

Abstract
Tobacco smoking is a well-known risk factor for both fibrogenesis and fibrotic progression; however, the mechanisms behind these processes remain enigmatic. RTKs (receptor tyrosine kinases) have recently been reported to drive profibrotic phenotypes in fibroblasts during pulmonary fibrosis (PF). Using a phospho-RTK array screen, we identified the RTK AXL as a top upregulated RTK in response to smoke. Both expression and signaling activity of AXL were indeed elevated in lung fibroblasts exposed to tobacco smoke, whereas no significant change to the levels of a canonical AXL ligand, Gas6 (growth arrest–specific 6), was seen upon smoke treatment. Notably, we found that smoke-exposed human lung fibroblasts exhibited highly proliferative and invasive activities and were capable of inducing fibrotic lung lesions in mice. Conversely, genetic suppression of AXL in smoke-exposed fibroblasts cells led to suppression of AXL downstream pathways and aggressive phenotypes. We further demonstrated that AXL interacted with MARCKS (myristoylated alanine-rich C kinase substrate) and cooperated with MARCKS in regulating downstream signaling activity and fibroblast invasiveness. Pharmacological inhibition of AXL with AXL-specific inhibitor R428 showed selectivity for smoke-exposed fibroblasts. In all, our data suggest that AXL is a potential marker for smoke-associated PF and that targeting of the AXL pathway is a potential therapeutic strategy in treating tobacco smoking–related PF.
Keywords: AXL, pulmonary fibrosis, R428, tobacco smoking, fibroblasts
Clinical Relevance
This study is the first to report activation of AXL receptor tyrosine kinase in response to tobacco smoke exposure, forming a fibrogenic molecular complex with MARCKS (myristoylated alanine-rich C kinase substrate) protein to promote pulmonary fibrosis. These findings provide evidence of a novel signaling complex as well as provide potential therapeutic avenues for pulmonary fibrosis.
Pulmonary fibrosis (PF), an interstitial lung disease (ILD), is marked by progressive scarring (fibrosis) of the interstitium due to excessive extracellular matrix (ECM) deposition and proliferative fibroblasts. Of the ILDs, idiopathic pulmonary fibrosis (IPF) is the most common (1–4). There is a dearth of effective antifibrotic therapies to stop the progression of PF, with only two U.S. Food and Drug Administration–approved therapeutics, pirfenidone and nintedanib, in clinical use. Unfortunately, both drugs are limited in therapeutic efficacy as they only improve the lung function in patients, with little to no effect on overall mortality (1, 2, 5). The use of steroid drugs has also been considered for IPF and other ILDs, but the lack of large randomized controlled monotherapy trials in addition to the significant morbidity of long-term usage have limited the use of steroids in treating PF (2, 6). These circumstances highlight the need for improved therapies as well as suitable biomarkers to guide clinical treatment of IPF and other ILDs.
The fibroblasts, especially the myofibroblasts, are the prima facie cell type in the progression of PF (7–10). Under normal conditions, fibroblasts display low activity, with little ECM production and deposition as well as low proliferation, motility, and invasiveness. However, fibroblasts are activated in PF and display upregulated activities. These proliferative cells form foci and display enhanced ECM production and/or deposition, disturbing the alveolar architecture (2, 11, 12). Additionally, the migratory and invasive capabilities of fibroblasts are increased under fibrotic conditions, enabling these cells to disseminate throughout the lung and worsen fibrosis. Although there is phenotypic heterogeneity in lung fibroblasts, the differential molecular signaling between PF fibroblasts and normal fibroblasts remain relatively unknown. One area of investigation that has gathered interest in recent years is the role of RTKs (receptor tyrosine kinases) as a major modulator of fibroblast activity, differentiation, and disease progression (13). In particular, AXL, an RTK in the TAM (Tyro-3, AXL, MerTK) family of receptors, has been recently implicated in playing a major role in driving profibrotic phenotypes in lung fibroblasts and contributing to fibrotic progression (14, 15), but how AXL activity is modulated in lung fibroblasts is yet to be elucidated.
Epidemiological studies have shown readily that ever-smokers, smokers with any sort of smoking history, are consistently overrepresented in PF cases (16–22). Not only is tobacco smoking a significant risk factor for PF, tobacco smoking has also been shown to have a detrimental effect on disease progression (16, 19, 21). In animal models of PF, tobacco smoke exposure is shown to promote a profibrotic milieu in the lung and potentiate bleomycin-induced lung fibrosis (23, 24). As compared with unexposed normal fibroblasts, fibroblasts exposed to tobacco smoke display behavioral differences and become more motile as well as having an increased collagen production capability (25, 26). These active fibroblasts participate in fibrotic progression through migrating throughout the lung and depositing ECM, worsening the fibrotic lesions in the lung. Despite these findings, the mechanisms of how tobacco smoke alters lung fibroblasts to a more profibrotic phenotype are currently not well understood.
Given the role of AXL and the prevalence of tobacco smoking in PF, AXL activity may also be implicated in driving the profibrotic phenotypes of fibroblasts exposed to tobacco smoke. In this study, we elucidated the role of tobacco smoke in activating AXL signaling, driving downstream fibroblast activities and fibrotic progression.
Methods
Reagents and Antibodies
All reagents and antibodies used in this study are described in the Supplementary Methods in the data supplement.
Cell Culture and Transfection
Human primary fibroblast cells were obtained from airway tissues provided from the UC Davis Medical Hospital with consent. The protocol for human tissue procurement and usage were periodically reviewed and approved by the University Human Subject Research Review Committee. Detailed experimental procedures for establishment of cell culture and siRNA transfections are described in the Supplementary Methods in the data supplement.
Exposure of Cultured Cells to Cigarette Smoke Extract
Cultures of cells were exposed to cigarette smoke extract (CSE) using a protocol similar to that previously described (27). Detailed experimental procedure for generation of CSE is described in the Supplementary Methods in the data supplement.
Cell Proliferation, Colony Formation, and Matrigel Transwell Invasion Assays
The biofunctional assays were performed as previously described (28–30) and detailed experimental procedures are described in the Supplementary Methods in the data supplement.
In Vivo Orthotopic Lung Implementation and Bleomycin-induced Lung Fibrosis
The lung fibrosis model by adoptive transfer of human pulmonary fibroblasts to severe combined immunodeficiency mice was modified from an established murine models of lung fibrosis and cancer (31–33). For the bleomycin-induced lung fibrosis model, C57BL/6J mice received saline or bleomycin intratracheally as previously described (30). Detailed experimental procedures are described in the Supplementary Methods in the data supplement. The present animal study has been approved by the Institutional Animal Care and Use Committees at UC Davis.
Human Phospho-RTK Array
The Human Phospho-RTK Array Kit (#ARY001B) was purchased from R&D Systems. We performed array screening according to the manufacturer’s protocol. The detailed experimental procedure is described in the Supplementary Methods in the data supplement.
Western Blot Analyses, IP, Quantitative Real-Time PCR, and ELISA
Detailed procedures for Western blots, IP, qRT-PCR, and ELISA assays are described in the Supplementary Methods in the data supplement.
Mouse Model of Cigarette Smoke Exposure and Immunofluorescent Staining
The paraffin-embedded specimens of mice exposed to filtered air (control) and environmental tobacco smoke were kindly provided by Dr. Kent E. Pinkerton (Center for Health and the Environment, UC Davis). Detailed procedures for the animal model of smoke exposure are described in the Supplementary Methods in the data supplement.
Statistical Analysis
Data analysis procedures are described in the Supplementary Methods in the data supplement.
Results
Smoke Exposure Potentiates Aggressive Phenotypes in Lung Fibroblasts
Severe lung fibrosis requires activated fibroblast phenotype characteristics such as increased proliferation and invasion (11). Given the profibrotic roles of tobacco smoke (23–26), we first characterized the influence of smoke exposure on the phenotypes of lung fibroblasts. Three normal lung fibroblast cell lines (Normal-1, -2, and -3) derived from nonsmoker donors, two normal lung fibroblast cell lines (Smoking Normal-1 and -2) derived from smokers without PF, and three lung fibroblasts (PF-1, -2, and -3) isolated from smokers with PF, as described previously (30), were subjected to colony formation and Matrigel transwell invasion assays, respectively. We observed stronger colony-forming ability (Figure 1A) and higher invasiveness (Figure 1B) in the two smoking normal (Smoking Normal-1 and -2) as well as further elevated activities in the three PF fibroblasts (PF-1, -2, and -3) as compared with normal lung fibroblasts. Furthermore, the fibroblastic foci lesions characterized by accumulation of α-smooth muscle actin (α-SMA)–positive fibroblasts were apparent in lung tissue from smokers with PF (Figure E1A in the data supplement). Patient information with pathological findings is included in Table E1. To determine that primary PF lung fibroblasts exhibit aggressiveness and unfavorable evolution in lung fibrosis, we performed a lung fibrosis model by adoptive transfer of primary human pulmonary fibroblasts to severe combined immunodeficiency mice that is modified from an established murine model of PF (31). Fibroblast cells were orthotopically inoculated into the left lobe of mice lung as we previously reported (28, 29), and we collected the lungs for histology and Masson's trichrome staining 60 days after inoculation. The lung architecture of mice inoculated with normal fibroblasts appeared normal, whereas mice bearing smoke-exposed PF fibroblasts showed disrupted architecture and extensive structural changes in the lungs, concomitant with fibroblastic lesions and deposited extracellular matrix (Figure 1C), implying that smoke-exposed PF fibroblasts were active in persisting, proliferating, and continuing collagen production and ECM deposition compared with normal fibroblasts.
