strabismus correction were judged to be serious TEAEs because of hospitalization for treatment (Supplementary Table 13, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41725/abstract).
Table 3.
Overview of treatment‐emergent adverse events*
|
Placebo (n = 220) |
DF‐HA (n = 220) |
|
|---|---|---|
| All events | 126 (57.3) | 134 (60.9) |
| Severity of events | ||
| Mild | 107 (48.6) | 114 (51.8) |
| Moderate | 19 (8.6) | 20 (9.1) |
| Severe | 0 (0) | 0 (0) |
| Death | 0 (0) | 0 (0) |
| Serious events | 1 (0.5) | 5 (2.3) |
| Events leading to study drug withdrawal | 2 (0.9) | 3 (1.4) |
| Common events (in ≥2% of patients) | ||
| Nasopharyngitis | 30 (13.6) | 37 (16.8) |
| Eczema | 2 (0.9) | 5 (2.3) |
| Arthralgia | 7 (3.2) | 10 (4.5) |
| Back pain | 7 (3.2) | 7 (3.2) |
| Myalgia | 1 (0.5) | 5 (2.3) |
| Osteoarthritis | 8 (3.6) | 7 (3.2) |
| Injection site joint pain | 6 (2.7) | 6 (2.7) |
| Contusion | 5 (2.3) | 4 (1.8) |
| Ligament sprain | 1 (0.5) | 7 (3.2) |
| Events of special interest | ||
| Events at injection site | 20 (9.1) | 19 (8.6) |
| Gastrointestinal disorders† | 0 (0) | 1 (0.5) |
| Cardiovascular disorders† | 3 (1.4) | 7 (3.2) |
| Renal dysfunction† | 1 (0.5) | 1 (0.5) |
| Anaphylactic reaction† | 6 (2.7) | 4 (1.8) |
| Hypersensitivity† | 12 (5.5) | 12 (5.5) |
Adverse events were classified based on the Medical Dictionary for Regulatory Activities (MedDRA) version. 21.1. Values are the number (%).
Standardized MedDRA query (broad scope) term. The term “gastrointestinal disorders” indicates gastrointestinal perforation, ulceration, hemorrhage, or obstruction. “Cardiovascular disorders” indicates acute cardiac failure, ischemic heart disease, or cardiac arrhythmias. “Renal dysfunction” indicates acute renal failure or chronic kidney disease.
TEAEs leading to study drug withdrawal were joint swelling and injection site pain in 1 subject each in the placebo group, and anaphylactic shock, anaphylactic reaction, and autonomic seizure (these 3 events were the serious TEAEs described above) in 1 subject each in the DF‐HA group. The incidence of TEAEs of special interest at the injection site was 20 (9.1%) in the placebo group and 19 (8.6%) in the DF‐HA group (Supplementary Table 14, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41725/abstract). The incidence of TEAEs associated with GI disorders was 1 (0.5%) in the DF‐HA group and was not reported in the placebo group; the incidence of TEAEs associated with CV disorders was 3 (1.4%) in the placebo group and 7 (3.2%) in the DF‐HA group; and the incidence of TEAEs associated with renal dysfunction was 1 (0.5%) in both groups (Supplementary Table 14, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41725/abstract). TEAEs associated with anaphylactic reaction and hypersensitivity were evaluated because serious TEAEs of anaphylaxis were observed in this study. The incidence of TEAEs associated with anaphylactic reaction was 6 (2.7%) in the placebo group and 4 (1.8%) in the DF‐HA group, and the incidence of TEAEs associated with hypersensitivity was 12 (5.5%) in both groups (Supplementary Table 14). Overall, there was no clear between‐group difference in the incidence of TEAEs.
There were no noteworthy changes from baseline in laboratory values and vital signs. The percentage of subjects who experienced worsening from baseline on target knee examination was low for each outcome measure in both the placebo and DF‐HA groups. The frequency did not increase with dose and was similar between groups (Supplementary Table 15, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41725/abstract). Some subjects were determined to have structurally “changed” (worsening) target knees on radiography, but there was no between‐group difference (Supplementary Table 16, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41725/abstract).
