

Abstract
Mesoionic imidazolylidenes are recognized as excellent electron‐donating ligands in organometallic and main group chemistry. However, these carbene ligands typically show poor π‐accepting properties. A computational analysis of 71 mesoionic imidazolylidenes that bear different aryl or heteroaryl substituents in C2 position was performed. The study has revealed that a diphenyltriazinyl (Dpt) substituent renders the corresponding carbene particularly π‐acidic. The computational results could be corroborated experimentally. A mesoionic imidazolylidene with a Dpt substituent was found to be a better σ‐donor and a better π‐acceptor compared to an Arduengo‐type N‐heterocyclic carbene. To demonstrate the utility of the new carbene, the ligand was used to stabilize a low‐valent paramagnetic tin compound.
Keywords: carbenes, computational analysis, mesoionic, sigma donor, pi acidity, radicals, tin
In silico screening and experimental verification: A mesoionic imidazolylidene ligand with a diphenyltriazinyl substituent in C2 position (Dpt‐MIC) was found to be a better π‐acceptor when compared to the standard N‐heterocyclic carbene IDipp or to a mesoionic carbene with a phenyl group in C2 position (Ph‐MIC). The utility of the new carbene Dpt‐MIC was evidenced by the synthesis of a low‐valent paramagnetic tin compound.

Introduction
Arduengo‐type N‐heterocyclic carbenes (NHCs, Figure 1a) are obtained by deprotonation of imidazolium salts at C2 position with strong bases.[1, 2] When the C2 position is protected by a substituent, deprotonation can occur at C4/C5. The resulting imidazolylidenes are referred to as abnormal N‐heterocyclic carbenes (aNHCs) or mesoionic carbenes (MICs, Figure 1a).[3, 4, 5, 6, 7, 8, 9, 10, 11, 12] Over the last decade, MICs have been used extensively as ligands for transition and main group metal complexes.[3, 4, 5, 6, 7, 8, 9, 10] Furthermore, MICs were employed for small molecule activation,[3, 4, 5, 6, 7, 8, 9, 10, 13, 14, 15, 16, 17] and as metal‐free catalysts for polymerization reactions.[3, 4, 5, 6, 7, 8, 9, 10, 18, 19, 20] The electronic properties of mesoionic imidazolylidenes have been examined experimentally[21, 22, 23, 24, 25, 26, 27] and theoretically.[28, 29, 30, 31] These studies have revealed that MICs display high‐lying HOMOs and high proton affinities. In line with these results, it was found that MICs are exceptionally good σ‐donor ligands (Figure 1b).
Figure 1.

a) Structures of Arduengo‐type N‐heterocyclic carbenes (NHCs), mesoionic imidazolylidenes (MICs), and cyclic alkyl amino carbenes (CAACs). b) Comparison of the π‐acceptor and σ‐donor properties of NHCs, MICs, and CAACs. c) Influence of the substituent in C2 position on the electronic properties of MICs.
Cyclic alkyl amino carbenes (CAACs) are analogues of NHCs, in which one of the flanking nitrogen atoms has been replaced by a carbon atom (Figure 1a). Following their discovery in 2005,[32] CAACs have received considerable attention.[33, 34, 35, 36, 37, 38] CAACs are also better σ‐donor ligands than normal NHCs, but less strong donors than MICs. A decisive feature of CAACs is the fact that they are significantly more π‐acidic than NHCs (Figure 1b). As a result, CAAC ligands are well‐suited to stabilize low‐valent transition metal complexes. For example, complexes of the general formula M(CAAC)2 have been reported for M=Zn, Mn, Fe, Co, Cu, Au, Pd, and Pt.[35, 36] Formally, these compounds can be described as M(0) complexes, but experimental and theoretical studies have shown that significant spin density can be found on the CAAC ligand. The diamagnetic Zn(CAAC)2, for example, is best described as a Zn(II) complex with reduced (CAAC).− ligands.[39] The good electron‐accepting properties of CAAC ligands have also enabled the preparation of low‐valent main group element compounds. A recent example is the synthesis of a Ge(I) radical containing a CAAC and a silylamide ligand.[40] Pronounced spin density is found at the CAAC ligand. Therefore, this compound can also be described as a Ge(II) bound to a (CAAC).− ligand.