Figure 1.
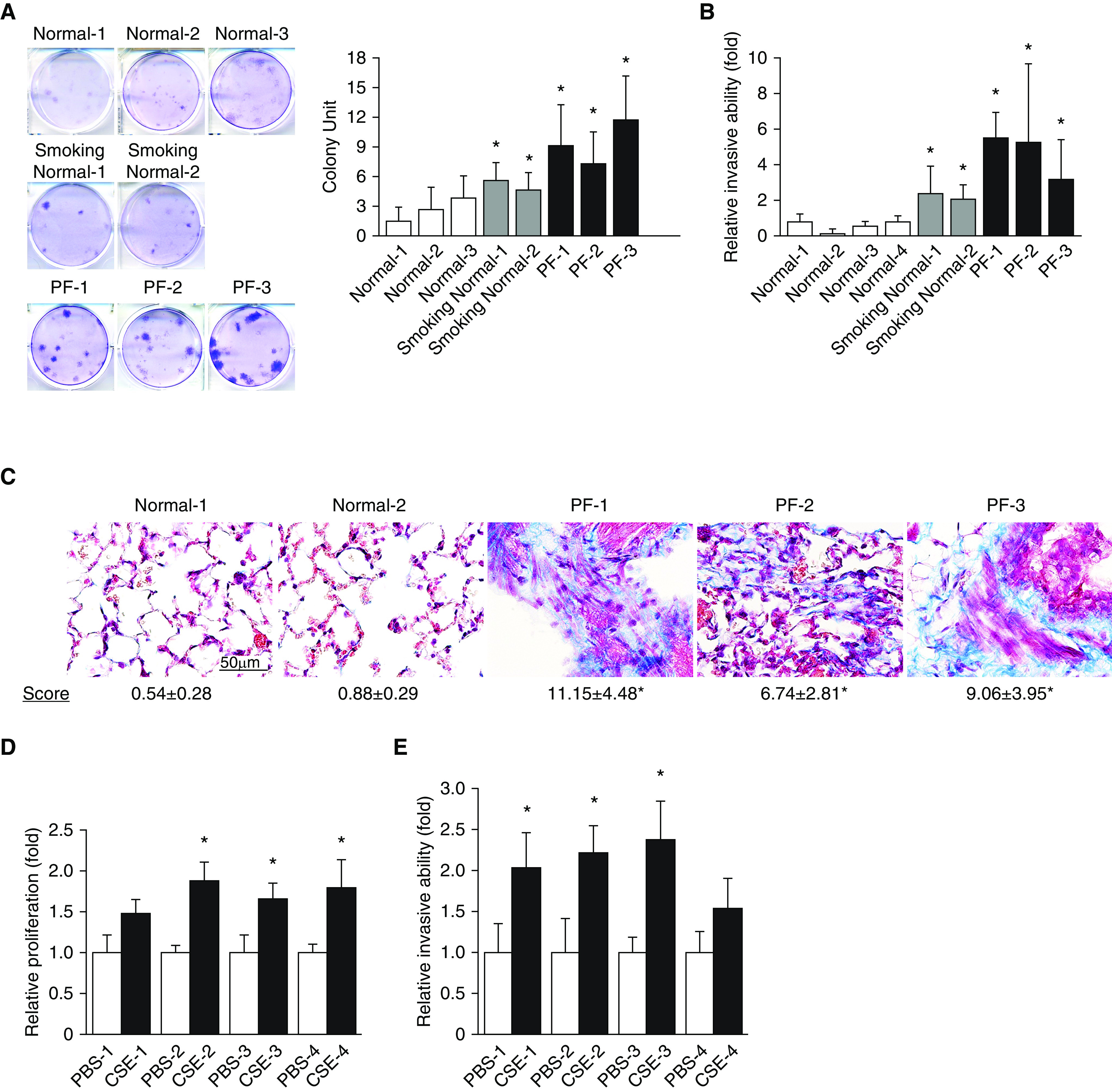
Aggressive nature of smoke-exposed fibroblasts in lung fibrosis. (A) Cells were seeded to grow for 9 days, and cell colonies were counted after crystal violet staining. Left, representative images of three independent experiments. Right, colony units expressed as means ± SD (n = 5). *P < 0.05 compared with Normal-1. (B) Comparison of invasion ability between normal, smoking normal, and PF cells as determined by Matrigel transwell invasion assays. Cells were plated on Transwells for 20 hours, and those that migrated to the lower chamber were fixed, stained, and counted using light microscopy. Data are expressed as means ± SD (n = 4). *P < 0.05 compared with Normal-1. (C) Representative Masson trichrome–stained sections of mouse lung implanted with indicated fibroblasts. Bottom, semiquantitative fibrosis scores from Masson trichrome–stained sections of mouse lung. Fibrosis score is expressed as the percentage of positive staining area per high-powered field. Analysis of 6–12 high-powered fields per lung was performed with ImageJ software. *P < 0.05. Scale bar, 50 μm. (D) Cells were seeded to grow for 72 hours and then subjected to MTS proliferation assays. *P < 0.05 compared with PBS-exposed counterpart. (E) Comparison of invasion capability between PBS- and CSE-exposed cells as determined by Matrigel transwell invasion assays. Data are expressed as means ± SD (n = 4). *P < 0.05 compared with PBS-exposed counterpart. CSE = cigarette smoke extract; PF = pulmonary fibrosis.
To elucidate whether PF fibroblasts are directly activated by smoke exposure, we selected apoptosis-resistant and proliferative populations in normal human fibroblasts receiving long-term CSE exposure; therefore, several CSE-exposed (or CSE-resistant) fibroblasts had been generated after exposure to 20% CSE for 4 weeks. We observed that the proliferative and invasive ability of the four CSE-treated fibroblasts is enhanced as compared with that of their PBS-treated control counterparts (Figures 1D and 1E). As such, exposure to compounds in cigarette smoke induced fibroblast activation and aggressiveness to promote PF, and the disease process could be halted if the aggressive nature of PF fibroblasts can be attenuated.
AXL Signalsomes Are Activated in Response to Smoke
Recent studies have demonstrated that RTKs can be major drivers influencing fibroblast proliferation and myofibroblast differentiation, contributing to fibrosis progression (13). To identify druggable smoke-associated signaling molecules to reduce fibroblast activation and differentiation, we therefore performed a phospho-RTK antibody array screen, which includes numerous kinases with available specific inhibitors. After comparing the array signal intensities between Normal-3 and PF-3 fibroblasts (Figure 2A) or between PBS-2 and CSE-2 fibroblasts (Figure 2B), we identified four top-ranked RTKs with upregulated tyrosine phosphorylation in response to tobacco smoke (Figure 2C). Of these RTKs, AXL receptor is the first to draw our attention given recent reports showing the importance of AXL activation in IPF lung fibroblasts (14, 15), and our observation of its activity being upregulated in two different conditions of smoke exposure.
Figure 2.

AXL and its signaling pathway are activated in smoke-associated lung fibroblasts. (A and B) Cell lysates from primary lung fibroblasts, Normal-3 and PF-3 cells (A), and selected cell lines, PBS-2 and CSE-2 cells (B), were subjected to human phospho-receptor tyrosine kinase (RTK) arrays. (C) Top four upregulated phospho-RTKs identified from RTK arrays in A and B. PF: PF-3, N: Normal-3, C: CSE-2, P: PBS-2. (D) Western blot analysis of phospho-AXL and downstream phospho-STAT3 and phospho-AKT in primary normal lung fibroblasts (Normal-1, -2, and -3) and primary lung fibroblasts isolated from smokers with pulmonary fibrosis (PF-1, -2, and -3). (E) Expression of AXL protein and its phosphorylation in PBS- and CSE-exposed lung fibroblast cells were determined by IB. HGFR = MET proto-oncogene receptor tyrosine kinase; IGF1R = insulin like growth factor 1 receptor; PDGFR = platelet derived growth factor receptor; RYK = receptor like tyrosine kinase; STAT = signal transducer and activator of transcription.
AXL expression and activation was demonstrated to be elevated in IPF and drove IPF fibroblast proliferation, migration, and differentiation (14, 15). Additionally, downstream pathways of AXL such as the PI3K/AKT (protein kinase B) and JAK (Janus kinase)/STAT (signal transducer and activator of transcription) pathways have also been shown to be upregulated in IPF (34, 35). To determine the significance of AXL in the context of smoke exposure, we next validated the results of the RTK array screen. As shown in Figures 2D and 2E, exposure to cigarette smoke elevated phospho-AXL levels in all tested primary PF series of lung fibroblast cell lines and CSE-exposed lung fibroblasts compared with those primary normal and PBS-treated fibroblasts, respectively. This is accompanied by an increase of AXL-mediated phosphorylation of STAT3 and AKT. In addition, through the analysis of the transcriptome data set GSE3320 (36), we found that several genes involved in AXL-mediated PI3K-AKT and JAK–STAT pathways are upregulated in lung cells from smokers (Figure 3), suggesting that AXL is a smoke-responsive molecule and may drive the aggressive nature of lung fibroblasts in lung fibrosis.