DISCUSSION
We achieved the primary objective of a significant improvement in WOMAC pain subscale score in subjects receiving DF‐HA compared to those receiving placebo for 12 weeks. Sensitivity analysis demonstrated the robustness of the results. Furthermore, the results were not influenced by demographic characteristics according to subgroup analysis, and there were no between‐group differences in demographic or other baseline characteristics. Similar to the primary outcome, most of the secondary outcomes indicated the superiority of 12 weeks of treatment with DF‐HA compared to placebo. These results confirmed the efficacy of DF‐HA and reproduced the results of our previous phase II study (18).
According to Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials (IMMPACT) recommendations (26), the clinical importance of a group difference should be evaluated by demonstrating a significant improvement in the primary end point relative to placebo, but also by multiple other factors. Responder analyses using the percentages of subjects with clinically meaningful pain reduction and subjects reporting different levels of response on ratings of treatment satisfaction are considered useful for interpreting between‐group differences (26). In this study, when the percentages of subjects with improved pain at multiple cutoffs were compared between groups, they were higher in the DF‐HA group than in the placebo group for all cutoffs at all time points. In addition, the OMERACT‐OARSI response rate was higher in the DF‐HA group than the placebo group at all time points. Improvement was demonstrated in ~50% of DF‐HA–treated subjects at week 1, and this was extended to >80% of DF‐HA–treated subjects at week 24.
Moreover, DF‐HA improved pain as well as physical function and global assessment. In a clinical trial conducted in patients with knee OA, assessment of pain, physical function, and global function were important outcomes (27). DF‐HA also improved the quality of life and decreased the use of acetaminophen as outcomes for chronic pain (28). These results suggest that DF‐HA improves multiple symptoms including pain in knee OA. Of note, in the present study DF‐HA decreased WOMAC pain subscale scores from as early as week 1, and this level of decrease was maintained for 4 weeks. This effect was extended to 24 weeks by injection once every 4 weeks.
However, WOMAC pain subscale scores were decreased from baseline after each injection in both the DF‐HA and placebo groups, and there were many responders in the placebo group. These results may be explained by the placebo effect. The large placebo effects related to IA injection are likely to be caused by the invasiveness of the administrative procedure and the physiologic response to liquid injected into the articular cavity (29, 30). In the present study, injections were administered frequently (6 times), which may have further increased the placebo effect (30). Indeed, a large placebo effect is always a concern in clinical trials using IA injection, and many trials in OA fail to show a significant difference between placebo and the study drug. In addition, the between‐group difference in WOMAC pain subscale scores was not significant at week 24, which may result from floor effects as well as large placebo effects. As described above, some benefits of DF‐HA were confirmed, but further studies are needed to evaluate their clinical importance.
Regarding safety, anaphylactic shock and anaphylactic reaction in 1 subject each were judged to be moderate and were resolved after pharmacotherapy. Because anaphylactic symptoms in response to DF‐HA may not be ruled out for clinical use, careful monitoring and immediate treatment with established anaphylaxis therapies will be required. No clinically important GI‐ or CV‐related TEAEs, which have been observed with oral NSAIDs and selective COX‐2 inhibitors, or clinically important renal dysfunction, were observed in the DF‐HA group. This promising safety profile may be related to the lower dose needed and reduced systemic exposure to DF that occurs when DF‐HA is administered via injection.
In a previous phase II study, joint inflammation at the injection site was observed in 1 subject. Therefore, we performed examination of the target knee to evaluate the safety of injection into the local joint. No clinical issues were revealed on target knee examination, demonstrating that AEs similar to those in the previous study did not occur in the present study despite the larger number of DF‐HA injections. No differences in the incidence of TEAEs at the injection site were observed between the treatment groups, whereas these local reactions have been reported for other HA preparations (31). Finally, no clinical issue was observed on radiography. Nevertheless, NSAIDs have been reported to be deleterious to joint cartilage (32). Although no particular concern was confirmed by the findings of this study, further studies are needed to determine the effects of DF‐HA on joint cartilage, since DF‐HA, after approval, would be the first IA preparation of NSAIDs to be injected into human knee cavities.
