

Abstract
Asthma is a common respiratory disease currently affecting more than 300 million worldwide and is characterized by airway inflammation, hyperreactivity, and remodeling. It is a heterogeneous disease consisting of corticosteroid-sensitive T-helper cell type 2–driven eosinophilic and corticosteroid-resistant, T-helper cell type 17-driven neutrophilic phenotypes. One pathway recently described to regulate asthma pathogenesis is cholesterol trafficking. Scavenger receptors, in particular SR-BI (scavenger receptor class B type I), are known to direct cellular cholesterol uptake and efflux. We recently defined SR-BI functions in pulmonary host defense; however, the function of SR-BI in asthma pathogenesis is unknown. To elucidate the role of SR-BI in allergic asthma, SR-BI–sufficient (SR-BI+/+) and SR-BI–deficient (SR-BI−/−) mice were sensitized (Days 0 and 7) and then challenged (Days 14, 15, and 16) with a house dust mite (HDM) preparation administered through oropharyngeal aspiration. Airway inflammation and cytokine production were quantified on Day 17. When compared with SR-BI+/+ mice, the HDM-challenged SR-BI−/− mice had increased neutrophils and pulmonary IL-17A production in BAL fluid. This augmented IL-17A production in SR-BI−/− mice originated from a non–T-cell source that included neutrophils and alveolar macrophages. Given that SR-BI regulates adrenal steroid hormone production, we tested whether the changes in SR-BI−/− mice were glucocorticoid dependent. Indeed, SR-BI−/− mice were adrenally insufficient during the HDM challenge, and corticosterone replacement decreased pulmonary neutrophilia and IL-17A production in SR-BI−/− mice. Taken together, these data indicate that SR-BI dampens pulmonary neutrophilic inflammation and IL-17A production in allergic asthma at least in part by maintaining adrenal function.
Keywords: asthma, IL-17A, SR-BI receptor, neutrophilia, adrenal insufficiency
Clinical Relevance
The data presented here indicate that SR-BI (scavenger receptor class B type I) dampens pulmonary neutrophilic inflammation and IL-17A production in allergic asthma at least in part by maintaining adrenal function.
Asthma impacts ∼300 million people worldwide, with at least 250,000 deaths occurring annually (1). It is characterized by recurring symptoms of reversible airflow obstruction, bronchial hyperresponsiveness, and airway inflammation (2). Increasingly, asthma is understood to be a heterogeneous lung disease consisting of corticosteroid-sensitive, T-helper cell type 2 (Th2)-associated eosinophilic phenotypes and corticosteroid-resistant, Th17-associated neutrophilic phenotypes (2, 3). Asthma research has predominantly focused on Th2-mediated mechanisms, whereas Th17 neutrophilic phenotypes have been less well studied. About 10% of patients who have neutrophilic inflammation and high airspace IL-17A concentrations are considered to have severe asthma, which traditionally responds poorly to corticosteroid treatment (4–9). Unfortunately, no specific treatments have been developed that specifically target neutrophilic, IL-17A–driven asthma.
Recent reports have shown that cholesterol trafficking, mediated through interactions between plasma lipoproteins (HDL [high-density lipoprotein] and apoA-I [apolipoprotein A-I]) and their receptors (ATP-binding cassette transporters ABCA1 and ABCG1), regulates Th17-driven neutrophilic asthma. Deficiencies of apoA-I, ABCA1, or ABCG1 increase the severity of airway inflammation in allergic asthma (10–12). Conversely, exogenous apoA-I and apoA-I mimetics attenuate airway inflammation in experimental asthma models (10, 13). Similarly, human studies have demonstrated a positive correlation between serum HDL concentrations and improved airway function in asthma (14–18).
Recently, we reported that SR-BI (scavenger receptor class B type I), a scavenger receptor that binds HDL and mediates cellular uptake of cholesterol ester from HDL, dampens the pulmonary innate immune response by decreasing neutrophil recruitment and increasing clearance of LPS from the airspace (19). SR-BI is ubiquitously expressed; however, its highest expression is in myeloid cells, endothelial cells, and steroidogenic tissues such as the adrenal glands (20–22). In the adrenal glands, SR-BI supplies exogenous cholesterol substrate for synthesis of glucocorticoids (20, 23–26). Previous studies have demonstrated increased inflammation and mortality from sepsis in SR-BI–deficient mice from insufficient adrenal glucocorticoid production (27, 28). These data indicate that SR-BI is critical to the innate immune response and stress-related adrenal responses. However, whether SR-BI plays a role in activation, maturation, and shaping of the adaptive immune response known to drive Th17-dependent neutrophilia and whether endogenous glucocorticoids can influence this response are currently unknown.
Herein, we demonstrate that allergen-sensitized and allergen-challenged SR-BI−/− mice exhibit adrenal insufficiency and have increased pulmonary neutrophilia and IL-17A production. Augmented IL-17A production in the airspace is due to increased IL-17A production by neutrophils and alveolar macrophages (aMϕ). Neutrophil depletion alone in SR-BI−/− mice with house dust mite (HDM)-induced asthma failed to reduce pulmonary IL-17A. However, when SR-BI−/− mice with HDM-induced asthma were given water supplemented with corticosteroids, the augmented pulmonary IL-17A and neutrophilia were reversed. These data suggest that SR-BI regulates Th17-driven neutrophilic asthma by maintaining systemic glucocorticoid production.
Methods
Animals
C57BL/6J (SR-BI+/+) and B6;129S2-Scarb1tm1Kri/J (SR-BI−/−) 9- to 12-week-old male mice were purchased from Jackson Laboratories and were bred in house. SR-BI−/− mice were backcrossed more than six generations with C57BL/6J mice before use. Previous experiments confirmed comparable pulmonary responses between littermate SR-BI+/+ mice and commercially available C57BL/6J mice (19). All experiments were performed in accordance with the Animal Welfare Act and U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals after review by the Animal Care and Use Committee of East Carolina University and The Ohio State University.
HDM Sensitization and Challenge
SR-BI+/+ and SR-BI−/− mice were randomly assigned to two different exposure groups: PBS or HDM and exposed as described previously (29). Briefly, mice were sensitized with 10 μg of an HDM solution (Dermatophagoides pteronyssinus, 28,750 endotoxin units/vial; catalog number XPB82D3A2.5, Greer Laboratories Inc.) in 50 μl of PBS via oropharyngeal (o.p.) aspiration on Days 0 and 7, which was followed by o.p. challenge on Days 14, 15, and 16 with 2 μg of the HDM preparation. In the control group, mice received 50 μl of PBS without the HDM preparation via o.p. aspiration on Days 0, 7, 14, 15, and 16. Mice were necropsied on Day 17. Right lung lobes were lavaged, and the left lung lobe was excised and incubated with the HDM preparation (10 μg/ml) in Dulbecco’s modified Eagle medium plus 10% FBS for 24 hours for cytokine analysis. For Figure 1, SR-BI+/+ mice were necropsied 4 hours and 24 hours after sensitization or challenge.
Figure 1.

Pulmonary SR-BI (scavenger receptor class B type I) expression is altered during the sensitization and challenge phases of house dust mite (HDM)-induced asthma. (A) Schematic of the HDM sensitization and challenge. SR-BI+/+ (C57BL/6J) mice were sensitized on Days 0 and 7 and challenged with an HDM preparation on Days 14, 15, and 16. SR-BI+/+ mice were killed 4 and 24 hours (hr) after the HDM sensitization and challenge. (B) At all time points, the right lung was lavaged, and total BAL cell counts were evaluated. (C) At all time points after the HDM sensitization and challenge, lung tissue was isolated to determine whether SR-BI expression had changed. Pulmonary Scarb1 (scavenger receptor B I) expression was determined by using real-time PCR, and expression was normalized to 18S. Data are presented as the mean ± SEM. Data are pooled from multiple experiments (n = 6 animals/group). *P < 0.05 and **P < 0.01.