Figure 3.

AXL pathway–associated upregulation of gene expression in lung cells from smokers. (A) Significant increases in the expression of genes related to AXL-mediated PI3K–AKT and JAK–STAT pathways were observed in lung cells from smokers versus nonsmokers by analysis of the GSE3320 data set. JAK = Janus kinase.
AXL Sustains Smoke-induced Signaling and Aggressiveness in Fibroblasts
The canonical activation pathway of AXL receptor is through the binding with its ligand, Gas6 (growth arrest–specific 6) (37). Thus, we investigated whether a change in Gas6 expression, the major ligand for AXL activation, is driving smoke-induced upregulation of AXL activity. Surprisingly, data from analysis of the two data sets (GDS534 and GDS3494) (38, 39) have shown no significant increase of Gas6 levels from current and former smokers compared with never-smokers, with a slight decrease observed instead (Figure 4A). Furthermore, our mRNA data from primary lung fibroblast cells and from cells treated with CSE also showed similar levels of Gas6 (Figure 4B). We further evaluated the secreted protein level of Gas6 and observed no significant changes between normal and smoking normal lung fibroblast cells as well as mostly no remarkable changes in our control and CSE-treated cells (Figure 4C). To evaluate the potential role of soluble AXL (sAXL) in modulating Gas6 activity, we also assessed the levels of sAXL in primary lung fibroblasts and in CSE-treated cells. Data from ELISA assays confirmed similar levels of sAXL in all primary normal cells as well as in CSE-treated cells (Figure 4D). These taken together suggest that Gas6 may play only a minor role in smoke-modulated AXL activation and indicate the possibility of a ligand (Gas6)-independent event for AXL activation upon smoke exposure.
Figure 4.
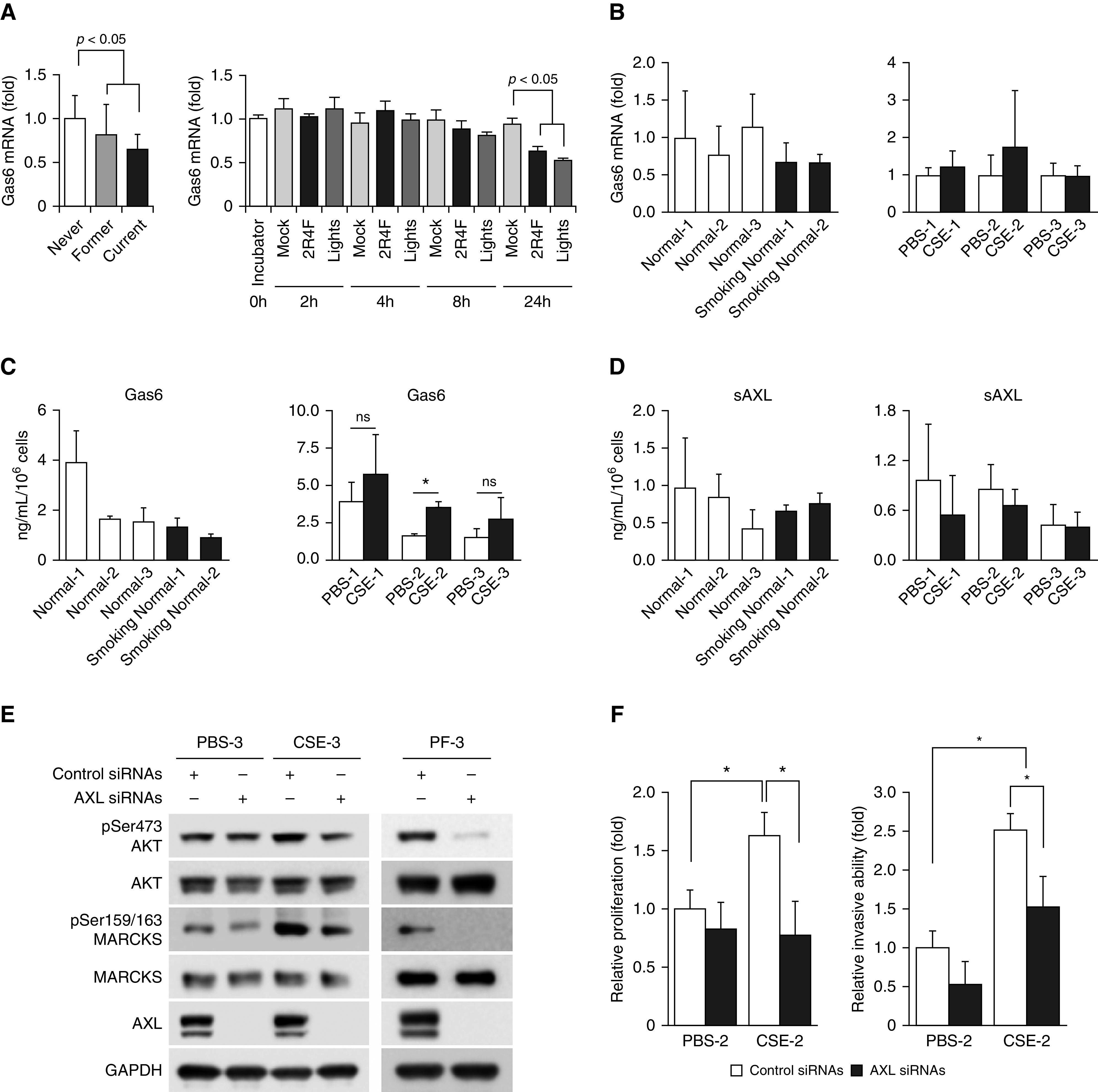
Knockdown of AXL attenuates AXL-mediated signaling molecules. (A) Left, normalized mRNA expression of Gas6 in lung tissues from smokers (former and current) versus nonsmokers (never) using the GDS534 data set. Right, Gas6 mRNA expression over time in response to tobacco smoke exposure (2R4F and Lights cigarettes) using the GDS3494 data set. (B) Real-time qRT-PCR analyses of Gas6 mRNA expression in normal and smoking normal primary fibroblasts (left) and in PBS- and smoke-treated (CSE) fibroblasts (right). Data are expressed as means ± SD (n = 3). (C) Left, secreted Gas6 levels from normal and smoking normal primary fibroblasts. Right, secreted Gas6 levels from PBS- and smoke-treated fibroblasts. Data are expressed as means ± SEM; *P < 0.05 versus PBS (n = 3). (D) Left, secreted sAXL levels from normal and smoking normal primary fibroblasts. Right, secreted sAXL levels from PBS- and smoke-treated fibroblasts. Data are expressed as means ± SEM (n = 3). (E and F) Genetic knockdown of AXL to downregulate AXL levels in cells. Multiple lung fibroblast cells as indicated were treated with nonspecific (control siRNAs) or AXL-specific siRNAs (AXL siRNAs); after 72 hours of transfection, cells were subjected to Western blot analyses (E), and two biofunctional assays (F), MTS proliferation assays (left) and Matrigel transwell invasion assays (right), respectively. Data are shown as mean ± SD; *P < 0.05 versus control siRNA (n = 4). Gas6 = growth arrest–specific 6; MARCKS = myristoylated alanine-rich C kinase substrate; sAXL = soluble AXL.
To determine if blockade of AXL activity inhibits smoke-induced signaling and aggressiveness in fibroblasts, we used an AXL-specific siRNA (AXL siRNA) to deplete endogenous AXL. We found that AXL knockdown suppressed the activities of several signaling molecules including AKT and MARCKS (myristoylated alanine-rich C kinase substrate), and this suppressive effect was more evident in the context of smoke exposure (Figure 4E). To confirm the contribution of AXL to fibroblast activation, PBS- and CSE-treated fibroblasts were transfected with control or AXL siRNAs and then subjected to in vitro biofunctional assays. As shown in Figure 4F (left), the proliferation ability of CSE-exposed fibroblasts after AXL silencing is attenuated to a level comparable with that of PBS-treated fibroblasts. Moreover, knockdown of AXL also decreased the number of invaded CSE-exposed fibroblasts (Figure 4F, right) in the transwell invasion assay. Therefore, a reversal or halt of fibrotic processes could potentially be achieved through AXL inhibition.
Smoke Stimulation Drives the Activation of the AXL–MARCKS Molecular Complex
The above results demonstrated the importance of AXL signaling in smoke-exposed fibroblast cells, so we next interrogated the molecular mechanism of how AXL activity is regulated in lung fibroblasts. We previously identified the signal molecule MARCKS as a druggable target in PF (30), and our current work had confirmed a positive association between phospho-MARCKS and AXL abundance (Figure 4E). Based on these observations, we presume that AXL cooperates with MARCKS to sustain activated phenotypes of fibroblasts in response to cigarette smoke. In a fibrosis mouse model, immunofluorescence data showed coexpression of AXL and MARCKS in α-SMA, a myofibroblast marker, positive cells (Figure 5A). Through analysis of the levels of phospho-AXL and phospho-MARCKS in a cigarette smoking mouse model, we demonstrated elevated expression of phospho-AXL and phospho-MARCKS in lung tissues with exposure to side-stream cigarette smoke (Figure 5B). The colocalization of phospho-AXL and phospho-MARCKS upon tobacco smoke exposure is also corroborated by co-IP analysis in which an interaction between AXL and phospho-MARCKS is enhanced in CSE-exposed fibroblasts (Figure 5C), indicating AXL–MARCKS as a smoke-specific molecular complex.