Some existing IA preparations are effective when injected once every 3 or 6 months. However, DF‐HA requires repeated injections every 4 weeks to maintain efficacy. This is a limitation of the practical use of DF‐HA. Furthermore, although not confirmed in this study, joint infection is a common risk of IA injection, and the risk is higher with more frequent administration compared with IA preparations administered once every 3 or 6 months. Given that serious safety issues caused by DF‐HA were confirmed in this study, it is necessary to compare the risk/benefit ratio with that of other IA preparations. In particular, a head‐to‐head study of DF‐HA versus an approved HA is required to determine whether there is a great enough benefit of DF‐HA to justify the monthly injection frequency versus the semiannual injection frequency of most HA preparations.
This study had some limitations. Data were collected from a limited population that only comprised Japanese patients, there were no data for the excluded patient population (e.g., patients with a BMI of ≥35.0 kg/m2), no information was obtained on combinations with other analgesics because use of other analgesics was prohibited, and the study did not evaluate safety with regard to joint tissues, including joint cartilage. Active treatment–controlled clinical trials are needed to evaluate the clinical usefulness of DF‐HA, and how long the effect of DF‐HA lasts after IA treatment is stopped should be investigated in future clinical studies. Additional safety data are needed, since the number of subjects in this study was relatively small, and the long‐term safety of DF‐HA treatment exceeding 24 weeks should be determined because the treatment term may be longer in actual clinical practice than that assessed in this study. In addition, evaluation of safety with regard to the joint tissue including joint cartilage is needed, using imaging techniques other than radiography by a central measurement.
In conclusion, DF‐HA, a conjugated compound with the advantages of IA HA and NSAIDs, promoted significant improvements in symptoms, with fast‐acting, long‐lasting efficacy in knee OA patients when injected once every 4 weeks. Anaphylactic reactions were observed, and further safety evaluation is needed. Although future studies are needed to further demonstrate its clinical usefulness, DF‐HA is expected to be a novel therapeutic agent fulfilling an unmet need for pharmacotherapy for knee OA.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Nishida had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Nishida, Kano, Nobuoka, Seo.
Acquisition of data
Kano, Nobuoka.
Analysis and interpretation of data
Seo.
ROLE OF THE STUDY SPONSOR
Seikagaku Corporation facilitated the study design, data collection, data analysis, interpretation of the data, and the writing of the manuscript. Ono Pharmaceutical Company, Ltd. provided writing assistance for the manuscript. Seikagaku Corporation and Ono Pharmaceutical Company, Ltd. reviewed and approved the manuscript prior to submission. Publication of this article was contingent upon approval by Seikagaku and Ono Pharmaceutical Company, Ltd. Medical writing and editorial support were provided by Dr. Kazuyoshi Masuda (ASCA Corporation, Chuo‐ku, Osaka, Japan) and funded by Seikagaku Corporation and Ono Pharmaceutical Company, Ltd.
Supporting information
Table S1‐S16
ACKNOWLEDGMENTS
The authors thank the patients who participated in the clinical trial, and also thank all study investigators, study coordinators, and other study personnel for their contributions to the study.
JAPIC identifier: JapicCTI‐173537.
Supported by Seikagaku Corporation and Ono Pharmaceutical Company, Ltd.
Dr. Nishida has received consulting fees, speaking fees, and/or honoraria from Eli Lilly, Kaken, Hisamitsu, Kyowa Hakko Kirin, and Asahi Kasei (less than $10,000 each) and from Seikagaku Corporation (more than $10,000) and has received research grants from Daiichi Sankyo, Chugai, Novartis, Pfizer, Eisai, and Zimmer Biomet. Mr Kano, Mr Nobuoka, and Dr. Seo own stock or stock options in Seikagaku Corporation.