Ovalbumin Sensitization and Challenge
SR-BI+/+ and SR-BI−/− mice were sensitized by o.p. instillation (100 μg in 50 μl) of OVA (ovalbumin) (Sigma-Aldrich) together with low-dose (0.1 μg) LPS (Sigma) as an adjuvant on Days 0 and 7, challenged with an aerosol of 1% OVA (Sigma) in saline on Day 14, and necropsied 48 hours after challenge (30). Right lungs were lavaged, and left lung tissue was incubated with OVA (10 μg/ml) in Dulbecco’s modified Eagle medium plus 10% FBS for 24 hours for cytokine analysis.
BAL and Cell Differential Counts
Mice were killed via an intraperitoneal injection of ketamine (100 mg/kg) and xylazine (10 mg/kg), and BAL was immediately performed as previously reported (31, 32). Total cells from lavages were counted using a hemocytometer (Hausser Scientific). Each sample was centrifuged onto slides using a Cytospin 4 centrifuge (Thermo Fisher Scientific) and stained with Kwik-Diff Stains (Thermo Fisher Scientific). Cell differential counts were determined by morphologic analysis using an evaluation of 200 cells/slide.
Cytokine Analysis
CXCL1, CXCL2, and CXCL5 concentrations in BAL fluid (BALF) were quantified by using an ELISA (R&D Systems). IL-2, IL-4, IL-5, IL-10, IL-12p70, GM-CSF, IFN-γ, TNF-α, and IL-17A in sample supernatants were measured by using a Multiplex ELISA (Bio-Rad) and were read using a MAGPIX instrument (Luminex).
Statistical Analysis
All data are presented as the mean ± SEM and were analyzed by one-way ANOVA or two-way ANOVA, with differences between groups assessed using Tukey post hoc tests. Graphs were generated and analyses were performed using GraphPad Prism 6 software (GraphPad Software). Differences were considered statistically significant at P < 0.05.
Results
SR-BI Expression in Lung Tissue Is Altered during Allergic Asthma
To determine whether allergic asthma altered pulmonary SR-BI expression, we measured lung tissue SR-BI expression after allergic asthma was induced by using the HDM preparation (Figure 1A). Unlavaged left lung tissue was collected from C57BL/6J (SR-BI+/+) mice after the HDM sensitization (4 and 24 h after Day 0) or challenge (4 and 24 h after Day 16). When compared with expression in naive mice, pulmonary SR-BI expression was unchanged during the HDM sensitization; however, 4 hours after the HDM challenge, SR-BI mRNA expression was increased (Figure 1C). Increased pulmonary SR-BI expression correlated with an increase in total cells in the airspace (Figure 1B), which included increases of neutrophils, lymphocytes, and eosinophils (see Figure E1 in the data supplement). At 24 hours after the HDM challenge, pulmonary SR-BI expression returned to baseline, whereas BALF cell counts remained elevated. Taken together, these data indicate that pulmonary SR-BI expression is increased during the allergen challenge.
SR-BI−/− Mice Have Exacerbated Pulmonary Neutrophilic Inflammation in Allergic Asthma
Next, we determined the degree of HDM-induced allergic airway inflammation in SR-BI–sufficient (SR-BI+/+) and SR-BI–deficient (SR-BI−/−) mice. Vehicle (PBS)-exposed SR-BI+/+ and SR-BI−/− mice did not have evidence of pulmonary inflammation (Figure 2). After the HDM sensitization and challenge, both SR-BI+/+ and SR-BI−/− exhibited increased BALF total leukocytes (Figure 2A) when compared with PBS-treated mice. However, the type of leukocytes differed between SR-BI+/+ and SR-BI−/− mice. In SR-BI−/− mice, relative to SR-BI+/+ mice, neutrophils and lymphocytes (Figures 2B and 2C), but not eosinophils or macrophages (Figures 2D and 2E), were significantly increased. HDM exposure enhanced pulmonary inflammation and airway mucous production (Periodic acid–Schiff staining) in both SR-BI+/+ and SR-BI−/− mice; however, the degree of inflammation and mucous production was not different between the strains (Figure 3).
Figure 2.
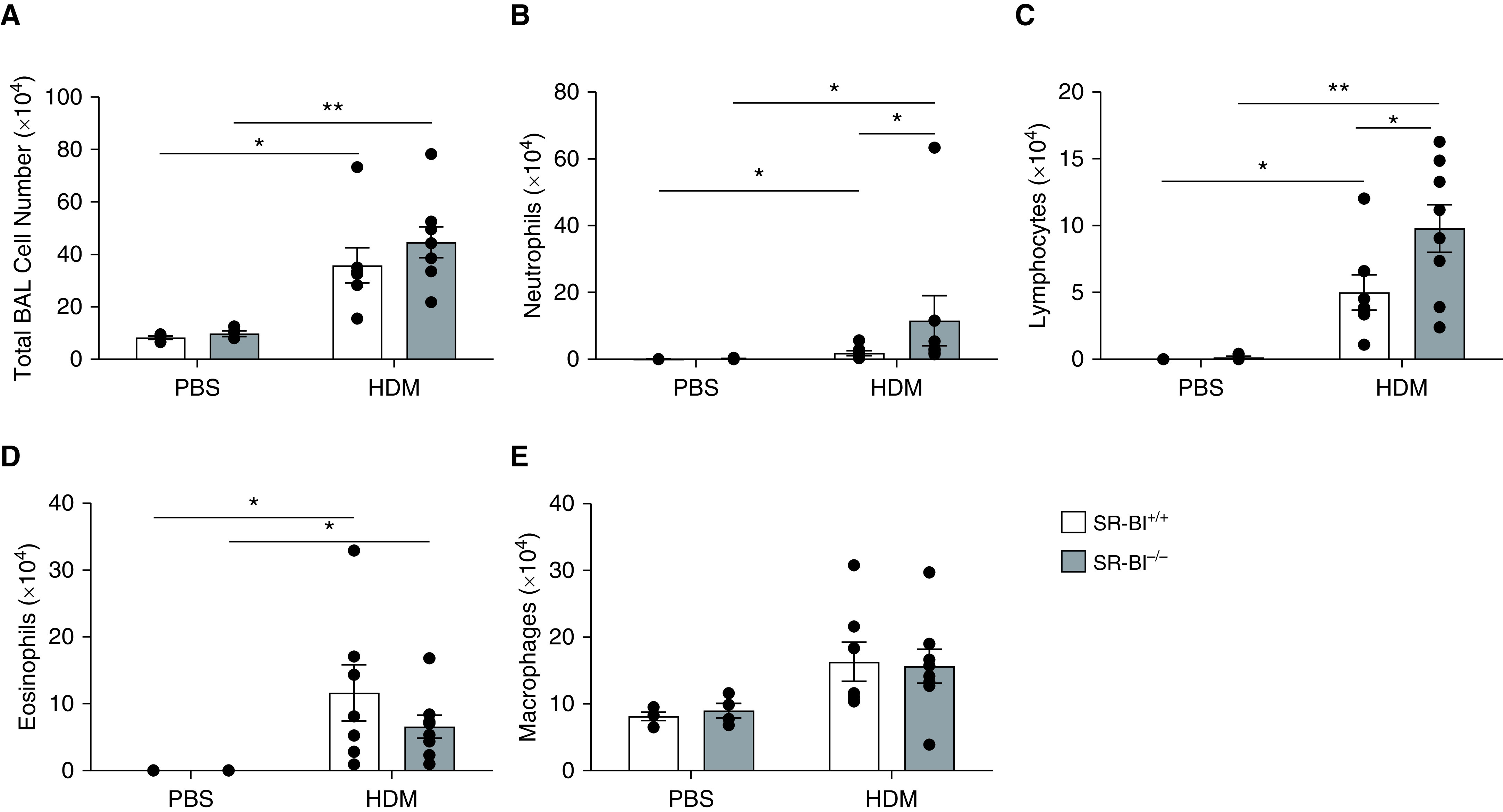
SR-BI−/− mice have increased pulmonary neutrophilia during allergic asthma. SR-BI+/+ (C57BL/6J) and SR-BI−/− mice were sensitized on Days 0 and 7 and challenged on Days 14, 15, and 16 with phosphate buffered saline (PBS) or an HDM preparation. Mice were killed 24 hr after the last challenge (on Day 17). The right lung from each mouse was lavaged to determine (A) the total BAL cell counts and the number of (B) neutrophils, (C) lymphocytes, (D) eosinophils, and (E) macrophages. Data are presented as the mean ± SEM. Data are pooled from multiple experiments (n = 4–8 animals/group). *P < 0.05 and **P < 0.01.