Figure 5.
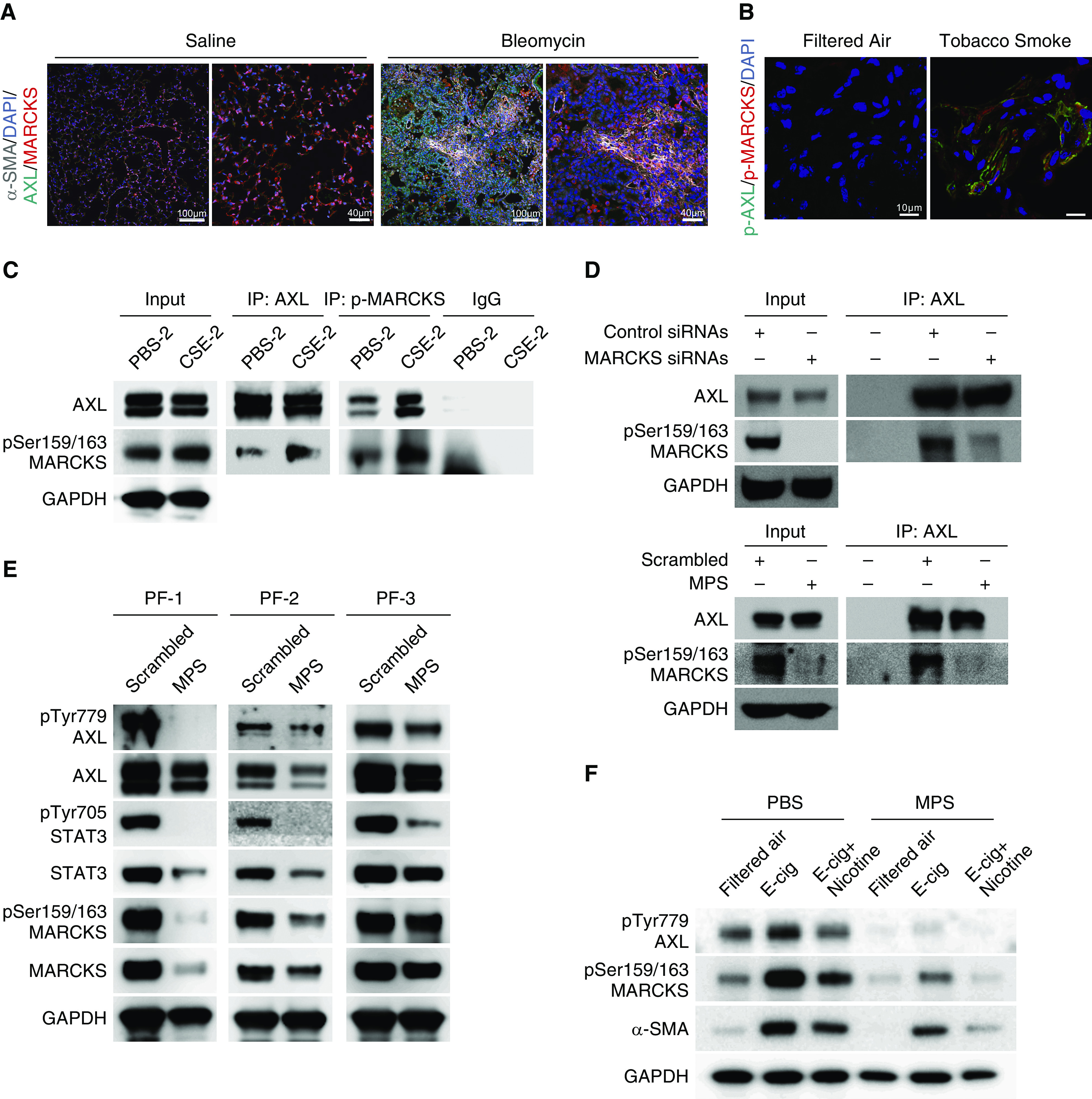
AXL is associated with MARCKS in activated lung fibroblasts. (A) Representative immunofluorescence images of α-SMA (gray color), AXL (green color), and MARCKS (red color) in lung tissues from mice exposed to saline or bleomycin. DAPI (blue color): nuclear stains. Scale bars, 40 μm and 100 μm. (B) Representative immunofluorescence images of phospho-AXL (green color) and phospho-MARCKS (red color) in lung tissues from mice with exposure to filtered air or tobacco smoke. DAPI (blue color): nucleus stains. Scale bars, 10 μm. (C) IP analysis of the association between endogenous AXL and phospho-MARCKS in PBS- and CSE-exposed cells. (D) Determination of the interaction between AXL and MARCKS in highly MARCKS-expressing A549 cells treated with nontargeting control siRNAs or MARCKS-specific siRNAs (top) or scrambled control peptide or MPS peptide (bottom) upon CSE exposure by co-IP assays. (E) Examination of phospho-AXL, phospho-MARCKS. and phospho-STAT3 levels in primary pulmonary fibrosis fibroblasts after incubation with PBS or MPS peptide (100 μM) for 48 hours by IB. (F) Effect of MPS treatment on phospho-AXL, phospho-MARCKS, and α-SMA levels in cells exposed to E-cig or E-cig plus nicotine analyzed by IB. α-SMA = α-smooth muscle actin; E-cig = electronic cigarette.
To test whether MARCKS inhibition can disrupt the AXL–MARCKS complex, our previously developed cell-permeable peptide, the MPS peptide, which targeted the MARCKS phosphorylation site domain (PSD) and inhibited MARCKS phosphorylation in cancers (29), and siRNAs targeting MARCKS were used to treat cells. Co-IP assays confirmed an interaction of AXL with MARCKS, and this interaction is decreased in MARCKS-knockdown cells (Figure 5D, top) as well as in MPS-treated cells (Figures 5D, bottom, and E2A). In addition to the disruption of complex formation, inhibiting phospho-MARCKS by treatment with MPS peptide or MARCKS-specific siRNAs downregulated AXL activity and its signaling in all tested primary PF fibroblasts (Figures 5E and E2B). We also treated human normal fibroblast cells with nicotine-containing and nicotine-free vapor extract from electronic cigarettes (e-cigarettes or E-cig) as shown in Figure 5F. An upregulation of phospho-AXL and phospho-MARCKS and a myofibroblast marker, α-SMA, was observed in the presence of E-cig vapors, whereas these molecules were attenuated by treatment with MPS peptide. Thus, cigarette smoke stimulates fibroblast activation, contributing to lung fibrosis progression through AXL–MARCKS signaling.
Targeting AXL Signaling Selectively Impairs the Viability of Smoke-exposed Fibroblasts
Owing to the determinant role of AXL signaling in the aggressive nature of all tested primary PF fibroblasts, targeting AXL activity with the selective inhibitor of AXL bemcentinib (R428), a small molecule kinase inhibitor undergoing phase II trials in treating various tumor types (40), may retard the fibrosis progression. MTT assays have confirmed that R428 treatment was more effective in decreasing cell viability of CSE-exposed fibroblasts, as compared with the treatment of PBS-treated fibroblasts (Figure 6A). Moreover, the half maximal inhibitory concentration of all tested smoke-exposed cells (CSE-1 to CSE-4) and primary PF fibroblasts (PF-1 to PF-3) was significantly lower than the half maximal inhibitory concentration of PBS-treated cells (PBS-1 to PBS-4) and primary normal fibroblasts (Figures 6B and 6C). Treatment with R428 for 24 hours also reduced the activity of AXL-downstream signaling molecules including AKT, Erk1/2, and STAT3 as well as the profibrotic marker α-SMA in smoke-exposed cells (Figure 6D), supporting the notion that AXL inhibition impairs the aggressiveness of lung fibroblasts. In view of an increase in AXL activity after exposure to smoke, we treated normal fibroblasts and smoke-exposed fibroblast cells with R428. Surprisingly, smoke-exposed cells displayed lower cell viability upon R428 treatment as compared with control cells without smoke exposure. Of note, cells receiving long-term smoke exposure (21 d) are more sensitive to this inhibitor (Figure 6E). This finding suggests that in smoke-exposed cells, there is selection for an apoptosis-resistant and more proliferative population, and these cells become addicted to AXL activity. Thus, AXL signaling may be a driver pathway supporting fibroblast cell survival and proliferation in response to tobacco smoke.
Figure 6.