Several other N‐heterocyclic carbene ligands with pronounced π‐acidity have been described in the literature,[5, 41, 42, 43, 44, 45, 46] including monoamido‐aminocarbenes (MAACs)[43, 44] and N,N′‐diamidocarbenes (DACs).[44, 45, 46] However, the π‐acidity of these carbenes goes along with a reduced σ‐donor capability, which is typically lower than what is found for standard NHC ligands.[41, 42]
Below, we report a mesoionic imidazolylidene ligand, Dpt‐MIC, which is more π‐acidic than the corresponding NHC ligand. The discovery of this ligand was enabled by a computational analysis of 71 mesoionic carbenes with different aryl and heteroaryl substituents in C2 position (Figure 1c). Dpt‐MIC is well suited to stabilize low‐valent compounds, as evidenced by the isolation of a neutral stannylene radical.
Results and Discussion
Previous investigations have shown that the substituent at C4 position of a mesoionic imidazolylidene ligand has a strong influence on the stability and the stereoelectronic properties of the MIC.[22, 23, 47, 48] The remote substituent at C2 position was thought to be less important.[23] We have recently studied MIC ligands featuring unusual diazoaryl substituents at C2.[49] These ′Azo‐MICs′ have low‐lying LUMO levels, and Au(I) and Rh(I) complexes with Azo‐MIC ligands were found to undergo reversible one‐electron reductions in cyclic voltammetry experiments. Intrigued by these results, we decided to investigate the electronic influence of the substituent in C2 position in more detail. In particular, we wanted to explore if it is possible to modulate the π‐accepting properties of MICs without compromising their good σ‐donor abilities.
First, we performed a computational analysis of 71 mesoionic imidazolylidenes with N‐xylyl wingtip groups and different aryl and heteroaryl substituents at C2 position (Scheme 1; for details see the Supporting Information). We focused on aromatic groups at C2 because we were interested to validate the theoretical results experimentally, and there are several procedures for synthesizing C2‐arylated imidazolium salts.[50, 51, 52, 53, 54] Following a geometry optimization at the PBE0(D3BJ)/def2‐SVP level,[55, 56, 57, 58, 59, 60] we then computed the HOMO energies of the carbenes. The values were expected to indicate the relative σ‐donor properties of the MICs (the absolute values of the HOMO energies can vary depending on the level of theory).
Scheme 1.

The electronic properties of 71 MICs with different aromatic substituents ′R′ were evaluated by computing the HOMO energies (correlation with the σ‐donor properties) and the 77Se NMR chemical sifts of Se adducts (correlation with the π‐acceptor properties).
The π‐accepting properties of singlet carbenes are often related to their LUMO energy levels.[3, 4, 5, 6, 7, 8, 9, 10] Instead of focusing on this frontier molecular orbital, we decided to compute a parameter which can directly be determined experimentally: the 77Se NMR chemical shift of Se adducts (Scheme 1). The shift of the 77Se NMR signal reflects the π‐accepting properties of the carbene, with higher values indicating a higher π‐acidity. This NMR‐based methodology was introduced by the group of Ganter in 2013.[42] Since then, and it has become a popular method for evaluating the π‐accepting properties of singlet carbenes.[21, 42, 47, 61, 62] The broad spectral range of 77Se NMR allows for a good discrimination of closely related carbenes. Furthermore, Se adducts of carbenes are easily accessible by the addition of elemental selenium to an in situ generated carbene. A potential drawback of this method is the fact that the chemical shift of the 77Se NMR signal can be influenced by the local environment of the Se atom (e. g. via non‐classical hydrogen bonding interactions).[61] This potential limitation was not of concern for our computational screening, because the local environment of the Se atom does not change for our compounds.
A summary of the computational results is given in Figure 2. The HOMO energies of the 71 MICs span a range from −4.84 eV to −5.66 eV. According to these values, they are all better σ‐donors than the corresponding NHC with N‐xylyl wingtip groups (HOMO at −6.01 eV; data not shown). The chemical shifts of the Se adducts range from +270 ppm to −74 ppm. It is interesting to note that there is a rather poor correlation between the HOMO energies and the δ(77Se) value (R=0.39). The lowest value with δ=−74 ppm is found for a MIC with an N‐methylpyrrolyl substituent. This compound also shows one of the highest HOMO energies (−4.84 eV). On the other hand, there are several compounds with a significantly higher δ(77Se) value compared to what is found for a MIC with a standard phenyl substituent (δ=−9 ppm). Two compounds stand out: a MIC with a tetrazinyl substituent (δ=270 ppm) and a MIC with a diphenyltriazinyl substituent (δ=218 ppm). The computational results suggested that these compounds should be significantly more π‐acidic than standard MICs with a phenyl substituent in C2 position. Consequently, they were chosen as synthetic targets for our experimental work.
