

Abstract
B-cell activation is increasingly linked to numerous fibrotic lung diseases, and it is well known that aggregates of lymphocytes form in the lung of many of these patients. Activation of B-cells by pattern recognition receptors (PRRs) drives the release of inflammatory cytokines, chemokines, and metalloproteases important in the pathophysiology of pulmonary fibrosis. However, the specific mechanisms of B-cell activation in patients with idiopathic pulmonary fibrosis (IPF) are poorly understood. Herein, we have demonstrated that B-cell activation by microbial antigens contributes to the inflammatory and profibrotic milieu seen in patients with IPF. B-cell stimulation by CpG and β-glucan via PRRs resulted in activation of mTOR-dependent and independent pathways. Moreover, we showed that the B-cell–secreted inflammatory milieu is specific to the inducing antigen and causes differential fibroblast migration and activation. B-cell responses to infectious agents and subsequent B-cell–mediated fibroblast activation are modifiable by antifibrotics, but each seems to exert a specific and different effect. These results suggest that, upon PRR activation by microbial antigens, B-cells can contribute to the inflammatory and fibrotic changes seen in patients with IPF, and antifibrotics are able to at least partially reverse these responses.
Keywords: B-lymphocytes, idiopathic pulmonary fibrosis, antifibrotics, pattern recognition receptor, fibrosis
Idiopathic pulmonary fibrosis (IPF) is a deadly disease that is characterized by the development of lung scaring resulting in progressive respiratory failure. The prognosis is extremely poor, with a median survival of 3–5 years from the time of diagnosis (1). To date there are only two U.S. Food and Drug Administration–approved drugs, nintedanib and pirfenidone, to treat IPF, which slow the progression of the disease in some patients (2, 3). Development of new drugs for IPF is challenged by its unknown etiology, although genetic defects in telomere genes as well as surfactant and host defense proteins such as TOLLIP, TLR3, and MUC5B have been described in a subgroup of patients (4, 5). These findings have led to support for the general belief that IPF is the result of a combination of injurious environmental exposures on a genetically predisposed individual resulting in defective tissue repair and fibrosis. However, the specific mechanisms of how environmental exposures and genetic predisposition influence the pathogenesis of IPF are not entirely understood.
Acute respiratory exacerbations (AEs) in IPF are life-threating events characterized by rapid deterioration of lung function and are shown to significantly impair the course of the disease and are associated with poor prognosis. AEs have been linked to infection, although an infectious organism cannot always be identified (6). However, microbiome studies suggest that patients with IPF have higher bacterial load than normal control subjects and that those with higher loads tend to have worse prognosis, suggesting that microbial antigens could play a role in the pathogenesis of IPF (7). Microbial antigens, also known as pathogen-associated molecular patterns (PAMPs), modulate the immune response by activating a variety of pattern recognition receptors (PRRs) expressed by innate immune cells, including B-cells; it is very plausible that acute or chronic stimulation of PRRs by microbial antigens may lead to alteration of the normal milieu into an inflammatory/profibrotic milieu, contributing to the development of AEs and fibrosis in the genetically predisposed host (8).
We and others have shown that activation of B-cells by PRRs from normal donors drives the release of inflammatory cytokines, chemokines, and metalloproteases implicated in the pathophysiology of fibrotic diseases, including IPF (9–12). However, the specific mechanisms of B-cell activation in IPF are poorly understood.
Hence, we have conducted investigations to determine the role of activated B-cells in fibrosis because B-cell activation is increasingly linked to numerous fibrotic lung diseases, and B-cells are known to form aggregates in the lungs of some of these patients (13, 14). Our results demonstrate that B-cells isolated from patients with IPF respond to microbial antigens (β-glucan and CpG) via PRRs by secreting an inflammatory and profibrotic milieu that promotes fibroblast migration and activation. B-cell–mediated fibroblast activation was modifiable by antifibrotics (nintedanib and pirfenidone). These results are novel, can direct new therapeutic strategies, and suggest that PRR regulation may be an effective potential way to affect fibrotic mechanisms in IPF.
Methods
Reagents and Antibodies
Full descriptions can be found in the Methods section of the data supplement.
Human B-Cells and Fibroblasts
Human blood samples from volunteers with IPF and normal volunteers were collected under the Mayo Clinic institutional review board–approved protocol (IRB#17–008088). Patients with IPF were recruited and consented from our interstitial lung disease clinic. The IPF blood samples included in our study were from patients not undergoing an exacerbation and who were not on antifibrotic or steroid treatments for at least 3 months before the collection of the blood sample. Baseline characteristics of the patients can be found in Table E1 in the data supplement. Normal fibroblasts and IPF fibroblasts were obtained from Lonza (Lonza Bioscience). See data supplement for further information.
Fibroblasts, B-Cell Isolation, and Culture
See data supplement for further information.
FACS for B-Lymphocyte Activation
Cells were counted, and 1 × 106 cells/tube were incubated on ice for 30 minutes with mixture of anti-CD19-FITC and anti-CD80-PE or anti-CD86-BV421. Flow cytometry data were analyzed and graphed using FlowJo software (BD Biosciences). For further details, see the data supplement Methods section.
Proliferation Assay
B-lymphocyte proliferation was determined by 3H-labeled thymidine incorporation during DNA synthesis. For further details, see the data supplement Methods section.
Lung Fibroblast Transwell Migration Assay
Primary lung fibroblasts were starved for 24 hours and seeded (10,000 cells/well) in the upper chamber of the Transwell with 8.0 μm pore-size polyester membrane (Corning) with the lower chambers containing differently conditioned media. The system was incubated at 37°C, 5% CO2 to allow the migration of cells through the membrane (8.0 μm). After 16 hours, media in the upper chamber and nonmigrated cells were removed using a cotton swab. Migrated cells on the bottom side of the Transwell were fixed and stained with Hema 3 staining kit (Thermo Fisher Scientific). Migrated cells were quantified using phase contrast microscopy.
Cytokines and IgM Detection
See the data supplement Methods section for further description.
Immunohistochemistry and Immunofluorescence
Formalin-fixed paraffin-embedded human lung tissue was obtained from the Mayo Clinic tissue registry. For further details, see the data supplement Methods section.
Preparation of Cell Lysates, Electrophoresis, IB, and Cell Viability
See the data supplement Methods section for a further description.
Statistical Analyses
All data are presented as the mean ± SEM, from at least three experiments from different biological donors. The data were first analyzed using one-way ANOVA and post hoc Sidak comparison test unless otherwise indicated. Statistical analysis was performed using GraphPad Prism, version 6 (GraphPad Software).
Results
B-Cells from Patients with IPF Secrete Proinflammatory and Profibrotic Proteins in Response to Microbial Antigens that Promote Fibroblast Migration and Activation
Peripheral B-cells from normal individuals respond to PRR activation by releasing inflammatory and profibrotic proteins such as IL-8, IL-6, and MMP-7 (9, 15). To evaluate whether B-cells from patients with IPF also responded to PRR activation, IL-8, IL-6, and MMP7 were measured in the cell supernatant from B-cells stimulated with CpG or β-glucan for 24 hours. CpG is an unmethylated synthetic oligodeoxynucleotide similar to those found in bacterial and fungal DNA, and β-glucans are carbohydrates found in the cell wall of many fungi (Aspergillus spp., Candida spp., and Pneumocystis) and bacteria (Pseudomonas and Streptococcus) (16). These two different infectious antigens are potent immunomodulators that activate B-lymphocytes via PRRs (TLR9 and dectin-1, respectively) without the need of T-cell participation (15, 17). To confirm that B-cells from IPF were responsive to CpG and β-glucan, IL-6, IL-8, and MMP-7 were measured in cell supernatant after B-cell stimulation (Figure 1). Similar to our published data in normal donors, patients with IPF responded to CpG by secreting IL-6 and MMP-7 (Figure 1A) and to β-glucan by secreting IL-6, MMP-7, and IL-8 (Figure 1B) (9, 15).