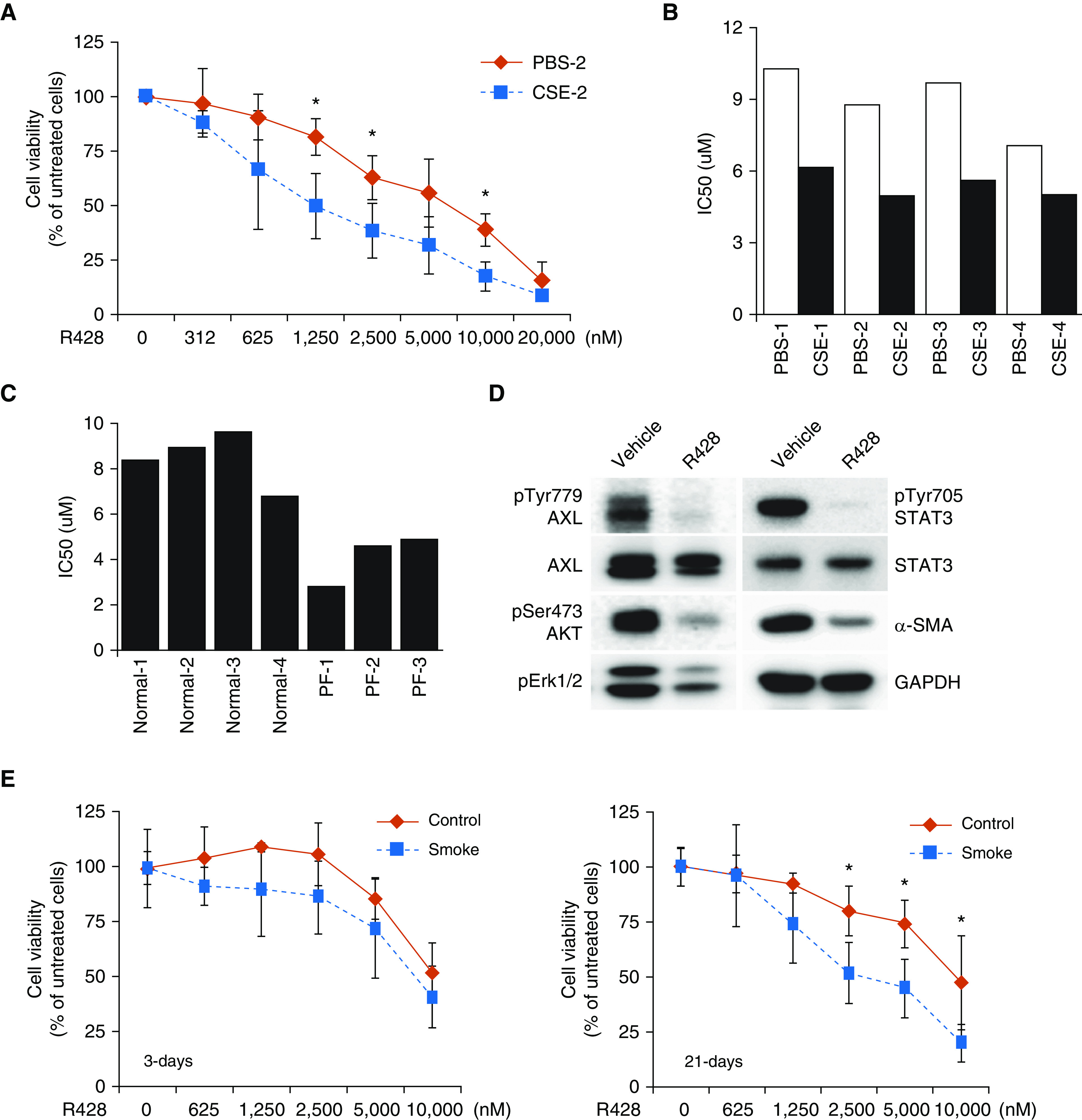
Pharmacologic inhibition of AXL by R428 treatment impairs lung fibroblast cell viability. (A) Cells were incubated with various concentrations of R428 (bemcentinib) for 72 hours and then subjected to MTT assays (n = 4). Data are expressed as means ± SD; *P < 0.05 versus PBS-2. (B and C) Lung fibroblasts were exposed to various concentrations of R428 as indicated above. After 72 hours of treatment, cell viability was determined by MTT assays. The IC50 value of R428 for all tested lung fibroblast cells was shown (B and C). (D) Smoke-exposed cells (CSE-2) were treated with 2.5 μM R428 for 24 hours and then subjected to Western blot analysis. (E) R428 treatment in cells with exposure to short-term and long-term CSE. Cells were exposed to PBS (control) or 10% CSE (smoke) for 3 days (left) or 21 days (right). These cells were subjected to the indicated doses of R428. After 72 hours of treatment, cell viability was determined by MTT assays. Data are expressed as means ± SD; *P < 0.05 versus control (n = 3). IC50 = half maximal inhibitory concentration.
Pharmacological Targeting of AXL Signaling Attenuates Experimental Smoke-related Pulmonary Fibrosis
Given the role of AXL signaling in promoting profibrotic phenotypes in response to tobacco smoke, we next evaluated the efficacy of AXL targeting in an experimental orthotopic model of PF. Briefly, immunodeficient mice were orthotopically injected with lung fibroblasts cells (PF-1 and -2) into both left and right lung lobes. After 4 weeks, mice were treated with either PBS or 10 mg/kg R428 for an additional 4 weeks. Histologically, R428-treated mice demonstrated reduced fibrosis compared with PBS control mice (Figure 7A). Additionally, we also stained for human-specific vimentin to evaluate the survival and dissemination of injected primary fibroblasts and demonstrated that R428-treated mice had reduced amounts of surviving fibroblasts compared with PBS-treated mice (Figure 7B). Lastly, R428-treated mice displayed overall decreased collagen content (Figure 7C) compared with controls. In summation, these demonstrate that targeting AXL activity can attenuate overall fibrosis in our experimental mouse fibrosis model.
Figure 7.
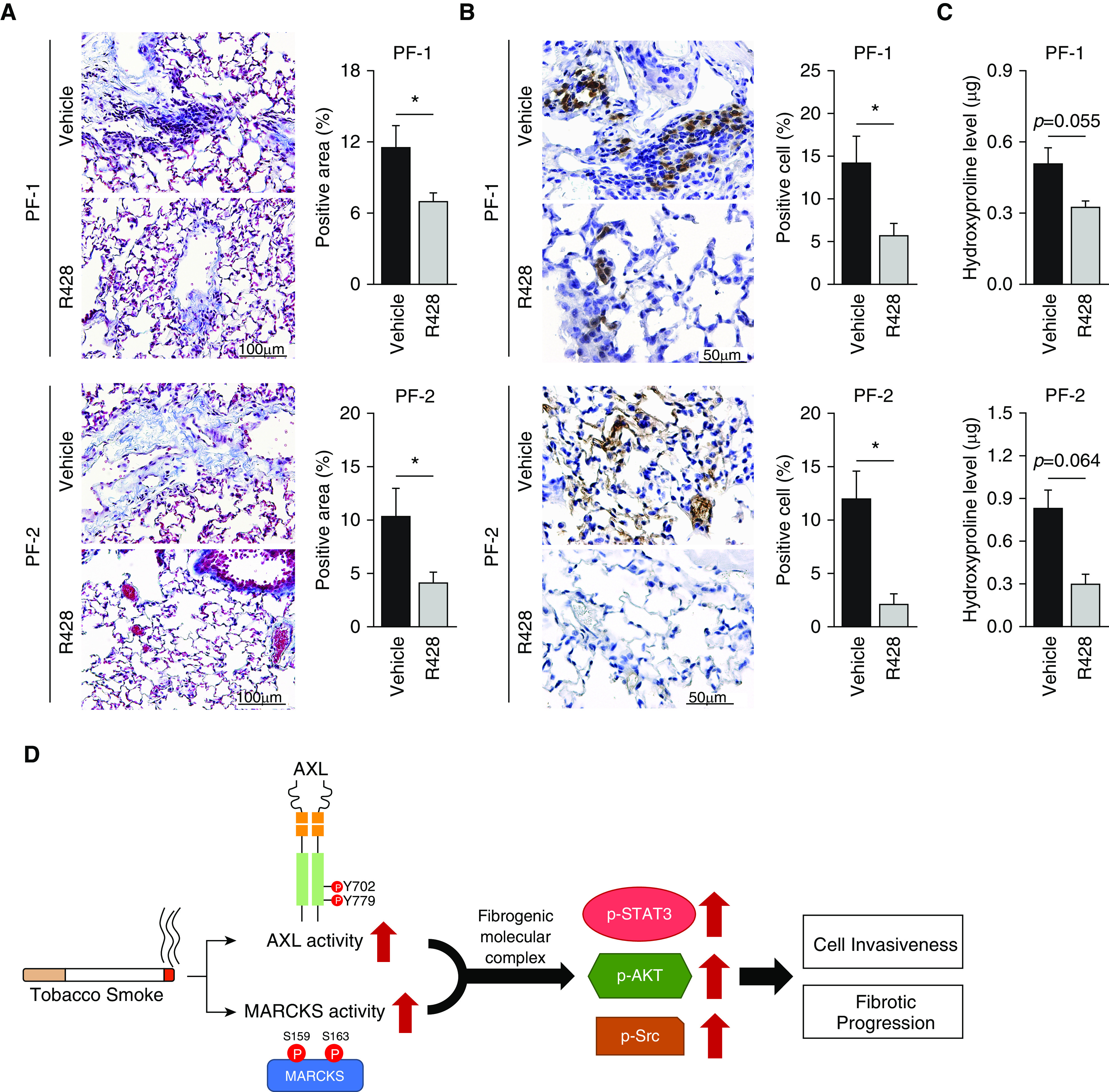
R428 reduces fibrosis in an experimental orthotopic model of pulmonary fibrosis. (A and C) Immunodeficient mice were orthotopically injected with 2.5 × 106 lung fibroblasts cells (PF-1 and -2) in Matrigel into both left and right lung lobes. After 4 weeks, mice were treated with 10 mg/kg AXL-specific inhibitor, R428, or PBS as a control for an additional 4 weeks (eight mice per PF-1 group and six mice per PF-2 group). (A) Representative images of Masson trichrome–stained sections of mouse lung with R428 or PBS treatment. Fibrosis scoring is expressed as the percentage of positive staining area per high-powered field. Analysis of 6–12 high-powered fields per lung was performed with ImageJ software. Data are expressed as means ± SEM; *P < 0.05. Scale bars, 100 μm. (B) Representative immunohistochemistry staining of human vimentin. Dissemination of fibroblast cells is expressed as percentage of positive cells per high-powered field. Analysis of 6–12 high-powered fields per lung was performed with ImageJ software. Data are expressed as means ± SEM; *P < 0.05. Scale bars, 50 μm. (C) Hydroxyproline level in the right lung of mice as described above was determined by a hydroxyproline ELISA assay. Data are expressed as means ± SEM; *P < 0.05. (D) MARCKS and AXL activity (p-MARCKS and p-AXL, respectively) is activated in fibroblast cells in response to tobacco smoke. Activation of these molecules form a fibrogenic molecular complex that propagates downstream signals (p-STAT3, p-AKT, and p-Src) driving cell invasiveness and fibrotic progression.
