

Abstract
Hemophilia A and B are rare X‐linked inherited bleeding disorders caused by complete or partial deficiency in or the absence of coagulation factors VIII and IX. Recurrent joint bleeding (hemarthrosis) is the most frequent clinical manifestation of severe hemophilia. Unless appropriately managed, even subclinical hemarthrosis can lead to the development of hemophilic arthropathy, a disabling condition characterized by joint remodelling, chronic pain, and a reduced quality of life, and eventually requires joint replacement. Given the lack of specific treatments to reduce blood‐induced synovitis, the prevention of bleeding is pivotal to the maintenance of joint health. Prophylactic coagulation factor replacement therapy using extended half‐life recombinant drugs has significantly improved patients' quality of life by reducing the burden of intravenous injections, and the more recent introduction of nonreplacement therapies such as subcutaneous emicizumab injections has improved treatment adherence and led to the greater protection of patients with hemophilia A. However, despite these advances, chronic arthropathy is still a significant problem. The introduction of point‐of‐care ultrasound imaging has improved the diagnosis of acute hemarthrosis and early hemophilic arthropathy, and allowed the better monitoring of progressive joint damage, but further research into the underlying mechanisms of the disease is required to allow the development of more targeted treatment. In the meantime, patient management should be based on the risk factors for the onset and progression of arthropathy of each individual patient, and all patients should be collaboratively cared for by multidisciplinary teams of hematologists, rheumatologists, orthopedic surgeons, and physiotherapists at comprehensive hemophilia treatment centers.
Keywords: hemarthrosis, hemophilia, hemophilic arthropathy, sub‐clinical joint bleeding
1. INTRODUCTION
Hemophilia A and B are rare, inherited X‐linked bleeding disorders caused by a complete or partial deficiency in coagulation factors VIII (FVIII) or IX (FIX). In children and adults with severe hemophilia (i.e., plasma FVIII or FIX levels of <1 U/dl), joint bleeding (hemarthrosis) is the most frequent clinical manifestation,1 but recent data have shown that it may also (although less frequently) occur in patients with moderate (plasma factor levels of 1–5 UI/dl) or mild disease (plasma factor levels of >5 UI/dl).2
Over the past 30 years, therapeutic advances have progressively improved the life expectancy and quality of life of hemophilia patients. The development and marketing of recombinant coagulation factors in the 1990s was crucial in overcoming limited supplies of plasma‐derived factors; the more recent implementation of techniques such as coagulation factor fusion with the crystallizable fragment of immunoglobulin G1 or albumin or the addition of polyethylene glycol3 has extended plasma factor half‐life and reduced the required number of intravenous injections; and the introduction of non‐factor replacement drugs such as subcutaneous emicizumab has greatly improved patients' quality of life and treatment adherence.4 The greater availability and convenience of such replacement and nonreplacement therapies has led to the wider use of prophylactic regimens that are effective in preventing clinically overt bleeding episodes, particularly when they are individualized on the basis of each patient's lifestyle and pharmacokinetic profile.5
These advances have also led to a change in the aim of treatment from increasing life expectancy to preventing joint damage and protecting against joint bleeding and hemophilic arthropathy.6 However, despite greater adherence to prophylactic regimens, a number of patients still experience breakthrough bleedings that are sometimes clinically overt but may also be subclinical, unrecognized, bleeding episodes that are associated with a risk of joint damage progression.7
Repeated episodes of hemarthrosis lead to joint remodelling and subsequent hemophilic arthropathy, the targets of which are the diarthrodial (synovial) joints (i.e., freely moving joints whose surfaces are covered by hyaline articular cartilage that interfaces with a film of viscous fluid secreted by the synovial membrane). This synovial fluid lines the joint capsule and consists of macrophage‐ and fibroblast‐like synoviocytes that normally form a few layers on the subintima with no basal membrane.8 A first episode of hemarthrosis typically occurs in hemophilic children after the age of 1 year, and mainly depends on the severity of the factor deficiency. The bleeding mainly affects large joints such as the knees, elbows, or ankles, with the weight‐bearing joints of the dominant side being frequently affected as children begin to walk.9, 10 The cartilage is progressively damaged by iron deposition and lysosomal enzymes and pro‐inflammatory cytokines produced by the inflamed synovium, which eventually leads to subarticular bone cyst formation. Repeated hemarthroses are responsible for the development of synovial hyperplasia and angiogenesis, with further bleeding occurring in the friable and thickened synovium. Joint bleeding stretches the joint capsule and ligaments and leads to joint instability, which is worsened because reduced joint motility from pain causes peri‐articular muscle weakness. In more advanced stages, the joint is grossly damaged by cartilage loss and subchondral bone sclerosis, which further limits movement and leads to crepitus and deformity. Soft‐tissue swelling and effusions are rare, and joint contracture occurs from muscle retraction and bone ankylosis, particularly if the muscles are weak. The level of pain varies and fluctuates, but may be severe.11
There is evidence that even a single episode of hemarthrosis can start the inflammatory process that leads to synovial thickening and irreversible angiogenesis, thus predisposing to recurrent bleeding.12 Prophylaxis decreases the incidence of bleeding at all ages, but joint motion can be preserved only if it is started before the age of 3 years.13 Prophylactic treatment is superior to episodic infusions in preventing joint disease and should therefore be started as soon as possible and needs to be continued throughout life.9 However, magnetic resonance imaging (MRI) data indicate that joint damage can still be identified in about 20% of clinically asymptomatic patients even if they are receiving prophylaxis,9 and it has been hypothesized that this due to subclinical bleeds silently leaking into clinically asymptomatic joints that are left untreated because they are not recognized.14
The aim of this review is to give an overview of our current knowledge of the mechanisms leading from hemarthrosis to hemophilic arthropathy, which is still the most important complication of hemophilia A and B, and describe future perspectives.