Figure 3.
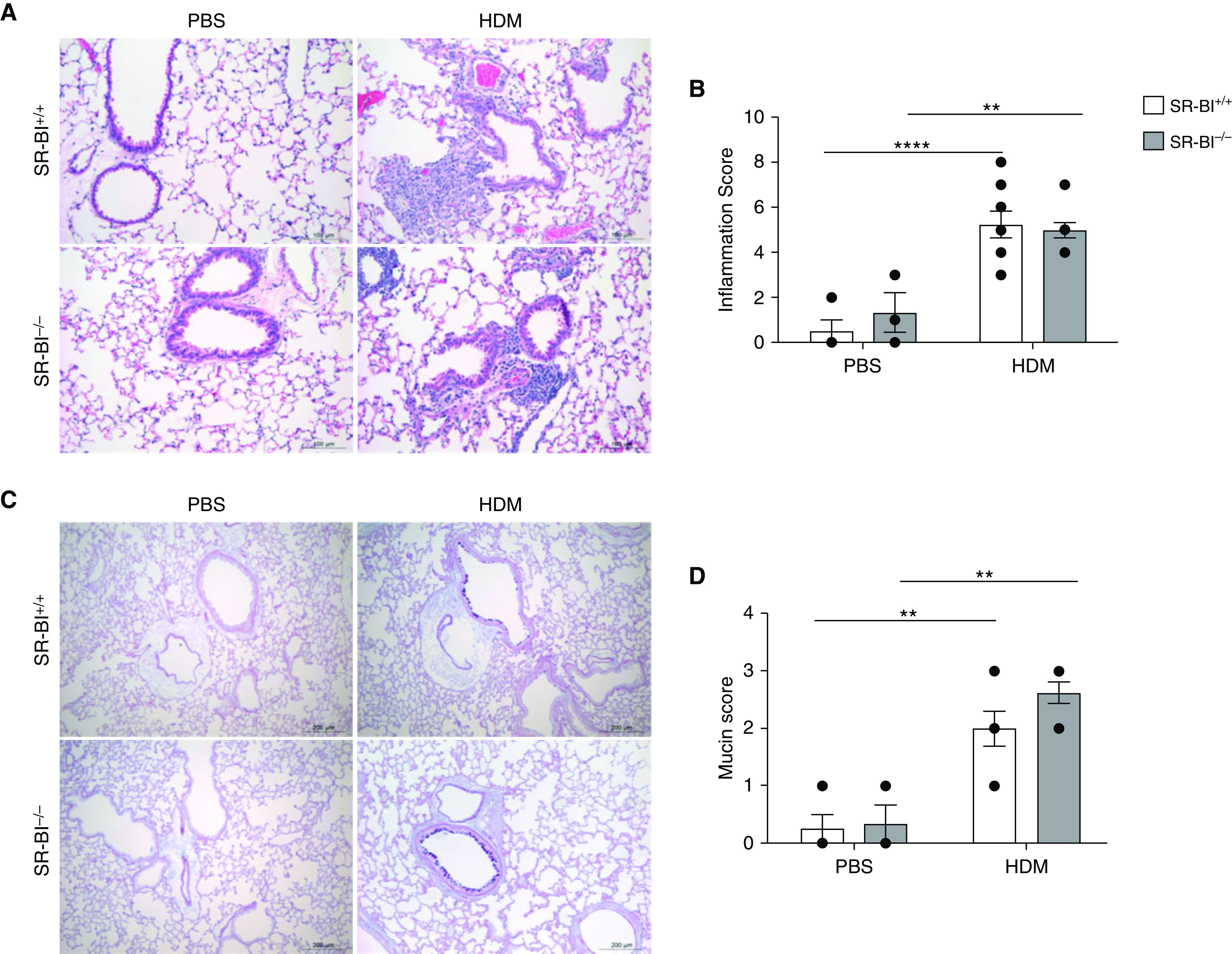
SR-BI deficiency does not lead to increased lung pathologic findings or mucous production during allergic asthma. SR-BI+/+ and SR-BI−/− mice were sensitized and challenged with either PBS or an HDM preparation. Mice were killed 24 hr after the last challenge, and the left lung from each mouse was fixed and paraffin embedded. (A) Representative images of hematoxylin and eosin–stained lung tissue. Scale bars, 100 μm. (B) Histologic characteristics were scored in a blinded fashion on the basis of perivascular and peribronchiolar inflammatory infiltrates. (C) Representative images of periodic acid–Schiff–stained lung tissue obtained from SR-BI+/+ and SR-BI−/− mice that had been sensitized and challenged with PBS or the HDM preparation. Scale bars, 200 μm. (D) Mucin production was scored in a blinded fashion. Data are presented as the mean ± SEM. Data are pooled from multiple experiments (n = 3–8 animals/group). **P < 0.01 and ****P < 0.0001.
IL-17A Production Is Increased in HDM-challenged SR-BI−/− Mice
To investigate potential mechanisms of SR-BI regulation of neutrophilic inflammation in allergic asthma, we measured Th1, Th2, and Th17 cytokines in lung tissue from HDM-challenged SR-BI+/+ and SR-BI−/− mice. The Th1-associated cytokine IFN-γ was present at relatively low concentrations in the lungs in HDM groups, and the concentrations were not significantly different between SR-BI+/+ and SR-BI−/− mice (Figure 4A). SR-BI+/+ and SR-BI−/− mice also had similar concentrations of the Th2 cytokines IL-4, IL-5, and IL-13 (Figures 4A and 4B). However, HDM-sensitized/challenged SR-BI−/− mice, when compared with SR-BI+/+ mice, exhibited increased production of IL-17A, GM-CSF, IL-2, and TNF-α (Figure 4A). Because HDM-sensitized/challenged SR-BI−/− mice had increased neutrophils, we measured amounts of the neutrophil chemoattractants CXCL1, CXCL2, and CXCL5 in the BALF. CXCL2 and CXCL5 did not demonstrate an HDM response, whereas CXCL1 was increased in the HDM groups compared with the PBS group but was not dependent on SR-BI (Figure E2).
Figure 4.

SR-BI−/− mice have a significant increase in pulmonary IL-17A production after the HDM sensitization and challenges. SR-BI+/+ (C57BL/6J) and SR-BI−/− mice were sensitized and challenged with an HDM preparation. Mice were killed 4 and 24 hr after the last challenge (on Day 17). (A) At 24 hr after the last challenge, the unlavaged left lung was collected, cultured ex vivo with complete Dulbecco’s modified Eagle medium, and stimulated in culture with the HDM preparation for 24 hr. Cytokine production in supernatants of HDM-stimulated lung tissue was measured by using the Bio-Plex Multiplex Immunoassay System kit. (B) At 4 hr after the last challenge, unlavaged lung tissue was isolated to determine whether IL-13 expression had changed. Pulmonary IL-13 expression was determined by using real-time PCR, and expression was normalized to 18S. Data are presented as the mean ± SEM. Data are pooled from multiple experiments (n = 7–8 animals/group). *P < 0.05.
SR-BI−/− Mice Have Increased Pulmonary Neutrophilic Inflammation and IL-17 Production after OVA Sensitization and Challenge
In another experimental allergic asthma model, we verified our finding by using OVA sensitization and challenge (10, 11, 30, 33, 34). Consistent with HDM-exposed mice, OVA-sensitized and OVA-challenged SR-BI−/− mice had increased airspace neutrophils (Figure 5A). BALF from OVA-sensitized and OVA-challenged SR-BI−/− mice, when compared with that from SR-BI+/+ mice, also had comparable concentrations of IL-4 and IL-5 but had significantly increased concentrations of IL-17A (Figure 5B). In contrast to HDM-exposed mice, OVA-exposed SR-BI−/− mice had no differences in TNF-α and GM-CSF levels, whereas G-CSF and CXCL5, chemokines known to be induced by IL-17 and to induce neutrophil accumulation in the lung, were increased (Figure 5B). These results suggest that the elevation in pulmonary IL-17 and neutrophils noted with SR-BI deficiency is observed across disparate experimental asthma models and that the mechanisms of excess neutrophil accumulation may differ across models.
