

Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has rapidly become a global pandemic. In addition to the acute pulmonary symptoms of coronavirus disease (COVID-19) (the disease associated with SARS-CoV-2 infection), pulmonary and distal coagulopathies have caused morbidity and mortality in many patients. Currently, the molecular pathogenesis underlying COVID-19–associated coagulopathies are unknown. Identifying the molecular basis of how SARS-CoV-2 drives coagulation is essential to mitigating short- and long-term thrombotic risks of sick and recovered patients with COVID-19. We aimed to perform coagulation-focused transcriptome analysis of in vitro infected primary respiratory epithelial cells, patient-derived bronchial alveolar lavage cells, and circulating immune cells during SARS-CoV-2 infection. Our objective was to identify transcription-mediated signaling networks driving coagulopathies associated with COVID-19. We analyzed recently published experimentally and clinically derived bulk or single-cell RNA sequencing datasets of SARS-CoV-2 infection to identify changes in transcriptional regulation of blood coagulation. We also confirmed that the transcriptional expression of a key coagulation regulator was recapitulated at the protein level. We specifically focused our analysis on lung tissue–expressed genes regulating the extrinsic coagulation cascade and the plasminogen activation system. Analyzing transcriptomic data of in vitro infected normal human bronchial epithelial cells and patient-derived bronchial alveolar lavage samples revealed that SARS-CoV-2 infection induces the extrinsic blood coagulation cascade and suppresses the plasminogen activation system. We also performed in vitro SARS-CoV-2 infection experiments on primary human lung epithelial cells to confirm that transcriptional upregulation of tissue factor, the extrinsic coagulation cascade master regulator, manifested at the protein level. Furthermore, infection of normal human bronchial epithelial cells with influenza A virus did not drive key regulators of blood coagulation in a similar manner as SARS-CoV-2. In addition, peripheral blood mononuclear cells did not differentially express genes regulating the extrinsic coagulation cascade or plasminogen activation system during SARS-CoV-2 infection, suggesting that they are not directly inducing coagulopathy through these pathways. The hyperactivation of the extrinsic blood coagulation cascade and the suppression of the plasminogen activation system in SARS-CoV-2–infected epithelial cells may drive diverse coagulopathies in the lung and distal organ systems. Understanding how hosts drive such transcriptional changes with SARS-CoV-2 infection may enable the design of host-directed therapeutic strategies to treat COVID-19 and other coronaviruses inducing hypercoagulation.
Keywords: SARS-CoV-2, COVID-19, coagulopathy, tissue factor, coagulation cascade
Clinical Relevance
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has rapidly become a global pandemic. In addition to the acute pulmonary symptoms of coronavirus disease (COVID-19) (the disease associated with SARS-CoV-2 infection), pulmonary and distal coagulopathies have caused morbidity and mortality in many patients. Currently, the molecular pathogenesis underlying COVID-19–associated coagulopathies is unknown. Identifying the molecular basis of how SARS-CoV-2 drives coagulation is essential to mitigating short- and long-term thrombotic risks of sick and recovered patients with COVID-19.
In December 2019, the novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) coronavirus emerged in Wuhan, China (1). It has since spread globally, causing societal shutdowns, with nearly 110 million global cases and 2.5 million deaths (2, 3). Severe cases are often complicated by acute respiratory distress syndrome (ARDS) and hyperinflammation, with many patients requiring mechanical ventilation and ICU admission because of hypoxia and pneumonia (4, 5). However, the pathology of COVID-19 also impacts organ systems and tissues beyond the lung, including the kidneys, gut, liver, and brain (6–9), and many of the most concerning distal pathologies have been associated with increased blood coagulation and clotting (8, 10–14).
Previous work has shown that increases in procoagulant biomarkers are associated with greater mortality rates for patients suffering acute lung injury (ALI) (15–17). In addition, modulation of blood coagulation and fibrinolysis have previously been proposed to treat ALI (18). Coagulopathies concomitant to ALI and ARDS have been hypothesized to emerge because of interactions of inflammation and the extrinsic coagulation cascade, much of which are through interaction with the vascular endothelium (19). Although some valid clinical reports have reported rare instances of coagulopathy complications occurring with influenza infection or influenza-like illness, case–control studies have not identified a significant association with these infections and pulmonary embolisms or deep vein thromboses (20, 21). Even among a study focusing only on the pulmonary pathology of patients with lethal 2009 pandemic swine flu in Brazil, 6 of 21 patients had microthrombi, and only four had pulmonary embolism (22). Coagulopathies have been observed in coronavirus infection at far greater rates compared with other viral infections driving pulmonary inflammation. For instance, a study of pulmonary pathology of 20 patients with lethal SARS-CoV in Toronto observed fibrin thrombi in 17 of 20 patients. Of these 20 emboli, 12 resulted in pulmonary infarction (23). From the extraordinarily large pool of SARS-CoV-2 clinical data, it is clear that severe COVID-19 pathology is associated with and partially driven by coagulopathies. One study from Tongji Hospital of Huazhong University found that disseminated intravascular coagulation occurred in 71.4% of patients, with elevated D-dimer, prothrombin time, and fibrinogen degradation products in the blood (24). A clinical study in the Netherlands reported in 31% of ICU patients with COVID-19 suffered coagulation complications, 81% of which were pulmonary emboli and 27% of which were deep vein thrombi (25). It has become standard practice for blood-thinning treatments to be administered prophylactically to minimize the risk of COVID-19–associated coagulopathies, and clinical trials to investigate the efficacy of common anticoagulants at mitigating COVID-19 are ongoing (26–28). (ClinicalTrials.gov identifiers: NCT04333407 and NCT04365309).
Blood coagulation is primarily regulated by three highly interconnected molecular signaling pathways, platelet activation, the coagulation cascade, and fibrinolysis (29–37). The extrinsic blood coagulation pathway is affected through the initial activity of the protein TF (tissue factor), which drives the cascading activation of several zymogen coagulation factors, including factor VII, factor V, and factor X. The result of the extrinsic coagulation cascade is the conversion of prothrombin into thrombin, which crosslinks fibrin into a mesh essential for clot formation. The activation of this zymogen cascade is similarly balanced by endogenously encoded inhibitors, including TF pathway inhibitor(TFPI) and APC (activated protein C). Plasmin suppresses coagulation via proteolytic degradation of this cross-linked fibrin mesh in blood clots.
Most hypotheses propose that COVID-19 coagulopathies are indirectly induced by acute inflammation and cytokine secretion associated with SARS-CoV-2, but the precise mechanisms underlying coagulopathies remain elusive (38, 39). Identifying the cellular source of signal transducers initially driving coagulopathy will be critical in understanding and mitigating SARS-CoV-2–induced coagulopathies. To this end, we performed a post hoc analysis of publicly available transcriptomics datasets of SARS-CoV-2–infected normal human bronchial epithelial cells (NHBEs), BAL fluid (BALF) from patients with COVID-19, and COVID-19 patient peripheral blood mononuclear cells (PBMCs), with the goal of identifying possible etiologies of SARS-CoV-2–induced coagulopathies (40–42). Our study demonstrates that changes to the lung epithelium directly caused by SARS-CoV-2 infection may directly contribute to the induction of coagulopathy seen in patients with COVID-19 via the modulation of the extrinsic coagulation cascade and plasminogen activation system. The altered transcriptional profile of the lung epithelium and increased production of TF protein as a result of SARS-CoV-2 infection is a likely contributor to COVID-19–associated coagulopathies in the lung and a possible contributor to systemic coagulopathies. Lung epithelial cells, as the primary target of SARS-CoV-2, may play a key role in the initiation of coagulopathies observed during COVID-19. Such changes likely occur upstream of or in concert with SARS-CoV-2–induced changes in lung endothelial cells, platelets, and immune cell–driven inflammation–thrombosis circuits that may also drive coagulopathies. These findings do not rule out coagulation defects driven by immune cells and the vascular endothelium but suggest that the lung epithelium as an additional factor driving coagulopathy in patients with COVID-19.