References
- 1.Kolasinski SL, Neogi T, Hochberg MC, Oatis C, Guyatt G, Block J, et al. 2019 American College of Rheumatology/Arthritis Foundation guideline for the management of osteoarthritis of the hand, hip, and knee. Arthritis Rheumatol 2020;72:220–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roos EM, Arden NK. Strategies for the prevention of knee osteoarthritis [review]. Nat Rev Rheumatol 2016;12:92–101. [DOI] [PubMed] [Google Scholar]
- 3.Hunter DJ, Bierma‐Zeinstra S. Osteoarthritis [review]. Lancet 2019;393:1745–59. [DOI] [PubMed] [Google Scholar]
- 4.Bannuru RR, Osani MC, Vaysbrot EE, Arden NK, Bennell K, Bierma‐Zeinstra SM, et al. OARSI guidelines for the non‐surgical management of knee, hip, and polyarticular osteoarthritis. Osteoarthritis Cartilage 2019;27:1578–89. [DOI] [PubMed] [Google Scholar]
- 5.American Academy of Orthopaedic Surgeons . Treatment of osteoarthritis of the knee–2nd edition: evidence‐based clinical practice guideline. May 2013. URL: https://www.aaos.org/globalassets/quality‐and‐practice‐resources/osteoarthritis‐of‐the‐knee/osteoarthritis‐of‐the‐knee‐2nd‐editiion‐clinical‐practice‐guideline.pdf. [DOI] [PubMed]
- 6.Gore M, Tai KS, Sadosky A, Leslie D, Stacey BR. Use and costs of prescription medications and alternative treatments in patients with osteoarthritis and chronic low back pain in community‐based settings. Pain Pract 2012;12:550–60. [DOI] [PubMed] [Google Scholar]
- 7.Schaffer D, Florin T, Eagle C, Marschner I, Singh G, Grobler M, et al. Risk of serious NSAID‐related gastrointestinal events during long‐term exposure: a systematic review. Med J Aust 2006;185:501–6. [DOI] [PubMed] [Google Scholar]
- 8.Bresalier RS, Sandler RS, Quan H, Bolognese JA, Oxenius B, Horgan K, et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med 2005;352:1092–102. [DOI] [PubMed] [Google Scholar]
- 9.Trelle S, Reichenbach S, Wandel S, Hildebrand P, Tschannen B, Villiger PM, et al. Cardiovascular safety of non‐steroidal anti‐inflammatory drugs: network meta‐analysis. BMJ 2011;342:c7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ungprasert P, Cheungpasitporn W, Crowson CS, Matteson EL. Individual non‐steroidal anti‐inflammatory drugs and risk of acute kidney injury: a systematic review and meta‐analysis of observational studies. Eur J Intern Med 2015;26:285–91. [DOI] [PubMed] [Google Scholar]
- 11.Deyle GD, Allen CS, Allison SC, Gill NW, Hando BR, Petersen EJ, et al. Physical therapy versus glucocorticoid injection for osteoarthritis of the knee. N Engl J Med 2020;382:1420–9. [DOI] [PubMed] [Google Scholar]
- 12.McAlindon TE, LaValley MP, Harvey WF, Price LL, Driban JB, Zhang M, et al. Effect of intra‐articular triamcinolone vs saline on knee cartilage volume and pain in patients with knee osteoarthritis: a randomized clinical trial. JAMA 2017;317:1967–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kompel AJ, Roemer FW, Murakami AM, Diaz LE, Crema MD, Guermazi A. Intra‐articular corticosteroid injections in the hip and knee: perhaps not as safe as we thought? Radiology 2019;293:656–63. [DOI] [PubMed] [Google Scholar]
- 14.Ghosh P, Guidolin D. Potential mechanism of action of intra‐articular hyaluronan therapy in osteoarthritis: are the effects molecular weight dependent? Semin Arthritis Rheum 2002;32:10–37. [DOI] [PubMed] [Google Scholar]
- 15.Altman RD, Dasa V, Takeuchi J. Review of the mechanism of action for Supartz FX in knee osteoarthritis. Cartilage 2018;9:11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshioka K, Kisukeda T, Zuinen R, Yasuda Y, Miyamoto K. Pharmacological effects of N‐[2‐[[2‐[2‐[(2,6‐dichlorophenyl)amino]phenyl]acetyl]oxy]ethyl]hyaluronamide (diclofenac etalhyaluronate, SI‐613), a novel sodium hyaluronate derivative chemically linked with diclofenac. BMC Musculoskelet Disord 2018;19:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kisukeda T, Onaya J, Yoshioka K. Effect of diclofenac etalhyaluronate (SI‐613) on the production of high molecular weight sodium hyaluronate in human synoviocytes. BMC Musculoskelet Disord 2019;20:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nishida Y, Kano K, Nobuoka Y, Seo T. Sustained‐release diclofenac conjugated to hyaluronate (diclofenac etalhyaluronate) for knee osteoarthritis: a randomized phase 2 study. Rheumatology (Oxford) 2021;60:1435–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.WOMAC . WOMAC 3.1 Knee and hip osteoarthritis index. URL: http://www.womac.com/womac/index.htm.