Discussion
Despite the breadth of work in PF, especially IPF, in the past few decades, the mechanisms driving fibrotic progression are still not completely understood and there is a lack of effective therapies and biomarkers for the disease for patients. Smoking has long been recognized as a significant risk factor for PF and plays a nontrivial role in propagating fibrosis, but the mechanisms have not yet fully explored (16–26, 41–43). Herein, we had identified a novel fibrogenic complex, the AXL–MARCKS molecular complex, where tobacco smoke–activated AXL promotes PF progression (Figure 7D). Through our studies, we had identified AXL as a critical RTK driving fibrotic activities in response to tobacco smoke exposure.
The fibroblasts residing in the histopathologic lesions of PF are characterized by increased ECM production and deposition, increased proliferative ability, and invasive capability (25, 26). Indeed, compared with fibroblasts isolated from normal lungs, fibroblast cells isolated from smoke-associated PF lungs display an “activated” phenotype as demonstrated by the increased colony formation and invasive abilities. Additionally, to investigate the profibrotic activities of these cells, we used an adoptive transfer model wherein we transferred PF and non-PF fibroblast cells into mouse lungs. PF fibroblasts displayed a marked capability to form pathological lesions of active fibroblasts and ECM deposition, resulting in altered lung architecture. In mice injected with non-PF cells, the fibroblasts did not form fibrotic lesions and little to no ECM deposition was noted. These in vivo results are consistent with the above in vitro findings and support the profibrotic role of tobacco smoking.
Tobacco smoke is an amalgamation of thousands of different compounds, many of them being toxicants. Studies have long implicated the adverse effects of tobacco smoking on the lung, with effects ranging from repeated alveolar damage to induction of inflammation as well as promoting proliferation and activation of lung cells. Tobacco smoking has been implicated as the causative factor in multiple ILDs such as respiratory bronchiolitis ILD, desquamative interstitial pneumonia, and smoking-related interstitial fibrosis (44). Clinical studies have indicated that smoking is an independent risk factor for IPF and is associated with a worse outcome in patients with IPF (16, 19, 21). Both in vitro and in vivo reports have also supported the concept of tobacco smoking in promoting lung fibrosis (23, 41, 42). In our study, we determined, for the first time to the best of our knowledge, the influence of tobacco smoke on lung fibroblasts through generation of CSE-resistant lung fibroblast cells. We have found that these cells demonstrate upregulated cell proliferation, invasion, and ECM deposition activities. Our findings fall in line with previous studies and support the concept that tobacco smoking can promote fibrosis through modulating fibroblast activities.
RTKs have been recently demonstrated to be major driving signaling molecules influencing fibroblast activities such as proliferation and differentiation, contributing to progression of fibrosis (13). In particular, AXL, a receptor in the TAM family of receptors, has recently been demonstrated to play a major role in IPF lung fibroblasts (14, 15). Although this contributes greatly to the understanding of the mechanisms of how IPF is driven, how AXL activity is modulated by tobacco smoking was left unexplored. To address this, we compared both primary normal fibroblasts and PF fibroblasts (isolated from patients with smoking history) as well as CSE-exposed (or CSE-resistant) fibroblast cells. By performing molecular analysis and biofunctional assays, we identified that both AXL activity and its signaling pathways are indeed elevated in response to tobacco smoke, potentially driving the profibrotic phenotype in fibroblast cells in PF.
AXL activation is canonically mediated by ligand binding and subsequent dimerization resulting in downstream phosphorylation and activation (45). The previous report of AXL’s role in IPF suggested that the AXL activation was due to an increase of its ligand Gas6, the prototypic ligand of AXL receptor (14). We therefore questioned whether the AXL activity in response to tobacco smoke was driven by Gas6. In light of the observations in both transcriptomic data sets and our CSE-exposed cells, we reason that this was not the case as Gas6 transcriptional levels were not significantly changed in the context of smoke. At the secreted protein levels, secreted Gas6 was generally also not significantly altered in CSE-exposed cells as well as in primary normal fibroblast cells isolated from individuals with smoking history (Figure 4C). Lastly, we also evaluated the levels of sAXL, a decoy receptor for Gas6 that can sequester Gas6 to prevent binding to cell surface AXL, to rule out its contribution in fibroblasts exposed to tobacco smoke. We found that sAXL levels were not appreciably altered in primary normal cells from individuals with smoking history or in CSE-exposed cells as compared with respective control cells. These taken together indicate that an increase of AXL activity in fibroblasts in response to smoke is likely not primarily driven by Gas6 and may instead be mediated through ligand (Gas6)-independent mechanisms. Several studies have found that AXL can be activated through ligand-independent mechanisms. For example, AXL overexpression can drive cell aggregation through binding of homophilic binding domains, leading to receptor activation. AXL homodimerization without ligand binding has also been demonstrated (45). Additionally, AXL has been shown to heterodimerize with other TAM and non-TAM receptors (45, 46). Although many factors have been proposed to drive ligand-independent activation of AXL from overexpression of AXL to reactive oxygen species, Gas6-indpendent activation of AXL remains very much unexplored.
We further inquired whether other receptors from the TAM receptor family were also altered in response to tobacco smoke, but we did not observe any appreciable changes in Tyro3 or MerTK phosphorylation levels in our RTK screening of our sample set (Figures 2A and 2B). Previous reports have indicated that Tyro3 is upregulated in IPF (14) and plays a role in modulating PF. Given that tobacco smoking is a significant risk factor for IPF (16–22, 47) and many patients have prior smoking history, it would not be illogical for tobacco smoke to influence Tyro3 levels and/or function. However, we did not observe changes in Tyro3 in our samples, perhaps indicating that Tyro3 upregulation is unique to IPF or that tobacco smoke in insufficient to drive changes in Tyro3 levels and activity.
Although we did observe increased AXL expression in some of the CSE-exposed cells and primary PF cells, the higher expression of total AXL compared with control and normal cells, respectively, was not consistent throughout and varied by cell or clone. However, the increase in phospho-AXL was consistent in both CSE-exposed cells and PF cells. This suggests that although cell aggregation or AXL homodimerization due to an increase of AXL abundance is a possibility, there are likely other mechanisms at play. Tobacco smoke exposure was shown to activate receptors independent of ligand. This has been demonstrated in the EGFR (epidermal growth factor receptor) (48), and a similar phenomenon may be at play in AXL. The role of Gas6 also cannot be completely ruled out as cells still express an endogenous amount of Gas6 and other cell types in the lung parenchyma may also be contributors of Gas6. However, in the context of smoke-exposed fibroblasts and PF fibroblasts and in reflection of our findings, the ligand-independent activation of AXL by tobacco smoke is a potential mechanism driving AXL activity.