Some of the results of these studies have been previously reported in the form of a preprint (bioRxiv, [07 July 2020] https://doi.org/10.1101/2020.07.06.182972).
Methods
Xiong and Colleagues—Bulk RNA Sequencing Analysis of BALF and PBMCs from Patients with SARS-CoV-2 Infection
Bulk BALF and PBMC transcriptomics data were generated through RNA sequencing (RNA-seq) of purified cells from SARS-CoV-2–infected patients as described in Xiong and colleagues (40). Raw sequencing data were accessed via the Chinese Academy of Science’s Genome Sequence Archive (GSA) (COVID+ BALF: GSA accession number CRP001417; PBMCs: GSA accession number CRA002390). Three BALF control samples isolated from healthy volunteers were downloaded from a publicly available National Center for Biotechnology Information (NCBI) dataset at sample accession numbers SRR10571724, SRR10571730, and SRR10571732 (43).
Blanco-Melo and Colleagues—Bulk RNA-Seq Analysis of SARS-CoV-2–infected NHBEs Cultured In Vitro
NHBEs were cultured under nondifferentiating conditions in bronchial epithelial growth media. SARS-CoV-2 isolate USA-WA1/2020 (NR-52281) was used to infect NHBEs (5 × 105) at a multiplicity of infection (MOI) of 2 for 24 hours and/or mock-infected in their culture media before RNA purification, library preparation, and sequencing, as described in as described in Blanco-Melo and colleagues (41). Raw sequencing data were accessed via the NCBI Gene Expression Omnibus (GEO) (accession number GSE63473).
Blanco-Melo and Colleagues—Bulk RNA-Seq Analysis of H1N1-infected NHBEs Cultured In Vitro
NHBEs were infected with A/Puerto Rico/8/1934 influenza A virus (IAV) at an MOI of 3 for 12 hours before RNA purification, library preparation, and sequencing, as described in Blanco-Melo and colleagues (41). Raw sequencing data were accessed via the NCBI GEO (accession number GSE63473).
Bulk RNA-Seq Analysis Pipeline
The analysis pipeline described below was used to analyze NHBE, PBMC, and BALF bulk RNA-seq data sets. Sequencing reads were downloaded in their raw fastq format. Read adapter and quality trimming was performed using Trim Galore!, and sequencing quality was confirmed using the FASTQC and MULTIQC (44, 45). Sequence alignment to the GRCh38 reference transcriptome was performed using Salmon, and differential gene expression analysis was performed using DESeq2 (46, 47). DESeq2-adjusted P values are produced by the Wald test and corrected for multiple hypothesis testing using the Benjamini-Hochberg method. See data supplement for full differential expression results of each bulk RNA-seq data set. (Data Files E1–E4) Gene plots and heat maps were generated in R using the pheatmap or ggplot2 R packages (48, 49).
SARS-CoV-2 Infection of NHBEs Cultured In Vitro for Quantification of TF Protein
NHBEs were cultured in Pneumacult-Ex media (#05008; STEMCELL) using the standard formulation recommended by the manufacturer. NHBEs (8.5 × 104 per well) were plated onto tissue culture–treated 24-well plates and grown to 85% confluence before infection. NHBEs were then infected with SARS-CoV-2 (USA-WA1/2020) at an MOI of 2 for 24 hours before supernatant and cell extract samples were collected for quantification of TF by ELISA. ELISA-based quantification of TF protein concentrations was performed using a Human Tissue Factor ELISA Kit (ab220653; Abcam) according to manufacturer’s instructions.
Liao and Colleagues—Single-Cell RNA-Seq Analysis of COVID-19 BALF
BALF samples were obtained from 13 patients with COVID-19 in Shenzhen Third People’s Hospital from January 2020 to February 2020. Patient disease severity was stratified as moderate, severe, or critical on the basis of the “Diagnosis and Treatment Protocol of COVID-19 by the National Health Commission of China.” (50) Single-cell RNA-seq libraries were generated using Chromium Single Cell V(D)J reagent kits (PN-1000006, PN-100014, PN-100020, and PN-1000005; 10x Genomics) according to the manufacturer’s protocol. Three healthy control BALF samples were also processed, and a fourth additional healthy control sample was included from the GEO (GSE128033). Refer to Liao and colleagues for further methodological detail (42). Single-cell RNA-seq data were accessed as a fully annotated Seurat R data file (rds format) deposited by the study authors at http://cells.ucsc.edu/covid19-balf/nCoV.rds. Quality control, visualization, and analysis were performed using Seurat, an R package for processing single-cell RNA-seq data.
Results
Coagulation Pathway Gene Expression in NHBEs Is Impacted by Infection with SARS-CoV-2
To determine the impact SARS-CoV-2 infection has on the coagulation cascade, we examined the transcriptional regulation of hemostasis and thrombosis, including the extrinsic coagulation pathway and the plasminogen activation system. We identified 12 differentially expressed genes that are part of the regulation of blood coagulation gene ontology (GO) term (GO:0030193) (Figure 1A).
Figure 1.
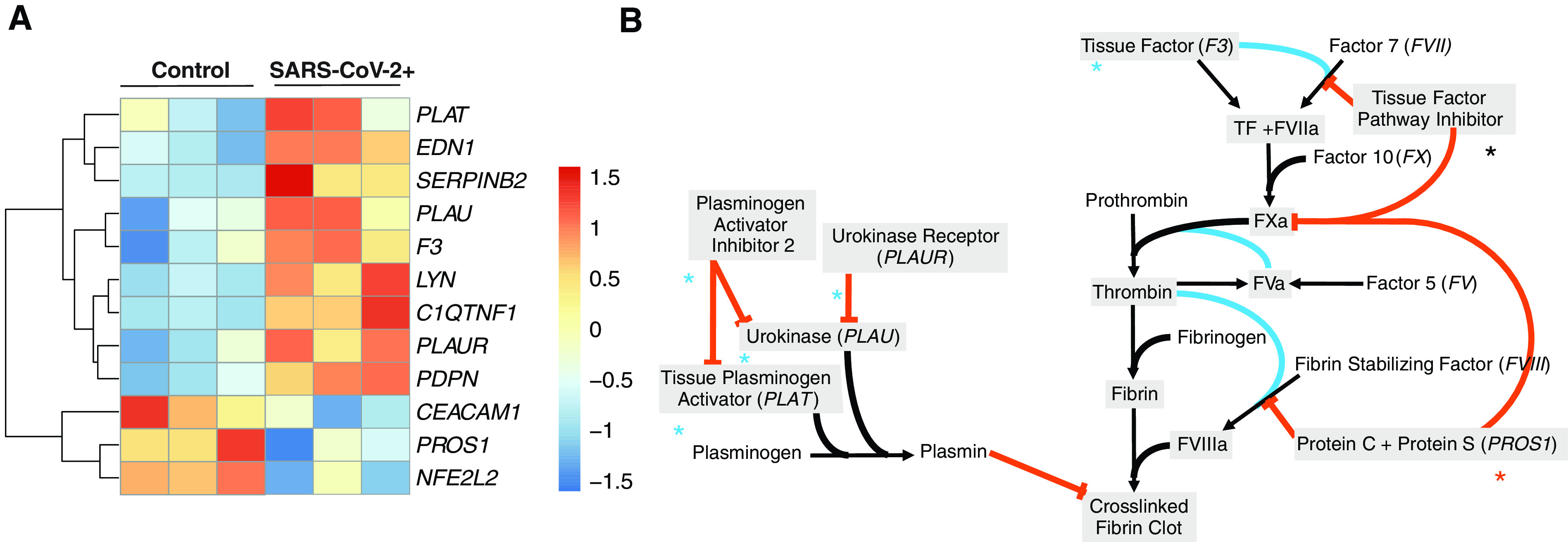
The gene expression profile of differentially expressed genes within the enriched regulation of blood coagulation gene ontology (GO) term for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)–infected NHBEs. (A) Heatmap of selected genes in the regulation of blood coagulation GO term. Bolded genes represent differentially expressed genes as calculated by DESeq2 (P.adj < 0.05). (B) Pathway map of the extrinsic blood coagulation cascade (right) and the plasminogen activation system (left) with overlaid expression values. Blue asterisks indicate upregulation, black asterisks indicate no change, and red asterisks indicate downregulation. NHBE = normal human bronchial epithelial cell; P.adj = P adjusted.