Figure 1.
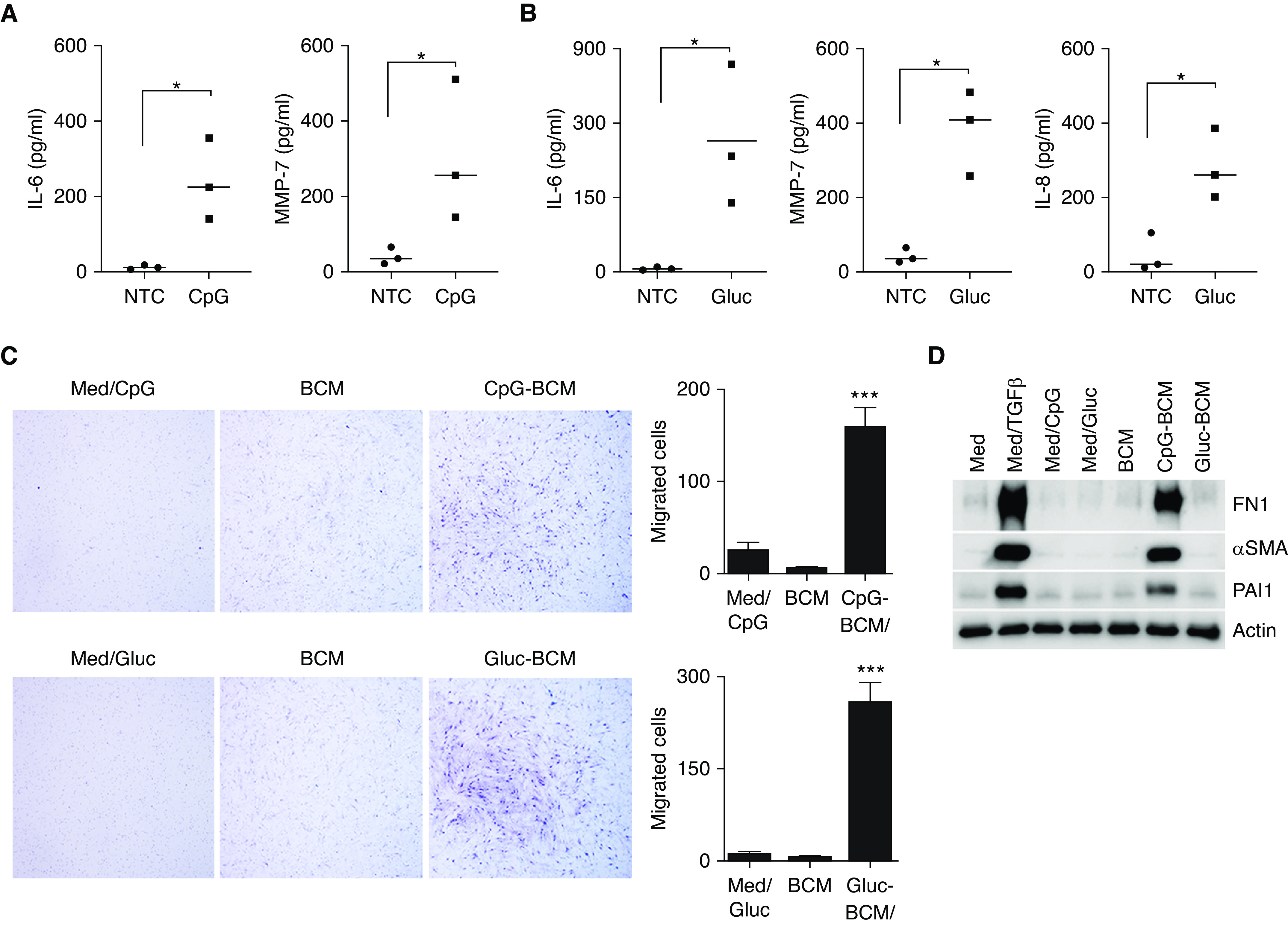
B-cells from patients with idiopathic pulmonary fibrosis (IPF) respond to microbial antigens by releasing a proinflammatory and profibrotic milieu that induced fibroblast migration and activation. (A and B) Peripheral B-cells from patients with IPF were either left unstimulated or were stimulated with CpG (A) or β-glucan (Gluc) (B), as indicated. IL-6, MMP-7, and IL-8 were measured in the cell supernatant by ELISA. (C) Images of quiescent IPF fibroblasts stained with Wright-Giemsa migrating toward B-cell–conditioned media (BCM). BCM was obtained from B-cell supernatant from unstimulated cells or B-cells stimulated with CpG (CpG-BCM) or Gluc (Gluc-BCM) for 48 hours. CpG and Gluc in regular media were used as controls. Migrated IPF fibroblasts were manually counted using ImageJ software. (D) Cell lysate of IPF fibroblasts stimulated with TGFβ, CpG, Gluc, CpG-BCM, or Gluc-BCM (as indicated) and immunoblotted for FN1 (fibronectin), αSMA (α-smooth muscle actin), PAI1 (plasminogen activator inhibitor-1), or β-actin. Data are representative of three independent experiments. *P < 0.05 and ***P < 0.001. Med = media; MMP-7 = matrix metallopeptidase-7; NTC = unstimulated B-cells; TGFβ = transforming growth factor β.
Because B-cells have been implicated in fibroblast migration in models of rheumatoid arthritis and scleroderma (18, 19), we sought to further elucidate the role of activated B-cells in fibroblast migration and activation. Hence, human primary fibroblasts derived from patients with IPF were cultured in the presence of supernatants from B-cells that were either untreated or stimulated with CpG or β-glucan. The supernatant from B-cells used to stimulate fibroblasts will be referred throughout the manuscript as “B-cell–conditioned media (BCM)”. We will use “CpG-BCM” or “β-glucan–BCM” if the supernatant was from B-cells that were stimulated with CpG or β-glucan, respectively. As shown in Figure 1C, migration was significantly increased after exposure of IPF fibroblasts to CpG-BCM or β-glucan–BCM compared with those exposed to BCM from unstimulated cells or fibroblasts activated directly with CpG (med/CpG) or β-glucan (med/gluc), suggesting that activated B-cells secrete factors that attract fibroblasts. Next, we determined whether fibroblasts exposed to CpG-BCM or β-glucan–BCM would also show an activated phenotype by assessing for an increase of the profibrotic proteins fibronectin, SMA (smooth muscle actin), and PAI1 (plasminogen activator inhibitor-1). As shown in Figure 1D, CpG-BCM did indeed promote an increased production of fibronectin, αSMA, and PAI1 by IPF fibroblasts, whereas β-glucan–BCM did not.
B-Cell Aggregates Are Present in the Fibrotic Areas of the Lungs of Patients with Usual Interstitial Pneumonia
Because our data showed that activated B-cells modulate fibroblast migration and activation and B-cell aggregates are known to occur in the lung after infection and injury, we next sought to investigate further the presence of B-lymphocytes in fibrotic lung tissue. We specifically looked at their location and spatial relationship with regard to fibroblasts and fibroblastic foci, the pathological hallmark of usual interstitial pneumonia (UIP). As shown in Figure 2, CD20-positive cells formed tight aggregates in the fibrotic areas, and many were also found adjacent to the fibroblastic foci (Figure 2A), suggesting a possible role of tissue B-cells in fibrosis. The selected cases are representative patients with clinical diagnosis of IPF and confirmed diagnosis of UIP by two expert pathologists. To further investigate the activation status of the B-cells present in the aggregates and because our published peripheral B-cell data showed activation of mTOR pathways upon CpG activation (9), we sought to determine the presence of phosphor-pS6, a marker for mTOR activation, in lung-resident B-cells. Like CpG-activated peripheral B-cells, lung B-cells showed activation of mTOR (as shown in Figure 2B), suggesting that these cells are not quiescent and raising the possibility of a role for the TLR-9 activation pathway in these cells.