Figure 2.
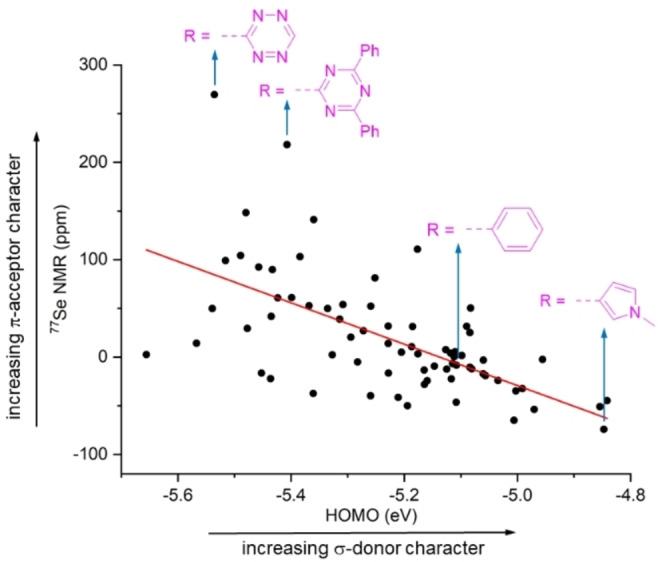
The calculated 77Se NMR chemical shifts versus the HOMO energies of Se‐MICs and MICs.
The functionalization of 1,3,5‐triazines is commonly achieved by nucleophilic substitution of chlorinated triazines.[63, 64] We envisioned that utilizing an NHC as nucleophile would allow for the preparation of an imidazolium salt with a triazinyl group in C2 position.[52, 53, 54] Indeed, addition of 1,3‐bis(2,6‐diisopropylphenyl)imidazol‐2‐ylidene (IDipp) to a solution of 2‐chloro‐4,6‐diphenyl‐1,3,5‐triazine in THF resulted in the formation of the imidazolium salt 1, which could be isolated in 80 % yield (Scheme 2). Unfortunately, we were not successful in preparing a tetrazolyl‐substituted imidazolium salt using a related methodology (for details, see Supporting Information, Scheme S6).
Scheme 2.

Synthesis of the imidazolium salt 1, the Rh complex 2, and the Se adduct 3. Reagents: i) KHMDS (1.05 equiv.) followed by [RhCl(CO)2]2 (0.5 equiv.), THF, −40 °C, 12 h. ii) KHMDS (1.05 equiv.) followed by Se (2 equiv.), THF, −40 °C, 12 h.
Colorless crystals of 1 were obtained from acetonitrile at −40 °C, and a single crystal X‐ray diffraction (XRD) analysis was performed (Figure 3).[65] The bond lengths observed for 1 are within the expected range (for details, see Supporting Information).
Figure 3.

Molecular structures of 1, 2 and 3 in the crystal. The thermal ellipsoids are at 50 % probability and hydrogen atoms are omitted for clarity.
Attempts to prepare the free carbene ligand Dpt‐MIC (Dpt=diphenyltriazinyl) by reaction of 1 with potassium bis(trimethylsilyl) amide (KHMDS) were not successful.[66, 67] The NMR spectrum of the reaction mixture recorded at room temperature was very complex, indicating decomposition of Dpt‐MIC. However, we were able to capture in situ generated Dpt‐MIC by coordination to RhCl(CO)2. The resulting Rh complex (2) was isolated in 85 % yield (Scheme 2). For comparison with our theoretical results, we have also prepared the Se adduct 3. Both compounds were characterized by NMR spectroscopy, high‐resolution mass spectrometry, and single crystal XRD (Figure 3). In addition, complex 2 was analyzed by FT‐IR spectroscopy.
Graphic representations of the structures of 2 and 3 in the crystal are depicted in Figure 3. As it was observed for the imidazolium salt 1, the triazinyl‐, the phenyl‐, and the imidazole rings adopt a co‐planar arrangement, suggesting extensive π‐conjugation. Such a co‐planar arrangement is not observed for MICs with phenyl substituents in C2 position. In these compounds, the acute angle between the planes defined by the imidazole ring and the phenyl ring lie between 37° and 50° (for details, see Supporting Information, Figure S14). For the Dpt substituent, the presence of a central triazine ring facilitates a co‐planar arrangement because steric interactions involving ortho H‐atoms are avoided.