Visualization of gene expression directionality on pathway maps for the extrinsic blood coagulation cascade and plasminogen activation pathway (Figure 1B and Table E1 in the data supplement), illustrate how infected respiratory epithelial cells may drive coagulopathy in COVID-19. Most notably, the extrinsic coagulation cascade master signal transducer, TF (F3), is significantly transcriptionally upregulated, whereas balancing inhibitory proteins are unmodified or significantly downregulated. In addition, although plasminogen-activating proteins are significantly upregulated, plasminogen-activating inhibitors and localizing receptors also increase. PDPN and EDN1 are also notably upregulated in the context of SARS-CoV-2 infection of NHBEs.
Upregulation of TF in NHBEs Infected with SARS-CoV-2
One epithelial expressed factor that is known to be a major driver of coagulopathy is increased expression of F3. F3 encodes the TF protein, which is the master regulator responsible for the initiation of the extrinsic coagulation cascade. F3 gene expression was found to be significantly upregulated in SARS-CoV-2–infected NHBEs, as performed by Blanco-Melo and colleagues (Figure 2A). To further confirm the upregulation of TF by NHBEs during SARS-CoV-2 infection at the protein level, we replicated the NHBE infection experiments performed by Blanco-Melo and colleagues as described in the methods section. The average amount of TF associated with the cellular fraction of NHBEs increased by 62.34%. (Figure 2B). Similarly, the average amount of TF that was released from NHBEs into the culture supernatant increased by 64.1% with SARS-CoV-2 infection (Figure 2C).
Figure 2.

(A) Violin plot depicting raw counts of reads mapping to key regulators of the extrinsic blood coagulation cascade in mock-infected and SARS-CoV-2–infected NHBEs as performed by Blanco-Melo and colleagues. Raw counts were normalized to library size in the DESeq2 software package. P.adj values for all differentially expressed genes were also calculated within DESeq2. Images were generated using GGPlot2 in the R studio environment. (B and C) ELISA measurements quantifying TF (tissue factor) in lysate and supernatant respectively. Samples were isolated at 24 hours after infection (multiplicity of infection 2) in NHBEs. Plotted values are the mean of two technical replicates ± SD. P values were determined using an unpaired two-tailed t test. CTRL = control.
NHBE Regulation of the Coagulation Cascade and Plasminogen with SARS-CoV-2 Infection
Extrinsic coagulation cascade signaling is regulated by the balance of TF with endogenously encoded inhibitors. The first inhibitor in this cascade is TFPI (TF pathway inhibitor), which encodes the TFPI protein. TFPI suppresses coagulation by inhibiting TF’s activation of coagulation factor VII, the first zymogen along the cascade. TFPI transcription is not significantly different in SARS-CoV-2–infected NHBEs (Figure 3A). TFPI is also responsible for inhibiting the activation of coagulation factor V via the inhibition of factor X. Significant increases in the amount of factor V have been correlated with greater COVID-19 disease severity and coagulopathy risk (50). Maintenance of homeostasis between TF and TFPI is essential to limit clotting. An increase of TF without corollary increases of TFPI could contribute to coagulopathies in patients with COVID-19 through unimpeded TF and coagulation factor X signaling (51).
Figure 3.
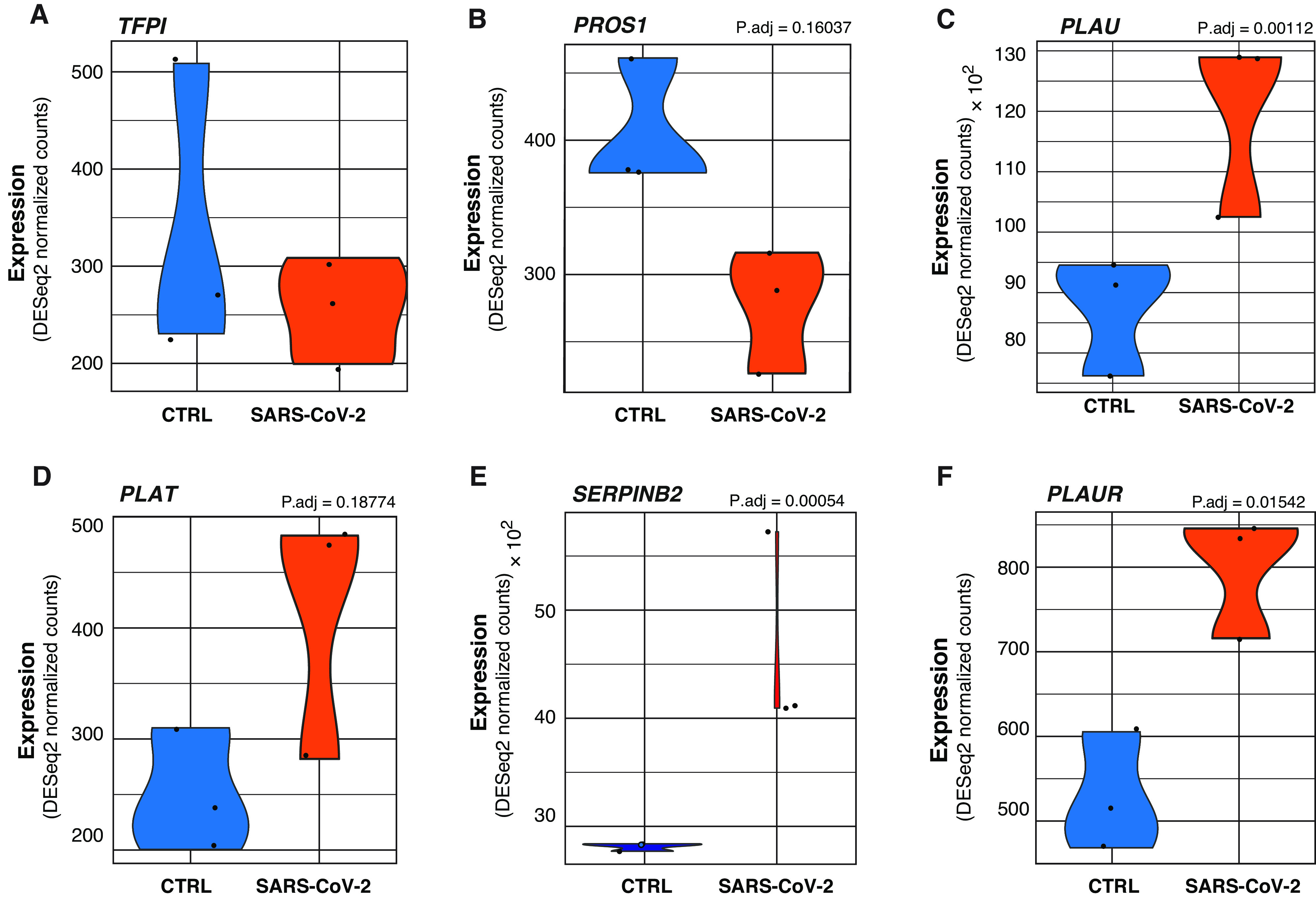
(A–F) Violin plots depicting raw counts of reads mapping to negative regulators of the extrinsic coagulation cascade and regulators of the plasminogen activation system in mock-infected and SARS-CoV-2–infected NHBEs. Raw counts were normalized to library size in the DESeq2 software package. P.adj values displayed for significant differences were also calculated within DESeq2. Images were generated using GGPlot2 in the R studio environment.