2. FACTORS CONTRIBUTING TO ARTHROPATHY
Multiple factors contribute to the development and progression of arthropathy in hemophilia patients, and account for the heterogeneity of the clinical phenotypes despite similar coagulation factor levels and prophylactic regimens.15
2.1. Genetic susceptibility
The main predisposing factors for recurrent hemarthrosis and arthropathy are plasma FVIII and FIX levels, which depend on the type of gene mutations. However, as patients with the same mutation often have different bleeding phenotypes, other genetic factors such as inherited thrombophilia may also be involved.16 Genetic susceptibility to more rapid joint damage progression also involves gene polymorphisms associated with an increased expression of pro‐inflammatory cytokines such as tumor necrosis factor (TNF)‐α. Lopez‐Jimenez et al. have reported that carriers of the TNFα‐308G>A variant have a larger number of subchondral cysts,17 platelet aggregation,18 and the innate immune response pathways that play a fundamental role in pathogen recognition and the activation of innate immunity such as nucleotide‐binding oligomerization domain‐containing protein 2, and toll‐like receptor 10 .19
2.2. Local factors
Susceptibility to spontaneous joint bleeds depends on the rich vascularization of synovial membranes, mechanical stress in weight‐bearing joints, and the local regulation of hemostasis, which is different from that of other tissues.20, 21 Human synovial membranes simultaneously have low levels of tissue factor (TF) and high levels of TF pathway inhibitor, which reduce the activation of the extrinsic coagulation pathway.20, 21 Furthermore, the synovial fluid of hemophilia patients with arthropathy contains high levels of thrombomodulin, a cofactor for the formation of anticoagulant activated protein C.22 Other hemostatic factors may be locally involved, such as thrombin‐activatable fibrinolysis inhibitor, the activation of which is decreased because of the inherent defect in thrombin generation during joint bleeding, thus leading to high levels of fibrinolytic activity and premature clot lysis.23, 24 Furthermore, hemarthrosis increases the number of synovial cells expressing urokinase plasminogen activator and plasminogen activator inhibitor 1, which has the net result of increasing synovial levels of plasmin, the effector protease of the fibrinolytic system.23 In addition to increasing susceptibility to bleeding, high urokinase plasminogen activator levels can have other detrimental biological effects, such as the stimulation of chemotaxis, angiogenesis, the proliferation of human synovial cells, and bone and cartilage damage.23, 25 Accordingly, it has been found that intra‐articular antiplasmin treatment is effective in reducing synovitis and cartilage damage in a mouse model of joint bleeding.26
2.3. Environmental factors
Several environmental factors may also contribute to accelerating the progression of hemophilic arthropathy: the prophylactic use of clotting factor replacement therapy, the type of replacement products or regimens, the frequency of treatment and joint bleeding, the presence of inhibitors, the type of lifestyle, and the body mass index.27, 28, 29, 30
Although the value of prophylaxis in decreasing the number of bleeds has been established,9 the optimal trough factor level for joint damage prevention is still debated. Because a longer time with FVIII levels of <1 IU/dl is associated with more bleeding, a trough FVIII level of >1% was once thought to be sufficient to protect joints from spontaneous bleeding,31 but den Uijl et al. have shown that patients with levels of ≥10% were still at albeit very low risk of spontaneous joint bleeding, which only disappeared at levels of ≥15%; furthermore, each 1% increase in plasma factor levels was associated with an 18% reduction in bleeding frequency.32 Recent recommendations by the European Directorate for the Quality of Medicines & Healthcare33 and the World Federation of Hemophilia34 suggest a target minimum trough level of 3% to 5% to preserve joint function. However, regular prophylaxis is not feasible for many patients worldwide, and so many of them still develop arthropathy9; furthermore, the availability of only standard half‐life factor products and a lack of access to extended half‐life products makes it difficult to maintain the recommended levels.
The presence of inhibitors promotes joint disease progression35 because of the unsatisfactory management of acute hemarthrosis with bypassing agents. Furthermore, the presence of an immune reaction to FVIII may have an unfavorable effect on the immunological milieu of joints.15 The recent introduction of nonfactor replacement drugs such as emicizumab provides more consistent protection, thus contributing to preserving joint health in patients with inhibitors,36 although long‐term study results are not yet available.
Physically active children are at higher risk of developing arthropathy,37 but regular physiotherapy and physical activity reduces the recurrence of joint bleeding by promoting joint stability. Early rehabilitation is strongly encouraged after the resolution of an acute episode of hemarthrosis.30, 38
Finally, other factors that are known to be associated with joint deterioration in osteoarthritis (OA), such as aging, a high body mass index, associated with increased joint weight loading, and altered joint biomechanics resulting from trauma and instability, may also be involved in the progression of hemophilic arthropathy.39, 40
3. MECHANISMS OF HEMOPHILIC ARTHROPATHY
The pathophysiology of hemophilic arthropathy has not yet been fully elucidated, especially in terms of the mechanisms associated with subclinical and early disease, and the development of intra‐articular inflammation. Recurrent joint bleeding leads to the two main consequences synovitis and osteochondral damage, which influence each other and contribute to the development of arthropathy (Figure 1).15, 41
FIGURE 1.
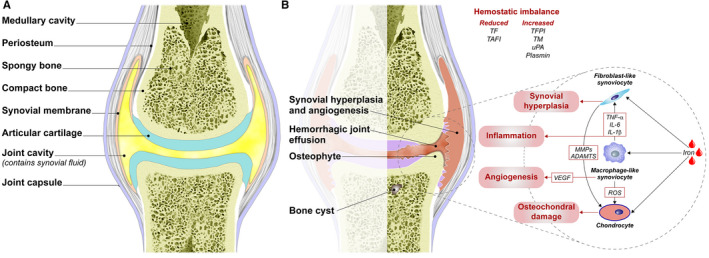
The structure of (A) a healthy joint and (B) a joint with hemophilic arthropathy. Recurrent hemarthrosis (represented by blood drops for the sake of simplicity) induces the proliferation (synovial hyperplasia) of fibroblast‐like synoviocytes and macrophage‐like synoviocytes that produce the vascular endothelial growth factor responsible for angiogenesis and vascular remodelling, and pro‐inflammatory cytokines such as tumor necrosis factor‐alpha, and interleukin‐6 and 1‐beta. These further amplify the fibroblast‐like synoviocyte proliferation and production of reactive oxygen species that induce chondrocyte apoptosis. Osteochondral damage is an inevitable consequence of the direct exposure of chondrocytes to iron, metalloproteinases, and a disintegrin and metalloproteinase with thrombospondin motifs that is produced by fibroblast‐like synoviocytes when stimulated by inflammation. ADAMTS, a disintegrin and metalloproteinase with thrombospondin motifs; IL‐1, interleukin‐1; IL‐6, interleukin‐6; MMPs, metalloproteinases; ROS, reactive oxygen species; TAFI, thrombin activatable fibrinolysis inhibitor; TF, tissue factor; TFPI, tissue factor pathway inhibitor; TM, thrombomodulin; TNF‐alpha, tumor necrosis factor‐alpha; uPA, urokinase plasminogen activator; VEGF, vascular endothelial growth factor
3.1. Synovitis
Depending on the stage of progression, hemophilic arthropathy shares some clinical and biological features with inflammatory diseases such as rheumatoid arthritis (RA)42 and degenerative joint diseases such as OA and hereditary hemochromatosis.43, 44 In patients with RA, synovitis is typically characterized by synovial hyperplasia (the proliferation of synovial cells and inflammatory cell infiltration) and a rich, newly formed and more permeable vascular network.45 In normal joints, after a single bleed from trauma, hemosiderin and ferritin are taken up by macrophage‐like synoviocytes and then returned to the circulation but, in patients with hemophilia, hemarthrosis is followed by the intracellular deposition of blood breakdown products in synovial macrophages and, in the case of ongoing or repeated bleeding, synovial clearing capacity is overwhelmed, and this leads to iron accumulation (hemosiderin) and synovial membrane hyperplasia.10, 44 In line with this, synovial membrane specimens from patients with early‐stage hemophilic arthropathy are characterized by inflammation, an increased number of synovial villi, and iron accumulation in macrophages that is greater than that observed in RA patients.46, 47