The Rh−C bond length in 2 (2.078(4) Å) is very similar to what was found for a RhCl(CO)2 complex with a mesoionic triazolylidene ligand (2.081(9) Å).[68] The Se−C bond length in 3 (1.857(2) Å) is likewise unremarkable, and comparable to what was observed for Se adducts of other C2‐arylated MICs (for details, see Figure S11).[21]
The IR bands of the CO groups of 2 can be used to calculate the Tolman electronic parameter (TEP),[69, 70] which allows to quantify the overall electron donating properties of a ligand. The average of the two CO bands of 2 is 2031 cm−1, which translates to a TEP of 2045 cm−1. This value suggests that Dpt‐MIC is a better electron donor than the normal NHC IDipp (TEP=2051 cm−1),[69] but weaker than C2‐phenylated MICs 2037–2039 cm−1).[21]
The TEP takes into account the σ‐donor and the π‐acceptor properties of a ligand, whereas the 77Se NMR chemical shift reflects selectively the π‐acidity. The experimental 77Se{1H} NMR spectrum of 3 shows a resonance at 147 ppm. This value is lower than what was predicted theoretically (218 ppm). However, it should be noted that the computed value is somewhat method‐dependent and is evaluated in the gas phase. Importantly, the experimental value is in line with the predicted trend: Dpt‐MIC is not only a better π‐acceptor than the C2‐phenylated MIC (δ (77Se)=−3 ppm),[21] but it is also more π‐acidic than the corresponding NHC (δ (77Se)=87 ppm)[42] (Figure 4).
Figure 4.

The experimental 77Se{1H} NMR chemical shift of 3 in comparison to what has been reported for the Se adduct of IDipp and for a C2‐phenylated MIC.
Ganter and co‐workers have suggested that the 1 J Se−C coupling constant reflects the σ‐donor properties of a carbene ligand.[41] For 3, a value of 223 Hz was determined, which is larger than what is found for C2‐phenylated MICs (211–214 Hz),[21] but smaller than what was reported for a standard NHC such as IMes (231 Hz).[41]
Overall, the experimental results are evidence that the Dpt group in C2 position has a unique effect on the electronic properties of the MIC ligand. Notably, Dpt‐MIC is a better σ‐donor and a better π‐acceptor than the commonly used N‐heterocyclic carbene IDipp. The π‐acidity of Dpt‐MIC cannot rival what is found for CAAC ligands (δ (77Se) ∼500 ppm), but the electronic properties of Dpt‐MIC are still exceptional for a mesoionic imidazolylidene ligand.
The good π‐acidity of Dpt‐MIC suggested that this ligand would be suited to stabilize low‐valent compounds. Therefore, we attempted the synthesis of a highly reduced Sn compound.
As precursor, we first prepared the carbene‐stabilized stannylene 4 by reaction of imidazolium salt 1 with two equivalents of KHMDS in THF at −40 °C, followed by addition of anhydrous SnCl2 (Scheme 3). The product was characterized by multinuclear NMR spectroscopy and by a single crystal XRD analysis (Figure 5).
Scheme 3.

Synthesis of stannylene 4.
Figure 5.
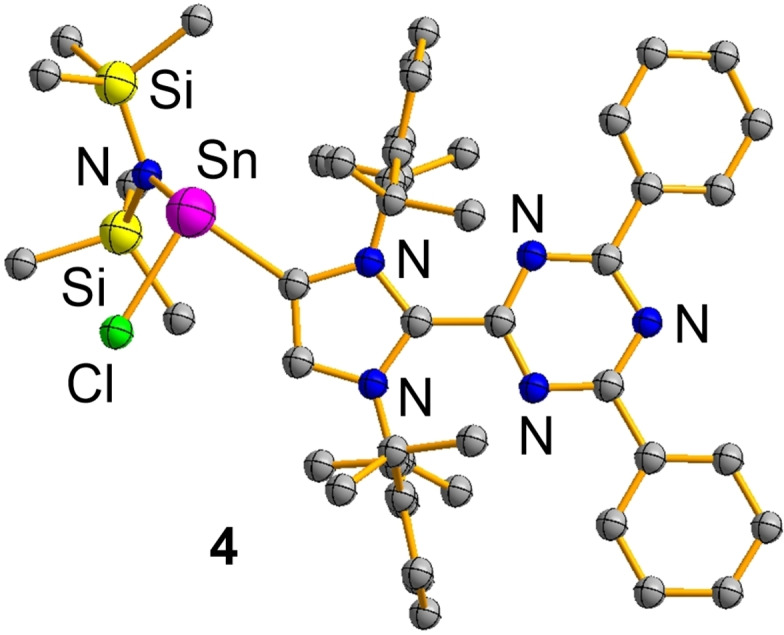
Molecular structure of 4 in the crystal. The thermal ellipsoids are at 50 % probability and hydrogen atoms are omitted for clarity.