Decreased Expression of PROS1 in NHBEs Infected with SARS-CoV-2
The PROS1 gene, which encodes protein S, was also downregulated in SARS-CoV-2–infected NHBEs (Figure 3B). The primary function of protein S is to antagonize the coagulation cascade by complexing with protein C. The complex (known as activated protein C) inhibits the maturation of procoagulation factors Va and VIIIa, suppressing thrombin maturation and activity. The activity of both protein C and protein S is required for this effect (30). Protein C also promotes TFPI activity (35). Interestingly, membrane-bound protein S also contributes to antiinflammatory efferocytic clearance by mediating membrane dynamics between macrophages and epithelial cells. Decreased lung epithelial PROS1 expression may further exacerbate COVID-19–related pathology through diverse mechanisms (52). Although protein S is canonically known to be produced in the liver together with protein C, biologically significant amounts of protein S are known to be produced in the lung, kidney, and gonads, where it play key roles in the regulation of tissue homeostasis (25).
In addition, within the regulation of blood coagulation GO term, several genes regulating the activity of plasminogen were identified. PLAU (encoding urokinase) and PLAT (encoding tissue plasminogen activator) are significantly increased in SARS-CoV-2 NHBEs (Figures 3C and 3D). Although PLAU is responsible for both plasminogen activating and tissue remodeling, research has also shown that upregulation of PLAU in the context of lung epithelia can induce increased TF expression and coagulation despite its plasminogen activation (36). Infected NHBEs also significantly upregulate SERPINB2, which encodes the protein PAI-2 (plasminogen activator inhibitor 2) (Figure 3E). PAI-2 inhibits urokinase and tissue plasminogen activator via proteolytic inactivation of plasminogen activators (53). PAI-2 is commonly cytoplasmic, but membrane-permeabilizing epithelial cell death in patients with COVID-19 may drive the secretion of cytoplasmic proteins such as PAI-2 during SARS-CoV-2 infection (54). The expression of PLAUR, a receptor localizing activated urokinase to the extracellular membrane, is also significantly increased in SARS-CoV-2–infected NHBEs (Figure 3F). The localized activity of PAI-2 may significantly inhibit the effect of PLAU/PLAUR in complex and thereby contribute to the formation of pulmonary embolisms and distal coagulopathies.
Regulation of Blood Coagulation by Cells Isolated from the BALF of Patients with COVID-19
We next hypothesized that NHBEs differentially expressed genes enriched in the regulation of blood coagulation (GO:0030193) GO term would be similarly active in BALF from patients with COVID-19. We expected to see this recapitulation of the observed NHBE phenotype in patient BALF because of the pulmonary epithelial cell fraction of cells in the BALF cell mixture. Plotting BALF expression data for this gene list revealed a clear and consistent pattern of transcriptional regulation as well (Figure 4A). In addition, plotting of the expression data of BALF differentially expressed genes in the regulation of blood coagulation GO term revealed further regulation of the coagulation cascade (Figure 4B).
Figure 4.

The gene expression profile of differentially enriched genes from RNA isolated from the BAL fluid (BALF) of patients with coronavirus disease (COVID-19). (A) Heatmap presentation of select differentially expressed genes presented in Figure 2 (NHBEs infected with SARS-CoV-2). The expression data presented in the heat map demonstrate the expression profile of those genes in BALF-derived samples. Bolded genes represent differentially expressed genes as calculated by DESeq2 (P.adj < 0.05). (B) Heatmap presentation of an expanded selection of all differentially enriched expressed genes within the enriched regulation of blood coagulation GO term (GO:0007596) from BALF-derived heathy control subjects and patient samples.
Many of these expression signatures recapitulate the activity of in vitro SARS-CoV-2–infected NHBEs. These include upregulation of procoagulation genes such as F3 (TF), SERPINA10, and SERPINB2 and downregulation of anticoagulation genes such as PROS1 and PLAUR, and PLAT. In addition, PROCR, encoding a receptor augmenting the inhibitory activity of protein S and protein C, was suppressed in BALF from patients with COVID-19. Unlike the NHBE expression profile, there is increased TFPI and PLAT expression in BALF from patients with COVID-19, indicating some antagonism of hypercoagulation. However, coagulopathies observed in some patients with COVID-19 indicate that this signaling can be insufficient to prevent hypercoagulation.
Analysis of Coagulation Pathway Gene Expression in PBMCs
To determine whether gene expression regulating the extrinsic coagulation cascade or plasminogen activation system was changed in circulating immune cells, we analyzed transcriptomes of purified PBMCs from patients with COVID-19 as described in Xiong and colleagues (40) PantherDB Functional enrichment analysis found no enrichment of genes regulating or effecting coagulation in PBMCs from patients with COVID-19 (Figure 5). The primary publication of these datasets describes expected induction of hyperinflammation and the immune cell death. We concluded it is unlikely that circulating immune cells during SARS-CoV-2 infection are driving coagulopathies via the coagulation cascade or plasminogen activation system.
Figure 5.

The gene expression profile of differentially enriched genes from RNA isolated from peripheral blood mononuclear cells (PBMCs) of patients with COVID-19. The genes included in this heatmap were identified as enriched in the regulation of blood coagulation GO term for NHBEs infected with SARS-CoV-2. No genes are bolded, as none were found to be differentially expressed as calculated by DESeq2 (P.adj < 0.05). The expression data presented in the heat map demonstrate the expression profile of these genes in PBMC-derived samples.
Infection of Human Lung Epithelial Cells with IAV Does Not Impact Coagulation Pathway Gene Expression
To determine whether these transcriptional changes are specific for SARS-CoV-2 or are more generalizable to respiratory viruses that infect the lung epithelium, we analyzed datasets from IAV-infected NHBE cells. Heatmap plotting of genes found to be differentially expressed in NHBEs during SARS-CoV-2 infection (Figure 1A) did not reveal notable signatures during IAV infection (Figure 6). Most critically, the master regulator of the coagulation cascade, TF (F3), is not differentially expressed in the context of IAV infection. This would significantly lessen the hemostatic impact of suppressed plasminogen activation. These findings are consistent with the lesser degree of coagulopathy associated with IAV in the clinic and further support the notion that coagulopathies during SARS-CoV-2 infection are independent of systemic inflammation common to both infections. COVID-19–associated coagulopathies may be triggered by changes in lung epithelial transcription uniquely induced by SARS-CoV-2 infection.
Figure 6.

The gene expression profile of differentially enriched genes from RNA isolated from NHBEs infected with A/Puerto Rico/8/1934 (PR8) influenza A virus (IAV). The genes included in this heatmap were identified as enriched in the regulation of blood coagulation GO term for NHBEs infected with SARS-CoV-2. The expression data presented in the heat map demonstrate the expression profile of these genes in NHBE cultures that are mock-infected or infected with PR8 IAV at a multiplicity of infection of 3. Bolded genes represent differentially expressed genes as calculated by DESeq2 (P.adj < 0.05).
Analysis of BALF from Patients with COVID-19 Single-Cell RNA-Seq
In vitro bulk RNA-seq data from SARS-CoV-2–infected NHBEs and BALF from patients with COVID-19 suggest that pulmonary epithelial cells contribute to the induction of COVID-19–associated coagulopathy through the expression of key genes regulating the extrinsic coagulation cascade and the plasminogen activation system. However, to further confirm these results, we analyzed single-cell RNA-seq of BALF samples from patients with COVID-19.
We accessed COVID-19 BALF single-cell RNA-seq data generated and published by Liao and colleagues (42) These data provided strong evidence that pulmonary epithelial cells in patients with COVID-19 are transcribing signals that likely contribute to diverse COVID-19–associated coagulopathies. (Figure 7) Figure 7A shows a uniform manifold approximation projection (UMAP) plot representing the cell-type–specific clustering of the Liao and colleagues’ data. The subset identified as epithelial cells comprised ~5% of the total cells sequenced and clustered away from all other immune cell subtypes identified. Figure 7B represents a dot plot depiction of expression data from this epithelial cell subset only, with patient samples stratified by disease severity. Figure 7C depicts the UMAP feature plots, highlighting the cell types expressing each gene and the intensity of that expression. From these data, we can conclude that lung epithelial cells in COVID-19 patients are significantly upregulating the expression of TF, the master regulator driving the extrinsic coagulation cascade. Furthermore, although there is an upregulation of the TF’s cognate inhibitor, TF pathway inhibitor, the degree of upregulation is smaller and restricted to fewer than half as many cells. In addition, during severe COVID-19, protein S expression is entirely lost in many pulmonary epithelial cells, where it is expressed at homeostasis. Cells that continue to express the transcript do so at much lower concentrations. These data also demonstrate that pulmonary epithelial cells are significantly upregulating the expression of proteins that suppress the anticoagulant effect of plasminogen. These changes include the upregulation of plasminogen activator inhibitors, such as SERPIN protease inhibitors, and the upregulation of PLAUR, a plasminogen urokinase-localizing protein. However, it is important to note that although these expression patterns were observed in vitro and in patient pulmonary epithelial cells, significant expression changes regarding the plasminogen activation system were also observed in macrophages and neutrophils that would likely also contribute to a procoagulative state.