Figure 2.

B-cell aggregates are present in fibrotic areas of the lungs of patients with pulmonary fibrosis and show mTOR activation. (A) Hematoxylin and eosin stain of lung tissue from three patients with clinical diagnosis of pulmonary fibrosis (1a–3a) showing pathologic features of usual interstitial pneumonia (UIP). CD20 stain (1b–3b) demonstrates B-cell aggregates (brown) neighboring fibroblastic foci (arrows). High-resolution computed tomography imaging of lung parenchyma of the respective patients demonstrate different patterns of fibrotic changes (1c–3c). (B) FFPE from patients with pathological findings of UIP underwent immunofluorescent staining for CD20 (red) and p-S6 (green). Scale bars, A, 500 µm; B, 500 µm;. FFPE = formalin-fixed, paraffin-embedded tissue.
Nintedanib and Pirfenidone Altered the Secretory Profile of B-Cells and Impaired Fibroblast Migration
Having shown the potential relevance of B-cells in fibroblast behavior, we next evaluated the possible effect of antifibrotics on B-cell activation. Nintedanib and pirfenidone are currently the only two drugs U.S. Food and Drug Administration approved for the treatment of patients with IPF and were therefore tested in our in vitro model. B-cells from patients with IPF were preincubated with either nintedanib or pirfenidone before stimulation with either CpG or β-glucan. Cytokines in the cell supernatant were then determined by ELISA. Remarkably, both antifibrotics significantly impaired cytokine secretion. Nintedanib decreased IL-6, IL-8, and MMP-7, whereas pirfenidone decreased MMP-7 but had little or no effect on IL-6 and IL-8 (Figure 3). The concentrations of nintedanib and pirfenidone used were chosen on the basis of their in vitro effect, lack of cell toxicity (Figure E1), and prior published data (20).
Figure 3.
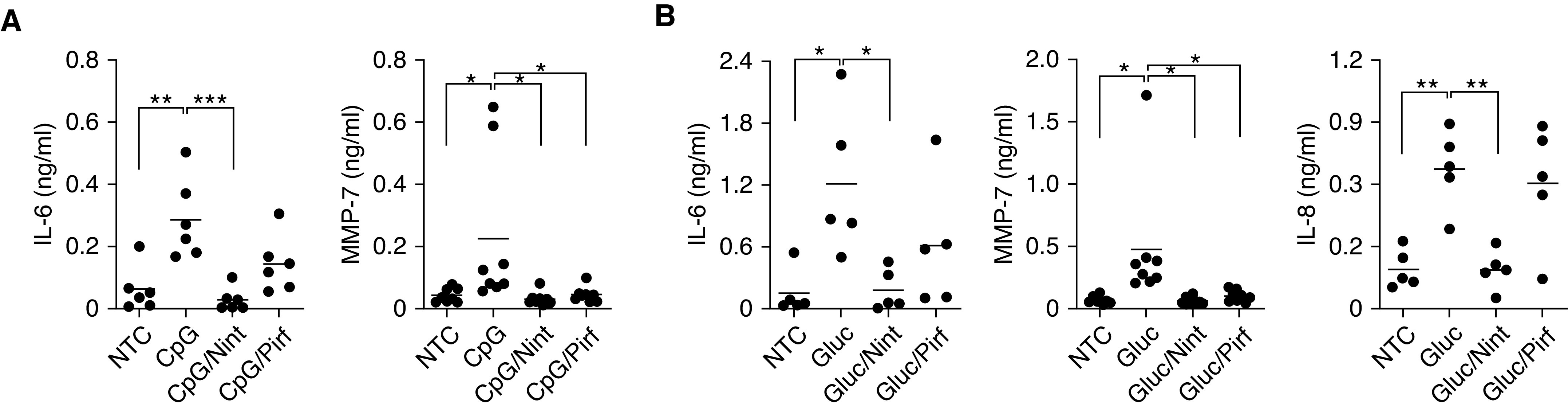
Antifibrotics alter the inflammatory milieu of activated B-cells from patients with IPF. (A and B) B-cells were either left unstimulated or stimulated with CpG (A) or Gluc (B) in the presence of 1 μM of Nint and 100 μM of Pirf, as indicated. IL-6, IL-8, and MMP-7 were measured by ELISA in the cell supernatants of peripheral B-cells from patients with IPF. *P < 0.05, **P < 0.01, and ***P < 0.001. Nint = nintedanib; ns = not significant (P > 0.05); Pirf = pirfenidone.
Because the pretreatment of B-cells with antifibrotics changed the cytokine milieu, induced by either CpG or β-glucan, we next sought to investigate the activation pathways involved. Our prior published data showed that CpG induced mTOR activation and β-glucan was mTOR independent but activated mitogen-activated protein kinases (MAPKs) (9, 12). We first tested the effect of rapamycin, a specific mTOR inhibitor, on IL-6 secretion by stimulated B-cells because IL-6 was induced by both CpG and β-glucan. As shown in Figure E2, pretreatment of B-cells with rapamycin resulted in impaired IL-6 secretion when B-cells were stimulated with CpG but not with β-glucan, suggesting that the mTOR pathway was crucial for CpG-mediated, but not β-glucan-mediated, IL-6 secretion. Next, we studied the effect of nintedanib and pirfenidone on the mTOR and MAPKs signaling pathways by looking at different protein expression using B-cells from patients with IPF. As shown in Figure 4, pretreatment of CpG-stimulated cells with nintedanib decreased mTOR activation, whereas pirfenidone had little or no effect on the mTOR signaling pathway (Figure 4A). When B-cells were stimulated with β-glucan, pretreatment with nintedanib decreased Src and JNK phosphorylation, whereas pretreatment with pirfenidone did not affect JNK or Src but decreased phospho-p38 (Figure 4B). Altogether, these data show that although both antifibrotics affected inflammatory and profibrotic cytokines, each one seemed to affect different signaling pathways that differed depending on the stimulating antigen.
Figure 4.

Nint interferes with mTOR and Src kinase signaling pathways, whereas Pirf affects p38 activation and has no effect on mTOR. (A) Phosphorylation of mTOR, S6, and 4EBP1 was detected by IB in the total cell lysates of CpG-stimulated B-cells in the presence of 1 μM Nint or 100 μM of Pirf, as indicated. Total mTOR, S6K, S6, and 4EBP1 were used as controls. (B) Phosphorylation of Src, JNK, and p38 was measured in the total cell lysates after Gluc stimulation of B-cells in the presence or absence of Nint and Pirf, as indicated. Total Src, JNK, and p38 were used as controls. B-cells were isolated from patients with IPF. Data are representative of at least three independent experiments.
To further understand the effect of antifibrotics on B-cells and because our prior studies showed that mTOR activation is important for CpG-mediated B-cell proliferation and IgM production (12), we investigated the effect of nintedanib and pirfenidone on B-cell proliferation and activation. As shown in Figure 5, nintedanib affected B-cell thymidine (3H) incorporation as well as the activation markers CD80 and CD86 and IgM secretion. In contrast, pirfenidone, at the concentrations tested, did not have an effect on B-cell proliferation or IgM secretion (Figures 5A and 5C).
Figure 5.