The Sn center displays the expected trigonal pyramidal geometry (Cl−Sn−C=87.64(15)°, Cl−Sn−N=97.20(15)°). The Sn−C bond length of 2.295(7) Å is close to what was reported for a SnCl2 complex with a MIC ligand featuring phenyl groups in C2 and C4 positions (Sn−C=2.308(9) Å).[71]
Next, we have reduced stannylene 4 with one equivalent of KC8 in THF at −40 °C. From the deep purple solution, we were able to isolate the neutral radical Sn(Dpt‐MIC)2{N(SiMe3)2} (5) in 45 % yield (Scheme 4).
Scheme 4.

Synthesis of neutral radical 5.
The structure of 5 was established by single crystal XRD. Two Dpt‐MIC ligands are coordinated to Sn, with bis(trimethylsilyl) amide occupying the third coordination site (Figure 6a). Apparently, the reduction had resulted in a ligand transfer reaction of Dpt‐MIC. Compound 5 has a net charge of zero, and it therefore features an unpaired electron. A close inspection of the crystallographic data suggests that the paramagnetic product should be described as a stannylene bound to one neutral Dpt‐MIC ligand and one anionic (Dpt‐MIC).− ligand. The two Sn−C bonds in 5 have nearly the same length (2.300(16) Å) and 2.293(16) Å). However, pronounced differences are observed for the imidazole and the triazine ring systems of the two Dpt‐MIC ligands (Figure 6b). The C45−C48 bond (1.420(2) Å) is substantially shorter than the C2−C6 bond (1.476(2) Å), indicating partial double bond character for the former. In contrast to the nearly equidistant C−N bonds of the triazinyl group connected to C2, a pronounced bond‐length alternation is observed for the triazine ring connected to C45. The data suggest that the electron is largely located on one of the two Dpt‐MIC ligands. This interpretation was substantiated by X‐band electron paramagnetic resonance (EPR) spectroscopy and by DFT calculations.[55, 56, 57, 58, 59, 60] The EPR spectrum of 5 in THF at room temperature corresponds to a single unpaired electron (spin doublet) with a characteristic hyperfine structure due to coupling to five 14N nuclei (g iso=2.0034, Figure 7a and Figure S7). The computational results are in line with this finding, and they show that the total spin density is localized on the heterocycles of one of the two Dpt‐MIC ligands (Figure 7b). Overall, the situation is reminiscent of what was found for several low‐valent CAAC complexes, where significant spin density at the carbene ligand was observed.[35, 36]
Figure 6.

a) Molecular structure of radical 5 in the crystal. The thermal ellipsoids are at 50 % probability and hydrogen atoms are omitted for clarity. b) Selected bond lengths for the central part of the Sn(Dpt‐MIC)2 unit.
Figure 7.

a) Simulated (red) and experimental (black) EPR spectrum of 5 (THF, RT). b) Calculated spin density map of 5 (isovalue=0.003).
Conclusion
A computational analysis of 71 mesoionic imidazolylidene ligands containing different aryl and heteroaryl substituents in C2 position was performed. The analysis has revealed that a particularly π‐acidic carbene is formed if a diphenyltriazinyl substituent is attached to C2. The computational results could be corroborated experimentally by synthesizing the imidazolium salt 1, which serves as a precursor for the carbene ligand Dpt‐MIC. The spectroscopy data for RhCl(CO)2 and Se adducts show that Dpt‐MIC is significantly more π‐acidic than a MIC ligand with a phenyl substituent in C2 position. Importantly, Dpt‐MIC is a better σ‐donor and a better π‐acceptor when comparted to the normal N‐heterocyclic carbene IDipp.[72] In that regard, Dpt‐MIC shows CAAC‐like behavior. To demonstrate the utility of Dpt‐MIC, we employed it to stabilize a low‐valent paramagnetic tin compound. In view of potential future applications, it is worth pointing out that the precursor of Dpt‐MIC can be obtained in one step from commercially available starting materials. Dpt‐MIC therefore represents an easily accessible mesoionic carbene with unique electronic properties.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
This work was supported by the École Polytechnique Fédérale de Lausanne (EPFL). We thank Dr. Euro Solari for mounting air‐sensitive single crystals.