Recurrent bleeding also causes the production of inflammatory cytokines such as TNF‐α, interferon‐γ, interleukin‐1β and interleukin‐6 that, by directly stimulating the expression of the receptor activator of nuclear factor‐κB (RANK) ligand (RANKL), further increase synovial membrane hyperplasia.7, 48, 49, 50 RA synovitis is typically characterized by leukocytic infiltration involving macrophages, lymphocytes, and plasma cells, which frequently form ectopic lymphoid aggregates.51, 52 However, such aggregates are not typical in the non‐inflammatory stages of OA or in hemophilic arthropathy, both of which are characterized by a higher degree of fibrosis.53, 54
Tissue macrophages can differentiate into an inflammatory subtype (M1), which expresses an array of inflammatory cytokines and has microbicidal functions, or into an anti‐inflammatory and pro‐healing subtype (M2).42, 45, 55 A transient increase in the pro‐inflammatory M1 response has been demonstrated in the synovial tissue of FVIII‐deficient mice 1 week after acute joint bleeding, followed by longer lasting up‐regulation of a reparative M2 response 2–4 weeks after the acute event. The pro‐inflammatory cytokines produced by M1 macrophages stimulate catabolic activity and H2O2 production by cartilage chondrocytes, thus leading to cartilage destruction56 and the poor elimination of iron from the joint.55 On the other hand, anti‐inflammatory M2 macrophages have stronger neo‐angiogenic properties than other macrophage subsets and may contribute to the abnormally greater vascularization and remodelling observed in hemophilic arthropathy patients.45 This view is further supported by the evidence that cells of monocyte/macrophage lineage in the synovial membrane also express vascular endothelial growth factor.57 Finally, macrophages from patients with hemophilia A have impaired macrophage colony stimulating factor (M‐CSF)‐mediated functions such as clot invasion, phagocytosis, wound infiltration, and the induction of a regenerative tissue response,58 and similar observations have been made in mice with hemophilia B.59 These findings may explain the delay in wound healing and tissue regeneration observed in patients with hemophilia as well as their susceptibility to recurrent hemarthrosis.58 It has also been shown that arthropathy patients have increased microvessel density in synovial tissue and increased serum vascular endothelial growth factor expression as a result of the inflammatory response and hypoxia induced by recurrent bleeding.57, 60
Fibroblast‐like synoviocytes, which play a key mechanistic role in RA by producing pro‐inflammatory cytokines and degrading enzymes such as matrix metalloproteinases, and have a marked tendency to invade and damage cartilage and the underlying bone,61 are also involved in the progression of hemophilic arthropathy. Recurrent joint bleeding and iron deposition induce their uncontrolled proliferation because of the increased expression of E3 ubiquitin‐protein ligase mdm2 and the proto‐oncogene c‐Myc, both of which are involved in regulating cell proliferation.50, 62
Hereditary hemochromatosis, a genetic disease characterized by hepcidin dysregulation and a high iron overload, is associated with a form of degenerative iron‐related arthropathy that presents with mild or absent synovitis and has clinical features similar to those of OA. In patients with hemochromatosis, iron is gradually accumulated as Fe3+, which is less toxic than the heme‐derived Fe2+ that is deposited together with circulating blood inflammatory cells during the course of hemarthrosis in hemophilia patients. Furthermore, the development of arthropathy in hemophilia patients starts with recurrent joint bleeding in early childhood, whereas hereditary hemochromatosis is characterized by more gradual iron accumulation and clinical symptoms that only become overt in later life. These differences account for the different pathological findings associated with hemochromatosis and hemophilic arthropathy.12
3.2. Osteochondral damage
In an early stage of arthropathy, osteochondral damage is probably the result of the direct effect of blood and iron on chondrocytes. Blood is toxic to human cartilage after only 2 days12 because the formation of hemosiderin and hydroxyl radicals leads to chondrocyte apoptosis and impaired renewal of the extracellular matrix. The growing cartilage of children is more susceptible to this kind of damage.63
Advanced stages of chronic synovitis further contribute to osteochondral damage because of the production of pro‐inflammatory cytokines and proteases,56 and altered local bone homeostasis from an imbalance in the RANK/RANKL/osteoprotegerin pathway eventually leads to increased bone resorption and the development of subchondral damage.64 The advanced stages of hemophilic arthropathy are characterized by quiescent fibrotic synovitis, geodes, and subchondral cysts, which are usually observed much later in the life of patients with degenerative arthropathies such as OA.54, 65 Before the introduction of biological disease‐modifying antirheumatic drugs (DMARDs), RA also frequently progressed to OA,44 although the degree of synovial fibrotic changes in the context of hemophilic arthropathy is greater than that observed in OA, thus indicating a disease‐specific expression of pro‐fibrotic factors such as connective tissue growth factor.46
4. IMAGING
In the past, joint damage was almost exclusively evaluated on the basis of X‐ray findings, but MRI is currently preferred because it allows the more precise detection of joint involvement in hemophilia patients. Furthermore, the more recent introduction of joint ultrasonography (US) in the clinical practice of hemophilia care has made it possible to identify early signs of arthropathy66 and the presence and site of bleeding in point‐of‐care settings.67
4.1. Radiography (X‐rays)
Early‐stage hemophilic arthropathy is radiologically characterized by soft‐tissue swelling, and the later stages by juxta‐articular osteoporosis, bone lesions with overgrowth of the epiphysis, and cartilage damage, which is first visualized as joint space narrowing and then as the complete loss of cartilage space and the associated formation of subchondral bone cysts, bone erosions, and joint profile irregularities.68 The most widely used scoring system for assessing the severity of joint damage by means of plain radiography is still the time‐honored Petterson score, but it is well known that radiography is insensitive to early changes.69
4.2. Magnetic resonance imaging and computed tomography
Magnetic resonance imaging allows an early evaluation of soft‐tissue involvement, subchondral cysts, and cartilage damage. Two main scoring systems have been proposed: the Denver score and the system coming from the international MRI expert working group of the International Prophylaxis Study Group, even though no international consensus was reached.70, 71 MRI is considered the gold standard for joint imaging but, in addition to its cost and unavailability in point‐of‐care settings, it has the further limitations that children may need to be sedated and it is less convenient in terms of multiple joint assessments and serial follow‐up examinations. Furthermore, although US and MRI are comparably sensitive in detecting synovial hypertrophy, MRI signal intensity may be unable to distinguish synovial fluid from synovitis, or bloody intra‐articular effusions from their nonbloody counterparts.72
Computed tomography is highly sensitive in detecting bone changes, but cannot provide detailed information concerning soft tissue involvement, and requires a large amount of ionizing radiation.