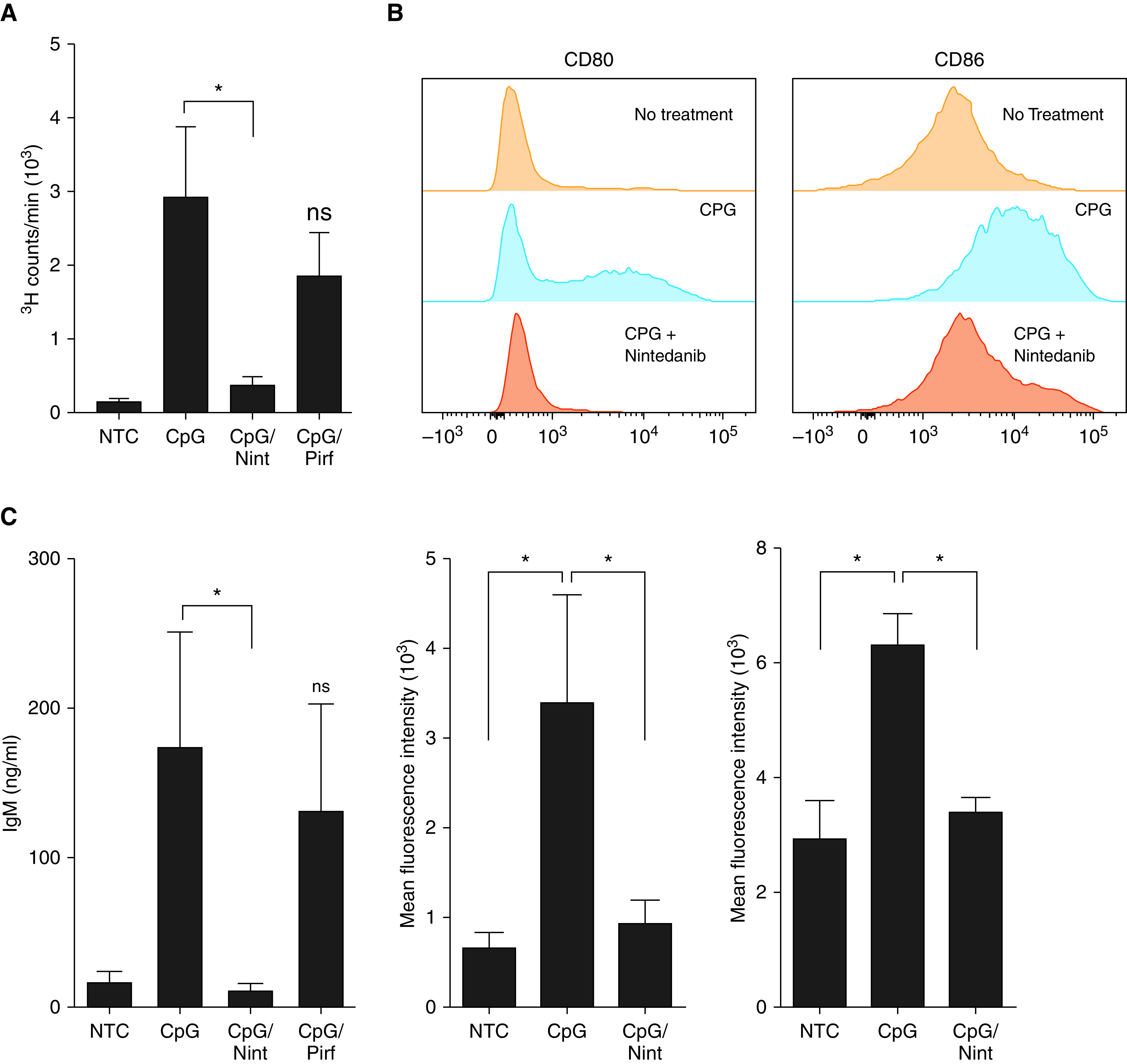
Nint impairs B-cell proliferation and activation. (A) 3H-thymidine incorporation was assessed in NTC and cells stimulated with CpG. Some cells were pretreated with 1 μM Nint or 100 μM Pirf for 1 hour before CpG stimulation (CpG/Nint and CpG/Pirf, respectively), as indicated. (B) Representative histograms of CD80 and CD86 on peripheral human B-cells. CD80 and CD86 were assessed in NTC, stimulated cells with CpG, and CpG-stimulated cells pretreated with nintenadib, as previously indicated. (C) IgM was measured by ELISA in the cell supernatants of B-cells of NTC, stimulated cells with CpG, and CpG-stimulated cells pretreated with 1 μM Nint or 100 μM Pirf for 1 hour before stimulation for a total of 5 days, as indicated. Data are representative of a typical experimental run of at least three separate experimental preparations, which were performed with different B-cell preparations. *P < 0.05. ns = not significant (P < 0.05).
Antifibrotics Decreased B-Cell–mediated Fibroblast Migration and Activation
IPF fibroblast migration increased upon incubation with CpG-BCM and β-glucan–BCM. Because the presence of antifibrotics altered B-cell cytokine milieu in the BCM, we next assessed fibroblast migration and activation by BCM from stimulated B-cells that were pretreated with antifibrotics. Interestingly, the migration of IPF and normal human fibroblasts was significantly decreased when fibroblasts were exposed to CpG-BCM or β-glucan–BCM from nintedanib-treated B-cells but not when exposed to CpG-BCM or β-glucan–BCM from B-cells pretreated with pirfenidone (Figures 6 and E3). Because CpG and nintedanib are soluble and can be carried over in the BCM and potentially affect fibroblast migration and function directly, supernatant from cell-free wells treated with CpG (Med/CpG) or nintedanib (med/nint) and with CpG-BCM with freshly added nintedanib (CpG-BCM/nit+), at the calculated carried over concentration were added to the fibroblasts as controls. As shown in Figures 6 and E3 and E4, no significant change in fibroblast migration was appreciated by either of the controls, suggesting that the effect seen in fibroblasts was secondary to the B-cell–secreted factors and not the small amounts of carryover nintedanib or CpG acting directly on the fibroblasts. Consistent with our prior observations, fibroblasts migration induced by CpG-BCM, but not β-glucan–BCM, also decreased in the presence of rapamycin (Figure E5).
Figure 6.