Z. Dong, J. T. Blaskovits, F. Fadaei-Tirani, R. Scopelliti, A. Sienkiewicz, C. Corminboeuf, K. Severin, Chem. Eur. J. 2021, 27, 11983.
Contributor Information
Prof. Clémence Corminboeuf, Email: clemence.corminboeuf@epfl.ch.
Prof. Kay Severin, Email: kay.severin@epfl.ch.
References
- 1.Hopkinson M. N., Richter C., Schedler M., Glorius F., Nature 2014, 510, 485–496. [DOI] [PubMed] [Google Scholar]
- 2.Hahn F. E., Jahnke M. C., Angew. Chem. Int. Ed. 2008, 47, 3122–3127; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 3166–3216. [Google Scholar]
- 3.Sau S. C., Hota P. K., Mandal S. K., Soleilhavoup M., Bertrand G., Chem. Soc. Rev. 2020, 49, 1233–1252. [DOI] [PubMed] [Google Scholar]
- 4.Vivancos Á., Segarra C., Albrecht M., Chem. Rev. 2018, 118, 9493–9586. [DOI] [PubMed] [Google Scholar]
- 5.Munz D., Organometallics 2018, 37, 275–289. [Google Scholar]
- 6.Schweinfurth D., Hettmanczyk L., Suntrup L., Sarkar B., Z. Anorg. Allg. Chem. 2017, 643, 554–584. [Google Scholar]
- 7.Ghadwal R. S., Dalton Trans. 2016, 45, 16081–16095. [DOI] [PubMed] [Google Scholar]
- 8.Albrecht M., Adv. Organomet. Chem. 2014, 62, 111–158. [Google Scholar]
- 9.Crabtree R. H., Coord. Chem. Rev. 2013, 257, 755–766. [Google Scholar]
- 10.Schuster O., Yang L., Raubenheimer H. G., Albrecht M., Chem. Rev. 2009, 109, 3445–3478. [DOI] [PubMed] [Google Scholar]
- 11.Aldeco-Perez E., Rosenthal A. J., Donnadieu B., Parmeswaran P., Frenking G., Bertrand G., Science 2009, 326, 556–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gründemann S., Kovacevic A., Albrecht M., Robert J. W. F., Crabtree H., Chem. Commun. 2001, 2274–2275. [DOI] [PubMed] [Google Scholar]
- 13.Sau S. C., Bhattacharjee R., Hota P. K., Vardhanpu P. K., Vijaykumar G., Govindarajan R., Datta A., Mandal S. K., Chem. Sci. 2019, 10, 1879–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hota P. K., Sau S. C., Mandal S. K., ACS Catal. 2018, 8, 11999–12003. [Google Scholar]
- 15.Eymann L. Y. M., Scopelliti R., Fadaei-Tirani F., Cecot G., Solari E., Severin K., Chem. Eur. J. 2018, 24, 7957–7963. [DOI] [PubMed] [Google Scholar]
- 16.Eymann L. Y. M., Scopelliti R., Fadaei-Tirani F., Cecot G., Solari E., Severin K., Chem. Commun. 2017, 53, 4331–4334. [DOI] [PubMed] [Google Scholar]
- 17.Thakur A., Vardhanapu P. K., Vijaykumar G., Hota P. K., Mandal S. K., Eur. J. Inorg. Chem. 2016, 913–920. [Google Scholar]
- 18.Bhunia M., Vijaykumar G., Adhikari D., Mandal S. K., Inorg. Chem. 2017, 56, 14459–14466. [DOI] [PubMed] [Google Scholar]
- 19.Sen T. K., Sau S. Ch., Mukherjee A., Hota P. K., Mandal S. K., Maity B., Koley D., Dalton Trans. 2013, 42, 14253–14260. [DOI] [PubMed] [Google Scholar]
- 20.Sen T. K., Sau S. Ch., Mukherjee A., Modak A., Mandal S. K., Koley D., Chem. Commun. 2011, 47, 11972–11974. [DOI] [PubMed] [Google Scholar]
- 21.Merschel A., Rottschäfer D., Neumann B., Stammler H. G., Ghadwal R. S., Organometallics 2020, 39, 1719–1729. [Google Scholar]
- 22.Ung G., Bertrand G., Chem. Eur. J. 2011, 17, 8269–8272. [DOI] [PubMed] [Google Scholar]
- 23.Heckenroth M., Neels A., Garnier M. G., Aebi P., Ehlers A. W., Albrecht N., Chem. Eur. J. 2009, 15, 9375–9386. [DOI] [PubMed] [Google Scholar]