Figure 7.
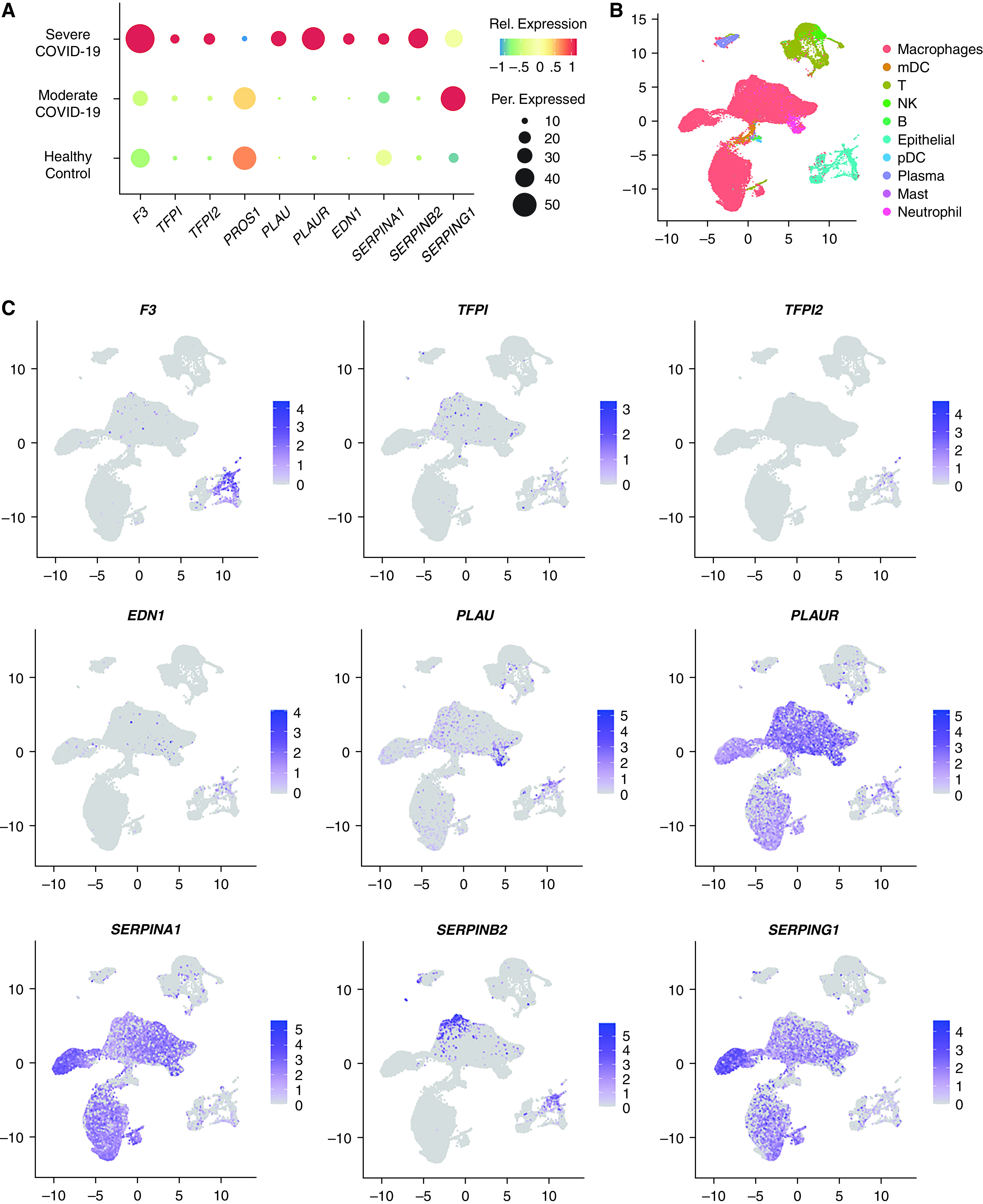
Single-cell RNA-seq analysis of select extrinsic coagulation cascade and plasminogen regulating genes in all epithelial cells from BALF samples described in Liao and colleagues (42). See Liao and colleagues for a full description of the epithelial cell markers used for identification. (A) Seurat-generated dot plot summarizing a selection of genes regulating coagulation. Dot color is representative of the degree of relative (Rel.) expression for each gene, and dot size is representative of the percent of cells expressing each gene. (B) Seurat-generated uniform manifold approximation projection (UMAP) plot depicting cell clustering for all identified cell types in Liao and colleagues. See Liao and colleagues for a full description of settings used for the Seurat UMAP computation. (C) Seurat-generated feature plots showing the distribution and Rel. expression of mRNA for selected genes regulating coagulation in cells isolated from BALF.
Discussion
The combined analysis of data from Blanco-Melo and colleagues, Xiong and colleagues, and Liao and colleagues collectively demonstrates how pulmonary epithelial cells may drive transcriptional responses promoting COVID-19–associated coagulopathies, including induction of the extrinsic coagulation cascade without compensatory inhibitory signals and the suppression of the plasminogen activation system via the upregulation of plasminogen inactivation proteins and localization factors. In vitro NHBE infection data from Blanco-Melo and colleagues further demonstrate that COVID-19 induced transcriptional changes driving the extrinsic coagulation cascade and suppressing the plasminogen activation system are not similarly induced during IAV infection. These data suggest the possibility coagulopathy in patients with COVID-19 may be more prevalent relative to patients with IAV because of the transcriptional changes SARS-CoV-2 induces during infection of the pulmonary epithelium. In addition, through replication of the infection model performed by Blanco-Melo and colleagues, we were able to demonstrate that the changes in mRNA detected through next-generation sequencing were consistent with an increase in the concentrations of secreted and cellular-bound TF during NHBE infection. The increase in TF protein concentrations was significantly greater than the detected increase in mRNA concentrations, providing further evidence that epithelial cell–derived TF is a likely contributor to COVID-19–associated coagulopathy in the pulmonary space. It is also possible that such epithelial-derived signals may contribute to COVID-19–associated coagulopathies beyond the lung, but further experimental evidence is needed to confirm a possible mechanistic link. Understanding these epithelial-derived signals and their interaction with other procoagulant signals that SARS-CoV-2 may induce (such as endothelial cell dysfunction, platelet hyperactivation, liver dysfunction, or inflammation–thrombosis circuits) will enable the scientific community to devise more effective ways to mitigate COVID-19 and other coronavirus-induced coagulopathies in the future.
There is mounting evidence that coagulation defects are a significant contributor to COVID-19 pathology. Reports of COVID-19–associated coagulopathies have proliferated globally, including acute pulmonary embolism in the microvasculature of the lung as well as cerebral-, renal-, and bowel-localized embolic disease (8, 11, 12). Acute pulmonary thromboembolism presents in 30% of patients with severe clinical COVID-19 by pulmonary computed tomographic angiography, which is also associated with elevation of serum D-dimer. D-dimer is a byproduct of fibrinolysis degrading cross-linked fibrin clots (13). Clinicians also reported that biomarkers of coagulation (clot strength, platelet, and fibrinogen contributions to clot strength and elevated D-dimer concentrations) are significantly increased with COVID-19–associated ARDS (14). A diverse spectrum of proinflammatory mediators shown to be dramatically upregulated in COVID-19 and other coronavirus pathologies are also known to contribute to TF-induced hypercoagulability (55). Recent work by Stefely and colleagues found marked increases in the concentrations of factor V activity associated with severe COVID-19 disease (50). Factor V was the most strongly associated parameter with disease severity across all measurements included in the study. Such increases directly indicate that the induction of the extrinsic coagulation cascade is a hallmark of severe COVID-19 disease, and the data aggregated here suggest that pulmonary epithelial cells are contributing factors in COVID-19–associated coagulopathies.