4.3. Ultrasonography
Joint US has many advantages over plain radiography because it does not use ionizing radiation and provides information concerning soft‐tissue involvement; it also has advantages over MRI because children do not need to be sedated, and is less time consuming, less expensive, and available in point‐of‐care settings. Di Minno et al. have shown that there is a good correlation between US and MRI findings, and that US is reliable in identifying abnormalities even in joints that patients do not report to be painful or swollen.73
Ultrasonography is very useful for identifying the tissue abnormalities responsible for joint pain in patients with hemophilia. It also detects the presence of joint bleeding rapidly and accurately, thus allowing the differential diagnosis of acute hemarthrosis and the monitoring of chronic arthropathy.74 In comparison with MRI, musculoskeletal US is much more sensitive in detecting bloody effusions (even as little as 5% of blood) and distinguishing them from nonbloody effusions,75 although its diagnostic sensitivity to blood clots and synovial hyperplasia is still not completely satisfactory and MRI may still be needed when this is clinically important.76
A recent international survey has found that the majority of hemophilia centers use US as a point‐of‐care means of detecting hemarthrosis rather than for serial arthropathy scanning.77 However, there is still a need for the standardization of algorithms that will help nonexpert operators in its point‐of‐care use.
Joint US can also be very important in patient education and professional training: its ability to show patients the status of their joints in real time can improve treatment adherence, and its use in the guidance of invasive intra‐articular procedures allows operators to define the best site of entry and needle positioning (see Videos S1 and S2).
Ultrasonography can also be used to detect osteochondral changes and, although the reflection of most of its beams from bone surfaces limits its penetrative ability and makes it impossible to assess deep changes, it clearly reveals marginal bone erosions, superficial subchondral cysts, and peripheral articular cartilage defects. Another possible limitation is that children's joints have a completely different US appearance from that of mature adult joints because of their thick immature cartilage and their age‐ or stage‐specific ossification centers and growth plates. In the absence of a pediatric joint atlas, these differences may make it difficult to interpret pediatric point‐of‐care US images.78, 79 Furthermore, although US is currently considered to be an accurate means of diagnosing arthropathy early, its sensitivity to changes developing during long‐term follow‐up still needs to be evaluated in prospective studies.80 Finally, it is still an examiner‐dependent technique, and some features are not considered as relevant by many authors, such as power Doppler examination for evaluating synovial vascularity and the presence of hemosiderin deposits. However, a number of scanning procedures and scoring systems have been proposed and some are currently used as outcome measures in clinical trials, such as the HEAD‐US and the Joint Tissue Activity and Damage Examination protocol66, 81, 82, 83, 84, 85, 86 (Table 1).
TABLE 1.
The most frequently used scoring systems and protocols for the ultrasound evaluation of hemophilic arthropathy
| First Author | Year | Synovial Membrane Hypertrophy | Power‐Doppler Signal | Hemosiderin Deposition | Articular Cartilage Damage (Partial or Full Loss of Thickness, Thinning) | Subchondral Bone Damage (Surface Irregularity, Bone Cysts, Erosions, and Osteophytes) | Investigated Joints |
|---|---|---|---|---|---|---|---|
| Klukovska | 2001 | Yes | Yes | No | Yes | Yes | Knee, ankle |
| Zukotynski | 2007 | Yes | Yes | Yes | Yes | Yes | Knee, ankle |
| Melchiorre | 2011 | Yes | Yes | Yes | Yes | Yes | Elbow, knee, ankle |
| Muça‐Peria | 2012 | Yes | Yes | No | Yes | Yes | Knee, ankle |
| Martinoli | 2013 | Yes | No | No | Yes | Yes | Elbow, knee, ankle |
| Doria | 2015 | Yes | No | Yes | No | Yes | Knee, ankle |
| Kandagaddala | 2019 | Yes | Yes | Yes | Yes | Yes | Knee, ankle |
| Volland | 2019 | Yes | Yes | No | Yes | Yes | Elbow, knee, ankle |
5. CHALLENGES IN THE TREATMENT OF HEMOPHILIC ARTHROPATHY
The mainstay of hemophilia treatment is replacement of the missing coagulation factor, which is used in order to prevent recurrent bleeding episodes such as hemarthrosis and the development of arthropathy. However, none of the currently available pharmacological treatments specifically targets synovitis, which is a known source of recurrent bleeding and the driver of hemophilic arthropathy. Blood aspiration from the joints at the time of acute hemarthrosis can rapidly resolve inflammation, but the benefit of this procedure remains controversial because of the lack of randomized controlled trials.87, 88 The same is true of intra‐articular corticosteroid treatment, which may be useful in selected patients,89 or conservative intra‐articular treatment with hyaluronic acid or platelet‐rich plasma, which may delay the onset and progression of osteochondral damage.90 A physiotherapy‐based program designed to restore joint function should be started shortly after acute joint bleeding,91 and the best option seems to be a program of isometric muscle exercises and stretching.38
Once it has become established, synovitis cannot be treated with factor replacement alone and it has been suggested TNF‐α blockade with monoclonal antibodies may be a means of controlling synovitis and potentially reducing joint bleeding.92 However, this cannot be considered an option until the pathophysiological mechanisms underlying the development and progression of hemophilic arthropathy have been defined, and the precise role of pro‐inflammatory cytokines has been established. Furthermore, no clinical trials addressing the efficacy of synthetic or biological DMARDs in controlling synovitis have yet been conducted partly because of the lack of sensitive outcome parameters, disease heterogeneity in small populations, and the long follow‐up required. To reduce the burden of inflammation that predisposes to recurrent bleeding by refuelling angiogenesis and synovial hyperplasia, patients with refractory disease may benefit from joint embolisation,93 or methods of synovial ablation such as chemical synoviorthesis with rifampicin or tetracyclines and radioisotopic synovectomy.94, 95
6. FUTURE PERSPECTIVES
The change from episodic treatment to prophylaxis was an important advance in hemophilia care; the next objective should be ensuring protection over time because preventing arthropathy depends on being able to prevent bleeding, which none of the available treatments is capable of doing consistently.96
The recent introduction of non‐replacement drugs such as emicizumab and the early clinical trials of gene therapy raise the question of whether these therapeutic options can equal the musculoskeletal effects of the current prophylactic regimens based on FVIII replacement products, particularly the preservation of growing joints in children. Some authors have suggested FVIII also play biological roles other than blood coagulation, but it is not clear whether these are due to FVIII itself or the improvement in thrombin generation. Similarly, it is still unclear whether the newly available products can enter the joint vasculature. The effects of extended half‐life products and non‐replacement therapy on joint preservation are currently being investigated in clinical trials, but these will require long‐term follow‐up, especially in the case of pediatric joints.
One of the main unmet needs in the management and follow‐up of hemophilic arthropathy is the current lack of serum and synovial biomarkers of disease activity, because their identification and validation would help to improve decision making. Studies of synovial tissue in patients with recent‐onset RA have found significant correlations between histological findings, transcriptomic profiles, and the clinical responses to therapeutic choices.97 Similarly, the availability of synovial biomarkers based on transcriptomic analysis may help to identify predictors of therapeutic responses and possibly new targets for the personalized management of hemophilic arthropathy.