- 24.Huynh H. V., Han Y., Jothibasu R., Yang J. A., Organometallics 2009, 28, 5395–5404. [Google Scholar]
- 25.Chianese A. R., Kovacevic A., Zeglis B. M., Faller J. W., Crabtree R. H., Organometallics 2004, 23, 2461–2468. [Google Scholar]
- 26.Beerhues J., Aberhan H., Streit T. N., Sarkar B., Organometallics 2020, 39, 4557–4564. [Google Scholar]
- 27.Beerhues J., Neubrand M., Sobottka S., Neuman N. I., Aberhan H., Chandra S., Sarkar B., Chem. Eur. J. 2021, 27, 6557–6568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andrada D. M., Holzmann N., Hamadi T., Frenking G., Beilstein J. Org. Chem. 2015, 11, 2727–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Phukan A. K., Guha A. K., Sarmah S., Dewhurst R. D., J. Org. Chem. 2013, 78, 11032–11039. [DOI] [PubMed] [Google Scholar]
- 30.Bernhammer J. C., Frison G., Huynh H. V., Chem. Eur. J. 2013, 19, 12892–12905. [DOI] [PubMed] [Google Scholar]
- 31.Gusev D. G., Organometallics 2009, 28, 6458–6461. [Google Scholar]
- 32.Lavallo V., Canac Y., Präsang C., Donnadieu B., Bertrand G., Angew. Chem. Int. Ed. 2005, 44, 5705–5709; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 5851–5855. [Google Scholar]
- 33.Jazzar R., Soleilhavoup M., Bertrand G., Chem. Rev. 2020, 120, 4141–4168. [DOI] [PubMed] [Google Scholar]
- 34.Kundu S., Sinhababu S., Chandrasekhar V., Roesky H. W., Chem. Sci. 2019, 10, 4727–4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paul U. S. D., Radius U., Eur. J. Inorg. Chem. 2017, 3362–3375. [Google Scholar]
- 36.Roy S., Mondal K. C., Roesky H. W., Acc. Chem. Res. 2016, 49, 357–369. [DOI] [PubMed] [Google Scholar]
- 37.Soleilhavoup M., Bertrand G., Acc. Chem. Res. 2015, 48, 256–266. [DOI] [PubMed] [Google Scholar]
- 38.Martin D. C., Soleilhavoup M., Bertrand G., Chem. Sci. 2013, 4, 3020–3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singh A. P., Samuel P. P., Roesky H. W., Schwarzer M. C., Frenking G., Sidhu N. S., Dittrich B., J. Am. Chem. Soc. 2013, 135, 7324–7329. [DOI] [PubMed] [Google Scholar]
- 40.Siddiqui M. M., Sarkar S. K., Sinhababu S., Ruth P. N., Herbst-Irmer R., Stalke D., Ghosh M., Fu M., Zhao L., Casanova D., Frenking G., Schwederski B., Kaim W., Roesky H. W., J. Am. Chem. Soc. 2019, 141, 1908–1912. [DOI] [PubMed] [Google Scholar]
- 41.Verlinden K., Buhl H., Frank W., Ganter C., Eur. J. Inorg. Chem. 2015, 2416–2425. [Google Scholar]
- 42.Liske A., Verlinden K., Buhl H., Schaper K., Ganter C., Organometallics 2013, 32, 5269–5272. [Google Scholar]
- 43.Benhamou L., Vujkovic N., César V., Gornitzka H., Lugan N., Lavigne G., Organometallics 2010, 29, 2616–2630. [Google Scholar]
- 44.Blake G. A., Moerdyk J. P., Bielawski C. W., Organometallics 2012, 31, 3373–3378. [Google Scholar]
- 45.Hudnall T. W., Bielawski C. W., J. Am. Chem. Soc. 2009, 131, 16039–16041. [DOI] [PubMed] [Google Scholar]
- 46.Braun M., Frank W., Ganter C., Organometallics 2012, 31, 1927–1934. [Google Scholar]
- 47.Merschel A., Glodde T., Neumann B., Stammler H. G., Ghadwal R. S., Angew. Chem. Int. Ed. 2021, 60, 2969–2973; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 3006–3010. [Google Scholar]
- 48.Jin H., Tan T. T. Y., Hahn F. E., Angew. Chem. Int. Ed. 2015, 54, 13811–13815; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 14016–14020. [Google Scholar]