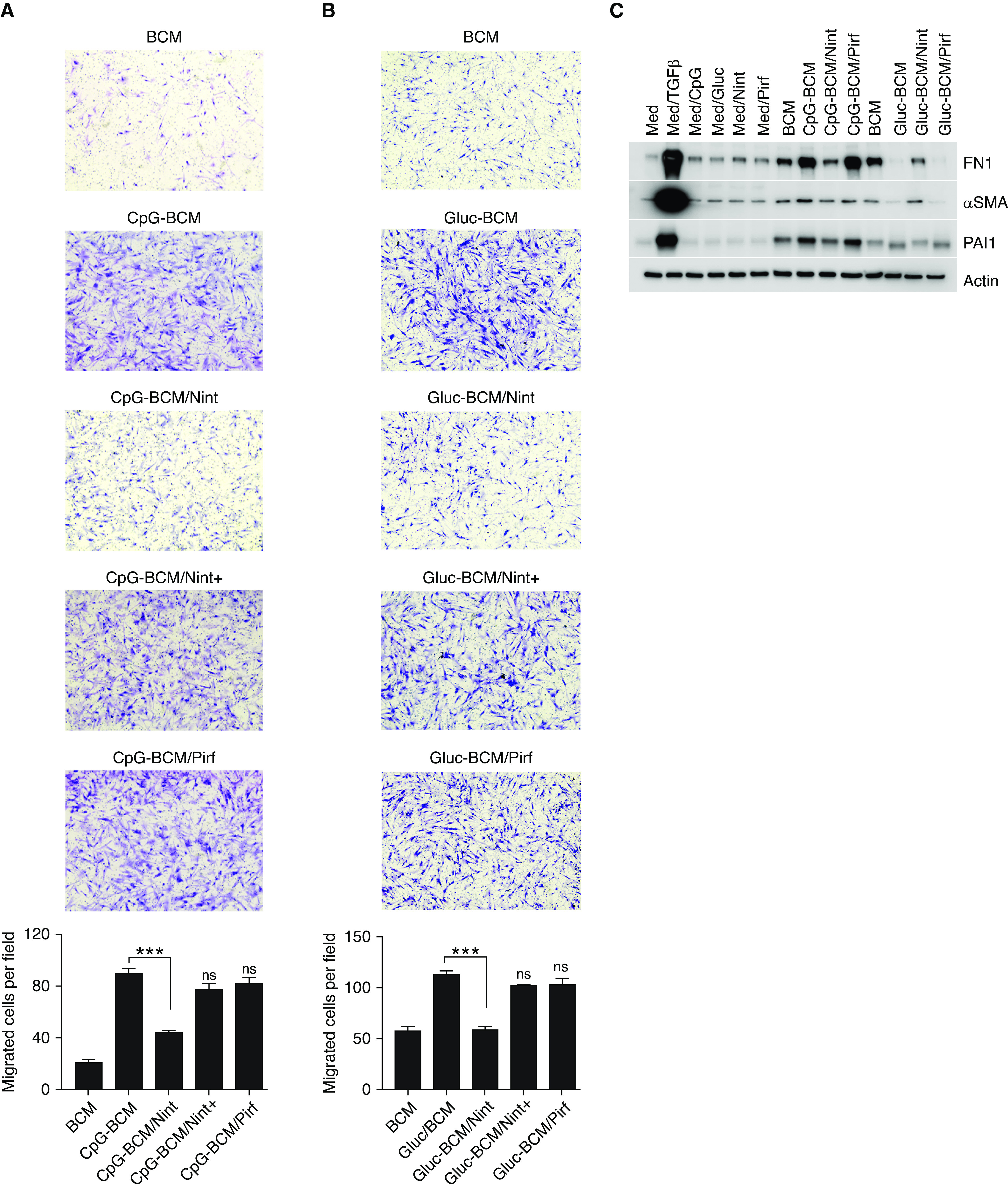
Treatment of B-cells with Nint impairs B-cell–driven fibroblast migration and activation. (A and B) Images of quiescent IPF fibroblasts stained with Wright-Giemsa stain migrating toward BCM, CPG-BCM, and Glu-BCM. Some BCMs were prepared by preincubating B-cells for 1 hour with 1 μM Nint or 100 μM Pirf before stimulation with CpG or β-glucan (CpG-BCM/Nint, CpG-BCM/Pirf, Gluc-BCM/Nint, and Gluc-BCM/Pirf), as indicated. Controls include regular medium (Med) and medium containing CpG, Gluc, Pirf, and Nint (Med/CpG, Med/Gluc, Med/Pirf and Med/Nint, respectively) and are shown in Figure E2. Nint was also added to the BCM only when the Transwell assay is set up to control for any Nint carryover (CpG-BCM/Nint+ and Gluc-BCM/Nint+). Each membrane was imaged and divided into four fields, migrated cells were counted in each field, and means are plotted as migrated cells per field. (C) Cell lysate of IPF fibroblasts stimulated with different BCMs (as indicated) and immunoblotted for FN1, αSMA, PAI1, or β-actin, as indicated. BCM was obtained as previously indicated (A and B). TGFβ was used as positive control. Data are representative of three separate experiments. ***P < 0.001.
Next, we assessed the effect of stimulated BCM (CpG-BCM and β-glucan–BCM) from cells pretreated with antifibrotics on fibroblast activation and showed that fibronectin, PAI1, and αSMA decreased in the presence of CpG-BCM from nintedanib- but not pirfenidone-treated B-lymphocytes (Figures 6C and E6), further suggesting the specific effect of each antifibrotic on B-cell–mediated fibroblast activation.
IL-6 and VEFGA Participate in IPF Fibroblast Migration and Activation
To understand what specific factors present in the BCM from stimulated B-cells caused fibroblast migration and activation and because IL-8 and IL-6 were both present in the BCM and have been shown to have chemoattractant properties, we further determined their migratory potential toward IPF fibroblasts. As shown in Figure 7A, IL-6, at the higher doses, showed chemoattractant properties on fibroblasts, whereas IL-8 did not. As the effect of IL-6 was modest, we hypothesized that other factors may be also contributing to the observed fibroblast migration. Therefore, we further screened at several growth factors with potential role in fibroblasts activation that could be affected by nintedanib, such as VEGFA. As shown in Figure 7B, VEGFA was increased in the B-cell supernatant upon CpG and β-glucan stimulation and was further decreased by nintedanib, whereas pirfenidone had little effect, suggesting that VEGFA present in the BCM may also play a role in fibroblast migration. Consistent with our prior observations, the use of rapamycin also decreased VEGFA secretion upon CpG, but not β-glucan, stimulation, further supporting the role of mTOR on CpG-mediated VEGFA production of B-cells (Figure E7A). Consequently, we next determined fibroblast migration in the presence of increasing concentrations of VEGFA and in the presence of a VEGFA inhibitor (axitinib). As shown in Figure 7C, fibroblast migration increased in the presence of recombinant VEGFA in a dose-dependent manner and decreased when axitinib was added (Figure E7B). In addition, migration was found to be impaired when fibroblasts were activated with CpG-BCM in the presence of axitinib, suggesting that the VEGFA present in the BCM of stimulated B-cells was indeed responsible for most of the fibroblast migration induced by activated B-cells (Figure 7C). Fibroblast activation, measured by an increase in fibronectin and PAI-1, was also noted upon VEGFA stimulation (Figure 7D).
Figure 7.