Figure 5.

SR-BI deficiency exacerbates pulmonary neutrophilia and IL-17A production after OVA (ovalbumin) sensitization and challenge. SR-BI+/+ and SR-BI−/− mice were sensitized by oropharyngeal instillation of OVA and LPS on Days 0 and 7 and then challenged with aerosolized OVA on Day 14. Mice were killed 48 hr after the OVA challenge. (A) The right lung was lavaged to determine the total BAL cell numbers and differential cell counts (neutrophils [PMNs], lymphocytes [Lymph], eosinophils [Eos], and macrophages [Mϕ]). (B) The unlavaged left lung was collected, cultured ex vivo, and restimulated in culture with OVA for 24 hr. Cytokine production in supernatants of OVA-stimulated lung tissue was measured by using the Bio-Plex Multiplex Immunoassay System kit. Data are presented as the mean ± SEM. n = 5 animals/group. *P < 0.05 and **P < 0.01. PMN = polymorphonuclear neutrophils.
Augmented IL-17A in Asthmatic SR-BI−/− Mice Does Not Only Originate from Th17 Cells
To better characterize the immune response in SR-BI−/− mice after the HDM challenge, we performed flow cytometry on HDM-exposed SR-BI+/+ and SR-BI−/− mice. After the HDM challenge, SR-BI+/+ and SR-BI−/− lungs had similar numbers of CD45+ cells, alveolar macrophages (aMϕ) (CD45+Ly6G−CD64+CD11b−CD11c+SiglecF+Autohigh), interstitial macrophages (CD45+Ly6G−CD64+CD11b+SiglecFlo), and CD4+ T cells (CD45+CD3+CD4+) (Figure 6A). When compared with neutrophils in SR-BI+/+ lung tissue, neutrophils (polymorphonuclear neutrophils [PMNs]) (CD45+Ly6G+) in SR-BI−/− lung tissue were significantly increased (Figure 6A). However, depleting neutrophils with anti-Gr1 antibodies during the HDM challenge did not decrease the excess IL-17A production in SR-BI−/− lungs (Figures E3A and E3B).
Figure 6.

Augmented IL-17A in asthmatic SR-BI−/− mice is not due to increased pulmonary T-helper cell type 17 cells. SR-BI+/+ (C57BL/6J) and SR-BI−/− mice were sensitized and challenged with an HDM preparation and killed 24 hr after the last HDM challenge. Lung tissue was digested and stained with antibodies to determine the number of (A) alveolar Mϕ (aMϕ; CD45+Ly6G−CD64+CD11b−CD11c+SiglecF+Autofluorescenthigh), iMϕ (CD45+Ly6G−CD64+CD11b+SiglecF+), neutrophils (PMNs; CD45+Ly6G+), and CD4+ T cells (CD45+CD4+) by using flow cytometry. (B) Representative contour plots depict CD4 and IL-17A expression in CD45+ cells. The percentage of CD4+ cells that produced IL-17A and the percentage of CD4− cells that produced IL-17A are shown in the plots as mean ± SEM. (C) The bar graphs represent the T-helper cell type 17 cell numbers in the lung and the MFI of IL-17A staining of CD4+ cells in the lung. (D) The bar graphs represent the IL-17 production in cells as the total percentage of and the total number of neutrophils present. Data are presented as the mean ± SEM. n = 5 animals/group. *P < 0.05 and **P < 0.01. iMϕ = interstitial Mϕ; MFI = mean fluorescent intensity.
Having observed higher IL-17A in allergen-sensitized and allergen-challenged SR-BI−/− mice, we investigated whether this difference was from increased T-cell polarization favoring conversion to Th17 cells. After HDM sensitization and challenge, SR-BI+/+ and SR-BI−/− lungs had similarly increased Th17 cell numbers (Figures 6B and 6C) and IL-17 mean fluorescent intensity in CD4+ T cells (Figure 6C), whereas SR-BI−/− mice had an increase in CD45+IL-17A+ cells, specifically neutrophils (Figure 6D). In addition, blocking IL-17 with anti-IL-17 antibodies during the HDM challenge decreased neutrophils in the airspace of SR-BI−/− mice (Figure E3C). Taken together, these findings suggest that IL-17A–producing immune cells are elevated in the lungs of SR-BI−/− mice after the HDM sensitization and challenge, with a preference being shown for non–T-cell sources of IL-17A.
Pulmonary IL-17A Production in SR-BI−/− Mice Is due to Adrenal Insufficiency
Previous studies demonstrated that SR-BI−/− mice exhibit stress-induced adrenal insufficiency (19, 23). In addition, previous studies from our laboratory indicate that augmented pulmonary neutrophilia in SR-BI−/− mice after bacterial pneumonia is caused in part by adrenal insufficiency (19). Therefore, we hypothesized that adrenal insufficiency contributes to the increased neutrophilic inflammation and IL-17A production in HDM-sensitized and HDM-challenged SR-BI−/− mice. To determine whether HDM-exposed SR-BI−/− mice were adrenally insufficient, we measured serum corticosterone concentrations. At baseline, SR-BI−/− mice did not have differences in serum corticosterone concentrations when compared with SR-BI+/+ mice (Figure 7A). Four hours after the last HDM challenge, SR-BI−/− mice had a significant decrease in serum corticosterone, indicating adrenal insufficiency (Figure 7A). To determine whether glucocorticoid supplementation can diminish the increased pulmonary neutrophilia and IL-17 production, we supplemented drinking water with corticosterone during the entire allergic asthma protocol. Corticosterone supplementation increased corticosterone serum concentrations in SR-BI−/− mice to concentrations equivalent to those in SR-BI+/+ mice (data not shown). Corticosterone replacement modestly decreased, although not by a statistically significant amount, the total BALF leukocyte counts in both strains after HDM challenge (Figure 7B). In SR-BI−/− mice, corticosterone replacement completely abolished the augmented pulmonary neutrophilia (Figure 7B) but did not alter eosinophilia (Figure E4). In addition, corticosterone replacement in SR-BI−/− mice did not change the production of IL-5 (Figure 7C) but did decrease IL-17A and GM-CSF production (Figure 7D). These data suggest that after allergic challenge, the enhanced IL-17A and neutrophilia in SR-BI−/− mice are due to adrenal insufficiency.
Figure 7.