It is important to note that many other cell types likely play key roles in the induction of COVID-19–associated coagulopathies. Much work is still needed to determine how epithelial cell–derived TF and other procoagulant signals induced by SARS-CoV-2 infection contribute to the induction of COVID-19 pulmonary coagulopathies. It also remains an open question whether epithelial cell–derived procoagulant signals contribute to the induction of systemic coagulopathy through distal interactions with other cell types. Other important players in regulating the coagulation cascade are vascular endothelial cells (56). The possibility of direct endothelial cell infection by SARS-CoV-2 has been proposed as a possible driver of hypercoagulation in COVID-19 (57). However, there has been a lack of confirmatory data from human patients regarding productive endothelial cell infection by SARS-CoV-2 over the course of the pandemic. For instance, immunohistochemical analysis failed to detect reactivity in postmortem staining of pulmonary endothelial cells from patients with COVID-19 (58). In addition, recent in vitro studies from two independent research teams found that primary endothelial cells and pluripotent stem cells differentiated into endothelial cells were not susceptible to direct infection with SARS-CoV-2 (59, 60). Indirect damage of endothelial cells by systemic inflammation or factors derived from infected epithelial cells during SARS-CoV-2–induced ALI is a much more likely driver of these signals. To our knowledge, there are currently no RNA-seq datasets with infected endothelial cell cultures or tissue available. However, such datasets would be invaluable in characterizing endothelial cell responses to epithelial-derived coagulation signals or endothelial induction of SARS-CoV-2–associated coagulopathies. In addition, significant amounts of coagulation regulatory proteins, including protein C and protein S, are synthesized in the liver (61). Liver coagulopathies and dysfunction have also been associated with COVID-19 (62, 63), and the possibility that liver dysfunction in COVID-19 is contributing to coagulation should also be investigated. Recent work has also shown that platelet activation may be involved in the induction of coagulopathy during COVID-19 (64). It is very likely that the induction of SARS-CoV-2–mediated coagulopathies is dependent on the interaction of several tissue types driving the extrinsic coagulation system and platelet activation while suppressing fibrinolysis via plasminogen.
Also, other molecular factors increased with SARS-CoV-2 infection, including phosphatidylserine exposure, IFN expression, ICAM expression, angiotensin II expression, and complement activation, are known to “decrypt” TF from its inactive form on the surface of tissue cells and endothelial cells (30). Such coagulation–inflammation–thrombosis circuit feedback loops, coupled with the multiple zymogen activation–mediated feedback loops within the extrinsic blood coagulation cascade, could significantly contribute to the induction of COVID-19 coagulopathy in patients (30).
Coagulation cascade induction by the epithelia is believed to be necessary during ARDS or ALI and may be protective (65). However, when it becomes dysregulated, it can be damaging. ARDS is often associated with increased biomarkers of coagulation and fibrinolysis. Furthermore, pulmonary edema fluids as well as plasma from patients with ALI contain lesser amounts of anticoagulant protein C and higher amounts of plasminogen activator inhibitors. Some proportion of these key coagulation inhibitors is likely secreted from epithelial and endothelial pulmonary cells and acting on the local environment in addition to proteins in the circulatory system (15–17).
Other genes such as PDPN and EDN1, which are known to drive vasoconstriction and disseminated intravascular coagulation, respectively, in infection contexts, are known to be significantly upregulated during SARS-CoV-2 (66–69). In the lung, podoplanin is expressed by epithelial cells, and endothelin is expressed by epithelial and endothelial cells (70–72).
The role of the lung epithelium in coagulation defects has not been fully explored; however, some prior research illuminates its possible contribution. Lung epithelial cell lines have been shown to have increased expression of TF after incubation with pulmonary edema fluid from patients with ARDS (73). In addition, mouse models demonstrate that lung epithelial-derived TF may play an important role in tissue protection during ALI caused by LPS (74). In vitro experiments with human epithelial cells indicate that TF may also contribute to basal cell survival (75). Taken together, these data suggest that although induction of the extrinsic coagulation cascade by lung epithelial cells may aid host responses during some stages of infection, SARS-CoV-2 infection can trigger changes that drive systemic and local coagulopathies.
Lytic-regulated cell death of respiratory epithelial cells, particularly via pyroptosis, may contribute to COVID-19 pathogenesis (76). During lytic cell death, intracellular contents typically isolated within cell membranes are released. Proteins such as TF, plasminogen-activating inhibitors, and procoagulant factors may be released into the pulmonary space or circulation because of COVID-19–induced lytic cell death of epithelial cells. Such factors likely drive paracrine signaling to nearby endothelial cells in the lung, which could further exacerbate coagulation systemically via the secretion of activated coagulation cascade zymogens and thrombin into the blood. It is also possible that such factors could also enter the blood stream directly near damaged endothelial tissues in the lung, potentially contributing to the induction of systemic coagulopathies observed in patients with severe COVID-19. Epithelial-derived hypercoagulation factors and plasminogen inhibitors may also drive local pulmonary hypercoagulation and exacerbate pulmonary tissue destruction during SARS-CoV-2 infection.
Further investigation of pulmonary endothelial, epithelial, and immune cell responses to SARS-CoV-2 will be essential for unraveling the mystery of COVID-19–induced coagulopathy. Identifying the cellular factors that drive SARS-CoV-2–induced coagulopathy is essential, both for understanding foundational SARS-CoV-2 biology and for optimizing clinical practices. Understanding the transcriptional regulation of the coagulation cascade in the lung epithelium, a particularly druggable target cell type, may help develop therapeutic strategies to mitigate this serious complication of coronavirus infection.
Acknowledgments
Acknowledgment
The authors thank Dr. Meredith Crane, Dr. Sharon Rounds, and Dr. Zhijin Wu for helpful comments and discussion on the manuscript. The authors also thank Eugene Foygelman for sharing his computational resources and hardware expertise with the authors during single-cell RNA sequencing analysis.