The more widespread use of joint US as a routine clinical examination in comprehensive care centers is expected to help optimize workflows, allow the prompt diagnosis of acute hemarthrosis, and improve early treatment. Furthermore, the use of point‐of‐care US in clinical practice and at the bedside should also help the regular monitoring of arthropathy progression once the standardization of US techniques has been completed.79 The results of ongoing studies using the HEAD‐US and Joint Tissue Activity and Damage Examination protocols as outcome measures will allow their prospective validation.66, 81, 82, 83, 84, 85, 86
Other developments include the currently ongoing evaluation of a patient self‐conducted US examination using a handheld device at home (e.g.. NCT04131920, NCT04550988, clinicaltrials.gov),98 and the growing spread of machine learning and artificial intelligence in various medical fields as this will further improve point‐of‐care US by aiding untrained operators to recognize pathological findings rapidly and may even help the development of self‐conducted US examinations.
7. CONCLUSIONS
Despite the growing availability of treatments aimed at preventing and stopping recurrent joint bleeding, blood‐induced arthropathy still has a considerable impact on the life of patients with hemophilia, and the many unclear aspects of this scourge require a better understanding of its underlying pathophysiological mechanisms to provide the evidence needed for the development of more targeted treatments. In the meanwhile, the prevention of arthropathy progression should be based on clotting factor replacement therapy, the use of nonreplacement drugs such as emicizumab, physical therapy, education, and motivation to undertake physical activity in the framework of constant patient referrals to multidisciplinary teams working at comprehensive hemophilia care centers.
CONFLICT OF INTEREST
Dr. Gualtierotti is a member of advisory boards of Biomarin, Pfizer, Bayer, and Takeda, and participates in educational seminars sponsored by Pfizer, Sobi, and Roche. Dr. Peyvandi is a member of advisory boards of Bioverativ, Grifols, Roche, Sanofi, Sobi, Spark, and Takeda. Dr. Solimeno has no disclosure to make.
AUTHOR CONTRIBUTION
Roberta Gualtierotti, Luigi Piero Solimeno, and Flora Peyvandi wrote and reviewed the manuscript.
Supporting information
Video S1
Video S2
ACKNOWLEDGMENTS
This review was partially supported by the Italian Ministry of Health ‐ Bando Ricerca Corrente, and partially financed by Italian 2017 "5x1000" fiscal contributions to the Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico in Milan. We would like to thank Dr Luigi Flaminio Ghilardini of the University of Milan for his support in designing the figure and processing the videos.
Gualtierotti R, Solimeno LP, Peyvandi F. Hemophilic arthropathy: Current knowledge and future perspectives. J Thromb Haemost. 2021;19:2112–2121. 10.1111/jth.15444
Manuscript Handled by: David Lillicrap
Final decision: David Lillicrap, 30 June 2021
REFERENCES
- 1.Bolton‐Maggs PH, Pasi KJ. Haemophilias A and B. Lancet. 2003;361:1801‐1809. [DOI] [PubMed] [Google Scholar]
- 2.Di Minno MN, Ambrosino P, Franchini M, Coppola A, Di Minno G. Arthropathy in patients with moderate hemophilia a: a systematic review of the literature. Semin Thromb Hemost. 2013;39:723‐731. [DOI] [PubMed] [Google Scholar]
- 3.Peyvandi F, Garagiola I, Biguzzi E. Advances in the treatment of bleeding disorders. J Thromb Haemost. 2016;14:2095‐2106. [DOI] [PubMed] [Google Scholar]
- 4.Mannucci PM. Hemophilia therapy: the future has begun. Haematologica. 2020;105:545‐553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collins PW. Personalized prophylaxis. Haemophilia. 2012;18(Suppl 4):131‐135. [DOI] [PubMed] [Google Scholar]
- 6.Manco‐Johnson MJ, Soucie JM, Gill JC, Joint Outcomes Committee of the Universal Data Collection USHTCN . Prophylaxis usage, bleeding rates, and joint outcomes of hemophilia, 1999 to 2010: a surveillance project. Blood. 2017;129(17):2368‐2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wojdasiewicz P, Poniatowski ŁA, Nauman P, et al. Cytokines in the pathogenesis of hemophilic arthropathy. Cytokine Growth Factor Rev. 2018;39:71‐91. [DOI] [PubMed] [Google Scholar]
- 8.Standring S. Gray's anatomy e‐book: the anatomical basis of clinical practice. London: Elsevier Health Sciences. 2015. [Google Scholar]
- 9.Manco‐Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357:535‐544. [DOI] [PubMed] [Google Scholar]
- 10.van Vulpen LFD, Holstein K, Martinoli C. Joint disease in haemophilia: pathophysiology, pain and imaging. Haemophilia. 2018;24(Suppl 6):44‐49. [DOI] [PubMed] [Google Scholar]
- 11.Arnold WD, Hilgartner MW. Hemophilic arthropathy. Current concepts of pathogenesis and management. J Bone Joint Surg Am. 1977;59:287‐305. [PubMed] [Google Scholar]
- 12.van Vulpen LF, van Meegeren ME, Roosendaal G, et al. Biochemical markers of joint tissue damage increase shortly after a joint bleed; an explorative human and canine in vivo study. Osteoarthritis Cartilage. 2015;23:63‐69. [DOI] [PubMed] [Google Scholar]
- 13.Manco‐Johnson MJ, Soucie JM, Gill JC. Prophylaxis usage, bleeding rates, and joint outcomes of hemophilia, 1999 to 2010: a surveillance project. Blood. 2017;129:2368‐2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Puetz J. Nano‐evidence for joint microbleeds in hemophilia patients. J Thromb Haemost. 2018;16:1914‐1917. [DOI] [PubMed] [Google Scholar]
- 15.Valentino LA, Hakobyan N, Enockson C, et al. Exploring the biological basis of haemophilic joint disease: experimental studies. Haemophilia. 2012;18:310‐318. [DOI] [PubMed] [Google Scholar]
- 16.Franchini M, Montagnana M, Targher G, et al. Interpatient phenotypic inconsistency in severe congenital hemophilia: a systematic review of the role of inherited thrombophilia. Semin Thromb Hemost. 2009;35:307‐312. [DOI] [PubMed] [Google Scholar]