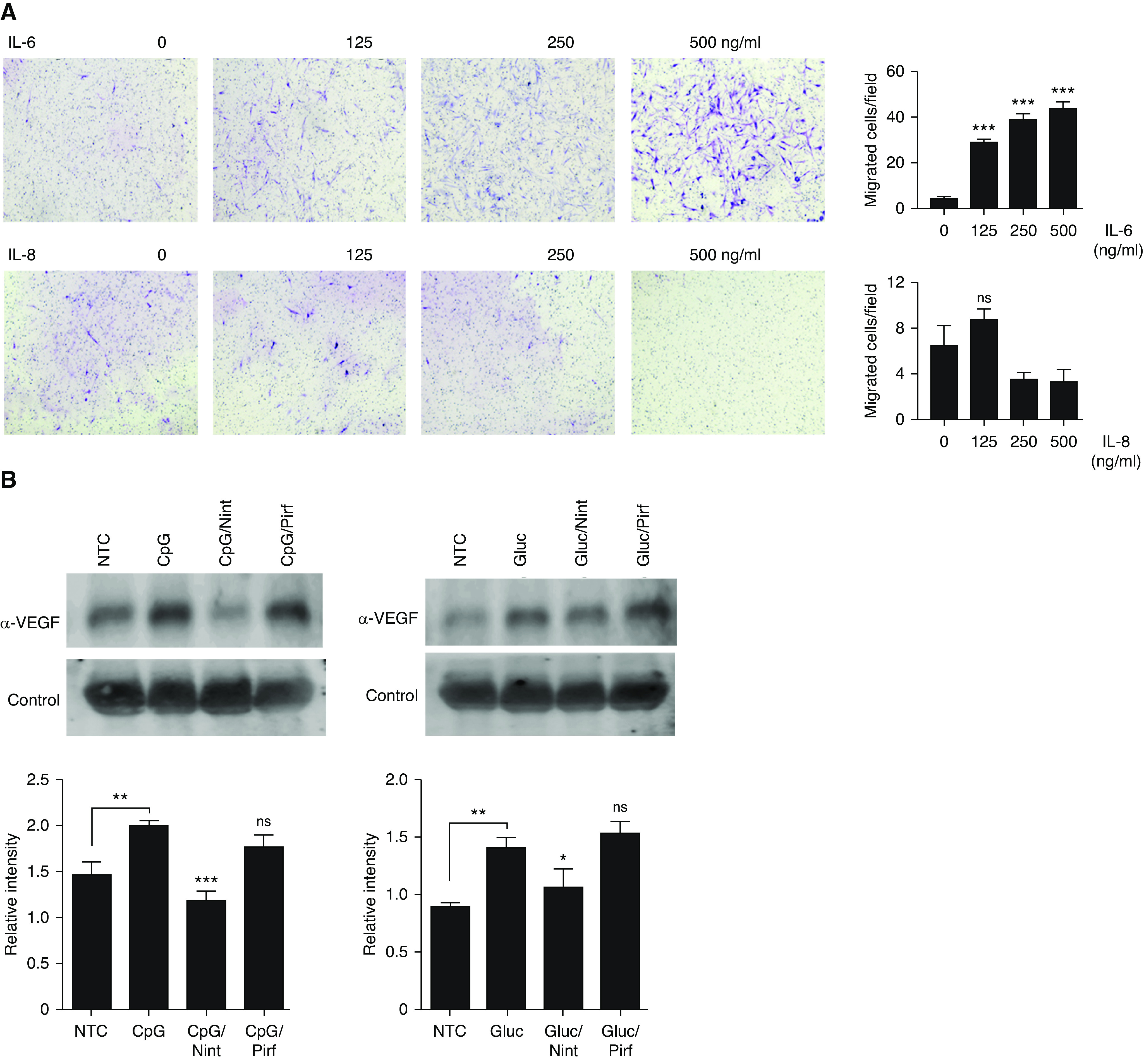
IL-6 and VEGFA participate in B-cell–mediated fibroblast migration and activation. (A) Images of quiescent IPF fibroblasts stained with Wright-Giemsa stain migrating toward different concentrations of recombinant IL-6 and IL-8 (A) or stimulated with different preparations of BCM, as indicated. Some fibroblasts were preincubated with 5 μM axitinib for 1 hour before the addition of the BCM, as indicated. (B) VEGFA secretion was measured by IB in the cell supernatant of NTC or stimulated cells with CpG and β-glucan for 24 hours as indicated. Some cells were pretreated with Nint and Pirf, as indicated. Nonspecific protein was used as loading control. Band intensities were quantified using ImageJ software version 1.48 (National Institutes of Health), and the relative intensities were calculated by normalizing the intensity of the target protein to that of the loading control. (C) Images of quiescent IPF fibroblasts stained with Wright-Giemsa stain migrating toward different concentrations of recombinant VEGFA or stimulated with different preparations of BCM, as indicated. (D) Cell lysates from IPF fibroblasts stimulated with different concentrations of IL-6 and VEGFA and immunoblotted for FN1, αSMA, PAI1, or β-actin, as indicated. Data are representative of three separate experiments. *P < 0.05, **P < 0.01, and ***P < 0.001. VEGFA = vascular endothelial growth factor A.
Discussion
Injury to the alveolar epithelial cell lining by environmental exposures is believed to be a major component of the initial insult in the development of pulmonary fibrosis. Repetitive microinjury of the alveoli results in the generation of a profibrotic milieu in which cytokines, chemokines, metalloproteases, and growth factors released by injured and apoptotic cells may orchestrate the recruitment of fibroblasts and other immune cells (21). This army of cells in the predisposed host is likely to generate aberrant healing, resulting in fibrosis and lung damage. The different genetic variants, senesce cells, and other unknown predisposing factors, together with a specific environmental injury, are likely to influence the different phenotypes seen in this devastating disease.
Among the different profibrotic and proinflammatory proteins, plasma concentrations of IL-8 and MMP-7 have been shown to correlate with poor outcome in patients with IPF (22–25). Furthermore, the extent of inflammation in the lung may also determine the rate of progression and respiratory functional decline measured by forced vital capacity (26), suggesting that fibrosis and inflammation are both contributors to the pathophysiology of this disease.
Herein, we have shown that in response to microbial antigens (CpG and β-glucan), B-cells from patients with IPF release a profibrotic and proinflammatory cocktail (MMP-7, IL-8, IL-6, and VEGFA) shown to be involved in the pathogenesis of fibrotic lung diseases. Our group have shown that B-cell responses to CpG and β-glucan are mediated by PRRs (9, 12, 15). Specifically, CpG preferentially signals via TLR9 and the mTOR pathway, whereas β-glucan stimulation results in the activation of dectin-1 and MAPKs and is independent of mTOR (9). The use of nintedanib and pirfenidone, the only two antifibrotic drugs available in the United States to treat patients with IPF outside of clinical trials, showed that they both affect B-cell activation and impaired cytokine, metalloprotease, and chemokine secretion. Interestingly, the effect of the antifibrotic drugs were slightly different from each other, suggesting that they regulate different pathways. Specifically, nintedanib decreased Src phosphorylation and interfered with mTOR activation, whereas pirfenidone decreased p38 phosphorylation and had little effect on the mTOR pathway. The fact that B-cells from patients with IPF responded to PRR activation in response to microbial antigens by secreting IL-6, IL-8, MMP-7, and VEGFA is novel and suggests that B-cells are likely to contribute to the general pool of serum proteins, especially during acute infectious exacerbations. Although further investigations in animal models and clinical trials are needed to further understand the role B-cell activation via PRRs during acute exacerbations of IPF, published microbiome data demonstrate that patients with IPF have increased bacterial burdens compared with control subjects, thus raising the hypothesis that chronic PRR stimulation may be a possible mechanism of injury in IPF (7).
The role of B-cells in fibrosis is controversial. However, the presence of lymphoid aggregates (also known as inducible bronchus-associated lymphoid tissue [iBALT]), although not viewed as a typical histologic finding of IPF, has been described in fibrotic lung diseases (13, 14). Their role is not clearly understood, and they are frequently looked at as merely bystanders. Herein, we have demonstrated the presence of B-cell aggregates in patients with biopsy-proven UIP. B-cells were located in areas of fibrosis and adjacent to fibroblastic foci, suggesting potential participation in lung fibrosis. B-cells showed constitutive mTOR activation compatible with an active state. Our in vitro data further show that CpG- and β-glucan–activated B-cells increased fibroblast migration and activation as measured by the increased production of fibronectin, αSMA, and PAI1, especially after CpG stimulation, suggesting cross-talk between B-cells and fibroblasts. The communication between B-cells and fibroblasts is very novel and poorly understood in patients with IPF; however, it has been recognized in autoimmune diseases such as rheumatoid arthritis and scleroderma (18, 27). Specifically, in a model of scleroderma, the interaction of activated B-cells with synovial fibroblasts has shown that B-cells modified the function of synovial fibroblasts, resulting in a more proinflammatory and aggressive phenotype. Although the exact role that iBALTs play in IPF is not clearly known, B-cells aggregate as part of the normal healing during infection but usually resolve after the infection is over. Occasionally, iBALTs can remain for several months and respond to lung antigens even if the antigens that triggered the initial stimuli are no longer present (28). It is therefore very likely that B-cell aggregates in the lungs of patients with IPF are part of an aberrant healing process. We speculate that PRR activation by both exogenous and endogenous ligands such as mitochondrial DNA (sterile inflammation), known to be a TLR9 ligand, could potentially trigger B-cell activation in the lung, contributing to worsening injury and even fibrosis, especially during episodes of acute exacerbations. The role of sterile inflammation is an attractive mechanism that is supported by newer data suggesting that damage-associated molecular patterns, such as mitochondrial DNA, can also activate inflammation via PRRs (29). Specifically, mitochondrial DNA has been shown to be increased in plasma and to mediate TLR9 activation in patients with IPF (30). Further investigations in this field may provide additional understanding on the role of PRR activation in the pathophysiology of lung fibrosis.