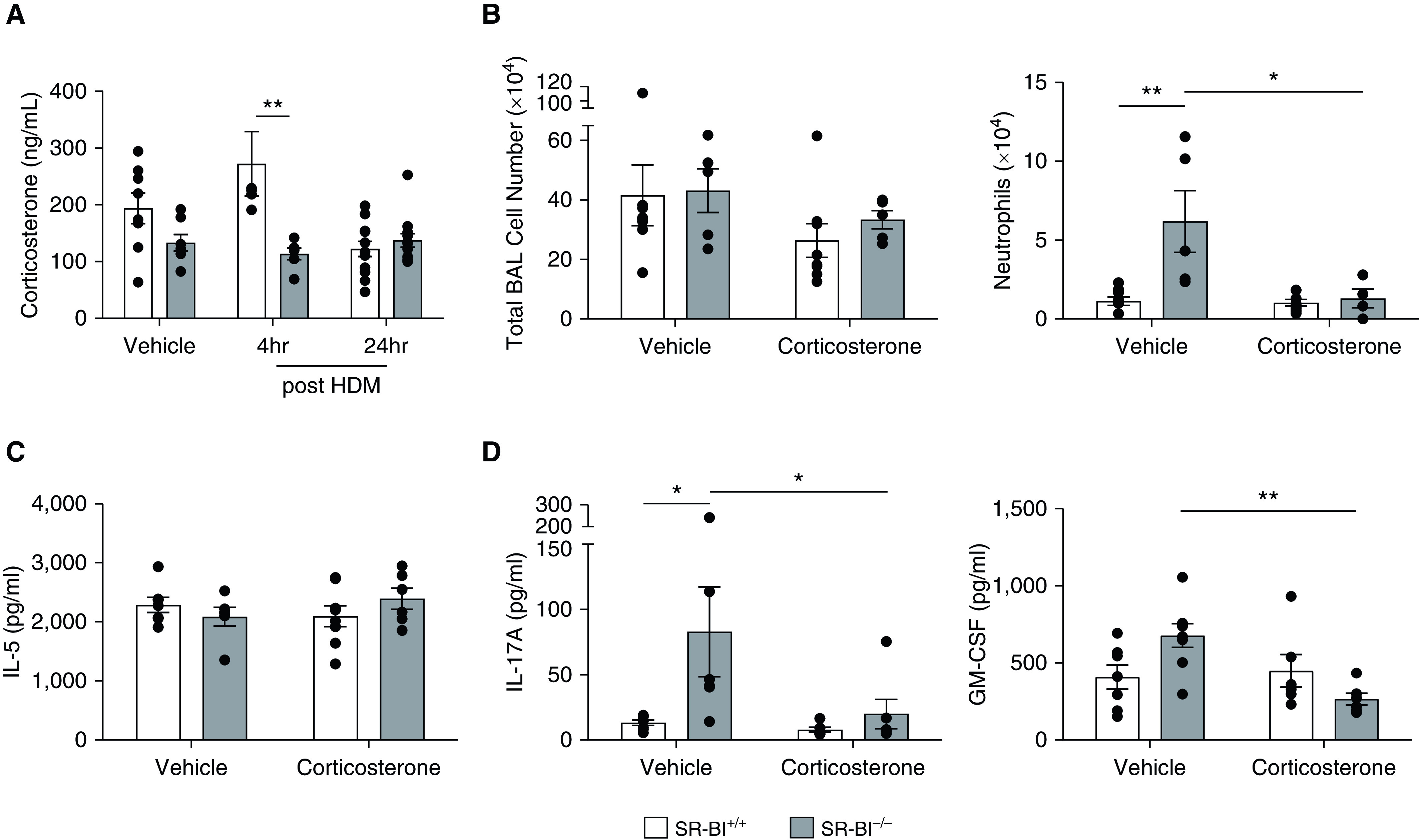
Enhanced pulmonary neutrophilia and IL-17A production in asthmatic SR-BI−/− mice are due to adrenal insufficiency. SR-BI+/+ (C57BL/6J) and SR-BI−/− mice were sensitized and challenged with an HDM preparation and killed 4 hr after the last challenge. (A) Serum corticosterone was measured by using an immunoassay. SR-BI+/+ and SR-BI−/− mice were given corticosterone in their drinking water (6.25 μg/ml) starting 24 hr before the HDM sensitization. Mice were then sensitized on Days 0 and 7 and challenged on Days 14, 15, and 16 with the HDM preparation. (B) The right lung from each mouse was lavaged to determine the total BAL cell counts and the number of neutrophils. The unlavaged left lung was collected, cultured ex vivo, and stimulated in culture with the HDM preparation for 24 hr. (C and D) Cytokine production in supernatants of HDM-stimulated lung tissue was measured by using a Bio-Plex Multiplex Immunoassay System kit. Data are presented as the mean ± SEM (n = 6–8 animals/group). *P < 0.05 and **P < 0.01. GM-CSF = granulocyte-macrophage colony-stimulating factor.
Discussion
Asthma is a heterogenous, chronic inflammatory lung disease characterized by airway inflammation and hyperresponsiveness (35). A growing body of literature suggests an association between cholesterol homeostasis and allergic asthma responses. In this study, we report that SR-BI, a receptor important in cholesterol ester uptake from HDL particles, is a novel regulator of airway neutrophil accumulation and IL-17 production during allergic asthma. In HDM-induced allergic asthma, pulmonary SR-BI expression is upregulated, whereas loss of SR-BI enhances airspace PMNs and IL-17A production, which was reversed with glucocorticoid supplementation. Taken together, these studies reveal SR-BI–dependent adrenal glucocorticoid production as an important endogenous endocrine regulator of asthma-induced pulmonary neutrophilia and IL-17.
SR-BI has been reported to be a critical regulator of the pulmonary innate immune responses to infectious and inflammatory stimuli (19, 28); however, it is currently unknown whether SR-BI influences allergic asthma pathogenesis. Previous studies have shown increased expression of other scavenger receptors (MARCO and SR-AI/II) in the lungs during experimental allergic asthma (36). Our data show that SR-BI expression is increased in the lung during the HDM-challenge phase of this model of allergic asthma, suggesting that SR-BI may exert a protective effect during allergic asthma exacerbations. Increased SR-BI expression correlated with neutrophil influx into the airspace. The mechanism driving the increased SR-BI expression observed in the present study is not clear. The increase in expression could reflect recruitment of SR-BI–expressing innate immune cells to the lung (e.g., neutrophils, dendritic cells, monocytes); in addition, or alternatively, it may reflect gene induction by damage-associated molecular patterns, pathogen-associated molecular patterns, and/or allergens present in the HDM preparation. SR-BI recognizes and binds pathogen-associated molecular patterns, such as LPS, commonly found in commercially available HDM preparations (37–40) and can initiate innate-immune-cell chemotaxis. In addition, HDM instillation induces damage-associated molecular patterns, such as oxidized lipids, which are known ligands of SR-BI (41). Lastly, SR-BI expression could be elevated in the lung during HDM exposure because of an increased cholesterol requirement, given that previous studies have noted that cholesterol is decreased in the airspace of mice sensitized to and challenged with OVA (10). Therefore, the increased pulmonary SR-BI expression could bind and clear debris and antigens while increasing pulmonary cholesterol, which ultimately limits allergic inflammation.
The protective role of SR-BI in experimental asthma, regarding its role in suppression of pulmonary neutrophilia and IL-17 in particular, was confirmed in two allergic asthma models. These data are consistent with other allergic asthma models in which cholesterol transport and/or metabolism are deficient. Among patients with atopic asthma, serum concentrations of HDL-C and apoA-I positively correlated with pulmonary function (15, 42). In experimental asthma models, allergen-challenged ApoA-I−/− mice exhibit increased pulmonary neutrophilia, CXCR2 ligands, and IL-17 (10). Furthermore, administration of apoA-I mimetic peptides mitigated murine allergen-induced neutrophilic airway inflammation (13). In the OVA model, mice lacking ABCG1 display increased Th17-dependent airway neutrophilic inflammation (12). Although the exact role that cholesterol plays in these asthma models is presently unclear, our study adds to this body of work, further supporting that cholesterol receptors and/or cholesterol uptake influences IL-17–dependent neutrophilia in asthma.
Our findings link SR-BI to augmented pulmonary IL-17, which appears to be independent of enhanced Th17 polarization, suggesting a non–T-cell IL-17 source. This was surprising, given that previous studies using models deficient in cholesterol transporters report enhanced Th17 polarization (12, 43) and that cholesterol uptake and metabolism influences Th17 regulatory-T-cell polarization (44–46). Given the lack of an increase in Th17 cells, other cell types must be considered for the increased IL-17 in SR-BI−/− mice. Multiple cell types have the capacity to produce IL-17A, including innate immune cells like macrophages and neutrophils (12, 47–49). However, neutrophil depletion did not affect IL-17A production. This suggests either that macrophages or other cells such as γδ T cells are the principal source of IL-17 or that anti-Gr1 treatment is not completely effective at abolishing IL-17–producing neutrophils. It has been previously reported that lipid metabolism can influence macrophage polarization and normal myeloid-cell development (50, 51). Therefore, it is possible that the excess IL-17 production in SR-BI deficiency is of myeloid origin and that augmented IL-17 production could be a result of altered macrophage polarization. However, we recognize that a limitation of this study is that we did not directly test T-cell depletion in this model to confirm that increased IL-17 concentrations noted in SR-BI–deficient mice are independent of T cells. Future studies will focus on the cell-specific expression of SR-BI and how this influences IL-17 production in the lung.