- 17.Lopez‐Jimenez JJ, Ortega‐Cervantes R, Luna‐Zaizar H, et al. Genetic biomarkers related to hemarthrosis, inflammation, and cartilage structure in pediatric patients with hemophilic arthropathy. Mol Genet Genomic Med. 2019;7:e979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ver Donck F, Downes K, Freson K. Strengths and limitations of high‐throughput sequencing for the diagnosis of inherited bleeding and platelet disorders. J Thromb Haemost. 2020;18:1839‐1845. [DOI] [PubMed] [Google Scholar]
- 19.Gomperts ED, Schwarz J, Donfield SM, et al. The importance of genetic factors for the development of arthropathy: a longitudinal study of children and adolescents with haemophilia A. Thromb Haemost. 2017;117:277‐285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Drake TA, Morrissey JH, Edgington TS. Selective cellular expression of tissue factor in human tissues. Implications for disorders of hemostasis and thrombosis. Am J Pathol. 1989;134:1087‐1097. [PMC free article] [PubMed] [Google Scholar]
- 21.Brinkmann T, Kahnert H, Prohaska W, Nordfang O, Kleesiek K. Synthesis of tissue factor pathway inhibitor in human synovial cells and chondrocytes makes joints the predilected site of bleeding in haemophiliacs. Eur J Clin Chem Clin Biochem. 1994;32:313‐317. [DOI] [PubMed] [Google Scholar]
- 22.Dargaud Y, Simpson H, Chevalier Y, et al. The potential role of synovial thrombomodulin in the pathophysiology of joint bleeds in haemophilia. Haemophilia. 2012;18:818‐823. [DOI] [PubMed] [Google Scholar]
- 23.Nieuwenhuizen L, Roosendaal G, Coeleveld K, et al. Haemarthrosis stimulates the synovial fibrinolytic system in haemophilic mice. Thromb Haemost. 2013;110:173‐183. [DOI] [PubMed] [Google Scholar]
- 24.Wyseure T, Cooke EJ, Declerck PJ, et al. Defective TAFI activation in hemophilia A mice is a major contributor to joint bleeding. Blood. 2018;132:1593‐1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fibbi G, Pucci M, Serni U, Cerinic MM, Del Rosso M. Antisense targeting of the urokinase receptor blocks urokinase‐dependent proliferation, chemoinvasion, and chemotaxis of human synovial cells and chondrocytes in vitro. Proc Assoc Am Physicians. 1998;110:340‐350. [PubMed] [Google Scholar]
- 26.Nieuwenhuizen L, Roosendaal G, Mastbergen SC, et al. Antiplasmin, but not amiloride, prevents synovitis and cartilage damage following hemarthrosis in hemophilic mice. J Thromb Haemost. 2014;12:237‐245. [DOI] [PubMed] [Google Scholar]
- 27.Wilding J, Zourikian N, Di Minno M, et al. Obesity in the global haemophilia population: prevalence, implications and expert opinions for weight management. Obes Rev. 2018;19:1569‐1584. [DOI] [PubMed] [Google Scholar]
- 28.Carpenter SL, Chrisco M, Johnson E. The effect of overweight and obesity on joint damage in patients with moderate or severe hemophilia. Blood. 2006;108:4064. [Google Scholar]
- 29.Iorio A, Marchesini E, Marcucci M, Stobart K, Chan AK. Clotting factor concentrates given to prevent bleeding and bleeding‐related complications in people with hemophilia A or B. Cochrane Database Syst Rev. 2011; (9):Cd003429. [DOI] [PubMed] [Google Scholar]
- 30.Srivastava A, Brewer AK, Mauser‐Bunschoten EP, et al. Guidelines for the management of hemophilia. Haemophilia. 2013;19:e1‐47. [DOI] [PubMed] [Google Scholar]
- 31.Collins PW, Blanchette VS, Fischer K, et al. Break‐through bleeding in relation to predicted factor VIII levels in patients receiving prophylactic treatment for severe hemophilia A. J Thromb Haemost. 2009;7:413‐420. [DOI] [PubMed] [Google Scholar]
- 32.Den Uijl IEM, Fischer K, van der Bom JG, Grobbee DE, Rosendaal FR, Plug I. Analysis of low frequency bleeding data: the association of joint bleeds according to baseline FVIII activity levels. Haemophilia. 2011;17:41‐44. [DOI] [PubMed] [Google Scholar]
- 33.Peyvandi F, Berger K, Seitz R, et al. Kreuth V initiative: European consensus proposals for treatment of haemophilia using standard products, extended half‐life coagulation factor concentrates and non‐replacement therapies. Haematologica. 2020;105(8):2038‐2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Srivastava A, Santagostino E, Dougall A, et al. WFH guidelines for the management of hemophilia, 3rd edition. Haemophilia. 2020;26(S6):1‐158. [DOI] [PubMed] [Google Scholar]
- 35.Morfini M, Haya S, Tagariello G, et al. European study on orthopaedic status of haemophilia patients with inhibitors. Haemophilia. 2007;13:606‐612. [DOI] [PubMed] [Google Scholar]
- 36.Ljung R, Auerswald G, Benson G, et al. Inhibitors in haemophilia A and B: management of bleeds, inhibitor eradication and strategies for difficult‐to‐treat patients. Eur J Haematol. 2019;102:111‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buzzard BM. Sports and hemophilia: antagonist or protagonist. Clin Orthop Relat Res. 1996;328:25‐30. [PubMed] [Google Scholar]
- 38.Boccalandro E, Mancuso ME, Riva S, et al. Ageing successfully with haemophilia: a multidisciplinary programme. Haemophilia. 2018;24:57‐62. [DOI] [PubMed] [Google Scholar]
- 39.Blagojevic M, Jinks C, Jeffery A, Jordan KP. Risk factors for onset of osteoarthritis of the knee in older adults: a systematic review and meta‐analysis. Osteoarthritis Cartilage. 2010;18:24‐33. [DOI] [PubMed] [Google Scholar]
- 40.Shapiro S, Makris M. Haemophilia and ageing. Br J Haematol. 2019;184:712‐720. [DOI] [PubMed] [Google Scholar]
- 41.Lafeber FP, Miossec P, Valentino LA. Physiopathology of haemophilic arthropathy. Haemophilia. 2008;14(Suppl 4):3‐9. [DOI] [PubMed] [Google Scholar]
- 42.Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol. 2013;229:176‐185. [DOI] [PubMed] [Google Scholar]
- 43.Roosendaal G, van Rinsum AC, Vianen ME, van den Berg HM, Lafeber FP, Bijlsma JW. Haemophilic arthropathy resembles degenerative rather than inflammatory joint disease. Histopathology. 1999;34:144‐153. [DOI] [PubMed] [Google Scholar]
- 44.Blobel CP, Haxaire C, Kalliolias GD, DiCarlo E, Salmon J, Srivastava A. Blood‐induced arthropathy in hemophilia: mechanisms and heterogeneity. Semin Thromb Hemost. 2015;41:832‐837. [DOI] [PubMed] [Google Scholar]