Antifibrotics have become the standard of care to treat patients with IPF, but little is known about their mechanism of action, especially on B-cells. Herein, we provide evidence that B-cell function is affected by nintedanib and pirfenidone, but each antifibrotic had a specific mechanism of action. Whereas nintedanib affected Src and mTOR phosphorylation in patients’ B-cells, pirfenidone decreased p38 and had little effect on mTOR. The effect of antifibrotics on B-cells further affected B-cell–mediated fibroblast responses depending on the source of B-cell stimulation. The effects of conditioned media from CpG-stimulated B-cells on fibroblasts was different than from that of β-glucan–stimulated B-cells, which were differentially affected by nintedanib and pirfenidone treatment. Specifically, fibroblasts exposed to CpG-BCM had an increased expression of fibronectin 1, αSMA, and PAI1, which were each reduced upon treatment with nintedanib. Conversely, fibroblasts exposed to β-glucan–BCM show decreased fibronectin 1 and αSMA but increased PAI1 expression, with nintedanib treatment resulting in an increase in fibronectin 1 and αSMA but decreasing PAI1 expression. Although pirfenidone modestly affected MMP-7 secretion by B-cells, it did not show much effect on B-cell–mediated fibroblast migration or activation. Compared with pirfenidone, nintedanib seem to have a broader effect on B-cells and B-cell–mediated fibroblast activation. It is possible that the doses of pirfenidone and nintedanib used in the experiments were not comparable. However, the concentrations used in our in vitro experiments were guided by cell toxicity and prior published data. Although further in vivo data are necessary to corroborate our findings, here we provide further insights in the complexity of the fibrotic mechanisms and showed in vitro evidence that although nintedanib can inhibit antifibrotic pathways, it can also increase others, as is the case of increasing PAI1 upon B-cell stimulation with β-glucan. Our data outline possible mechanisms by which combination therapy that targets several fibrotic pathways may be an alternative approach in selected patients. Further investigations are therefore necessary to fully understand the role of combination therapy on these pathways and the effects in patients with IPF.
Lastly, we identified VEGFA as one of the main players in B-cell–induced fibroblast migration and activation. VEGFA is primarily known for its angiogenesis properties, although newer evidence suggests a further role in regulating fibroblast activation (31). Though the exact role of VEGFA in the lung has not been fully defined, compartmentalization of VEGFA within the alveolar space seems to be key for maintaining normal lung function. It is believed that processes that disrupt the intact alveolar capillary membrane, such as acute respiratory distress syndrome (ARDS), alter VEGFA compartmentalization, resulting in lung injury and fibrosis. The role of VEGF in lung fibrosis and ARDS is further supported by the presence of VEGF in fibrotic lung tissue and by the fact that VEGFA polymorphisms have been associated with increased severity and mortality in ARDS (31, 32). Our observations that VEGFA was present in the BCM from stimulated B-cells led us to further investigate its role in fibroblast migration. As axitinib decreased fibroblast migration induced by recombinant VEGFA and activated BCM containing VEGFA increased fibroblast migration, this further supports the notion that VEGFA is important in B-cell–mediated fibroblast activation. Nintedanib decreased VEGFA secretion from activated B-cells, which also resulted in decreased fibroblast activation. These data suggest that the decrease in fibroblast migration and activation seen by nintedanib was in part due to its effect on VEGFA secretion. Pirfenidone, however, did not seem to significantly affect VEGFA secretion by B-cells.
At the molecular level, the ratio of free versus bound VEGFA seems to be important. Free VEGFA promotes a proangiogenic state, whereas VEGFA bound to several cytokines results in inhibition of its activity. VEGFA can also bind to soluble VEGF receptor-1. Interestingly, MMP-7 has been shown to increase VEGFA bioavailability by degradation of the soluble VEGF receptor-1, allowing free VEGFA to signal via VEGF receptor-2 in endothelial cells (33). MMP-7 is increased in patients with IPF and associates with severity of disease and poor prognosis (11). Our observations that microbial antigens induced VEGFA secretion by B-cells as well as MMP-7, in addition to our data that VEGFA drives IPF fibroblasts migration and activation, further supports the hypothesis that VEGFA plays a role in the acute injury process. We further hypothesized that B-cell–secreted VEGFA and MMP-7 are part of the normal healing of the lung, which involves fibroblast migration and activation, especially after an acute infectious process. Dysregulation of this healing process in patients with IPF may further contribute to the fibroproliferative response of the lung and the development of fibrosis, especially during acute exacerbations of IPF, in which B-cells activated by infectious antigens may contribute to the exudative and fibroproliferative process. Although further in vivo studies are needed, our observations are a step forward in the understanding of the role of B-cells in the pathophysiology of lung fibrosis.
In summary, we have shown that pirfenidone and nintedanib have different mechanisms of action and exert differential effects on B-cells and B-cell–derived fibroblast behavior. Our data further show the relevance of PRRs in B-cell activation and subsequent role on fibroblast modulation and emphasizes the roles of IL-8, MMP-7, IL-6, and VEGFA in fibroblast migration and activation. Our in vitro data also suggest that nintedanib may be of help during the acute inflammatory process because of its in vitro antiinflammatory properties, and this warrants further investigation in in vivo models and clinical trials.
Footnotes
Supported by the U.S. National Institutes of Health (grants K08HL112849 and R03HL144427) and by funds from the Annenberg to E.M.C. and the Hurvis Foundation to E.M.C.