We had previously demonstrated the role of MARCKS, a master regulator of the PI3K/AKT pathway, in modulating fibroblast activities such as invasiveness in IPF fibroblasts (30). Because the PI3K/AKT pathway is the major signaling downstream to AXL activation, we questioned whether the signal molecule MARCKS also played a role in tobacco smoke–mediated fibroblast invasiveness and AXL signaling. As we expected, genetic knockdown of AXL reduced MARCKS signaling (Figure 4E). We next found coexpression of AXL and MARCKS in α-SMA–positive cells in bleomycin-treated mouse lungs (Figure 5A). Furthermore, we identified that phospho-AXL and phospho-MARCKS are coexpressed into smoke-treated mouse lung (Figure 5B), indicating that there may be an interaction between the two molecules. Protein interaction between AXL and MARCKS was demonstrated through reciprocal IP (Figure 5C), and this interaction was disrupted by both MARCKS or AXL targeting (Figure 5D). Surprisingly, targeting of MARCKS activity through a MARCKS inhibitor, MPS peptide, also decreased AXL phosphorylation. Targeting of either MARCKS or AXL demonstrated downregulation of phosphorylation of both proteins as well as attenuated downstream signaling (Figures 4E, 5E, and 6D). Taken as a whole, this indicates that MARCKS and AXL form a molecular complex in response to tobacco smoke that drives downstream signaling, ultimately driving a profibrotic phenotype in smoke-exposed cells. Currently, it is unclear if there is a distinct hierarchy in the signaling activities of MARCKS and AXL. There is a need to identify the exact interaction sites between MARCKS and AXL. Because MPS targets the phosphorylation of MARCKS PSD and was able to disrupt the interaction between MARCKS and AXL, it would not be surprising to identify the AXL interaction site in the PSD of MARCKS. This is, however, beyond the scope of this study but requires further investigation.
In addition to tobacco smoke, AXL activity was also increased in the presence of e-cigarette smoke. E-cigarettes operate through vaporizing a liquid carrier containing nicotine and other compounds and have been shown to have some similar effects as tobacco smoke (49). We observed an increase in MARCKS and AXL activity in response to this vapor, suggesting that a similar mechanism may be at play in e-cigarette smoke exposure. Given that tobacco smoke upregulated AXL signaling to activate fibroblasts, we further explored if we could target these activated fibroblasts through the inhibition of AXL activity. Pharmacological studies show the selectivity of an AXL-specific inhibitor, R428 (bemcentinib), in fibroblasts exposed to tobacco smoke, especially long-term smoke exposure (21 days or longer), whereas no significant sensitizing effect was noted in normal fibroblasts treated with R428. These findings suggest that there may be a shift to dependence on the AXL signaling pathway as exposure to tobacco smoke continues. Assessment of the efficacy of AXL inhibition in attenuating smoke-related lung fibrosis in mice was also explored and the results support our notion that targeting AXL with the inhibitor R428 was effective in reducing overall fibrosis (Figure 7). In addition, we have found that the MARCKS–AXL signaling complex is activated in response to tobacco smoke and that targeting this pathway is a potential therapeutic avenue. As we observe that AXL activity is upregulated in PF fibroblasts from patients with a smoking history, both AXL expression and activity could potentially serve as a biomarker for smoking-associated PF. This marker could assist in predicting the fibroblast activity in patients and may help inform therapeutic choices.
In summary, we demonstrate that tobacco smoking is able to modulate profibrotic phenotypes in fibroblasts and promote fibrotic progression. Our findings suggest that AXL cooperatively interacts with activated MARCKS to form a fibrogenic molecular complex as a potential mechanism of this process and that this AXL activity may be independent of Gas6. Much is still unknown concerning the mechanism of AXL activation in fibroblasts in response to smoke, but our findings offer evidence of AXL activation as a potential key pathway linking the fibrotic process with tobacco smoking. This pathway is of interest in addressing the challenges of halting PF progression and presents as a promising target and potential marker for smoke-related PF.
Acknowledgments
Acknowledgment
The authors thank Dr. Kent E. Pinkerton (Center for Health and the Environment, UC Davis) and Dr. Reen Wu (Department of Internal Medicine, UC Davis) for their advice and assistance with smoke exposure studies, as well as the UC Davis Comprehensive Cancer Center Biorepository (University of California at Davis, Davis, CA) for pathology support.
Footnotes
Supported by the California University of California Office of The President Tobacco-Related Disease Research Program grants 27KT-0004, 28IR-0061, and T31DT1849, and the U.S. National Institutes of Health grant National Heart, Lung, and Blood Institute R01HL146802.
Author Contributions: Conception and design: C.-H.C. Development of methodology: C.-H.C. Acquisition of data (provided animals and provided facilities, etc.): D.C.Y., S.G., J.-M.L., S.-J.C., and C.-H.C. Analysis and interpretation of data (e.g., statistical analysis, biostatistics, and computational analysis): J.-M.L., S.-W.H., and W.-H.C. Writing, review, and/or revision of the manuscript: D.C.Y., W.-H.C., and C.-H.C. Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): S.-W.H., W.-H.C., and C.-H.C. Study supervision: C.-H.C.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2020-0303OC on March 17, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Ley B, Collard HR, King TE., Jr Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;183:431–440. doi: 10.1164/rccm.201006-0894CI. [DOI] [PubMed] [Google Scholar]
- 2. Martinez FJ, Collard HR, Pardo A, Raghu G, Richeldi L, Selman M, et al. Idiopathic pulmonary fibrosis. Nat Rev Dis Primers. 2017;3:17074. doi: 10.1038/nrdp.2017.74. [DOI] [PubMed] [Google Scholar]
- 3. Nalysnyk L, Cid-Ruzafa J, Rotella P, Esser D. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur Respir Rev. 2012;21:355–361. doi: 10.1183/09059180.00002512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thannickal VJ, Toews GB, White ES, Lynch JP, III, Martinez FJ. Mechanisms of pulmonary fibrosis. Annu Rev Med. 2004;55:395–417. doi: 10.1146/annurev.med.55.091902.103810. [DOI] [PubMed] [Google Scholar]
- 5. Kreuter M, Bonella F, Wijsenbeek M, Maher TM, Spagnolo P. Pharmacological treatment of idiopathic pulmonary fibrosis: current approaches, unsolved issues, and future perspectives. BioMed Res Int. 2015;2015:329481. doi: 10.1155/2015/329481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bjoraker JA, Ryu JH, Edwin MK, Myers JL, Tazelaar HD, Schroeder DR, et al. Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1998;157:199–203. doi: 10.1164/ajrccm.157.1.9704130. [DOI] [PubMed] [Google Scholar]
- 8. Katzenstein AL, Myers JL. Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification. Am J Respir Crit Care Med. 1998;157:1301–1315. doi: 10.1164/ajrccm.157.4.9707039. [DOI] [PubMed] [Google Scholar]
- 9. Kuhn C, McDonald JA. The roles of the myofibroblast in idiopathic pulmonary fibrosis: ultrastructural and immunohistochemical features of sites of active extracellular matrix synthesis. Am J Pathol. 1991;138:1257–1265. [PMC free article] [PubMed] [Google Scholar]
- 10. Wynn TA. Integrating mechanisms of pulmonary fibrosis. J Exp Med. 2011;208:1339–1350. doi: 10.1084/jem.20110551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jordana M, Schulman J, McSharry C, Irving LB, Newhouse MT, Jordana G, et al. Heterogeneous proliferative characteristics of human adult lung fibroblast lines and clonally derived fibroblasts from control and fibrotic tissue. Am Rev Respir Dis. 1988;137:579–584. doi: 10.1164/ajrccm/137.3.579. [DOI] [PubMed] [Google Scholar]
- 12. Raghu G, Chen YY, Rusch V, Rabinovitch PS. Differential proliferation of fibroblasts cultured from normal and fibrotic human lungs. Am Rev Respir Dis. 1988;138:703–708. doi: 10.1164/ajrccm/138.3.703. [DOI] [PubMed] [Google Scholar]
- 13. Grimminger F, Günther A, Vancheri C. The role of tyrosine kinases in the pathogenesis of idiopathic pulmonary fibrosis. Eur Respir J. 2015;45:1426–1433. doi: 10.1183/09031936.00149614. [DOI] [PubMed] [Google Scholar]
- 14. Espindola MS, Habiel DM, Narayanan R, Jones I, Coelho AL, Murray LA, et al. Targeting of tam receptors ameliorates fibrotic mechanisms in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2018;197:1443–1456. doi: 10.1164/rccm.201707-1519OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rangarajan S, Lee JS. The taming of the idiopathic pulmonary fibrosis myofibroblast: one step closer? Am J Respir Crit Care Med. 2018;197:1377–1378. doi: 10.1164/rccm.201802-0222ED. [DOI] [PubMed] [Google Scholar]