Footnotes
Supported by National Institute of General Medical Science Centers of Biomedical Research Excellence Award P20GM109035, National Heart Lung Blood Institute grant 1R01HL126887-01A1 (A.M.J.), National Institute of Allergy and Infectious Disease grant R37 AI067497 (K.A.F.), Brown University COVID-19 Seed Award (A.M.J.), the University of Massachusetts COVID19 Pandemic Research Fund Pilot Grant (K.A.F.), Emergent Ventures Mercatus Center–George Mason University FAST grant awards #2170 and #2227 (A.M.J.), and Brown Molecular Biology, Cell Biology, and Biochemistry T32 (National Institute of General Medical Science) T32GM007601-40 (E.S.F.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author Contributions: E.S.F.: substantial contribution to study design, analysis of transcriptional data, and drafting of the manuscript. A.M.J.: substantial contribution to study design and drafting of the manuscript. Y.C.: experimental data generation and analysis and manuscript drafting for important intellectual content. K.A.F.: manuscript drafting for important intellectual content and study design of experimental data.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2020-0453OC on March 19, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. China Novel Coronavirus Investigating and Research Team. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dong E, Du H, Gardner L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect Dis. 2020;20:533–534. doi: 10.1016/S1473-3099(20)30120-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ayittey FK, Ayittey MK, Chiwero NB, Kamasah JS, Dzuvor C. Economic impacts of Wuhan 2019-nCoV on China and the world. J Med Virol. 2020;92:473–475. doi: 10.1002/jmv.25706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bialek S, Gierke R, Hughes M, McNamara LA, Pilishvili T, Skoff T. CDC COVID-19 Response Team. Coronavirus disease 2019 in children - United States, February 12-April 2, 2020. MMWR Morb Mortal Wkly Rep. 2020;69:422–426. doi: 10.15585/mmwr.mm6914e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Naicker S, Yang CW, Hwang SJ, Liu BC, Chen JH, Jha V. The novel coronavirus 2019 epidemic and kidneys. Kidney Int. 2020;97:824–828. doi: 10.1016/j.kint.2020.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang C, Shi L, Wang FS. Liver injury in COVID-19: management and challenges. Lancet Gastroenterol Hepatol. 2020;5:428–430. doi: 10.1016/S2468-1253(20)30057-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bhayana R, Som A, Li MD, Carey DE, Anderson MA, Blake MA, et al. Abdominal imaging findings in COVID-19: preliminary observations. Radiology. 2020;297:E207–E215. doi: 10.1148/radiol.2020201908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mao L, Jin H, Wang M, Hu Y, Chen S, He Q, et al. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol. 2020;77:683–690. doi: 10.1001/jamaneurol.2020.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Oxley TJ, Mocco J, Majidi S, Kellner CP, Shoirah H, Singh IP, et al. Large-vessel stroke as a presenting feature of Covid-19 in the young. N Engl J Med. 2020;382:e60. doi: 10.1056/NEJMc2009787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grillet F, Behr J, Calame P, Aubry S, Delabrousse E. Acute pulmonary embolism associated with COVID-19 pneumonia detected with pulmonary CT angiography. Radiology. 2020;296:E186–E188. doi: 10.1148/radiol.2020201544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lushina N, Kuo JS, Shaikh HA. Pulmonary, cerebral, and renal thromboembolic disease in a patient with COVID-19. Radiology. 2020;296:E181–E183. doi: 10.1148/radiol.2020201623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Leonard-Lorant I, Delabranche X, Severac F, Helms J, Pauzet C, Collange O, et al. Acute pulmonary embolism in patients with COVID-19 at CT angiography and relationship to D-dimer levels. Radiology. 2020;296:E189–E191. doi: 10.1148/radiol.2020201561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ranucci M, Ballotta A, Dedda UD, Bayshnikova E, Poli MD, Resta M, et al. The procoagulant pattern of patients with COVID-19 acute respiratory distress syndrome. J Thromb Haemost. 2020;18:1747–1751. doi: 10.1111/jth.14854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McClintock D, Zhuo H, Wickersham N, Matthay MA, Ware LB. Biomarkers of inflammation, coagulation and fibrinolysis predict mortality in acute lung injury. Crit Care. 2008;12:R41. doi: 10.1186/cc6846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Oddo M, Schaller M-D, Feihl F, Ribordy V, Liaudet L. From evidence to clinical practice: effective implementation of therapeutic hypothermia to improve patient outcome after cardiac arrest. Crit Care Med. 2006;34:1865–1873. doi: 10.1097/01.CCM.0000221922.08878.49. [DOI] [PubMed] [Google Scholar]
- 17. Sapru A, Wiemels JL, Witte JS, Ware LB, Matthay MA. Acute lung injury and the coagulation pathway: potential role of gene polymorphisms in the protein C and fibrinolytic pathways. Intensive Care Med. 2006;32:1293–1303. doi: 10.1007/s00134-006-0223-5. [DOI] [PubMed] [Google Scholar]
- 18. Sebag SC, Bastarache JA, Ware LB. Therapeutic modulation of coagulation and fibrinolysis in acute lung injury and the acute respiratory distress syndrome. Curr Pharm Biotechnol. 2011;12:1481–1496. doi: 10.2174/138920111798281171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Frantzeskaki F, Armaganidis A, Orfanos SE. Immunothrombosis in acute respiratory distress syndrome: cross talks between inflammation and coagulation. Respiration. 2017;93:212–225. doi: 10.1159/000453002. [DOI] [PubMed] [Google Scholar]
- 20. van Wissen M, Keller TT, Ronkes B, Gerdes VE, Zaaijer HL, van Gorp EC, et al. Influenza infection and risk of acute pulmonary embolism. Thromb J. 2007;5:16. doi: 10.1186/1477-9560-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Smeeth L, Cook C, Thomas S, Hall AJ, Hubbard R, Vallance P. Risk of deep vein thrombosis and pulmonary embolism after acute infection in a community setting. Lancet. 2006;367:1075–1079. doi: 10.1016/S0140-6736(06)68474-2. [DOI] [PubMed] [Google Scholar]
- 22. Mauad T, Hajjar LA, Callegari GD, da Silva LF, Schout D, Galas FR, et al. Lung pathology in fatal novel human influenza A (H1N1) infection. Am J Respir Crit Care Med. 2010;181:72–79. doi: 10.1164/rccm.200909-1420OC. [DOI] [PubMed] [Google Scholar]
- 23. Hwang DM, Chamberlain DW, Poutanen SM, Low DE, Asa SL, Butany J. Pulmonary pathology of severe acute respiratory syndrome in Toronto. Mod Pathol. 2005;18:1–10. doi: 10.1038/modpathol.3800247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tang N, Bai H, Chen X, Gong J, Li D, Sun Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thromb Haemost. 2020;18:1094–1099. doi: 10.1111/jth.14817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Klok FA, Kruip MJHA, van der Meer NJM, Arbous MS, Gommers DAMPJ, Kant KM, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. 2020;191:145–147. doi: 10.1016/j.thromres.2020.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–1062. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yao Y, Cao J, Wang Q, Shi Q, Liu K, Luo Z, et al. D-dimer as a biomarker for disease severity and mortality in COVID-19 patients: a case control study. J Intensive Care. 2020;8:49. doi: 10.1186/s40560-020-00466-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Griffin DO, Jensen A, Khan M, Chin J, Chin K, Saad J, et al. Pulmonary embolism and increased levels of d-dimer in patients with coronavirus disease. Emerg Infect Dis. 2020;26:1941–1943. doi: 10.3201/eid2608.201477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mackman N, Tilley RE, Key NS. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler Thromb Vasc Biol. 2007;27:1687–1693. doi: 10.1161/ATVBAHA.107.141911. [DOI] [PubMed] [Google Scholar]
- 30. Chu AJ. Tissue factor, blood coagulation, and beyond: an overview. Int J Inflamm. 2011;2011:367284. doi: 10.4061/2011/367284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Grover SP, Mackman N. Tissue factor: an essential mediator of hemostasis and trigger of thrombosis. Arterioscler Thromb Vasc Biol. 2018;38:709–725. doi: 10.1161/ATVBAHA.117.309846. [DOI] [PubMed] [Google Scholar]
- 32. Pieters M, Wolberg AS. Fibrinogen and fibrin: an illustrated review. Res Pract Thromb Haemost. 2019;3:161–172. doi: 10.1002/rth2.12191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Maroney SA, Ellery P, Martinez ND, Zogg M, Weiler H, Mast AE. Hyperactivatable protein C expression partially rescues the embryonic lethality of TFPI null mice but surviving mice develop severe hydrocephalus. Blood. 2015;126:422. [Google Scholar]
- 34. Hackeng TM, Maurissen LFA, Castoldi E, Rosing J. Regulation of TFPI function by protein S. J Thromb Haemost. 2009;7:165–168. doi: 10.1111/j.1538-7836.2009.03363.x. [DOI] [PubMed] [Google Scholar]
- 35. Hackeng TM, Seré KM, Tans G, Rosing J. Protein S stimulates inhibition of the tissue factor pathway by tissue factor pathway inhibitor. Proc Natl Acad Sci USA. 2006;103:3106–3111. doi: 10.1073/pnas.0504240103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shetty S, Bhandary YP, Shetty SK, Velusamy T, Shetty P, Bdeir K, et al. Induction of tissue factor by urokinase in lung epithelial cells and in the lungs. Am J Respir Crit Care Med. 2010;181:1355–1366. doi: 10.1164/rccm.200901-0015OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ye S, Goldsmith EJ. Serpins and other covalent protease inhibitors. Curr Opin Struct Biol. 2001;11:740–745. doi: 10.1016/s0959-440x(01)00275-5. [DOI] [PubMed] [Google Scholar]
- 38. Willyard C. Coronavirus blood-clot mystery intensifies. Nature. 2020;581:250. doi: 10.1038/d41586-020-01403-8. [DOI] [PubMed] [Google Scholar]
- 39. McGonagle D, O’Donnell JS, Sharif K, Emery P, Bridgewood C. Immune mechanisms of pulmonary intravascular coagulopathy in COVID-19 pneumonia. Lancet Rheumatol. 2020;2:e437–e445. doi: 10.1016/S2665-9913(20)30121-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xiong Y, Liu Y, Cao L, Wang D, Guo M, Jiang A, et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg Microbes Infect. 2020;9:761–770. doi: 10.1080/22221751.2020.1747363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Møller R, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181:1036–1045, e9. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med. 2020;26:842–844. doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- 43. Michalovich D, Rodriguez-Perez N, Smolinska S, Pirozynski M, Mayhew D, Uddin S, et al. Obesity and disease severity magnify disturbed microbiome-immune interactions in asthma patients. Nat Commun. 2019;10:5711. doi: 10.1038/s41467-019-13751-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andrews S. FastQC: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- 45. Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32:3047–3048. doi: 10.1093/bioinformatics/btw354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wickham H. ggplot2: Elegant graphics for data analysis. ISBN 978-3-319-24277-4. New York: Springer-Verlag; 2016 [accessed 2020 Jun 30]. Available from: https://cran.r-project.org/web/packages/ggplot2/citation.html.