Author Contributions: Concept and design: M.F.A., G.F.S., D.K.A., T.J.K., V.P.V.K., T.P., and E.M.C. Data acquisition: M.F.A., A.M.E., G.F.S., D.K.A., H.D., V.P.V.K., and E.M.C. Data interpretation/analysis: M.F.A., A.M.E., G.F.S., H.D., V.P.V.K., M.-C.A., E.S.Y., A.H.L., T.P., and E.M.C. Manuscript drafting: M.F.A., V.P.V.K., T.P., and E.M.C. Manuscript review: all authors.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2020-0387OC on March 9, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071–2082. doi: 10.1056/NEJMoa1402584. [DOI] [PubMed] [Google Scholar]
- 3. King TE, Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083–2092. doi: 10.1056/NEJMoa1402582. [DOI] [PubMed] [Google Scholar]
- 4. Seibold MA, Wise AL, Speer MC, Steele MP, Brown KK, Loyd JE, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. 2011;364:1503–1512. doi: 10.1056/NEJMoa1013660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Noth I, Zhang Y, Ma SF, Flores C, Barber M, Huang Y, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med. 2013;1:309–317. doi: 10.1016/S2213-2600(13)70045-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Collard HR, Moore BB, Flaherty KR, Brown KK, Kaner RJ, King TE, Jr, et al. Idiopathic Pulmonary Fibrosis Clinical Research Network Investigators. Acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2007;176:636–643. doi: 10.1164/rccm.200703-463PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Molyneaux PL, Cox MJ, Willis-Owen SA, Mallia P, Russell KE, Russell AM, et al. The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2014;190:906–913. doi: 10.1164/rccm.201403-0541OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Desai O, Winkler J, Minasyan M, Herzog EL. The role of immune and inflammatory cells in idiopathic pulmonary fibrosis. Front Med (Lausanne) 2018;5:43. doi: 10.3389/fmed.2018.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ali MF, Dasari H, Van Keulen VP, Cornec D, Vasmatzis G, Peikert T, et al. Microbial antigens stimulate metalloprotease-7 secretion in human B-lymphocytes using mTOR-dependent and independent pathways. Sci Rep. 2017;7:3869. doi: 10.1038/s41598-017-04199-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gill SE, Nadler ST, Li Q, Frevert CW, Park PW, Chen P, et al. Shedding of syndecan-1/CXCL1 complexes by matrix metalloproteinase 7 functions as an epithelial checkpoint of neutrophil activation. Am J Respir Cell Mol Biol. 2016;55:243–251. doi: 10.1165/rcmb.2015-0193OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rosas IO, Richards TJ, Konishi K, Zhang Y, Gibson K, Lokshin AE, et al. MMP1 and MMP7 as potential peripheral blood biomarkers in idiopathic pulmonary fibrosis. PLoS Med. 2008;5:e93. doi: 10.1371/journal.pmed.0050093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ali MF, Dasari H, Van Keulen VP, Carmona EM. Canonical stimulation of the NLRP3 inflammasome by fungal antigens links innate and adaptive B-lymphocyte responses by modulating IL-1β and IgM production. Front Immunol. 2017;8:1504. doi: 10.3389/fimmu.2017.01504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Atkins SR, Turesson C, Myers JL, Tazelaar HD, Ryu JH, Matteson EL, et al. Morphologic and quantitative assessment of CD20+ B cell infiltrates in rheumatoid arthritis-associated nonspecific interstitial pneumonia and usual interstitial pneumonia. Arthritis Rheum. 2006;54:635–641. doi: 10.1002/art.21758. [DOI] [PubMed] [Google Scholar]
- 14. Rangel-Moreno J, Hartson L, Navarro C, Gaxiola M, Selman M, Randall TD. Inducible bronchus-associated lymphoid tissue (iBALT) in patients with pulmonary complications of rheumatoid arthritis. J Clin Invest. 2006;116:3183–3194. doi: 10.1172/JCI28756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ali MF, Driscoll CB, Walters PR, Limper AH, Carmona EM. β-Glucan-Activated human B lymphocytes participate in innate immune responses by releasing proinflammatory cytokines and stimulating neutrophil chemotaxis. J Immunol. 2015;195:5318–5326. doi: 10.4049/jimmunol.1500559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mennink-Kersten MA, Ruegebrink D, Verweij PE. Pseudomonas aeruginosa as a cause of 1,3-beta-D-glucan assay reactivity. Clin Infect Dis. 2008;46:1930–1931. doi: 10.1086/588563. [DOI] [PubMed] [Google Scholar]
- 17. Takeshita F, Leifer CA, Gursel I, Ishii KJ, Takeshita S, Gursel M, et al. Cutting edge: role of Toll-like receptor 9 in CpG DNA-induced activation of human cells. J Immunol. 2001;167:3555–3558. doi: 10.4049/jimmunol.167.7.3555. [DOI] [PubMed] [Google Scholar]
- 18. Störch H, Zimmermann B, Resch B, Tykocinski LO, Moradi B, Horn P, et al. Activated human B cells induce inflammatory fibroblasts with cartilage-destructive properties and become functionally suppressed in return. Ann Rheum Dis. 2016;75:924–932. doi: 10.1136/annrheumdis-2014-206965. [DOI] [PubMed] [Google Scholar]
- 19. Kang YM, Kim SY, Kang JH, Han SW, Nam EJ, Kyung HS, et al. LIGHT up-regulated on B lymphocytes and monocytes in rheumatoid arthritis mediates cellular adhesion and metalloproteinase production by synoviocytes. Arthritis Rheum. 2007;56:1106–1117. doi: 10.1002/art.22493. [DOI] [PubMed] [Google Scholar]
- 20. Knüppel L, Ishikawa Y, Aichler M, Heinzelmann K, Hatz R, Behr J, et al. Inhibition of Collagen Fibril Assembly. A novel antifibrotic mechanism of nintedanib and pirfenidone. Am J Respir Cell Mol Biol. 2017;57:77–90. doi: 10.1165/rcmb.2016-0217OC. [DOI] [PubMed] [Google Scholar]
- 21. Kolahian S, Fernandez IE, Eickelberg O, Hartl D. Immune mechanisms in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2016;55:309–322. doi: 10.1165/rcmb.2016-0121TR. [DOI] [PubMed] [Google Scholar]
- 22. Yang L, Herrera J, Gilbertsen A, Xia H, Smith K, Benyumov A, et al. IL-8 mediates idiopathic pulmonary fibrosis mesenchymal progenitor cell fibrogenicity. Am J Physiol Lung Cell Mol Physiol. 2018;314:L127–L136. doi: 10.1152/ajplung.00200.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ziegenhagen MW, Zabel P, Zissel G, Schlaak M, Müller-Quernheim J. Serum level of interleukin 8 is elevated in idiopathic pulmonary fibrosis and indicates disease activity. Am J Respir Crit Care Med. 1998;157:762–768. doi: 10.1164/ajrccm.157.3.9705014. [DOI] [PubMed] [Google Scholar]
- 24. Song JW, Do KH, Jang SJ, Colby TV, Han S, Kim DS. Blood biomarkers MMP-7 and SP-A: predictors of outcome in idiopathic pulmonary fibrosis. Chest. 2013;143:1422–1429. doi: 10.1378/chest.11-2735. [DOI] [PubMed] [Google Scholar]
- 25. Tzouvelekis A, Herazo-Maya JD, Slade M, Chu JH, Deiuliis G, Ryu C, et al. Validation of the prognostic value of MMP-7 in idiopathic pulmonary fibrosis. Respirology. 2016 doi: 10.1111/resp.12920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Balestro E, Calabrese F, Turato G, Lunardi F, Bazzan E, Marulli G, et al. Immune inflammation and disease progression in idiopathic pulmonary fibrosis. PLoS One. 2016;11:e0154516. doi: 10.1371/journal.pone.0154516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. François A, Chatelus E, Wachsmann D, Sibilia J, Bahram S, Alsaleh G, et al. B lymphocytes and B-cell activating factor promote collagen and profibrotic markers expression by dermal fibroblasts in systemic sclerosis. Arthritis Res Ther. 2013;15:R168. doi: 10.1186/ar4352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hwang JY, Randall TD, Silva-Sanchez A. Inducible bronchus-associated lymphoid tissue: taming inflammation in the lung. Front Immunol. 2016;7:258. doi: 10.3389/fimmu.2016.00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ryu C, Sun H, Gulati M, Herazo-Maya JD, Chen Y, Osafo-Addo A, et al. Extracellular mitochondrial DNA is generated by fibroblasts and predicts death in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2017;196:1571–1581. doi: 10.1164/rccm.201612-2480OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Barratt SL, Blythe T, Jarrett C, Ourradi K, Shelley-Fraser G, Day MJ, et al. Differential expression of VEGF-axxx isoforms is critical for development of pulmonary fibrosis. Am J Respir Crit Care Med. 2017;196:479–493. doi: 10.1164/rccm.201603-0568OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Barratt SL, Flower VA, Pauling JD, Millar AB. VEGF (Vascular Endothelial Growth Factor) and fibrotic lung disease. Int J Mol Sci. 2018;19:1269. doi: 10.3390/ijms19051269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ito TK, Ishii G, Saito S, Yano K, Hoshino A, Suzuki T, et al. Degradation of soluble VEGF receptor-1 by MMP-7 allows VEGF access to endothelial cells. Blood. 2009;113:2363–2369. doi: 10.1182/blood-2008-08-172742. [DOI] [PubMed] [Google Scholar]