IL-17 has been shown to induce neutrophil infiltration into the airspace via the generation of the CXCR2 ligands CXCL1, CXCL2, CXCL5, and G-CSF (30, 52). In addition, GM-CSF promotes the polarization of Th17 cells and the production of IL-17A (53–55). In our study, we observed no differences in pulmonary production of CXCR2 ligands in SR-BI−/− mice after the HDM sensitization and challenge, whereas GM-CSF was increased in conjunction with an increase in IL-17. GM-CSF has been shown to be produced by the airway epithelium during allergic asthma as a result of NF-κB activation (56). SR-BI suppresses inflammation through inhibiting the production of cytokines from macrophages via the TLR-4 and NF-κB pathway (57). GM-CSF promotes the differentiation, proliferation, and function of aMϕ (58–60). In a previous allergic asthma study using OVA, BALF IL-17 was principally produced by aMϕ rather than by Th17 cells (48). Therefore, our data could reveal an important link between macrophage SR-BI and GM-CSF that leads to IL-17 production.
Lastly, the data presented here indicate that excess IL-17 and neutrophilia in SR-BI deficiency can be mitigated with corticosterone supplementation. SR-BI–deficient mice are reportedly adrenally insufficient upon challenge (19, 61, 62). This adrenal insufficiency accelerates the maturation of bone-marrow neutrophils, exacerbating neutrophil influx at sites of inflammation (63). Consistent with these prior results, the HDM-challenged SR-BI−/− mice had decreased serum corticosterone and augmented pulmonary neutrophilia, which were reversed with corticosterone supplementation. This is consistent with data that indicate that exogenous glucocorticoids have been shown to inhibit PMN chemotaxis (64). Interestingly, glucocorticoid supplementation also reversed pulmonary IL-17 and GM-CSF production. Exogenous glucocorticoids have previously been shown to influence T-cell polarization away from a Th2 phenotype and have recently been shown to influence T-cell polarization away from a Th17 phenotype (65, 66). In addition, recent studies examining endogenous production of glucocorticoids in sterile lung injury and inflammation models have shown that pulmonary neutrophilia is mediated with no influence on Th2-derived cytokines (67). This is intriguing and may reveal a novel role for glucocorticoids that is dependent on the model and disease. Alternatively, it may reveal that exogenous versus endogenous glucocorticoids have differential roles regarding pulmonary immune responses. Taken together, these data indicate that endogenous glucocorticoids may influence IL-17 production by non-T cells and may influence neutrophil recruitment in certain lung injury and inflammation models and diseases.
In conclusion, we provide evidence for a novel protective role of SR-BI in IL-17–dependent neutrophilic asthma, whereby this scavenger receptor suppresses pulmonary neutrophilia by supporting adrenal function. A deficiency in this mechanism may potentially contribute to the pathogenesis of neutrophilic asthma. Given that adrenal insufficiency has been associated with long-term use of inhaled steroids (68, 69), we speculate that adrenal hormone production influences the IL-17 production of myeloid cells in inflamed tissues such as the asthmatic lung. Future investigations will be needed to address the cell-specific contributions of SR-BI in pulmonary IL-17 production as well as the role of SR-BI in human neutrophilic asthma. Overall, these data indicate that augmenting SR-BI expression and/or function may be a potential therapeutic strategy for neutrophilic asthma.
Acknowledgments
Acknowledgment
The authors thank Joani Zary (East Carolina University), Dr. Hideki Nakano (National Institute of Environmental Health Sciences), and Ralph Wilson (National Institute of Environmental Health Sciences) for technical assistance and thank the East Carolina University Brody School of Medicine Flow Cytometry Core.
Footnotes
Supported by a Brody School of Medicine internal seed bridge grant (S.W.R.), National Institutes of Health, National Institute of Environmental Health Sciences grants Z01-ES102005 (M.B.F.) and ZIA-ES102025-09 (D.N.C.), and National Institutes of Health Center for Human Health and the Environment grant P30ES025128.
Author Contributions: S.W.R., D.N.C., M.B.F., and K.M.G.: concept and design. S.W.R., S.V., B.K.-B., M.X.H., B.L., J.H.M., S.Y.T., and R.M.T.: acquisition of data. S.W.R., S.V., B.K.-B., K.D.-R., M.X.H., B.L., J.H.M., S.Y.T., D.A.T., and K.M.G.: analysis and interpretation. All authors: drafting of manuscript and evaluation for important intellectual content.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2020-0007OC on February 26, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Croisant S. Epidemiology of asthma: prevalence and burden of disease. Adv Exp Med Biol. 2014;795:17–29. doi: 10.1007/978-1-4614-8603-9_2. [DOI] [PubMed] [Google Scholar]
- 2. Fahy JV. Type 2 inflammation in asthma: present in most, absent in many. Nat Rev Immunol. 2015;15:57–65. doi: 10.1038/nri3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Woodruff PG, Modrek B, Choy DF, Jia G, Abbas AR, Ellwanger A, et al. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med. 2009;180:388–395. doi: 10.1164/rccm.200903-0392OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bruijnzeel PLB, Uddin M, Koenderman L. Targeting neutrophilic inflammation in severe neutrophilic asthma: can we target the disease-relevant neutrophil phenotype? J Leukoc Biol. 2015;98:549–556. doi: 10.1189/jlb.3VMR1214-600RR. [DOI] [PubMed] [Google Scholar]
- 5. Chung KF. Neutrophilic asthma: a distinct target for treatment? Lancet Respir Med. 2016;4:765–767. doi: 10.1016/S2213-2600(16)30232-6. [DOI] [PubMed] [Google Scholar]
- 6. Fahy JV. Asthma was talking, but we weren’t listening: missed or ignored signals that have slowed treatment progress. Ann Am Thorac Soc. 2016;13:S78–S82. doi: 10.1513/AnnalsATS.201508-515MG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fahy JV. Eosinophilic and neutrophilic inflammation in asthma: insights from clinical studies. Proc Am Thorac Soc. 2009;6:256–259. doi: 10.1513/pats.200808-087RM. [DOI] [PubMed] [Google Scholar]
- 8. Fahy JV, Kim KW, Liu J, Boushey HA. Prominent neutrophilic inflammation in sputum from subjects with asthma exacerbation. J Allergy Clin Immunol. 1995;95:843–852. doi: 10.1016/s0091-6749(95)70128-1. [DOI] [PubMed] [Google Scholar]
- 9. Ordoñez CL, Shaughnessy TE, Matthay MA, Fahy JV. Increased neutrophil numbers and IL-8 levels in airway secretions in acute severe asthma: clinical and biologic significance. Am J Respir Crit Care Med. 2000;161:1185–1190. doi: 10.1164/ajrccm.161.4.9812061. [DOI] [PubMed] [Google Scholar]