- 45.Cooke EJ, Zhou JY, Wyseure T, et al. Vascular permeability and remodelling coincide with inflammatory and reparative processes after joint bleeding in factor VIII‐deficient mice. Thromb Haemost. 2018;118:1036‐1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang J, Leong NL, Khalique U, Phan TM, Lyons KM, Luck JV Jr. Connective tissue growth factor (CTGF/CCN2) in haemophilic arthropathy and arthrofibrosis: a histological analysis. Haemophilia. 2016;22:e527‐e536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morris CJ, Blake DR, Wainwright AC, Steven MM. Relationship between iron deposits and tissue damage in the synovium: an ultrastructural study. Ann Rheum Dis. 1986;45:21‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sen D, Chapla A, Walter N, Daniel V, Srivastava A, Jayandharan GR. Nuclear factor (NF)‐κB and its associated pathways are major molecular regulators of blood‐induced joint damage in a murine model of hemophilia. J Thromb Haemost. 2013;11:293‐306. [DOI] [PubMed] [Google Scholar]
- 49.Manetti M, Linari S, Romano E, et al. TNF‐α/TNF‐R system may represent a crucial mediator of proliferative synovitis in hemophilia A. J Clin Med. 2019;8:939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wen FQ, Jabbar AA, Chen YX, Kazarian T, Patel DA, Valentino LA. c‐myc proto‐oncogene expression in hemophilic synovitis: in vitro studies of the effects of iron and ceramide. Blood. 2002;100:912‐916. [DOI] [PubMed] [Google Scholar]
- 51.Baeten D, Demetter P, Cuvelier C, et al. Comparative study of the synovial histology in rheumatoid arthritis, spondyloarthropathy, and osteoarthritis: influence of disease duration and activity. Ann Rheum Dis. 2000;59:945‐953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krenn V, Morawietz L, Burmester GR, et al. Synovitis score: discrimination between chronic low‐grade and high‐grade synovitis. Histopathology. 2006;49:358‐364. [DOI] [PubMed] [Google Scholar]
- 53.Haraoui B, Pelletier JP, Cloutier JM, Faure MP, Martel‐Pelletier J. Synovial membrane histology and immunopathology in rheumatoid arthritis and osteoarthritis. In vivo effects of antirheumatic drugs. Arthritis Rheum. 1991;34:153‐163. [DOI] [PubMed] [Google Scholar]
- 54.Solimeno L, Goddard N, Pasta G, et al. Management of arthrofibrosis in haemophilic arthropathy. Haemophili. 2010;16(Suppl 5):115‐120. [DOI] [PubMed] [Google Scholar]
- 55.Nieuwenhuizen L, Schutgens RE, Coeleveld K, et al. Hemarthrosis in hemophilic mice results in alterations in M1–M2 monocyte/macrophage polarization. Thromb Res. 2014;133:390‐395. [DOI] [PubMed] [Google Scholar]
- 56.Roosendaal G, Vianen ME, Wenting MJ, et al. Iron deposits and catabolic properties of synovial tissue from patients with haemophilia. J Bone Joint Surg Br. 1998;80:540‐545. [DOI] [PubMed] [Google Scholar]
- 57.Acharya SS, Kaplan RN, Macdonald D, Fabiyi OT, DiMichele D, Lyden D. Neoangiogenesis contributes to the development of hemophilic synovitis. Blood. 2011;117:2484‐2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Knowles LM, Kagiri D, Bernard M, Schwarz EC, Eichler H, Pilch J. Macrophage polarization is deregulated in haemophilia. Thromb Haemost. 2019;119:234‐245. [DOI] [PubMed] [Google Scholar]
- 59.Hoffman M, Harger A, Lenkowski A, Hedner U, Roberts HR, Monroe DM. Cutaneous wound healing is impaired in hemophilia B. Blood. 2006;108:3053‐3060. [DOI] [PubMed] [Google Scholar]
- 60.Bhat V, Olmer M, Joshi S, et al. Vascular remodeling underlies rebleeding in hemophilic arthropathy. Am J Hematol. 2015;90:1027‐1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bartok B, Firestein GS. Fibroblast‐like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev. 2010;233:233‐255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hakobyan N, Kazarian T, Jabbar AA, Jabbar KJ, Valentino LA. Pathobiology of hemophilic synovitis I: overexpression of mdm2 oncogene. Blood. 2004;104:2060‐2064. [DOI] [PubMed] [Google Scholar]
- 63.Roosendaal G, Tekoppele JM, Vianen ME, van den Berg HM, Lafeber FP, Bijlsma JW. Articular cartilage is more susceptible to blood induced damage at young than at old age. J Rheumatol. 2000;27:1740‐1744. [PubMed] [Google Scholar]
- 64.Melchiorre D, Milia AF, Linari S, et al. RANK‐RANKL‐OPG in hemophilic arthropathy: from clinical and imaging diagnosis to histopathology. J Rheumatol. 2012;39:1678‐1686. [DOI] [PubMed] [Google Scholar]
- 65.Martel‐Pelletier J, Barr AJ, Cicuttini FM, et al. Osteoarthritis. Nat Rev Dis Primers. 2016;2:16072. [DOI] [PubMed] [Google Scholar]
- 66.Martinoli C, Della Casa Alberighi O, Di Minno G, et al. Development and definition of a simplified scanning procedure and scoring method for haemophilia early arthropathy detection with ultrasound (HEAD‐US). Thromb Haemost. 2013;109:1170‐1179. [DOI] [PubMed] [Google Scholar]
- 67.Ceponis A, Wong‐Sefidan I, Glass CS, von Drygalski A. Rapid musculoskeletal ultrasound for painful episodes in adult haemophilia patients. Haemophilia. 2013;19:790‐798. [DOI] [PubMed] [Google Scholar]
- 68.Lan HH, Eustace SJ, Dorfman D. Hemophilic arthropathy. Radiol Clin North Am. 1996;34:446‐450. [PubMed] [Google Scholar]
- 69.Pettersson H, Ahlberg A, Nilsson IM. A radiologic classification of hemophilic arthropathy. Clin Orthop Relat Res. 1980;149:153‐159. [PubMed] [Google Scholar]
- 70.Lundin B, Pettersson H, Ljung R. A new magnetic resonance imaging scoring method for assessment of haemophilic arthropathy. Haemophilia. 2004;10:383‐389. [DOI] [PubMed] [Google Scholar]
- 71.Doria AS, Lundin B, Kilcoyne RF, et al. Reliability of progressive and additive MRI scoring systems for evaluation of haemophilic arthropathy in children: expert MRI working group of the international prophylaxis study group. Haemophilia. 2005;11:245‐253. [DOI] [PubMed] [Google Scholar]
- 72.Lundin B, Berntorp E, Pettersson H, et al. Gadolinium contrast agent is of limited value for magnetic resonance imaging assessment of synovial hypertrophy in hemophiliacs. Acta Radiol. 2007;48(5):520‐530. [DOI] [PubMed] [Google Scholar]
- 73.Di Minno MN, Iervolino S, Soscia E, et al. Magnetic resonance imaging and ultrasound evaluation of "healthy" joints in young subjects with severe haemophilia A. Haemophilia. 2013;19:e167‐e173. [DOI] [PubMed] [Google Scholar]