- 49.Chadwick F. M., Curchod B. F. E., Scopelliti R., Fadaei-Tirani F., Solari E., Severin K., Angew. Chem. Int. Ed. 2019, 58, 1764–1767; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1778–1781. [Google Scholar]
- 50.Ho N. K. T., Neumann B., Stammler H.-G., da Silva V. H. M., Watanable D. G., Braga A. A. C., Ghadwal R. S., Dalton Trans. 2017, 46, 12027–12031. [DOI] [PubMed] [Google Scholar]
- 51.Ghadwal R. S., Reichmann S. O., Herbst-Irmer R., Chem. Eur. J. 2015, 21, 4247–4251. [DOI] [PubMed] [Google Scholar]
- 52.Kim Y., Lee Y., Chem. Commun. 2016, 52, 10922–10925. [DOI] [PubMed] [Google Scholar]
- 53.Mallah E., Kuhn N., Maichle-Mößmer C., Steinmann M., Stöbele M., Zeller K.-P., Z. Naturforsch. 2009, 64b, 1176–1182. [Google Scholar]
- 54.Kuhn N., Fahl J., Boese R., Henkel G., Z. Naturforsch. B 1998, 53, 881–886. [Google Scholar]
- 55.Perdew J. P., Burke K., Ernzerhof M., Phys. Rev. Lett. 1996, 77, 3865–3868. [DOI] [PubMed] [Google Scholar]
- 56.Adamo C., Barone V., J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar]
- 57.Grimme S., Antony J., Ehrlich S., Krieg H., J. Chem. Phys. 2010, 132, 154104. [DOI] [PubMed] [Google Scholar]
- 58.Grimme S., Ehrlich S., Goerigk L., J. Comput. Chem. 2011, 32, 1456–1465. [DOI] [PubMed] [Google Scholar]
- 59.Weigend F., Ahlrichs R., Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- 60.For a detailed description of computational methods, see the Supporting Information.
- 61.Junor G. P., Lorkowski J., Weinstein C. M., Jazzar R., Pietraszuk C., Bertrand G., Angew. Chem. Int. Ed. 2020, 59, 22028–22033; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 22212–22217. [Google Scholar]
- 62.Saab M., Nelson D. J., Tzouras N. V., Bayrakdar T. A. C. A., Nolan S. P., Narhra F., Hecke K. V., Dalton Trans. 2020, 49, 12068–12081. [DOI] [PubMed] [Google Scholar]
- 63.Giacomelli G., Porcheddu A., De Luca L., Curr. Org. Chem. 2004, 8, 1497–1519. [Google Scholar]
- 64.Blotny G., Tetrahedron 2006, 62, 9507–9522. [Google Scholar]
- 65.Deposition Numbers 2002574 (for 1), 2064706 (for 2), 2064707 (for 3), 2064708 (for 4), and 2064709 (for 5) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.”.
- 66.Rottschäfer D., Glodde T., Neumann B., Stammler H. G., Ghadwal R. S., Chem. Commun. 2020, 56, 2027–2030. [DOI] [PubMed] [Google Scholar]
- 67.Aldeco-Perez E., Rosenthal A. J., Donnadieu B., Parameswaran P., Frenking G., Bertrand G., Science 2009, 326, 556–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bidal Y. D., Santoro O., Melaimi O., Cordes D. B., Slawin A. M. Z., Bertrand G., Cazin C. S. J., Chem. Eur. J. 2016, 22, 9404–9409. [DOI] [PubMed] [Google Scholar]
- 69.Huynh H. V., Chem. Rev. 2018, 118, 9457–9492. [DOI] [PubMed] [Google Scholar]
- 70.Tolman C. A., J. Am. Chem. Soc. 1970, 92, 2953–2956. [Google Scholar]
- 71.Singh A. P., Samuel P. P., Modal K. C., Roesky H. W., Dittrich B., Organometallics 2013, 32, 354–357. [Google Scholar]
- 72.The π-acidity is slightly lower than what is found for the saturated standard N-heterocyclic carbene SIDipp (181 ppm). See refs [41] and [42].
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information