- 10. Dai C, Yao X, Keeran KJ, Zywicke GJ, Qu X, Yu ZX, et al. Apolipoprotein A-I attenuates ovalbumin-induced neutrophilic airway inflammation via a granulocyte colony-stimulating factor-dependent mechanism. Am J Respir Cell Mol Biol. 2012;47:186–195. doi: 10.1165/rcmb.2011-0322OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dai C, Yao X, Vaisman B, Brenner T, Meyer KS, Gao M, et al. ATP-binding cassette transporter 1 attenuates ovalbumin-induced neutrophilic airway inflammation. Am J Respir Cell Mol Biol. 2014;51:626–636. doi: 10.1165/rcmb.2013-0264OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Draper DW, Gowdy KM, Madenspacher JH, Wilson RH, Whitehead GS, Nakano H, et al. ATP binding cassette transporter G1 deletion induces IL-17-dependent dysregulation of pulmonary adaptive immunity. J Immunol. 2012;188:5327–5336. doi: 10.4049/jimmunol.1101605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yao X, Dai C, Fredriksson K, Dagur PK, McCoy JP, Qu X, et al. 5A, an apolipoprotein A-I mimetic peptide, attenuates the induction of house dust mite-induced asthma. J Immunol. 2011;186:576–583. doi: 10.4049/jimmunol.1001534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Barochia AV, Gordon EM, Kaler M, Cuento RA, Theard P, Figueroa DM, et al. High density lipoproteins and type 2 inflammatory biomarkers are negatively correlated in atopic asthmatics. J Lipid Res. 2017;58:1713–1721. doi: 10.1194/jlr.P077776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barochia AV, Kaler M, Cuento RA, Gordon EM, Weir NA, Sampson M, et al. Serum apolipoprotein A-I and large high-density lipoprotein particles are positively correlated with FEV1 in atopic asthma. Am J Respir Crit Care Med. 2015;191:990–1000. doi: 10.1164/rccm.201411-1990OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cirillo DJ, Agrawal Y, Cassano PA. Lipids and pulmonary function in the third National Health and Nutrition Examination Survey. Am J Epidemiol. 2002;155:842–848. doi: 10.1093/aje/155.9.842. [DOI] [PubMed] [Google Scholar]
- 17. Gordon EM, Figueroa DM, Barochia AV, Yao X, Levine SJ. High-density lipoproteins and apolipoprotein A-I: potential new players in the prevention and treatment of lung disease. Front Pharmacol. 2016;7:323. doi: 10.3389/fphar.2016.00323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rastogi D, Fraser S, Oh J, Huber AM, Schulman Y, Bhagtani RH, et al. Inflammation, metabolic dysregulation, and pulmonary function among obese urban adolescents with asthma. Am J Respir Crit Care Med. 2015;191:149–160. doi: 10.1164/rccm.201409-1587OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gowdy KM, Madenspacher JH, Azzam KM, Gabor KA, Janardhan KS, Aloor JJ, et al. Key role for scavenger receptor B-I in the integrative physiology of host defense during bacterial pneumonia. Mucosal Immunol. 2015;8:559–571. doi: 10.1038/mi.2014.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. 1996;271:518–520. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 21. Cao G, Garcia CK, Wyne KL, Schultz RA, Parker KL, Hobbs HH. Structure and localization of the human gene encoding SR-BI/CLA-1: evidence for transcriptional control by steroidogenic factor 1. J Biol Chem. 1997;272:33068–33076. doi: 10.1074/jbc.272.52.33068. [DOI] [PubMed] [Google Scholar]
- 22. Murao K, Terpstra V, Green SR, Kondratenko N, Steinberg D, Quehenberger O. Characterization of CLA-1, a human homologue of rodent scavenger receptor BI, as a receptor for high density lipoprotein and apoptotic thymocytes. J Biol Chem. 1997;272:17551–17557. doi: 10.1074/jbc.272.28.17551. [DOI] [PubMed] [Google Scholar]
- 23. Hoekstra M, van der Sluis RJ, Van Eck M, Van Berkel TJC. Adrenal-specific scavenger receptor BI deficiency induces glucocorticoid insufficiency and lowers plasma very-low-density and low-density lipoprotein levels in mice. Arterioscler Thromb Vasc Biol. 2013;33:e39–e46. doi: 10.1161/ATVBAHA.112.300784. [DOI] [PubMed] [Google Scholar]
- 24. Mavridou S, Venihaki M, Rassouli O, Tsatsanis C, Kardassis D. Feedback inhibition of human scavenger receptor class B type I gene expression by glucocorticoid in adrenal and ovarian cells. Endocrinology. 2010;151:3214–3224. doi: 10.1210/en.2009-1302. [DOI] [PubMed] [Google Scholar]
- 25. Rigotti A, Miettinen HE, Krieger M. The role of the high-density lipoprotein receptor SR-BI in the lipid metabolism of endocrine and other tissues. Endocr Rev. 2003;24:357–387. doi: 10.1210/er.2001-0037. [DOI] [PubMed] [Google Scholar]
- 26. Vergeer M, Korporaal SJ, Franssen R, Meurs I, Out R, Hovingh GK, et al. Genetic variant of the scavenger receptor BI in humans. N Engl J Med. 2011;364:136–145. doi: 10.1056/NEJMoa0907687. [DOI] [PubMed] [Google Scholar]
- 27. Cai L, Ji A, de Beer FC, Tannock LR, van der Westhuyzen DR. SR-BI protects against endotoxemia in mice through its roles in glucocorticoid production and hepatic clearance. J Clin Invest. 2008;118:364–375. doi: 10.1172/JCI31539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gilibert S, Galle-Treger L, Moreau M, Saint-Charles F, Costa S, Ballaire R, et al. Adrenocortical scavenger receptor class B type I deficiency exacerbates endotoxic shock and precipitates sepsis-induced mortality in mice. J Immunol. 2014;193:817–826. doi: 10.4049/jimmunol.1303164. [DOI] [PubMed] [Google Scholar]
- 29. House JS, Li H, DeGraff LM, Flake G, Zeldin DC, London SJ. Genetic variation in HTR4 and lung function: GWAS follow-up in mouse. FASEB J. 2015;29:323–335. doi: 10.1096/fj.14-253898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wilson RH, Whitehead GS, Nakano H, Free ME, Kolls JK, Cook DN. Allergic sensitization through the airway primes Th17-dependent neutrophilia and airway hyperresponsiveness. Am J Respir Crit Care Med. 2009;180:720–730. doi: 10.1164/rccm.200904-0573OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tighe RM, Birukova A, Yaeger MJ, Reece SW, Gowdy KM. Euthanasia- and lavage-mediated effects on bronchoalveolar measures of lung injury and inflammation. Am J Respir Cell Mol Biol. 2018;59:257–266. doi: 10.1165/rcmb.2017-0357OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang X, Katwa P, Podila R, Chen P, Ke PC, Rao AM, et al. Multi-walled carbon nanotube instillation impairs pulmonary function in C57BL/6 mice. Part Fibre Toxicol. 2011;8:24. doi: 10.1186/1743-8977-8-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Eisenbarth SC, Piggott DA, Huleatt JW, Visintin I, Herrick CA, Bottomly K. Lipopolysaccharide-enhanced, toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. J Exp Med. 2002;196:1645–1651. doi: 10.1084/jem.20021340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang P, Thevenot P, Saravia J, Ahlert T, Cormier SA. Radical-containing particles activate dendritic cells and enhance Th17 inflammation in a mouse model of asthma. Am J Respir Cell Mol Biol. 2011;45:977–983. doi: 10.1165/rcmb.2011-0001OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Levy BD, Noel PJ, Freemer MM, Cloutier MM, Georas SN, Jarjour NN, et al. Future research directions in asthma: an NHLBI working group report. Am J Respir Crit Care Med. 2015;192:1366–1372. doi: 10.1164/rccm.201505-0963WS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Arredouani MS, Franco F, Imrich A, Fedulov A, Lu X, Perkins D, et al. Scavenger receptors SR-AI/II and MARCO limit pulmonary dendritic cell migration and allergic airway inflammation. J Immunol. 2007;178:5912–5920. doi: 10.4049/jimmunol.178.9.5912. [DOI] [PubMed] [Google Scholar]
- 37. Baranova IN, Souza AC, Bocharov AV, Vishnyakova TG, Hu X, Vaisman BL, et al. Human SR-BI and SR-BII potentiate lipopolysaccharide-induced inflammation and acute liver and kidney injury in mice. J Immunol. 2016;196:3135–3147. doi: 10.4049/jimmunol.1501709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cai L, Wang Z, Meyer JM, Ji A, van der Westhuyzen DR. Macrophage SR-BI regulates LPS-induced pro-inflammatory signaling in mice and isolated macrophages. J Lipid Res. 2012;53:1472–1481. doi: 10.1194/jlr.M023234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Daan de Boer J, Roelofs JJ, de Vos AF, de Beer R, Schouten M, Hommes TJ, et al. Lipopolysaccharide inhibits Th2 lung inflammation induced by house dust mite allergens in mice. Am J Respir Cell Mol Biol. 2013;48:382–389. doi: 10.1165/rcmb.2012-0331OC. [DOI] [PubMed] [Google Scholar]