- 20.Pocock SJ, Simon R. Sequential treatment assignment with balancing for prognostic factors in the controlled clinical trial. Biometrics 1975;31:103–15. [PubMed] [Google Scholar]
- 21.Pham T, van Der Heijde D , Lassere M, Altman RD, Anderson JJ, Bellamy N, et al. Outcome variables for osteoarthritis clinical trials: the OMERACT‐OARSI set of responder criteria. J Rheumatol 2003;30:1648–54. [PubMed] [Google Scholar]
- 22.Fukuhara S, Bito S, Green J, Hsiao A, Kurokawa K. Translation, adaptation, and validation of the SF‐36 Health Survey for use in Japan. J Clin Epidemiol 1998;51:1037–44. [DOI] [PubMed] [Google Scholar]
- 23.Fukuhara S, Ware JE Jr, Kosinski M, Wada S, Gandek B. Psychometric and clinical tests of validity of the Japanese SF‐36 Health Survey. J Clin Epidemiol 1998;51:1045–53. [DOI] [PubMed] [Google Scholar]
- 24.Fukuhara S, Suzukamo Y. Manual of SF‐36v2 Japanese version, Kyoto: Institute for Health Outcomes and Process Evaluation Research, 2004. [Google Scholar]
- 25.Hurst NP, Kind P, Ruta D, Hunter M, Stubbings A. Measuring health‐related quality of life in rheumatoid arthritis: validity, responsiveness and reliability of EuroQol (EQ‐5D). Br J Rheumatol 1997;36:551–9. [DOI] [PubMed] [Google Scholar]
- 26.Dworkin RH, Turk DC, McDermott MP, Peirce‐Sandner S, Burke LB, Cowan P, et al. Interpreting the clinical importance of group differences in chronic pain clinical trials: IMMPACT recommendations. Pain 2009;146:238–44. [DOI] [PubMed] [Google Scholar]
- 27.McAlindon TE, Driban JB, Henrotin Y, Hunter DJ, Jiang GL, Skou ST, et al. OARSI clinical trials recommendations: design, conduct, and reporting of clinical trials for knee osteoarthritis. Osteoarthritis Cartilage 2015;23:747–60. [DOI] [PubMed] [Google Scholar]
- 28.Committee for Medicinal Products for Human Use . Guideline on the clinical development of medicinal products intended for the treatment of pain. European Medicines Agency. December 2016. URL: https://www.ema.europa.eu/en/documents/scientific‐guideline/guideline‐clinical‐development‐medicinal‐products‐intended‐treatment‐pain‐first‐version_en.pdf.
- 29.Finniss DG, Kaptchuk TJ, Miller F, Benedetti F. Biological, clinical, and ethical advances of placebo effects [review]. Lancet 2010;375:686–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang W, Robertson J, Jones AC, Dieppe PA, Doherty M. The placebo effect and its determinants in osteoarthritis: meta‐analysis of randomised controlled trials. Ann Rheum Dis 2008;67:1716–23. [DOI] [PubMed] [Google Scholar]
- 31.Bannuru RR, Osani M, Vaysbrot EE, McAlindon TE. Comparative safety profile of hyaluronic acid products for knee osteoarthritis: a systematic review and network meta‐analysis. Osteoarthritis Cartilage 2016;24:2022–41. [DOI] [PubMed] [Google Scholar]
- 32.Brandt K. The role of analgesics in the management of osteoarthritis pain [review]. Am J Ther 2000;7:75–90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1‐S16