- 74.Keshava SN, Gibikote S, Doria AS. Imaging evaluation of hemophilia: musculoskeletal approach. Semin Thromb Hemost. 2015;41:880‐893. [DOI] [PubMed] [Google Scholar]
- 75.Nguyen S, Lu X, Ma Y, Du J, Chang EY, von Drygalski A. Musculoskeletal ultrasound for intra‐articular bleed detection: a highly sensitive imaging modality compared with conventional magnetic resonance imaging. J Thromb Haemost. 2018;16:490‐499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.von Drygalski A, Moore RE, Nguyen S, et al. Advanced hemophilic arthropathy: sensitivity of soft tissue discrimination with musculoskeletal ultrasound. J Ultrasound Med. 2018;37:1945‐1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ignas DM, Doria AS, von Drygalski A, et al. Use of ultrasound for assessment of musculoskeletal disease in persons with haemophilia: results of an international prophylaxis study group global survey. Haemophilia. 2020;26:685‐693. [DOI] [PubMed] [Google Scholar]
- 78.Soliman M, Daruge P, Dertkigil SSJ, et al. Imaging of haemophilic arthropathy in growing joints: pitfalls in ultrasound and MRI. Haemophilia. 2017;23(5):660‐672. [DOI] [PubMed] [Google Scholar]
- 79.Bakeer N, Shapiro AD. Merging into the mainstream: the evolution of the role of point‐of‐care musculoskeletal ultrasound in hemophilia. F1000Res. 2019;8:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ligocki CC, Abadeh A, Wang KC, Adams‐Webber T, Blanchette VS, Doria AS. A systematic review of ultrasound imaging as a tool for evaluating haemophilic arthropathy in children and adults. Haemophilia. 2017;23:598‐612. [DOI] [PubMed] [Google Scholar]
- 81.Zukotynski K, Jarrin J, Babyn PS, et al. Sonography for assessment of haemophilic arthropathy in children: a systematic protocol. Haemophilia. 2007;13:293‐304. [DOI] [PubMed] [Google Scholar]
- 82.Melchiorre D, Linari S, Innocenti M, et al. Ultrasound detects joint damage and bleeding in haemophilic arthropathy: a proposal of a score. Haemophilia. 2011;17:112‐117. [DOI] [PubMed] [Google Scholar]
- 83.Klukowska A, Czyrny Z, Laguna P, Brzewski M, Serafin‐Krol MA, Rokicka‐Milewska R. Correlation between clinical, radiological and ultrasonographical image of knee joints in children with haemophilia. Haemophilia. 2001;7:286‐292. [DOI] [PubMed] [Google Scholar]
- 84.Kandagaddala M, Sundaramoorthy M, Keshava SN, et al. A new and simplified comprehensive ultrasound protocol of haemophilic joints: the Universal Simplified Ultrasound (US‐US) protocol. Clin Radiol. 2019;74:897.e9‐897.e16. [DOI] [PubMed] [Google Scholar]
- 85.Volland LM, Zhou JY, Barnes RFW, et al. Development and reliability of the joint tissue activity and damage examination for quantitation of structural abnormalities by musculoskeletal ultrasound in hemophilic joints. J Ultrasound Med. 2019;38:1569‐1581. [DOI] [PubMed] [Google Scholar]
- 86.Doria AS, Keshava SN, Mohanta A, et al. Diagnostic accuracy of ultrasound for assessment of hemophilic arthropathy: MRI correlation. AJR Am J Roentgenol. 2015;204:W336‐W347. [DOI] [PubMed] [Google Scholar]
- 87.De la Corte‐Rodriguez H, Rodriguez‐Merchan EC, Alvarez‐Roman MT, Martin‐Salces M, Romero‐Garrido JA, Jimenez‐Yuste V. Accelerating recovery from acute hemarthrosis in patients with hemophilia: the role of joint aspiration. Blood Coagul Fibrinolysis. 2019;30:111‐119. [DOI] [PubMed] [Google Scholar]
- 88.Manners PJ, Price P, Buurman D, Lewin B, Smith B, Cole CH. Joint aspiration for acute hemarthrosis in children receiving factor VIII prophylaxis for severe hemophilia: 11‐year safety data. J Rheumatol. 2015;42:885‐890. [DOI] [PubMed] [Google Scholar]
- 89.Martin EJ, Cooke EJ, Ceponis A, et al. Efficacy and safety of point‐of‐care ultrasound‐guided intra‐articular corticosteroid joint injections in patients with haemophilic arthropathy. Haemophilia. 2017;23:135‐143. [DOI] [PubMed] [Google Scholar]
- 90.Li TY, Wu YT, Chen LC, Cheng SN, Pan RY, Chen YC. An exploratory comparison of single intra‐articular injection of platelet‐rich plasma vs hyaluronic acid in treatment of haemophilic arthropathy of the knee. Haemophilia. 2019;25:484‐492. [DOI] [PubMed] [Google Scholar]
- 91.Mejia‐Carvajal C, Hakobyan N, Enockson C, Valentino LA. The impact of joint bleeding and synovitis on physical ability and joint function in a murine model of haemophilic synovitis. Haemophilia. 2008;14:119‐126. [DOI] [PubMed] [Google Scholar]
- 92.Melchiorre D, Morfini M, Linari S, Zignego AL, Innocenti M, Matucci CM. Anti‐TNF‐α therapy prevents the recurrence of joint bleeding in haemophilia and arthritis. Rheumatology (Oxford). 2014;53:576‐578. [DOI] [PubMed] [Google Scholar]
- 93.Obaji S, Jones C, Yates A, et al. Selective angiographic embolization for recurrent elbow and knee haemarthroses in haemophilia: a retrospective case series. Haemophilia. 2015;21:e226‐e228. [DOI] [PubMed] [Google Scholar]
- 94.Rodriguez‐Merchan EC, De la Corte‐Rodriguez H, Jimenez‐Yuste V. Radiosynovectomy in haemophilia: long‐term results of 500 procedures performed in a 38‐year period. Thromb Res. 2014;134:985‐990. [DOI] [PubMed] [Google Scholar]
- 95.Ali T, Abou Fakher FH, Schved JF. Chemical vs. radioactive synoviorthesis for treatment of chronic haemophilic synovitis: Syrian experience. Haemophilia. 2016;22:e573‐e575. [DOI] [PubMed] [Google Scholar]
- 96.Valentino LA, Khair K. Prophylaxis for hemophilia A without inhibitors: treatment options and considerations. Expert Rev Hematol. 2020;13:731‐743. [DOI] [PubMed] [Google Scholar]
- 97.Humby F, Lewis M, Ramamoorthi N, et al. Synovial cellular and molecular signatures stratify clinical response to csDMARD therapy and predict radiographic progression in early rheumatoid arthritis patients. Ann Rheum Dis. 2019;78:761‐772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.von Drygalski A, Pasta G, de la Corte‐Rodriguez H. Ultrasound and patient self‐imaging in hemophilia. Haemophilia. 2021;27:e298‐e301. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1
Video S2