- 49.Kolde R. pheatmap: Pretty Heatmaps. R package version 1.0.12. 2019 [accessed 2020 Jun 30]. Available from: https://CRAN.R-project.org/package=pheatmap.
- 50. Stefely JA, Christensen BB, Gogakos T, Sullivan JKC, Montgomery GG, Barranco JP, et al. Marked factor V activity elevation in severe COVID-19 is associated with venous thromboembolism. Am J Hematol. 2020;95:1522–1530. doi: 10.1002/ajh.25979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Crawley JT, Lane DA. The haemostatic role of tissue factor pathway inhibitor. Arterioscler Thromb Vasc Biol. 2008;28:233–242. doi: 10.1161/ATVBAHA.107.141606. [DOI] [PubMed] [Google Scholar]
- 52. Kawano M, Nagata S. Efferocytosis and autoimmune disease. Int Immunol. 2018;30:551–558. doi: 10.1093/intimm/dxy055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Myöhänen H, Vaheri A. Regulation and interactions in the activation of cell-associated plasminogen. Cell Mol Life Sci. 2004;61:2840–2858. doi: 10.1007/s00018-004-4230-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA. 2020;323:1061–1069. doi: 10.1001/jama.2020.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chu AJ. Tissue factor mediates inflammation. Arch Biochem Biophys. 2005;440:123–132. doi: 10.1016/j.abb.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 56. Szotowski B, Antoniak S, Poller W, Schultheiss HP, Rauch U. Procoagulant soluble tissue factor is released from endothelial cells in response to inflammatory cytokines. Circ Res. 2005;96:1233–1239. doi: 10.1161/01.RES.0000171805.24799.fa. [DOI] [PubMed] [Google Scholar]
- 57. Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395:1417–1418. doi: 10.1016/S0140-6736(20)30937-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schaefer IM, Padera RF, Solomon IH, Kanjilal S, Hammer MM, Hornick JL, et al. In situ detection of SARS-CoV-2 in lungs and airways of patients with COVID-19. Mod Pathol. 2020;33:2104–2114. doi: 10.1038/s41379-020-0595-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ahmetaj-Shala B, Peacock TP, Baillon L, Swann OC, Gashaw H, Barclay WS, et al. Resistance of endothelial cells to SARS-CoV-2 infection in vitro [preprint] bioRxiv 2020. [accessed 2020 Nov 9]. Available from: https://www.biorxiv.org/content/10.1101/2020.11.08.372581v1
- 60. Yang L, Han Y, Nilsson-Payant BE, Gupta V, Wang P, Duan X, et al. A human pluripotent stem cell-based platform to study SARS-CoV-2 tropism and model virus infection in human cells and organoids. Cell Stem Cell. 2020;27:125–136, e7. doi: 10.1016/j.stem.2020.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Padda IS, Patel P, Citla Sridhar D.Treasure Island, FL: StatPearls Publishing; 2020. [PubMed] [Google Scholar]
- 62. Jothimani D, Venugopal R, Abedin MF, Kaliamoorthy I, Rela M. COVID-19 and the liver. J Hepatol. 2020;73:1231–1240. doi: 10.1016/j.jhep.2020.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hundt MA, Deng Y, Ciarleglio MM, Nathanson MH, Lim JK. Abnormal liver tests in COVID-19: a retrospective observational cohort study of 1,827 patients in a major U.S. hospital network. Hepatology. 2020;72:1169–1176. doi: 10.1002/hep.31487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhang S, Liu Y, Wang X, Yang L, Li H, Wang Y, et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J Hematol Oncol. 2020;13:120. doi: 10.1186/s13045-020-00954-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chambers RC. Role of coagulation cascade proteases in lung repair and fibrosis. Eur Respir J Suppl. 2003;44:33s–35s. doi: 10.1183/09031936.03.00001003. [DOI] [PubMed] [Google Scholar]
- 66. Pepke-Zaba J, Morrell NW. The endothelin system and its role in pulmonary arterial hypertension (PAH) Thorax. 2005;60:443–444. doi: 10.1136/thx.2004.031724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Carpenter TC, Stenmark KR. Endothelin receptor blockade decreases lung water in young rats exposed to viral infection and hypoxia. Am J Physiol Lung Cell Mol Physiol. 2000;279:L547–L554. doi: 10.1152/ajplung.2000.279.3.L547. [DOI] [PubMed] [Google Scholar]
- 68. Hofer CC, Woods PS, Davis IC. Infection of mice with influenza A/WSN/33 (H1N1) virus alters alveolar type II cell phenotype. Am J Physiol Lung Cell Mol Physiol. 2015;308:L628–L638. doi: 10.1152/ajplung.00373.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Henry PJ, Carr MJ, Goldie RG, Jeng AY. The role of endothelin in mediating virus-induced changes in endothelinB receptor density in mouse airways. Eur Respir J. 1999;14:92–97. doi: 10.1034/j.1399-3003.1999.14a16.x. [DOI] [PubMed] [Google Scholar]
- 70. Henry PJ. Endothelin receptor distribution and function in the airways. Clin Exp Pharmacol Physiol. 1999;26:162–167. doi: 10.1046/j.1440-1681.1999.03010.x. [DOI] [PubMed] [Google Scholar]
- 71. Astarita JL, Acton SE, Turley SJ. Podoplanin: emerging functions in development, the immune system, and cancer. Front Immunol. 2012;3:283. doi: 10.3389/fimmu.2012.00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Fagan KA, McMurtry IF, Rodman DM. Role of endothelin-1 in lung disease. Respir Res. 2001;2:90–101. doi: 10.1186/rr44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bastarache JA, Wang L, Geiser T, Wang Z, Albertine KH, Matthay MA, et al. The alveolar epithelium can initiate the extrinsic coagulation cascade through expression of tissue factor. Thorax. 2007;62:608–616. doi: 10.1136/thx.2006.063305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Shaver CM, Grove BS, Putz ND, Clune JK, Lawson WE, Carnahan RH, et al. Regulation of alveolar procoagulant activity and permeability in direct acute lung injury by lung epithelial tissue factor. Am J Respir Cell Mol Biol. 2015;53:719–727. doi: 10.1165/rcmb.2014-0179OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ahmad S, Ahmad A, Rancourt RC, Neeves KB, Loader JE, Hendry-Hofer T, et al. Tissue factor signals airway epithelial basal cell survival via coagulation and protease-activated receptor isoforms 1 and 2. Am J Respir Cell Mol Biol. 2013;48:94–104. doi: 10.1165/rcmb.2012-0189OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Yap JKY, Moriyama M, Iwasaki A. Inflammasomes and pyroptosis as therapeutic targets for COVID-19. J Immunol. 2020;205:307–312. doi: 10.4049/jimmunol.2000513. [DOI] [PMC free article] [PubMed] [Google Scholar]