- 16. Antoniou KM, Hansell DM, Rubens MB, Marten K, Desai SR, Siafakas NM, et al. Idiopathic pulmonary fibrosis: outcome in relation to smoking status. Am J Respir Crit Care Med. 2008;177:190–194. doi: 10.1164/rccm.200612-1759OC. [DOI] [PubMed] [Google Scholar]
- 17. Callahan SJ, Xia M, Murray S, Flaherty KR. Clinical characteristics in patients with asymmetric idiopathic pulmonary fibrosis. Respir Med. 2016;119:96–101. doi: 10.1016/j.rmed.2016.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kishaba T, Nagano H, Nei Y, Yamashiro S. Clinical characteristics of idiopathic pulmonary fibrosis patients according to their smoking status. J Thorac Dis. 2016;8:1112–1120. doi: 10.21037/jtd.2016.03.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Oh CK, Murray LA, Molfino NA. Smoking and idiopathic pulmonary fibrosis. Pulm Med. 2012;2012:808260. doi: 10.1155/2012/808260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Poletti V, Ravaglia C, Buccioli M, Tantalocco P, Piciucchi S, Dubini A, et al. Idiopathic pulmonary fibrosis: diagnosis and prognostic evaluation. Respiration. 2013;86:5–12. doi: 10.1159/000353580. [DOI] [PubMed] [Google Scholar]
- 21. Taskar V, Coultas D. Exposures and idiopathic lung disease. Semin Respir Crit Care Med. 2008;29:670–679. doi: 10.1055/s-0028-1101277. [DOI] [PubMed] [Google Scholar]
- 22. Vassallo R, Ryu JH. Smoking-related interstitial lung diseases. Clin Chest Med. 2012;33:165–178. doi: 10.1016/j.ccm.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 23. Cisneros-Lira J, Gaxiola M, Ramos C, Selman M, Pardo A. Cigarette smoke exposure potentiates bleomycin-induced lung fibrosis in Guinea pigs. Am J Physiol Lung Cell Mol Physiol. 2003;285:L949–L956. doi: 10.1152/ajplung.00074.2003. [DOI] [PubMed] [Google Scholar]
- 24. Ko JW, Shin NR, Park SH, Lee IC, Ryu JM, Kim HJ, et al. Silibinin inhibits the fibrotic responses induced by cigarette smoke via suppression of TGF-β1/Smad 2/3 signaling. Food Chem Toxicol. 2017;106:424–429. doi: 10.1016/j.fct.2017.06.016. [DOI] [PubMed] [Google Scholar]
- 25. Kanaji N, Basma H, Nelson A, Farid M, Sato T, Nakanishi M, et al. Fibroblasts that resist cigarette smoke-induced senescence acquire profibrotic phenotypes. Am J Physiol Lung Cell Mol Physiol. 2014;307:L364–L373. doi: 10.1152/ajplung.00041.2014. [DOI] [PubMed] [Google Scholar]
- 26. Milara J, Serrano A, Peiró T, Artigues E, Gavaldà A, Miralpeix M, et al. Aclidinium inhibits cigarette smoke-induced lung fibroblast-to-myofibroblast transition. Eur Respir J. 2013;41:1264–1274. doi: 10.1183/09031936.00017712. [DOI] [PubMed] [Google Scholar]
- 27. Thai P, Statt S, Chen CH, Liang E, Campbell C, Wu R. Characterization of a novel long noncoding RNA, SCAL1, induced by cigarette smoke and elevated in lung cancer cell lines. Am J Respir Cell Mol Biol. 2013;49:204–211. doi: 10.1165/rcmb.2013-0159RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen CH, Thai P, Yoneda K, Adler KB, Yang PC, Wu R. A peptide that inhibits function of Myristoylated Alanine-Rich C Kinase Substrate (MARCKS) reduces lung cancer metastasis. Oncogene. 2014;33:3696–3706. doi: 10.1038/onc.2013.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen CH, Statt S, Chiu CL, Thai P, Arif M, Adler KB, et al. Targeting myristoylated alanine-rich C kinase substrate phosphorylation site domain in lung cancer: mechanisms and therapeutic implications. Am J Respir Crit Care Med. 2014;190:1127–1138. doi: 10.1164/rccm.201408-1505OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang DC, Li JM, Xu J, Oldham J, Phan SH, Last JA, et al. Tackling MARCKS-PIP3 circuit attenuates fibroblast activation and fibrosis progression. FASEB J. 2019;33:14354–14369. doi: 10.1096/fj.201901705R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pierce EM, Carpenter K, Jakubzick C, Kunkel SL, Flaherty KR, Martinez FJ, et al. Therapeutic targeting of CC ligand 21 or CC chemokine receptor 7 abrogates pulmonary fibrosis induced by the adoptive transfer of human pulmonary fibroblasts to immunodeficient mice. Am J Pathol. 2007;170:1152–1164. doi: 10.2353/ajpath.2007.060649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen CH, Chang WH, Su KY, Ku WH, Chang GC, Hong QS, et al. HLJ1 is an endogenous Src inhibitor suppressing cancer progression through dual mechanisms. Oncogene. 2016;35:5674–5685. doi: 10.1038/onc.2016.106. [DOI] [PubMed] [Google Scholar]
- 33. Chen C-H, Chuang S-M, Yang M-F, Liao J-W, Yu S-L, Chen JJW. A novel function of YWHAZ/β-catenin axis in promoting epithelial-mesenchymal transition and lung cancer metastasis. Mol Cancer Res. 2012;10:1319–1331. doi: 10.1158/1541-7786.MCR-12-0189. [DOI] [PubMed] [Google Scholar]
- 34. Chakraborty D, Šumová B, Mallano T, Chen CW, Distler A, Bergmann C, et al. Activation of STAT3 integrates common profibrotic pathways to promote fibroblast activation and tissue fibrosis. Nat Commun. 2017;8:1130. doi: 10.1038/s41467-017-01236-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Conte E, Gili E, Fruciano M, Korfei M, Fagone E, Iemmolo M, et al. PI3K p110γ overexpression in idiopathic pulmonary fibrosis lung tissue and fibroblast cells: in vitro effects of its inhibition. Lab Invest. 2013;93:566–576. doi: 10.1038/labinvest.2013.6. [DOI] [PubMed] [Google Scholar]
- 36. Harvey BG, Heguy A, Leopold PL, Carolan BJ, Ferris B, Crystal RG. Modification of gene expression of the small airway epithelium in response to cigarette smoking. J Mol Med (Berl) 2007;85:39–53. doi: 10.1007/s00109-006-0103-z. [DOI] [PubMed] [Google Scholar]
- 37. Zhu C, Wei Y, Wei X. AXL receptor tyrosine kinase as a promising anti-cancer approach: functions, molecular mechanisms and clinical applications. Mol Cancer. 2019;18:153. doi: 10.1186/s12943-019-1090-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jorgensen E, Stinson A, Shan L, Yang J, Gietl D, Albino AP. Cigarette smoke induces endoplasmic reticulum stress and the unfolded protein response in normal and malignant human lung cells. BMC Cancer. 2008;8:229. doi: 10.1186/1471-2407-8-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Spira A, Beane J, Shah V, Liu G, Schembri F, Yang X, et al. Effects of cigarette smoke on the human airway epithelial cell transcriptome. Proc Natl Acad Sci USA. 2004;101:10143–10148. doi: 10.1073/pnas.0401422101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Myers SH, Brunton VG, Unciti-Broceta A. Axl inhibitors in cancer: a medicinal chemistry perspective. J Med Chem. 2016;59:3593–3608. doi: 10.1021/acs.jmedchem.5b01273. [DOI] [PubMed] [Google Scholar]
- 41. Checa M, Hagood JS, Velazquez-Cruz R, Ruiz V, García-De-Alba C, Rangel-Escareño C, et al. Cigarette smoke enhances the expression of profibrotic molecules in alveolar epithelial cells. PLoS One. 2016;11:e0150383. doi: 10.1371/journal.pone.0150383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Guan S, Liu Q, Han F, Gu W, Song L, Zhang Y, et al. Ginsenoside Rg1 ameliorates cigarette smoke-induced airway fibrosis by suppressing the TGF- β1/Smad pathway in vivo and in vitro. BioMed Res Int. 2017;2017:6510198. doi: 10.1155/2017/6510198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hubbard R, Lewis S, Richards K, Johnston I, Britton J. Occupational exposure to metal or wood dust and aetiology of cryptogenic fibrosing alveolitis. Lancet. 1996;347:284–289. doi: 10.1016/s0140-6736(96)90465-1. [DOI] [PubMed] [Google Scholar]
- 44. Kumar A, Cherian SV, Vassallo R, Yi ES, Ryu JH. Current concepts in pathogenesis, diagnosis, and management of smoking-related interstitial lung diseases. Chest. 2018;154:394–408. doi: 10.1016/j.chest.2017.11.023. [DOI] [PubMed] [Google Scholar]
- 45. Korshunov VA. Axl-dependent signalling: a clinical update. Clin Sci (Lond) 2012;122:361–368. doi: 10.1042/CS20110411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vouri M, Croucher DR, Kennedy SP, An Q, Pilkington GJ, Hafizi S. Axl-EGFR receptor tyrosine kinase hetero-interaction provides EGFR with access to pro-invasive signalling in cancer cells. Oncogenesis. 2016;5:e266. doi: 10.1038/oncsis.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1997;155:242–248. doi: 10.1164/ajrccm.155.1.9001319. [DOI] [PubMed] [Google Scholar]
- 48. Khan EM, Lanir R, Danielson AR, Goldkorn T. Epidermal growth factor receptor exposed to cigarette smoke is aberrantly activated and undergoes perinuclear trafficking. FASEB J. 2008;22:910–917. doi: 10.1096/fj.06-7729com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Reinikovaite V, Rodriguez IE, Karoor V, Rau A, Trinh BB, Deleyiannis FW-B, et al. The effects of electronic cigarette vapour on the lung: direct comparison to tobacco smoke. Eur Respir J. 2018;51:1701661. doi: 10.1183/13993003.01661-2017. [DOI] [PubMed] [Google Scholar]