- 40. Guo L, Zheng Z, Ai J, Huang B, Li X-A. Hepatic scavenger receptor BI protects against polymicrobial-induced sepsis through promoting LPS clearance in mice. J Biol Chem. 2014;289:14666–14673. doi: 10.1074/jbc.M113.537258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chan TK, Loh XY, Peh HY, Tan WNF, Tan WSD, Li N, et al. House dust mite-induced asthma causes oxidative damage and DNA double-strand breaks in the lungs. J Allergy Clin Immunol. 2016;138:84–96, e1. doi: 10.1016/j.jaci.2016.02.017. [DOI] [PubMed] [Google Scholar]
- 42. Fessler MB, Massing MW, Spruell B, Jaramillo R, Draper DW, Madenspacher JH, et al. Novel relationship of serum cholesterol with asthma and wheeze in the United States. J Allergy Clin Immunol. 2009;124:967–974, e1–e15. doi: 10.1016/j.jaci.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yao X, Fredriksson K, Yu ZX, Xu X, Raghavachari N, Keeran KJ, et al. Apolipoprotein E negatively regulates house dust mite-induced asthma via a low-density lipoprotein receptor-mediated pathway. Am J Respir Crit Care Med. 2010;182:1228–1238. doi: 10.1164/rccm.201002-0308OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med. 2014;20:1327–1333. doi: 10.1038/nm.3704. [DOI] [PubMed] [Google Scholar]
- 45. Howie D, Cobbold SP, Adams E, Ten Bokum A, Necula AS, Zhang W, et al. Foxp3 drives oxidative phosphorylation and protection from lipotoxicity. JCI Insight. 2017;2:e89160. doi: 10.1172/jci.insight.89160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Howie D, Ten Bokum A, Necula AS, Cobbold SP, Waldmann H. The role of lipid metabolism in T lymphocyte differentiation and survival. Front Immunol. 2018;8:1949. doi: 10.3389/fimmu.2017.01949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chesné J, Braza F, Mahay G, Brouard S, Aronica M, Magnan A. IL-17 in severe asthma. Where do we stand? Am J Respir Crit Care Med. 2014;190:1094–1101. doi: 10.1164/rccm.201405-0859PP. [DOI] [PubMed] [Google Scholar]
- 48. Song C, Luo L, Lei Z, Li B, Liang Z, Liu G, et al. IL-17-producing alveolar macrophages mediate allergic lung inflammation related to asthma. J Immunol. 2008;181:6117–6124. doi: 10.4049/jimmunol.181.9.6117. [DOI] [PubMed] [Google Scholar]
- 49. Taylor PR, Roy S, Leal SM, Jr, Sun Y, Howell SJ, Cobb BA, et al. Activation of neutrophils by autocrine IL-17A-IL-17RC interactions during fungal infection is regulated by IL-6, IL-23, RORγt and dectin-2. Nat Immunol. 2014;15:143–151. doi: 10.1038/ni.2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Feingold KR, Shigenaga JK, Kazemi MR, McDonald CM, Patzek SM, Cross AS, et al. Mechanisms of triglyceride accumulation in activated macrophages. J Leukoc Biol. 2012;92:829–839. doi: 10.1189/jlb.1111537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Riffelmacher T, Clarke A, Richter FC, Stranks A, Pandey S, Danielli S, et al. Autophagy-dependent generation of free fatty acids is critical for normal neutrophil differentiation. Immunity. 2017;47:466–480.e5. doi: 10.1016/j.immuni.2017.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mei J, Liu Y, Dai N, Hoffmann C, Hudock KM, Zhang P, et al. Cxcr2 and Cxcl5 regulate the IL-17/G-CSF axis and neutrophil homeostasis in mice. J Clin Invest. 2012;122:974–986. doi: 10.1172/JCI60588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Codarri L, Gyülvészi G, Tosevski V, Hesske L, Fontana A, Magnenat L, et al. RORγt drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2011;12:560–567. doi: 10.1038/ni.2027. [DOI] [PubMed] [Google Scholar]
- 54. El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, et al. The encephalitogenicity of TH17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. 2011;12:568–575. doi: 10.1038/ni.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Laan M, Prause O, Miyamoto M, Sjöstrand M, Hytönen AM, Kaneko T, et al. A role of GM-CSF in the accumulation of neutrophils in the airways caused by IL-17 and TNF-α. Eur Respir J. 2003;21:387–393. doi: 10.1183/09031936.03.00303503. [DOI] [PubMed] [Google Scholar]
- 56. Sheller JR, Polosukhin VV, Mitchell D, Cheng DS, Peebles RS, Blackwell TS. Nuclear factor kappa B induction in airway epithelium increases lung inflammation in allergen-challenged mice. Exp Lung Res. 2009;35:883–895. doi: 10.3109/01902140903019710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Guo L, Song Z, Li M, Wu Q, Wang D, Feng H, et al. Scavenger receptor BI protects against septic death through its role in modulating inflammatory response. J Biol Chem. 2009;284:19826–19834. doi: 10.1074/jbc.M109.020933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Akagawa KS, Kamoshita K, Tokunaga T. Effects of granulocyte-macrophage colony-stimulating factor and colony-stimulating factor-1 on the proliferation and differentiation of murine alveolar macrophages. J Immunol. 1988;141:3383–3390. [PubMed] [Google Scholar]
- 59. Chen BDM, Mueller M, Chou TH. Role of granulocyte/macrophage colony-stimulating factor in the regulation of murine alveolar macrophage proliferation and differentiation. J Immunol. 1988;141:139–144. [PubMed] [Google Scholar]
- 60. Shibata Y, Berclaz P-Y, Chroneos ZC, Yoshida M, Whitsett JA, Trapnell BC. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity. 2001;15:557–567. doi: 10.1016/s1074-7613(01)00218-7. [DOI] [PubMed] [Google Scholar]
- 61. Hoekstra M, Meurs I, Koenders M, Out R, Hildebrand RB, Kruijt JK, et al. Absence of HDL cholesteryl ester uptake in mice via SR-BI impairs an adequate adrenal glucocorticoid-mediated stress response to fasting. J Lipid Res. 2008;49:738–745. doi: 10.1194/jlr.M700475-JLR200. [DOI] [PubMed] [Google Scholar]
- 62. Hoekstra M, Ye D, Hildebrand RB, Zhao Y, Lammers B, Stitzinger M, et al. Scavenger receptor class B type I-mediated uptake of serum cholesterol is essential for optimal adrenal glucocorticoid production. J Lipid Res. 2009;50:1039–1046. doi: 10.1194/jlr.M800410-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Cavalcanti DMH, Lotufo CMC, Borelli P, Tavassi AMC, Pereira ALM, Markus RP, et al. Adrenal deficiency alters mechanisms of neutrophil mobilization. Mol Cell Endocrinol. 2006;249:32–39. doi: 10.1016/j.mce.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 64. Caramori G, Adcock I. Anti-inflammatory mechanisms of glucocorticoids targeting granulocytes. Curr Drug Targets Inflamm Allergy. 2005;4:455–463. doi: 10.2174/1568010054526331. [DOI] [PubMed] [Google Scholar]
- 65. Falcón-Beas C, Tittarelli A, Mora-Bau G, Tempio F, Pérez C, Hevia D, et al. Dexamethasone turns tumor antigen-presenting cells into tolerogenic dendritic cells with T cell inhibitory functions. Immunobiology. 2019;224:697–705. doi: 10.1016/j.imbio.2019.05.011. [DOI] [PubMed] [Google Scholar]
- 66. McKinley L, Alcorn JF, Peterson A, Dupont RB, Kapadia S, Logar A, et al. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J Immunol. 2008;181:4089–4097. doi: 10.4049/jimmunol.181.6.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Henriquez A, House J, Miller DB, Snow SJ, Fisher A, Ren H, et al. Adrenal-derived stress hormones modulate ozone-induced lung injury and inflammation. Toxicol Appl Pharmacol. 2017;329:249–258. doi: 10.1016/j.taap.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Choi IS, Sim DW, Kim SH, Wui JW. Adrenal insufficiency associated with long-term use of inhaled steroid in asthma. Ann Allergy Asthma Immunol. 2017;118:66–72. doi: 10.1016/j.anai.2016.10.002. [DOI] [PubMed] [Google Scholar]
- 69. Haida M, Ogawa K, Hashiguchi A, Inoue T. Adrenal insufficiency in asthma treated with high/low systemic corticosteroids in the past and in those who sucessfully discontinued systemic corticosteroids after treatment with omalizmab. Eur Respir J. 2017;50:PA3590. [Google Scholar]